 Tridib Das
Tridib Das Zhongli Chen
Zhongli Chen Rudi W. Hendriks
Rudi W. Hendriks Mirjam Kool
Mirjam Kool- Department of Pulmonary Medicine, Erasmus MC, Rotterdam, Netherlands
Immune cell activation is a stringently regulated process, as exaggerated innate and adaptive immune responses can lead to autoinflammatory and autoimmune diseases. Perhaps the best-characterized molecular pathway promoting cell activation is the nuclear factor-κB (NF-κB) signaling pathway. Stimulation of this pathway leads to transcription of numerous pro-inflammatory and cell-survival genes. Several mechanisms tightly control NF-κB activity, including the key regulatory zinc finger (de)ubiquitinating enzyme A20/tumor necrosis factor α-induced protein 3 (TNFAIP3). Single nucleotide polymorphisms (SNPs) in the vicinity of the TNFAIP3 gene are associated with a spectrum of chronic systemic inflammatory diseases, indicative of its clinical relevance. Mice harboring targeted cell-specific deletions of the Tnfaip3 gene in innate immune cells such as macrophages spontaneously develop autoinflammatory disease. When immune cells involved in the adaptive immune response, such as dendritic cells or B-cells, are targeted for A20/TNFAIP3 deletion, mice develop spontaneous inflammation that resembles human autoimmune disease. Therefore, more knowledge on A20/TNFAIP3 function in cells of the immune system is beneficial in our understanding of autoinflammation and autoimmunity. Using the aforementioned mouse models, novel A20/TNFAIP3 functions have recently been described including control of necroptosis and inflammasome activity. In this review, we discuss the function of the A20/TNFAIP3 enzyme and its critical role in various innate and adaptive immune cells. Finally, we discuss the latest findings on TNFAIP3 SNPs in human autoinflammatory and autoimmune diseases and address that genotyping of TNFAIP3 SNPs may guide treatment decisions.
Introduction
Autoinflammatory and autoimmune diseases share a spectrum of chronic immune system disorders (1). Autoinflammatory diseases are rare and occur due to innate immune cell dysfunction with increased cytokines such as interleukin (IL)-1β and tumor necrosis factor (TNF) α (2, 3). In contrast, autoimmune diseases are caused by adaptive immune cell dysfunction and affect millions of people worldwide (4). Self-reactive T-cells and/or autoreactive antibodies facilitate responses against harmless tissue (5). Essential for development of these diseases is the activation status of immune cells, wherein nuclear factor-κB (NF-κB) plays a key role. NF-κB activation is tightly controlled by several mechanisms, including the key regulatory (de)ubiquitinating enzyme A20 or tumor necrosis factor α-induced protein 3 (TNFAIP3) (6). Genetic studies have demonstrated the association of TNFAIP3 single nucleotide polymorphisms (SNPs) with multiple human diseases (7), such as systemic lupus erythematosus (SLE) (8–10), rheumatoid arthritis (RA) (9), and Crohn’s disease (CD) (11, 12). A20/TNFAIP3 regulates crucial stages in immune cell homeostasis, such as NF-κB activation and apoptosis. Recently, new functions have become apparent, including the control of necroptosis and inflammasome activity (13–15). Here, we review the latest understanding of A20/TNFAIP3 as a key regulator of immune signaling and its cell-specific role in the pathogenesis of autoinflammation and autoimmunity as demonstrated in murine models.
NF-κB Pathway
NF-κB Activation
An important and well-characterized signaling pathway of immune cell activation is the NF-κB pathway (7), which is activated through canonical or non-canonical cascades (16). The canonical pathway is triggered by several pattern recognition receptors (PRRs), such as toll-like receptors (TLRs) and nucleotide oligomerization domain (NOD)-like receptors (NLRs) and cytokine receptors, such as TNF receptor (TNFR) and IL-1 receptor (16). PRRs are essential within the innate immune response in defense against invading pathogens. In addition, T-cell receptor (TCR) or B-cell receptor (BCR) triggering, crucial in the adaptive immune response, also leads to NF-κB activation (17). In total, five NF-κB family members have been identified thus far, termed p65 (RelA), RelB, c-Rel, NF-κB1, and NF-κB2 (18). These five members can form homo- or heterodimers and distinctive NF-κB dimers bind different DNA-binding sites, resulting in cytokine release, enhanced cell survival, proliferation, differentiation, and changes in metabolism (18, 19).
Regulation of NF-κB Activity
Several regulatory mechanisms control NF-κB signaling to maintain tissue homeostasis. One of the proteins that terminate NF-κB signaling is A20/TNFAIP3 (6). A20/TNFAIP3 regulates protein ubiquitination, an important post-translational modification (6). Ubiquitination is reversible and tightly controlled by opposing actions of ubiquitin ligases and deubiquitinases (DUBs) (20). Several ubiquitin chains are known, each having specific functions. Lysine (K)48-linked polyubiquitin chains target a protein for proteasomal degradation, whereas K63-linked or linear polyubiquitin chains stabilize protein–protein interactions important for downstream signaling molecules (16). Interestingly, A20/TNFAIP3 has both ligase and DUB activity to perform both K48 ubiquitination and K63 deubiquitination (6).
A20/TNFAIP3
A20/TNFAIP3 Protein Structure
In 1990, A20/TNFAIP3 was identified as a primary response gene after TNFα exposure in endothelial cells (21, 22). The structure of A20/TNFAIP3 reveals its dual function (Figure 1A). First, the N-terminal OTU domain houses the C103 catalytic cysteine site, responsible for K63 deubiquitination (6, 23). Second, the C-terminal ZnF4 domain adds K48 ubiquitin to target proteins for degradation (6). Both domains cooperate to inhibit NF-κB signaling (24). Finally, A20/TNFAIP3 ZnF7 binds linear polyubiquitin, which aids to suppress NF-κB activation (25, 26). To achieve adequate function, A20/TNFAIP3 must bind either target or accessory proteins. The OTU domain binds the target protein TNFR-associated factors (TRAF), while the C-terminus binds accessory molecules such as A20-binding protein (ABIN1 and ABIN2), Tax1 Binding Protein 1 (TAX1BP1) and NF-κB essential modulator (NEMO) (27). These accessory molecules function as adaptor proteins and localize A20/TNFAIP3 near polyubiquitin chains (28–31) [reviewed in Ref. (27, 32)].
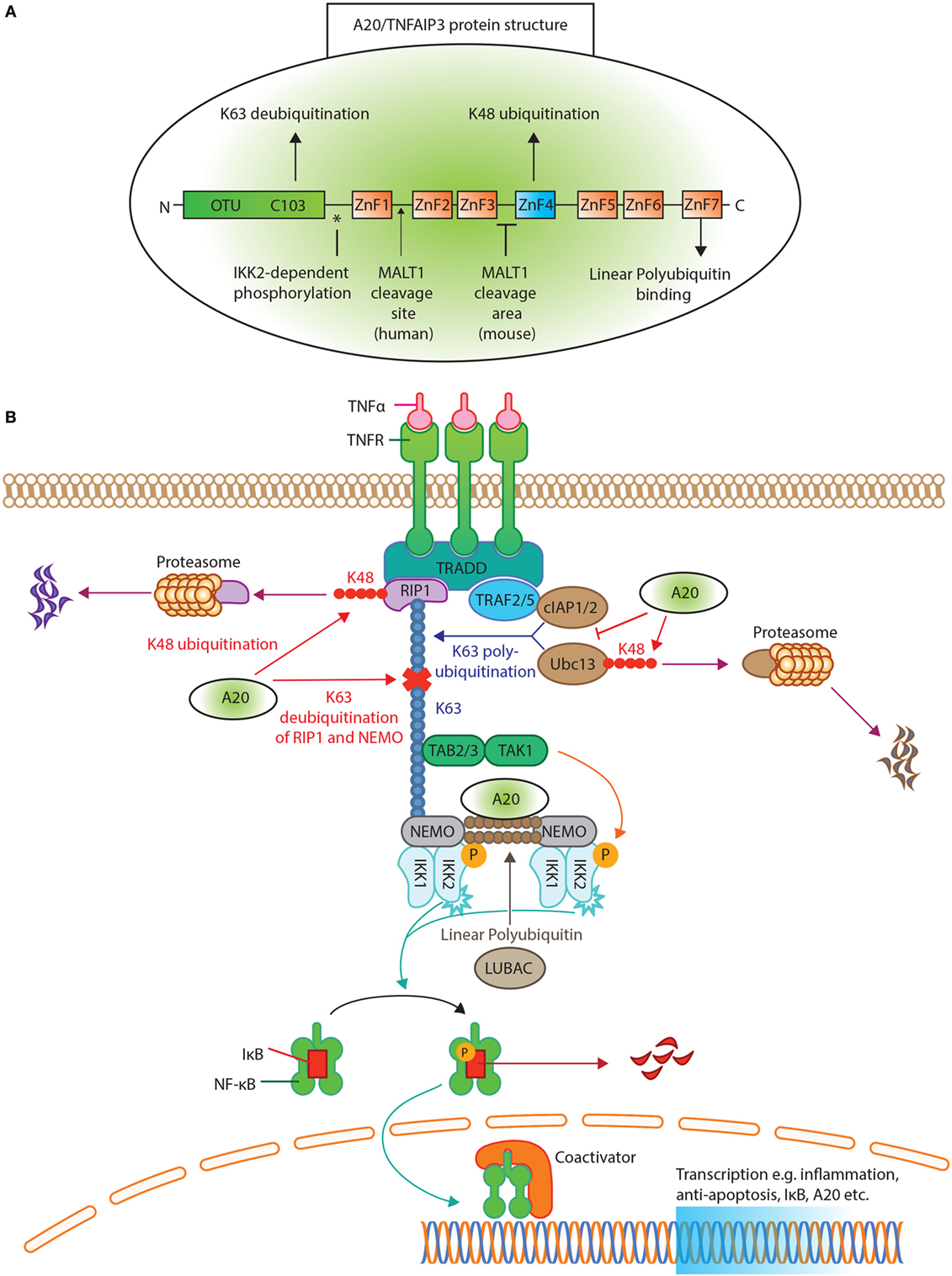
Figure 1. A20/tumor necrosis factor α-induced protein 3 (TNFAIP3) protein structure and function in tumor necrosis factor receptor (TNFR) induced NK-κB inhibition. (A) The protein structure of A20/TNFAIP3. The N-terminus contains the ovarian tumor (OTU) domain, with the C103 cysteine site of K63 deubiquitination. The seven zinc fingers (ZnF) are illustrated, where ZnF4 has K48-ubiquitinating activity and ZnF7 can bind linear polyubiquitin. The asterisk (*) indicates the site of IκB kinase (IKK)2-dependent phosphorylation. An arrow indicates where MALT1 cleaves human A20/TNFAIP3 (after Arginine 439), while for murine A20/TNFAIP3 it is only known that MALT1 cleaves A20/TNFAIP3 between ZnF3 and ZnF4. (B) TNFR activation of the NF-κB pathway. Ligand TNFα binds the TNFR receptor and allows binding of TNFR1-associated death domain protein to the TNFR. This recruits receptor-interacting serine/threonine-protein kinase 1 (RIP1) and TNFR-associated factor (TRAF)2 or TRAF5 to form the TNFR complex. RIP1 is K63 polyubiquitinated by E2-E3 ubiquitin-conjugating enzyme (Ubc)13 and cellular inhibitor of apoptosis protein (cIAP)1/2. The polyubiquitin acts as a scaffold for TAB2/TAB3 and NF-kappa-B essential modulator (NEMO) to recruit the transforming growth factor beta-activated kinase 1 (TAK1)-TAB 2/3 complex. TAK1 phosphorylates and activates the IKK, composed of IKK1(α), IKK2(β), and NEMO. The linear ubiquitin chain assembly complex (LUBAC) was shown to generate linear polyubiquitin on NEMO (and also RIP1), recruiting and stabilizing another IKK–NEMO complex. IKK2, phosphorylates IκB, allowing IκB K48 polyubiquitination and consequently degrading by proteasomes, thereby releasing NF-κB to translocate to the nucleus. A20/TNFAIP3 acts in different levels of the pathway. A20/TNFAIP3 removes K63-linked polyubiquitin chains from RIP1 and NEMO, thereby disrupting downstream signals. In addition, A20/TNFAIP3 adds K48-linked polyubiquitin chains to RIP1 and Ubc13, thus targeting them for proteasomal destruction. Beyond (de)ubiquitinating mechanisms, A20/TNFAIP3 also destabilizes Ubc13 interaction with cIAP1/2, thereby preventing new K63-ubiquitinating activity. The ZnF7 of A20/TNFAIP3 binds linear ubiquitin, thereby accelerating the dissociation of LUBAC and IKK/NEMO, resulting in NF-κB termination.
Function of A20/TNFAIP3 in the TNFR Signaling Pathway
The multiple functions of A20/TNFAIP3 in NF-κB regulation are most apparent in the TNFR signaling pathway (Figure 1B). Briefly, TNFα binding to TNFR recruits receptor-interacting serine/threonine-protein kinase 1 (RIP1) and TRAF2/TRAF5 to shape the TNFR complex (33, 34). RIP1 is K63 polyubiquitinated by ubiquitin-conjugating enzyme (Ubc)13 and cellular inhibitor of apoptosis protein (cIAP)1/2. RIP1–polyubiquitin is a scaffold to recruit NEMO and transforming growth factor beta-activated kinase 1 (TAK1)-TAB2/3 complex (27). The linear ubiquitin chain assembly complex (LUBAC) produces linear polyubiquitin on NEMO, recruiting and stabilizing another IκB kinase (IKK)-NEMO complex (35, 36) (Figure 1B). TAK1 phosphorylates and activates IKK, containing IKK2, that finally phosphorylates IκB (37, 38). Phosphorylated IκB will be K48 polyubiquitinated and degraded (19), thereby releasing NF-κB (16) leading to its nuclear translocation.
To terminate NF-κB activation, A20/TNFAIP3 removes K63–polyubiquitin chains from RIP1 and NEMO (Figure 1B), thereby disrupting interactions with downstream proteins (6, 30). Furthermore, A20/TNFAIP3 adds K48 polyubiquitin chains to RIP1 and Ubc13, leading to their degradation (6, 39). A20/TNFAIP3 also destabilizes Ubc13 interaction with cIAP1/2 to prevent new K63-ubiquitinating activity (40). Finally, the ZnF7 domain of A20/TNFAIP3 binds linear ubiquitin, resulting in dissociation of LUBAC and IKK/NEMO (25, 35) and thus inhibits IKK phosphorylation (41).
Regulation of A20/TNFAIP3 Expression and Function
A20/TNFAIP3’s expression and function are controlled at several levels, e.g., transcriptional, post-transcriptional, and post-translational. During steady state, A20/TNFAIP3 is minimally present in several cell types (27) due to repression by downstream regulatory element antagonist modulator (DREAM) (42). Transcriptional activation of the TNFAIP3 gene is facilitated by two NF-κB binding sites in the TNFAIP3 promoter (43). TNFAIP3 promotor activity is also controlled by regulators of cell-intrinsic energy homeostasis such as estrogen-related receptor α (ERRα) (44), linking energy homeostasis to cell activation. The stability of the TNFAIP3 transcript is regulated by mRNA-binding proteins [e.g., ROQUIN (Rc3h1) (45)] and micro-(mi)RNAs, such as miR-125b, miR-19b, and miR-29c (46–48). Interestingly, one of the downstream targets of NF-κB is miR-125b, which thereby prolongs NF-κB activity (47). ROQUIN destabilizes TNFAIP3 mRNA, leading to lower A20/TNFAIP3 protein expression (45), and mutated ROQUIN is known to induce autoimmunity in mice (49). Post-translationally, A20/TNFAIP3 protein function is improved by IKK2-dependent phosphorylation (50) (Figure 1A), which enhances K63 deubiquitination and K48 ubiquitination (51). Also, cell-extrinsic factors control A20/TNFAIP3 protein stability, e.g., high glucose levels target A20/TNFAIP3 for proteasomal degradation and/or reactive oxygen species (ROS) inactivate its deubiquitinating activity (52–54). Especially, the latter is important in RA, in which elevated ROS plays a pathogenic role (55, 56), possibly by inhibiting A20/TNFAIP3 function. Finally, unlike most cell types, resting T-cells constitutively express high levels of A20/TNFAIP3 protein (57), which is degraded after activation by paracaspase MALT1 to facilitate NF-κB translocation (58) (Figure 1A).
Immune Cell-Specific Deletion of A20/Tnfaip3 in Mice
A20/TNFAIP3 is critical in inflammation regulation, as mice with germ-line A20/Tnfaip3-deletion developed severe multiorgan inflammation and cachexia, resulting in early death (59). Conditional A20/Tnfaip3-floxed alleles enabled lineage-specific Tnfaip3-deletion and study of cell-specific contributions to autoinflammation and autoimmunity (60).
A20/TNFAIP3 Function in Myeloid Cells
To evaluate the role of A20/TNFAIP3 in myeloid cells, Tnfaip3fl/fl mice were crossed with lysozyme M (LysM)-cre Tg mice (61), generating Tnfaip3LysM mice (13, 60, 62, 63). The LysM-cre promoter is expressed in ~95–99% of macrophages and neutrophils and ~15% of splenic dendritic cells (DCs) (61). Tnfaip3LysM-KO mice developed enthesitis (62) and paw inflammation (63). While hallmarks of RA comprising increased Th17-cells and serum anti-collagen type II antibodies (anti-CII) were present in Tnfaip3LysM-KO mice, T and B cells were dispensable for paw inflammation (63). Rather, paw inflammation in Tnfaip3LysM-KO mice depended on IL-1β (13), suggestive of an autoinflammatory disease such as Still’s disease or juvenile idiopathic arthritis. In vitro cultured Tnfaip3-deficient macrophages produced increased amounts of IL-1β, IL-6, IL-18, and TNFα compared to control macrophages (13, 63). IL-1β and IL-18 release is regulated by the NLRP3 inflammasome (64), which is pathogenic in autoinflammatory diseases such as Cryopyrin-associated autoinflammatory syndrome (CAPS) (3, 65). A20/TNFAIP3 directly controls the activity of the NLRP3 inflammasome in macrophages (13, 66).
Next, interferon (IFN)γ or IL-6-induced JAK-STAT signaling is implicated in autoinflammatory diseases (3), which is also regulated by A20/TNFAIP3 (62). Tnfaip3-deficient macrophages had elevated STAT1-dependent gene transcription, leading to enhanced chemokine (C–X–C motif) ligand (CXCL)9 and CXCL10 production (62). Pharmacologic JAK-STAT inhibition by tofacitinib in Tnfaip3LysM-KO mice resulted in reduced enthesitis (62), which is a treatment option for several autoinflammatory diseases (3).
In short, in macrophages, A20/TNFAIP3 regulates IL-1β/IL-18 release by controlling NLRP3 inflammasome activity and CXCL9/CXCL10 production through STAT1 signaling. Both pathways are essential in controlling the autoinflammatory arthritis phenotype. However, a role for neutrophils and/or DCs in the pathogenesis of arthritis cannot be excluded.
Function of A20/TNFAIP3 in DCs
DCs play a crucial role in immune homeostasis and arise in two main subsets, comprising conventional DCs type 1 or 2 (cDC1s, cDC2s) and plasmacytoid DCs (pDCs) (67). When activated, cDCs induce antigen-specific adaptive immune responses and pDCs control anti-viral responses (67). During inflammation, monocyte-derived DCs (moDCs) are recruited to inflammatory sites (68). To characterize A20/TNFAIP3 function in DCs in vivo, CD11c-cre-mediated (69) targeting was used in mice (70–72). Tnfaip3CD11c-KO mice had perturbed splenic DC homeostasis as cDC1s, cDC2s, and pDCs were drastically reduced, while moDCs were increased (71). In vivo loss of cDCs and pDCs in Tnfaip3CD11c-KO mice suggested that A20/TNFAIP3 supports their survival. However, in vitro generated granulocyte-macrophage colony-stimulating factor (GM-CSF) bone marrow-derived Tnfaip3-deficient DCs were more resistant to apoptosis due to upregulated anti-apoptotic molecules (71). This discrepancy might be caused by contaminating macrophages in GM-CSF cultures (73). GM-CSF-cultured DCs from Tnfaip3CD11c-KO mice exhibited an activated phenotype, shown by increased co-stimulatory molecules (e.g., CD80/CD86) and cytokine expression of IL-6, TNFα (70, 71), IL-1β, and IL-10 (71). In the pathogenesis of SLE, pDCs are pathogenic by secreting type I interferons (74), but increased type I interferon by activated pDCs was observed only in vitro (70).
To maintain peripheral tolerance, antigens derived from apoptotic cells are normally not presented in an immunogenic manner to T-cells (75). Strikingly, in vitro Tnfaip3-deficient DCs present these antigens to T-cells and induce T-cell activation (71) leading to a break of tolerance. In vitro apoptotic cell-pulsed DCs produce T-cell differentiating cytokines IL-12 and IL-23, leading to increased Th1-cell and Th17-cell differentiation, respectively, in Tnfaip3CD11c-KO mice (70, 71, 76). Surprisingly, three independent studies with Tnfaip3CD11c-KO mice generated different spontaneous phenotypes, i.e., inflammatory bowel disease (IBD) (70), systemic autoimmunity resembling SLE (71), and multiorgan inflammation (72). Serum IL-6 was elevated in mice developing SLE or IBD (70, 71), while both TNFα and IFNγ were significantly increased in mice with multiorgan inflammation (72). As IL-6 depletion ameliorated murine colitis and SLE development (77–80), IL-6 might directly have contributed to IBD and SLE development in Tnfaip3CD11c-KO mice. While CD is recently considered an autoinflammatory disease (81), T-cells were essential for colitis development in Tnfaip3CD11c-KO mice (70). SLE patients have increased anti-dsDNA autoantibodies (82), which were also observed in Tnfaip3CD11c-KO mice (71). The diversity of phenotypes observed in Tnfaip3CD11c-KO mice might be due to environmental differences, such as microbiota (70, 83), as antibiotics reduced IBD in Tnfaip3CD11c-KO mice (76).
Summarizing, the expression of co-stimulatory molecules, pro-inflammatory cytokines such as IL-6, and anti-apoptotic proteins in DCs is controlled by A20/TNFAIP3. A20/TNFAIP3 in DCs functions to maintain in vivo T-cell and B-cell homeostasis, thereby preventing spontaneous autoinflammation.
A20/TNFAIP3 Functions in T-Cells
A20/TNFAIP3 is known to regulate TCR/CD28-mediated NF-κB activation and TCR-mediated survival (84–86) and is highly expressed in naïve T-cells (57). A20/TNFAIP3’s influence on T-cell homeostasis has been examined using mature T cell (maT)-cre and Cd4-cre mice, targeting both CD8+ T-cells and CD4+ T-cells (14, 15, 87). Tnfaip3-deletion efficiency differs between Tnfaip3maT and Tnfaip3CD4 mice. In Tnfaip3maT-KO mice, ~80% of CD8+ T-cells and ~30% of CD4+ T-cells are affected (88), whereas in Tnfaip3CD4-KO mice, ~100% of both CD8+ and CD4+ T-cells are targeted (89). Targeted T-cells from both mouse strains showed an activated phenotype (14, 87), but only Tnfaip3maT-KO mice developed inflammatory lung and liver infiltrates with increased proportions of CD8+ T-cells (87). TCR-stimulated CD8+ T-cells from Tnfaip3maT-KO mice had enhanced IL-2 and IFNγ production in vitro which correlated with in vivo increased serum IFNγ (87). Serum TNFα and IL-17 were also elevated in Tnfaip3maT-KO mice (87). Since both IFNγ and TNFα are hepatotoxic factors (90–92), these cytokines likely mediated liver inflammation.
Differences in T-cell-specific Tnfaip3 deletion between the two mouse strains could indicate that either CD8+ T-cells drive inflammation in Tnfaip3maT-KO mice or CD4+ T-cells have increased regulatory function in Tnfaip3CD4-KO mice. Indeed, regulatory T cell (Treg) proportions were increased in Tnfaip3CD4-KO mice, because of a reduced IL-2 dependence for their development (93). In vitro activated CD4+ T-cells from Tnfaip3CD4-KO mice died quicker than wild-type T-cells (14, 15), due to A20/TNFAIP3’s control on necroptosis (14) and autophagy (15). Necroptosis is RIPK3-dependent programmed cell death (94). Increased necroptosis in A20/Tnfaip3-deficient CD4+ T-cells impaired Th1 and Th17-cell differentiation in vitro (14). Interestingly, perinatal death of Tnfaip3KO mice was greatly delayed by RIPK3 deficiency, implying that A20/TNFAIP3 may control necroptosis in other cell types (14), such as CD8+ T-cells (95). Preventing necroptosis did not fully restore survival of A20/Tnfaip3-deficient CD4+ T-cells (14), which could be attributed to autophagy, a lysosomal degradation pathway necessary for survival after TCR stimulation (96). Autophagy is regulated by mechanistic target of rapamycin (mTOR), which is increased in Tnfaip3-deficient CD4+ T-cells after TCR stimulation (15). Consequently, treatment with an mTOR inhibitor improves survival by enhancing autophagy (15). mTOR inhibitors are effective in murine SLE and RA (97), but should not be used in patients with A20/TNFAIP3 alterations, as it may improve pathogenic T-cell survival.
In conclusion, in CD4+ T-cells, A20/TNFAIP3 regulates necroptosis and autophagy. In contrast to conventional Th-cells, Treg development is restricted by A20/TNFAIP3. In CD8+ T-cells, A20/TNFAIP3 regulates necroptosis, IL-2, and IFNγ release, of which IFNγ might have contributed to a further undefined lung and liver inflammatory phenotype in Tnfaip3maT-KO mice.
A20/TNFAIP3 Function in B-Cells
B-cell homeostasis demands proper integration of TLR, BCR, and CD40-derived signals, all leading to NF-κB activation and controlled by A20/TNFAIP3 (98, 99). Using CD19-cre-driven Tnfaip3-ablation in mice (100–102), B-cell-specific function of A20/TNFAIP3 was examined. In vitro activated Tnfaip3-deficient B-cells exhibited exaggerated activation as assessed by CD80 and CD95 expression (101, 102) and IL-6 production (100, 102). B-cell numbers in Tnfaip3CD19-KO mice are increased in secondary lymphoid organs (100–102), most likely due to increased anti-apoptotic protein B-cell lymphoma-extra large (Bcl-x) expression (102). Already in 6-week-old Tnfaip3CD19-KO mice, elevated numbers of germinal center B-cells and plasma cells in spleen and peripheral lymph nodes were observed (100–102). Tnfaip3CD19-KO mice developed autoreactive immunoglobulins, including anti-dsDNA antibodies (100–102) and glomerular immunoglobulin deposits (102), features also observed in SLE patients. Surprisingly, no malignancies developed in Tnfaip3CD19-KO mice (100, 102), which might have been expected as A20/TNFAIP3 also functions as a tumor suppressor gene in B-cell lymphomas (103–105).
Summarizing, A20/TNFAIP3 in B-cells controls co-stimulatory molecule expression, IL-6 production, and Bcl-x survival protein expression, thereby preventing autoreactive B-cells formation resulting in an autoimmune SLE phenotype.
A20/TNFAIP3 in Autoinflammatory and Autoimmune Patients
TNFAIP3 is one of the few genes that has been linked by genome-wide association studies (GWAS) to multiple immune diseases (106, 107). The list of common coding and non-coding variants (SNPs) in the vicinity of the TNFAIP3 gene region associated with autoimmune conditions keeps expanding, with recently reported associations with autoimmune hepatitis (AIH) (108, 109), primary biliary cirrhosis (110) and colitis ulcerosa (111). Since a comprehensive overview of SNPs within and around the TNFAIP3 gene has been provided elsewhere (7), we focus on a selection of SNPs with known different functional, clinical, and therapeutical consequences (Figure 2). We also discuss a recently described monogenic disease “Haplo-insufficiency of A20 (HA20)” (112), which clearly illustrates the importance of functional A20/TNFAIP3 protein expression levels (Figure 2).
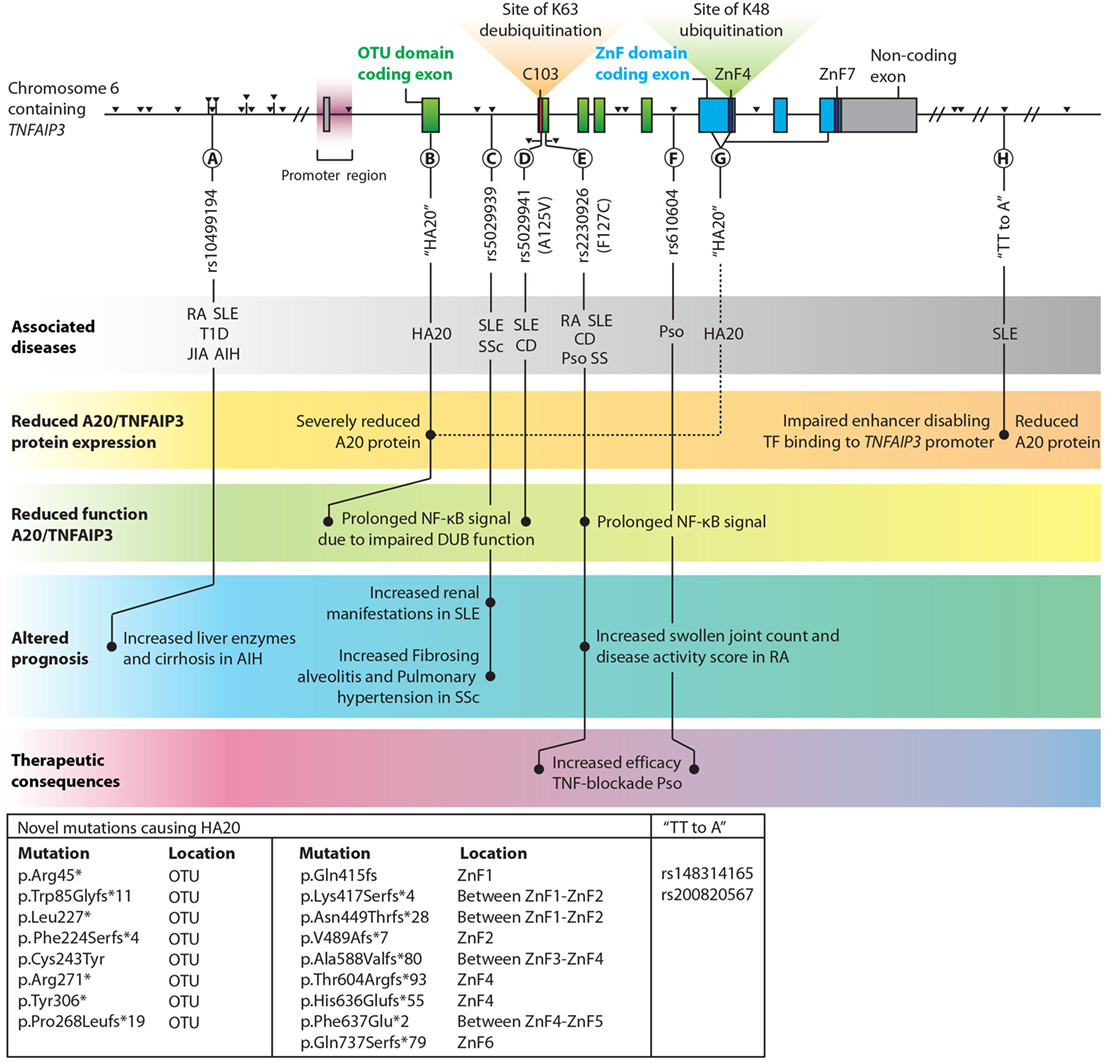
Figure 2. Overview of single nucleotide polymorphisms (SNPs) and novel haplo-insufficiency of A20 (HA20) mutations in the proximity of TNFAIP3 which are highlighted in this review. TNFAIP3 gene SNPs, adapted with permission from: Springer Nature, Nature Reviews Immunology, A. Ma & B.A. Malynn © 2012 (7). Exons contributing to the OTU domain are depicted in green, and exons forming the zinc finger (ZnF) domains are blue. Non-coding exons are gray. The catalytic C103 site, ZnF4, and ZnF7 are highlighted. Black triangles indicate all known SNPs in the TNFAIP3 gene with associations with autoimmune diseases. Among the various documented SNPs/novel mutations, several lead to (1) reduced A20/TNFAIP3 protein level, (2) reduced A20/TNFAIP3 efficiency, (3) altered disease prognosis, or (4) therapeutic implications and are thus highlighted in this figure (A–H). Known associations with (autoinflammatory and autoimmune) diseases for SNPs are indicated in the top gray bar. Multiple novel mutations causing “HA20” and two SNPs termed “TT>A” (associated with SLE) are listed in the box in the lower left corner. The reported p.Gln415fs mutation (113) should be reported as p.Lys417Serfs*4 to stay consistent with Human Genome Variation Society nomenclature (114). Abbreviations: OTU, ovarian tumor; ZnF, zinc finger; TF, transcription factors; TNFAIP3, tumor necrosis factor α-induced protein 3; HA20, haploinsufficiency of A20; AIH, autoimmune hepatitis; SLE, systemic lupus erythematosus; SSc, systemic sclerosis; RA, rheumatoid arthritis; T1D, type 1 Diabetes; JIA, juvenile idiopathic arthritis; CD, Crohn’s disease; Pso, psoriasis; SS, Sjögren syndrome.
TNFAIP3 SNPs and Novel Mutations Affecting A20/TNFAIP3 Expression and Function
Reduced TNFAIP3 mRNA expression was observed in peripheral blood mononuclear cells (PBMCs) in SLE and RA patients (115–117) and in disease-affected organs, e.g., in colon or skin biopsies from CD and psoriasis patients compared to healthy tissues (118–120). In RA synovium, reduced A20/TNFAIP3 protein expression was detected compared to non-autoimmune osteoarthritic synovium (121). SNPs near the TNFAIP3 gene can result in reduced A20/TNFAIP3 mRNA expression and consequently protein concentrations. For instance, specific SNPs associated with SLE (“TT>A”, Figure 2H) are situated in an enhancer region of the TNFAIP3 gene and hamper DNA looping, resulting in reduced TNFAIP3 mRNA expression (122) and reduced A20/TNFAIP3 protein expression in B-cells (8).
Recently, novel rare familial TNFAIP3 mutations (Figures 2B,G) causing HA20 have been described (112). These mutations lead to severely reduced functional A20/TNFAIP3 protein expression (112, 123). HA20 is a dominantly inherited disease caused by high-penetrance heterozygous germ line (mostly nonsense or frameshift) mutations in TNFAIP3 (112). Previously, A20/TNFAIP3 loss-of-function mutations were only identified as somatic variants in lymphomas (105) [reviewed in Ref. (124)]. HA20-associated mutations were first reported in seven unrelated families with an early-onset inflammatory disease resembling the common polygenic Behçet disease (112). Some patients diagnosed with Behçet-like disease were found to have similar HA20 mutations (125, 126). Recently, in a Japanese cohort the majority (59%) of HA20 patients did not fulfill the criteria of Behçet disease (127). In this study, a genotype–phenotype correlation was not observed (127). However, careful evaluation of clinical characteristics can aid diagnosing patients with HA20 or Behçet disease (128). Autoimmune diseases such as autoimmune lymphoproliferative syndrome (ALPS) and SLE were additionally recognized in HA20 patients (113, 123, 127). Excess Th17-cell differentiation was also observed in HA20 patients (127). All HA20 patients identified thus far have a strong inflammatory signature as demonstrated by elevated levels of many pro-inflammatory cytokines (e.g., IL-1β, IL-6, TNFα, IL-17, and IFNγ) and most patients respond to treatment with cytokine inhibitors (anti-TNF and anti-IL-1) (112, 127, 128). Interestingly, Tnfaip3+/− mice do not have an overt inflammatory phenotype despite elevated inflammatory cytokines (e.g., IL-1β and IL-6) in serum (129) and brain (130). Nevertheless, Tnfaip3+/− mice are more susceptible to experimental psoriasis (120) and atherosclerosis (129), but these specific symptoms are not commonly reported for HA20. Increased NLRP3 activity was detected in PBMCs of HA20 patients after LPS stimulation, leading to elevated IL-1β (112). Transfection of mutant-truncated A20/TNFAIP3 prolonged NF-κB activation due to reduced deubiquitinating function (112) (Figure 2B). PBMCs of a patient with HA20 also demonstrated prolonged NF-κB activation (112, 123). Mutant-truncated A20/TNFAIP3 proteins do not exert a dominant-negative effect on protein function, and this indicates that sustained NF-κB activation in HA20 is due to haploinsufficiency rather than an aberrant protein function (112). It remains unclear whether missense high penetrance mutations may have a different impact on A20/TNFAIP3 function.
Two SNPs, rs5029941 (A125V) and rs2230926 (F127C), are located in close proximity of each other near the C103 catalytic site in the OTU domain and result in non-synonymous coding changes in the A20/TNFAIP3 protein (Figures 2D,E). The rs2230926 (F127C) SNP, associated with multiple autoimmune diseases (Figure 2E), hampers A20/TNFAIP3 function after TNFα stimulation (10). The SNP location within the OTU domain (Figure 2E) suggests that the K63-deubiquitinating efficacy is decreased, although this was not evaluated. The A125V mutation (Figure 2D) results in reduced DUB activity and was shown to impair A20-mediated degradation and deubiquitination of TRAF2 (131). Although the A125V mutation was associated with protection from SLE, surprisingly the same allele was associated with increased risk of IBD (131).
In conclusion, specific SNPs functionally alter A20/TNFAIP3 expression or function, and HA20 is a disease with generalized inflammation due to severely reduced functional A20/TNFAIP3 protein expression.
TNFAIP3 SNPs Affecting Disease Progression and Treatment in Patients
Common, presumably hypomorphic, variants in TNFAIP3 can have clinical consequences. For instance, lower TNFAIP3 mRNA expression in PBMCs correlates with SLE disease activity as susceptibility to lupus nephritis is increased (115). SLE or SSc patients with an intron SNP (Figure 2C) predisposes for increased risk for either renal involvement (132) or aggravated disease with fibrosing alveolitis and pulmonary hypertension (133). Similarly, RA patients with a previously described functional SNP (Figure 2E) had more swollen joints and increased disease activity scores (DAS28) compared to RA patients without this SNP, indicating worse clinical prognosis (9, 117). Finally, AIH patients with an upstream SNP (Figure 2A) harbored increased liver enzymes and more cirrhosis at disease presentation compared to patients without this SNP (109). These findings illustrate that within autoimmune patients certain SNPs around the TNFAIP3 gene predispose a worse clinical prognosis.
Analysis of TNFAIP3 SNPs might guide treatment choices, e.g., with TNF-blocking therapy. For RA and CD patients, reduced TNFAIP3 mRNA in PBMCs or colonic biopsies, respectively, is correlated with effective TNF-blocking therapy (118, 134). Psoriasis patients harboring specific TNFAIP3 SNPs (Figures 2E,F) respond more effectively to TNF blockade (135). This indicates that TNFAIP3 SNP analysis before TNF-blocking therapy initiation is worthwhile to perform in several autoimmune diseases and may be more practical than evaluating TNFAIP3 mRNA expression.
Treatment of Autoinflammation and Autoimmunity
Knowledge from cell-specific targeting studies in mice illustrate that loss of A20/TNFAIP3 results in either autoinflammation or autoimmunity. The pathophysiologic distinction between these conditions has therapeutic implications. Autoinflammatory diseases such as Still’s disease, Behçet’s disease, and most cases of HA20 are well treated with IL-1 blockade, which has only marginal effect in autoimmune diseases including RA (136). Autoinflammation may also underlie other chronic disorders such as atherosclerosis, as these patients benefit from anti-IL-1 therapy (137, 138). In contrast, autoimmune disorders (e.g., SLE) have a strong contribution of IL-6 highlighted by successful anti-IL-6 treatment (139). This is in line with mouse studies in which innate cell activation (e.g., Tnfaip3LysM-KO mice) leads to increased IL-1β (13) and adaptive immune cell activation (e.g., Tnfaip3CD19-KO mice) leads to enhanced IL-6 (70, 71, 100, 102). In line with the adaptive nature of the disease, several autoimmune diseases also improve after treatments targeting adaptive immune cells [e.g., T-cell suppression using cyclosporine (140, 141) or B-cell depletion using Rituximab] (142).
Conclusion
Control of immune system activation is crucial to prevent both autoinflammation and autoimmunity. A20/TNFAIP3 hereby plays an important role in several innate and adaptive immune cells. Through analysis of cell-specific deletion of A20/Tnfaip3 in mice, it became apparent that innate myeloid cells require A20/TNFAIP3 to suppress autoinflammation, while the development of autoimmunity is primarily controlled by A20/TNFAIP3 in DCs and B-cells. In addition, novel functions of A20/TNFAIP3 on inflammasome activity and necroptosis are uncovered. It would be of great value to examine in patient material cell-specific profiles of A20/TNFAIP3 and its effector function. The direct consequence of many SNPs on A20/TNFAIP3 is yet unknown. However, it is becoming increasingly clear that specific TNFAIP3 SNPs can alter A20/TNFAIP3 function, can affect its expression level, or are associated with poor clinical outcomes. Finally, future studies on TNFAIP3 SNPs to predict therapeutic effectivity would greatly benefit patient health care to obtain personalized therapy.
Author Contributions
All the authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This project was supported by grants from the Dutch Arthritis Association (12-2-410) and European Framework program 7 (FP7-MC-CIG grant 304221).
References
1. McGonagle D, McDermott MF. A proposed classification of the immunological diseases. PLoS Med (2006) 3(8):e297. doi:10.1371/journal.pmed.0030297
2. McDermott MF, Aksentijevich I. The autoinflammatory syndromes. Curr Opin Allergy Clin Immunol (2002) 2(6):511–6. doi:10.1097/00130832-200212000-00006
3. Peckham D, Scambler T, Savic S, McDermott MF. The burgeoning field of innate immune-mediated disease and autoinflammation. J Pathol (2017) 241(2):123–39. doi:10.1002/path.4812
4. Ngo ST, Steyn FJ, McCombe PA. Gender differences in autoimmune disease. Front Neuroendocrinol (2014) 35(3):347–69. doi:10.1016/j.yfrne.2014.04.004
5. Ma WT, Chang C, Gershwin ME, Lian ZX. Development of autoantibodies precedes clinical manifestations of autoimmune diseases: a comprehensive review. J Autoimmun (2017) 83:95–112. doi:10.1016/j.jaut.2017.07.003
6. Wertz IE, O’Rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S, et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature (2004) 430(7000):694–9. doi:10.1038/nature02794
7. Ma A, Malynn BA. A20: linking a complex regulator of ubiquitylation to immunity and human disease. Nat Rev Immunol (2012) 12(11):774–85. doi:10.1038/nri3313
8. Adrianto I, Wen F, Templeton A, Wiley G, King JB, Lessard CJ, et al. Association of a functional variant downstream of TNFAIP3 with systemic lupus erythematosus. Nat Genet (2011) 43(3):253–8. doi:10.1038/ng.766
9. Moaaz M, Mohannad N. Association of the polymorphisms of TRAF1 (rs10818488) and TNFAIP3 (rs2230926) with rheumatoid arthritis and systemic lupus erythematosus and their relationship to disease activity among Egyptian patients. Cent Eur J Immunol (2016) 41(2):165–75. doi:10.5114/ceji.2016.60991
10. Musone SL, Taylor KE, Lu TT, Nititham J, Ferreira RC, Ortmann W, et al. Multiple polymorphisms in the TNFAIP3 region are independently associated with systemic lupus erythematosus. Nat Genet (2008) 40(9):1062–4. doi:10.1038/ng.202
11. Nair RP, Duffin KC, Helms C, Ding J, Stuart PE, Goldgar D, et al. Genome-wide scan reveals association of psoriasis with IL-23 and NF-kappaB pathways. Nat Genet (2009) 41(2):199–204. doi:10.1038/ng.311
12. Indhumathi S, Rajappa M, Chandrashekar L, Ananthanarayanan PH, Thappa DM, Negi VS. TNFAIP3 and TNIP1 polymorphisms confer psoriasis risk in South Indian Tamils. Br J Biomed Sci (2015) 72(4):168–73. doi:10.1080/09674845.2015.11665748
13. Vande Walle L, Van Opdenbosch N, Jacques P, Fossoul A, Verheugen E, Vogel P, et al. Negative regulation of the NLRP3 inflammasome by A20 protects against arthritis. Nature (2014) 512(7512):69–73. doi:10.1038/nature13322
14. Onizawa M, Oshima S, Schulze-Topphoff U, Oses-Prieto JA, Lu T, Tavares R, et al. The ubiquitin-modifying enzyme A20 restricts ubiquitination of the kinase RIPK3 and protects cells from necroptosis. Nat Immunol (2015) 16(6):618–27. doi:10.1038/ni.3172
15. Matsuzawa Y, Oshima S, Takahara M, Maeyashiki C, Nemoto Y, Kobayashi M, et al. TNFAIP3 promotes survival of CD4 T cells by restricting MTOR and promoting autophagy. Autophagy (2015) 11(7):1052–62. doi:10.1080/15548627.2015.1055439
16. Catrysse L, Vereecke L, Beyaert R, van Loo G. A20 in inflammation and autoimmunity. Trends Immunol (2014) 35(1):22–31. doi:10.1016/j.it.2013.10.005
17. Siebenlist U, Brown K, Claudio E. Control of lymphocyte development by nuclear factor-kappaB. Nat Rev Immunol (2005) 5(6):435–45. doi:10.1038/nri1629
18. Hoesel B, Schmid JA. The complexity of NF-kappaB signaling in inflammation and cancer. Mol Cancer (2013) 12:86. doi:10.1186/1476-4598-12-86
19. Hayden MS, Ghosh S. NF-kappaB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev (2012) 26(3):203–34. doi:10.1101/gad.183434.111
20. Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem (1998) 67:425–79. doi:10.1146/annurev.biochem.67.1.425
21. Opipari AW Jr, Boguski MS, Dixit VM. The A20 cDNA induced by tumor necrosis factor alpha encodes a novel type of zinc finger protein. J Biol Chem (1990) 265(25):14705–8.
22. Dixit VM, Green S, Sarma V, Holzman LB, Wolf FW, O’Rourke K, et al. Tumor necrosis factor-alpha induction of novel gene products in human endothelial cells including a macrophage-specific chemotaxin. J Biol Chem (1990) 265(5):2973–8.
23. Makarova KS, Aravind L, Koonin EV. A novel superfamily of predicted cysteine proteases from eukaryotes, viruses and Chlamydia pneumoniae. Trends Biochem Sci (2000) 25(2):50–2. doi:10.1016/S0968-0004(99)01530-3
24. Lu TT, Onizawa M, Hammer GE, Turer EE, Yin Q, Damko E, et al. Dimerization and ubiquitin mediated recruitment of A20, a complex deubiquitinating enzyme. Immunity (2013) 38(5):896–905. doi:10.1016/j.immuni.2013.03.008
25. Verhelst K, Carpentier I, Kreike M, Meloni L, Verstrepen L, Kensche T, et al. A20 inhibits LUBAC-mediated NF-kappaB activation by binding linear polyubiquitin chains via its zinc finger 7. EMBO J (2012) 31(19):3845–55. doi:10.1038/emboj.2012.240
26. Tokunaga F, Nishimasu H, Ishitani R, Goto E, Noguchi T, Mio K, et al. Specific recognition of linear polyubiquitin by A20 zinc finger 7 is involved in NF-kappaB regulation. EMBO J (2012) 31(19):3856–70. doi:10.1038/emboj.2012.241
27. Shembade N, Harhaj EW. Regulation of NF-kappaB signaling by the A20 deubiquitinase. Cell Mol Immunol (2012) 9(2):123–30. doi:10.1038/cmi.2011.59
28. Shembade N, Harhaj NS, Liebl DJ, Harhaj EW. Essential role for TAX1BP1 in the termination of TNF-alpha-, IL-1- and LPS-mediated NF-kappaB and JNK signaling. EMBO J (2007) 26(17):3910–22. doi:10.1038/sj.emboj.7601823
29. Oshima S, Turer EE, Callahan JA, Chai S, Advincula R, Barrera J, et al. ABIN-1 is a ubiquitin sensor that restricts cell death and sustains embryonic development. Nature (2009) 457(7231):906–9. doi:10.1038/nature07575
30. Mauro C, Pacifico F, Lavorgna A, Mellone S, Iannetti A, Acquaviva R, et al. ABIN-1 binds to NEMO/IKKgamma and co-operates with A20 in inhibiting NF-kappaB. J Biol Chem (2006) 281(27):18482–8. doi:10.1074/jbc.M601502200
31. Iha H, Peloponese JM, Verstrepen L, Zapart G, Ikeda F, Smith CD, et al. Inflammatory cardiac valvulitis in TAX1BP1-deficient mice through selective NF-kappaB activation. EMBO J (2008) 27(4):629–41. doi:10.1038/emboj.2008.5
32. Verstrepen L, Verhelst K, van Loo G, Carpentier I, Ley SC, Beyaert R. Expression, biological activities and mechanisms of action of A20 (TNFAIP3). Biochem Pharmacol (2010) 80(12):2009–20. doi:10.1016/j.bcp.2010.06.044
33. Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell (2003) 114(2):181–90. doi:10.1016/S0092-8674(03)00521-X
34. Brenner D, Blaser H, Mak TW. Regulation of tumour necrosis factor signalling: live or let die. Nat Rev Immunol (2015) 15(6):362–74. doi:10.1038/nri3834
35. Tokunaga F. Linear ubiquitination-mediated NF-kappaB regulation and its related disorders. J Biochem (2013) 154(4):313–23. doi:10.1093/jb/mvt079
36. Tokunaga F, Sakata S, Saeki Y, Satomi Y, Kirisako T, Kamei K, et al. Involvement of linear polyubiquitylation of NEMO in NF-kappaB activation. Nat Cell Biol (2009) 11(2):123–32. doi:10.1038/ncb1821
37. Chen ZJ, Parent L, Maniatis T. Site-specific phosphorylation of IkappaBalpha by a novel ubiquitination-dependent protein kinase activity. Cell (1996) 84(6):853–62. doi:10.1016/S0092-8674(00)81064-8
38. Alkalay I, Yaron A, Hatzubai A, Orian A, Ciechanover A, Ben-Neriah Y. Stimulation-dependent I kappa B alpha phosphorylation marks the NF-kappa B inhibitor for degradation via the ubiquitin-proteasome pathway. Proc Natl Acad Sci U S A (1995) 92(23):10599–603. doi:10.1073/pnas.92.23.10599
39. Bosanac I, Wertz IE, Pan B, Yu C, Kusam S, Lam C, et al. Ubiquitin binding to A20 ZnF4 is required for modulation of NF-kappaB signaling. Mol Cell (2010) 40(4):548–57. doi:10.1016/j.molcel.2010.10.009
40. Shembade N, Ma A, Harhaj EW. Inhibition of NF-kappaB signaling by A20 through disruption of ubiquitin enzyme complexes. Science (2010) 327(5969):1135–9. doi:10.1126/science.1182364
41. Skaug B, Chen J, Du F, He J, Ma A, Chen ZJ. Direct, noncatalytic mechanism of IKK inhibition by A20. Mol Cell (2011) 44(4):559–71. doi:10.1016/j.molcel.2011.09.015
42. Tiruppathi C, Soni D, Wang DM, Xue J, Singh V, Thippegowda PB, et al. The transcription factor DREAM represses the deubiquitinase A20 and mediates inflammation. Nat Immunol (2014) 15(3):239–47. doi:10.1038/ni.2823
43. Krikos A, Laherty CD, Dixit VM. Transcriptional activation of the tumor necrosis factor alpha-inducible zinc finger protein, A20, is mediated by kappa B elements. J Biol Chem (1992) 267(25):17971–6.
44. Yuk JM, Kim TS, Kim SY, Lee HM, Han J, Dufour CR, et al. Orphan nuclear receptor ERRalpha controls macrophage metabolic signaling and A20 expression to negatively regulate TLR-induced inflammation. Immunity (2015) 43(1):80–91. doi:10.1016/j.immuni.2015.07.003
45. Murakawa Y, Hinz M, Mothes J, Schuetz A, Uhl M, Wyler E, et al. RC3H1 post-transcriptionally regulates A20 mRNA and modulates the activity of the IKK/NF-kappaB pathway. Nat Commun (2015) 6:7367. doi:10.1038/ncomms8367
46. Gantier MP, Stunden HJ, McCoy CE, Behlke MA, Wang D, Kaparakis-Liaskos M, et al. A miR-19 regulon that controls NF-kappaB signaling. Nucleic Acids Res (2012) 40(16):8048–58. doi:10.1093/nar/gks521
47. Kim SW, Ramasamy K, Bouamar H, Lin AP, Jiang D, Aguiar RC. MicroRNAs miR-125a and miR-125b constitutively activate the NF-kappaB pathway by targeting the tumor necrosis factor alpha-induced protein 3 (TNFAIP3, A20). Proc Natl Acad Sci U S A (2012) 109(20):7865–70. doi:10.1073/pnas.1200081109
48. Wang CM, Wang Y, Fan CG, Xu FF, Sun WS, Liu YG, et al. miR-29c targets TNFAIP3, inhibits cell proliferation and induces apoptosis in hepatitis B virus-related hepatocellular carcinoma. Biochem Biophys Res Commun (2011) 411(3):586–92. doi:10.1016/j.bbrc.2011.06.191
49. Vinuesa CG, Cook MC, Angelucci C, Athanasopoulos V, Rui L, Hill KM, et al. A RING-type ubiquitin ligase family member required to repress follicular helper T cells and autoimmunity. Nature (2005) 435(7041):452–8. doi:10.1038/nature03555
50. Hutti JE, Turk BE, Asara JM, Ma A, Cantley LC, Abbott DW. IkappaB kinase beta phosphorylates the K63 deubiquitinase A20 to cause feedback inhibition of the NF-kappaB pathway. Mol Cell Biol (2007) 27(21):7451–61. doi:10.1128/MCB.01101-07
51. Wertz IE, Newton K, Seshasayee D, Kusam S, Lam C, Zhang J, et al. Phosphorylation and linear ubiquitin direct A20 inhibition of inflammation. Nature (2015) 528(7582):370–5. doi:10.1038/nature16165
52. Kulathu Y, Garcia FJ, Mevissen TE, Busch M, Arnaudo N, Carroll KS, et al. Regulation of A20 and other OTU deubiquitinases by reversible oxidation. Nat Commun (2013) 4:1569. doi:10.1038/ncomms2567
53. Lee JG, Baek K, Soetandyo N, Ye Y. Reversible inactivation of deubiquitinases by reactive oxygen species in vitro and in cells. Nat Commun (2013) 4:1568. doi:10.1038/ncomms2532
54. Shrikhande GV, Scali ST, da Silva CG, Damrauer SM, Csizmadia E, Putheti P, et al. O-glycosylation regulates ubiquitination and degradation of the anti-inflammatory protein A20 to accelerate atherosclerosis in diabetic ApoE-null mice. PLoS One (2010) 5(12):e14240. doi:10.1371/journal.pone.0014240
55. Di Dalmazi G, Hirshberg J, Lyle D, Freij JB, Caturegli P. Reactive oxygen species in organ-specific autoimmunity. Auto Immun Highlights (2016) 7(1):11. doi:10.1007/s13317-016-0083-0
56. Kienhofer D, Boeltz S, Hoffmann MH. Reactive oxygen homeostasis – the balance for preventing autoimmunity. Lupus (2016) 25(8):943–54. doi:10.1177/0961203316640919
57. Tewari M, Wolf FW, Seldin MF, O’Shea KS, Dixit VM, Turka LA. Lymphoid expression and regulation of A20, an inhibitor of programmed cell death. J Immunol (1995) 154(4):1699–706.
58. Coornaert B, Baens M, Heyninck K, Bekaert T, Haegman M, Staal J, et al. T cell antigen receptor stimulation induces MALT1 paracaspase-mediated cleavage of the NF-kappaB inhibitor A20. Nat Immunol (2008) 9(3):263–71. doi:10.1038/ni1561
59. Lee EG, Boone DL, Chai S, Libby SL, Chien M, Lodolce JP, et al. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science (2000) 289(5488):2350–4. doi:10.1126/science.289.5488.2350
60. Maizels N. Genome engineering with Cre-loxP. J Immunol (2013) 191(1):5–6. doi:10.4049/jimmunol.1301241
61. Clausen BE, Burkhardt C, Reith W, Renkawitz R, Forster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res (1999) 8(4):265–77. doi:10.1023/A:1008942828960
62. De Wilde K, Martens A, Lambrecht S, Jacques P, Drennan MB, Debusschere K, et al. A20 inhibition of STAT1 expression in myeloid cells: a novel endogenous regulatory mechanism preventing development of enthesitis. Ann Rheum Dis (2016) 76(3):585–92. doi:10.1136/annrheumdis-2016-209454
63. Matmati M, Jacques P, Maelfait J, Verheugen E, Kool M, Sze M, et al. A20 (TNFAIP3) deficiency in myeloid cells triggers erosive polyarthritis resembling rheumatoid arthritis. Nat Genet (2011) 43(9):908–12. doi:10.1038/ng.874
64. Agostini L, Martinon F, Burns K, McDermott MF, Hawkins PN, Tschopp J. NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity (2004) 20(3):319–25. doi:10.1016/S1074-7613(04)00046-9
65. de Torre-Minguela C, Mesa Del Castillo P, Pelegrin P. The NLRP3 and pyrin inflammasomes: implications in the pathophysiology of autoinflammatory diseases. Front Immunol (2017) 8:43. doi:10.3389/fimmu.2017.00043
66. Duong BH, Onizawa M, Oses-Prieto JA, Advincula R, Burlingame A, Malynn BA, et al. A20 restricts ubiquitination of pro-interleukin-1beta protein complexes and suppresses NLRP3 inflammasome activity. Immunity (2015) 42(1):55–67. doi:10.1016/j.immuni.2014.12.031
67. Steinman RM, Hemmi H. Dendritic cells: translating innate to adaptive immunity. Curr Top Microbiol Immunol (2006) 311:17–58. doi:10.1007/3-540-32636-7_2
68. Naik SH, Metcalf D, van Nieuwenhuijze A, Wicks I, Wu L, O’Keeffe M, et al. Intrasplenic steady-state dendritic cell precursors that are distinct from monocytes. Nat Immunol (2006) 7(6):663–71. doi:10.1038/ni1340
69. Caton ML, Smith-Raska MR, Reizis B. Notch-RBP-J signaling controls the homeostasis of CD8- dendritic cells in the spleen. J Exp Med (2007) 204(7):1653–64. doi:10.1084/jem.20062648
70. Hammer GE, Turer EE, Taylor KE, Fang CJ, Advincula R, Oshima S, et al. Expression of A20 by dendritic cells preserves immune homeostasis and prevents colitis and spondyloarthritis. Nat Immunol (2011) 12(12):1184–93. doi:10.1038/ni.2135
71. Kool M, van Loo G, Waelput W, De Prijck S, Muskens F, Sze M, et al. The ubiquitin-editing protein A20 prevents dendritic cell activation, recognition of apoptotic cells, and systemic autoimmunity. Immunity (2011) 35(1):82–96. doi:10.1016/j.immuni.2011.05.013
72. Xuan NT, Wang X, Nishanth G, Waisman A, Borucki K, Isermann B, et al. A20 expression in dendritic cells protects mice from LPS-induced mortality. Eur J Immunol (2015) 45(3):818–28. doi:10.1002/eji.201444795
73. Helft J, Bottcher J, Chakravarty P, Zelenay S, Huotari J, Schraml BU, et al. GM-CSF mouse bone marrow cultures comprise a heterogeneous population of CD11c(+)MHCII(+) macrophages and dendritic cells. Immunity (2015) 42(6):1197–211. doi:10.1016/j.immuni.2015.05.018
74. Crow MK. Advances in understanding the role of type I interferons in systemic lupus erythematosus. Curr Opin Rheumatol (2014) 26(5):467–74. doi:10.1097/BOR.0000000000000087
75. Cummings RJ, Barbet G, Bongers G, Hartmann BM, Gettler K, Muniz L, et al. Different tissue phagocytes sample apoptotic cells to direct distinct homeostasis programs. Nature (2016) 539(7630):565–9. doi:10.1038/nature20138
76. Liang J, Huang HI, Benzatti FP, Karlsson AB, Zhang JJ, Youssef N, et al. Inflammatory Th1 and Th17 in the intestine are each driven by functionally specialized dendritic cells with distinct requirements for MyD88. Cell Rep (2016) 17(5):1330–43. doi:10.1016/j.celrep.2016.09.091
77. Cash H, Relle M, Menke J, Brochhausen C, Jones SA, Topley N, et al. Interleukin 6 (IL-6) deficiency delays lupus nephritis in MRL-Faslpr mice: the IL-6 pathway as a new therapeutic target in treatment of autoimmune kidney disease in systemic lupus erythematosus. J Rheumatol (2010) 37(1):60–70. doi:10.3899/jrheum.090194
78. Atreya R, Mudter J, Finotto S, Mullberg J, Jostock T, Wirtz S, et al. Blockade of interleukin 6 trans signaling suppresses T-cell resistance against apoptosis in chronic intestinal inflammation: evidence in crohn disease and experimental colitis in vivo. Nat Med (2000) 6(5):583–8. doi:10.1038/75068
79. Jacob N, Stohl W. Cytokine disturbances in systemic lupus erythematosus. Arthritis Res Ther (2011) 13(4):228. doi:10.1186/ar3349
80. Neurath MF. Current and emerging therapeutic targets for IBD. Nat Rev Gastroenterol Hepatol (2017) 14(5):269–78. doi:10.1038/nrgastro.2016.208
81. Ciccarelli F, De Martinis M, Ginaldi L. An update on autoinflammatory diseases. Curr Med Chem (2014) 21(3):261–9. doi:10.2174/09298673113206660303
82. Holman HR, Kunkel HG. Affinity between the lupus erythematosus serum factor and cell nuclei and nucleoprotein. Science (1957) 126(3265):162–3. doi:10.1126/science.126.3265.162
83. Hammer GE, Ma A. Molecular control of steady-state dendritic cell maturation and immune homeostasis. Annu Rev Immunol (2013) 31:743–91. doi:10.1146/annurev-immunol-020711-074929
84. Duwel M, Welteke V, Oeckinghaus A, Baens M, Kloo B, Ferch U, et al. A20 negatively regulates T cell receptor signaling to NF-kappaB by cleaving Malt1 ubiquitin chains. J Immunol (2009) 182(12):7718–28. doi:10.4049/jimmunol.0803313
85. Malewicz M, Zeller N, Yilmaz ZB, Weih F. NF kappa B controls the balance between Fas and tumor necrosis factor cell death pathways during T cell receptor-induced apoptosis via the expression of its target gene A20. J Biol Chem (2003) 278(35):32825–33. doi:10.1074/jbc.M304000200
86. Shi L, Chen S, Lu Y, Wang X, Xu L, Zhang F, et al. Changes in the MALT1-A20-NF-kappaB expression pattern may be related to T cell dysfunction in AML. Cancer Cell Int (2013) 13(1):37. doi:10.1186/1475-2867-13-37
87. Giordano M, Roncagalli R, Bourdely P, Chasson L, Buferne M, Yamasaki S, et al. The tumor necrosis factor alpha-induced protein 3 (TNFAIP3, A20) imposes a brake on antitumor activity of CD8 T cells. Proc Natl Acad Sci U S A (2014) 111(30):11115–20. doi:10.1073/pnas.1406259111
88. Roncagalli R, Hauri S, Fiore F, Liang Y, Chen Z, Sansoni A, et al. Quantitative proteomics analysis of signalosome dynamics in primary T cells identifies the surface receptor CD6 as a Lat adaptor-independent TCR signaling hub. Nat Immunol (2014) 15(4):384–92. doi:10.1038/ni.2843
89. Lee PP, Fitzpatrick DR, Beard C, Jessup HK, Lehar S, Makar KW, et al. A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival. Immunity (2001) 15(5):763–74. doi:10.1016/S1074-7613(01)00227-8
90. Bae HR, Leung PS, Tsuneyama K, Valencia JC, Hodge DL, Kim S, et al. Chronic expression of interferon-gamma leads to murine autoimmune cholangitis with a female predominance. Hepatology (2016) 64(4):1189–201. doi:10.1002/hep.28641
91. Morita M, Watanabe Y, Akaike T. Protective effect of hepatocyte growth factor on interferon-gamma-induced cytotoxicity in mouse hepatocytes. Hepatology (1995) 21(6):1585–93. doi:10.1016/0270-9139(95)90463-8
92. Toyonaga T, Hino O, Sugai S, Wakasugi S, Abe K, Shichiri M, et al. Chronic active hepatitis in transgenic mice expressing interferon-gamma in the liver. Proc Natl Acad Sci U S A (1994) 91(2):614–8. doi:10.1073/pnas.91.2.614
93. Fischer JC, Otten V, Kober M, Drees C, Rosenbaum M, Schmickl M, et al. A20 restrains thymic regulatory T cell development. J Immunol (2017) 199(7):2356–65. doi:10.4049/jimmunol.1602102
94. He S, Wang L, Miao L, Wang T, Du F, Zhao L, et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell (2009) 137(6):1100–11. doi:10.1016/j.cell.2009.05.021
95. Just S, Nishanth G, Buchbinder JH, Wang X, Naumann M, Lavrik I, et al. A20 curtails primary but augments secondary CD8+ t cell responses in intracellular bacterial infection. Sci Rep (2016) 6:39796. doi:10.1038/srep39796
96. McLeod IX, Jia W, He YW. The contribution of autophagy to lymphocyte survival and homeostasis. Immunol Rev (2012) 249(1):195–204. doi:10.1111/j.1600-065X.2012.01143.x
97. Thomson AW, Turnquist HR, Raimondi G. Immunoregulatory functions of mTOR inhibition. Nat Rev Immunol (2009) 9(5):324–37. doi:10.1038/nri2546
98. Bekeredjian-Ding I, Jego G. Toll-like receptors – sentries in the B-cell response. Immunology (2009) 128(3):311–23. doi:10.1111/j.1365-2567.2009.03173.x
99. Ferch U, Zum Buschenfelde CM, Gewies A, Wegener E, Rauser S, Peschel C, et al. MALT1 directs B cell receptor-induced canonical nuclear factor-kappaB signaling selectively to the c-Rel subunit. Nat Immunol (2007) 8(9):984–91. doi:10.1038/ni1493
100. Chu Y, Vahl JC, Kumar D, Heger K, Bertossi A, Wojtowicz E, et al. B cells lacking the tumor suppressor TNFAIP3/A20 display impaired differentiation and hyperactivation and cause inflammation and autoimmunity in aged mice. Blood (2011) 117(7):2227–36. doi:10.1182/blood-2010-09-306019
101. Hovelmeyer N, Reissig S, Xuan NT, Adams-Quack P, Lukas D, Nikolaev A, et al. A20 deficiency in B cells enhances B-cell proliferation and results in the development of autoantibodies. Eur J Immunol (2011) 41(3):595–601. doi:10.1002/eji.201041313
102. Tavares RM, Turer EE, Liu CL, Advincula R, Scapini P, Rhee L, et al. The ubiquitin modifying enzyme A20 restricts B cell survival and prevents autoimmunity. Immunity (2010) 33(2):181–91. doi:10.1016/j.immuni.2010.07.017
103. Honma K, Tsuzuki S, Nakagawa M, Tagawa H, Nakamura S, Morishima Y, et al. TNFAIP3/A20 functions as a novel tumor suppressor gene in several subtypes of non-Hodgkin lymphomas. Blood (2009) 114(12):2467–75. doi:10.1182/blood-2008-12-194852
104. Novak U, Rinaldi A, Kwee I, Nandula SV, Rancoita PM, Compagno M, et al. The NF-{kappa}B negative regulator TNFAIP3 (A20) is inactivated by somatic mutations and genomic deletions in marginal zone lymphomas. Blood (2009) 113(20):4918–21. doi:10.1182/blood-2008-08-174110
105. Schmitz R, Hansmann ML, Bohle V, Martin-Subero JI, Hartmann S, Mechtersheimer G, et al. TNFAIP3 (A20) is a tumor suppressor gene in Hodgkin lymphoma and primary mediastinal B cell lymphoma. J Exp Med (2009) 206(5):981–9. doi:10.1084/jem.20090528
106. Dieguez-Gonzalez R, Calaza M, Perez-Pampin E, Balsa A, Blanco FJ, Canete JD, et al. Analysis of TNFAIP3, a feedback inhibitor of nuclear factor-kappaB and the neighbor intergenic 6q23 region in rheumatoid arthritis susceptibility. Arthritis Res Ther (2009) 11(2):R42. doi:10.1186/ar2650
107. Vereecke L, Beyaert R, van Loo G. Genetic relationships between A20/TNFAIP3, chronic inflammation and autoimmune disease. Biochem Soc Trans (2011) 39(4):1086–91. doi:10.1042/BST0391086
108. de Boer YS, van Gerven NM, Zwiers A, Verwer BJ, van Hoek B, van Erpecum KJ, et al. Genome-wide association study identifies variants associated with autoimmune hepatitis type 1. Gastroenterology (2014) 147(2):443.e–52.e. doi:10.1053/j.gastro.2014.04.022
109. Xu E, Cao H, Lin L, Liu H. rs10499194 polymorphism in the tumor necrosis factor-alpha inducible protein 3 (TNFAIP3) gene is associated with type-1 autoimmune hepatitis risk in Chinese Han population. PLoS One (2017) 12(4):e0176471. doi:10.1371/journal.pone.0176471
110. Cordell HJ, Han Y, Mells GF, Li Y, Hirschfield GM, Greene CS, et al. International genome-wide meta-analysis identifies new primary biliary cirrhosis risk loci and targetable pathogenic pathways. Nat Commun (2015) 6:8019. doi:10.1038/ncomms9019
111. Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature (2012) 491(7422):119–24. doi:10.1038/nature11582
112. Zhou Q, Wang H, Schwartz DM, Stoffels M, Park YH, Zhang Y, et al. Loss-of-function mutations in TNFAIP3 leading to A20 haploinsufficiency cause an early-onset autoinflammatory disease. Nat Genet (2016) 48(1):67–73. doi:10.1038/ng.3459
113. Takagi M, Ogata S, Ueno H, Yoshida K, Yeh T, Hoshino A, et al. Haploinsufficiency of TNFAIP3 (A20) by germline mutation is involved in autoimmune lymphoproliferative syndrome. J Allergy Clin Immunol (2017) 139(6):1914–22. doi:10.1016/j.jaci.2016.09.038
114. Aksentijevich I, Zhou Q. NF-kappaB pathway in autoinflammatory diseases: dysregulation of protein modifications by ubiquitin defines a new category of autoinflammatory diseases. Front Immunol (2017) 8:399. doi:10.3389/fimmu.2017.00399
115. Li D, Wang L, Fan Y, Song L, Guo C, Zhu F, et al. Down-regulation of A20 mRNA expression in peripheral blood mononuclear cells from patients with systemic lupus erythematosus. J Clin Immunol (2012) 32(6):1287–91. doi:10.1007/s10875-012-9764-2
116. Wang Z, Zhang Z, Yuan J, Li LI. Altered TNFAIP3 mRNA expression in peripheral blood mononuclear cells from patients with rheumatoid arthritis. Biomed Rep (2015) 3(5):675–80. doi:10.3892/br.2015.486
117. Zhu L, Wang L, Wang X, Zhou L, Liao Z, Xu L, et al. Characteristics of A20 gene polymorphisms and clinical significance in patients with rheumatoid arthritis. J Transl Med (2015) 13:215. doi:10.1186/s12967-015-0566-1
118. Arsenescu R, Bruno ME, Rogier EW, Stefka AT, McMahan AE, Wright TB, et al. Signature biomarkers in Crohn’s disease: toward a molecular classification. Mucosal Immunol (2008) 1(5):399–411. doi:10.1038/mi.2008.32
119. Bruno ME, Rogier EW, Arsenescu RI, Flomenhoft DR, Kurkjian CJ, Ellis GI, et al. Correlation of biomarker expression in colonic mucosa with disease phenotype in Crohn’s disease and ulcerative colitis. Dig Dis Sci (2015) 60(10):2976–84. doi:10.1007/s10620-015-3700-2
120. Aki A, Nagasaki M, Malynn BA, Ma A, Kagari T. Hypomorphic A20 expression confers susceptibility to psoriasis. PLoS One (2017) 12(6):e0180481. doi:10.1371/journal.pone.0180481
121. Elsby LM, Orozco G, Denton J, Worthington J, Ray DW, Donn RP. Functional evaluation of TNFAIP3 (A20) in rheumatoid arthritis. Clin Exp Rheumatol (2010) 28(5):708–14.
122. Wang S, Wen F, Tessneer KL, Gaffney PM. TALEN-mediated enhancer knockout influences TNFAIP3 gene expression and mimics a molecular phenotype associated with systemic lupus erythematosus. Genes Immun (2016) 17(3):165–70. doi:10.1038/gene.2016.4
123. Duncan CJA, Dinnigan E, Theobald R, Grainger A, Skelton AJ, Hussain R, et al. Early-onset autoimmune disease due to a heterozygous loss-of-function mutation in TNFAIP3 (A20). Ann Rheum Dis (2017). doi:10.1136/annrheumdis-2016-210944
124. Malynn BA, Ma A. A20 takes on tumors: tumor suppression by an ubiquitin-editing enzyme. J Exp Med (2009) 206(5):977–80. doi:10.1084/jem.20090765
125. Ohnishi H, Kawamoto N, Seishima M, Ohara O, Fukao T. A Japanese family case with juvenile onset Behcet’s disease caused by TNFAIP3 mutation. Allergol Int (2017) 66(1):146–8. doi:10.1016/j.alit.2016.06.006
126. Shigemura T, Kaneko N, Kobayashi N, Kobayashi K, Takeuchi Y, Nakano N, et al. Novel heterozygous C243Y A20/TNFAIP3 gene mutation is responsible for chronic inflammation in autosomal-dominant Behcet’s disease. RMD Open (2016) 2(1):e000223. doi:10.1136/rmdopen-2015-000223
127. Kadowaki T, Ohnishi H, Kawamoto N, Hori T, Nishimura K, Kobayashi C, et al. Haploinsufficiency of A20 causes autoinflammatory and autoimmune disorders. J Allergy Clin Immunol (2017). doi:10.1016/j.jaci.2017.10.039
128. Aeschlimann FA, Batu ED, Canna SW, Go E, Gul A, Hoffmann P, et al. A20 haploinsufficiency (HA20): clinical phenotypes and disease course of patients with a newly recognised NF-kB-mediated autoinflammatory disease. Ann Rheum Dis (2018). doi:10.1136/annrheumdis-2017-212403
129. Wolfrum S, Teupser D, Tan M, Chen KY, Breslow JL. The protective effect of A20 on atherosclerosis in apolipoprotein E-deficient mice is associated with reduced expression of NF-kappaB target genes. Proc Natl Acad Sci U S A (2007) 104(47):18601–6. doi:10.1073/pnas.0709011104
130. Guedes RP, Csizmadia E, Moll HP, Ma A, Ferran C, da Silva CG. A20 deficiency causes spontaneous neuroinflammation in mice. J Neuroinflammation (2014) 11:122. doi:10.1186/1742-2094-11-122
131. Lodolce JP, Kolodziej LE, Rhee L, Kariuki SN, Franek BS, McGreal NM, et al. African-derived genetic polymorphisms in TNFAIP3 mediate risk for autoimmunity. J Immunol (2010) 184(12):7001–9. doi:10.4049/jimmunol.1000324
132. Bates JS, Lessard CJ, Leon JM, Nguyen T, Battiest LJ, Rodgers J, et al. Meta-analysis and imputation identifies a 109 kb risk haplotype spanning TNFAIP3 associated with lupus nephritis and hematologic manifestations. Genes Immun (2009) 10(5):470–7. doi:10.1038/gene.2009.31
133. Dieude P, Guedj M, Wipff J, Ruiz B, Riemekasten G, Matucci-Cerinic M, et al. Association of the TNFAIP3 rs5029939 variant with systemic sclerosis in the European Caucasian population. Ann Rheum Dis (2010) 69(11):1958–64. doi:10.1136/ard.2009.127928
134. Koczan D, Drynda S, Hecker M, Drynda A, Guthke R, Kekow J, et al. Molecular discrimination of responders and nonresponders to anti-TNF alpha therapy in rheumatoid arthritis by etanercept. Arthritis Res Ther (2008) 10(3):R50. doi:10.1186/ar2419
135. Tejasvi T, Stuart PE, Chandran V, Voorhees JJ, Gladman DD, Rahman P, et al. TNFAIP3 gene polymorphisms are associated with response to TNF blockade in psoriasis. J Invest Dermatol (2012) 132(3 Pt 1):593–600. doi:10.1038/jid.2011.376
136. Dayer JM, Oliviero F, Punzi L. A brief history of IL-1 and IL-1 Ra in rheumatology. Front Pharmacol (2017) 8:293. doi:10.3389/fphar.2017.00293
137. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med (2017) 377(12):1119–31. doi:10.1056/NEJMoa1707914
138. Ridker PM, MacFadyen JG, Thuren T, Everett BM, Libby P, Glynn RJ. Effect of interleukin-1beta inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet (2017) 390(10105):1833–42. doi:10.1016/S0140-6736(17)32247-X
139. Illei GG, Shirota Y, Yarboro CH, Daruwalla J, Tackey E, Takada K, et al. Tocilizumab in systemic lupus erythematosus: data on safety, preliminary efficacy, and impact on circulating plasma cells from an open-label phase I dosage-escalation study. Arthritis Rheum (2010) 62(2):542–52. doi:10.1002/art.27221
140. Kronke M, Leonard WJ, Depper JM, Arya SK, Wong-Staal F, Gallo RC, et al. Cyclosporin A inhibits T-cell growth factor gene expression at the level of mRNA transcription. Proc Natl Acad Sci U S A (1984) 81(16):5214–8. doi:10.1073/pnas.81.16.5214
141. Szamel M, Bartels F, Resch K. Cyclosporin A inhibits T cell receptor-induced interleukin-2 synthesis of human T lymphocytes by selectively preventing a transmembrane signal transduction pathway leading to sustained activation of a protein kinase C isoenzyme, protein kinase C-beta. Eur J Immunol (1993) 23(12):3072–81. doi:10.1002/eji.1830231205
Keywords: A20, tumor necrosis factor α-induced protein 3, NF-κB, ubiquitination, autoinflammation, autoimmune disease, mouse models, single nucleotide polymorphisms
Citation: Das T, Chen Z, Hendriks RW and Kool M (2018) A20/Tumor Necrosis Factor α-Induced Protein 3 in Immune Cells Controls Development of Autoinflammation and Autoimmunity: Lessons from Mouse Models. Front. Immunol. 9:104. doi: 10.3389/fimmu.2018.00104
Received: 03 November 2017; Accepted: 12 January 2018;
Published: 21 February 2018
Edited by:
Massimo Gadina, National Institute of Arthritis and Musculoskeletal and Skin Diseases, United StatesReviewed by:
Michael Francis McDermott, University of Leeds, United KingdomIvona Aksentijevich, National Human Genome Research Institute (NIH), United States
Copyright: © 2018 Das, Chen, Hendriks and Kool. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mirjam Kool, m.kool@erasmusmc.nl