 Henry Y. Lu1,2
Henry Y. Lu1,2 Bradly M. Bauman3
Bradly M. Bauman3 Swadhinya Arjunaraja3
Swadhinya Arjunaraja3 Batsukh Dorjbal3
Batsukh Dorjbal3 Joshua D. Milner4
Joshua D. Milner4 Andrew L. Snow3†
Andrew L. Snow3† Stuart E. Turvey1,2*†
Stuart E. Turvey1,2*†- 1Department of Pediatrics, British Columbia Children's Hospital, The University of British Columbia, Vancouver, BC, Canada
- 2Experimental Medicine Program, Faculty of Medicine, The University of British Columbia, Vancouver, BC, Canada
- 3Department of Pharmacology and Molecular Therapeutics, Uniformed Services University of the Health Sciences, Bethesda, MD, United States
- 4Laboratory of Allergic Diseases, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD, United States
The caspase recruitment domain family member 11 (CARD11 or CARMA1)—B cell CLL/lymphoma 10 (BCL10)—MALT1 paracaspase (MALT1) [CBM] signalosome complex serves as a molecular bridge between cell surface antigen receptor signaling and the activation of the NF-κB, JNK, and mTORC1 signaling axes. This positions the CBM complex as a critical regulator of lymphocyte activation, proliferation, survival, and metabolism. Inborn errors in each of the CBM components have now been linked to a diverse group of human primary immunodeficiency diseases termed “CBM-opathies.” Clinical manifestations range from severe combined immunodeficiency to selective B cell lymphocytosis, atopic disease, and specific humoral defects. This surprisingly broad spectrum of phenotypes underscores the importance of “tuning” CBM signaling to preserve immune homeostasis. Here, we review the distinct clinical and immunological phenotypes associated with human CBM complex mutations and introduce new avenues for targeted therapeutic intervention.
Introduction
Inborn errors of immunity or primary immunodeficiency diseases (PIDs) are a group of ~350 genetic disorders that are characterized by defects in immune system development and/or function (1). Defining the genetic and molecular basis of these diseases has not only benefitted the affected individuals but has greatly enhanced our understanding of the fundamental factors that regulate human immunity. A powerful example of the value of studying patients with PIDs is nuclear factor kappa B (NF-κB)—a critical transcription factor that facilitates lymphocyte activation, proliferation and survival. Aberrant signaling in the NF-κB pathway is associated with inflammatory diseases (2), malignancy (3), autoimmunity (4), and immunodeficiency (5). With the description of patients with monogenic immune disorders affecting various components of this signaling cascade, we now have an improved understanding of how NF-κB is positively and negatively regulated.
In the past decade, the assembly of the caspase recruitment domain family member 11 (CARD11 or CARMA1)—B cell CLL/lymphoma 10 (BCL10)—MALT1 paracaspase (MALT1) [CBM] signalosome complex has emerged as a critical step in the antigen-dependent activation of NF-κB in B and T lymphocytes (6). Major landmarks in the understanding of CBM function first came from oncology. These advances included the characterization of mutant BCL10 and MALT1 proteins caused by chromosomal translocations leading to constitutive/aberrant NF-κB signaling in lymphoma (7), the discovery of CARD11 and its ability to interact with and regulate BCL10 (8), and the identification of mutant CARD11 isoforms affecting the coiled-coil domain that activate NF-κB, constituting ~10% of activated B cell-like diffuse large B cell lymphomas (9, 10). These findings, along with the generation and study of Card11−/− (11–13), Bcl10−/− (14, 15), and Malt1−/− (16) mice, collectively positioned the CBM complex as a central modulator of lymphocyte signaling through the regulation of the NF-κB, c-Jun N-terminal kinase (JNK), and mechanistic target of rapamycin complex (mTORC1) pathways (Figures 1, 2).
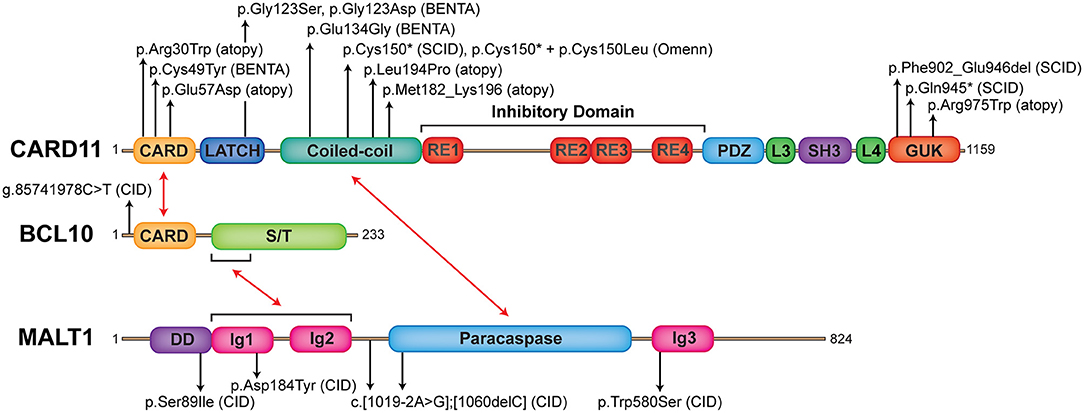
Figure 1. The landscape of human germline mutations causing CBM-opathies. Schematic representation of protein domains found in CARD11, BCL10, and MALT1. Red arrows indicate interactions between domains. Annotated are confirmed mutations causing CBM-opathies and where they localize to on the protein. CARD, caspase recruitment domain; RE, repressive element; L, linker; PDZ, postsynaptic density protein (PSD95), Drosophila disc large tumor suppressor (Dlg1), zonula occludens-1 protein (zo-1) domain; SH3, SRC homology 3 (SH3); GUK, guanylate kinase domain; S/T, serine/threonine; DD, death domain; Ig, immunoglobulin-like domain.
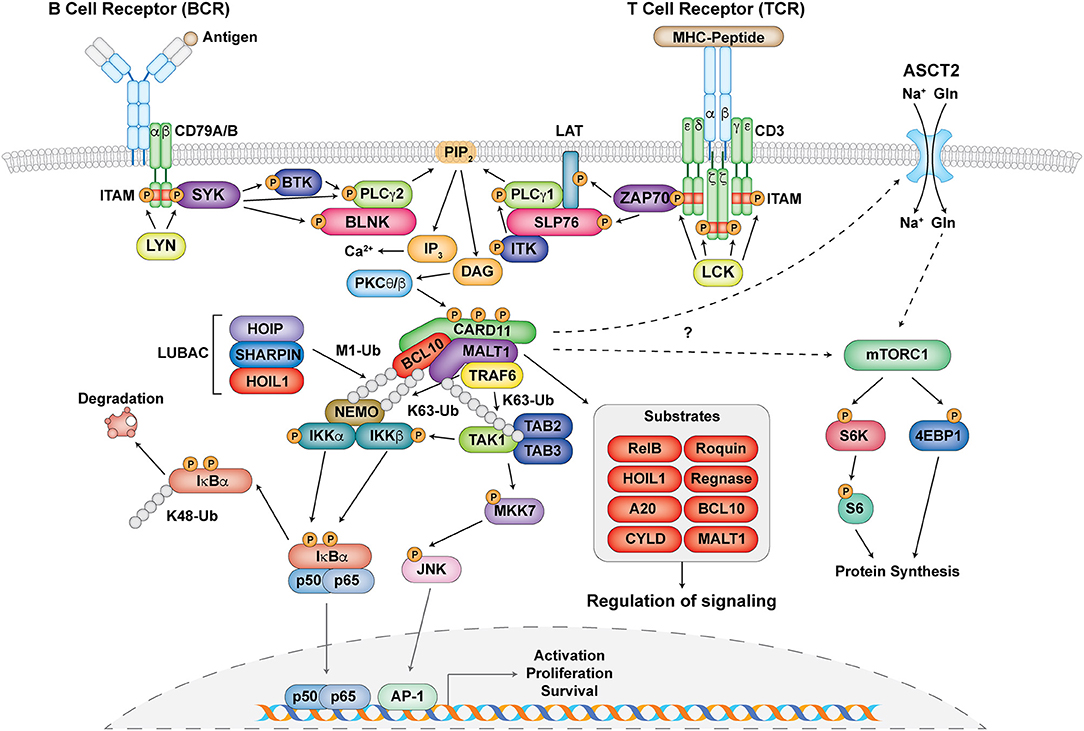
Figure 2. The central role of the CBM complex in BCR- and TCR- signaling. Schematic representation of proximal antigen receptor signaling events in the BCR and TCR, activation and assembly of the CBM complex, and downstream targets and effects of CBM activation. Gray circles represent ubiquitin chains.
The importance of the CBM complex in adaptive immunity was experimentally established by the fact that B and T cells from mice deficient in Card11, Bcl10, or Malt1 all displayed impaired cellular activation and proliferation, aberrant cytokine secretion, and blocks in cell differentiation, resulting in diminished serum immunoglobulin levels. These experimental observations were then validated in the intact human system by the recent discovery of individuals suffering from profound immune defects [i.e., combined immunodeficiency (CID) and severe combined immunodeficiency (SCID)] involving germline loss-of-function (LOF) mutations in CARD11 (17–19), BCL10 (20), and MALT1 (21–23, 24) (Figure 1). While human deficiency of each of the CBM components has some unique defining clinical features (e.g., gastrointestinal inflammation seen in MALT1 deficiency or susceptibility to Pneumocystis jirovecii pneumonia (PJP) typical for CARD11 deficiency), as testament to their highly synergistic activities, many phenotypic manifestations are shared across these CBM deficiencies. In particular, some unifying features of CBM PIDs include: CID/SCID occurring in the context of generally normal total B and T cell numbers, a predominantly naïve phenotype in peripheral blood lymphocytes, impaired T cell proliferation, and compromised antigen receptor-induced NF-κB activation.
Recent discoveries have now moved beyond relatively simple LOF mutations, and there is now an interesting spectrum of additional clinical phenotypes attributed to CARD11 mutations (25), with gain-of-function mutations causing “B cell Expansion with NF-κB and T cell Anergy” (BENTA) disease (26–30), hypomorphic dominant-interfering mutations causing combined immunodeficiency with atopic disease “CARD11-associated Atopy with Dominant Interference of NF-κB Signaling” (CADINS) (31, 32), and loss-of-function mutations with somatic reversion associated with Omenn syndrome (19) (Figure 1).
In this review, we will illustrate the current understanding of CBM-mediated activation of the NF-κB, JNK, and mTORC1 pathways in lymphocytes, and highlight the diverse and rapidly expanding clinical and immunological phenotypes of “CBM-opathies.”
The CBM Complex in Antigen Receptor Signaling
Proximal Antigen Receptor Signaling
Upon antigen recognition, the CBM complex is primarily involved in signal transduction downstream of antigen receptors leading to the activation of NF-κB, JNK, and mTORC1 in lymphocytes (33–35) (Figure 2). Signaling following B cell receptor (BCR) and T cell receptor (TCR) activation is highly symmetrical and begins with the phosphorylation of immunoreceptor tyrosine-based activation motifs (ITAMs) found on the CD79A/CD79B chains of the BCR and the ζ-chains of the TCR complex by Src family tyrosine kinases LYN and lymphocyte-specific protein tyrosine kinase (LCK), respectively (33, 36). This facilitates the recruitment and phosphorylation of the spleen tyrosine kinase (Syk) family tyrosine kinases SYK (for BCR) and zeta-chain-associated protein kinase 70 (ZAP70) (for TCR) (33, 36) (Figure 2). From here, a collection of adaptor, phospholipase, and kinase proteins come together to form signalosomes, including B cell linker protein (BLNK) and Bruton tyrosine kinase (BTK) for the BCR and SH2 domain containing leukocyte protein of 76 kDa (SLP76), linker of activated T cells (LAT), and IL-2 inducible T cell kinase (ITK) for the TCR. This assembly ultimately culminates in the activation of phospholipase Cγ1 (PLCγ1) for the TCR, PLCγ2 for the BCR, and phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K) for both (37, 38) (Figure 2).
CBM Assembly
Phosphorylated PLCγ1 and PLCγ2 mediate the hydrolysis of phosphatidylinositol 4,5 biphosphate (PIP2) to synthesize the second messengers diacylglycerol (DAG) and inositol-1,4,5-triphosphate (IP3) (37, 38). While IP3 induces calcium influx, DAG activates protein kinase C (PKC) θ (in T cells) and PKCβ (in B cells) (Figure 2). PKCθ/β act to phosphorylate a series of serine sites along the CARD11 inhibitory domain, the first of several post-translational modifications required for the assembly of the CBM complex (39, 40). CARD11 converts to an open conformation, making it accessible for BCL10-MALT1 binding. BCL10, which constitutively associates with MALT1 through serine/threonine-rich and immunoglobulin-like domain interactions, respectively (7, 41), binds to CARD11 through caspase recruitment domain (CARD)-CARD domain interactions (42) (Figure 1). MALT1 can also bind directly to CARD11 through the interaction of its paracaspase domain and the coiled-coil domain of CARD11 (43). These initial events nucleate the formation of higher order structures consisting of branched BCL10 filaments sheathed with MALT1, allowing for MALT1 oligomerization and activation, and the cooperative recruitment and incorporation of tumor necrosis factor receptor-associated factor 6 (TRAF6) (41, 42).
Signaling to NF-κB
Canonical NF-κB activation is mediated by the activation of the IκB kinase (IKK) complex, which consists of two catalytic subunits IKKα and IKKβ and a regulatory subunit NF-κB essential modulator (NEMO, also known as IKKγ) (5). After the assembly of the CBM complex, various ubiquitination events occur in order to facilitate the phosphorylation and activation of the IKK complex (35) (Figure 2). MALT1 contains binding motifs for the E3 ubiquitin ligase TRAF6 and acts as a scaffold to facilitate the oligomerization and activation of TRAF6 (44, 45). It is thought that TRAF6 and BCL10, as well as other factors such as the ubiquitin conjugating enzyme UBC13, mediate the K63-linked ubiquitination of various proteins including MALT1 and NEMO (35, 46–48). In addition to K63-linked ubiquitination, M1-linked linear ubiquitin chains are also ligated to NEMO and BCL10 through the linear ubiquitin chain assembly complex (LUBAC), which consists of heme-oxidized IRP2 ubiquitin ligase 1 (HOIL1), HOIL1-interacting protein (HOIP), and SHANK-associated RH domain interacting protein (SHARPIN) (49–51) (Figure 2). These two types of ubiquitination collectively mediate optimal recruitment and phosphorylation/activation of the IKK complex (5).
The phosphorylation of IKKα/β after antigen receptor ligation is thought to be principally mediated by TGFβ-activated kinase 1 (TAK1) and its associated adaptor proteins TAB2/3 (44, 52, 53). It is speculated that since both TAB2/3 and NEMO can specifically recognize K63-linked ubiquitination (47, 54), these two complexes are brought in close proximity to each other, thus facilitating optimal IKK complex activation (34). NF-κB inhibitor alpha (IκBα) in resting cells normally exists in a complex with the NF-κB subunits p50 and p65, which prevents them from becoming activated. The activated IKK complex can phosphorylate IκBα, which causes it to undergo K48-linked ubiquitination and degradation by the proteasome. This allows NF-κB to translocate into the nucleus to initiate target gene transcription (5) (Figure 2).
Signaling to JNK
Another arm of antigen receptor signaling that the CBM complex mediates is the c-Jun N-terminal kinase (JNK) pathway (Figure 2) [reviewed in (55)]. This process is usually activated by successive phosphorylation events mediated by mitogen-activated protein kinases (MAPKs) (56), with TAK1 also playing a critical role in JNK activation (57, 58). This highlights a cross-talk mechanism in NF-κB and JNK signaling. Although regulation of this process by the CBM complex is not as well understood as the NF-κB pathway, it is thought that TAK1 associates with CARD11-mediated BCL10 oligomers, MKK7 gets recruited, and the selective phosphorylation of JNK2 occurs (59) (Figure 2). This ultimately leads to the accumulation and phosphorylation of c-Jun, which regulates lymphocyte proliferation as part of the AP-1 transcription factor complex.
Signaling to mTORC1
The mechanistic target of rapamycin (mTOR) kinase is a PI3K-related kinase (PIKK) represented by two distinct catalytic protein complexes, mTOR Complex 1 (mTORC1) and 2 (mTORC2), which have different mechanisms of activation and signaling (60). In particular, mTORC1 can be activated by TCR and CD28 co-stimulation, environmental stimuli such as growth factors and stressors, and nutrients such as amino acids (61). It is critical for regulating T cell growth and proliferation (62) as well as T helper 1 (Th1) and Th17 differentiation (63). Recent studies have demonstrated that the CBM complex regulates TCR-mediated glutamine uptake and the subsequent activation of the mTOR pathway independent of NF-κB (32, 64, 65). Following TCR stimulation, CBM components associate with and mediate the upregulation of the alanine-serine-cysteine transporter 2 (ASCT2) glutamine transporter at the cell surface (32, 64) (Figure 2). MALT1 also has the ability to associate with mTOR, and its paracaspase activity mediates the phosphorylation of the ribosomal protein S6, a target of mTORC1, ultimately impacting metabolic programming (65) (Figure 2). However, the exact molecular mechanism by which the CBM complex links TCR signals to glutamine uptake and the activation of mTOR is unknown and requires further study.
The CARD Family
The CARMA/CARD protein family consists of CARD9, CARD10 (or CARMA3), CARD11 (or CARMA1), and CARD14 (or CARMA2) (66). These scaffold proteins are evolutionarily conserved, structurally homologous, mostly membrane-associated (with the exception of CARD9) and have varying patterns of expression in the body. Mutations in this family of proteins have been implicated in different pathological states, including CARD9 deficiency increasing susceptibility to fungal infections [reviewed in (67)] and CARD14 mutations being linked to increased susceptibility to psoriasis (68, 69) and the skin disease pityriasis rubra pilaris (70).
Each CARD protein participates in its own “CBM” complex with BCL10 and MALT1 in different cell types to facilitate downstream signaling events leading to NF-κB activation (66). CARD11 mediates the activation of lymphocytes by the antigen receptors BCR and TCR and facilitates natural killer (NK) cell activation downstream of ITAM-coupled receptors such as natural killer group 2, member D (NKG2D), NK1.1, Ly49D, and Ly49H (71, 72). CARD14 is expressed in the placenta and skin tissue (69) and can mediate NF-κB activation in keratinocytes possibly downstream of Dectin-1 (73). CARD10 is broadly expressed in non-hematopoietic cells and serves as a link between G-protein coupled receptors (GPCRs) and NF-κB (74). CARD9 is mostly expressed by myeloid cells (macrophages, dendritic cells, neutrophils) and transduces signals emanating from C-type lectin receptors (Dectin-1, Dectin-2, Mincle) and ITAM-associated receptors (FcRγ, DAP12) (67). Although CARD9 deficiency also causes immunodeficiency, this has been covered in detail very recently in the “CARMA Proteins: Playing a Hand of Four CARDs” Research Topic (67) and thus, we will be focusing on immune defects caused by mutations in CARD11.
CARD11
Role of CARD11 in Immunity
CARD11 is a ~130 kDa protein originally discovered by bioinformatics screens (8, 75). It is primarily expressed in hematopoietic tissue and lymphocytes and is crucial for antigen receptor signaling (11, 76). CARD11 is a classic scaffold molecule comprised of various defined domains, including CARD, LATCH, coiled-coil (CC), inhibitory, postsynaptic density protein (PSD95), Drosophila disc large tumor suppressor (Dlg1), zonula occludens-1 protein (zo-1) (PDZ), SRC homology 3 (SH3), and guanylate kinase (GUK) domains (Figure 1). Initial insight into CARD11 function came from in vitro studies on leukemia cell lines, which demonstrated its essential role in NF-κB activation downstream of the TCR complex (77–79). Following this, various CARD11 mouse models were generated, including the Card11−/− mouse and CARD11 “unmodulated” (Card11un/un) mouse (11–13, 80) (compared in Table 1).
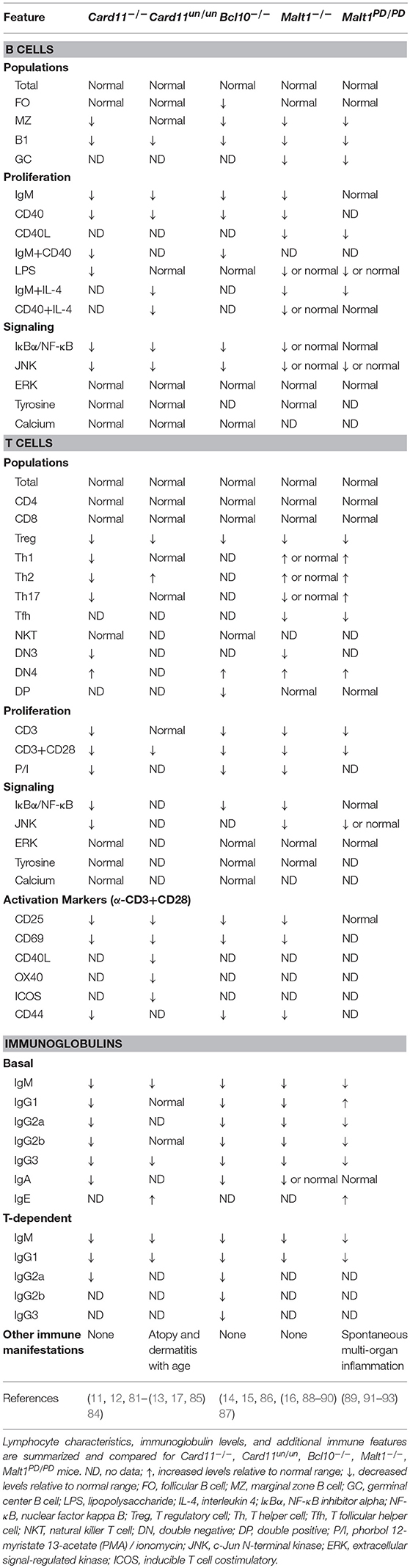
Table 1. Overview of CBM-deficient mouse models.
Card11-deficient mice were found to be immunodeficient and displayed defective antigen receptor-induced NF-κB and JNK signaling, impaired B and T cell proliferation, and decreased expression of immune activation markers; however, ERK and p38 activation remained intact. Lymphocyte numbers were generally normal, but some abnormalities were present: decreased B1 and marginal zone (MZ) B cells, decreased Th2 and Th17 cells, and absent regulatory T cells (Tregs) (11, 12, 81–84). In addition, both immunoglobulin levels and antibody responses were impaired. These studies highlighted the essential role of CARD11 in mediating B cell development, Treg development, and antibody production.
The Card11un/un mouse, in contrast, possessed a single ENU mutagenesis-induced point mutation in the coiled-coil domain of Card11, which did not impact CARD11 protein expression but conveyed a hypomorphic effect on NF-κB and JNK signaling (13). Similar to the Card11−/− mice, Card11un/un lymphocytes displayed impaired proliferation and upregulation of activation markers after stimulation (13). Surprisingly, Card11un/un mice did not exhibit overt pathology but developed spontaneous atopy and dermatitis with age. Accordingly, despite some immunoglobulins being decreased, IgE was significantly elevated (13, 85) and this was paired with elevated Th2 cells, diminished Tregs, and unchanged Th1 and Th17 cells (85). These mice highlighted a possible role for CARD11 in the pathogenesis of allergic disease and tolerance. Indeed, a genome-wide association study identified CARD11 as a susceptibility locus for atopic dermatitis (94) and it was found that CARD11 is important for both Th2 polarization and allergic airway disease (95, 96).
Of the three CBM components, CARD11 mutations have been associated with the most diverse phenotypes. Human germline CARD11 mutations cause a broad (and expanding) range of clinical phenotypes including SCID, CID and atopy, and BENTA—with more being characterized (summarized in Tables 2, 4–7).
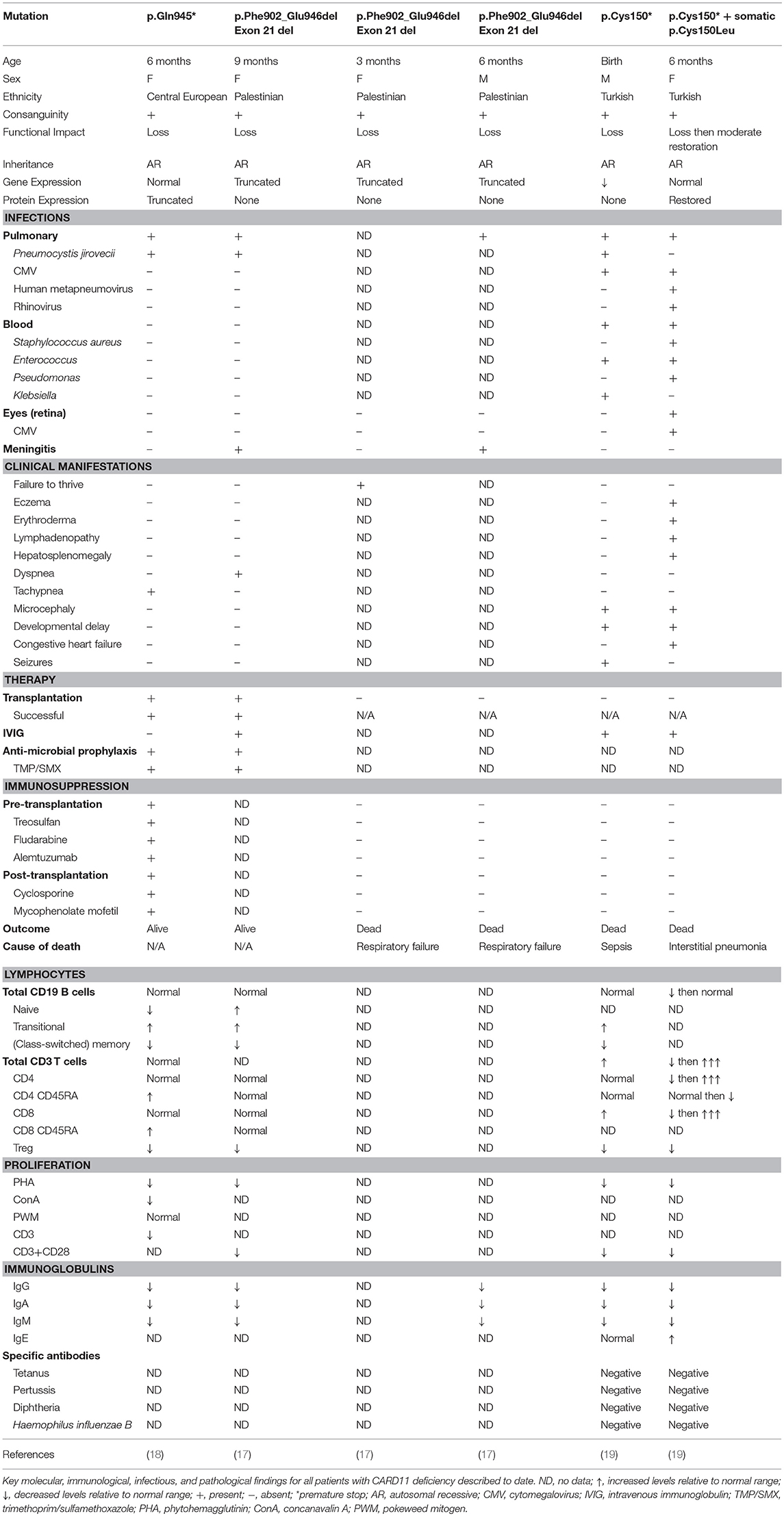
Table 2. Clinical and laboratory phenotype of human CARD11 deficiency.
Biallelic Loss-of-Function CARD11 Mutations Causing SCID
Germline homozygous loss-of-function (LOF) mutations in CARD11 are associated with SCID (OMIM 615206) (17–19) (Table 2). To date, there have been three reported cases of complete CARD11 deficiency and they were discovered by whole exome sequencing (WES) or directed Sanger sequencing, with mutations localizing to either the CC (19) or GUK (17, 18) domains. Patients were of different ethnicities but all were born to consanguineous parents: a Palestinian girl with p.Phe902_Glu946del mutations/exon 21 deletion (17), a Central European girl with p.Gln945* mutations (18), and a Turkish boy with p.Cys150* mutations (19). The p.Cys150* homozygous boy also had an older sister with the same mutation, but she additionally had a second site somatic reversion (p.Cys150Leu), which led to Omenn syndrome (19).
Patients typically presented within the first year of life (3–15 months) with severe respiratory tract infections/pneumonia caused by Pneumocystis jirovecii (PJP) and abnormal serum immunoglobulin levels that progressed to panhypogammaglobulinemia (3/3 patients). Development was generally normal, and patients did not have any overt organ pathology. All patients generally had normal numbers of total B and T cells, κ-deleted receptor excision circle (KREC)/T cell receptor excision circle (TREC) values (where available), and recent thymic emigrants (Table 2). However, detailed immune profiling revealed a developmental block in B cells, with increased transitional B cells and decreased (class-switched) memory B cells (3/3 patients). All patients had severely diminished Tregs (3/3 patients) and a predominantly naïve T cell phenotype (2/2 patients). Functional evaluation of these patients revealed absent/severely diminished lymphocyte activation of NF-κB and decreased T cell proliferation in response to antigen receptor stimulation (3/3 patients).
Fuchs and colleagues described a Turkish girl homozygous for p.C150* who had a somatic second site reversion (p.C150L), which restored some CARD11 protein expression and function in her lymphocytes (19). Consequently, this young girl had a very different clinical course from the fully CARD11-deficient patients. She presented at the age of 5 months with features of Omenn syndrome, including severe eczema that progressed to generalized erythroderma, lymphadenopathy, and hepatosplenomegaly (Table 2). She experienced multiple bouts of sepsis caused by both bacteria and viruses (Staphylococcus aureus, Enterococcus, Pseudomonas) and eventually succumbed to viral pneumonia positive for human metapneumovirus, rhinovirus, and cytomegalovirus (CMV). Immune investigations revealed progressive T cell lymphoproliferation, massive infiltration of highly proliferative T cells in the skin, oligoclonal T cell expansion, and elevated IgE. Interestingly, the patient also had some features that overlapped with fully-deficient patients, including progressive panhypogammaglobulinemia and reduced T cell proliferation (Table 2). Surprisingly, this somatic reversion led to only a partial restoration of NF-κB activity without causing a gain-of-function (GOF) effect. It is thought that the somatic reversion occurred only in some founder T cells, giving them a proliferative advantage over the fully CARD11-deficient T cells. In the context of specific immune triggers such as viral infections, these T cell clones expanded, perturbing immune homeostasis in the absence of Tregs, and ultimately caused skin infiltration, progressive eczema and erythroderma, lymphadenopathy, and hepatosplenomegaly. This case highlights the differential requirements for NF-κB signaling between T cell ontogeny and maintenance.
Diagnosis and Treatment of CARD11 Deficiency
The first cases of CARD11 deficiency were diagnosed by WES (17, 18), while the latter two were found by directed Sanger sequencing based on suspicion and similarity to the first two cases (19). CARD11 deficiency should be considered in patients who present in early life with respiratory tract infections most commonly caused by Pneumocystis jirovecii (PJP) (3/3 patients), impaired B cell development with increased transitional and decreased memory (3/3 patients), predominantly naïve T cells (2/2 patients), impaired T cell proliferation (3/3 patients), and panhypogammaglobulinemia (3/3 patients) (Table 3). Specific testing for NF-κB phosphorylation, IκBα degradation, and IL-2 secretion, while often only available in the research setting, will show decreased/absent response. In the appropriate clinical context, sequencing of the CARD11 gene should help confirm the diagnosis, but depending on the nature of any genetic variants identified, functional tests of the CBM may be required to make a definitive diagnosis (97).
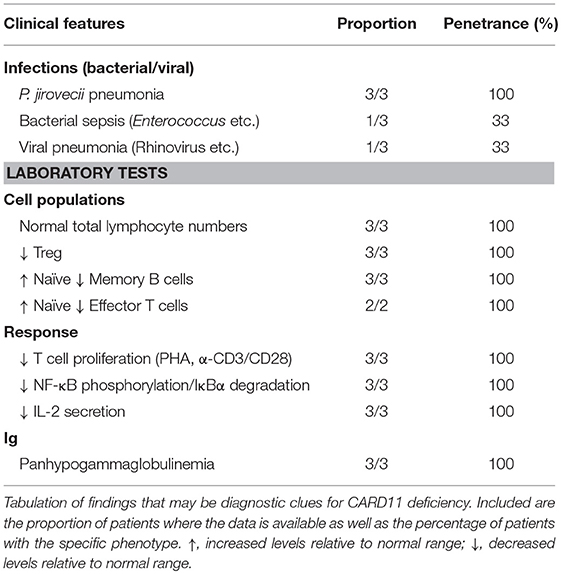
Table 3. “Red flags” suggestive of human CARD11 deficiency (LOF CARD11 mutations).
In addition, patients presenting with features of Omenn syndrome should also be evaluated for CARD11 deficiency (with possible somatic second site reversion) (19). Although there has only been a single patient described with this phenotype to date, suggestive clinical features include erythroderma, lymphadenopathy, hepatosplenomegaly, T cell lymphocytosis, diminished naïve T cells, eosinophilia, and elevated IgE. Analysis of TCR rearrangement would possibly detect oligoclonality. Sequencing CARD11 in cells from different sites of the body may show different genotypes (LOF vs. LOF+somatic reversion).
In general, CARD11 deficiency is fatal within the first 2 years of life (3/5 died). Infection-related respiratory failure was the most common cause of death (2/5 patients) (17) and one died of sepsis (19). The two patients who survived (17, 18) received successful hematopoietic stem cell transplants. One survivor received bone marrow-derived stem cells from a human leukocyte antigen (HLA)-identical brother who was a heterozygous carrier of the CARD11 mutation (17), emphasizing that CARD11 haploinsufficiency is not pathological. The second survivor received peripheral blood hematopoietic stem cells from an HLA-matched unrelated donor (18). Overall, these results indicate that patients with confirmed CARD11 deficiency should be considered for curative hematopoietic stem cell transplantation as soon as possible following diagnosis. While awaiting transplant, CARD11-deficient patients should receive immunoglobulin replacement therapy and PJP prophylaxis.
Dominant Negative Loss-of-Function CARD11 Mutations Causing CID, Atopy, and Novel Phenotypes
Germline heterozygous LOF mutations in CARD11 are associated with severe atopic disease and CID with a susceptibility to infections (OMIM 617638) (31, 32). These LOF mutations dominantly interfere with wild-type (WT) CARD11 and signaling to NF-κB and mTORC1, thus explaining the observed autosomal dominant inheritance pattern. To date, five distinct dominant negative (DN) LOF CARD11 mutations have been linked to disease in 12 patients (Figure 1). Although the cohort of known CARD11 DN patients and associated clinical phenotypes is expanding rapidly (98) (Table 4), the cardinal feature noted in ~90% of patients is severe atopic disease, encompassing symptoms of immediate hypersensitivity (allergic rhinitis, food allergy) and/or allergic inflammation (atopic dermatitis, eosinophilic esophagitis), specific allergens notwithstanding (99). Importantly, as discussed in the “Role of CARD11 in Immunity” section, similar atopic phenotypes were described in unmodulated (Card11un/un) mice harboring a hypomorphic mutation in Card11 (13, 85) and CARD11 has previously been identified as a risk locus for atopic dermatitis in a Japanese genome-wide association study (94).
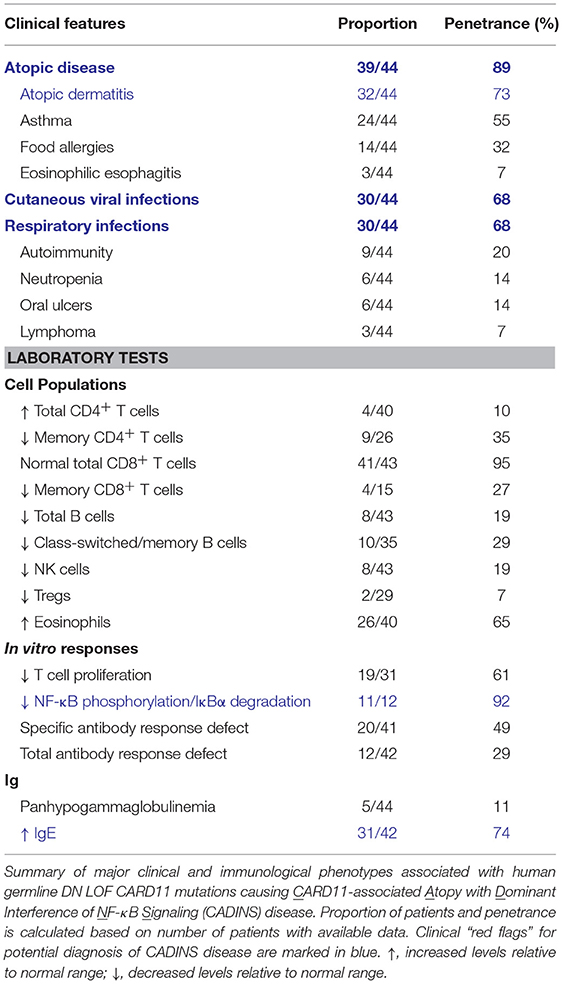
Table 4. “Red flags” suggestive of CADINS disease (DN LOF CARD11 mutations).
Heterozygous CARD11 mutations were initially identified by WES in eight patients with severe atopic dermatitis (32). These mutations included three missense mutations encoding p.Glu57Asp, p.Leu194Pro, and p.Arg975Trp and one in-frame mutation encoding a 14-amino acid insertion (p.Met183_Lys196). These patients generally possessed features of recalcitrant atopic dermatitis with elevated serum IgE levels and eosinophilia, with severity waning with age in certain patients. Most patients also presented with respiratory distress associated with recurrent pulmonary infections and pneumonias, as well as viral skin infections (e.g., molluscum, eczema herpeticum). Four additional patients were subsequently described with missense mutations encoding p.Arg30Trp, leading to multiorgan atopy, autoimmunity, and a prominent susceptibility to infections (31).
These mutations affected several different domains of the CARD11 protein, with 2 in the CARD (p.Arg30Trp and p.Glu57Asp), 2 in the CC (p.Leu194Pro and p.Met183_Lys196), and 1 in the GUK (p.Arg975Trp) domain (31, 32). Patient T cells and mutant plasmid-transfected Jurkat T leukemia cell lines demonstrated that each variant impaired TCR-induced NF-κB activation by disrupting WT CARD11 signaling. Subsequent studies of an expanded patient cohort have identified at least 10 additional DN mutations primarily found in the CARD and CC domains, where they are most likely to impede CBM complex assembly by thwarting BCL10 and MALT1 binding and oligomerization (98). In addition to NF-κB blockade, many (but not all) of these mutations also reduced TCR-mediated mTORC1 activation, ostensibly by preventing optimal glutamine uptake through upregulation of the ASCT2 transporter (32). These TCR signaling defects not only resulted in impaired T cell proliferation and induction of cell surface activation markers, but also contributed to a Th2-skewed CD4+ T cell phenotype, with enhanced secretion of IL-4 and IL-13 and decreased IFN-γ production. Intriguingly, in vitro culture of patient T cells with excess glutamine partially restored T cell proliferation and IFN-γ secretion in the presence of cytokines that trigger NF-κB (e.g., IL-1/TNF) and signal transducer and activator of transcription (STAT3) (e.g., IL-6) activation independent to the TCR (32). These findings suggest that both NF-κB and mTORC1 signaling defects contribute to atopic predisposition and disease pathology, even though diagnostic readouts of impaired mTORC1 signaling (e.g., ribosomal protein S6 phosphorylation) can be variable and difficult to detect experimentally. In contrast to Card11un/un mice, the frequency and suppressive function of Tregs is normal in almost all CARD11 DN patients tested to date, suggesting important mechanistic discrepancies between mouse and human (31, 32).
Diagnosis and Treatment of Disease Caused by DN CARD11 Mutations
The recent identification of many additional patients harboring CARD11 DN variants provides a clearer picture of the full phenotypic spectrum of disease (98) (Table 4). Clinically, patients with germline DN CARD11 mutations most often present in early childhood with atopy, cutaneous viral infections, and recurrent respiratory infections. These signs and symptoms occur in an autosomal dominant manner with high penetrance and no gender bias. However, subsets of patients may also present with hypogammaglobulinemia and specific antibody deficiency (SAD), neutropenia, oral ulcers, autoimmunity (e.g., alopecia), or lymphoma. Diagnostic tests include: (i) assaying for a NF-κB signaling defect in response to TCR or PMA stimulation (reduced p65 phosphorylation, IκB degradation) and (ii) sequencing CARD11 to identify rare/novel variants, with CARD and CC domain variants having the highest likelihood of being pathogenic. Ultimately, functional testing (e.g., Jurkat T cell transfections) is highly recommended to confirm DN LOF activity for any novel variants found.
Based on these collective clinical and experimental findings, we propose to classify this disorder as CARD11-associated Atopy with Dominant Interference of NF-κB Signaling (CADINS) disease. Although more work is required to mechanistically connect faulty CARD11 signaling to the various phenotypes of CADINS disease, defects in both T and B cell function explain CID in these patients and underscore the essential role of CARD11 in governing peripheral lymphocyte differentiation and effective humoral immune responses. Continued mechanistic studies will also inform future clinical management and therapeutic strategies for these patients. Although glutamine supplementation may offer the simplest intervention (see “Novel Therapeutic Insights Emerging From Our Understanding of Human CBM-opathies” section), newer biologics targeting Th2 cytokine signaling (e.g., dupilumab, mepolizumab) or IgE directly (omalizumab) may be useful in ameliorating atopic disease (100). In contrast to CARD11 deficiency, hematopoietic stem cell transplantation should only be considered in the most severe pediatric cases, since symptoms may improve with age.
Gain-of-Function CARD11 Mutations Causing BENTA
B cell Expansion with NF-κB and T-cell Anergy (BENTA) is a congenital lymphoproliferative and immunodeficiency disorder caused by heterozygous GOF CARD11 mutations (OMIM 616452) (26). The first patient diagnosed with BENTA disease was initially reported in 1971, and presented with splenomegaly and persistent B cell lymphocytosis that worsened with splenectomy and resembled chronic lymphocytic leukemia (CLL) (101). This patient eventually developed monoclonal CLL around age 44 and received a curative hematopoietic stem cell transplant from his sister (26). His two daughters presented with frequent sinopulmonary and ear infections and were found to exhibit splenomegaly and marked B cell lymphocytosis in infancy (26). RNA-Seq analyses of this first patient and his two daughters revealed a novel heterozygous missense mutation (p.Glu134Gly), located in the N-terminal portion of the CC domain of CARD11. A fourth unrelated patient that presented with similar symptoms was simultaneously identified with unique heterozygous missense mutations located in the LATCH domain (p.Gly123Ser) (26). Since the initial description of BENTA disease in these four patients in 2012, GOF CARD11 mutations associated with BENTA have been identified in >25 additional patients (27–30, 102–104) (Figure 1, Table 5).
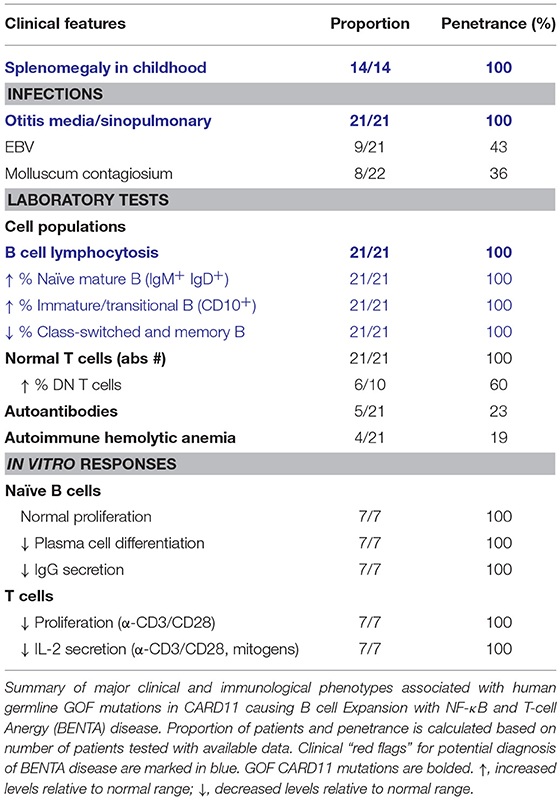
Table 5. “Red flags” suggestive of BENTA disease (GOF CARD11 mutations).
The primary hallmark of BENTA disease is polyclonal B cell lymphocytosis in early childhood paired with splenomegaly and lymphadenopathy. Pediatric patients possess excessive accumulation of immature transitional (CD10+CD24hiCD38hi) and mature naïve (IgM+IgD+) polyclonal B cells, with very low percentages of circulating memory and class-switched B cells. Circulating naïve and transitional B cell counts typically decrease into adulthood, likely reflecting reduced output of immature B cells from the bone marrow. Conversely, patient T cell numbers are usually normal, unless chronic viral infection (e.g., EBV) is present. Histologic analyses of lymphoid tissues reveal follicular hyperplasia with an impressive expansion of naïve IgD+ B cells in mantle zones, but normal numbers and distribution of CD3+ T cells (26). Aside from selective B cell lymphocytosis, BENTA patients also exhibit features of primary immunodeficiency. All BENTA patients experienced frequent ear and sinus infections in early life, and opportunistic viral infections such as molluscum contagiosum and JC/BK virus are noted in some patients. Chronic EBV infection with moderate viremia is also found in ~50% of BENTA patients (104).
Similar to specific antibody deficiency (SAD) (105), poor humoral immune responses are observed in most BENTA patients in response to T cell-independent vaccines such as pneumococcal and meningococcal polysaccharide vaccines, even with repeated boosts. Some patients also fail to mount lasting protective titers to T cell-dependent conjugate vaccines for pneumococcal bacteria (i.e., Prevnar), varicella-zoster virus (VZV), or measles. Low serum IgM and IgA levels are noted in some patients, with IgG being variable. In vitro studies of naïve patient B cells demonstrated impaired B cell differentiation into plasmablasts and long-lived plasma cells, consistent with poor IgG secretion in culture (106). These defects could be explained in part by a failed induction of specific factors required for plasma cell commitment, including BLIMP-1 and XBP-1. Conversely, in mice, ectopic expression of GOF CARD11 variants in activated B cells promoted the transient expansion of self-reactive plasmablasts and autoantibody production (107). This discrepancy may be due to differences in mouse and human B cell differentiation requirements or may reflect the need for in vivo cytokines that were not provided in vitro. Nevertheless, profound apoptosis resistance was readily observed in both mouse and human B cells expressing GOF CARD11 variants and may be the most likely driver of B cell lymphocytosis in BENTA disease. Surprisingly, BENTA patient T cells are generally hyporesponsive in culture with poor proliferation and reduced IL-2 secretion (26). T cell function can largely be rescued by stronger stimulation or IL-2 supplementation in vitro, implying a mild state of anergy in BENTA T cells. Although autoantibodies are detected in a few patients, autoimmune disease symptoms are not common in BENTA patients, perhaps reflecting underlying B and T cell differentiation defects.
Most of the germline GOF CARD11 mutations described in BENTA patients (p.Cys49Tyr, p.Gly123Ser, p.Gly123Asp, p.Phe130Ile, p.Glu134Gly) are also found as somatic GOF CARD11 mutations in diffuse large B cell lymphoma (DLBCL) and other lymphoid malignancies (9, 108). In fact, knockdown of CARD11 effectively kills DLBCL cell lines harboring GOF CARD11 mutations, underscoring the connection between enhanced CBM signaling and B cell growth and survival. Remarkably, these single GOF mutations can disrupt the auto-inhibition of CARD11 conferred by several repressive elements within the inhibitory linker domain (109, 110). This allows CARD11 to adopt an open, active conformation and drive constitutive NF-κB activation via spontaneous aggregation, unimpeded recruitment of BCL10/MALT1, and IKKα/β phosphorylation, in the absence of antigen receptor engagement (26, 28, 109, 110). Indeed, spontaneous CARD11 aggregation and elevated NF-κB signaling is also observed in B and T cells from BENTA patients (26). In addition, the ectopic expression of BENTA-associated CARD11 mutants in B and T cell lines results in the spontaneous assembly of large protein aggregates including CARD11, BCL10, MALT1, and phosphorylated IKKα/β, which induces constitutive NF-κB signaling independent of antigen receptor ligation (26, 28).
Despite highly congruent signaling pathways emanating from the TCR and BCR, constitutive activation of canonical NF-κB driven by GOF CARD11 mutations in BENTA disease leads to surprisingly distinct functional consequences in B and T cells. However, the mechanisms behind this dichotomy in B and T cell phenotypes remain unclear. Previous studies using conditional transgenic mice offer tantalizing parallels; while B cell-specific expression of constitutively active IKKβ (caIKKβ) promotes survival and proliferation even in the absence of B cell activating factor (BAFF) (111), restricted transgenic expression of caIKKβ renders murine T cells anergic and more susceptible to apoptosis, consistent with poor responses to bacterial infections (112). Collectively, studies of BENTA patients to date indicate that constitutive NF-κB can also lead to combined immunodeficiency, albeit less severe than patients harboring (DN) LOF CARD11 mutations. We therefore speculate that intrinsic B cell defects in BENTA disease most likely contribute to impaired humoral immunity and frequent infections with extracellular bacteria, while mildly anergic T cells could make BENTA patients more susceptible to certain viral infections.
Diagnosis and Treatment of BENTA
Patients presenting with splenomegaly, selective B cell lymphocytosis, and frequent sinopulmonary infections early in life should raise suspicion for BENTA disease. Sequencing of CARD11 may find variants, particularly in the LATCH or CC domains. It is recommended that novel variants be confirmed experimentally (e.g., B or T cell line transfections to look for constitutive NF-κB activation) and cross-referenced to reported somatic mutations in lymphoma using Catalogue of Somatic Mutations in Cancer (COSMIC) (113) or related databases of oncogenic mutations.
At present, BENTA patients are clinically managed with supportive therapy, and minimal therapeutic interventions are available. Polyclonal B cell lymphocytosis in BENTA disease may predispose patients to B cell malignancies later in life. However, only two patients with confirmed malignancy have been reported to date: B cell CLL at ~44 years in the original index patient (26), and another with Hodgkin's lymphoma at ~50 years of age. Still, BENTA patients should be regularly monitored for B cell clonal outgrowth using flow cytometry and IgH heavy chain rearrangement analyses. The presence of EBV viremia may also heighten the risk of B cell lymphomagenesis.
Removal of the spleen is generally not recommended, given that circulating B cell counts rose dramatically in 3 patients after splenectomy, and splenectomy itself may put the patient at increased risk for certain bacterial infections (26, 28). One patient with a p.Gly123Asp mutation and an exceptionally high number of peripheral B cells after splenectomy was treated with methotrexate for 4 years to restrain B cell counts and reduce the risk of stroke (28). Rituximab was effective in both this patient and another with respiratory distress and excessive lymphocytic nodules in her lungs. However, the utility of B cell depleting agents for BENTA should be evaluated on a case-by-case basis and may not be necessary as B cell lymphocytosis wanes over time. Intravenous or subcutaneous immunoglobulin therapy has also been administered in a few patients during childhood to control infections. Interestingly, MALT1 protease inhibitors, which specifically constrain CBM signaling output without completely blocking NF-κB activation, could be an attractive targeted treatment option for certain BENTA patients (see Novel Therapeutic Insights Emerging From Our Understanding of Human CBM-opathies section). These inhibitors are currently being explored for treating B cell lymphomas and autoimmune diseases (114–117).
BCL10
Role of BCL10 in Immunity
BCL10 was originally identified from a recurrent breakpoint (1p22) in mucosa-associated lymphoid tissue (MALT) B cell lymphomas possessing the t(1;14)(p22;q32) translocation, which caused BCL10 to be overexpressed (118). This observation, in combination with the finding that BCL10 could potently induce NF-κB activation (119, 120), highlighted its involvement in NF-κB signaling. BCL10 is a ~27 kDa protein, which contains an N-terminal CARD domain and C-terminal serine/threonine rich region (Figure 1). Through CARD-CARD interactions, BCL10 can oligomerize with other CARD-containing proteins, including CARD9, CARD10, CARD11, and CARD14 (8, 75, 121–123) as well as MALT1 (7) to form various CBM complexes. These complexes collectively regulate both innate and adaptive immune processes in various cell types, although its lymphoid role is the main focus here. Like CARD11, BCL10 also undergoes various post-translational modifications that regulate CBM assembly and signaling and can form high order filamentous structures [reviewed in (124)]. The generation of Bcl10−/− mice defined key physiological roles for BCL10, particularly its essential role in antigen receptor signaling (Table 1) (14, 15, 86, 87).
Deletion of Bcl10 in mice causes partial embryonic lethality (1/3 die) caused by issues with neural tube closure during development (14). Aside from this particular phenotype, Bcl10−/− mice were immunodeficient and generally resembled Card11−/− mice. They possessed normal total numbers of B and T cells, but decreased numbers of Tregs, natural killer T (NKT), B1, and MZ B cells (15, 86). Lymphocytes lacking Bcl10 failed to activate NF-κB, secrete pro-inflammatory cytokines, proliferate effectively in response to antigen receptor stimulation, and upregulate activation markers (14). Similar to CARD11-deficient mice, Bcl10−/− mice also had panhypogammaglobulinemia and impaired T-dependent humoral responses (15).
Interestingly, it was recently demonstrated that BCL10 also contributes to glutamine uptake via the ASCT2 transporter (as mentioned in the “Signaling to mTORC1” section), and together with CARD11 and MALT1, governs Th1 and Th17 polarization independent of the NF-κB pathway (64). However, another group found that BCL10 was dispensable in the phosphorylation of S6 (65). Thus, BCL10 contribution to this pathway is controversial and requires further investigation.
Loss-of-Function BCL10 Mutations Causing Combined Immunodeficiency
A single case of BCL10 deficiency has been identified in a consanguineous Amerindian boy from Ecuador caused by homozygosity for a germline loss-of-function mutation (OMIM 616098) (20). The patient exhibited features of CID and immune dysregulation. WES discovered a homozygous splice site mutation (g.85741978C>T;IVS1+1G>A) affecting the invariant first nucleotide of intron 1 (donor site for splicing), which led to absent mRNA and protein expression. The patient had a complex clinical course, including respiratory infections positive for influenza A/B, adenovirus, respiratory syncytial virus, gastroenteritis, otitis, oral candidiasis and diaper dermatitis from Candida albicans superinfection, recurrent diarrhea positive for Campylobacter jejuni, adenovirus, and Clostridium difficile at different times, acute gastroenteritis positive for adenovirus, chronic colitis, and suspected encephalitis (Table 6). The patient eventually died due to respiratory failure.
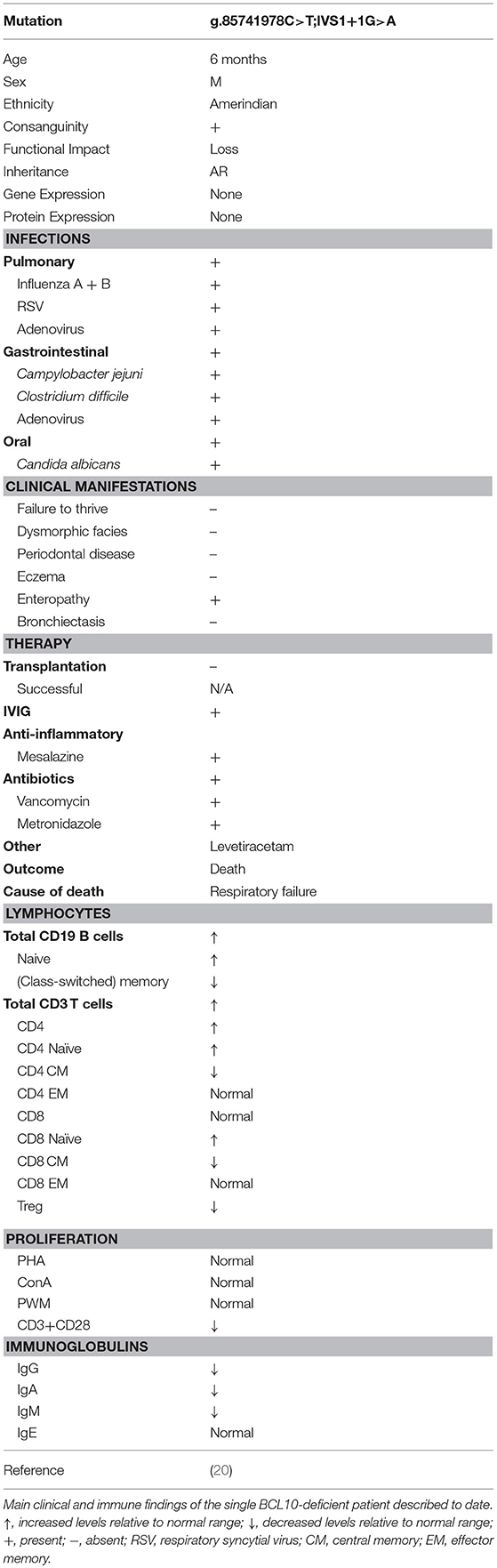
Table 6. Clinical and laboratory phenotype of human BCL10 deficiency (LOF BCL10 mutations).
Lymphocyte counts were generally normal, but B and T cells mostly displayed a naïve phenotype with an associated reduction in memory B and T cells and a profound absence of Tregs (Table 6). In keeping with the naïve phenotype, the patient also displayed hypogammaglobulinemia. Interestingly, in contrast to murine studies, patient myeloid cells responded normally to innate ligands (125–127), while fibroblasts displayed impaired NF-κB activation in response to Toll-like receptor (TLR)2/6, TLR4, and Dectin-1 stimulation as measured by NF-κB nuclear translocation and cytokine secretion (20). In addition, patient T cells displayed an impaired proliferative response to antigen receptor ligation (but not mitogen stimulation), and this was paired with a significant reduction in the expression of activation markers ICOS and CD25. Contrary to murine studies (14), CD69 expression was upregulated normally by T cells.
Diagnosis and Treatment of BCL10 Deficiency
BCL10 deficiency should be considered if a patient is found to have broad immune defects/CID affecting both innate (fibroblasts) and adaptive immunity (B and T cells), especially if a patient presents with severe inflammatory gastrointestinal (GI) and respiratory disease. Diagnostic clues include hypogammaglobulinemia, absent Tregs, and the presence of mostly naïve B and T cells with reduced memory compartments. Sequencing of BCL10 is likely to confirm a diagnosis, although functional assessment of novel BCL10 variants may be needed to definitively link the variant to the clinical phenotype. At this time, since only a single patient has been described and he died at the age of three from respiratory failure, validated treatment options remain unclear. However, based on our understanding of BCL10 biology, an allogeneic hematopoietic stem cell transplant would be anticipated to restore immune function by normalizing BCL10 protein expression and function in cells of hematopoietic origin.
MALT1
Role of MALT1 in Immunity
MALT1 paracaspase (also known as mucosa-associated lymphoid tissue lymphoma translocation protein 1) was first identified from MALT lymphomas possessing the chromosomal breakpoint t(11;18)(q21;q21) (128–131). This led to the formation of an oncogenic fusion protein of MALT1 with inhibitor of apoptosis (IAP2) called API2-MALT1. It was later found that API2-MALT1 was capable of interacting with BCL10 and potently inducing NF-κB activation (7).
MALT1 is a ~92 kDa protein, which consists of an N-terminal death domain (DD), three immunoglobulin-like domains (Ig), and a caspase-like (paracaspase) domain (Figure 1). As mentioned in the BCL10 section, MALT1 exists in a complex with BCL10 (7), and together they associate with a variety of CARD proteins in response to stimulation to form the family of CBM complexes. This makes MALT1 an important regulator of both innate and adaptive immunity. Initially believed to act mostly as a scaffold for the recruitment of other NF-κB signaling proteins (e.g., TRAF6), MALT1 is now appreciated to also have important proteolytic activity (at mostly arginine residues), allowing it to cleave substrates involved in the regulation of NF-κB, JNK, mTORC1, and more [reviewed in (114, 132)] (Figure 2). There are currently a total of ten validated MALT1 paracaspase substrates: A20 (133), BCL10 (134), CYLD (135), RelB (136), Regnase-1 (137, 138), Roquin-1/2 (138), MALT1 (139), HOIL1 (140–142), NIK (143), and LIMA1α (144), with more likely to be discovered. By cleaving these substrates, MALT1 can positively regulate canonical NF-κB (A20), JNK (CYLD), DNA binding of RelA and c-Rel (RelB), and mRNA stability (Regnase-1 and Roquin-1/2). However, MALT1 protease activity may also negatively regulate NF-κB activity (HOIL1). How MALT1 paracaspase activity fine-tunes immune function in different cellular contexts is an area of intense research activity.
In order to better understand the physiological roles of MALT1, two important murine models have been generated: the Malt1−/− mouse and the MALT1 paracaspase dead/mutated mouse (Malt1PD/PD) (Table 1). Malt1−/− mice share many features with Card11−/− and Bcl10−/− mice, including having generally normal total numbers of B and T cells, diminished innate B cells (MZ and B1), severely impaired Treg numbers, and panhypogammaglobulinemia paired with compromised T-dependent antibody responses (16, 88–91). In response to stimulation, both B and T cells proliferate poorly, with T cells being more profoundly impacted as measured by the activation of NF-κB, JNK, p38, and the upregulation of activation markers. In addition, Malt1−/− mice had absent germinal center B cells with an associated decrease in T follicular helper (Tfh) cells. Together, these studies confirmed that MALT1 is an essential regulator of T cell activation and Treg development. The contribution of MALT1 to B cell activation remains less clear.
Malt1PD/PD mice on the other hand, had intact MALT1 protein expression and scaffolding activity, but abrogated paracaspase function (89, 91–93). Surprisingly, these mice developed spontaneous multi-organ inflammation, including autoimmune gastritis, which was not seen in MALT1-deficient mice. Interestingly, immune findings between the two mouse models were quite similar, including decreased MZ, B1 cells, Tregs, and proliferation, although these phenotypes were less pronounced in Malt1PD/PD mice. In contrast to its scaffolding function, protease activity was mostly dispensable for NF-κB and JNK activation. Interestingly, Malt1PD/PD mice possessed expanded Th1, Th2, Th17 phenotypes, CD4+ and CD8+ effector T cells, and elevated IFN-γ, IgE, and IgG. These studies highlighted the unique contributions of MALT1 scaffolding and paracaspase functions in signaling and lymphocyte development; for example, IKK and JNK activation were dependent on the scaffolding role rather than protease activity. In particular, it seems proteolytic activity is important for the development of anti-inflammatory Tregs as well as controlling excessive IFN-γ secretion and accumulation of effector T cells (93). More studies are needed to understand the exact factors and cell populations mediating this inflammatory phenotype. It is possible that this inflammatory phenotype may be mediated in part by the lack of Tregs and the inability of MALT1 to cleave HOIL1 to turn off NF-κB activation (114).
Loss-of-Function MALT1 Mutations Causing Combined Immunodeficiency
Germline loss-of-function mutations in MALT1 cause CID (OMIM 615468). To date, six cases of MALT1 deficiency have been reported (21–24) (Figure 1 and Table 7). This includes a 4-year-old girl and 2-year-old boy who were both homozygous for the p.Ser89Ile mutation (21), a 15-year-old Kurdish-Canadian girl homozygous for the p.Trp580Ser mutation (22), a 1-month-old boy who was compound heterozygous for c.[1019-2A>G];[1060delC] mutations (23), and a 4-year-old boy and 7-year-old girl who were both homozygous for the p.Asp184Tyr mutation (24). All patients were identified by next generation sequencing techniques; the majority were found by WES (5/6 patients) and one case was discovered by whole genome sequencing (21). Most patients were born to consanguineous parents (4/6 patients) and possessed homozygous mutations, while one patient possessed de novo compound heterozygous mutations (23). These mutations span the length of the MALT1 protein, including the DD (21), the first Ig-like domain (24), the paracaspase domain (23), and the third Ig-like domain (22) and generally caused no MALT1 protein to be expressed (Figure 1).
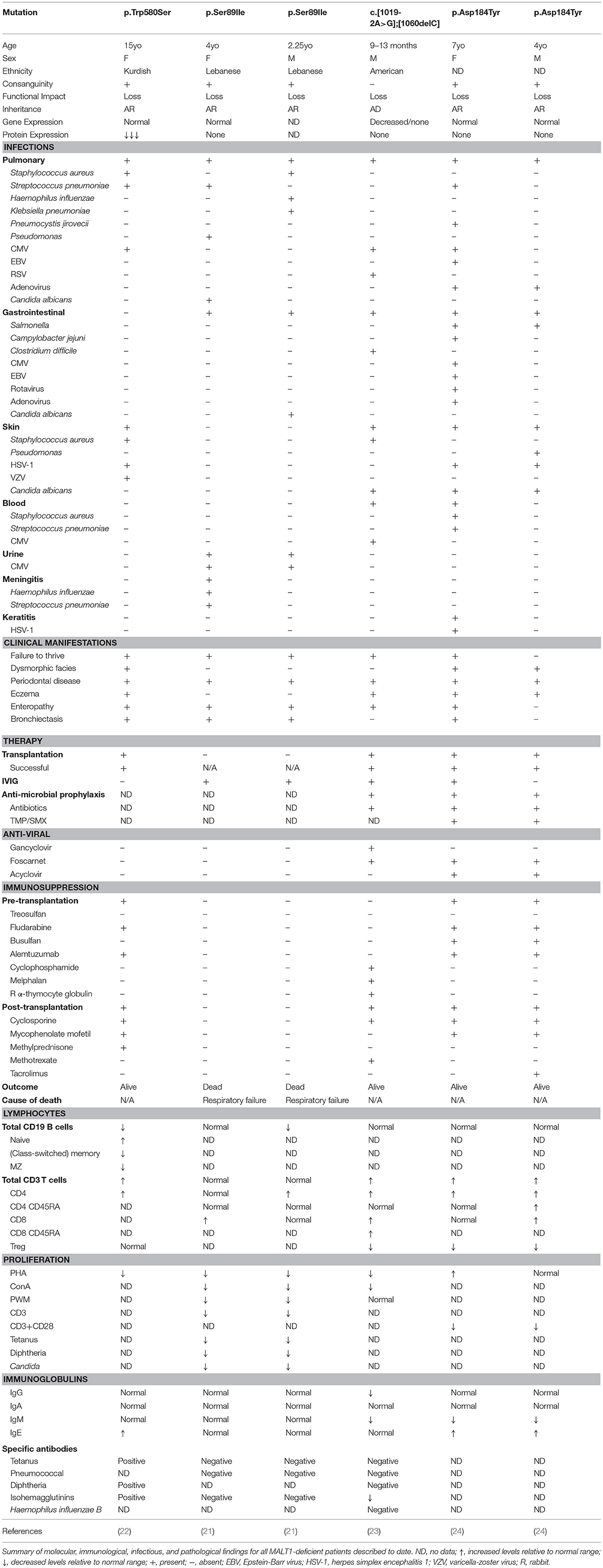
Table 7. Clinical and laboratory phenotype of human MALT1 deficiency (LOF MALT1 mutations).
MALT1 deficiency is characterized by recurrent sinopulmonary infections, enteropathy, eczema, periodontal disease, and failure to thrive (6). Indeed, patients typically presented with recurrent bacterial, viral, and fungal infections affecting the lungs (6/6 patients), skin (3/6 patients), and GI tract (3/6 patients) (Table 7). However, some patients experienced bloodborne infections, including one patient who had Staphylococcus aureus and Streptococcus pneumoniae bacteremia (24) and another who had CMV viremia (23). One of the patients also had meningitis positive for Streptococcus pneumoniae and Haemophilus influenzae (21).
Periodontal disease (6/6 patients) was common to all patients, with many developing aphthous ulcers, cheilitis, gingivitis, and thrush (21–24, 145). In addition, both dermatitis (4/6 patients) and inflammatory GI disease (5/6 patients) were frequently reported findings. Consequently, significant T cell infiltration in the skin and or the GI tract was found in biopsies (21–24). Developmentally, half of the patients had abnormal facial features (although these may be related to inflammatory changes affecting the oral cavity) (22, 24) and the majority had failure to thrive (5/6 patients).
Some patients had additional unique presentations of disease. The patient carrying the p.Trp580Ser was found to have very low bone density and suffered from fractures due to low-impact injuries (22). She also recurrently generated granulation tissue on her vocal cords, larynx, and external auditory canal. In addition, the two patients carrying the p.Ser89Ile mutation also developed mastoiditis (21).
Immunophenotyping of these MALT1-deficient patients found generally normal (21, 23, 24) or decreased (21, 22) B cell numbers. Interestingly, in contrast to other patients, the p.Trp580Ser mutation was associated with a developmental block in their B cell compartment characterized by absent MZ B cells, reduced transitional and class-switched memory B cells, and elevated naïve B cells (22). Despite relatively normal B cell populations, only the p.Ser89Ile siblings had normal serum immunoglobulin titers (21), while half of the patients possessed diminished IgM (23, 24) and elevated IgE (22, 24). On the other hand, CD3+ and CD4+ T cells were found to be expanded in most patients (4/6 patients) with the exception of the p.Ser89Ile siblings (21) who were within the normal range. CD8+ T cells were mostly elevated (3/5 patients) or within the normal range. Similar to CARD11- and BCL10-deficient patients, these patients also generally had diminished Tregs (3/4 patients). All MALT1-deficient patient T cells showed impaired proliferation in response to PHA or α-CD3/CD28 stimulation. In line with impaired T cell responses, most patients also possessed poor vaccine antibody titers (3/4 patients) (21–23).
Biochemical characterization of patient cells demonstrated completely abrogated NF-κB phosphorylation and/or IκBα degradation, along with diminished IL-2 secretion (21–24). In addition, McKinnon et al. was also able to demonstrate impaired paracaspase activity as measured by BCL10 cleavage (22). Using these MALT1-deficient patient cells, the same group discovered the novel MALT1 substrate HOIL1 (140). This defined a novel negative regulatory role for MALT1 in NF-κB signaling, where, by cleaving HOIL1, linear ubiquitination-mediated signaling and inflammation is decreased/turned off. It is possible that in MALT1-deficient patients, the loss of MALT1 proteolytic activity on HOIL1 leads to an accumulation in linear ubiquitination, resulting in unrestricted NF-κB activation and chronic inflammation, thus contributing to the exaggerated skin and mucosal inflammation seen in MALT1-deficient patients (140).
Diagnosis and Treatment of MALT1 Deficiency
All cases of MALT1 deficiency described to date have been discovered by next generation sequencing. However, based on this small cohort of patients, there are some diagnostic clues that raise suspicion for MALT1 deficiency (Table 8). Specifically, MALT1 deficiency should be considered in patients who present with the majority of the following: (i) severe recurrent sinopulmonary infections positive for bacteria or viruses, (ii) severe inflammatory GI disease, (iii) eczematous rash, (iv) severe periodontal disease, and (v) failure to thrive. Diagnostic testing “red flags” include finding relatively normal lymphocyte and B cell numbers, expanded CD3+ and CD4+ T cell subsets, impaired T cell proliferation, and compromised NF-κB phosphorylation, IκBα degradation, and IL-2 secretion.
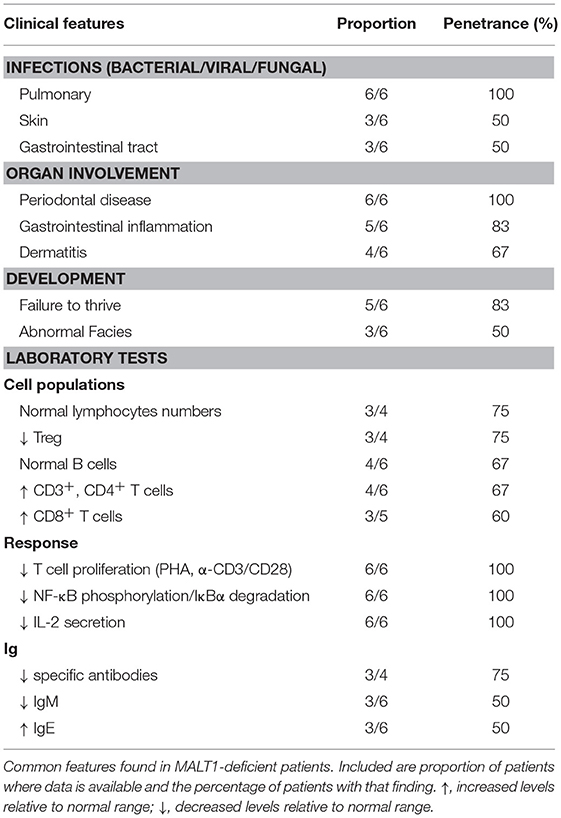
Table 8. “Red flags” suggestive of human MALT1 deficiency.
MALT1 deficiency can be cured by hematopoietic stem cell transplantation (4/6 patients received successful transplants) (23, 24, 146). Highlighting the value of curative transplantation, the siblings homozygous for the p.Ser89Ile were not transplanted and they continued to experience persistent infections until their eventual deaths due to respiratory failure at the ages of 7 and 13.5 years (21). Successful donor choices have included: bone marrow from an HLA-matched sibling (p.Trp580Ser patient) (146); peripheral stem cells from two unrelated 10/10 HLA-matched donors (p.Asp184Tyr siblings) (24); and peripheral stem cells from an unrelated 9/10 HLA-matched donor (c.[1019-2A>G];[1060delC] patient) (23). Despite successful engraftment being achieved in these patients, there were some noteworthy post-transplantation complications. The p.Trp580Ser patient experienced a range of infections including CMV, Epstein-Barr virus (EBV), VZV, and herpes simplex virus-1 (HSV-1), adenovirus viremia, Staphylococcus aureus bacteremia, Klebsiella pneumoniae pneumonia, extended spectrum beta-lactamase positive Streptococcus pneumoniae and Escherichia coli, BK virus-associated hemorrhagic cystitis, and rotavirus-associated gastroenteritis (146). The c.[1019-2A>G];[1060delC] patient developed diarrhea and CMV viremia (23). The p.Asp184Tyr siblings developed transient CMV viremia and the younger brother developed an adenovirus infection as well as bacterial pneumonia (24).
CBM Mutations in Relation to Other Primary Atopic TCR-Mediated Disorders
Germline MALT1 and CARD11 mutations can be considered primary atopic disorders in that they are associated with early-onset, severe atopic disease, amongst other comorbidities (99). Primary atopic disorders are often associated with primary immunodeficiency, most commonly caused by mutations in cytokine signaling or disruptions in TCR signaling or repertoire. Atopy is hypothesized to be caused by the propensity of naïve CD4 T cells to skew toward Th2 differentiation when relatively weak TCR signals are delivered (147). While CBM-associated mutations are the most directly linked to TCR signaling, other disruptions which can indirectly impact TCR signaling to cause atopic disease include actin cytoskeleton remodeling genes such as dedicator of cytokinesis 8 (DOCK8) deficiency (148), Wiskott-Aldrich Syndrome (WAS) protein interacting protein (WIP) deficiency (149), Wiskott-Aldrich Syndrome (150), actin related protein 2/3 (ARP2/3) complex mutations (151–153) and, potentially, ZAP70 deficiency (154, 155). The clinical presentation of DOCK8 deficiency is somewhat similar to CBM mutation-associated atopy, though infectious and neoplastic manifestations are more severe. WIP-, WAS- and ARP2/3-associated disease have more systemic manifestations including thrombocytopenia likely due to more broad protein expression patterns. In addition, the clearest TCR repertoire defect associated with atopic disease is Omenn syndrome, which is caused by hypomorphic mutations in most genes associated with SCID [including one CARD11-deficient case (19)], and results in an oligoclonal expansion of CD4 T cells. This, in turn, leads to severe dermatitis, elevated IgE, eosinophilia, and lymphoproliferation.
Comparing and Contrasting CBM-Opathies: Unanswered Questions
In adaptive immunity, the CBM complex functions in a highly synergistic manner. In line with this, CARD11-, BCL10-, and MALT1-deficient patients share many features, including having CID/SCID with normal total B and T cell numbers, aberrant B and T cell subsets, little-to-no Tregs, impaired T cell proliferation, and recurrent bacterial/viral infections (17–24). As a group, patients with these CBM-opathies have established that the CBM complex is a critical regulator of human Treg development and tolerance; however, the exact mechanisms by which this occurs are not completely understood. In murine models, it is thought that the ability of the CBM complex to modulate TCR signal strength and transduce signals downstream of the IL-2R contribute significantly to this process (33, 88, 89, 92, 93, 156).
While CARD11 is mostly restricted to hematopoietic cells, BCL10 and MALT1 have much broader cellular expression and associate with other CARD proteins downstream of a diverse assortment of receptors (66). Thus, individual CBM-opathies each have their own unique features (Figure 3). For example, CARD11-deficient patients characteristically exhibit panhypogammaglobulinemia, which is not present in MALT1 or BCL10 deficiency. In contrast, all CBM-deficient mice have panhypogammaglobulinemia. This demonstrates both important differences between mouse models and human patients as well as an incomplete understanding of how the CBM complex regulates antibody production. Another clinical feature that varies between the CBM-opathies is susceptibility to Pneumocystis jirovecii pneumonia (PJP). PJP is a very common infection in CARD11 deficiency (reported in 75% of identified patients) but is not a reported pathogen in MALT1 and BCL10 deficiency. It is still not known why Pneumocystis jirovecii seems to preferentially infect CARD11-deficient patients.
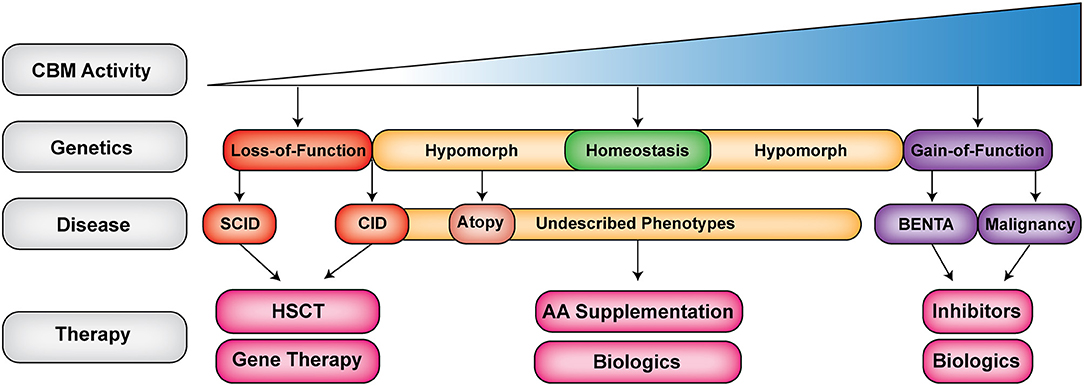
Figure 3. The expanding clinical spectrum of CBM-opathies. Shown is a gradient of CBM activity caused by germline mutations. Activity ranges from absent to hyperactive CBM activity. Red indicates loss-of-function (LOF) mutations (CARD11, BCL10, and MALT1 deficiencies), yellow indicates hypomorphic mutations that do not completely abrogate signaling leading to combined immunodeficiency and atopy as well as novel emerging phenotypes (DN LOF CARD11), purple indicates gain-of-function (GOF) mutations that can lead to BENTA (GOF CARD11) or malignancy (somatic GOF in CBM). Biologics refer to antibodies, which target cells, cytokines or cell surface receptors. SCID, severe combined immunodeficiency; CID, combined immunodeficiency; AA, amino acid; HSCT, hematopoietic stem cell transplantation.
Moving beyond infections, both MALT1- and BCL10-deficient patients presented simultaneously with immunodeficiency and immune dysregulation, where in addition to recurrent infections, they also developed inflammatory gastrointestinal disease (20–24). These features were not present in CARD11-deficient patients, nor were they found in Card11−/−, Malt1−/−, and Bcl10−/− mice. In fact, the pharmacological inhibition of MALT1 in dextran sulfate solution (DSS)-induced colitis was found to be protective through the inhibition of NF-κB and NLRP3 inflammasome activation in macrophages (157), and a T cell-dependent transfer model of autoimmune colitis found that Malt1−/− T cells were unable to induce colitis (92). However, it is important to note that Malt1PD/PD mice developed inflammatory gastrointestinal disease, and this was associated with an expansion of Th1, Th2, and Th17, with increased inflammatory cytokines and IgE, which was not present in the Malt1−/− mice (89, 91–93). It is tempting to speculate that the diminished Tregs, dysregulated tolerance, and lack of MALT1 paracaspase regulation collectively mediated the inflammatory phenotype in patients. In addition, both MALT1-deficient and BCL10-deficient patients displayed elevated CD3+ and CD4+ T cells, but it is not known whether there was any skewing in Th1 and Th2 responses (as is the case in Malt1PD/PD mice), which could be significant pro-inflammatory cytokine secretors, thus contributing to pathology. It should also be noted that MALT1 and BCL10 have wider expression than CARD11 and some non-hematopoietic cells in these patients could be contributing to the gastrointestinal pathology.
GOF and DN LOF CARD11 mutations give rise to considerably different phenotypes from CBM-deficiencies, with GOF manifesting chiefly as selective B cell lymphocytosis and DN LOF leading to severe atopic disease. Deeper mechanistic studies are needed to address several outstanding questions, including (i) how GOF CARD11 mutations dampen T cell responsiveness, (ii) whether GOF CARD11 mutations enhance JNK and mTORC1 signaling, and how this contributes to differential T and B cell responses, (iii) if/how DN CARD11 mutations ultimately skew Th2 responses via decreased NF-κB signaling and/or restricted CARD11-dependent glutamine uptake, and (iv) whether DN CARD11 mutations affect B cell intrinsic signaling, including the fate of class-switched IgE+ B cells. Overall, shared phenotypes in BENTA and CADINS patients (e.g., poor antibody responses, increased respiratory and skin infections) emphasize the requirement for properly “tuned” CBM signaling to ensure proper B and T cell differentiation in response to antigens in order to maintain immune homeostasis.
Novel Therapeutic Insights Emerging From Our Understanding of Human CBM-Opathies
CBM-opathies have been invaluable in enhancing our understanding of how dysregulated CBM complex signaling contributes to the pathogenesis of various diseases including immunodeficiency, atopic disease (94), autoimmunity (158), and malignancies (159). Given the role of the CBM complex in a range of human pathologies, there is considerable interest in developing and studying therapeutics that can target/ameliorate these diseases. In the realm of cancer, targeting either the CBM complex or the catalytic function of MALT1 have been the methods of choice (160). In particular, MALT1 inhibitors have received a great deal of attention for their specificity and efficiency. These inhibitors may eventually be promising options for treating cancers and diseases that have a lymphoproliferative component, including BENTA (104). However, given the central position of the CBM complex in signaling, inhibition should be approached with caution. Here, LOF mutations in individual CBM components and the recent characterization of Malt1PD/PD mice have been uniquely informative in highlighting possible side effects that can arise from the therapeutic inhibition of the CBM complex, including decreasing Tregs and tolerance (114).
Currently, the treatment of complete CARD11, BCL10, and MALT1 deficiencies relies upon hematopoietic stem cell transplantation in order to functionally normalize immune function (with immunoglobulin replacement and prophylactic antimicrobials used as supportive therapy). Without transplantation, the survival rate is very low. Moving forward, autologous gene therapy may be an attractive therapeutic option, whereby patient hematopoietic stem cells could be “corrected” by genetic approaches (e.g., viral transduction or CRISPR/Cas9 editing) and re-infused to give rise to a normal immune system (161). In support of this approach, transplantation outcomes have been quite good for CBM deficiency patients (17, 146) and the artificial expression of WT genes in patient cells is able to rescue NF-κB activation (22). Further proof of concept studies in mice or patient stem cells will have to be done to determine efficacy and safety.
The initial description of Card11un/un mice (13) paired with the recent discovery of DN LOF mutations in CARD11 causing atopy and immunodeficiency (31, 32), implicated the CBM complex in the pathogenesis of allergic disease. Affected patients were found to have decreased upregulation of ASCT2 and impaired mTORC1 signaling, which is thought to contribute to Th2 skewing (32). Since it was previously shown that impaired mTORC1 signaling and Th1 differentiation could be rescued by glutamine supplementation (64), Ma et al. tested whether glutamine supplementation could rescue the phenotype of patient cells. Interestingly, this was able to partially rescue signaling defects (32). This demonstrated that modulating immune metabolism through amino acid supplementation could be useful for therapy. Indeed, glutamine supplementation is currently being explored in low birth weight infants for the reduction of atopic dermatitis and has shown some success (162).
Concluding Remarks
The CBM complex is an essential molecular bridge linking cell surface antigen receptor signaling with downstream activation of NF-κB, JNK, and mTORC1. This makes it a critical regulator of lymphocyte activation, differentiation, proliferation, maintenance, and metabolism. Since the discovery of germline loss-of-function mutations in CARD11 causing SCID just 5 years ago (17), ~48 patients with genetically confirmed CBM-opathies have been described. Germline mutations in this complex have led to an impressive spectrum of diseases, ranging from CID/SCID to CID with atopy to BENTA disease (Figure 3). The detailed study of these rare patients with CBM-opathies has provided unique insights into how the CBM complex regulates human immune reactivity and tolerance. Ultimately, the discovery and characterization of more CBM-opathies will not only benefit the affected patients but will broadly inform any future therapeutic targeting of these signaling pathways in cancer, autoimmunity, and allergic disease.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported in part by the Canadian Institutes of Health Research (CIHR), Genome British Columbia (SIP007 to ST), the National Institutes of Health (NIH) (R21AI109187 to AS), the Canadian Allergy, Asthma and Immunology Foundation, and the BC Children's Hospital Foundation. ST also holds the Aubrey J. Tingle Professorship in Pediatric Immunology and is a clinical scholar of the Michael Smith Foundation for Health Research. HL is supported by the Shaughnessy Hospital Volunteer Society Fellowship in Healthcare, Theodore E Arnold Fellowship, and University of British Columbia Faculty of Medicine Graduate Award.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Picard C, Bobby Gaspar H, Al-Herz W, Bousfiha A, Casanova JL, Chatila T, et al. International union of immunological societies: 2017 primary immunodeficiency diseases committee report on inborn errors of immunity. J Clin Immunol. (2018) 38:96–128. doi: 10.1007/s10875-017-0464-9
2. Tak PP, Firestein GS. NF-kappaB: a key role in inflammatory diseases. J Clin Invest. (2001) 107:7–11. doi: 10.1172/JCI11830
3. Karin M. NF-kappaB as a critical link between inflammation and cancer. Cold Spring Harb Perspect Biol. (2009) 1:a000141. doi: 10.1101/cshperspect.a000141
4. Sun SC, Chang JH, Jin J. Regulation of nuclear factor-kappaB in autoimmunity. Trends Immunol. (2013) 34:282–9. doi: 10.1016/j.it.2013.01.004
5. Zhang Q, Lenardo MJ, Baltimore D. 30 Years of NF-kappaB: A Blossoming of relevance to human pathobiology. Cell (2017) 168:37–57. doi: 10.1016/j.cell.2016.12.012
6. Turvey SE, Durandy A, Fischer A, Fung SY, Geha RS, Gewies A, et al. The CARD11-BCL10-MALT1 (CBM) signalosome complex: Stepping into the limelight of human primary immunodeficiency. J Allergy Clin Immunol. (2014) 134:276–84. doi: 10.1016/j.jaci.2014.06.015
7. Lucas PC, Yonezumi M, Inohara N, McAllister-Lucas LM, Abazeed ME, Chen FF, et al. Bcl10 and MALT1, independent targets of chromosomal translocation in malt lymphoma, cooperate in a novel NF-kappa B signaling pathway. J Biol Chem. (2001)276:19012–9. doi: 10.1074/jbc.M009984200
8. Gaide O, Martinon F, Micheau O, Bonnet D, Thome M, Tschopp J. Carma1, a CARD-containing binding partner of Bcl10, induces Bcl10 phosphorylation and NF-kappaB activation. FEBS Lett. (2001) 496:121–7. doi: 10.1016/S0014-5793(01)02414-0
9. Lenz G, Davis RE, Ngo VN, Lam L, George TC, Wright GW, et al. Oncogenic CARD11 mutations in human diffuse large B cell lymphoma. Science. (2008) 319:1676–9. doi: 10.1126/science.1153629
10. Davis RE, Ngo VN, Lenz G, Tolar P, Young RM, Romesser PB, et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature. (2010) 463:88–92. doi: 10.1038/nature08638
11. Hara H, Wada T, Bakal C, Kozieradzki I, Suzuki S, Suzuki N, et al. The MAGUK family protein CARD11 is essential for lymphocyte activation. Immunity. (2003)18:763–75. doi: 10.1016/S1074-7613(03)00148-1
12. Egawa T, Albrecht B, Favier B, Sunshine MJ, Mirchandani K, O'Brien W, et al. Requirement for CARMA1 in antigen receptor-induced NF-kappa B activation and lymphocyte proliferation. Curr Biol. (2003) 13:1252–8. doi: 10.1016/S0960-9822(03)00491-3
13. Jun JE, Wilson LE, Vinuesa CG, Lesage S, Blery M, Miosge LA, et al. Identifying the MAGUK protein Carma-1 as a central regulator of humoral immune responses and atopy by genome-wide mouse mutagenesis. Immunity. (2003) 18:751–62. doi: 10.1016/S1074-7613(03)00141-9
14. Ruland J, Duncan GS, Elia A, del Barco Barrantes I, Nguyen L, Plyte S, et al. Bcl10 is a positive regulator of antigen receptor-induced activation of NF-kappaB and neural tube closure. Cell. (2001) 104:33–42. doi: 10.1016/S0092-8674(01)00189-1
15. Xue L, Morris SW, Orihuela C, Tuomanen E, Cui X, Wen R, et al. Defective development and function of Bcl10-deficient follicular, marginal zone and B1 B cells. Nat Immunol. (2003)4:857–65. doi: 10.1038/ni963
16. Ruefli-Brasse AA, French DM, Dixit VM. Regulation of NF-kappaB-dependent lymphocyte activation and development by paracaspase. Science. (2003) 302:1581–4. doi: 10.1126/science.1090769
17. Stepensky P, Keller B, Buchta M, Kienzler AK, Elpeleg O, Somech R, et al. Deficiency of caspase recruitment domain family, member 11 (CARD11), causes profound combined immunodeficiency in human subjects. J Allergy Clin Immunol. (2013) 131:477–85 e1. doi: 10.1016/j.jaci.2012.11.050
18. Greil J, Rausch T, Giese T, Bandapalli OR, Daniel V, Bekeredjian-Ding I, et al. Whole-exome sequencing links caspase recruitment domain 11 (CARD11) inactivation to severe combined immunodeficiency. J Allergy Clin Immunol. (2013) 131:1376–83 e3. doi: 10.1016/j.jaci.2013.02.012
19. Fuchs S, Rensing-Ehl A, Pannicke U, Lorenz MR, Fisch P, Jeelall Y, et al. Omenn syndrome associated with a functional reversion due to a somatic second-site mutation in CARD11 deficiency. Blood (2015) 126:1658–69. doi: 10.1182/blood-2015-03-631374
20. Torres JM, Martinez-Barricarte R, Garcia-Gomez S, Mazariegos MS, Itan Y, Boisson B, et al. Inherited BCL10 deficiency impairs hematopoietic and nonhematopoietic immunity. J Clin Invest. (2014) 124:5239–48. doi: 10.1172/JCI77493
21. Jabara HH, Ohsumi T, Chou J, Massaad MJ, Benson H, Megarbane A, et al. A homozygous mucosa-associated lymphoid tissue 1 (MALT1) mutation in a family with combined immunodeficiency. J Allergy Clin Immunol. (2013) 132:151–8. doi: 10.1016/j.jaci.2013.04.047
22. McKinnon ML, Rozmus J, Fung SY, Hirschfeld AF, Del Bel KL, Thomas L, et al. Combined immunodeficiency associated with homozygous MALT1 mutations. J Allergy Clin Immunol. (2014)133:1458–62, 62 e1-7. doi: 10.1016/j.jaci.2013.10.045
23. Punwani D, Wang H, Chan AY, Cowan MJ, Mallott J, Sunderam U, et al. Combined immunodeficiency due to MALT1 mutations, treated by hematopoietic cell transplantation. J Clin Immunol. (2015) 35:135–46. doi: 10.1007/s10875-014-0125-1
24. Charbit-Henrion F, Jeverica AK, Begue B, Markelj G, Parlato M, Avcin SL, et al. Deficiency in mucosa-associated lymphoid tissue lymphoma translocation 1: a novel cause of IPEX-Like Syndrome. J Pediatr Gastroenterol Nutr. (2017) 64:378–84. doi: 10.1097/MPG.0000000000001262
25. Biggs CM, Lu HY, Turvey SE. Monogenic immune disorders and severe atopic disease. Nat Genet. (2017) 49:1162–3. doi: 10.1038/ng.3925
26. Snow AL, Xiao W, Stinson JR, Lu W, Chaigne-Delalande B, Zheng L, et al. Congenital B cell lymphocytosis explained by novel germline CARD11 mutations. J Exp Med. (2012) 209:2247–61. doi: 10.1084/jem.20120831
27. Buchbinder D, Stinson JR, Nugent DJ, Heurtier L, Suarez F, Sukumar G, et al. Mild B-cell lymphocytosis in patients with a CARD11 C49Y mutation. J Allergy Clin Immunol. (2015) 136:819–21 e1. doi: 10.1016/j.jaci.2015.03.008
28. Brohl AS, Stinson JR, Su HC, Badgett T, Jennings CD, Sukumar G, et al. Germline CARD11 mutation in a patient with severe congenital B Cell Lymphocytosis. J Clin Immunol. (2015) 35:32–46. doi: 10.1007/s10875-014-0106-4
29. Gupta M, Aluri J, Desai M, Lokeshwar M, Taur P, Lenardo M, et al. Clinical, Immunological, and molecular findings in four cases of B Cell expansion with NF-κB and T Cell anergy disease for the first time from India. Front Immunol. (2018) 9:1049. doi: 10.3389/fimmu.2018.01049
30. Outinen T, Syrjanen J, Rounioja S, Saarela J, Kaustio M, Helminen M. Constant B cell lymphocytosis since early age in a patient with CARD11 mutation: a 20-year follow-up. Clin Immunol. (2016) 165:19–20. doi: 10.1016/j.clim.2016.02.002
31. Dadi H, Jones TA, Merico D, Sharfe N, Ovadia A, Schejter Y, et al. Combined immunodeficiency and atopy caused by a dominant negative mutation in caspase activation and recruitment domain family member 11 (CARD11). J Allergy Clin Immunol. (2018) 141:1818–30 e2. doi: 10.1016/j.jaci.2017.06.047
32. Ma CA, Stinson JR, Zhang Y, Abbott JK, Weinreich MA, Hauk PJ, et al. Germline hypomorphic CARD11 mutations in severe atopic disease. Nat Genet. (2017) 49:1192–201. doi: 10.1038/ng.3898
33. Roche MI, Ramadas RA, Medoff BD. The role of CARMA1 in T cells. Crit Rev Immunol. (2013) 33:219–43. doi: 10.1615/CritRevImmunol.2013007056
34. Meininger I, Krappmann D. Lymphocyte signaling and activation by the CARMA1-BCL10-MALT1 signalosome. Biol Chem. (2016) 397:1315–33. doi: 10.1515/hsz-2016-0216
35. Hu H, Sun SC. Ubiquitin signaling in immune responses. Cell Res. (2016) 26:457–83. doi: 10.1038/cr.2016.40
36. Bojarczuk K, Bobrowicz M, Dwojak M, Miazek N, Zapala P, Bunes A, et al. B-cell receptor signaling in the pathogenesis of lymphoid malignancies. Blood Cells Mol Dis. (2015) 55:255–65. doi: 10.1016/j.bcmd.2015.06.016
37. Woyach JA, Johnson AJ, Byrd JC. The B-cell receptor signaling pathway as a therapeutic target in CLL. Blood (2012) 120:1175–84. doi: 10.1182/blood-2012-02-362624
38. Huse M. The T-cell-receptor signaling network. J Cell Sci. (2009) 122:1269–73. doi: 10.1242/jcs.042762
39. Matsumoto R, Wang D, Blonska M, Li H, Kobayashi M, Pappu B, et al. Phosphorylation of CARMA1 plays a critical role in T Cell receptor-mediated NF-kappaB activation. Immunity (2005) 23:575–85. doi: 10.1016/j.immuni.2005.10.007
40. Sommer K, Guo B, Pomerantz JL, Bandaranayake AD, Moreno-Garcia ME, Ovechkina YL, et al. Phosphorylation of the CARMA1 linker controls NF-kappaB activation. Immunity (2005) 23:561–74. doi: 10.1016/j.immuni.2005.09.014
41. Qiao Q, Yang C, Zheng C, Fontan L, David L, Yu X, et al. Structural architecture of the CARMA1/Bcl10/MALT1 signalosome: nucleation-induced filamentous assembly. Mol Cell (2013) 51:766–79. doi: 10.1016/j.molcel.2013.08.032
42. David L, Li Y, Ma J, Garner E, Zhang X, Wu H. Assembly mechanism of the CARMA1-BCL10-MALT1-TRAF6 signalosome. Proc Natl Acad Sci USA. (2018) 115:1499–504. doi: 10.1073/pnas.1721967115
43. Che T, You Y, Wang D, Tanner MJ, Dixit VM, Lin X. MALT1/paracaspase is a signaling component downstream of CARMA1 and mediates T cell receptor-induced NF-kappaB activation. J Biol Chem. (2004) 279:15870–6. doi: 10.1074/jbc.M310599200
44. Sun L, Deng L, Ea CK, Xia ZP, Chen ZJ. The TRAF6 ubiquitin ligase and TAK1 kinase mediate IKK activation by BCL10 and MALT1 in T lymphocytes. Mol Cell (2004) 14:289–301. doi: 10.1016/S1097-2765(04)00236-9
45. Noels H, van Loo G, Hagens S, Broeckx V, Beyaert R, Marynen P, et al. A Novel TRAF6 binding site in MALT1 defines distinct mechanisms of NF-kappaB activation by API2middle dotMALT1 fusions. J Biol Chem. (2007) 282:10180–9. doi: 10.1074/jbc.M611038200
46. Zhou H, Wertz I, O'Rourke K, Ultsch M, Seshagiri S, Eby M, et al. Bcl10 activates the NF-kappaB pathway through ubiquitination of NEMO. Nature (2004) 427:167–71. doi: 10.1038/nature02273
47. Oeckinghaus A, Wegener E, Welteke V, Ferch U, Arslan SC, Ruland J, et al. Malt1 ubiquitination triggers NF-kappaB signaling upon T-cell activation. EMBO J. (2007) 26:4634–45. doi: 10.1038/sj.emboj.7601897
48. Shambharkar PB, Blonska M, Pappu BP, Li H, You Y, Sakurai H, et al. Phosphorylation and ubiquitination of the IkappaB kinase complex by two distinct signaling pathways. EMBO J. (2007) 26:1794–805. doi: 10.1038/sj.emboj.7601622
49. Yang Y, Schmitz R, Mitala J, Whiting A, Xiao W, Ceribelli M, et al. Essential role of the linear ubiquitin chain assembly complex in lymphoma revealed by rare germline polymorphisms. Cancer Discov. (2014) 4:480–93. doi: 10.1158/2159-8290.CD-13-0915
50. Dubois SM, Alexia C, Wu Y, Leclair HM, Leveau C, Schol E, et al. A catalytic-independent role for the LUBAC in NF-kappaB activation upon antigen receptor engagement and in lymphoma cells. Blood (2014) 123:2199–203. doi: 10.1182/blood-2013-05-504019
51. Satpathy S, Wagner SA, Beli P, Gupta R, Kristiansen TA, Malinova D, et al. Systems-wide analysis of BCR signalosomes and downstream phosphorylation and ubiquitylation. Mol Syst Biol. (2015) 11:810. doi: 10.15252/msb.20145880
52. Sato S, Sanjo H, Tsujimura T, Ninomiya-Tsuji J, Yamamoto M, Kawai T, et al. TAK1 is indispensable for development of T cells and prevention of colitis by the generation of regulatory T cells. Int Immunol. (2006) 18:1405–11. doi: 10.1093/intimm/dxl082
53. Schuman J, Chen Y, Podd A, Yu M, Liu HH, Wen R, et al. A critical role of TAK1 in B-cell receptor-mediated nuclear factor kappaB activation. Blood (2009) 113:4566–74. doi: 10.1182/blood-2008-08-176057
54. Wu CJ, Ashwell JD. NEMO recognition of ubiquitinated Bcl10 is required for T cell receptor-mediated NF-kappaB activation. Proc Natl Acad Sci USA. (2008) 105:3023–8. doi: 10.1073/pnas.0712313105
55. Blonska M, Lin X. CARMA1-mediated NF-kappaB and JNK activation in lymphocytes. Immunol Rev. (2009) 228:199–211. doi: 10.1111/j.1600-065X.2008.00749.x
56. Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell (2000) 103:239–52. doi: 10.1016/S0092-8674(00)00116-1
57. Liu HH, Xie M, Schneider MD, Chen ZJ. Essential role of TAK1 in thymocyte development and activation. Proc Natl Acad Sci USA. (2006) 103:11677–82. doi: 10.1073/pnas.0603089103
58. Wan YY, Chi H, Xie M, Schneider MD, Flavell RA. The kinase TAK1 integrates antigen and cytokine receptor signaling for T cell development, survival and function. Nat Immunol. (2006) 7:851–8. doi: 10.1038/ni1355
59. Blonska M, Pappu BP, Matsumoto R, Li H, Su B, Wang D, et al. The CARMA1-Bcl10 signaling complex selectively regulates JNK2 kinase in the T cell receptor-signaling pathway. Immunity (2007) 26:55–66. doi: 10.1016/j.immuni.2006.11.008
60. Saxton RA, Sabatini DM. mTOR Signaling in growth, metabolism, and disease. Cell (2017) 169:361–71. doi: 10.1016/j.cell.2017.03.035
61. Pollizzi KN, Powell JD. Regulation of T cells by mTOR: the known knowns and the known unknowns. Trends Immunol. (2015) 36:13–20. doi: 10.1016/j.it.2014.11.005
62. Yang K, Chi H. mTOR and metabolic pathways in T cell quiescence and functional activation. Semin Immunol. (2012) 24:421–8. doi: 10.1016/j.smim.2012.12.004
63. Chi H. Regulation and function of mTOR signalling in T cell fate decisions. Nat Rev Immunol. (2012) 12:325–38. doi: 10.1038/nri3198
64. Nakaya M, Xiao Y, Zhou X, Chang JH, Chang M, Cheng X, et al. Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity (2014) 40:692–705. doi: 10.1016/j.immuni.2014.04.007
65. Hamilton KS, Phong B, Corey C, Cheng J, Gorentla B, Zhong X, et al. T cell receptor-dependent activation of mTOR signaling in T cells is mediated by Carma1 and MALT1, but not Bcl10. Sci Signal. (2014) 7:ra55. doi: 10.1126/scisignal.2005169
66. Blonska M, Lin X. NF-kappaB signaling pathways regulated by CARMA family of scaffold proteins. Cell Res. (2011) 21:55–70. doi: 10.1038/cr.2010.182
67. Drummond RA, Franco LM, Lionakis MS. Human CARD9: a critical molecule of fungal immune surveillance. Front Immunol. (2018) 9:1836. doi: 10.3389/fimmu.2018.01836
68. Jordan CT, Cao L, Roberson ED, Duan S, Helms CA, Nair RP, et al. Rare and common variants in CARD14, encoding an epidermal regulator of NF-kappaB, in psoriasis. Am J Hum Genet. (2012) 90:796–808. doi: 10.1016/j.ajhg.2012.03.013
69. Jordan CT, Cao L, Roberson ED, Pierson KC, Yang CF, Joyce CE, et al. PSORS2 is due to mutations in CARD14. Am J Hum Genet. (2012) 90:784–95. doi: 10.1016/j.ajhg.2012.03.012
70. Fuchs-Telem D, Sarig O, van Steensel MA, Isakov O, Israeli S, Nousbeck J, et al. Familial pityriasis rubra pilaris is caused by mutations in CARD14. Am J Hum Genet. (2012) 91:163–70. doi: 10.1016/j.ajhg.2012.05.010
71. Malarkannan S, Regunathan J, Chu H, Kutlesa S, Chen Y, Zeng H, et al. Bcl10 plays a divergent role in NK cell-mediated cytotoxicity and cytokine generation. J Immunol. (2007) 179:3752–62. doi: 10.4049/jimmunol.179.6.3752
72. Gross O, Grupp C, Steinberg C, Zimmermann S, Strasser D, Hannesschlager N, et al. Multiple ITAM-coupled NK-cell receptors engage the Bcl10/Malt1 complex via Carma1 for NF-kappaB and MAPK activation to selectively control cytokine production. Blood (2008) 112:2421–8. doi: 10.1182/blood-2007-11-123513
73. Van Nuffel E, Schmitt A, Afonina IS, Schulze-Osthoff K, Beyaert R, Hailfinger S. CARD14-Mediated activation of paracaspase MALT1 in keratinocytes: implications for psoriasis. J Invest Dermatol. (2017) 137:569–75. doi: 10.1016/j.jid.2016.09.031
74. Sun J. CARMA3: A novel scaffold protein in regulation of NF-kappaB activation and diseases. World J Biol Chem. (2010) 1:353–61. doi: 10.4331/wjbc.v1.i12.353
75. Bertin J, Wang L, Guo Y, Jacobson MD, Poyet JL, Srinivasula SM, et al. CARD11 and CARD14 are novel caspase recruitment domain (CARD)/membrane-associated guanylate kinase (MAGUK) family members that interact with BCL10 and activate NF-kappa B. J Biol Chem. (2001) 276:11877–82. doi: 10.1074/jbc.M010512200
76. Rawlings DJ, Sommer K, Moreno-Garcia ME. The CARMA1 signalosome links the signalling machinery of adaptive and innate immunity in lymphocytes. Nat Rev Immunol. (2006) 6:799–812. doi: 10.1038/nri1944
77. Wang D, You Y, Case SM, McAllister-Lucas LM, Wang L, DiStefano PS, et al. A requirement for CARMA1 in TCR-induced NF-kappa B activation. Nat Immunol. (2002) 3:830–5. doi: 10.1038/ni824
78. Gaide O, Favier B, Legler DF, Bonnet D, Brissoni B, Valitutti S, et al. CARMA1 is a critical lipid raft-associated regulator of TCR-induced NF-kappa B activation. Nat Immunol. (2002) 3:836–43. doi: 10.1038/ni830
79. Pomerantz JL, Denny EM, Baltimore D. CARD11 mediates factor-specific activation of NF-kappaB by the T cell receptor complex. EMBO J. (2002) 21:5184–94. doi: 10.1093/emboj/cdf505
80. Newton K, Dixit VM. Mice lacking the CARD of CARMA1 exhibit defective B lymphocyte development and impaired proliferation of their B and T lymphocytes. Curr Biol. (2003) 13:1247–51. doi: 10.1016/S0960-9822(03)00458-5
81. Medoff BD, Sandall BP, Landry A, Nagahama K, Mizoguchi A, Luster AD, et al. Differential requirement for CARMA1 in agonist-selected T-cell development. Eur J Immunol. (2009) 39:78–84. doi: 10.1002/eji.200838734
82. Molinero LL, Cubre A, Mora-Solano C, Wang Y, Alegre ML. T cell receptor/CARMA1/NF-kappaB signaling controls T-helper (Th) 17 differentiation. Proc Natl Acad Sci USA. (2012) 109:18529–34. doi: 10.1073/pnas.1204557109
83. Blonska M, Joo D, Zweidler-McKay PA, Zhao Q, Lin X. CARMA1 controls Th2 cell-specific cytokine expression through regulating JunB and GATA3 transcription factors. J Immunol. (2012) 188:3160–8. doi: 10.4049/jimmunol.1102943
84. Pappu BP, Lin X. Potential role of CARMA1 in CD40-induced splenic B cell proliferation and marginal zone B cell maturation. Eur J Immunol. (2006) 36:3033–43. doi: 10.1002/eji.200535663
85. Altin JA, Tian L, Liston A, Bertram EM, Goodnow CC, Cook MC. Decreased T-cell receptor signaling through CARD11 differentially compromises forkhead box protein 3-positive regulatory versus T(H)2 effector cells to cause allergy. J Allergy Clin Immunol. (2011) 127:1277–85 e5. doi: 10.1016/j.jaci.2010.12.1081
86. Schmidt-Supprian M, Tian J, Grant EP, Pasparakis M, Maehr R, Ovaa H, et al. Differential dependence of CD4+CD25+ regulatory and natural killer-like T cells on signals leading to NF-kappaB activation. Proc Natl Acad Sci USA. (2004) 101:4566–71. doi: 10.1073/pnas.0400885101
87. Jost PJ, Weiss S, Ferch U, Gross O, Mak TW, Peschel C, et al. Bcl10/Malt1 signaling is essential for TCR-induced NF-kappaB activation in thymocytes but dispensable for positive or negative selection. J Immunol. (2007) 178:953–60. doi: 10.4049/jimmunol.178.2.953
88. Ruland J, Duncan GS, Wakeham A, Mak TW. Differential requirement for Malt1 in T and B cell antigen receptor signaling. Immunity (2003) 19:749–58. doi: 10.1016/S1074-7613(03)00293-0
89. Bornancin F, Renner F, Touil R, Sic H, Kolb Y, Touil-Allaoui I, et al. Deficiency of MALT1 paracaspase activity results in unbalanced regulatory and effector T and B cell responses leading to multiorgan inflammation. J Immunol. (2015) 194:3723–34. doi: 10.4049/jimmunol.1402254
90. Brustle A, Brenner D, Knobbe CB, Lang PA, Virtanen C, Hershenfield BM, et al. The NF-kappaB regulator MALT1 determines the encephalitogenic potential of Th17 cells. J Clin Invest. (2012) 122:4698–709. doi: 10.1172/JCI63528
91. Yu JW, Hoffman S, Beal AM, Dykon A, Ringenberg MA, Hughes AC, et al. MALT1 protease activity is required for innate and adaptive immune responses. PLoS ONE (2015) 10:e0127083. doi: 10.1371/journal.pone.0127083
92. Jaworski M, Marsland BJ, Gehrig J, Held W, Favre S, Luther SA, et al. Malt1 protease inactivation efficiently dampens immune responses but causes spontaneous autoimmunity. EMBO J. (2014) 33:2765–81. doi: 10.15252/embj.201488987
93. Gewies A, Gorka O, Bergmann H, Pechloff K, Petermann F, Jeltsch KM, et al. Uncoupling Malt1 threshold function from paracaspase activity results in destructive autoimmune inflammation. Cell Rep. (2014) 9:1292–305. doi: 10.1016/j.celrep.2014.10.044
94. Hirota T, Takahashi A, Kubo M, Tsunoda T, Tomita K, Sakashita M, et al. Genome-wide association study identifies eight new susceptibility loci for atopic dermatitis in the Japanese population. Nat Genet. (2012) 44:1222–6. doi: 10.1038/ng.2438
95. Medoff BD, Seed B, Jackobek R, Zora J, Yang Y, Luster AD, et al. CARMA1 is critical for the development of allergic airway inflammation in a murine model of asthma. J Immunol. (2006) 176:7272–7. doi: 10.4049/jimmunol.176.12.7272
96. Ramadas RA, Roche MI, Moon JJ, Ludwig T, Xavier RJ, Medoff BD. CARMA1 is necessary for optimal T cell responses in a murine model of allergic asthma. J Immunol. (2011) 187:6197–207. doi: 10.4049/jimmunol.1101348
97. Lu HY, Sharma M, Biggs CM, Huang YH, Shopsowitz KE, Frosk P, et al. The importance of functional validation after next-generation sequencing: evaluation of a novel CARD11 variant. Pediatr Allergy Immunol. (2018) 29:663–8. doi: 10.1111/pai.12930
98. Dorjbal B, Stinson JR, Ma CA, Weinreich MA, Miraghazadeh B, Hartberger JM, et al. (in press). Hypomorphic CARD11 mutations associated with diverse immunologic phenotypes with or without atopic disease. J. Allergy Clin. Immunol. doi: 10.1016/j.jaci.2018.08.013
99. Lyons JJ, Milner JD. Primary atopic disorders. J Exp Med. (2018) 215:1009–22. doi: 10.1084/jem.20172306
100. Manka LA, Wechsler ME. New biologics for allergic diseases. Expert Rev Clin Immunol. (2018) 14:285–96. doi: 10.1080/1744666X.2018.1459188
101. Darte JM, McClure PD, Saunders EF, Weber JL, Donohue WL. Congenital lymphoid hyperplasia with persistent hyperlymphocytosis. N Engl J Med. (1971) 284:431–2. doi: 10.1056/NEJM197102252840807
102. Arjunaraja S, Snow AL. Gain-of-function mutations and immunodeficiency: at a loss for proper tuning of lymphocyte signaling. Curr Opin Allergy Clin Immunol. (2015) 15:533–8. doi: 10.1097/ACI.0000000000000217
103. Buchbinder D, Sassoon A. A case of bad Carma! Blood (2017) 129:1737. doi: 10.1182/blood-2016-12-756007
104. Arjunaraja S, Angelus P, Su HC, Snow AL. Impaired control of epstein-barr virus infection in B-cell expansion with NF-kappaB and T-Cell anergy disease. Front Immunol. (2018) 9:198. doi: 10.3389/fimmu.2018.00198
105. Perez E, Bonilla FA, Orange JS, Ballow M. Specific antibody deficiency: controversies in diagnosis and management. Front Immunol. (2017) 8:586. doi: 10.3389/fimmu.2017.00586
106. Arjunaraja S, Nose BD, Sukumar G, Lott NM, Dalgard CL, Snow AL. Intrinsic plasma cell differentiation defects in B cell expansion with NF-kappaB and T Cell anergy patient B cells. Front Immunol. (2017) 8:913. doi: 10.3389/fimmu.2017.00913
107. Jeelall YS, Wang JQ, Law HD, Domaschenz H, Fung HK, Kallies A, et al. Human lymphoma mutations reveal CARD11 as the switch between self-antigen-induced B cell death or proliferation and autoantibody production. J Exp Med. (2012) 209:1907–17. doi: 10.1084/jem.20112744
108. Chan W, Schaffer TB, Pomerantz JL. A quantitative signaling screen identifies CARD11 mutations in the CARD and LATCH domains that induce Bcl10 ubiquitination and human lymphoma cell survival. Mol Cell Biol. (2013) 33:429–43. doi: 10.1128/MCB.00850-12
109. Jattani RP, Tritapoe JM, Pomerantz JL. Intramolecular interactions and regulation of cofactor binding by the four repressive elements in the caspase recruitment domain-containing protein 11 (CARD11) inhibitory domain. J Biol Chem. (2016) 291:8338–48. doi: 10.1074/jbc.M116.717322
110. Jattani RP, Tritapoe JM, Pomerantz JL. Cooperative control of caspase recruitment domain-containing protein 11 (CARD11) signaling by an unusual array of redundant repressive elements. J Biol Chem. (2016) 291:8324–36. doi: 10.1074/jbc.M115.683714
111. Sasaki Y, Derudder E, Hobeika E, Pelanda R, Reth M, Rajewsky K, et al. Canonical NF-kappaB activity, dispensable for B cell development, replaces BAFF-receptor signals and promotes B cell proliferation upon activation. Immunity (2006) 24:729–39. doi: 10.1016/j.immuni.2006.04.005
112. Krishna S, Xie D, Gorentla B, Shin J, Gao J, Zhong XP. Chronic activation of the kinase IKKbeta impairs T cell function and survival. J Immunol. (2012) 189:1209–19. doi: 10.4049/jimmunol.1102429
113. Forbes SA, Beare D, Boutselakis H, Bamford S, Bindal N, Tate J, et al. COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res. (2017) 45:D777–83. doi: 10.1093/nar/gkw1121
114. Demeyer A, Staal J, Beyaert R. Targeting MALT1 Proteolytic activity in immunity, inflammation and disease: Good or Bad? Trends Mol Med. (2016) 22:135–50. doi: 10.1016/j.molmed.2015.12.004
115. Lee CH, Bae SJ, Kim M. Mucosa-associated lymphoid tissue lymphoma translocation 1 as a novel therapeutic target for rheumatoid arthritis. Sci Rep. (2017) 7:11889. doi: 10.1038/s41598-017-12349-9
116. Saba NS, Wong DH, Tanios G, Iyer JR, Lobelle-Rich P, Dadashian EL, et al. MALT1 inhibition is efficacious in both naive and ibrutinib-resistant chronic lymphocytic leukemia. Cancer Res. (2017) 77:7038–48. doi: 10.1158/0008-5472.CAN-17-2485
117. Bardet M, Unterreiner A, Malinverni C, Lafossas F, Vedrine C, Boesch D, et al. The T-cell fingerprint of MALT1 paracaspase revealed by selective inhibition. Immunol Cell Biol. (2018) 96:81–99. doi: 10.1111/imcb.1018
118. Willis TG, Jadayel DM, Du MQ, Peng H, Perry AR, Abdul-Rauf M, et al. Bcl10 is involved in t(1;14)(p22;q32) of MALT B cell lymphoma and mutated in multiple tumor types. Cell (1999) 96:35–45. doi: 10.1016/S0092-8674(00)80957-5
119. Costanzo A, Guiet C, Vito P. c-E10 is a caspase-recruiting domain-containing protein that interacts with components of death receptors signaling pathway and activates nuclear factor-kappaB. J Biol Chem. (1999) 274:20127–32. doi: 10.1074/jbc.274.29.20127
120. Koseki T, Inohara N, Chen S, Carrio R, Merino J, Hottiger MO, et al. CIPER, a novel NF kappaB-activating protein containing a caspase recruitment domain with homology to Herpesvirus-2 protein E10. J Biol Chem. (1999) 274:9955–61. doi: 10.1074/jbc.274.15.9955
121. Bertin J, Guo Y, Wang L, Srinivasula SM, Jacobson MD, Poyet JL, et al. CARD9 is a novel caspase recruitment domain-containing protein that interacts with BCL10/CLAP and activates NF-kappa B. J Biol Chem. (2000) 275:41082–6. doi: 10.1074/jbc.C000726200
122. Wang L, Guo Y, Huang WJ, Ke X, Poyet JL, Manji GA, et al. Card10 is a novel caspase recruitment domain/membrane-associated guanylate kinase family member that interacts with BCL10 and activates NF-kappa B. J Biol Chem. (2001) 276:21405–9. doi: 10.1074/jbc.M102488200
123. McAllister-Lucas LM, Inohara N, Lucas PC, Ruland J, Benito A, Li Q, et al. Bimp1, a MAGUK family member linking protein kinase C activation to Bcl10-mediated NF-kappaB induction. J Biol Chem. (2001) 276:30589–97. doi: 10.1074/jbc.M103824200
124. Gehring T, Seeholzer T, Krappmann D. BCL10 – Bridging CARDs to immune activation. Front Immunol. (2018) 9:1539. doi: 10.3389/fimmu.2018.01539
125. Liu Y, Dong W, Chen L, Xiang R, Xiao H, De G, et al. BCL10 mediates lipopolysaccharide/toll-like receptor-4 signaling through interaction with Pellino2. J Biol Chem. (2004) 279:37436–44. doi: 10.1074/jbc.M400241200
126. Gross O, Gewies A, Finger K, Schafer M, Sparwasser T, Peschel C, et al. Card9 controls a non-TLR signalling pathway for innate anti-fungal immunity. Nature (2006) 442:651–6. doi: 10.1038/nature04926
127. Gringhuis SI, Wevers BA, Kaptein TM, van Capel TM, Theelen B, Boekhout T, et al. Selective C-Rel activation via Malt1 controls anti-fungal T(H)-17 immunity by dectin-1 and dectin-2. PLoS Pathog. (2011) 7:e1001259. doi: 10.1371/journal.ppat.1001259
128. Dierlamm J, Baens M, Wlodarska I, Stefanova-Ouzounova M, Hernandez JM, Hossfeld DK, et al. The apoptosis inhibitor gene API2 and a novel 18q gene, MLT, are recurrently rearranged in the t(11;18)(q21;q21) associated with mucosa-associated lymphoid tissue lymphomas. Blood (1999) 93:3601–9.
129. Akagi T, Motegi M, Tamura A, Suzuki R, Hosokawa Y, Suzuki H, et al. A novel gene, MALT1 at 18q21, is involved in t(11;18) (q21;q21) found in low-grade B-cell lymphoma of mucosa-associated lymphoid tissue. Oncogene (1999) 18:5785–94. doi: 10.1038/sj.onc.1203018
130. Morgan JA, Yin Y, Borowsky AD, Kuo F, Nourmand N, Koontz JI, et al. Breakpoints of the t(11;18)(q21;q21) in mucosa-associated lymphoid tissue (MALT) lymphoma lie within or near the previously undescribed gene MALT1 in chromosome 18. Cancer Res. (1999) 59:6205–13.
131. Rosebeck S, Rehman AO, Lucas PC, McAllister-Lucas LM. From MALT lymphoma to the CBM signalosome: three decades of discovery. Cell Cycle (2011) 10:2485–96. doi: 10.4161/cc.10.15.16923
132. Afonina IS, Elton L, Carpentier I, Beyaert R. MALT1–a universal soldier: multiple strategies to ensure NF-kappaB activation and target gene expression. FEBS J. (2015) 282:3286–97. doi: 10.1111/febs.13325
133. Coornaert B, Baens M, Heyninck K, Bekaert T, Haegman M, Staal J, et al. T cell antigen receptor stimulation induces MALT1 paracaspase-mediated cleavage of the NF-kappaB inhibitor A20. Nat Immunol. (2008) 9:263–71. doi: 10.1038/ni1561
134. Rebeaud F, Hailfinger S, Posevitz-Fejfar A, Tapernoux M, Moser R, Rueda D, et al. The proteolytic activity of the paracaspase MALT1 is key in T cell activation. Nat Immunol. (2008) 9:272–81. doi: 10.1038/ni1568
135. Staal J, Driege Y, Bekaert T, Demeyer A, Muyllaert D, Van Damme P, et al. T-cell receptor-induced JNK activation requires proteolytic inactivation of CYLD by MALT1. EMBO J. (2011) 30:1742–52. doi: 10.1038/emboj.2011.85
136. Hailfinger S, Nogai H, Pelzer C, Jaworski M, Cabalzar K, Charton JE, et al. Malt1-dependent RelB cleavage promotes canonical NF-kappaB activation in lymphocytes and lymphoma cell lines. Proc Natl Acad Sci USA. (2011) 108:14596–601. doi: 10.1073/pnas.1105020108
137. Uehata T, Iwasaki H, Vandenbon A, Matsushita K, Hernandez-Cuellar E, Kuniyoshi K, et al. Malt1-induced cleavage of regnase-1 in CD4(+) helper T cells regulates immune activation. Cell (2013) 153:1036–49. doi: 10.1016/j.cell.2013.04.034
138. Jeltsch KM, Hu D, Brenner S, Zoller J, Heinz GA, Nagel D, et al. Cleavage of roquin and regnase-1 by the paracaspase MALT1 releases their cooperatively repressed targets to promote T(H)17 differentiation. Nat Immunol. (2014) 15:1079–89. doi: 10.1038/ni.3008
139. Baens M, Bonsignore L, Somers R, Vanderheydt C, Weeks SD, Gunnarsson J, et al. MALT1 auto-proteolysis is essential for NF-kappaB-dependent gene transcription in activated lymphocytes. PLoS ONE (2014) 9:e103774. doi: 10.1371/journal.pone.0103774
140. Klein T, Fung SY, Renner F, Blank MA, Dufour A, Kang S, et al. The paracaspase MALT1 cleaves HOIL1 reducing linear ubiquitination by LUBAC to dampen lymphocyte NF-kappaB signalling. Nat Commun. (2015) 6:8777. doi: 10.1038/ncomms9777
141. Douanne T, Gavard J, Bidere N. The paracaspase MALT1 cleaves the LUBAC subunit HOIL1 during antigen receptor signaling. J Cell Sci. (2016) 129:1775–80. doi: 10.1242/jcs.185025
142. Elton L, Carpentier I, Staal J, Driege Y, Haegman M, Beyaert R. MALT1 cleaves the E3 ubiquitin ligase HOIL-1 in activated T cells, generating a dominant negative inhibitor of LUBAC-induced NF-kappaB signaling. FEBS J. (2016) 283:403–12. doi: 10.1111/febs.13597
143. Rosebeck S, Madden L, Jin X, Gu S, Apel IJ, Appert A, et al. Cleavage of NIK by the API2-MALT1 fusion oncoprotein leads to noncanonical NF-kappaB activation. Science (2011) 331:468–72. doi: 10.1126/science.1198946
144. Nie Z, Du MQ, McAllister-Lucas LM, Lucas PC, Bailey NG, Hogaboam CM, et al. Conversion of the LIMA1 tumour suppressor into an oncogenic LMO-like protein by API2-MALT1 in MALT lymphoma. Nat Commun. (2015) 6:5908. doi: 10.1038/ncomms6908
145. Tsang P, Derkson G, Priddy R, Junker AK, Slots J, Larjava H. Severe periodontitis in a 5-year-old girl with hyperimmunoglobulin E syndrome. Pediatr Dent. (2005) 27:68–73.
146. Rozmus J, McDonald R, Fung SY, Del Bel KL, Roden J, Senger C, et al. Successful clinical treatment and functional immunological normalization of human MALT1 deficiency following hematopoietic stem cell transplantation. Clin Immunol. (2016) 168:1–5. doi: 10.1016/j.clim.2016.04.011
147. Milner JD. TCR Signaling abnormalities in human Th2-associated atopic disease. Front Immunol. (2018) 9:719. doi: 10.3389/fimmu.2018.00719
148. Zhang Q, Davis JC, Lamborn IT, Freeman AF, Jing H, Favreau AJ, et al. Combined immunodeficiency associated with DOCK8 mutations. N Engl J Med. (2009) 361:2046–55. doi: 10.1056/NEJMoa0905506
149. Lanzi G, Moratto D, Vairo D, Masneri S, Delmonte O, Paganini T, et al. A novel primary human immunodeficiency due to deficiency in the WASP-interacting protein WIP. J Exp Med. (2012) 209:29–34. doi: 10.1084/jem.20110896
150. Ochs HD. Mutations of the Wiskott-Aldrich Syndrome Protein affect protein expression and dictate the clinical phenotypes. Immunol Res. (2009) 44:84–8. doi: 10.1007/s12026-008-8084-3
151. Kuijpers TW, Tool ATJ, van der Bijl I, de Boer M, van Houdt M, de Cuyper IM, et al. Combined immunodeficiency with severe inflammation and allergy caused by ARPC1B deficiency. J Allergy Clin Immunol. (2017) 140:273–7 e10. doi: 10.1016/j.jaci.2016.09.061
152. Kahr WH, Pluthero FG, Elkadri A, Warner N, Drobac M, Chen CH, et al. Loss of the Arp2/3 complex component ARPC1B causes platelet abnormalities and predisposes to inflammatory disease. Nat Commun. (2017) 8:14816. doi: 10.1038/ncomms14816
153. Somech R, Lev A, Lee YN, Simon AJ, Barel O, Schiby G, et al. Disruption of Thrombocyte and T Lymphocyte Development by a Mutation in ARPC1B. J Immunol. (2017) 199:4036–45. doi: 10.4049/jimmunol.1700460
154. Turul T, Tezcan I, Artac H, de Bruin-Versteeg S, Barendregt BH, Reisli I, et al. Clinical heterogeneity can hamper the diagnosis of patients with ZAP70 deficiency. Eur J Pediatr. (2009) 168:87–93. doi: 10.1007/s00431-008-0718-x
155. Karaca E, Karakoc-Aydiner E, Bayrak OF, Keles S, Sevli S, Barlan IB, et al. Identification of a novel mutation in ZAP70 and prenatal diagnosis in a Turkish family with severe combined immunodeficiency disorder. Gene. (2013) 512:189–93. doi: 10.1016/j.gene.2012.10.062
156. Lee AJ, Wu X, Cheng H, Zhou X, Cheng X, Sun SC. CARMA1 regulation of regulatory T cell development involves modulation of interleukin-2 receptor signaling. J Biol Chem. (2010) 285:15696–703. doi: 10.1074/jbc.M109.095190
157. Liu W, Guo W, Hang N, Yang Y, Wu X, Shen Y, et al. MALT1 inhibitors prevent the development of DSS-induced experimental colitis in mice via inhibiting NF-kappaB and NLRP3 inflammasome activation. Oncotarget (2016) 7:30536–49. doi: 10.18632/oncotarget.8867
158. International Multiple Sclerosis Genetics C, Wellcome Trust Case Control C, Sawcer S, Hellenthal G, Pirinen M, Spencer CC, et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature. (2011) 476:214–9. doi: 10.1038/nature10251
159. Juilland M, Thome M. Role of the CARMA1/BCL10/MALT1 complex in lymphoid maliignancies. Curr Opin Hematol. (2016) 23:402–9. doi: 10.1097/MOH.0000000000000257
160. Yang C, David L, Qiao Q, Damko E, Wu H. The CBM signalosome: potential therapeutic target for aggressive lymphoma? Cytokine Growth Factor Rev. (2014) 25:175–83. doi: 10.1016/j.cytogfr.2013.12.008
161. Thrasher AJ, Williams DA. Evolving gene therapy in primary immunodeficiency. Mol Ther. (2017) 25:1132–41. doi: 10.1016/j.ymthe.2017.03.018
Keywords: CBM complex, CARD11, MALT1, BCL10, combined immunodeficiency, severe combined immunodeficiency, primary atopic disease, BENTA
Citation: Lu HY, Bauman BM, Arjunaraja S, Dorjbal B, Milner JD, Snow AL and Turvey SE (2018) The CBM-opathies—A Rapidly Expanding Spectrum of Human Inborn Errors of Immunity Caused by Mutations in the CARD11-BCL10-MALT1 Complex. Front. Immunol. 9:2078. doi: 10.3389/fimmu.2018.02078
Received: 21 July 2018; Accepted: 22 August 2018;
Published: 19 September 2018.
Edited by:
Elissa Deenick, Garvan Institute of Medical Research, AustraliaReviewed by:
Ruben Martinez-Barricarte, Rockefeller University, United StatesJolan Eszter Walter, University of South Florida, United States
Copyright © 2018 Lu, Bauman, Arjunaraja, Dorjbal, Milner, Snow and Turvey. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stuart E. Turvey, sturvey@cw.bc.ca
†These authors have contributed equally to this work