 Sarah M. Carpanini
Sarah M. Carpanini Megan Torvell
Megan Torvell Bryan Paul Morgan
Bryan Paul Morgan- 1UK Dementia Research Institute, Cardiff University, Cardiff, United Kingdom
- 2Division of Infection and Immunity, School of Medicine, Systems Immunity Research Institute, Cardiff University, Cardiff, United Kingdom
The complement system plays critical roles in development, homeostasis, and regeneration in the central nervous system (CNS) throughout life; however, complement dysregulation in the CNS can lead to damage and disease. Complement proteins, regulators, and receptors are widely expressed throughout the CNS and, in many cases, are upregulated in disease. Genetic and epidemiological studies, cerebrospinal fluid (CSF) and plasma biomarker measurements and pathological analysis of post-mortem tissues have all implicated complement in multiple CNS diseases including multiple sclerosis (MS), neuromyelitis optica (NMO), neurotrauma, stroke, amyotrophic lateral sclerosis (ALS), Alzheimer's disease (AD), Parkinson's disease (PD), and Huntington's disease (HD). Given this body of evidence implicating complement in diverse brain diseases, manipulating complement in the brain is an attractive prospect; however, the blood-brain barrier (BBB), critical to protect the brain from potentially harmful agents in the circulation, is also impermeable to current complement-targeting therapeutics, making drug design much more challenging. For example, antibody therapeutics administered systemically are essentially excluded from the brain. Recent protocols have utilized “Trojan horse” techniques to transport therapeutics across the BBB or used osmotic shock or ultrasound to temporarily disrupt the BBB. Most research to date exploring the impact of complement inhibition on CNS diseases has been in animal models, and some of these studies have generated convincing data; for example, in models of MS, NMO, and stroke. There have been a few recent clinical trials of available anti-complement drugs in CNS diseases associated with BBB impairment, for example the use of the anti-C5 monoclonal antibody (mAb) eculizumab in NMO, but for most CNS diseases there have been no human trials of anti-complement therapies. Here we will review the evidence implicating complement in diverse CNS disorders, from acute, such as traumatic brain or spine injury, to chronic, including demyelinating, neuroinflammatory, and neurodegenerative diseases. We will discuss the particular problems of drug access into the CNS and explore ways in which anti-complement therapies might be tailored for CNS disease.
Introduction
The Central Nervous System (CNS) as a Distinct Environment
The CNS was, for a long time, considered an immunologically privileged organ because the brain and spinal cord are protected from circulating inflammagens by the BBB. The BBB is a specialized membrane comprised of endothelial cells with tight junctions, vascular pericytes and perivascular glia (Figure 1A), which cooperate to form a selectively permeable barrier, protecting the CNS from fluctuating ion concentrations and circulating neurotransmitters, macromolecules, large proteins such as complement, and pathogens (1). However, isolation of the CNS is not absolute and there are a number of pathways by which systemic inflammation can be communicated to the CNS [reviewed; (2)]. Indeed, the recent demonstration of a CNS lymphatic system further undermines the concept of brain immunological privilege (3).
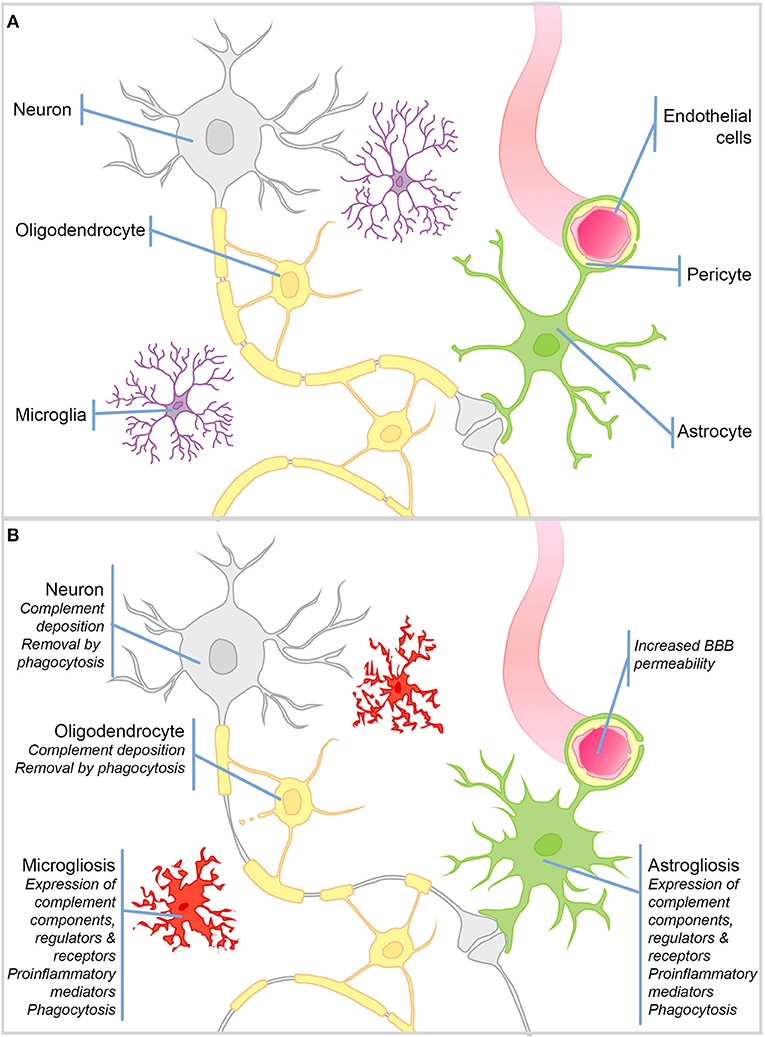
Figure 1. Schematic representation of cell types in the brain and their responses to injury. (A) Schematic representation of the cell types in the healthy brain. (B) During CNS injury and disease the BBB is compromised. There is significant microgliosis and astrogliosis, characterized by glial cell proliferation, upregulation of complement components, regulators and receptors, proinflammatory mediators, and active phagocytosis. Complement protein expression/deposition are increased on neurons and oligodendrocytes tagging them for removal by phagocytosis and driving neurodegeneration and demyelination.
The healthy BBB forms early in development and restricts the infiltration of circulating immune cells into the brain parenchyma; hence, the dominant immune cells of the brain are the resident macrophage population—microglia (Figure 1B). This self-renewing (4), yolk sac-derived population develops within the CNS (5–7) and differs in many respects from macrophage populations found in the periphery (8–10). Compared to tissue macrophages, microglia are relatively “immune suppressed” due to expression of receptors for soluble signals in the extracellular milieu, for example, β2 adrenergic receptor binding of noradrenaline (11, 12), and signals delivered through direct contact with surrounding neurons, including CD200R, CX3CR1 (13, 14). The downstream signaling of such receptors suppresses the production of proinflammatory mediators and encourages a neuroprotective microglial phenotype. Resting microglia are relatively sessile, ramified cells; their numerous highly motile protrusions sample the entire brain every few hours (15, 16). Upon stimulation, these protrusions are withdrawn to create ameboid microglia that are migratory and upregulate expression of proinflammatory mediators and activating receptors involved in pattern recognition and phagocytosis (Figure 1B). This activated phenotype, if not kept in check, can cause havoc in the vulnerable CNS. More recently, it has been recognized that there are brain region-specific subpopulations of microglia with different responses to triggers and varying degrees of immune-vigilance (17, 18). There also exist resident non-microglial populations in the healthy brain including perivascular, meningeal and choroid plexus macrophages, which are capable of responding to noxious stimuli. In addition, during pathology blood borne macrophages and other immune cells are recruited to the injured brain as a result of increased BBB permeability. Astrocytes are a neglected cell type, despite the fact that they comprise ~70% of the cells in the brain, where they form syncytial networks around neurons. During health their primary role is homeostatic; they provide neurons with energy and neurotrophic support, and buffer ion and neurotransmitter concentrations [Reviewed elsewhere by Sofroniew and Vinters (19)—Figure 1A]. During inflammation, astrocytes demonstrate their immune-competence; they produce proinflammatory cytokines, are capable of phagocytosis, and can even present antigens to adaptive immune cells; however, acquisition of these immune roles is often associated with loss of homeostatic functions [(19)—Figure 1B].
Importantly for the subject matter of this review, there is compelling evidence that, during inflammation, not only microglia and astrocytes, but also neurons, oligodendrocytes, and endothelial cells in the brain, can express complement components, receptors, and regulators.
The Complement System
Complement is recognized as an important branch of the innate immune system, providing the first line of defense against microorganisms. As complement is the subject of this issue, we will confine ourselves to a brief summary (represented in Figure 2). Complement comprises multiple recognition molecules that detect and bind target surfaces and recruit a cascade of protease enzymes and substrates, resulting in: (1) production of potent anaphylatoxins that attract and activate phagocytes; (2) formation of the lytic membrane attack complex (MAC); (3) target opsonization for phagocytosis and destruction (Figure 2). Three activation pathways, classical, lectin and alternative, converge on a common final pathway. The classical pathway is initiated by the C1 complex binding to antibody/antigen aggregates; the lectin pathway is triggered by binding of mannose-binding lectin (MBL) or ficolins to carbohydrate epitopes on targets; the alternative pathway is better considered as an amplification loop that is engaged regardless of the initial trigger. The activation pathways converge at the central C3 and C5 convertases, which generate potent anaphylatoxins C3a and C5a, C3b to opsonize surfaces facilitating phagocytosis and C5b to initiate MAC formation. The complement pathway mediates many of its effects through specific receptors on cells and is tightly controlled by regulators present on cells and in plasma, as discussed below (20).
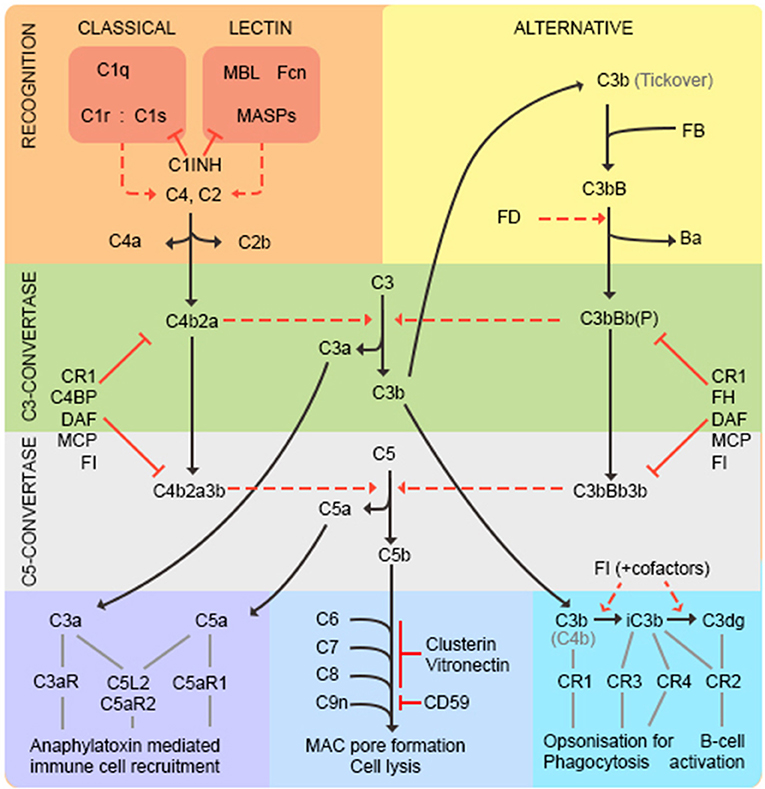
Figure 2. The complement pathway. The classical pathway is activated through antibody/antigen recognition by C1q in complex with C1r and C1s. The proteases C1r and C1s cleave C4 and C2 to generate the C3 convertase C4b2a regulated by complement receptor 1 (CR1), C4 binding protein (C4BP), decay accelerating factor (DAF), membrane cofactor protein (MCP), and factor I (FI). The lectin pathway is triggered by binding of mannose-binding lectin (MBL) or ficolins (FCN) to carbohydrate epitopes on targets. The MBL-associated serine proteases (MASPs) then cleave C4 and C2 to generate the C3-convertase as in the classical pathway. C1-inhibitor (C1INH) functions as a regulator to prevent excessive activation of both classical and lectin pathways. The alternative pathway is better considered as an amplification loop. C3b binds factor B (FB) to form C3bB. FB is cleaved by Factor D (FD) to form the C3bBb C3-convertase stabilized by properdin (P). This process is regulated by CR1, FI, factor H (FH), DAF and MCP. At this point the pathways converge—both C3-convertases cleave C3 to generate the anaphylatoxin C3a, and more C3b that binds to form the C5-convertases (C4b2a3b and C3bBb3b) that cleave C5 into C5a and C5b. C3a and C5a are potent anaphylatoxins that act through their respective receptors (C3aR, C5aR1, C5L2, and C5aR2) to recruit immune cells. C5b binds C6, C7, C8 (inhibited by vitronectin and clusterin) and multiple copies of C9 (inhibited by CD59) to form the lytic membrane attack complex (MAC). C3b opsonizes targets for phagocytosis and B-cell activation; C3b decays to iC3b then C3dg catalyzed by FI in the presence of cofactors (CR1, MCP, FH, C4BP).
The majority of complement proteins are predominantly synthesized in the liver (21, 22); however, it is becoming increasingly clear that complement proteins and their cognate receptors and regulators are expressed throughout the CNS. Most studies to date have utilized primary brain cell cultures and relevant cell lines and have identified complement expression at messenger RNA (mRNA) and/or protein level. Human primary oligodendrocytes expressed mRNA for all the components of the classical and terminal pathways and protein was detected for most of these (23). Human oligodendrocyte cells (HOG cell line) expressed the membrane complement regulators CD59, decay accelerating factor (DAF) and membrane cofactor protein (MCP) and secreted C1-inhibitor (C1INH), Vitronectin and Clusterin, whereas human astrocyte-derived cell lines expressed the same membrane regulators and the important C3/C5 fragment receptors complement receptor 1 (CR1) and C5a receptor (C5aR) (24, 25). Cultured microglia from human post-mortem brain (normal and Shy-Drager's syndrome) constitutively expressed mRNA for C1qB and C3 while C4 was expressed upon interferon (IFN)-γ stimulation (26). C4, C9, C1q, FH, C1INH, C3, C6, and Factor B (FB) were expressed in human neuronal cells in vitro whereas primary rat cerebellar granule cells expressed mRNA for C4, C1q, and C3 upon differentiation (27). Additionally, complement expression can be upregulated in disease; for example, C3, C1r and C1s expression was increased in primary microglia and astrocyte cultures from post-mortem brain upon exposure to cytokines associated with AD (28). Expression of C1INH, C1s, C1q and C3 mRNA was detected in AD and control brain extracts (29). Additionally, C1q, C3 and C4 gene expression was reported in primary microglia from AD patients (30). Cumulatively, the evidence suggests that nearly all complement proteins, regulators, and receptors are expressed in the CNS, and many are upregulated by inflammatory signals; it is therefore likely that a functional complement system is present in the CNS independent of peripheral complement.
Roles of Complement in CNS Development
Complement proteins are involved in both prenatal and postnatal development of the healthy brain. In Xenopus embryos, morpholino knockdown of C3a receptor (C3aR) or blocking antibody against C3a administered during neural tube formation cause loss of neural crest cell organization, demonstrating a role for C3a and its receptor in the migration of neural crest cells (31). Central lectin pathway components Mannan-binding lectin associated serine protease (MASP)-1 and MASP-2 are highly expressed in the developing mouse brain with MASP (and C3) knockout mice showing defects in neuronal migration suggesting critical roles for complement activation in CNS development (32).
Complement also plays key roles in postnatal brain development. In humans and rodents, removal of redundant connections by synaptic pruning during childhood and early adult life is crucial for optimal brain function (33, 34). Using the developing rodent visual system as a model for synaptic pruning, it was shown that C1q and C3 (likely C3b/iC3b) localize to, and tag, specific synapses in the dorsal lateral geniculate nucleus (dLGN) for removal during development (35). C3 deficient (−/−) mice had improved hippocampal-dependent learning and memory (36), and failed to show the age-associated synapse loss observed in wild type animals (37), suggesting that complement is detrimental to synapse health. However, in a finding illustrating the dual nature of complement, C1q−/−, C3−/−, and C4−/− mice all showed defects in synaptic pruning in the CNS that, in the former, associated with increased susceptibility to epilepsy (35, 38, 39). Furthermore, the C3b/iC3b receptor complement receptor 3 (CR3) is expressed on the surface of microglia and CR3−/− mice showed defects in microglial engulfment of synapses, suggesting a collaboration between complement and microglia in synapse elimination (40). Taken together, these data highlight a critical involvement of the classical pathway in refinement of synapse networks during normal development.
Complement and Neuroinflammation in CNS Disorders—Identifying Druggable Targets
As has been thoroughly reviewed elsewhere (41), inflammation in general and neuroinflammation in particular, is a double-edged sword, evolved to fight infection and restore or maintain homeostasis but, when uncontrolled, capable of wreaking havoc. The aim is therefore not to stop inflammation but encourage a protective rather than destructive profile and prompt resolution of inflammation. Given the important roles of complement in the developing brain, in defense against infection and in maintaining homeostasis, there may be situations where enhancing complement activity may be of benefit; however, in the context of CNS pathology, complement dysregulation leading to over-activation, has deleterious consequences and contributes to neuroinflammation.
Thus, the complement system offers an attractive drug target for these diseases that are currently without effective therapies. Drugging the complement pathway in the periphery is well researched and this knowledge could potentially be extrapolated to the CNS.
CNS disorders can be divided into acute, for example traumatic injury and stroke, and chronic, for example, demyelinating and neurodegenerative disorders, dependent on the causation, severity and duration. In the sections below, we will present evidence for the role of complement in both acute and chronic neurological disorders. We will not include stroke because this is discussed elsewhere in this issue and will also omit detailed discussion of the demyelinating diseases MS and NMO for brevity and because these have been well reviewed elsewhere (42, 43). Our overall aim is to provide insight into how complement therapeutics might impact these problematic diseases.
Complement Proteins in Acute Neurological Disorders—Traumatic Brain and Spinal Cord Injury
Traumatic Brain Injury (TBI)
TBI is classified as an injury to the brain due to trauma to the head via an external force; this can occur as a result of road traffic accidents, falls, sporting injuries or assaults; consequently, TBI is the major cause of brain injury and death in young adults in the Western world. TBI can cause diffuse or focal damage to the brain tissue and blood vessels depending on the type of injury. Subsequent to this primary injury, the BBB becomes compromised and there is a huge influx of cells, inflammatory mediators and plasma proteins, including complement proteins, that drive the delayed secondary inflammation, which is the major determinant of clinical outcome and thus recovery and survival (44). Human post-mortem TBI studies have shown increased expression of C3 and FB in brain and CSF (45) and both axonal and astrocytic expression of Clusterin (46). Increased soluble C5b-9 (terminal complement complex; TCC) levels were found in CSF after TBI, positively correlating with the degree of BBB damage (47), and further increased in response to secondary insults (oxygen deprivation/seizures) (48).
A wide array of TBI models are utilized in animal research, including cryoinjury, controlled cortical impact and standardized weight drop [models are reviewed elsewhere; (44)]. Increased C3 deposition, Clusterin and MAC deposition were observed alongside increased microglial and astrocytic activation markers after cortical contusion in the rat (49). Serum proteomics in Sprague-Dawley rats after “severe” deep cortical impact reported increased C9 and FB within the first few days after TBI (50). Complement deficient mouse models have been used to identify the impact of complement on neuropathology after TBI (Table 1). C3−/− and C5−/− mice showed reduced neutrophil extravasation upon traumatic brain cryoinjury (51); C4−/− mice, but not C3−/− or C1q−/− mice showed reduced motor deficits and tissue damage following controlled cortical impact (52). In the same TBI model, CR2/CR1−/− mice showed improved outcome with decreased mortality, neuronal cell death, C3 deposition, astrogliosis, and microgliosis (53). FB−/− mice also showed reduced cell death in TBI with increased anti-apoptotic and decreased pro-apoptotic markers (54). In one study, MBL−/− mice were protected from neurological injury following TBI (70); in contrast, another reported that MBL−/− mice showed increased levels of degenerating neurons in the hippocampus CA3 region and impaired performance in non-spatial learning tasks (71). Despite several such inconsistencies, the studies to date suggest that deficiencies of individual complement proteins of the classical, alternative or terminal pathway improves outcome after TBI. However, all these studies used rodent models where the relevant protein is knocked out systemically from embryogenesis. Thus, it is important to identify whether anti-complement therapeutics administered immediately post-injury can have a similar beneficial effect, and to define the “therapeutic window of opportunity” for intervention post-injury.
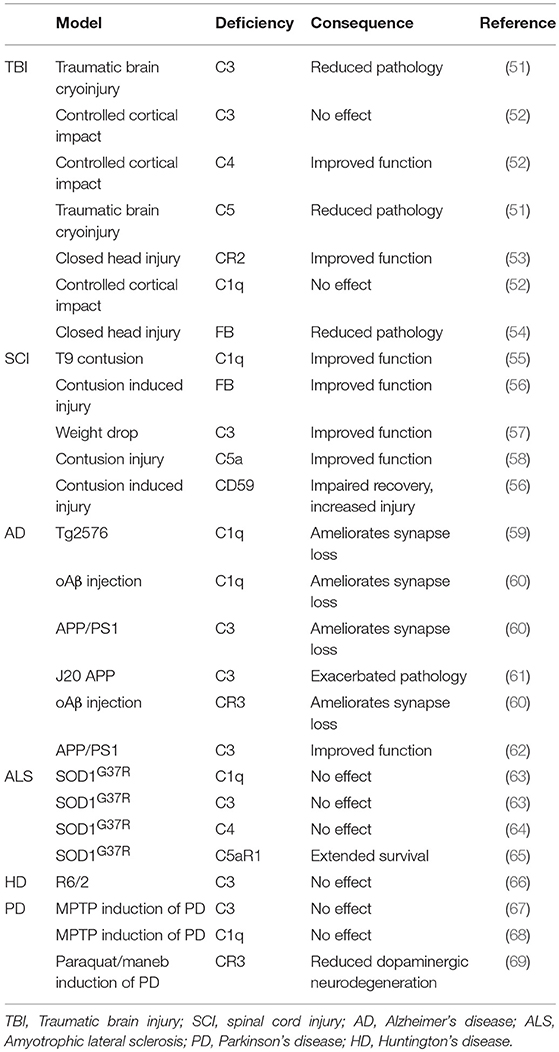
Table 1. Consequence of complement deficiency on outcome of neurodegenerative disease.
The majority of therapeutic studies in TBI models have focused on targeting the C3 convertase as a central player of all three activation pathways (Table 2). Administration of soluble CR1 (sCR1) pre- and post-TBI in rats reduced neutrophil accumulation in the injured brain (72). Administration of vaccinia virus complement control protein (VCP), an inhibitor of C3 activation, improved performance in spatial memory tasks after TBI (77). In mice, systemic inhibition of C3 via administration of Crry-Ig (recombinant chimeric Complement receptor 1-related protein Y (Crry) fused to mouse IgG1 Fc) 1 and 24 h post-TBI ameliorated neuronal damage in hippocampus and improved neurological outcome (87). Transgenic mice expressing soluble Crry (sCrry) specifically in astrocytes were protected in closed head injury TBI, with reduced C3 deposition, decreased BBB damage and improved neurological scores (88). Intravenous (iv) administration of C1INH 10 min post TBI reduced cognitive deficits and brain lesion size (89) and, in a separate study, improved motor scores, reduced cognitive dysfunction and reduced injury volume (90). Alternative pathway inhibition with systemically administered anti-FB reduced neuronal damage after TBI in mice (73). Lectin pathway inhibition using a multivalent MBL ligand improved functional and pathological outcome measures in a mouse TBI model (76). Terminal pathway inhibition using either the tick-derived C5 inhibitor OmCI or C6 antisense oligonucleotide decreased neuropathology and promoted recovery in severe closed head injury (74), and targeted inhibition of the terminal pathway using a CD59-CRIg hybrid that localized to areas of C3b/iC3b deposition in the injured brain was strongly neuroprotective in the same model (75). Recently, to determine which component or pathway of complement should be targeted for most efficient protection in TBI, three hybrid proteins, all containing CR2 to target to areas of complement activation, CR2-CD59 (inhibition of MAC), CR2-Crry (all complement pathways) and CR2-FH (alternative pathway), were compared; the latter two were most effective, demonstrating important roles for early activation products, both opsonins and anaphylatoxins (91).
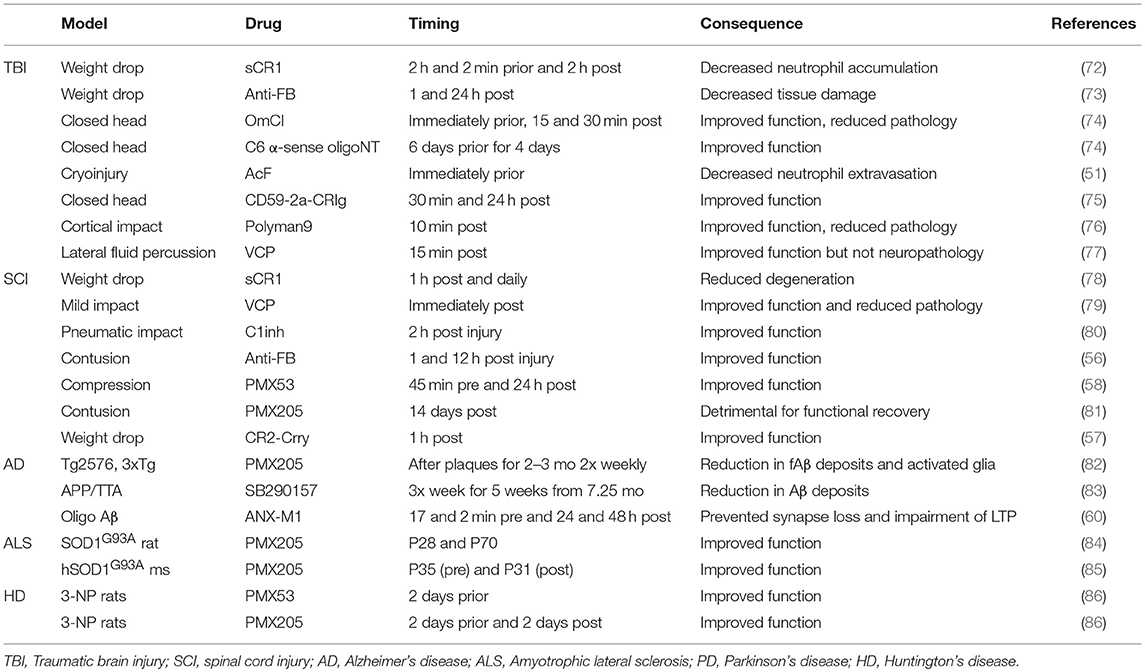
Table 2. Consequence of pharmacological complement inhibition on outcome of neurodegenerative disease.
Spinal Cord Injury (SCI)
SCI can be caused by sudden traumatic insults that crush or sever the cord, or non-traumatic injuries, for instance, triggered by cancer, arthritis or infection and usually compressing sections of cord; here we will restrict discussion to traumatic causes. SCI results in dysfunction and sometimes complete loss of function below the lesion site. Symptoms are often life-long and, since SCI is most common in under-30s, is associated with huge personal and health-care costs (https://www.spinal-research.org/). In traumatic SCI the primary pathology is caused by a mechanical force directly damaging the neural tissue—this primary insult is difficult to protect against. However, post-injury inflammation, with infiltration of immune cells and production of pro-inflammatory mediators, results in secondary pathology in adjacent areas characterized by oedema, ischemia, and excitotoxicity (92). There is considerable blood-spinal cord barrier (BSCB) damage and resultant inflammation in this secondary phase; despite this, studies of complement in SCI are scarce. Early human studies showed elevated C3, C4, and C5 levels in plasma of patients post-SCI suggestive of an acute phase response (93). As with TBI, rodent models of SCI vary widely; many different approaches to inducing injury have been taken, including weight drop, contusion, compression, laceration, and chemical injection. Complement proteins were deposited at sites of SCI in rodents; C1q, FB, C4, and TCC expression all increased at and around the injury within 24-h post-SCI and remained high up to 6 weeks (94, 95), and FH and Clusterin were elevated in lesioned neurons and oligodendrocytes (96). In a less severe weight-drop contusion SCI mouse model, C3 (likely C3b/iC3b) was deposited in white matter at the site of injury at 4-h and more widely in adjacent white and grey matter at 12- and 24-h, returning to baseline by 3 days post-injury (57).
C3−/− mice were significantly protected in contusion-induced SCI with reduced lesion size, necrosis, demyelination, and neutrophil infiltration, improved locomotor score and accelerated recovery (57); C1q−/− mice showed decreased lesion volume and improvements in locomotion and fine motor control compared to controls in the same model (55). Mice deficient in FB subjected to contusion SCI showed accelerated recovery of locomotion, marked improvements in macroscopic tissue integrity, and reduced demyelination, C3 and C9 deposition and infiltration of neutrophils and macrophages compared to controls (56), while mice deficient in the terminal pathway inhibitor CD59 showed increased pathology with loss of myelin structure, scarring and vacuolation, hemorrhage, neutrophil and macrophage infiltration, and TCC deposition (56). C5aR−/− mice showed acute but not long-term improvements in functional recovery (97). In vitro studies showed that C1q-treatment increased cortical neurite length on myelin by inhibition of growth cone repulsion by myelin associated glycoprotein (MAG); however, comparison of C1q KO and C1q WT mice in a peripheral conditioning lesion model of SCI showed no differences in axon length, lesion volume or scarring, although C1q deficiency was associated with increased axonal turning (98).
There are currently no proven therapies for SCI, unsurprising given the many obstacles in promoting re-wiring of axons and remyelination. Preventing or reducing the inflammation-driven secondary phase offers opportunity; indeed, methylprednisolone is the only currently available treatment though its effectiveness is unclear (99–101). There have been a few studies of anti-complement agents in SCI rodent models and the majority of these have utilized iv administration, possible because of BSCB disruption post-injury. Injection of the C3 convertase inhibitor VCP into the injured spinal cord in a rat SCI model, restored spinal cord tissue integrity, reduced macrophage and microglial activation and improved acute motor deficits (79, 102); iv administration of recombinant sCR1 in mice 1 h post-SCI and daily thereafter reduced neuron swelling, degeneration, necrosis and neutrophil infiltration and improved recovery (78); iv C1INH 2-h post-SCI in rats improved motor recovery, reduced lesion volume and leukocyte infiltration (80). Alternative pathway inhibition with iv anti-FB mAb accelerated recovery and reduced lesion size in the same model (56). In a contusion SCI mouse model, iv administered CR2-Crry localized to the lesion site, improved locomotor deficits and reduced necrosis, demyelination, and neutrophil infiltration (57); because CR2-Crry targets specifically areas of pathology, it is bioavailable in the SCI when given at a dose that does not influence circulating complement activity, reducing the risk of infections and other undesirable effects of systemic complement inhibition. Another strategy to reduce infection risk is to target C5a, a potent chemoattractant, or its receptor C5aR1 (CD88), a G protein-coupled receptor (GPCR) expressed on granulocytes monocytes/macrophages peripherally and on astrocytes and microglia (and at low level neurons and oligodendrocytes) in CNS (103, 104). Two small cyclic peptide C5aR1 antagonists PMX53 and PMX205 (86, 105) have been tested in SCI models; iv administration of PMX53 improved functional recovery, and reduced macrophage/microglial numbers, expression of pro-inflammatory cytokines IL-1β and TNF-α and astrogliosis in mice compared with controls (58). In a rat SCI model, the impact of PMX205 administration was dependent on timing post-injury and was linked to the sequence of immune cell recruitment to the site; whereas early administration accelerated recovery, late administration inhibited the macrophage/microglial response and slowed functional recovery and re-myelination following injury, further emphasizing the importance of timing interventions (81). Mice treated with C5aR antagonist [hydrocinnamate—(OpdChaWR)] showed acute but not long term improvements in functional recovery (97). Additionally, bone marrow chimeric mice lacking peripheral but not central C5aR showed no differences from control mice. Together these data suggest an initially detrimental role of C5aR followed by a delayed neuroprotective role, likely mediated by CNS resident cells.
Taken together, these studies indicate that inhibition of classical/lectin and/or alternative pathways or specific effectors like C5a can be efficacious in SCI. Timing of interventions may be crucial to avoid impacting beneficial clearance roles of complement. Terminal pathway inhibition has not been tested in the models but the impact of CD59 deficiency noted above suggests that this is a viable target.
Complement Proteins in Chronic Neurological Disorders—Demyelination and Neurodegeneration
Inflammation was noted early as a feature of chronic brain disease; indeed, astrogliosis and microgliosis were included in the original descriptions of AD by Alois Alzheimer over a century ago. Despite this, a classification divide emerged with diseases like MS considered inflammatory while diseases like AD were considered degenerative. The artificial nature of this divide has become clear in recent years with the realization that there are many shared features. The evidence implicating inflammation as a driver of pathology in chronic neurodegenerative diseases is now substantial and includes genetic studies identifying inflammatory risk genes (106), clinical studies demonstrating that long-term treatment with non-steroidal anti-inflammatory drugs (NSAIDs) is protective in humans (107) and mouse models (108) and the observation that systemic infections and inflammation increase the risk and/or rate of progression of dementia (109–111). Complement, the focus of this review, goes hand-in-hand with inflammation and represents a potential driver of chronic CNS diseases.
Alzheimer's Disease (AD)
AD is the leading cause of dementia affecting almost 50 million people worldwide, a number projected to increase to 150 million by 2050 (https://www.alz.co.uk/research/statistics). AD is characterized by two hallmark pathologies; amyloid-β (Aβ) plaques and neurofibrillary tangles comprising hyperphosphorylated tau. Recent studies have implicated complement in AD pathogenesis. Genome wide association studies identified single nucleotide polymorphisms (SNPs) associated with risk of late-onset AD in genes encoding complement proteins Clusterin (CLU) and CR1 (CR1) (106, 112, 113). Biomarker studies have identified complement proteins and activation products in plasma and/or CSF that distinguish AD from controls and predict risk of progression to AD (114–117). Immunohistochemistry (IHC) of post-mortem AD brain revealed complement proteins and activation products decorating plaques and tangles. In particular, classical pathway proteins C1q, C3, and C4 co-localized with amyloid fibrils, Aβ deposits and neurofibrillary tangles, notably in temporal cortex, amygdala, and hippocampus, in AD brain (118–120). The terminal pathway activation marker TCC was abundant in AD cortex in association with aggregated Aβ, neurofibrillary tangles and neuropil threads (121). Cells expressing C5a receptors C5aR1 and C5L2 were associated with neurofibrillary tangles, neuropil threads, and dystrophic neurites in AD plaques in hippocampus and frontal cortex (122). A weakness of these IHC studies is that they are performed on post-mortem brain, inevitably end-stage disease, and do not provide insight into early disease or disease progression. A large-scale microarray study of young, healthy old and AD brains identified marked changes in complement expression with ageing, and elevated expression of C4A, C4B, C3aR1, C5aR1, CFHR1, and CLU in AD compared to age-matched controls; C1q binding protein (C1qBP) expression decreased in AD (123). Increased C1q expression in brain with ageing (healthy or AD) has been robustly replicated (59, 124, 125).
Mechanisms of complement activation in the AD brain have been studied in vitro and in animal models. Aβ fibrils activate and consume complement classical and alternative pathways in vitro and generate C3a,C5a, and TCC (119, 126). C5a administration resulted in death of primary mouse neurons in culture; this could be blocked by addition of C5aR1 antagonist PMX53, demonstrating that C5a (acting via C5aR1) is sufficient to induce neuronal cell death in vitro (127). Animal models have underpinned the majority of research into roles and mechanisms of complement in AD. Most mouse models mimic the rare early-onset forms of AD in which single gene mutations have been identified rather than the common polygenic late onset AD, and thus individually mimic only certain aspects of the disease; it is therefore unsurprising that different models yield different and often contradictory results. Despite these reservations, these mouse models have aided understanding of AD pathology. Broadly, models can be divided into three groups: Aβ pathology; Tau pathology; both. These mouse models recapitulate many of the pathologies found in AD brain; for example, in the PS/APP model fibrillar Aβ plaques form and C1q localizes to these plaques (128). Back-crossing AD mouse models onto complement deficiencies has been used to determine the role of complement in the pathophysiology of AD (Table 1). Deficiency of C1q (classical pathway) in Tg2576 (Aβ pathology) mice reduced glial activation and synaptic loss without influencing Aβ load compared with controls (59). Genetic deletion of C1q, C3 or CR3, all of which are required for effective opsonization and phagocytosis of synapses, reduced microglial numbers and synapse loss when crossed to two different Aβ models (J20 and APP/PS1); further, when Aβ fibrils were injected directly into brain, C1q deficiency protected from toxicity (60). In contrast, C1q deficiency in the 3xTG model exacerbated neurodegeneration because of a loss of C1q-triggered expression of neuronal survival pathways (129). Others showed that C3 deficiency improved performance on learning and memory tests and decreased microglia and astrocyte number associated with plaques in an Aβ model (APP/PS1) (62); in contrast, C3 deficiency was associated with increased amyloid burden, decreased neuronal staining and activated glia in the J20 (Aβ) model (61). Despite the evidence noted above that Tau pathology is associated with complement, there was a dearth of studies in Tau models; two recent publications have changed this. Administration of blocking anti-C1q antibodies in a mouse Tau model (P301S) inhibited microglial synapse loss and rescued synapse density (130), while C3aR deletion attenuated neuroinflammation and reduced synaptic deficits and neurodegeneration in the PS19 Tau model (131).
As is clear from the anti-C1q experiment described above, mouse models also offer a way to test in vivo the impact of complement therapeutics on disease (Table 2). The C5aR1 antagonist PMX205 decreased amyloid and tau deposits, reduced activated glia and improved cognition in two Aβ models (Tg2576 and 3xTg) (82). Levels of C1q and C3 were unchanged upon PMX205 treatment, suggesting that their physiological functions are preserved. As noted above, blocking antibody against C1q [(ANX-M1/ANX005); Annexon Biosciences] protected from synapse loss in Aβ models and reduced toxicity of Aβ fibrils injected into the lateral ventricles (60); this agent showed no toxicity, even at high doses (200 mg/kg) and has proceeded to clinical trials (132). A note of caution in the use of anti-complement agents comes from a study of C3 inhibitor sCrry administered to an Aβ model (hAPP × TGFβ1) which resulted in increased Aβ deposition and neuronal degeneration (133).
The evidence—genetic, clinical, and from models—implicating complement as a driver of pathology in AD is compelling. A complicating factor is that complement may also have protective roles in clearing debris in early disease. Improved understanding of the time course of complement involvement may identify therapeutic windows where complement inhibitors will improve outcome.
Amyotrophic Lateral Sclerosis (ALS)
ALS, also known as Lou Ghering's disease, is an adult onset neurodegenerative disease, usually fatal within 2–5 years of onset (134). ALS is caused by progressive loss of upper and lower (α) motor neurons (135), resulting in denervation of neuromuscular junctions in the peripheral nervous system, progressive muscle weakness, atrophy, spasticity, respiratory failure, and ultimately paralysis and death. Approximately 90% of ALS cases are sporadic and 10% familial. Causative missense point mutations have been identified in superoxide dismutase (SOD1), TAR DNA binding protein (TDP-43), fused-in-sarcoma-protein (FUS), and chromosome 9 open reading frame 72 (C9orf72). The only currently available treatment for ALS is Riluzole, an ion channel blocker and inhibitor of glutamate release which modestly increases survival (136, 137).
Neuroinflammation is a consistent feature of ALS with abundant reactive microglia and astrocytes and T-cell infiltration observed (138). IHC identified increased C1q protein in motor cortex and spinal cord of ALS post-mortem tissue; C3 activation fragments and TCC were also demonstrated in areas of pathology (139, 140). C3c labeled astrocyte-like cells in the former study whereas C1q and C3d co-localized with neurons, astrocytes and microglia, and TCC primarily microglia, in the latter. Others described C4d and TCC staining of degenerating neurons and glia in ALS motor cortex and spinal cord (141) and C5aR1 upregulation in areas of pathology (142). C3d and C4d were also found on oligodendroglia and degenerating neurites, surrounded by CR4-positive microglia, in spinal cord and motor cortex (141, 143). C1q, C3, and TCC were present on motor end-plates in intercostal muscles in ALS donors even early in the disease process (144); DAF and CD59 were upregulated at the end-plates, perhaps reflecting a response to complement activation and TCC/MAC deposition. TCC immunoreactivity at end-plates negatively correlated with α-bungarotoxin staining, implicating TCC/MAC in loss of end-plates (144). In myasthenia gravis, end-plate destruction is dependent on complement activation and MAC formation (145), supporting a causative role in ALS.
The source of complement in ALS pathology is unclear; the BBB is disrupted in the disease (146); however, local biosynthesis likely also contributes. In situ hybridization demonstrated upregulated C1qb and CLU mRNA in areas most affected by neurodegeneration (147); more recently, increased C1q and C4 expression by glial cells was demonstrated in ALS cord white matter (140) indicating a local source of complement. Complement expression positively associated with increased infiltration of dendritic cells and CD8+ T-lymphocytes from the periphery (140, 141). Biomarkers also implicate complement. Complement activation products C3c and C4d were present in CSF and correlated with disease severity scores (148–150). Levels of C5a and TCC were significantly elevated in ALS plasma, and leukocytes from ALS patients had increased surface (C5aR1-bound) C5a (151). These biomarker findings strongly support a role for complement dysregulation in ALS patients; however, the nature and location of complement protein deposition in different studies was contradictory, perhaps due to differences in disease stage or comorbidities.
Numerous rodent models of ALS have been generated based on known causative mutations in SOD1, responsible for ~10% of familial ALS. Rodents over-expressing human mutant SOD1G93A recapitulate key neuropathological and functional hallmarks of ALS, characterized by lumbar motor neuron loss which correlates with progressive motor deficits and ultimately paralysis, and by inflammatory changes including robust astrogliosis, microgliosis, and BBB-disruption (152–154). Complement dysregulation is apparent from increased expression and deposition of C1q, C4, FB, C3 activation products and TCC, increased expression of C5aR1, and reduced expression of complement regulators DAF and CD59 (64, 84, 144, 154–156). Complement deposition has also been observed in sciatic nerves (64) and at the neuromuscular junction (156) in ALS models, consistent with the concept that complement contributes to nerve terminal destruction in ALS. In the TDP43Q331K mouse model, progressive motor deficits, astrogliosis, and microgliosis correlated with complement dysregulation in the spinal cord; expression of C1qB, C4 and C3 was elevated and DAF mRNA reduced in the lumbar spinal cord and in tibialis anterior muscle of TDP43Q331K mice compared with controls (157). Immunofluorescence confirmed markedly increased C1q and C5aR1 in motor neurons and microglia.
Surprisingly, C1q deletion in SOD1G37R ALS mice exacerbated synaptic loss at end-stage and it was implied that this was a consequence of increased microglial phagocytosis; however, C1q deletion did not significantly affect disease onset, progression, or survival and had no effect on global astrogliosis, microgliosis, or neuronal loss (63) (Table 1). Deletion of the gene encoding C4, which is necessary for activation of both the classical and lectin pathways, significantly reduced the number of activated macrophages found in sciatic nerves of mSOD1G93A mice but again failed to influence the disease course (64). C3 deletion also failed to affect overall survival or motor neuron loss in SOD1G93A ALS mice (63); the finding that deletion of C3, central to all complement pathways, fails to rescue disease has provoked the suggestion that complement does not contribute to ALS disease progression (at least in this model). The demonstration that anti-complement drugs ameliorate disease in a similar model contradicts this suggestion (Table 2). Oral administration of C5aR1 antagonist PMX205, even when given in established disease, reduced weight loss and motor deficit scores, slowed disease progression and enhanced survival times in SOD1G93A rats and mice (84, 85). These functional improvements were associated with reduced astrocyte proliferation, reduced influx of proinflammatory monocytes and granulocytes and an increase in the CD4+: CD8+ T-cell ratio, consistent with the reported neuroprotective role of CD4+-T cells in ALS (158). The same authors showed that deficiency of C5aR1 (upregulated in human and rodent ALS) extended survival in SOD1G93A mice (65). Taken together, these data strongly implicate the C5a/C5aR1 axis in disease and identify it as a target for therapy in ALS.
Huntington's Disease (HD)
HD is an autosomal dominant, inherited neurodegenerative disease characterized by progressive motor symptoms, psychiatric disturbances, and dementia. It is caused by expansion of a three-base-pair (CAG) repeat (39–121 repeats vs. normal range 8–39 repeats) in exon 1 of the HTT gene that translates into a polyglutamine tract at the N-terminus of the protein. This results in a polyglutamine length-dependent misfolding and accumulation of huntingtin protein in the striatum and cortex (layers 3, 5, and 6) followed by neuronal loss in these areas which spreads to the hippocampus (159, 160). Neuropathology of HD is graded based on Vonsattel staging (161) dependent on the severity of neuronal loss, astrogliosis, and brain atrophy. Precisely how the huntingtin trinucleotide expansions result in neuronal death and associated gliosis remain unclear. Microglial activation can be demonstrated by PET scanning even in early disease and correlates with disease severity (11C-raclopride binding) (162); indeed, even in pre-symptomatic gene carriers, microglial activation was present and correlated with striatal neuronal dysfunction and with risk of developing HD within 5 years (163).
HD post-mortem tissue showed abundant reactive astrogliosis and microgliosis and intranuclear ubiquitin positive inclusions in the caudate and temporal lobes (164). IHC showed that neurons, astrocytes and myelin sheaths in the HD caudate and striatum were immunoreactive for C1q, C4, C3 and neo-epitopes in iC3b and TCC (164). Expression of mRNA encoding early complement components C1q (c-chain), C1r, C3, and C4, complement regulators C1INH, Clusterin, MCP, DAF and CD59, and complement receptors C3a and C5a was upregulated in the HD striatum. Early disease stages did not stain for complement suggesting that early neuronal damage precedes local complement synthesis and activation. Microarray analysis in HD post-mortem tissue demonstrated increased expression of complement components C4A, C4B and C3, most significantly in the most affected areas, caudate nucleus, and motor cortex (165). Unbiased proteomic profiling revealed 18 proteins that were differentially expressed in HD plasma, several of which are involved in the innate immune system; Clusterin, C7 and C9 increased with disease severity (166).
Early animal models of HD utilized toxin-mediated striatal lesions; for example, Lewis rats given intracerebral 3-nitropropionic acid (3-NP), an inhibitor of the mitochondrial citric acid cycle, developed striatal lesions, weight loss, gait disturbances, dystonia and ataxia, thus reproducing some of the pathological and clinical characteristics of HD (86). Oral administration of C5aR antagonist (PMX53 or PMX205) reduced weight loss and motor deficits, even when given post-toxin administration, whereas NSAID, ibuprofen, and a TNF-α inhibitor (infliximab) had no significant functional impact, suggesting that ability to rescue these deficits hinged on the complement pathway per se rather than neuroinflammation in general (Table 2). 3-NP treatment caused lesions with robust neuronal death and neutrophil infiltration and surrounded by C5aR-, C3-, and C9-positive glia. C5aR blockade reduced lesion volume and number; lesions contained fewer apoptotic cells and astrocytes and were no longer surrounded by complement-positive glia. While these data were a helpful proof of concept (and this was the first paper demonstrating that PMX53 and PMX205 cross the BBB), the model used is extremely artificial, acute and invasive, unlike the chronic, cumulative dysfunction seen in HD.
R6/2 transgenic mice provide a more realistic HD model; these mice express exon 1 of the human huntingtin gene, including a pathological trinucleotide repeat; they develop progressive behavioral and neuropathological deficits, including synaptic loss, but do not develop neuronal loss and fail to demonstrate upregulation of complement proteins (66). It is, therefore, unsurprising that C3 deficiency did not alter disease progression in this model. C5aR was the only complement molecule upregulated in the model and it remains undetermined whether targeting the C5a-C5aR1 axis would be beneficial.
Parkinson's Disease (PD)
PD is characterized by loss of dopaminergic neurons in the substantia nigra and deposits of the protein α-synuclein that form the pathological hallmarks of the disease, Lewy bodies. Patients present with resting tremor, bradykinesia, and rigidity. Complement activation has been associated with α-synuclein and Lewy bodies in Parkinson's disease; in vitro studies demonstrated that the disease-associated splice variant α-synuclein 112, but not the full length protein, cause activation of complement (167). In vivo, C3d, C4d, C7 and C9 localization in Lewy bodies was reported in one study (168), although this was not recapitulated in a separate study (169). More recently, deposition of iC3b and C9 in Lewy bodies and melanized neurons was reported; iC3b immunoreactivity increased with normal ageing and was further elevated in PD vs. age-matched controls (170). A correlation was described between the ratios of C3/Aβ42 or FH/Aβ42 in CSF and severity of Parkinson's disease motor and cognitive symptoms, but not with absolute levels of C3 or FH (171).
Although there are many mouse models of PD, drug or neurotoxin induced, or genetic, none fully replicates the human disease (172). A few studies have explored roles of complement in these models; absence of C3 in mice did not protect against depletion of dopaminergic neurons in the toxin-induced MPTP model (67) (Table 1). There was an increase of C1q in relevant brain regions in this model but C1q deficiency did not protect from disease (68). A very recent study identified a role for CR3 in activation of the microglial NADPH oxidase (Nox2) and subsequent neurodegeneration in a toxin-induced PD model; CR3 knockout mice were protected from dopaminergic neuron loss and motor dysfunction, suggesting that complement opsonization and CR3 engagement contribute to the disease process (69).
Targeting Complement in Neurological Disease
Getting Therapeutics Into the CNS
Having made the case above for an involvement of complement in acute neurological injuries and neurodegenerative diseases, attention naturally turns to therapeutic significance. There is a huge and growing complement therapeutics industry with a myriad of new drugs emerging; however, to date CNS targets have been largely ignored (173). Drug delivery is a major limiting factor for CNS therapies that needs to be considered when designing therapeutics for treating neurological conditions. The BBB precludes passive entry of molecules larger than ~400 kDa thus occluding entry of macromolecules, including antibody and protein therapeutics. In TBI and SCI the BBB is impaired to some degree, enabling drugs to access the injured areas (47). Treatment options to access the CNS in diseases where the BBB remains intact include both invasive and non-invasive techniques [reviewed in (174, 175)]. Historically, access of drugs to the CNS involved disruption or damage to the BBB or the use of pharmacological agents to increase its permeability; however, in many cases this resulted in widespread neuronal damage and an associated inflammatory response. Less damaging ways of opening the BBB include the use of focused ultrasound waves of low intensity that cause local and temporary disruption to the BBB and administration of “osmotic shock” agents (176). Pharma companies have designed a host of other strategies to deliver therapies, including Trojan horse delivery, use of viral vectors, nanoparticles and chimeric peptides, expanded on below.
Trojan horse technologies involve the creation of fusion proteins that lock the drug to a delivery component that utilizes receptors in the BBB, such as the insulin receptor and transferrin receptor, to enable bidirectional transport into and out of the brain. As an example of this concept, a recombinant anti-Aβ single chain fV antibody (fragment variable region only) fused to a fAb fragment of an anti-insulin receptor mAb bound the insulin receptor at the BBB, was transcytosed across the barrier enabling it to access and recognized Aβ within the brain and was then shuttled out again with its Aβ cargo for disposal (177). Anti-complement therapeutic antibodies, of which there are many in the clinic or in development, could be similarly piggy-backed into the brain to inhibit complement.
Small, hydrophobic molecules can cross the BBB via lipid-mediated diffusion. As an example, oral administration of the small molecule NLRP3 inhibitor MCC950 in PD mouse models reduced nigrostriatal dopaminergic degeneration, motor deficits and accumulation of α-synuclein through inhibiting inflammasome activation (178). Several small-molecule complement inhibitors are in development but, with the exception of the anti-C5aR1 molecules PMX53 and PMX205 described above, these have yet to be assessed for BBB permeability.
Targeting Complement in CNS
Eculizumab, a humanized anti-C5 antibody, is the lead anti-complement drug but to date has only been approved for two rare disorders, paroxysmal nocturnal hemoglobinuria (PNH) and atypical hemolytic uremic syndrome (aHUS) and recently for Myasthenia Gravis. In a recent small trial of eculizumab in NMO, a demyelinating disease characterized by BBB disruption and inflammation/degeneration of the optic nerve and spinal cord (www.clinicaltrials.gov NCT00904826), treatment reduced the number of neurological episodes (179). This study has raised the prospect of using eculizumab for other inflammatory CNS diseases, although the BBB is likely a much greater hurdle in these other conditions and it is unclear whether they can be treated systemically. There is an urgent need to apply rational drug design for targeting complement activation in the CNS to obtain effective treatments with low side-effects and costs; for example, there is little point in designing an anti-C1q therapeutic to be administered systemically for a CNS disease given that C1q exists throughout the body and in the circulation at micromolar levels—extremely high drug doses would be required to have any impact at the desired site in the CNS even if the drug is BBB penetrant. Given the ubiquitous expression of complement proteins throughout the body and the role of complement proteins in fighting infection and maintaining homeostasis, anti-complement therapeutics at these doses would likely have consequences throughout the body. Rather, therapies should target areas of pathology, as described above for the fusion proteins linking CR2 (localizes to C3 activation products in tissues) with a complement regulator, or target complexes, for example MAC, which exist at much lower concentrations and are found only in areas of pathology.
A common stumbling block in designing treatments for neuroinflammatory disorders is timing. Despite early conceptions about the fixed nature of post-mitotic neurons in the CNS, there is a great deal of redundancy and flexibility in neuronal circuits. Networks are hence able to compensate for a huge amount of cell loss through synaptic plasticity so that, by the time patients present at the clinic with symptoms, major neuronal death has already occurred. Inflammation as a cause and consequence of this neuronal death occurs early and, unlike in acute conditions, fails to resolve because the primary stimulus, cell death, accumulation of toxic proteins or mitochondrial dysfunction, persists. Thus, effective treatments that aim to halt or slow disease progression must be administered early—likely before symptoms are apparent—and must prevent further neuronal cell death and encourage resolution of inflammation. Evidence from studies of the impact of NSAIDs on neurodegeneration support the idea that early and long-term treatment is protective but treatment post-onset fails (reviewed by 106). Early intervention requires ways of identifying those at high risk; genetic studies have identified polygenic signals that include many inflammatory genes and are highly predictive of risk of AD (106), and inflammatory biomarkers may also help predict risk (114). For large scale screening of pre-symptomatic populations expensive interventions such as brain imaging or CSF sampling are not practicable; in contrast, plasma offers an attractive source of biomarkers, although the levels of inflammatory markers in plasma may not reflect inflammation in the CNS. Simple and highly predictive plasma biomarkers are emerging and are likely to transform treatment of AD and other neurodegenerative diseases in the near future (180).
With regard to anti-complement therapies, it is likely that different approaches will be needed for different diseases; identifying for each disease when complement is activated, which pathways are activated and what the consequences are will be essential for effective interventions. Studies to date have been restricted to models and have focused on targeting the C3 convertases, central to all three activation pathways and thus a blunt tool likely to impact immune defense and other beneficial functions. The implication of C5a/C5aR in several CNS diseases offers the prospect of more targeted therapy with less risk of iatrogenic disease, although the systemic impact of long-term inhibition of the C5a/C5aR axis are uncertain. Evidence from models obtained either by complement gene deletion (Table 1) or anti-complement therapies (Table 2) has been helpful but are often contradictory, studies reporting both positive and negative impacts in similar models; this likely results from differences in timing and precise nature of the intervention and highlights once again the need for a thorough understanding of the underpinning biology prior to human studies.
Concluding Remarks
Therapy of acute neurological injury and neurodegenerative diseases represent a major therapeutic challenge. Most of the diseases described above currently have no effective therapies and new approaches are desperately needed. Although there are some common features, notably inflammation and complement activation, the described diseases are very heterogeneous, even within disease labels—AD is not a single disease! Patient stratification, for example, selecting patients with high inflammatory markers and evidence of ongoing complement activation for anti-complement drug therapy, will be necessary for successful trails in the future; this requires better biomarkers. For most of the diseases, proof of concept for new approaches to therapy is stymied by the lack of good models; critically, agents that are effective in current models usually fail in human trails (https://www.nature.com/articles/d41586-018-05722-9). For AD, models that better reflect the human disease are now available and may help overcome this issue. Switching off complement systemically will impact immune defense; while this may not be an issue for short-term therapy in acute conditions, in chronic diseases requiring life-long treatment it is a major consideration. Despite all these problems, inflammation and complement activation present tractable targets in neuroinjury and neurodegenerative disease and deserve investment into basic understanding and the development of CNS-targeting anti-inflammatory and anti-complement drugs.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by the UK Dementia Research Institute.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank members of the Cardiff Complement Biology Group and DRI Cardiff for helpful discussion and reviewing aspects of this work.
References
1. Ueno M, Chiba Y, Murakami R, Matsumoto K, Kawauchi M, Fujihara R. Blood–brain barrier and blood–cerebrospinal fluid barrier in normal and pathological conditions. Brain Tumor Pathol. (2016) 33:89–96. doi: 10.1007/s10014-016-0255-7
2. Galea I, Bechmann I, Perry VH. What is immune privilege (not)? Trends Immunol. (2007) 28:12–8. doi: 10.1016/j.it.2006.11.004
3. Louveau A, Herz J, Alme MN, Salvador AF, Dong MQ, Viar KE, et al. CNS lymphatic drainage and neuroinflammation are regulated by meningeal lymphatic vasculature. Nat Neurosci. (2018) 21:1380–91. doi: 10.1038/s41593-018-0227-9
4. Askew K, Li K, Olmos-Alonso A, Garcia-Moreno F, Liang Y, Richardson P, et al. Coupled proliferation and apoptosis maintain the rapid turnover of microglia in the adult brain. Cell Rep. (2017) 18:391–405. doi: 10.1016/j.celrep.2016.12.041
5. Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. (2010) 330:841–5. doi: 10.1126/science.1194637
6. Hoeffel G, Chen J, Lavin Y, Low D, Almeida FF, See P, et al. C-Myb(+) erythro-myeloid progenitor-derived fetal monocytes give rise to adult tissue-resident macrophages. Immunity. (2015) 42:665–78. doi: 10.1016/j.immuni.2015.03.011
7. Sheng J, Ruedl C, Karjalainen K. Most tissue-resident macrophages except microglia are derived from fetal hematopoietic stem cells. Immunity. (2015) 43:382–93. doi: 10.1016/j.immuni.2015.07.016
8. Hickman SE, Kingery ND, Ohsumi TK, Borowsky ML, Wang LC, Means TK, et al. The microglial sensome revealed by direct RNA sequencing. Nat Neurosci. (2013) 16:1896–905. doi: 10.1038/nn.3554
9. London A, Cohen M, Schwartz M. Microglia and monocyte-derived macrophages: functionally distinct populations that act in concert in CNS plasticity and repair. Front Cell Neurosci. (2013) 7:34. doi: 10.3389/fncel.2013.00034
10. Zarruk JG, Greenhalgh AD, David S. Microglia and macrophages differ in their inflammatory profile after permanent brain ischemia. Exp Neurol. (2018) 301:120–32. doi: 10.1016/j.expneurol.2017.08.011
11. O'Sullivan JB, Ryan KM, Curtin NM, Harkin A, Connor TJ. Noradrenaline reuptake inhibitors limit neuroinflammation in rat cortex following a systemic inflammatory challenge: implications for depression and neurodegeneration. Int J Neuropsychopharmacol. (2009) 12:687–99. doi: 10.1017/S146114570800967X
12. Heneka MT, Nadrigny F, Regen T, Martinez-Hernandez A, Dumitrescu-Ozimek L, Terwel D, et al. Locus ceruleus controls Alzheimer's disease pathology by modulating microglial functions through norepinephrine. Proc Natl Acad Sci USA. (2010) 107:6058–63. doi: 10.1073/pnas.0909586107
13. Hoek RM, Ruuls SR, Murphy CA, Wright GJ, Goddard R, Zurawski SM, et al. Down-regulation of the macrophage lineage through interaction with OX2 (CD200). Science. (2000) 290:1768–71. doi: 10.1126/science.290.5497.1768
14. Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, et al. Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci. (2006) 9:917–24. doi: 10.1038/nn1715
15. Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. (2005) 308:1314–8. doi: 10.1126/science.1110647
16. Hanisch U-K, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. (2007) 10:1387–94. doi: 10.1038/nn1997
17. Grabert K, Michoel T, Karavolos MH, Clohisey S, Baillie JK, Stevens MP, et al. Microglial brain region-dependent diversity and selective regional sensitivities to aging. Nat Neurosci. (2016) 19:504–16. doi: 10.1038/nn.4222
18. Gosselin D, Skola D, Coufal NG, Holtman IR, Schlachetzki JCM, Sajti E, et al. An environment-dependent transcriptional network specifies human microglia identity. Science. (2017) 356:6344. doi: 10.1126/science.aal3222
19. Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. (2010) 119:7–35. doi: 10.1007/s00401-009-0619-8
20. Ricklin D, Mastellos DC, Reis ES, Lambris JD. The renaissance of complement therapeutics. Nat Rev Nephrol. (2018) 14:26–47. doi: 10.1038/nrneph.2017.156
21. Alper CA, Johnson AM, Birtch AG, Moore FD. Human C'3: evidence for the liver as the primary site of synthesis. Science. (1969) 163:286–8. doi: 10.1126/science.163.3864.286
22. Lubbers R, van Essen MF, van Kooten C, Trouw LA. Production of complement components by cells of the immune system. Clin Exp Immunol. (2017) 188:183–94. doi: 10.1111/cei.12952
23. Hosokawa M, Klegeris A, Maguire J, McGeer PL. Expression of complement messenger RNAs and proteins by human oligodendroglial cells. Glia. (2003) 42:417–23. doi: 10.1002/glia.10234
24. Gasque P, Chan P, Fontaine M, Ischenko A, Lamacz M, Gotze O, et al. Identification and characterization of the complement C5a anaphylatoxin receptor on human astrocytes. J Immunol. (1995) 155:4882–9.
25. Gasque P, Morgan BP. Complement regulatory protein expression by a human oligodendrocyte cell line: cytokine regulation and comparison with astrocytes. Immunology. (1996) 89:338–47. doi: 10.1046/j.1365-2567.1996.d01-756.x
26. Walker DG, Kim SU, McGeer PL. Complement and cytokine gene expression in cultured microglial derived from postmortem human brains. J Neurosci Res. (1995) 40:478–93. doi: 10.1002/jnr.490400407
27. Thomas A, Gasque P, Vaudry D, Gonzalez B, Fontaine M. Expression of a complete and functional complement system by human neuronal cells in vitro. Int Immunol. (2000) 12:1015–23. doi: 10.1093/intimm/12.7.1015
28. Veerhuis R, Janssen I, De Groot CJ, Van Muiswinkel FL, Hack CE, Eikelenboom P. Cytokines associated with amyloid plaques in Alzheimer's disease brain stimulate human glial and neuronal cell cultures to secrete early complement proteins, but not C1-inhibitor. Exp Neurol. (1999) 160:289–99. doi: 10.1006/exnr.1999.7199
29. Veerhuis R, Janssen I, Hack CE, Eikelenboom P. Early complement components in Alzheimer's disease brains. Acta Neuropathol. (1996) 91:53–60.
30. Walker DG, McGeer PL. Complement gene expression in human brain: comparison between normal and Alzheimer disease cases. Brain Res Mol Brain Res. (1992) 14:109–16. doi: 10.1016/0169-328X(92)90017-6
31. Carmona-Fontaine C, Theveneau E, Tzekou A, Tada M, Woods M, Page KM, et al. Complement fragment C3a controls mutual cell attraction during collective cell migration. Dev Cell. (2011) 21:1026–37. doi: 10.1016/j.devcel.2011.10.012
32. Gorelik A, Sapir T, Haffner-Krausz R, Olender T, Woodruff TM, Reiner O. Developmental activities of the complement pathway in migrating neurons. Nat Commun. (2017) 8:15096. doi: 10.1038/ncomms15096
33. Goda Y, Davis GW. Mechanisms of synapse assembly and disassembly. Neuron. (2003) 40:243–64. doi: 10.1016/S0896-6273(03)00608-1
34. Hua JY, Smith SJ. Neural activity and the dynamics of central nervous system development. Nat Neurosci. (2004) 7:327–32. doi: 10.1038/nn1218
35. Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, et al. The classical complement cascade mediates CNS synapse elimination. Cell. (2007) 131:1164–78. doi: 10.1016/j.cell.2007.10.036
36. Perez-Alcazar M, Daborg J, Stokowska A, Wasling P, Björefeldt A, Kalm M, et al. Altered cognitive performance and synaptic function in the hippocampus of mice lacking C3. Exp Neurol. (2014) 253:154–64. doi: 10.1016/j.expneurol.2013.12.013
37. Shi Q, Colodner KJ, Matousek SB, Merry K, Hong S, Kenison JE, et al. Complement C3-deficient mice fail to display age-related hippocampal decline. J Neurosci. (2015) 35:13029–42. doi: 10.1523/JNEUROSCI.1698-15.2015
38. Chu Y, Jin X, Parada I, Pesic A, Stevens B, Barres B, et al. Enhanced synaptic connectivity and epilepsy in C1q knockout mice. Proc Natl Acad Sci USA. (2010) 107:7975–80. doi: 10.1073/pnas.0913449107
39. Sekar A, Bialas AR, de Rivera H, Davis A, Hammond TR, Kamitaki N, et al. Schizophrenia risk from complex variation of complement component 4. Nature. (2016) 530:177–83. doi: 10.1038/nature16549
40. Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. (2012) 74:691–705. doi: 10.1016/j.neuron.2012.03.026
41. DiSabato DJ, Quan N, Godbout JP. Neuroinflammation: the devil is in the details. J Neurochem. (2016) 139(Suppl. 2):136–53. doi: 10.1111/jnc.13607
42. Pilch KS, Spaeth PJ, Yuki N, Wakerley BR. Therapeutic complement inhibition: a promising approach for treatment of neuroimmunological diseases. Expert Rev Neurother. (2017) 17:579–91. doi: 10.1080/14737175.2017.1282821
43. Tatomir A, Talpos-Caia A, Anselmo F, Kruszewski AM, Boodhoo D, Rus V, et al. The complement system as a biomarker of disease activity and response to treatment in multiple sclerosis. Immunol Res. (2017) 65:1103–9. doi: 10.1007/s12026-017-8961-8
44. Hammad A, Westacott L, Zaben M. The role of the complement system in traumatic brain injury: a review. J Neuroinflamm. (2018) 15:24. doi: 10.1186/s12974-018-1066-z
45. Kossmann T, Stahel PF, Morganti-Kossmann MC, Jones JL, Barnum SR. Elevated levels of the complement components C3 and factor B in ventricular cerebrospinal fluid of patients with traumatic brain injury. J Neuroimmunol. (1997) 73:63–9. doi: 10.1016/S0165-5728(96)00164-6
46. Troakes C, Smyth R, Noor F, Maekawa S, Killick R, King A, et al. Clusterin expression is upregulated following acute head injury and localizes to astrocytes in old head injury. Neuropathology. (2017) 37:12–24. doi: 10.1111/neup.12320
47. Stahel PF, Morganti-Kossmann MC, Perez D, Redaelli C, Gloor B, Trentz O, et al. Intrathecal levels of complement-derived soluble membrane attack complex (sC5b-9) correlate with blood-brain barrier dysfunction in patients with traumatic brain injury. J Neurotrauma. (2001) 18:773–81. doi: 10.1089/089771501316919139
48. Bellander B-M, Olafsson IH, Ghatan PH, Skejo HPB, Hansson L-O, Wanecek M, et al. Secondary insults following traumatic brain injury enhance complement activation in the human brain and release of the tissue damage marker S100B. Acta Neurochirurgica. (2011) 153:90–100. doi: 10.1007/s00701-010-0737-z
49. Bellander BM, von Holst H, Fredman P, Svensson M. Activation of the complement cascade and increase of clusterin in the brain following a cortical contusion in the adult rat. J Neurosurg. (1996) 85:468–75. doi: 10.3171/jns.1996.85.3.0468
50. Thelin EP, Just D, Frostell A, Häggmark-Månberg A, Risling M, Svensson M, et al. Protein profiling in serum after traumatic brain injury in rats reveals potential injury markers. Behav Brain Res. (2018) 340:71–80. doi: 10.1016/j.bbr.2016.08.058
51. Sewell DL, Nacewicz B, Liu F, Macvilay S, Erdei A, Lambris JD, et al. Complement C3 and C5 play critical roles in traumatic brain cryoinjury: blocking effects on neutrophil extravasation by C5a receptor antagonist. J Neuroimmunol. (2004) 155:55–63. doi: 10.1016/j.jneuroim.2004.06.003
52. You Z, Yang J, Takahashi K, Yager PH, Kim HH, Qin T, et al. Reduced tissue damage and improved recovery of motor function after traumatic brain injury in mice deficient in complement component C4. J Cereb Blood Flow Metab. (2007) 27:1954–64. doi: 10.1038/sj.jcbfm.9600497
53. Neher MD, Rich MC, Keene CN, Weckbach S, Bolden AL, Losacco JT, et al. Deficiency of complement receptors CR2/CR1 in Cr2(-)/(-) mice reduces the extent of secondary brain damage after closed head injury. J Neuroinflammation. (2014) 11:95. doi: 10.1186/1742-2094-11-95
54. Leinhase I, Holers VM, Thurman JM, Harhausen D, Schmidt OI, Pietzcker M, et al. Reduced neuronal cell death after experimental brain injury in mice lacking a functional alternative pathway of complement activation. BMC Neurosci. (2006) 7:55. doi: 10.1186/1471-2202-7-55
55. Galvan MD, Luchetti S, Burgos AM, Nguyen HX, Hooshmand MJ, Hamers FP, et al. Deficiency in complement C1q improves histological and functional locomotor outcome after spinal cord injury. J Neurosci. (2008) 28:13876–88. doi: 10.1523/JNEUROSCI.2823-08.2008
56. Qiao F, Atkinson C, Kindy MS, Shunmugavel A, Morgan BP, Song H, et al. The alternative and terminal pathways of complement mediate post-traumatic spinal cord inflammation and injury. Am J Pathol. (2010) 177:3061–70. doi: 10.2353/ajpath.2010.100158
57. Qiao F, Atkinson C, Song H, Pannu R, Singh I, Tomlinson S. Complement plays an important role in spinal cord injury and represents a therapeutic target for improving recovery following trauma. Am J Pathol. (2006) 169:1039–47. doi: 10.2353/ajpath.2006.060248
58. Li L, Xiong Z-Y, Qian ZM, Zhao T-Z, Feng H, Hu S, et al. Complement C5a is detrimental to histological and functional locomotor recovery after spinal cord injury in mice. Neurobiol Dis. (2014) 66:74–82. doi: 10.1016/j.nbd.2014.02.008
59. Fonseca MI, Zhou J, Botto M, Tenner AJ. Absence of C1q leads to less neuropathology in transgenic mouse models of Alzheimer's disease. J Neurosci. (2004) 24:6457–65. doi: 10.1523/JNEUROSCI.0901-04.2004
60. Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. (2016) 352:712–6. doi: 10.1126/science.aad8373
61. Maier M, Peng Y, Jiang L, Seabrook TJ, Carroll MC, Lemere CA. Complement C3 deficiency leads to accelerated amyloid beta plaque deposition and neurodegeneration and modulation of the microglia/macrophage phenotype in amyloid precursor protein transgenic mice. J Neurosci. (2008) 28:6333–41. doi: 10.1523/JNEUROSCI.0829-08.2008
62. Shi Q, Chowdhury S, Ma R, Le KX, Hong S, Caldarone BJ, et al. Complement C3 deficiency protects against neurodegeneration in aged plaque-rich APP/PS1 mice. Sci Transl Med. (2017) 9:aaf6295. doi: 10.1126/scitranslmed.aaf6295
63. Lobsiger CS, Boillée S, Pozniak C, Khan AM, McAlonis-Downes M, Lewcock JW, et al. C1q induction and global complement pathway activation do not contribute to ALS toxicity in mutant SOD1 mice. Proc Natl Acad Sci USA. (2013) 110:E4385–92. doi: 10.1073/pnas.1318309110
64. Chiu IM, Phatnani H, Kuligowski M, Tapia JC, Carrasco MA, Zhang M, et al. Activation of innate and humoral immunity in the peripheral nervous system of ALS transgenic mice. Proc Natl Acad Sci USA. (2009) 106:20960–5. doi: 10.1073/pnas.0911405106
65. Woodruff TM, Lee JD, Noakes PG. Role for terminal complement activation in amyotrophic lateral sclerosis disease progression. Proc Natl Acad Sci USA. (2014) 111:E3–4. doi: 10.1073/pnas.1321248111
66. Larkin PB, Muchowski PJ. Genetic deficiency of complement component 3 does not alter disease progression in a mouse model of Huntington's disease. J Huntingtons Dis. (2012) 1:107–18. doi: 10.3233/JHD-2012-120021
67. Liang Y, Li S, Guo Q, Zhang Y, Wen C, Zou Q, et al. Complement 3-deficient mice are not protected against MPTP-induced dopaminergic neurotoxicity. Brain Res. (2007) 1178:132–40. doi: 10.1016/j.brainres.2007.08.033
68. Depboylu C, Schorlemmer K, Klietz M, Oertel WH, Weihe E, Hoglinger GU, et al. Upregulation of microglial C1q expression has no effects on nigrostriatal dopaminergic injury in the MPTP mouse model of Parkinson disease. J Neuroimmunol. (2011) 236:39–46. doi: 10.1016/j.jneuroim.2011.05.006
69. Hou L, Wang K, Zhang C, Sun F, Che Y, Zhao X, et al. Complement receptor 3 mediates NADPH oxidase activation and dopaminergic neurodegeneration through a Src-Erk-dependent pathway. Redox Biol. (2018) 14:250–60. doi: 10.1016/j.redox.2017.09.017
70. Longhi L, Orsini F, De Blasio D, Fumagalli S, Ortolano F, Locatelli M, et al. Mannose-binding lectin is expressed after clinical and experimental traumatic brain injury and its deletion is protective. Crit Care Med. (2014) 42:1910–8. doi: 10.1097/CCM.0000000000000399
71. Yager PH, You Z, Qin T, Kim HH, Takahashi K, Ezekowitz AB, et al. Mannose binding lectin gene deficiency increases susceptibility to traumatic brain injury in mice. J Cereb Blood Flow Metab. (2008) 28:1030–9. doi: 10.1038/sj.jcbfm.9600605
72. Kaczorowski SL, Schiding JK, Toth CA, Kochanek PM. Effect of soluble complement receptor-1 on neutrophil accumulation after traumatic brain injury in rats. J Cereb Blood Flow Metab. (1995) 15:860–4. doi: 10.1038/jcbfm.1995.107
73. Leinhase I, Rozanski M, Harhausen D, Thurman JM, Schmidt OI, Hossini AM, et al. Inhibition of the alternative complement activation pathway in traumatic brain injury by a monoclonal anti-factor B antibody: a randomized placebo-controlled study in mice. J Neuroinflammation. (2007) 4:13. doi: 10.1186/1742-2094-4-13
74. Fluiter K, Opperhuizen AL, Morgan BP, Baas F, Ramaglia V. Inhibition of the membrane attack complex of the complement system reduces secondary neuroaxonal loss and promotes neurologic recovery after traumatic brain injury in mice. J Immunol. (2014) 192:2339–48. doi: 10.4049/jimmunol.1302793
75. Ruseva MM, Ramaglia V, Morgan BP, Harris CL. An anticomplement agent that homes to the damaged brain and promotes recovery after traumatic brain injury in mice. Proc Natl Acad Sci USA. (2015) 112:14319–24. doi: 10.1073/pnas.1513698112
76. De Blasio D, Fumagalli S, Longhi L, Orsini F, Palmioli A, Stravalaci M, et al. Pharmacological inhibition of mannose-binding lectin ameliorates neurobehavioral dysfunction following experimental traumatic brain injury. J Cereb Blood Flow Metab. (2017) 37:938–50. doi: 10.1177/0271678X16647397
77. Hicks RR, Keeling KL, Yang MY, Smith SA, Simons AM, Kotwal GJ. Vaccinia virus complement control protein enhances functional recovery after traumatic brain injury. J Neurotrauma. (2002) 19:705–14. doi: 10.1089/08977150260139093
78. Li LM, Li JB, Zhu Y, Fan GY. Soluble complement receptor type 1 inhibits complement system activation and improves motor function in acute spinal cord injury. Spinal Cord. (2010) 48:105–11. doi: 10.1038/sc.2009.104
79. Reynolds DN, Smith SA, Zhang YP, Mengsheng Q, Lahiri DK, Morassutti DJ, et al. Vaccinia virus complement control protein reduces inflammation and improves spinal cord integrity following spinal cord injury. Ann NY Acad Sci. (2004) 1035:165–78. doi: 10.1196/annals.1332.011
80. Tei R, Kaido T, Nakase H, Sakaki T. Protective effect of C1 esterase inhibitor on acute traumatic spinal cord injury in the rat. Neurol Res. (2008) 30:761–7. doi: 10.1179/174313208X284241
81. Beck KD, Nguyen HX, Galvan MD, Salazar DL, Woodruff TM, Anderson AJ. Quantitative analysis of cellular inflammation after traumatic spinal cord injury: evidence for a multiphasic inflammatory response in the acute to chronic environment. Brain. (2010) 133:433–47. doi: 10.1093/brain/awp322
82. Fonseca MI, Ager RR, Chu SH, Yazan O, Sanderson SD, LaFerla FM, et al. Treatment with a C5aR antagonist decreases pathology and enhances behavioral performance in murine models of Alzheimer's disease. J Immunol. (2009) 183:1375–83. doi: 10.4049/jimmunol.0901005
83. Lian H, Litvinchuk A, Chiang AC, Aithmitti N, Jankowsky JL, Zheng H. Astrocyte-microglia cross talk through complement activation modulates amyloid pathology in mouse models of Alzheimer's disease. J Neurosci. (2016) 36:577–89. doi: 10.1523/JNEUROSCI.2117-15.2016
84. Woodruff TM, Costantini KJ, Crane JW, Atkin JD, Monk PN, Taylor SM, et al. The complement factor C5a contributes to pathology in a rat model of amyotrophic lateral sclerosis. J Immunol. (2008) 181:8727–34. doi: 10.4049/jimmunol.181.12.8727
85. Lee JD, Kumar V, Fung JNT, Ruitenberg MJ, Noakes PG, Woodruff TM. Pharmacological inhibition of complement C5a-C5a1 receptor signalling ameliorates disease pathology in the hSOD1G93A mouse model of amyotrophic lateral sclerosis. Br J Pharmacol. (2017) 174:689–99. doi: 10.1111/bph.13730
86. Woodruff TM, Crane JW, Proctor LM, Buller KM, Shek AB, de Vos K, et al. Therapeutic activity of C5a receptor antagonists in a rat model of neurodegeneration. FASEB J. (2006) 20:1407–17. doi: 10.1096/fj.05-5814com
87. Leinhase I, Schmidt OI, Thurman JM, Hossini AM, Rozanski M, Taha ME, et al. Pharmacological complement inhibition at the C3 convertase level promotes neuronal survival, neuroprotective intracerebral gene expression, and neurological outcome after traumatic brain injury. Exp Neurol. (2006) 199:454–64. doi: 10.1016/j.expneurol.2006.01.033
88. Rancan M, Morganti-Kossmann MC, Barnum SR, Saft S, Schmidt OI, Ertel W, et al. Central nervous system-targeted complement inhibition mediates neuroprotection after closed head injury in transgenic mice. J Cereb Blood Flow Metab. (2003) 23:1070–4. doi: 10.1097/01.WCB.0000084250.20114.2C
89. Longhi L, Perego C, Zanier ER, Ortolano F, Bianchi P, Stocchetti N, et al. Neuroprotective effect of C1-inhibitor following traumatic brain injury in mice. Acta Neurochir Suppl. (2008) 102:381–4. doi: 10.1007/978-3-211-85578-2_73
90. Longhi L, Perego C, Ortolano F, Zanier ER, Bianchi P, Stocchetti N, et al. C1-inhibitor attenuates neurobehavioral deficits and reduces contusion volume after controlled cortical impact brain injury in mice. Crit Care Med. (2009) 37:659–65. doi: 10.1097/CCM.0b013e318195998a
91. Alawieh A, Langley EF, Weber S, Adkins D, Tomlinson S. Identifying the role of complement in triggering neuroinflammation after traumatic brain injury. J Neurosci. (2018). doi: 10.1523/JNEUROSCI.2197-17.2018. [Epub ahead of print].
92. Kwon BK, Tetzlaff W, Grauer JN, Beiner J, Vaccaro AR. Pathophysiology and pharmacologic treatment of acute spinal cord injury. Spine J. (2004) 4:451–64. doi: 10.1016/j.spinee.2003.07.007
93. Rebhun J, Madorsky JG, Glovsky MM. Proteins of the complement system and acute phase reactants in sera of patients with spinal cord injury. Ann Allergy. (1991) 66:335–8.
94. Liu L, Tornqvist E, Mattsson P, Eriksson NP, Persson JK, Morgan BP, et al. Complement and clusterin in the spinal cord dorsal horn and gracile nucleus following sciatic nerve injury in the adult rat. Neuroscience. (1995) 68:167–79. doi: 10.1016/0306-4522(95)00103-P
95. Anderson AJ, Robert S, Huang W, Young W, Cotman CW. Activation of complement pathways after contusion-induced spinal cord injury. J Neurotrauma. (2004) 21:1831–46. doi: 10.1089/neu.2004.21.1831
96. Anderson AJ, Najbauer J, Huang W, Young W, Robert S. Upregulation of complement inhibitors in association with vulnerable cells following contusion-induced spinal cord injury. J Neurotrauma. (2005) 22:382–97. doi: 10.1089/neu.2005.22.382
97. Brennan FH, Gordon R, Lao HW, Biggins PJ, Taylor SM, Franklin RJM, et al. The complement receptor C5aR controls acute inflammation and astrogliosis following spinal cord injury. J Neurosci. (2015) 35:6517–31. doi: 10.1523/JNEUROSCI.5218-14.2015
98. Peterson SL, Nguyen HX, Mendez OA, Anderson AJ. Complement protein C1q modulates neurite outgrowth in vitro and spinal cord axon regeneration in vivo. J Neurosci. (2015) 35:4332–49. doi: 10.1523/JNEUROSCI.4473-12.2015
99. Fiore C, Inman DM, Hirose S, Noble LJ, Igarashi T, Compagnone NA. Treatment with the neurosteroid dehydroepiandrosterone promotes recovery of motor behavior after moderate contusive spinal cord injury in the mouse. J Neurosci Res. (2004) 75:391–400. doi: 10.1002/jnr.10821
100. Hall ED, Springer JE. Neuroprotection and acute spinal cord injury: a reappraisal. NeuroRx. (2004) 1:80–100. doi: 10.1602/neurorx.1.1.80
101. Tsai EC, Tator CH. Neuroprotection and regeneration strategies for spinal cord repair. Curr Pharmaceut Design. (2005) 11:1211–22. doi: 10.2174/1381612053507404
102. McKenzie R, Kotwal GJ, Moss B, Hammer CH, Frank MM. Regulation of complement activity by vaccinia virus complement-control protein. J Infect Dis. (1992) 166:1245–50. doi: 10.1093/infdis/166.6.1245
103. Barnum SR. Complement in central nervous system inflammation. Immunol Res. (2002) 26:7–13. doi: 10.1385/IR:26:1-3:007
104. Woodruff TM, Ager RR, Tenner AJ, Noakes PG, Taylor SM. The role of the complement system and the activation fragment C5a in the central nervous system. Neuromolecular Med. (2010) 12:179–92. doi: 10.1007/s12017-009-8085-y
105. Woodruff TM, Pollitt S, Proctor LM, Stocks SZ, Manthey HD, Williams HM, et al. Increased potency of a novel complement factor 5a receptor antagonist in a rat model of inflammatory bowel disease. J Pharmacol Exp Ther. (2005) 314:811–7. doi: 10.1124/jpet.105.086835
106. Lambert J-C, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet. (2013) 45:1452. doi: 10.1038/ng.2802
107. Etminan M, Gill S, Samii A. Effect of non-steroidal anti-inflammatory drugs on risk of Alzheimer's disease: systematic review and meta-analysis of observational studies. BMJ. (2003) 327:128. doi: 10.1136/bmj.327.7407.128
108. Lim GP, Yang F, Chu T, Chen P, Beech W, Teter B, et al. Ibuprofen suppresses plaque pathology and inflammation in a mouse model for Alzheimer's disease. J Neurosci. (2000) 20:5709–14. doi: 10.1523/JNEUROSCI.20-15-05709.2000
109. Engelhart MJ, Geerlings MI, Meijer J, Kiliaan A, Ruitenberg A, van Swieten JC, et al. Inflammatory proteins in plasma and the risk of dementia: the rotterdam study. Arch Neurol. (2004) 61:668–72. doi: 10.1001/archneur.61.5.668
110. Dunn N, Mullee M, Perry VH, Holmes C. Association between dementia and infectious disease: evidence from a case-control study. Alzheimer Dis Assoc Disord. (2005) 19:91–4. doi: 10.1097/01.wad.0000165511.52746.1f
111. Holmes C, Colm C, Zotova E, Woolford J, Dean C, Kerr S, et al. Systemic inflammation and disease progression in Alzheimer disease. Neurology. (2009) 73:768–74. doi: 10.1212/WNL.0b013e3181b6bb95
112. Lambert J-C, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat Genet. (2009) 41:1094–9. doi: 10.1038/ng.439
113. Karch CM, Goate AM. Alzheimer's disease risk genes and mechanisms of disease pathogenesis. Biol Psychiatry. (2015) 77:43–51. doi: 10.1016/j.biopsych.2014.05.006
114. Hakobyan S, Harding K, Aiyaz M, Hye A, Dobson R, Baird A, et al. Complement biomarkers as predictors of disease progression in Alzheimer's disease. J Alzheimers Dis. (2016) 54:707–16. doi: 10.3233/JAD-160420
115. Hu WT, Watts KD, Tailor P, Nguyen TP, Howell JC, Lee RC, et al. CSF complement 3 and factor H are staging biomarkers in Alzheimer's disease. Acta Neuropathol Commun. (2016) 4:14–14. doi: 10.1186/s40478-016-0277-8
116. Morgan AR, Touchard S, O'Hagan C, Sims R, Majounie E, Escott-Price V, et al. The correlation between inflammatory biomarkers and polygenic risk score in Alzheimer's disease. J Alzheimers Dis. (2017) 56:25–36. doi: 10.3233/JAD-160889
117. Rasmussen KL, Nordestgaard BG, Frikke-Schmidt R, Nielsen SF. An updated Alzheimer hypothesis: complement C3 and risk of Alzheimer's disease-a cohort study of 95,442 individuals. Alzheimers Dement. (2018) 14:1589–601. doi: 10.1016/j.jalz.2018.07.223
118. Ishii T, Haga S. Immuno-electron-microscopic localization of complements in amyloid fibrils of senile plaques. Acta Neuropathol. (1984) 63:296–300. doi: 10.1007/BF00687336
119. Rogers J, Cooper NR, Webster S, Schultz J, McGeer PL, Styren SD, et al. Complement activation by beta-amyloid in Alzheimer disease. Proc Natl Acad Sci USA. (1992) 89:10016–20. doi: 10.1073/pnas.89.21.10016
120. Veerhuis R, van der Valk P, Janssen I, Zhan SS, Van Nostrand WE, Eikelenboom P. Complement activation in amyloid plaques in Alzheimer's disease brains does not proceed further than C3. Virchows Arch. (1995) 426:603–10. doi: 10.1007/BF00192116
121. Webster S, Lue LF, Brachova L, Tenner AJ, McGeer PL, Terai K, et al. Molecular and cellular characterization of the membrane attack complex, C5b-9, in Alzheimer's disease. Neurobiol Aging. (1997) 18:415–21. doi: 10.1016/S0197-4580(97)00042-0
122. Fonseca MI, McGuire SO, Counts SE, Tenner AJ. Complement activation fragment C5a receptors, CD88 and C5L2, are associated with neurofibrillary pathology. J Neuroinflammation. (2013) 10:25. doi: 10.1186/1742-2094-10-25
123. Cribbs DH, Berchtold NC, Perreau V, Coleman PD, Rogers J, Tenner AJ, et al. Extensive innate immune gene activation accompanies brain aging, increasing vulnerability to cognitive decline and neurodegeneration: a microarray study. J Neuroinflammation. (2012) 9:179. doi: 10.1186/1742-2094-9-179
124. Zhou J, Fonseca MI, Pisalyaput K, Tenner AJ. Complement C3 and C4 expression in C1q sufficient and deficient mouse models of Alzheimer's disease. J Neurochem. (2008) 106:2080–92. doi: 10.1111/j.1471-4159.2008.05558.x
125. Stephan AH, Madison DV, Mateos JM, Fraser DA, Lovelett EA, Coutellier L, et al. A dramatic increase of C1q protein in the CNS during normal aging. J Neurosci. (2013) 33:13460–74. doi: 10.1523/JNEUROSCI.1333-13.2013
126. Bradt BM, Kolb WP, Cooper NR. Complement-dependent proinflammatory properties of the Alzheimer's disease beta-peptide. J Exp Med. (1998) 188:431–8. doi: 10.1084/jem.188.3.431
127. Hernandez MX, Namiranian P, Nguyen E, Fonseca MI, Tenner AJ. C5a increases the injury to primary neurons elicited by fibrillar amyloid beta. ASN Neuro. (2017) 9:1759091416687871. doi: 10.1177/1759091416687871
128. Matsuoka Y, Picciano M, Malester B, LaFrancois J, Zehr C, Daeschner JM, et al. Inflammatory responses to amyloidosis in a transgenic mouse model of Alzheimer's disease. Am J Pathol. (2001) 158:1345–54. doi: 10.1016/S0002-9440(10)64085-0
129. Benoit ME, Hernandez MX, Dinh ML, Benavente F, Vasquez O, Tenner AJ. C1q-induced LRP1B and GPR6 proteins expressed early in Alzheimer disease mouse models, are essential for the C1q-mediated protection against amyloid-β neurotoxicity. J Biol Chem. (2013) 288:654–65. doi: 10.1074/jbc.M112.400168
130. Dejanovic B, Huntley MA, De Maziere A, Meilandt WJ, Wu T, Srinivasan K, et al. Changes in the synaptic proteome in tauopathy and rescue of tau-induced synapse loss by C1q antibodies. Neuron. (2018) 100:1322–36.e7. doi: 10.1016/j.neuron.2018.10.014
131. Litvinchuk A, Wan YW, Swartzlander DB, Chen F, Cole A, Propson NE, et al. Complement C3aR inactivation attenuates tau pathology and reverses an immune network deregulated in tauopathy models and Alzheimer's disease. Neuron. (2018) 100:1337–53.e5. doi: 10.1016/j.neuron.2018.10.031
132. Lansita JA, Mease KM, Qiu H, Yednock T, Sankaranarayanan S, Kramer S. Nonclinical development of ANX005: a humanized anti-C1q antibody for treatment of autoimmune and neurodegenerative diseases. Int J Toxicol. (2017) 36:449–62. doi: 10.1177/1091581817740873
133. Wyss-Coray T, Yan F, Lin AH-T, Lambris JD, Alexander JJ, Quigg RJ, et al. Prominent neurodegeneration and increased plaque formation in complement-inhibited Alzheimer's mice. Proc Natl Acad Sci USA. (2002) 99:10837–42. doi: 10.1073/pnas.162350199
134. Cleveland DW, Rothstein JD. From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat Rev Neurosci. (2001) 2:806–19. doi: 10.1038/35097565
135. Bruijn LI, Miller TM, Cleveland DW. Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu Rev Neurosci. (2004) 27:723–49. doi: 10.1146/annurev.neuro.27.070203.144244
136. Kretschmer BD, Kratzer U, Schmidt WJ. Riluzole, a glutamate release inhibitor, and motor behavior. Naunyn Schmiedebergs Arch Pharmacol. (1998) 358:181–90. doi: 10.1007/PL00005241
137. Wang SJ, Wang KY, Wang WC. Mechanisms underlying the riluzole inhibition of glutamate release from rat cerebral cortex nerve terminals (synaptosomes). Neuroscience. (2004) 125:191–201. doi: 10.1016/j.neuroscience.2004.01.019
138. McGeer PL, McGeer EG. Inflammatory processes in amyotrophic lateral sclerosis. Muscle Nerve. (2002) 26:459–70. doi: 10.1002/mus.10191
139. Donnenfeld H, Kascsak RJ, Bartfeld H. Deposits of IgG and C3 in the spinal cord and motor cortex of ALS patients. J Neuroimmunol. (1984) 6:51–7. doi: 10.1016/0165-5728(84)90042-0
140. Sta M, Sylva-Steenland RM, Casula M, de Jong JM, Troost D, Aronica E, et al. Innate and adaptive immunity in amyotrophic lateral sclerosis: evidence of complement activation. Neurobiol Dis. (2011) 42:211–20. doi: 10.1016/j.nbd.2011.01.002
141. Kawamata T, Akiyama H, Yamada T, McGeer PL. Immunologic reactions in amyotrophic lateral sclerosis brain and spinal cord tissue. Am J Pathol. (1992) 140:691–707.
142. Lee JD, Lee JY, Taylor SM, Noakes PG, Woodruff TM. Innate immunity in ALS. In: Maurer MH, editor. Amyotrophic Lateral Sclerosis. Croatia: InTech (2012). p. 393–412.
143. Yamada T, Akiyama H, McGeer PL. Complement-activated oligodendroglia: a new pathogenic entity identified by immunostaining with antibodies to human complement proteins C3d and C4d. Neurosci Lett. (1990) 112:161–6. doi: 10.1016/0304-3940(90)90196-G
144. Bahia El Idrissi N, Bosch S, Ramaglia V, Aronica E, Baas F, Troost D. Complement activation at the motor end-plates in amyotrophic lateral sclerosis. J Neuroinflammation. (2016) 13:72. doi: 10.1186/s12974-016-0538-2
145. Tüzün E, Christadoss P. Complement associated pathogenic mechanisms in myasthenia gravis. Autoimmun Rev. (2013) 12:904–11. doi: 10.1016/j.autrev.2013.03.003
146. Rodrigues MC, Hernandez-Ontiveros DG, Louis MK, Willing AE, Borlongan CV, Sanberg PR, et al. Neurovascular aspects of amyotrophic lateral sclerosis. Int Rev Neurobiol. (2012) 102:91–106. doi: 10.1016/B978-0-12-386986-9.00004-1
147. Grewal RP, Morgan TE, Finch CE. C1qB and clusterin mRNA increase in association with neurodegeneration in sporadic amyotrophic lateral sclerosis. Neurosci Lett. (1999) 271:65–7. doi: 10.1016/S0304-3940(99)00496-6
148. Annunziata P, Volpi N. High levels of C3c in the cerebrospinal fluid from amyotrophic lateral sclerosis patients. Acta Neurol Scand. (1985) 72:61–4. doi: 10.1111/j.1600-0404.1985.tb01548.x
149. Apostolski S, Nikolic J, Bugarski-Prokopljevic C, Miletic V, Pavlovic S, Filipovic S. Serum and CSF immunological findings in ALS. Acta Neurol Scand. (1991) 83:96–8. doi: 10.1111/j.1600-0404.1991.tb04656.x
150. Tsuboi Y, Yamada T. Increased concentration of C4d complement protein in CSF in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. (1994) 57:859–61. doi: 10.1136/jnnp.57.7.859
151. Mantovani S, Gordon R, Macmaw JK, Pfluger CM, Henderson RD, Noakes PG, et al. Elevation of the terminal complement activation products C5a and C5b-9 in ALS patient blood. J Neuroimmunol. (2014) 276:213–8. doi: 10.1016/j.jneuroim.2014.09.005
152. Boillee S, Vande Velde C, Cleveland DW. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron. (2006) 52:39–59. doi: 10.1016/j.neuron.2006.09.018
153. Da Cruz S, Cleveland DW. Understanding the role of TDP-43 and FUS/TLS in ALS and beyond. Curr Opin Neurobiol. (2011) 21:904–19. doi: 10.1016/j.conb.2011.05.029
154. Lee JD, Kamaruzaman NA, Fung JN, Taylor SM, Turner BJ, Atkin JD, et al. Dysregulation of the complement cascade in the hSOD1G93A transgenic mouse model of amyotrophic lateral sclerosis. J Neuroinflammation. (2013) 10:119. doi: 10.1186/1742-2094-10-119
155. Lobsiger CS, Boillee S, Cleveland DW. Toxicity from different SOD1 mutants dysregulates the complement system and the neuronal regenerative response in ALS motor neurons. Proc Natl Acad Sci USA. (2007) 104:7319–26. doi: 10.1073/pnas.0702230104
156. Heurich B, El Idrissi NB, Donev RM, Petri S, Claus P, Neal J, et al. Complement upregulation and activation on motor neurons and neuromuscular junction in the SOD1 G93A mouse model of familial amyotrophic lateral sclerosis. J Neuroimmunol. (2011) 235:104–9. doi: 10.1016/j.jneuroim.2011.03.011
157. Lee JD, Levin SC, Willis EF, Li R, Woodruff TM, Noakes PG. Complement components are upregulated and correlate with disease progression in the TDP-43(Q331K) mouse model of amyotrophic lateral sclerosis. J Neuroinflammation. (2018) 15:171. doi: 10.1186/s12974-018-1217-2
158. Beers DR, Henkel JS, Zhao W, Wang J, Appel SH. CD4+ T cells support glial neuroprotection, slow disease progression, and modify glial morphology in an animal model of inherited ALS. Proc Natl Acad Sci USA. (2008) 105:15558–63. doi: 10.1073/pnas.0807419105
159. Graveland GA, Williams RS, DiFiglia M. Evidence for degenerative and regenerative changes in neostriatal spiny neurons in Huntington's disease. Science. (1985) 227:770–3. doi: 10.1126/science.3155875
160. Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. (1996) 87:493–506. doi: 10.1016/S0092-8674(00)81369-0
161. Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EPJr. Neuropathological classification of Huntington's disease. J Neuropathol Exp Neurol. (1985) 44:559–77. doi: 10.1097/00005072-198511000-00003
162. Pavese N, Gerhard A, Tai YF, Ho AK, Turkheimer F, Barker RA, et al. Microglial activation correlates with severity in Huntington disease: a clinical and PET study. Neurology. (2006) 66:1638–43. doi: 10.1212/01.wnl.0000222734.56412.17
163. Tai YF, Pavese N, Gerhard A, Tabrizi SJ, Barker RA, Brooks DJ, et al. Microglial activation in presymptomatic Huntington's disease gene carriers. Brain. (2007) 130(Pt 7):1759–66. doi: 10.1093/brain/awm044
164. Singhrao SK, Neal JW, Morgan BP, Gasque P. Increased complement biosynthesis by microglia and complement activation on neurons in Huntington's disease. Exp Neurol. (1999) 159:362–76. doi: 10.1006/exnr.1999.7170
165. Hodges A, Strand AD, Aragaki AK, Kuhn A, Sengstag T, Hughes G, et al. Regional and cellular gene expression changes in human Huntington's disease brain. Hum Mol Genet. (2006) 15:965–77. doi: 10.1093/hmg/ddl013
166. Dalrymple A, Wild EJ, Joubert R, Sathasivam K, Björkqvist M, Petersén Å, et al. Proteomic profiling of plasma in Huntington's disease reveals neuroinflammatory activation and biomarker candidates. J Proteome Res. (2007) 6:2833–40. doi: 10.1021/pr0700753
167. Klegeris A, McGeer PL. Complement activation by islet amyloid polypeptide (IAPP) and alpha-synuclein 112. Biochem Biophys Res Commun. (2007) 357:1096–9. doi: 10.1016/j.bbrc.2007.04.055
168. Yamada T, McGeer PL, McGeer EG. Lewy bodies in Parkinson's disease are recognized by antibodies to complement proteins. Acta Neuropathol. (1992) 84:100–4. doi: 10.1007/BF00427222
169. Rozemuller AJ, Eikelenboom P, Theeuwes JW, Jansen Steur EN, de Vos RA. Activated microglial cells and complement factors are unrelated to cortical Lewy bodies. Acta Neuropathol. (2000) 100:701–8. doi: 10.1007/s004010000225
170. Loeffler DA, Camp DM, Conant SB. Complement activation in the Parkinson's disease substantia nigra: an immunocytochemical study. J Neuroinflammation. (2006) 3:29. doi: 10.1186/1742-2094-3-29
171. Wang Y, Hancock AM, Bradner J, Chung KA, Quinn JF, Peskind ER, et al. Complement 3 and factor h in human cerebrospinal fluid in Parkinson's disease, Alzheimer's disease, and multiple-system atrophy. Am J Pathol. (2011) 178:1509–16. doi: 10.1016/j.ajpath.2011.01.006
172. Antony PM, Diederich NJ, Balling R. Parkinson's disease mouse models in translational research. Mamm Genome. (2011) 22:401–19. doi: 10.1007/s00335-011-9330-x
173. Morgan BP, Harris CL. Complement, a target for therapy in inflammatory and degenerative diseases. Nat Rev Drug Discov. (2015) 14:857–77. doi: 10.1038/nrd4657
174. Kasinathan N, Jagani HV, Alex AT, Volety SM, Rao JV. Strategies for drug delivery to the central nervous system by systemic route. Drug Deliv. (2015) 22:243–57. doi: 10.3109/10717544.2013.878858
175. Lu CT, Zhao YZ, Wong HL, Cai J, Peng L, Tian XQ. Current approaches to enhance CNS delivery of drugs across the brain barriers. Int J Nanomedicine. (2014) 9:2241–57. doi: 10.2147/IJN.S61288
176. van Tellingen O, Yetkin-Arik B, de Gooijer MC, Wesseling P, Wurdinger T, de Vries HE. Overcoming the blood-brain tumor barrier for effective glioblastoma treatment. Drug Resist Updat. (2015) 19:1–12. doi: 10.1016/j.drup.2015.02.002
177. Boado RJ, Zhang Y, Zhang Y, Xia C-F, Pardridge WM. Fusion antibody for Alzheimer's disease with bidirectional transport across the blood– brain barrier and Aβ Fibril disaggregation. Bioconjugate Chem. (2007) 18:447–55. doi: 10.1021/bc060349x
178. Gordon R, Albornoz EA, Christie DC, Langley MR, Kumar V, Mantovani S, et al. Inflammasome inhibition prevents alpha-synuclein pathology and dopaminergic neurodegeneration in mice. Sci Transl Med. (2018) 10:eaah4066. doi: 10.1126/scitranslmed.aah4066
179. Pittock SJ, Lennon VA, McKeon A, Mandrekar J, Weinshenker BG, Lucchinetti CF, et al. Eculizumab in AQP4-IgG-positive relapsing neuromyelitis optica spectrum disorders: an open-label pilot study. Lancet Neurol. (2013) 12:554–62. doi: 10.1016/S1474-4422(13)70076-0
Keywords: complement, therapeutics, CNS, neurodegeneration, injury
Citation: Carpanini SM, Torvell M and Morgan BP (2019) Therapeutic Inhibition of the Complement System in Diseases of the Central Nervous System. Front. Immunol. 10:362. doi: 10.3389/fimmu.2019.00362
Received: 05 December 2018; Accepted: 12 February 2019;
Published: 04 March 2019.
Edited by:
John D. Lambris, University of Pennsylvania, United StatesReviewed by:
Marcela Pekna, University of Gothenburg, SwedenMaria-Grazia De Simoni, Istituto Di Ricerche Farmacologiche Mario Negri, Italy
Copyright © 2019 Carpanini, Torvell and Morgan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Megan Torvell, torvellm@cardiff.ac.uk
†These authors have contributed equally to this work