 Khalid Rashid
Khalid Rashid Isha Akhtar-Schaefer1
Isha Akhtar-Schaefer1 Thomas Langmann
Thomas Langmann- 1Laboratory for Experimental Immunology of the Eye, Department of Ophthalmology, Faculty of Medicine and University Hospital Cologne, University of Cologne, Cologne, Germany
- 2Center for Molecular Medicine Cologne, Cologne, Germany
The retina is a complex tissue with multiple cell layers that are highly ordered. Its sophisticated structure makes it especially sensitive to external or internal perturbations that exceed the homeostatic range. This necessitates the continuous surveillance of the retina for the detection of noxious stimuli. This task is mainly performed by microglia cells, the resident tissue macrophages which confer neuroprotection against transient pathophysiological insults. However, under sustained pathological stimuli, microglial inflammatory responses become dysregulated, often worsening disease pathology. In this review, we provide an overview of recent studies that depict microglial responses in diverse retinal pathologies that have degeneration and chronic immune reactions as key pathophysiological components. We also discuss innovative immunomodulatory therapy strategies that dampen the detrimental immunological responses to improve disease outcome.
Introduction
The retina is part of the central nervous system with more than 60 distinct retinal cell types, high level organization, and a evolutionarily conserved structure (1, 2). The mammalian retina contains three distinct glia cells types; Müller cells, astrocytes, and microglia (3, 4). Müller cells are specialized radial cells that span all retinal layers and account for about 90% of the retinal glia population (3, 5). They support functioning and metabolism of retinal neurons by releasing trophic factors, recycling neurotransmitter glutamate, and controlling extracellular ion homeostasis (6). Retinal astrocytes perform similar functions to Müller cells, ranging from neurotrophic, metabolic and mechanical support of neurons, and maintenance of the blood retinal barrier (3, 7). Retinal astrocytes are however almost entirely restricted to the nerve fiber layer, and to a lesser extent to the ganglion cell layer (7, 8). Microglia, the third glial cell type, represent the resident tissue macrophages and play important roles in retinal homeostasis, recovery from injury and progression of disease (9, 10).
As early as 1897, Spanish neuroscientist Ramón y Cajal began to appreciate that there were more cell types in the brain than just neurons and astrocytes (11, 12). In pursuit of this hypothesis, he developed the gold chloride sublimate staining approach that labeled astrocytes and other cells that he named the “third element” (12, 13). Del Río-Hortega, another Spanish neuroscientist later separated the cells in the “third element,” becoming the first scientist to describe microglia as distinct cellular entity (12, 14). He described their ramified morphology in the normal brain using elegant drawings and provided evidence of their morphological transformations during tissue brain pathology (12, 14). These tremendous findings have withstood the test of time and remain highly relevant today.
In the normal retina, microglia cells are located in the plexiform layers where they exhibit elaborate ramified processes responsible for immune surveillance of the retina (4). Noxious insults in the retina such as oxidative stress, hypoxia or inherited mutations trigger microglia reactivity manifested by amoeboid morphology, increased proliferation and migration to the sites of injury (10, 15). While this initial “constructive” inflammatory response can rapidly enhance tissue repair and return to homeostasis, sustained microglial inflammatory responses can instigate severe alterations in retinal integrity, and aggravate neuronal demise (16). This review therefore offers a comprehensive overview on the dual role of microglia in the retina, examining their contribution in various retinal neurodegenerative disorders and in aging and parainflammation. In addition, we discuss recent strategies that have been shown to suppress microglial inflammatory responses in the retina and prevent neuronal cell death.
Origin and Maintenance of Microglia in the Retina
Earlier attempts to find answers to the uncertainties existing around the origin and maintenance of retinal microglia were made by transplanting bone marrow (BM) derived cells from eGFP-transgenic mice into irradiated normal adult mice (17). Retinal flat mounts of the recipient mice were found to contain BM derived eGFP+ cells as early as 8-weeks post transplantation, and by the 6th month, all retinal myeloid cells were eGFP+ (17). Using CX3CR1-GFP/+ mice as BM donors, where GFP expression is restricted to monocyte lineage cells, a separate study observed recruitment of monocyte-derived cells into the retinal tissue 4 weeks post-transplantation (18). Of note, both studies observed that under steady state conditions, retinal microglia exhibited very low proliferation rates (17, 18). However, in a separate study where mice were irradiated and injected with eGFP+ BM cells prior to induction of retinal injury, only a minute number of donor cells were observed in the uninjured normal retina up to 12 months following BM transplantation (19). In contrast, a massive recruitment of BM derived cells into the retina was observed following injury (19). The authors partially attributed the low recruitment of donor cells to the uninjured eye to shielding eyes and heads of mice prior to irradiation (19). They argued that in the previous experiments, irradiation could have unintentionally damaged photoreceptors and enhanced migration of BM-derived cells to the injured retina (19). Indeed, additional factors not mentioned by the authors, such as large amounts of circulating cytokines, CNS vascular changes, and temporary disruptions of the blood-brain barrier alterations may confound interpretation of results following irradiation (20, 21).
Therefore, to further analyse the contribution of circulating BM derived progenitors on the turnover of resident microglia in the CNS, a separate study employed parabiosis experiments (22). Parabiosis, the surgical joining of two organisms, allowed for the sharing of the blood circulation between wild type and GFP transgenic mice without affecting their blood-brain nor the blood-retinal barrier (21–23). The obtained results revealed that under steady-state conditions, CNS microglia were a closed system with the capacity to self-renew and were not replenished by BM derived cells (22). Indeed, it was later established that microglia were derived from primitive myeloid progenitors originating from the yolk sac and that postnatal hematopoietic progenitors did not significantly contribute to microglia homeostasis (24).
A subsequent study (25) demonstrated that the brain harbored latent nestin+ microglial progenitors throughout the CNS (25). Pharmacological depletion of microglia using selective CSF1R inhibitor PLX3397 triggered rapid proliferation of nestin+ cells throughout the CNS, which later differentiated into microglia and repopulated the entire brain within 1 week of inhibitor cessation (25). Interestingly, this regeneration mirrored some aspects of normal development, as embryonic stem cells require a nestin+ stage on their way to becoming microglia (25, 26). However, subsequent microglia ablation studies have strongly disputed the presence of nestin+ microglia progenitor cells and instead provide evidence that microglia repopulation following pharmacological or genetic depletion occurs solely from residual non-depleted internal pools which exert massive proliferation and transiently express nestin (27–29). Similarly, retinas depleted of microglia using selective CSF1R inhibitor PLX5622 were not repopulated from nestin+ precursors (30, 31). Rather, repopulated retinal microglia exhibited dual extra retinal origins; first from residual microglia in the optic nerve and secondly from ciliary body/iris macrophages (30). Residual optic nerve microglia repopulated the retina along the center-to-periphery axis, while macrophages in the ciliary body/iris repopulated the retina along the periphery and accounted for around 15% of the repopulated microglia (30). Of note, these findings uncovered for the first time radial migratory routes of retinal microglia and demonstrated the presence of peripheral macrophage-derived microglia which were significantly less ramified than their central counterparts (30). In a parallel study, it was shown that microglia repopulation in the retina was regulated via neuronal-microglial crosstalk in the form of CX3CL1-CX3CR1 signaling which potentiated microglia proliferation and morphological maturation (31). Most importantly, the latter study demonstrated that that the repopulated cells fully restored microglia functions in the retina including immunosurveillance and synaptic maintenance (31).
Microglia in the Healthy Retina
Microglial cells play active roles in maintaining the normal structure and functioning of the retina. During retinal development, microglia cells are largely confined to the ganglion cell layer and inner plexiform layer, where they phagocytose cellular corpses of excessively produced retinal ganglion cells (RGCs) early in development (32). Moreover, in the early postnatal stages when there is a robust synaptic remodeling, microglia are involved in pruning of weak presynaptic terminals of RGCs (33). This process occurs in a complement C3-CR3-dependent mechanism, where activated C3 (iC3b/C3b) selectively labels the weak RGCs terminals triggering a C3-receptor dependent phagocytosis pathway in microglia (4, 33). Importantly, microglial dependent apoptosis of RGCs and the elimination of costly neural connections deemed unfit for proper functioning plays a crucial role in the normal postnatal development of the retina and cortical visual areas (32, 33).
Following the development phase, microglia cells move to occupy the inner and outer plexiform layers where they adopt a quiescent phenotype characterized by very small somata and extensively ramified filopodia-like processes (4). They form a mosaic network of evenly distributed non-overlapping cells that provide immunological surveillance to the entire retina by the continuous movement of their processes (4, 34). It is also possible that the dynamic activity of the filopodial structures serves a housekeeping function, enabling microglia eliminate accumulated waste metabolic products and cellular debris in the retinal microenvironment (35). In addition, the extensive processes facilitate close relationships with other retinal cells such as the neurons, promoting the maintenance of their synaptic structures and neurotransmission (3, 36). Indeed, sustained microglia depletion in the retina has been shown to result in the degeneration of photoreceptor synapses with subsequent progressive decline in the magnitude of electroretinographic responses to light stimuli (36).
Cross-talk between microglia and other retinal cells is enabled by the assortment of cell surface molecules on the microglial cell membrane (4, 37). These surface proteins are key regulators of tissue homeostasis that limit unnecessary microglia activation in the healthy retina (9). CD200-CD200R interaction is an important example that induces a negative inhibitory signal to restrain microglia from tissue damaging activation (38, 39). CD200 (previously known as OX2) is a broadly expressed membrane glycoprotein in ganglion cells, photoreceptors, vascular endothelium and the retinal pigment epithelium (RPE), while its receptor, CD200R, is predominantly expressed in retinal microglia (4, 9, 40). Studies on mice lacking CD200 have reported heightened pro-inflammatory responses in experimental animal models of uveoretinitis and exudative form of age related macular degeneration (38–40). Conversely, enhancement of CD200R signaling ameliorates pathological outcome in optic nerve injury and experimental autoimmune uveoretinitis animal models, suggesting that CD200-CD200R interaction could be an exploitable avenue for therapeutic intervention (39, 41).
Sialic acids, covalently linked to cellular membrane proteins and lipids, also contribute to the inhibition of the innate immune system in the CNS including the retina (42, 43). The interaction between polysialic acid chains (PSA) present on the glycocalyx of healthy neurons and Sialic acid binding immunoglobulin-like lectin 11 (Siglec11) receptor of microglia restricts unwanted microglial activation in the brain and retina (42, 44). The inhibitory signals are propagated via immunoreceptor tyrosine-based inhibitory motifs (ITIM) contained in the cytosolic domain of Siglec-11 in microglia (43). Therefore, given its immunomodulatory capacity, several studies have reported Siglec-11-ITIM mediated neuroprotective effects which will be discussed further later in this review (42, 44, 45).
A further neuroimmune regulator is CX3CL1 (fractalkine), a constitutively expressed neuron derived chemokine which binds to its sole receptor CX3CR1 present in microglia and maintains them in a quiescent mode (46). Indeed, numerous studies have demonstrated the immunomodulatory and neuroprotective effects of CX3CL1-CX3CR1 interaction in the brain and retina (46, 47). For instance, transplantation of mesenchymal stem cells engineered to secrete CX3CR1 in the subretinal space inhibited microglial activation and expression of pro-inflammatory factors in light-induced retinal degeneration in rats (48). In contrast, deletion of CX3CR1 in an rd10 retinal degeneration mouse model augmented microglial activation and infiltration into the photoreceptor layers with concomitant increase in photoreceptor demise (49). Remarkably, delivery of exogenous CX3CL1 to the rd10 mouse eye significantly slowed down the tempo of photoreceptor demise, underscoring CX3CL1-CX3CR1 signaling axis as an influential regulator of microglial activity (49).
Microglia also exchange functionally significant signals with Müller cells both in health and disease states, and this bidirectional communication can act as mediator of neuron-microglia crosstalk [Figure 1; (50, 51)]. Indeed, previous studies had demonstrated that certain microglia-derived neurotrophic factors such as BDNF and CNTF were neuroprotective for photoreceptor cells despite these cells not expressing their receptors (52–55). Subsequent studies later established that microglia derived neurotrophic factors interact with Müller cells and induce or inhibit the release of secondary factors including basic fibroblast growth factor (bFGF), leukemia inhibitory factor (LIF) and glial cell line-derived neurotrophic factor (GDNF) that could act directly on photoreceptors and mediate survival or apoptosis during stress conditions in the retina (56–60). Recent investigations have also revealed additional microglia-Müller cell cross-talk via translocator protein (TSPO; 18 kDa) signaling axis, where Müller cells release TSPO's endogenous ligand, diazepam binding inhibitor (DBI) protein, which binds microglial TSPO and suppresses microglial activation during retinal pathology (61).
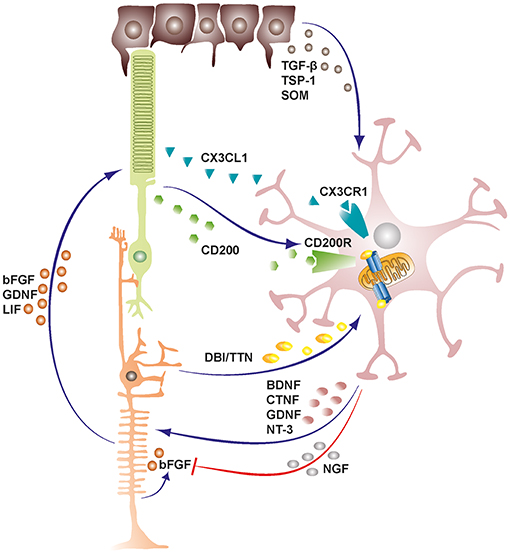
Figure 1. Schematic representation depicting cellular cross-talk between retinal cells and microglia. Retinal cells constantly communicate with microglia via various soluble factors and receptors to restrain microglia from tissue damaging activation and maintain them in a quiescent protective state. The bidirectional communicational between microglia and Müller cells can act as a mediator of neuron-microglia crosstalk. Microglia derived factors may either induce or inhibit release of secondary factors from Müller cells. TGF-β, transforming growth factor beta; TSP-1, thrombospondin-1; SOM, somatostatin; DBI, diazepine binding inhibitor; TTN, triakontatetraneuropeptide; BDNF, brain derived neurotrophic factor; CTNF, ciliary neurotrophic factor; GDNF, glial cell line-derived neurotrophic factor; NT-3, neurotrophin-3; NGF, nerve growth factor; bFGF, basic fibroblast growth factor; LIF, leukemia inhibitory factor.
As a result of the heterocellular signaling and crosstalk in the retina, microglia cells are rendered extremely sensitive to slight changes in their environment, demanding efficient inhibitory mechanisms to maintain them in a quiescent neuroprotective state (4). RPE cells prominently support the maintenance of an immunosuppressive intraocular environment by secreting inhibitory factors into the subretinal space between the neural retina-RPE interface (62). This blocks the undesirable infiltration of microglia cells and other mononuclear phagocytes into the photoreceptor layers and subretinal space, rendering these regions devoid of immune cells in the healthy retina (10, 63). Conversely, in both wet and dry forms of age related macular degeneration, there is an accumulation of mononuclear phagocytes in the subretinal space where they may contribute to disease pathogenesis (63). Some of the inhibitory factors released by RPE cells into the subretinal space include transforming growth factor-β (TGF-β), thrombospondin-1 (TSP-1) and somatostatin (SOM) (64–66).
Microglia in the Diseased Retina
A sustained chronic pro-inflammatory environment is an important common denominator of retinal degenerative diseases and neurological disorders that affect vision (10). Neuroinflammatory responses in the retina are orchestrated by microglial cells which constitute the resident immune cell population (67). Under acute conditions, microglia-mediated neuroinflammation promotes neuroprotection and regenerative processes and facilitates a rapid return to tissue homeostasis (68, 69). However, under chronic conditions where the insult persists over time, such as in retinal degenerative disorders, microglia become pathologically activated and release exaggerated amounts of inflammatory mediators that promote tissue damage and disease exacerbation (16, 68, 70). Consequently, modulation of microglial reactivity has emerged over the years as a promising therapeutic approach to attenuate neuronal demise and potentially slow down the onset and progression of retinal degenerative diseases. Therefore, the next sections will discuss the role microglia-related mechanisms in major retinal degenerative diseases like age-related macular degeneration, hereditary retinopathies, glaucoma, diabetic retinopathy as well as in aging. In addition, promising regimens that have shown potent immunomodulatory effects in animal models of retinal diseases and/or in human tissue culture as well as patient studies will be discussed.
Microglia in Age Related Macular Degeneration
AMD, with a global prevalence of ~170 million people, refers to a progressive deterioration of the macula (71). It is a neurodegenerative disease with a multifactorial etiology and is currently considered the leading cause of debilitating vision loss among the elderly (72). Early clinical features of the disease includes fundoscopic manifestation of drusen between the RPE and Bruch's Membrane and fundus autofluorescence that derives primarily from accumulation of lipofuscin granules in the RPE (73, 74). Unfortunately, 15% of patients presenting with early AMD symptoms will advance to the late stages of the disease which progresses in two main forms; (I) Neovascular or “wet” AMD characterized by the ingrowth of immature leaky choroidal blood vessel through the RPE and into the avascular outer retina and (II) Geographic atrophy or “dry” AMD characterized by deterioration of the RPE and associated support functions around the macula with concomitant loss of the overlying photoreceptors (75, 76). While effective treatments involving the inhibition of vascular endothelial growth factor (VEGF) exist for wet AMD, no truly effective form of treatment is available for dry AMD form which affects the majority of patients (77).
There is substantial evidence to indicate the involvement of inflammation and dysregulated innate immunity in the pathogenesis of AMD (73, 75, 78). Dysregulated innate immunity in AMD is associated with complement factors, inflammasome activation, and reactive microglia (71). Wet AMD patients for instance show elevated levels of complement fragments C3a and Ba as well as a wide range of cytokines in their aqueous humor (73, 79). Similarly, soft drusen from AMD donors contain bioactive fragments of complement components C3a and C5a which can induce VEGF expression and predispose patients to choroidal neovascularization (CNV) (80). Drusen from AMD donors can induce inflammasome activation in myeloid and mononuclear cells, suggesting that macular drusen in AMD is a potent pro-inflammatory stimulus (81). Indeed, the widespread accumulation of drusen components as seen in AMD is a prominent chemoattractant stimulus for microglia cells which results in their translocation to the subretinal space (82–84).
Consistently, enlarged amoeboid microglia have been found in the vicinity of RPE cells overlying drusen in retinal sections from dry AMD patients (70). In the outer retina, reactive microglia or microglia-derived factors can induce NLRP3 inflammasome activation in RPE cells with concomitant secretion of the proinflammatory cytokine IL-1β (85–87). NLRP3 inflammasome activation can subsequently induce RPE degeneration via caspase-1-mediated pyroptosis, contributing to AMD pathology (88). Notably, human ocular tissue sections from geographic atrophy or neovascular AMD donors exhibit NLRP3 inflammasome activation (88). Eventually, RPE degeneration causes secondary photoreceptor cell death, resulting in loss of visual function (75). Moreover, the pro-inflammatory environment fostered by accumulating subretinal microglia can directly induce death of nearby photoreceptors (89). We have indeed demonstrated on several instances that conditioned medium from reactive human and murine microglial cells triggers caspase-mediated photoreceptor cell death (85, 90). These findings therefore strongly suggest that microglia reactivity is a driving force in photoreceptor degeneration and progression of retinal degenerative disorders.
Indeed, the role of microglial reactivity in severity of disease has been clearly demonstrated in several mouse models of AMD. However, it is important to note that conclusions drawn from these studies are limited in nature since rodents lack an anatomical macula. Nonetheless, these models effectively mimic distinct features of human pathology including accumulation of immune cells such as macrophages or microglia in the subretinal space, photoreceptor degeneration, choroidal neovascularization, funduscopically visible drusen-like lesions, and RPE degeneration (91). We will therefore discuss a few of these models here to highlight the crucial role played by microglia in AMD pathogenesis. However, for a more detailed discussion on the animal models of AMD, the reader is directed to other articles (91–93).
Bright white light for example can be used to mimic the photoreceptor cell death and retinal degeneration witnessed in human AMD patients (94). Intense light causes bleaching of rhodopsin and excessive phototransduction signaling, resulting in photoreceptor apoptosis in an AP-1 dependent manner (94, 95). This is followed by a robust migration of microglia to the outer retina, making the light-damage model useful for studying physiological as well as pathological consequences of reactive microglia accumulation in the subretinal space as seen in human AMD patients (70). Of note is that microglial activation states and morphological changes are early events that precede recruitment to the outer retina following light induced damage (96). This recruitment of microglia to the outer retina following light damage is mediated in-part by chemokines (C-C motif) ligand 2 (Ccl2) (97) and (C-X3-C motif) ligand 1 (Cx3cl1/ fractalkine) (98) and by the complement anaphylatoxin receptor C5aR (99). Once in the subretinal space, microglia and other mononuclear phagocytes enhance their phagocytic capacity and produce high levels of pro-inflammatory factors including IL-1β, TNF-α, IL-6, CCL2, and iNOS (100). An overt consequence of this massive recruitment of amoeboid microglia in the subretinal space is a severe thinning of the outer nuclear layer as a result of photoreceptor degeneration via pro-inflammatory and phagocytosis mechanisms (16, 61, 89).
Another useful model for dry AMD is generated by immunizing mice with carboxyethylpyrrole (CEP)-adducted proteins (101). CEP is an oxidation product of docosahexaenoic acid (DHA) and is present in the eyes of AMD patients and in their serum at higher levels than in age-matched non-AMD controls (91, 101). This model mimics many aspects of the dry AMD pathology observed in human patients, including presence of sub-RPE drusen like deposits, RPE degeneration and infiltration of phagcoytes around hypertrophied photoreceptors and degenerating RPE (102). However in this model, RPE atrophy is more pronounced in regions devoid of immune cells in their vicinity, suggesting that subretinal mononuclear phagocytes are not involved in initiating RPE pathology, but may rather exert beneficial effects by removing debris released from apoptotic corpses of RPE cells (102). Indeed, some of the infiltrating macrophages found in the subretinal space contained melanin pigment, confirming RPE engulfment (102). This model could therefore be useful in understanding what processes or factors tips the scales toward neurotoxic damaging phagocytes in subretinal microglia.
To study microglial immune responses in wet AMD, a laser induced bruch's membrane photocoagulation model is used (71). Briefly, laser photocoagulation results in the rupture of Bruch's membrane, leading to the ingrowth of immature leaky blood vessels into the outer avascular retina (103). A local inflammatory reaction is triggered at the laser burn site and is accompanied by a rapid recruitment and accumulation of amoeboid microglia and other mononuclear phagocytes (104–106). The recruited mononuclear phagocytes produce high levels of pro-inflammatory cytokines and the pro-angiogenic factor VEGF, worsening disease progression and severity (104, 105). Indeed, blocking the VEGF receptor signaling significantly inhibits microglia accumulation in the laser spots with concomitant reduction in CNV (107). Conversely, blocking ifnar1/IFN-β or CX3CR1 signaling drastically enhances microglia reactivity and recruitment to the laser lesion site, exacerbating the development of CNV lesions in mice (104, 105).
Microglia in Hereditary Retinopathies
Hereditary degenerations of the human retina are a diverse group of clinically and genetically heterogeneous blinding diseases with more than 260 causal genes identified to date (108). Inherited retinopathies are mostly monogenic, with the causal mutations predominantly occurring in genes expressed in photoreceptors and RPE (4, 109). The most prevalent and severe form of inherited retinopathies is retinitis pigmentosa (RP), where vision loss is brought about by primary degeneration of rods, followed by the degeneration of cones (110). Microglia in human RP patients become reactive in response to signals from degenerating rods and migrate to the photoreceptor layers (70). These bloated microglia in the outer retina of RP patients engage in the phagocytosis of rod debris, as evidenced by their rhodopsin intracellular inclusions (70). In addition to phagocytosis of degenerate cells (70), proposed these reactive microglia in the outer retina exacerbate photoreceptor cell death including adjacent healthy cones by secreting pro-inflammatory neurotoxic factors. Using animal models that recapitulate aspects of human disease, scientists have proven this hypothesis and demonstrated accumulation of reactive microglia in the degenerating retinas that produce high levels pro-inflammatory cytokines and chemokines aggravating photoreceptor demise.
The rodless mouse, discovered approximately 93 years ago by the medical geneticist Keeler, was the first ever reported murine model of retinal degeneration (111). This natural mouse line carries a nonsense mutation in the Pde6b gene that codes for the β-subunit of cGMP phosphodiesterase (PDE), and mimics to a great extent the RP in human patients bearing mutations in the human homolog of the gene (112). Rd1 mice display an early onset and a rapid rate of photoreceptor degeneration, such that by 4 weeks of age, only a single layer of photoreceptors consisting predominantly of cone photoreceptors is left in the outer nuclear layer (113). Microglia cells are present in the outer nuclear layer of the photoreceptors by post-natal day 10 where they exhibit a highly proliferative capacity (114). Of note is that their distribution coincides with the spatiotemporal pattern of photoreceptor cell death, implying that microglial cells are intimately engaged with the degenerative process and are not mere bystanders of disease (114). During their migration to the outer nuclear layer of the photoreceptors, microglia lose their filopodial-like processes, and by the time they emerge in the subretinal space, they are fully transformed into amoeboid phagocytes whose pseudopodia can often be seen (114). These bloated infiltrating microglia have been shown to secrete high levels of TNF-α, chemokines CCL2 (alias MCP-1) and (CCL5, alias RANTES) accentuating the ongoing photoreceptor demise (115).
In the retinal degeneration 10 (rd10) mouse which harbors a missense mutation in the Pde6b gene but displays a much slower onset of retinal degeneration, a similar infiltration of microglia into the outer nuclear layer is observed at around P21 as the number of apoptotic rods increase (16). Interestingly, microglia activation in rd10 mice, indicated by the upregulation of microglia specific genes Cx3cr1, Aif1, Irf8, C1qc is already apparent at P12 before the onset of neurodegeneration (116). Upon activation, microglia migrate toward the degenerating photoreceptors and transition into amoeboid cells containing multiple phagosomes where they produce large amounts of the pro-inflammatory cytokine IL-1β and potentiate photoreceptor apoptosis (16). In addition, the reactive outer retina microglia engage in indiscriminate phagocytosis of stressed but living photoreceptors, aggravating neuronal demise, and disease severity (16). Of note is that the number of infiltrating microglia is significantly reduced in ccr2−/− rd10 mice, indicating that the ccr2/ccl2 signaling pathway plays a crucial role in mobilizing mononuclear phagocytes into the degenerating photoreceptor layer (117). Moreover, the reduced microglial infiltration was also associated with an increase in retinal thickness and function, strongly affirming microglia's involvement in inducing degenerative changes during retinal pathology (117).
In addition to the commonly used rd1 and rd10 mutants, rd7 and rd8 mutant mouse strains also display widespread microglia activation (118). In the rd7 mice where retinal degeneration occurs as a result of a spontaneous mutation in the Nr2e3 gene (119), reactive microglia are found under abnormal foldings (retinal rosettes) that develop in the outer nuclear layer and inner segments of photoreceptors (118). These reactive microglia phagocytose large amounts of debris between photoreceptor and RPE layers, resulting in the accumulation of lysosomes containing autofluorescent material (118). Activated-lysosome-laden microglia, which appear as autofluorescent dots in fundus images, secrete high amounts of pro-inflammatory cytokines IL-1β, IL-6, and TNF-α thereby accelerating retinal degeneration (118). Similarly, in the rd8 mice bearing a mutation in the Crb1 gene, increased amounts of microglia/macrophages positive for pro-inflammatory markers CD16, MHC-II, and iNOS accumulate in the subretinal space when compared to age matched wild type controls (120). This enhanced accumulation of reactive microglia overexpressing complement (C3 and CFB) and pro-inflammatory (TNF-α and NFκB) genes, is a specific response to degenerative changes that occur in the rd8 retina including development of retinal dystrophic lesions (120, 121).
Microglia in Diabetic Retinopathies
Diabetic retinopathy (DR) is a severe ocular complication of diabetes mellitus and is also the leading cause of blindness among the working age populations of industrialized regions (122). DR can be clinically divided into two forms; (i) an early non-proliferative form (NPDR) characterized by increased vascular permeability, retinal microvasculature degeneration, basement membrane thickening, and loss of pericytes in the retinal capillaries (ii) an advanced proliferative form (PDR) involving pathological neovascularization, vitreous hemorrhage, and retinal scars and detachment (123). Based on vascular abnormalities such as acellular capillaries, microscopic capillary loss, and microaneurysms, DR has traditionally been regarded as a classical microvascular disease (122). However, in recent years, numerous studies have highlighted the crucial role played by inflammation in the pathogenesis of DR (124–126).
Early indications of inflammation involvement in the pathogenesis of DR came from a study reporting a low incidence and high regression rate of DR when diabetic patients were treated with salicylates for rheumatoid arthritis complications (127). Since then, ample evidence exists to show that DR is a low grade chronic inflammatory condition characterized by leukostasis, RPE, and endothelial cell damage and associated blood retinal barrier (BRB) alteration. Subsequently, these events compromise the immune suppressive environment in the retina resulting in increased expression of pro-inflammatory factors (124, 128, 129). Indeed, several clinical studies have shown that the levels of many pro-inflammatory cytokines including TNF-α, IL-1β, IL-6, and IL-8 are elevated in the vitreous of DR patients (124, 128). In addition, persistent hyperglycaemia results in increased polyol and hexosamine pathways flux, inducing cellular oxidative stress and the generation of advanced glycation or lipoxidation end products (AGEs or ALEs) (122, 130). High levels of pro-inflammatory cytokines and the accumulation of AGEs and ALEs compromise cellular physiology and induce microglia activation (131, 132). Furthermore, oxidative stress induced by the noxious hyperglycaemic environment can trigger NFκB-mediated inflammatory responses in retinal microglia (133).
In human DR, hypertrophic and amoeboid microglia are present at different stages of the disease (134). In the NPDR form, there is a moderate increase in the number of reactive microglia which are mostly clustered around perivascular vessels and fresh hemorrhages in microaneurysms (135). In the intermediate pre-proliferative form, there is a dramatic increase in reactive microglia which cluster around cotton-wool spots and dilated vessels (135). Lastly in the PDR form, a marked increase in reactive microglia number is seen in the ganglion cell layer and around new immature blood vessels in the nerve fiber layer and the optic nerve head where the proliferative process is most prominent (135). Strikingly, reactive microglia in human DR are closely associated with perivascular compartments where they are postulated to exacerbate vascular permeability by propagating inflammatory responses (135, 136).
Similarly in murine models, changes to retinal microglia are a prominent feature of diabetic retinopathy (137, 138). In the Ins2Akita mouse model which harbors a spontaneous missense mutation in the insulin 2 gene, discrete pockets containing bloated microglia with shorter less branched processes can be observed in the diseased retina (138). Moreover, the normal laminar arrangement of the retinal microglia is markedly disrupted in these mice from 10 weeks of age (137). These DR induced microglia morphological alterations might result partly from disruptions in microglial Cx3cr1 signaling, as the detrimental morphological abnormalities are exacerbated in the absence of Cx3cr1 signaling (137).
In the Goto-Kakizaki (GK) inbred rat model of type II diabetes, numerous Iba1+ amoeboid microglia/macrophages overexpressing inducible form of nitric oxide synthase (iNOS) can be found all over the retina and in the subretinal space following 12 months of hyperglycaemia (139). The enhanced trafficking of microglia/macrophages to the subretinal space at week 12 is correlated with a reduction in the number of “tunnel-like” invaginations which mediate a physiological transcellular pathway in RPE cells (139). Alteration of this transcellular pathway and subsequent subretinal accumulation of activated microglia/macrophages induces morphological abnormalities in the outer retina including disorganization of photoreceptor outer segments (139). Notably, intraocular injection of a protein kinase C zeta (PKCζ) inhibitor suppresses iNOS expression in microglia/macrophages and impairs their recruitment to the subretinal space (139).
Microglia reaction is also elicited in pharmacologically induced diabetes where toxic glucose analogs alloxan and streptozotocin (STZ) are used to preferentially destroy pancreatic beta cells and induce a state of insulin-dependent diabetes (134, 140, 141). Starting at 4 months of age, an upsurge of microglia is observed in the different retinal layers and especially the outer plexiform and ganglion cell layer in STZ treated rats (134). There is also a significant shift in microglia phenotype toward activated amoeboid cells with hypertrophied cell bodies (134, 142). As the disease progresses, microglia in the outer plexiform layer extend their processes into the outer nuclear layer, with some CD11b positive microglia occasionally being found in the photoreceptor layers and subretinal space (134). Interestingly in alloxan treated mice, microglia morphological changes including shortened processes and hypertrophied cellular somas precede neuronal apoptosis and BRB breakdown in this model, implying that microglia cells play an important role in the onset and development of DR (141).
Microglia in Glaucoma
Glaucoma refers to a distinctive group of optic neuropathies characterized by the progressive demise of retinal ganglion cells (RGCs) and their axons, thinning of the retinal nerve fiber layer and cupping of the optic disc (143). Glaucoma, estimated to affect ~79.6 million people by 2020, is the most frequent cause of irreversible blindness worldwide (144, 145). Major risk factors for the development and progression of glaucoma include old age, race, ocular and systemic hypertension, diabetes, and high myopia (146–148). To date, the only proven therapeutic approach to prevent development and slow down disease progression is the lowering of intraocular pressure (IOP) via drug treatment, laser therapy or surgery (51, 143). However, despite effectiveness of the intraocular pressure-lowering approach which eliminates the primary source of injury, evidence exists of a secondary degeneration that persists, affecting neighboring neurons and continuing the pathological process (149–151). An important mechanism that has been proposed to provoke such secondary degeneration of the RGCs in optic nerve disorders is the dysregulation of innate immunity and the associated neurodegenerative inflammatory responses (152).
Consistent with this hypothesis, aqueous humor from glaucoma patients have been shown in several studies to contain higher levels of pro-inflammatory cytokines and chemokines, suggesting that neuroinflammatory processes play a key role in glaucomatous neurodegeneration (153–156). Microglia, the resident tissue macrophages, are the principal sources of pro-inflammatory mediators in the retina and have been shown in both human and experimentally induced glaucoma in rodents to be central players in perpetuating the neuroinflammatory process in glaucoma (4, 157–159). In human glaucoma eyes, clusters of large amoeboid, reactive microglia gather around the compressed lamina cribrosa and its surrounding blood vessels, forming concentric circles (159). Enlarged reactive microglia, occurring either singly or in clusters, are also found in the parapapillary chorioretinal region (where the RPE and the bruch's membrane terminate) of glaucomatous optic nerve heads (ONH) (159). Proteomic and subsequent immunohistochemical analysis detected abundant expression of toll like receptor 2 (TLR2), TLR4, and TLR7 in microglia of diseased patients, implicating TLR signaling as a pathomechanism in glaucomatous neurodegeneration (160). Moreover, these reactive microglia were shown to express abundant levels of matrix metalloproteinases (MMP) 1, MMP-2, MMP-3, and MMP-14, pro-inflammatory factors TNF-α and NOS-2 and the anti-inflammatory cytokine TGF-β (158). The high expression of TGF-β suggests that microglia attempt to downregulate the degenerative reactions in the glaucomatous ONH, but as the disease progresses, their contribution turns detrimental leading to neurodegeneration and degradation of the extracellular matrix (158).
Murine models of glaucoma where increased ocular hypertension occurs naturally or is experimentally induced using genetic manipulation or surgical procedures have been instrumental in understanding microglia's role in disease pathogenesis (161). Using DBA/2J (D2) mice, an established model of chronic inherited glaucoma carrying spontaneous mutations in TYRP1 (tyrosinase-related protein 1) and GPNMB (glycosylated protein nmb), microglia activation and proliferation was shown to occur at stages prior to overt RGCs neurodegeneration (162, 163). These findings provide evidence supporting an active involvement of microglial activation in the onset and progression of RGCs neurodegeneration in the glaucomatous retina (162, 163). Microglia mediate the neurodegeneration of RGCs, at least in-part, by secreting high amounts of TNF-α (164). Consistently, mice deficient for TNF-R1 are significantly protected against RGCs loss following optic nerve crush when compared to control animals with a functional TNF death receptor signaling (164). Moreover, both TNF-α and its receptor TNF-R1 are upregulated in glaucomatous retinas in the RGCs and retinal glial cells, respectively, suggesting a direct contribution of the TNF-α signaling cascade in optic nerve degeneration in glaucoma (165, 166). Notably, blocking TNF-α activity with Etanercept, a clinically approved agent, inhibits microglial inflammation with concomitant attenuation of axonal degeneration in the optic nerve and RGC demise (167).
Nitric oxide (NO) synthesized by microglia also plays a role in the pathophysiology of glaucoma (168, 169). Increased levels of iNOS and NO have been shown to occur in the ONH of glaucomatous patients and experimental animal models of glaucoma (168–171). Importantly, pharmacological inhibition of NO synthesis using timolol and aminoguanidine in rats with elevated IOP drastically suppresses the degeneration of RGCs, showing that NO-mediated mechanisms contribute to neuronal cell death during glaucoma (169, 172).
Increased IL-6 expression has also been reported as a key component of the retinal microglia response in glaucoma, although the available evidence is contradictory (173–175). Early in-vitro experiments conducted on primary glia cultures isolated from rat retina demonstrated that microglia, and not astrocytes were the primary sources of IL-6 following elevated pressure conditions (175). Interestingly, microglial secreted IL-6 was shown to significantly abate apoptotic cell death in RGCs injured by elevated pressure, suggesting a beneficial effect of this cytokine in countering proapoptotic signals in the glaucomatous retina (174). In contrast, other investigations have reported that while an IL6 deficiency promotes an anti-inflammatory, pro-survival retinal environment, it also induces an exaggerated TNF-α response in the presence of glaucomatous stressors such as ocular hypertension (176). Moreover, IL6−/− mice are protected from IOP induced structural degeneration of the optic nerve and subsequent decrease in vision acuity, suggesting that IL-6 may play a specific role in the progression of RGC axonopathy and vision loss (173). Further studies are therefore needed to delineate ambiguities and lack of clarity in reported outcomes prior to the development of IL-6 as a therapeutic target.
Targeting Microglia for the Treatment of Retinal Degenerative Diseases
Microglia participates in both physiological and pathophysiological functions in the retina (4). Therefore, despite the fact that neuroinflammatory responses from overly reactive microglia play a critical role in the onset and progression of retinal degenerative disorders, complete blocking of retinal microglial functions would result to undesirable effects (36). Hence, valid immunotherapeutic approaches for the treatment of retinal degeneration should be those that inhibit dysregulated microglial-mediated pro-inflammatory responses and/or simultaneously enhance their beneficial neuroprotective functions. In the remainder of this review, we discuss promising therapeutic strategies that have been used to modulate microgliosis and improve disease outcome during retinal pathologies.
Polysialic Acid Receptors
In the vertebrate nervous system, healthy neuronal cells carry a polysialic acid (PolySia) cap on their glycocalyx made up of α2-8-glycosidically linked N-acetylneuraminic acid residues and which are attached by means of Hildebrandt et al. (177) and Schnaar et al. (178). The glycosylation state of healthy neurons is vigilantly monitored by cell surface carbohydrate binding receptors on microglia referred to sialic acid-binding immunoglobulin-like lectins (Siglecs) (43). Neural cell-adhesion molecule (NCAM), involved in cell–cell interactions and cell–extracellular-matrix adhesion, is the most prominent neuronal protein modified by PolySia (179). Polysialylated neuronal cell adhesion molecule (PolySia-NCAM) binds a CD33-related, primate lineage-specific Siglec-11 receptor on microglia, and this interaction serves to limit microglia immune responses and maintaining them in a quiescent neuroprotective state [Figure 2; (43)]. The inhibitory signaling of Siglecs is mediated through conserved ITIM-domains contained in their cytosolic tails (180). Upon ligand binding, ITIM is phosphorylated by Src-kinases, leading to a subsequent recruitment of protein-tyrosine phosphatases Src-homology region 2 domain-containing phosphatase 1 (SHP1) and SHP2 (180). Once recruited, SHPs dephosphorylate activated tyrosine residues on immunoreceptor tyrosine-based activation motif (ITAM), inhibiting Syk kinase mediated inflammatory signaling, ROS production and phagocytosis (180–183). Indeed, the interaction of ectopically expressed human Siglec-11 in mouse microglia with PolySia residues on neurons potently downregulates LPS-induced pro-inflammatory gene transcription and phagocytic capacity in microglia, confirming that ITIM-signaling impedes the inflammatory, and phagocytosis-associated ITAM-Syk cascade (181).
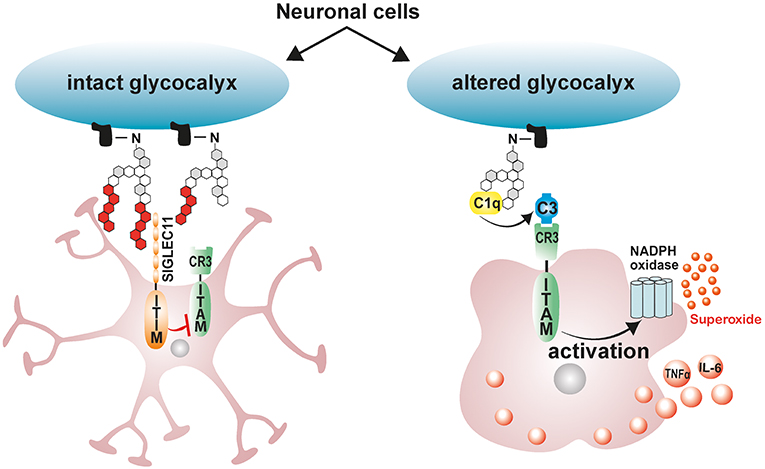
Figure 2. Schematic model depicting the immunomodulatory effects of neuronal polysialic acids on microglia. Healthy neuronal cells have an intact glycocalyx displaying a polysialic acid (PolySia) cap. The PolySia cap is recognized and bound by Siglec-11, an inhibitory ITIM-signaling receptor. This interaction between neuronal PolySia and Siglec-11 inhibits microglia activation and maintains retinal homeostasis. However, during pathological conditions, neuronal glycocalyx is altered, leading to degradation of sialic acid caps. The exposed neuronal glycocalyx is then opsonized by complement component C1q, leading to subsequent recognition by ITAM-containing complement receptor 3 (CR3) in microglia and activation of inflammatory signaling and ROS production. ITIM, immunoreceptor tyrosine-based inhibition motif; ITAM, immunoreceptor tyrosine-based activation motif.
Under pathological conditions such as elevated inflammation and oxidative stress, degradation of sialic acid caps occurs leaving neurons and other cells with a damaged glycocalyx (184–186). During such pathological conditions, soluble sialic acid residues actually accumulate in serum signifying the removal of sialic acid caps from glycoproteins (184, 185). Desialylated neuronal glycocalyx are then opsonized by complement component C1q, leading to their subsequent phagocytosis and clearance by the activating-signaling receptor complement receptor 3 (CR3) in microglia/macrophages (43). Consistently, blockage of CD11b as part of the complement receptor 3 (CR3) attenuates desialylated neurite phagocytosis by macrophages (187). However, excessive C1q mediated microglial phagocytosis of desialylated neurons would exacerbate ongoing neuronal loss in retinal degenerative disorders (188). This phenomenon is clearly observed in glaucoma mouse models where C1q localizes with neuronal synapses early on in the disease, and blocking neuronal opsonization by depleting C1q protects mice from glaucoma induced neurodegeneration (188). These findings therefore suggest that treatment with exogenous polysialic acids may be neuroprotective by limiting disease induced microglial inflammatory responses and phagocytic capacity.
Indeed, treatment of human macrophages challenged with LPS or amyloid-β1−42 with nanomolar concentrations of low molecular weight polySia with an average degree of polymerization 20 (polySia avDP20) significantly reduces pro-inflammatory gene transcription, inflammation-induced phagocytosis, and oxidative burst (44). Similarly in the retina, treatment with polySia avDP20 was shown to significantly reduce microglial activation, vascular leakage and production of deleterious ROS in humanized transgenic mice expressing SIGLEC-11 following laser-induced choroidal neovascularization (42). Interestingly, higher doses of polySia avDP20 were shown to block alternative complement activation and membrane attack complex formation in the diseased retina but in a SIGLEC-11 independent manner (42).
Translocator Protein Ligands
Translocator protein (18 kDa; TSPO), formerly referred to as the peripheral benzodiazepine receptor (PBR), is a highly conserved 5α-helical transmembrane protein located on the outer mitochondrial membrane (OMM) (189). TSPO exhibits a high constitutive expression in steroidogenic tissues such as adrenal glands, gonads and the placenta, but is very weakly expressed in the normal healthy brain (190, 191). However, during neuropathology, there is a strong increase in TSPO protein expression in the brain which colocalizes predominantly with activated microglia (190, 192, 193). This strong upregulation of TSPO in the brain from a very low baseline led to the development of numerous TSPO positron-emission tomography (PET) ligands for the non-invasive imaging of neuroinflammation (194, 195). Similarly, during retinal inflammation and disease, there is a strong upregulation of TSPO in reactive migratory microglia, and this induction accurately marks the extent and duration of retinal inflammation (190, 193). Interestingly, as activated microglia upregulate TSPO expression during retinal inflammation and disease, astrocytes, and Müller cells upregulate the production and secretion of Diazepam binding inhibitor (DBI), a 9 kDa endogenous TSPO protein ligand (61). DBI is then taken up by microglia cells, and the binding of DBI to microglial TSPO serves to limit the magnitude of microglial inflammatory responses [Figure 3; (61)]. Secreted DBI can also be cleaved extracellularly by acidic endopeptidases into its biologically active cleavage product triakontatetraneuropeptide (TTN) (196). TTN interaction with retinal microglial TSPO similarly suppresses inflammatory responses and facilitates a return of activated microglia to baseline quiescence (61, 196). Exploiting this endogenous immunomodulatory pathway, we tested the ability of a synthetic and highly specific TSPO ligand, XBD173 (AC-5216, emapunil), to influence microglial reactivity in the acute white light-induced retinal degeneration mouse model (193). White light exposure is an environmental risk factor that contributes to the faster onset and progression of human retinal degeneration disorders such as AMD and Retinitis Pigmentosa (197–200). Consistently, rodents exposed to bright white light experience a synchronized burst of photoreceptor cell death and thinning of the outer nuclear layer (94, 95, 193). Photoreceptor demise is brought about by rhodopsin bleaching and excessive phototransduction signaling, which creates an intracellular death signal that translocates transcription factor AP-1 to the nucleus to mediate photoreceptor apoptosis (59, 94, 95). Using this model, we demonstrated that the TSPO ligand XBD173 markedly inhibited accumulation of amoeboid microglia in the outer retina with concomitant preservation of the outer nuclear layer (193). Moreover, XBD173 efficiently suppressed pro-inflammatory induced gene expression and morphological transition in microglia with concomitant reduction in microglial neurotoxicity on cultured photoreceptors (190). However, the precise mechanisms involved in the TSPO ligands neuroprotective effects remain largely unknown, but likely involves, at least in part, enhanced steroidogenesis.
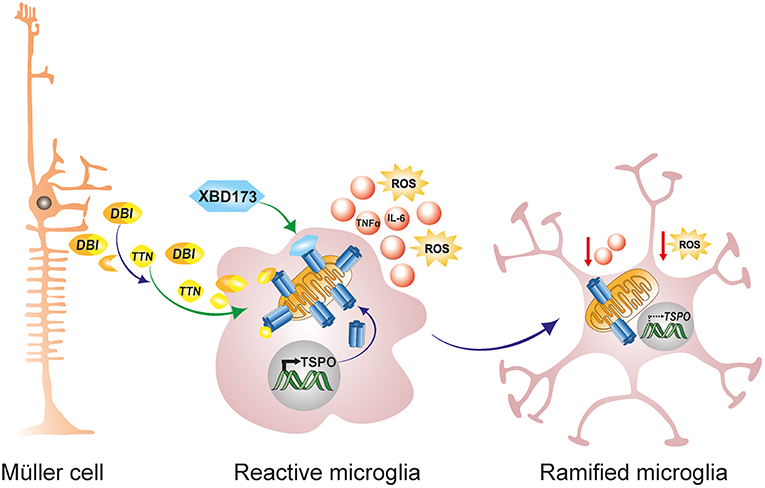
Figure 3. Endogenous and exogenous TSPO ligands dampen microglial activation. During retinal pathophysiology, microglia upregulate TSPO expression while Müller cells simultaneously upregulate the production and secretion of the endogenous TSPO ligand DBI. Secreted DBI can subsequently be cleaved extracellularly into the biologically active product TTN. Extracellular DBI and TTN are taken up by microglia, and their binding to the TSPO receptor serves to limit the magnitude of microglial inflammatory responses. DBI, diazepine binding inhibitor; TTN, triakontatetraneuropeptide; TSPO, translocator protein 18 kDa.
Indeed, the most studied physiological function of TSPO involves the translocation of cholesterol from the outer to the inner mitochondrial membrane as a rate limiting step for steroidogenesis (189, 201). TSPO's implication in steroidogenesis has been based upon its localization to the OMM, presence of a high affinity cholesterol recognition amino acid consensus (CRAC) motif, high expression in steroidogenic cells and the ability of TSPO ligands to stimulate steroid synthesis (202–204). In our own in-vitro experiments in the lab, we have demonstrated that treatment of microglia cells with XBD173 induces pregnenolone synthesis, and that inhibition of this process with aminoglutethimide partially abolishes XBD173 therapeutic effects (190). Similarly, the endogenous ligand TTN induces synthesis of pregnenolone derived Dehydroepiandrosterone (DHEA), inhibiting microglia-mediated proinflammatory responses (61). Outside the retina, DBI and synthetic TSPO binding ligands stimulates cholesterol translocation to the inner mitochondrial membrane increasing pregnenolone formation by isolated adrenocortical, Leydig and glia cell mitochondria (205, 206). In addition, gene silencing to inhibit DBI expression in Leydig cells was shown to significantly suppress hormone-induced steroidogenesis but not adenylate cyclase nor cholesterol side chain cleavage (P450SCC) enzyme activities (207). Of note is that the absence of TSPO in mutant rats causes increased neutral lipid accumulation in the adrenal glands and testis, reduced circulating testosterone and undetectable levels of the neurosteroid allopregnanolone (208). Together, these findings strongly suggest that TSPO and its ligands regulate steroid and neurosteroid levels.
Steroid hormones have subsequently been shown to attenuate neuroinflammatory reactions via autocrine and paracrine signaling mechanisms (209). Binding of steroids to their cytoplasmic and nuclear bound receptors results in the rapid inhibition of pro-inflammatory gene transcription via both genomic and non-genomic mechanisms (210). In the retina, a synthetic progesterone, norgestrel, exerts potent neuroprotective effects in the rd10 mouse model of human RP by reducing the extent of neuronal apoptosis with concurrent preservation of the photoreceptor layer (211). Norgestrel mediated rescue of stressed photoreceptors was shown to be mediated by the pleiotropic cytokine LIF, the pro-survival growth factor bFGF and the fractalkine-CX3CR1 signaling cascade (211–213). In addition, norgestrel directly targets reactive microglia in the rd10 mouse, attenuating their pro-inflammatory gene expression and NO production and significantly lessening microglial neurotoxicity on photoreceptor like cells in vitro (214). Moreover, the steroid hormone 17β-estradiol (βE2), which can be synthesized in the brain from testosterone, has been shown to protect rats against light-induced retinal degeneration by upregulating the NRF2-antioxidant pathway and reducing ROS production (215). In summary, TSPO ligands, working in part via inducing neurosteroidogenesis, present as attractive therapeutic regimens to dampen inflammatory responses from overly reactive microglia during retinal degenerative diseases.
Interferon-β
Interferon-β (IFN-β) belongs to the type I IFN family which are best known for their ability to induce a cellular antimicrobial state during a viral or bacterial infection (216, 217). IFN-β also possesses strong immunomodulatory properties, making it a first-line immunotherapy against relapsing remitting Multiple Sclerosis (MS) (217). IFN-β mediates neuroprotection against MS, an inflammatory and demyelinating CNS disorder characterized by multifocal brain lesions, by recruiting microglia to the lesion sites, enhancing their phagocytic capacity and accelerating clearance of accumulated myelin debris (218–220). Conversely, experimental autoimmune encephalomyelitic (EAE) mice which mimic the disease process of MS develop an exacerbated disease course accompanied by microglial hyperactivation and increased lethality in the absence of myeloid IFN-β signaling (221). Therefore, based on the immunomodulatory properties of IFN-β in MS neuropathology, we hypothesized that IFN-β may confer neuroprotection against chronic inflammation observed in neovascular age related macular degeneration (AMD) and thereby improve disease outcome.
We used the laser-induced choroidal neovascularization (CNV) mouse model that mimics features of exudative AMD to test this hypothesis (103). Briefly, laser photocoagulation results in the rupture of the Bruch's membrane, leading to the ingrowth of immature leaky blood vessels from the choroid to the subretinal space (103). Of note is that Bruch's membrane rupture is accompanied by rapid recruitment of myeloid cells to the lesion sites with concomitant elevation of proinflammatory and angiogenic factors, triggering the formation and growth of new blood vessels from the choroid (75, 222). Using this model, we demonstrated that IFN-β strongly inhibits microglia/macrophage activation and recruitment and induces a transition of microglia morphology toward a neuroprotective ramified with more cellular processes (104). Moreover, IFN-β treated animals showed considerable improvement in disease outcome signified by a reduction in vascular leakage and neoangiogenesis (104). These results corroborated those of a previous study which showed that local administration of IFN-β exacerbated wound healing of retinal lesions produced by laser photocoagulation in rabbits (223). Conversely, global or microglia specific conditional deletion of IFN-β/IFNAR1 signaling in mice resulted in an exacerbated disease marked by reactive microgliosis and enhanced vascular leakage (104). These findings strongly suggest that Ifnar1/IFN-β signaling, particularly in microglia, prevents chronic inflammation and pathological neovascularization in the retina and could be therapeutically targeted in wet AMD and other retinal inflammatory disorders. Remarkably, IFN-β treatment was reported to completely reverse subfoveal neovascularization and choroiditis in an MS patient, underscoring the importance of IFN-β in the management of ocular inflammation and neovascularization (224).
Several mechanisms have been proposed for IFN-β immunomodulatory and anti-angiogenic effects. One such mechanism involves the IFN-β induced expression of suppressor of cytokine signaling 1 (SOCS1) and SOCS3 (225, 226). IFN-β and other type I IFNs activate the transcription of SOCS1 and SOCS3 as part of a negative feedback loop to curb prolonged signaling by pro-inflammatory cytokines via the JAK-STAT pathway (227). Once produced, SOCS1 and SOCS3 are recruited to cytokine receptors where they inhibit the catalytic activity of JAK tyrosine kinases and negatively regulate inflammatory responses (228, 229). This phenomenon has been clearly demonstrated in the retinas of experimental autoimmune uveoretinitis (EAU) mice lacking SOCS3 in myeloid cells (230). These retinas showed elevated levels of pro-angiogenic factor VEGF-A and pro-inflammatory cytokines IL-1β, TNF-α, and IFN-γ (230). Consequently, myeloid-specific SOCS3 deficient mice displayed exaggerated retinal degeneration and accelerated retinal angiogenesis compared to their wildtype counterparts (230). In contrast, overexpression of SOCS1 was shown to protect retinal cells against staurosporine and H2O2-induced apoptosis (231). Notably, the overexpression SOCS1 in EAU mice significantly lessens disease severity by suppressing inflammatory chemokine expression and inhibiting recruitment of inflammatory cells into the retina (231).
IFN-β is also known to mediate its immunomodulatory effects via the noncanonical activation of the PI3K–AKT–mTOR pathway (232, 233). A recent study indeed revealed a central role of IFN-β signaling in the regulation of the PI3K–AKT pathway, by demonstrating that neurons from Ifnb−/− mice exhibited striking reductions in Pi3K and Akt mRNA and protein levels (234). Moreover, phosphorylated Pi3k and Akt, which signify active signaling, were even more pronouncedly reduced in the IFN-β incompetent neurons (234). Following activation, PI3K–AKT–mTOR pathway has been shown to exert anti-inflammatory and neuroprotective effects (215, 235). Activation of mTORC1, downstream of PI3K–AKT induces the phosphorylation of STAT3 at Ser727, thereby enhancing expression of the counter regulator of inflammatory signaling, SOCS3 (236). Of note, the pharmacological blockade of Pi3k-Akt pathway abolishes the antioxidative and neuroprotective effects conferred by the steroid hormone βE2 following light induced retinal degeneration in rats (215). Similarly, in LPS activated primary rat microglia, pharmacological blockade of the Pi3k-Akt-mTOR pathway significantly enhances inflammatory cyclooxygenase-2 (COX-2) activity and elevates production of associated downstream prostanoids PGE2 and PGD2 (237, 238).
Furthermore, IFN-β mediated activation of the PI3-AKT pathway negatively regulates the activity of the pro-inflammatory enzyme glycogen synthase kinase-3 (GSK-3) (239). GSK-3 is a critical mediator of inflammatory responses in the CNS and has been shown to downregulate production of anti-inflammatory cytokine IL-10 and promote that of pro-inflammatory cytokines TNF-α, IFN-γ, and IL-6 in microglia and macrophages (239, 240). Activation of AKT downstream of IFN-β-PI3 causes inhibition of GSK-3 by serine phosphorylation, negatively regulating inflammatory responses in the CNS [Figure 4; (239)]. Notably, treatment of rd10 mice with a small molecule inhibitor of GSK-3 suppresses pro-inflammatory responses in the retina with concomitant reduction in photoreceptor demise and preservation of visual function (241). However, further research in this area is necessary to delineate the contributions of this pathway to the immunomodulatory effects of IFN-β on microglia during retinal inflammation and disease.
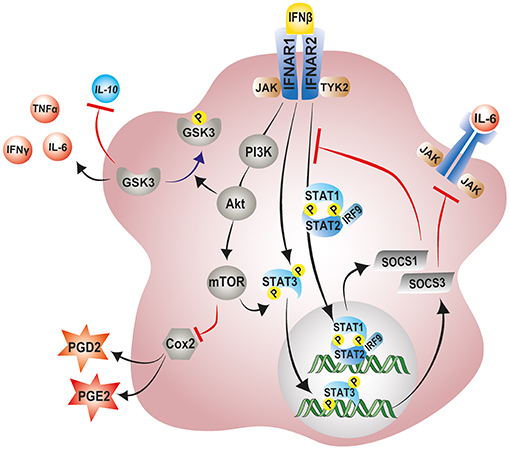
Figure 4. Modulation of microglial inflammatory responses by IFN-β signaling. IFN-β ligation to its receptor complex, IFNAR, triggers activation of the associated tyrosine kinases JAK1 and TYK2 and the subsequent phosphorylation of STAT1, STAT2, and STAT3 transcription factors. Phosphorylated STAT1 and STAT2 can recruit IRF-9 to the form the trimolecular complex STAT1–STAT2–IRF9 (ISGF3). Phosphorylated STAT3, and ISGF3 translocate to the nucleus and induce the transcription of various interferon-stimulated genes including SOCS1 and SOCS3 as part of a negative feedback loop. SOCS1 and SOCS3 inhibit excessive signaling by pro-inflammatory cytokines via the JAK-STAT pathway. IFN-β can also non-canonically activate the PI3K–AKT–mTOR pathway and suppress microglial inflammatory responses. mTORC1, downstream of PI3K–AKT can phosphorylate and activate STAT3, thereby enhancing expression of SOCS3. Activation of AKT downstream of IFN-β-PI3 inhibits GSK-3 activity through phosphorylation, negatively regulating GSK-3 mediated inflammatory responses, and promoting IL-10 expression. JAK1, Janus kinase 1; TYK2, tyrosine kinase 2; STAT, signal transducer and activator of transcription; IRF9, interferon regulatory factor 9; PI3K, phosphoinositide 3-kinase; AKT, protein Kinase B; mTOR, mammalian target of rapamycin; GSK-3, glycogen synthase kinase 3.
Minocycline
Minocycline is a highly lipophilic, semi-synthetic tetracycline derivative which is mainly used in the treatment of acne vulgaris and rheumatoid arthritis (242). However, due to its ability to readily penetrate the CNS and its profound anti-inflammatory properties, minocycline has recently emerged as a powerful immunomodulatory drug that is well-suited for CNS disorders (242, 243). Minocycline blocks microglial activation in response to a variety of inflammatory stimuli by inhibiting Toll-like receptor 2 (TLR2) and TLR4 signaling as well as several MAP kinases including p38, c-Jun-N-terminal activated protein kinase (JNK) 1/2, and extracellular signal regulated kinase (ERK) 1/2 (244, 245). In addition, minocycline inhibits NFκB transcriptional activity by suppressing the degradation of inhibitor of kappa B-alpha (IκBα) (244, 246).
Consistent with its anti-inflammatory properties, minocycline benefits during retinal pathophysiology have been confirmed in several experimental models. In a light-induced retinal degeneration mouse model, minocycline treatment was shown to significantly inhibit photoreceptor apoptosis and preserve retinal structure (89). Moreover, minocycline potently inhibited microgliosis and the accumulation of reactive amoeboid microglia in the subretinal space following light induced retinal damage (89). In an rd10 mouse model of human RP, treatment with minocycline significantly reduced microglia-mediated photoreceptor apoptosis, improving retinal structure and function (247). In a rat model of diabetic retinopathy, minocycline treatment represses the release of pro-inflammatory cytokines IL-1β, and TNF-α with concomitant reduction in caspase-3 mediated apoptosis in the retina (248). Similarly, in a streptozotocin-induced rat model of DR, minocycline treatment inhibited the abnormal expression of poly (ADP-ribose) polymerase 1 (PARP1), a chromatin-associated enzyme that promotes proinflammatory responses in glial cells, with concurrent reduction in the number of apoptotic cells in the diabetic retina (249). In the DBA/2J chronic mouse model of glaucoma, minocycline inhibited microglial activation, and increased the fraction of microglia in a ramified neuroprotective state (250). Of note, minocycline improved RGC axonal transport and integrity in the glaucomatous retina (250).
Minocycline protective effects have also been examined in the human retina (251). Retinitis pigmentosa mediated decline in visual field was shown to be reversed upon long term minocycline treatment (251). In a NIH proof of concept clinical study involving patients with diabetic macula edema (NTC01120899), a vision threatening form of DR, treatment with oral minocycline reduced abnormal vascular permeability and leakage with concomitant improvement in vision acuity (252). Taken together, these findings demonstrate the broad range immunomodulatory and neuroprotective effects of minocycline and underscore the importance of further testing to establish this antibiotic as an immunotherapy against retinal pathologies.
Conclusion
Studies using rodent models in the past decade have been instrumental in understanding the role of microglial responses in retinal pathologies. Ample evidence generated from these studies shows unequivocally that microglia reactivity and chronic inflammation is a common hallmark of various retinal pathologies, and that pharmacological targeting of overly reactive microglia ameliorates disease pathogenesis. However, why and how immunologic checkpoints become overwhelmed resulting in dysregulation of microglial inflammatory responses remains poorly understood. Therefore, prior to development of microglial-based therapeutic approaches, further studies are needed to understand the etiological factors and molecular mechanisms that propagate microglial responses during retinal degeneration. In addition, owing to the numerous overlapping insults that predispose retinal pathologies, it will be prudent to develop microglia based therapeutic approaches that will complement, or even synergize, other retinal therapeutic approaches.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This study was funded by the Velux Foundation, the German Academic Exchange Service (DAAD), and the Deutsche Forschungsgemeinschaft (FOR2240 project 6).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Gollisch T, Meister M. Eye smarter than scientists believed: neural computations in circuits of the retina. Neuron. (2010) 65:150–64. doi: 10.1016/j.neuron.2009.12.009
2. Matsuda T, Cepko CL. Electroporation and RNA interference in the rodent retina in vivo and in vitro. Proc Natl Acad Sci USA. (2004) 101:16–22. doi: 10.1073/pnas.2235688100
3. Vecino E, Rodriguez FD, Ruzafa N, Pereiro X, Sharma SC. Glia–neuron interactions in the mammalian retina. Prog Retin Eye Res. (2016) 51:1–40. doi: 10.1016/j.preteyeres.2015.06.003
4. Karlstetter M, Scholz R, Rutar M, Wong WT, Provis JM, Langmann T. Retinal microglia: just bystander or target for therapy? Prog Retin Eye Res. (2015) 45:30–57. doi: 10.1016/j.preteyeres.2014.11.004
5. Goldman D. Müller glial cell reprogramming and retina regeneration. Nat Rev Neurosci. (2014) 15:431–42. doi: 10.1038/nrn3723
6. Reichenbach A, Bringmann A. New functions of Müller cells. Glia. (2013) 61:651–78. doi: 10.1002/glia.22477
7. Ramírez JM, Triviño A, Ramírez AI, Salazar JJ. Organization and function of astrocytes in human retina. In: Castellano B, González B, Nieto-Sampedro M, editors. Understanding Glial Cells. Boston, MA: Springer US. p. 47–62. doi: 10.1007/978-1-4615-5737-1_3
8. Fernández-Sánchez L, Lax P, Campello L, Pinilla I, Cuenca N. Astrocytes and Müller cell alterations during retinal degeneration in a transgenic rat model of retinitis pigmentosa. Front Cell Neurosci. (2015) 9:484. doi: 10.3389/fncel.2015.00484
9. Langmann T. Microglia activation in retinal degeneration. J Leukoc Biol. (2007) 81:1345–51. doi: 10.1189/jlb.0207114
10. Chen M, Xu H. Parainflammation, chronic inflammation, and age-related macular degeneration. J Leukoc Biol. (2015) 98:713–25. doi: 10.1189/jlb.3RI0615-239R
11. Ramon y Cajal. Algo sobre la significacion fisiologica de la neuroglia. Rev Trimest Microgr. (1897) 2:33–47.
12. Sierra A, de Castro F, del Río-Hortega J, Rafael Iglesias-Rozas J, Garrosa M, Kettenmann H. The “Big-Bang” for modern glial biology: translation and comments on Pío del Río-Hortega 1919 series of papers on microglia. Glia. (2016) 64:1801–40. doi: 10.1002/glia.23046
13. Ramon y Cajal. Contribucion al conocimiento de la neuroglia del cerebro humano. Trab Lab Invest Biol Univer Madrid. (1913) 11:215–315.
14. Río-Hortega. “Tercer elemento” de Los Centros Nerviosos. II. Intervencion de la microglia en los procesos patologicos (Cellulas en bastocito y cuerpos granulo-adiposos). Bol Soc Esp Biol. (1919) 9:91–103.
15. Masuda T, Shimazawa M, Hara H. Retinal diseases associated with oxidative stress and the effects of a free radical scavenger (Edaravone). Oxid Med Cell Longev. (2017) 2017:9208489. doi: 10.1155/2017/9208489
16. Zhao L, Zabel MK, Wang X, Ma W, Shah P, Fariss RN, et al. Microglial phagocytosis of living photoreceptors contributes to inherited retinal degeneration. EMBO Mol Med. (2015) 7:1179–97. doi: 10.15252/emmm.201505298
17. Xu H, Chen M, Mayer EJ, Forrester JV, Dick AD. Turnover of resident retinal microglia in the normal adult mouse. Glia. (2007) 55:1189–98. doi: 10.1002/glia.20535
18. Kezic J, McMenamin PG. Differential turnover rates of monocyte-derived cells in varied ocular tissue microenvironments. J Leukoc Biol. (2008) 84:721–9. doi: 10.1189/jlb.0308166
19. Kaneko H, Nishiguchi KM, Nakamura M, Kachi S, Terasaki H. Characteristics of bone marrow–derived microglia in the normal and injured retina. Investig Opthalmol Vis Sci. (2008) 49:4162. doi: 10.1167/iovs.08-1738
20. Diserbo M, Agin A, Lamproglou I, Mauris J, Staali F, Multon E, Amourette C. Blood-brain barrier permeability after gamma whole-body irradiation: an in vivo microdialysis study. Can J Physiol Pharmacol. (2002) 80:670–8. doi: 10.1139/y02-070
21. Ransohoff RM. Microgliosis: the questions shape the answers. Nat Neurosci. (2007) 10:1507–9. doi: 10.1038/nn1207-1507
22. Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FMV. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci. (2007) 10:1538–43. doi: 10.1038/nn2014
23. Kamran P, Sereti K-I, Zhao P, Ali SR, Weissman IL, Ardehali R. Parabiosis in mice: a detailed protocol. J Vis Exp. (2013) e50556. doi: 10.3791/50556
24. Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. (2010) 330:841–5. doi: 10.1126/science.1194637
25. Elmore MRP, Najafi AR, Koike MA, Dagher NN, Spangenberg EE, Rice RA, et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron. (2014) 82:380–97. doi: 10.1016/j.neuron.2014.02.040
26. Hughes EG, Bergles DE. Hidden progenitors replace microglia in the adult brain. Neuron. (2014) 82:253–5. doi: 10.1016/j.neuron.2014.04.010
27. Zhan L, Krabbe G, Du F, Jones I, Reichert MC, Telpoukhovskaia M, et al. Proximal recolonization by self-renewing microglia re-establishes microglial homeostasis in the adult mouse brain. PLoS Biol. (2019) 17:e3000134. doi: 10.1371/journal.pbio.3000134
28. Bruttger J, Karram K, Wörtge S, Regen T, Marini F, Hoppmann N, et al. Genetic cell ablation reveals clusters of local self-renewing microglia in the mammalian central nervous system. Immunity. (2015) 43:92–106. doi: 10.1016/j.immuni.2015.06.012
29. Huang Y, Xu Z, Xiong S, Sun F, Qin G, Hu G, et al. Repopulated microglia are solely derived from the proliferation of residual microglia after acute depletion. Nat Neurosci. (2018) 21:530–40. doi: 10.1038/s41593-018-0090-8
30. Huang Y, Xu Z, Xiong S, Qin G, Sun F, Yang J, et al. Dual extra-retinal origins of microglia in the model of retinal microglia repopulation. Cell Discov. (2018) 4:9. doi: 10.1038/s41421-018-0011-8
31. Zhang Y, Zhao L, Wang X, Ma W, Lazere A, Qian H-H, et al. Repopulating retinal microglia restore endogenous organization and function under CX3CL1-CX3CR1 regulation. Sci Adv. (2018) 4:eaap8492. doi: 10.1126/sciadv.aap8492
32. Bodeutsch N, Thanos S. Migration of phagocytotic cells and development of the murine intraretinal microglial network: an in vivo study using fluorescent dyes. Glia. (2000) 32:91–101. doi: 10.1002/1098-1136(200010)32:1<91::AID-GLIA90>3.0.CO;2-X
33. Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. (2012) 74:691–705. doi: 10.1016/j.neuron.2012.03.026
34. Damani MR, Zhao L, Fontainhas AM, Amaral J, Fariss RN, Wong WT. Age-related alterations in the dynamic behavior of microglia. Aging Cell. (2011) 10:263–76. doi: 10.1111/j.1474-9726.2010.00660.x
35. Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. (2005) 308:1314–8. doi: 10.1126/science.1110647
36. Wang X, Zhao L, Zhang J, Fariss RN, Ma W, Kretschmer F, et al. Requirement for microglia for the maintenance of synaptic function and integrity in the mature retina. J Neurosci. (2016) 36:2827–42. doi: 10.1523/JNEUROSCI.3575-15.2016
37. Kierdorf K, Prinz M. Factors regulating microglia activation. Front Cell Neurosci. (2013) 7:44. doi: 10.3389/fncel.2013.00044
38. Broderick C, Hoek RM, Forrester JV, Liversidge J, Sedgwick JD, Dick AD. Constitutive retinal CD200 expression regulates resident microglia and activation state of inflammatory cells during experimental autoimmune uveoretinitis. Am J Pathol. (2002) 161:1669–77. doi: 10.1016/S0002-9440(10)64444-6
39. Copland DA, Calder CJ, Raveney BJE, Nicholson LB, Phillips J, Cherwinski H, et al. Monoclonal antibody-mediated CD200 receptor signaling suppresses macrophage activation and tissue damage in experimental autoimmune uveoretinitis. Am J Pathol. (2007) 171:580–8. doi: 10.2353/ajpath.2007.070272
40. Horie S, Robbie SJ, Liu J, Wu W-K, Ali RR, Bainbridge JW, et al. CD200R signaling inhibits pro-angiogenic gene expression by macrophages and suppresses choroidal neovascularization. Sci Rep. (2013) 3:3072. doi: 10.1038/srep03072
41. Huang R, Lan Q, Chen L, Zhong H, Cui L, Jiang L, et al. CD200Fc attenuates retinal glial responses and RGCs apoptosis after optic nerve crush by modulating CD200/CD200R1 interaction. J Mol Neurosci. (2018) 64:200–10. doi: 10.1007/s12031-017-1020-z
42. Karlstetter M, Kopatz J, Aslanidis A, Shahraz A, Caramoy A, Linnartz-Gerlach B, et al. Polysialic acid blocks mononuclear phagocyte reactivity, inhibits complement activation, and protects from vascular damage in the retina. EMBO Mol Med. (2017) 9:154–66. doi: 10.15252/emmm.201606627
43. Linnartz-Gerlach B, Mathews M, Neumann H. Sensing the neuronal glycocalyx by glial sialic acid binding immunoglobulin-like lectins. Neuroscience. (2014) 275:113–24. doi: 10.1016/j.neuroscience.2014.05.061
44. Shahraz A, Kopatz J, Mathy R, Kappler J, Winter D, Kapoor S, et al. Anti-inflammatory activity of low molecular weight polysialic acid on human macrophages. Sci Rep. (2015) 5:16800. doi: 10.1038/srep16800
45. Wielgat P, Braszko JJ. The participation of sialic acids in microglia–neuron interactions. Cell Immunol. (2012) 273:17–22. doi: 10.1016/j.cellimm.2011.12.002
46. Wolf Y, Yona S, Kim K-W, Jung S. Microglia, seen from the CX3CR1 angle. Front Cell Neurosci. (2013) 7:26. doi: 10.3389/fncel.2013.00026
47. Zieger M, Ahnelt PK, Uhrin P. CX3CL1 (Fractalkine) protein expression in normal and degenerating mouse retina: in vivo studies. PLoS ONE. (2014) 9:e106562. doi: 10.1371/journal.pone.0106562
48. Huang L, Xu W, Xu G. Transplantation of CX3CL1-expressing mesenchymal stem cells provides neuroprotective and immunomodulatory effects in a rat model of retinal degeneration. Ocul Immunol Inflamm. (2013) 21:276–85. doi: 10.3109/09273948.2013.791925
49. Zabel MK, Zhao L, Zhang Y, Gonzalez SR, Ma W, Wang X, et al. Microglial phagocytosis and activation underlying photoreceptor degeneration is regulated by CX3CL1-CX3CR1 signaling in a mouse model of retinitis pigmentosa. Glia. (2016) 64:1479–91. doi: 10.1002/glia.23016
50. Wang M, Wong WT. Microglia-Mller cell interactions in the retina. In: Ash J, Grimm C, Hollyfield J, Anderson R, LaVail M, Bowes Rickman C, editors. Recent Advances in Retinal Degeneration. Advances in Experimental Medicine and Biology, Vol. 801. New York, NY: Springer (2014) p. 333–8. doi: 10.1007/978-1-4614-3209-8_42
51. Madeira MH, Boia R, Santos PF, Ambrósio AF, Santiago AR. Contribution of microglia-mediated neuroinflammation to retinal degenerative diseases. Mediat Inflamm. (2015) 2015:673090. doi: 10.1155/2015/673090
52. Ugolini G, Cremisi F, Maffei L. TrkA, TrkB and p75 mRNA expression is developmentally regulated in the rat retina. Brain Res. (1995) 704:121–4. doi: 10.1016/0006-8993(95)01191-9
53. Kirsch M, Lee M-Y, Meyer V, Wiese A, Hofmann H-D. Evidence for multiple, local functions of ciliary neurotrophic factor (CNTF) in retinal development: expression of CNTF and its receptor and in vitro effects on target cells. J Neurochem. (2002) 68:979–90. doi: 10.1046/j.1471-4159.1997.68030979.x
54. Wilson RB, Kunchithapautham K, Rohrer B. Paradoxical role of BDNF: BDNF ± retinas are protected against light damage-mediated stress. Invest Ophthalmol Vis Sci. (2007) 48:2877–86. doi: 10.1167/iovs.06-1079
55. Wen R, Song Y, Liu Y, Li Y, Zhao L, Laties AM. CNTF negatively regulates the phototransduction machinery in rod photoreceptors: implication for light-induced photostasis plasticity. In: Anderson RE, LaVail MM, Hollyfield JG, editors. Recent Advances in Retinal Degeneration Advances in Experimental Medicine and Biology, Vol. 613. New York, NY: Springer. (2008). p. 407–13. doi: 10.1007/978-0-387-74904-4_48
56. Harada T, Harada C, Nakayama N, Okuyama S, Yoshida K, Kohsaka S, et al. Modification of glial-neuronal cell interactions prevents photoreceptor apoptosis during light-induced retinal degeneration. Neuron. (2000) 26:533–41. doi: 10.1016/S0896-6273(00)81185-X
57. Harada T, Harada C, Kohsaka S, Wada E, Yoshida K, Ohno S, et al. Microglia-Müller glia cell interactions control neurotrophic factor production during light-induced retinal degeneration. J Neurosci. (2002) 22:9228–36. doi: 10.1523/JNEUROSCI.22-21-09228.2002
58. Wang M, Ma W, Zhao L, Fariss RN, Wong WT. Adaptive Müller cell responses to microglial activation mediate neuroprotection and coordinate inflammation in the retina. J Neuroinflammation. (2011) 8:173. doi: 10.1186/1742-2094-8-173
59. Wenzel A, Grimm C, Samardzija M, Reme CE. Molecular mechanisms of light-induced photoreceptor apoptosis and neuroprotection for retinal degeneration. Prog Retin Eye Res. (2005) 24:275–306. doi: 10.1016/j.preteyeres.2004.08.002
60. Shen W, Zhu L, Lee S-R, Chung SH, Gillies MC. Involvement of NT3 and P75NTR in photoreceptor degeneration following selective Müller cell ablation. J Neuroinflammation. (2013) 10:137. doi: 10.1186/1742-2094-10-137
61. Wang M, Wang X, Zhao L, Ma W, Rodriguez IR, Fariss RN, et al. Macroglia-microglia interactions via TSPO signaling regulates microglial activation in the mouse retina. J Neurosci. (2014) 34:3793–806. doi: 10.1523/JNEUROSCI.3153-13.2014
62. Zamiri P, Sugita S, Streilein JW. Immunosuppressive properties of the pigmented epithelial cells and the subretinal space. In: Niederkorn JY, Kaplan HJ, editors. Immune Response and the Eye. Basel: KARGER. p. 86–93. doi: 10.1159/000099259
63. Sennlaub F, Auvynet C, Calippe B, Lavalette S, Poupel L, Hu SJ, et al. CCR2(+) monocytes infiltrate atrophic lesions in age-related macular disease and mediate photoreceptor degeneration in experimental subretinal inflammation in Cx3cr1 deficient mice. EMBO Mol Med. (2013) 5:1775–93. doi: 10.1002/emmm.201302692
64. Pfeffer BA, Flanders KC, Guérin CJ, Danielpour D, Anderson DH. Transforming growth factor beta 2 is the predominant isoform in the neural retina, retinal pigment epithelium-choroid and vitreous of the monkey eye. Exp Eye Res. (1994) 59:323–33. doi: 10.1006/exer.1994.1114
65. Miyajima-Uchida H, Hayashi H, Beppu R, Kuroki M, Fukami M, Arakawa F, et al. Production and accumulation of thrombospondin-1 in human retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. (2000) 41:561–7. Available online at: https://iovs.arvojournals.org/article.aspx?articleid=2199849
66. Zamiri P, Masli S, Streilein JW, Taylor AW. Pigment epithelial growth factor suppresses inflammation by modulating macrophage activation. Investig Opthalmology Vis Sci. (2006) 47:3912–8. doi: 10.1167/iovs.05-1267
67. Karlstetter M, Ebert S, Langmann T. Microglia in the healthy and degenerating retina : insights from novel mouse models. Immunobiology. (2010) 215:685–91. doi: 10.1016/j.imbio.2010.05.010
68. Gupta N, Shyamasundar S, Patnala R, Karthikeyan A, Arumugam TV, Ling E-A, et al. Recent progress in therapeutic strategies for microglia-mediated neuroinflammation in neuropathologies. Expert Opin Ther Targets. (2018) 22:765–81. doi: 10.1080/14728222.2018.1515917
69. Bellver-Landete V, Bretheau F, Mailhot B, Vallières N, Lessard M, Janelle M-E, et al. Microglia are an essential component of the neuroprotective scar that forms after spinal cord injury. Nat Commun. (2019) 10:518. doi: 10.1038/s41467-019-08446-0
70. Gupta N, Brown KE, Milam AH. Activated microglia in human retinitis pigmentosa, late-onset retinal degeneration, and age-related macular degeneration. Exp Eye Res. (2003) 76:463–71. doi: 10.1016/S0014-4835(02)00332-9
71. Ambati J, Atkinson JP, Gelfand BD. Immunology of age-related macular degeneration. Nat Rev Immunol. (2013) 13:438–51. doi: 10.1038/nri3459
72. Pennington KL, DeAngelis MM. Epidemiology of age-related macular degeneration (AMD): associations with cardiovascular disease phenotypes and lipid factors. Eye Vis. (2016) 3:34. doi: 10.1186/s40662-016-0063-5
73. Fritsche LG, Fariss RN, Stambolian D, Abecasis GR, Curcio CA, Swaroop A. Age-related macular degeneration: genetics and biology coming together. Annu Rev Genomics Hum Genet. (2014) 15:151–71. doi: 10.1146/annurev-genom-090413-025610
74. Tong Y, Ben Ami T, Hong S, Heintzmann R, Gerig G, Ablonczy Z, et al. Hyperspectral autofluorescence imaging of drusen and retinal pigment epithelium in donor eyes with age-related macular degeneration. Retina. (2016) 36 (Suppl. 1):S127–36. doi: 10.1097/IAE.0000000000001325
75. Ambati J, Fowler BJ. Mechanisms of age-related macular degeneration. Neuron. (2012) 75:26–39. doi: 10.1016/j.neuron.2012.06.018
76. Chichagova V, Hallam D, Collin J, Zerti D, Dorgau B, Felemban M, et al. Cellular regeneration strategies for macular degeneration: past, present and future. Eye. (2018) 32:946–71. doi: 10.1038/s41433-018-0061-z
77. Taskintuna I, Elsayed MEAA, Schatz P. Update on clinical trials in dry age-related macular degeneration. Middle East Afr J Ophthalmol. (2016) 23:13–26. doi: 10.4103/0974-9233.173134
78. Schick T, Steinhauer M, Aslanidis A, Altay L, Karlstetter M, Langmann T, et al. Local complement activation in aqueous humor in patients with age-related macular degeneration. Eye. (2017) 31:810–3. doi: 10.1038/eye.2016.328
79. Jonas JB, Tao Y, Neumaier M, Findeisen P. Cytokine concentration in aqueous humour of eyes with exudative age-related macular degeneration. Acta Ophthalmol. (2012) 90:e381–8. doi: 10.1111/j.1755-3768.2012.02414.x
80. Nozaki M, Raisler BJ, Sakurai E, Sarma JV, Barnum SR, Lambris JD, et al. Drusen complement components C3a and C5a promote choroidal neovascularization. Proc Natl Acad Sci USA. (2006) 103:2328–33. doi: 10.1073/pnas.0408835103
81. Doyle SL, Campbell M, Ozaki E, Salomon RG, Mori A, Kenna PF, et al. NLRP3 has a protective role in age-related macular degeneration through the induction of IL-18 by drusen components. Nat Med. (2012) 18:791–8. doi: 10.1038/nm.2717
82. Indaram M, Ma W, Zhao L, Fariss RN, Rodriguez IR, Wong WT. 7-Ketocholesterol increases retinal microglial migration, activation and angiogenicity: a potential pathogenic mechanism underlying age-related macular degeneration. Sci Rep. (2015) 5:9144. doi: 10.1038/srep09144
83. Rodriguez IR, Clark ME, Lee JW, Curcio CA. 7-ketocholesterol accumulates in ocular tissues as a consequence of aging and is present in high levels in drusen. Exp Eye Res. (2014) 128:151–5. doi: 10.1016/j.exer.2014.09.009
84. Penfold PL, Madigan MC, Gillies MC, Provis JM. Immunological and aetiological aspects of macular degeneration. Prog Retin Eye Res. (2001) 20:385–414. doi: 10.1016/S1350-9462(00)00025-2
85. Madeira MH, Rashid K, Ambrósio AF, Santiago AR, Langmann T. Blockade of microglial adenosine A2A receptor impacts inflammatory mechanisms, reduces ARPE-19 cell dysfunction and prevents photoreceptor loss in vitro. Sci Rep. (2018) 8:2272. doi: 10.1038/s41598-018-20733-2
86. Nebel C, Aslanidis A, Rashid K, Langmann T. Activated microglia trigger inflammasome activation and lysosomal destabilization in human RPE cells. Biochem Biophys Res Commun. (2017) 484:681–6. doi: 10.1016/j.bbrc.2017.01.176
87. Ma W, Zhao L, Fontainhas AM, Fariss RN, Wong WT. Microglia in the mouse retina alter the structure and function of retinal pigmented epithelial cells: a potential cellular interaction relevant to AMD. PLoS ONE. (2009) 4:e7945. doi: 10.1371/journal.pone.0007945
88. Tseng WA, Thein T, Kinnunen K, Lashkari K, Gregory MS, D'Amore PA, et al. NLRP3 inflammasome activation in retinal pigment epithelial cells by lysosomal destabilization: implications for age-related macular degeneration. Investig Opthalmol Vis Sci. (2013) 54:110–20. doi: 10.1167/iovs.12-10655
89. Scholz R, Sobotka M, Caramoy A, Stempfl T, Moehle C, Langmann T. Minocycline counter-regulates pro-inflammatory microglia responses in the retina and protects from degeneration. J Neuroinflammation. (2015) 12:209. doi: 10.1186/s12974-015-0431-4
90. Wiedemann J, Rashid K, Langmann T. Resveratrol induces dynamic changes to the microglia transcriptome, inhibiting inflammatory pathways and protecting against microglia-mediated photoreceptor apoptosis. Biochem Biophys Res Commun. (2018) 501:239–45. doi: 10.1016/j.bbrc.2018.04.223
91. Pennesi ME, Neuringer M, Courtney RJ. Animal models of age related macular degeneration. Mol Aspects Med. (2012) 33:487–509. doi: 10.1016/j.mam.2012.06.003
92. Zeiss CJ. Review paper: animals as models of age-related macular degeneration. Vet Pathol. (2010) 47:396–413. doi: 10.1177/0300985809359598
93. Fletcher EL, Jobling AI, Greferath U, Mills SA, Waugh M, Ho T, et al. Studying age-related macular degeneration using animal models. Optom Vis Sci. (2014) 91:878–86. doi: 10.1097/OPX.0000000000000322
94. Grimm C, Remé CE. Light damage as a model of retinal degeneration. In: Weber B, Langmann T, editors. Methods in Molecular Biology. Clifton, NJ; Totowa, NJ: Humana Press. p. 87–97. doi: 10.1007/978-1-62703-080-9_6
95. Wenzel A, Grimm C, Marti A, Kueng-Hitz N, Hafezi F, Niemeyer G, et al. c-fos controls the “private pathway” of light-induced apoptosis of retinal photoreceptors. J Neurosci. (2000) 20:81–8. doi: 10.1523/JNEUROSCI.20-01-00081.2000
96. Santos AM, MartÃ-n-Oliva D, Ferrer-MartÃ-n RM, Tassi M, Calvente R, Sierra A, et al. Microglial response to light-induced photoreceptor degeneration in the mouse retina. J Comp Neurol. (2010) 518:477–92. doi: 10.1002/cne.22227
97. Rutar M, Natoli R, Provis JM. Small interfering RNA-mediated suppression of Ccl2 in Müller cells attenuates microglial recruitment and photoreceptor death following retinal degeneration. J Neuroinflammation. (2012) 9:761. doi: 10.1186/1742-2094-9-221
98. Zhang M, Xu G, Liu W, Ni Y, Zhou W. Role of fractalkine/CX3CR1 interaction in light-induced photoreceptor degeneration through regulating retinal microglial activation and migration. PLoS ONE. (2012) 7:e35446. doi: 10.1371/journal.pone.0035446
99. Song D, Sulewski ME, Wang C, Song J, Bhuyan R, Sterling J, et al. Complement C5a receptor knockout has diminished light-induced microglia/macrophage retinal migration. Mol Vis. (2017) 23:210–18.
100. Guillonneau X, Eandi CM, Paques M, Sahel J-A, Sapieha P, Sennlaub F. On phagocytes and macular degeneration. Prog Retin Eye Res. (2017) 61:98–128. doi: 10.1016/j.preteyeres.2017.06.002
101. Hollyfield JG. Age-related macular degeneration: the molecular link between oxidative damage, tissue-specific inflammation and outer retinal disease: the Proctor lecture. Invest Ophthalmol Vis Sci. (2010) 51:1275–81. doi: 10.1167/iovs.09-4478
102. Hollyfield JG, Perez VL, Salomon RG. A hapten generated from an oxidation fragment of docosahexaenoic acid is sufficient to initiate age-related macular degeneration. Mol Neurobiol. (2010) 41:290–8. doi: 10.1007/s12035-010-8110-z
103. Lambert V, Lecomte J, Hansen S, Blacher S, Gonzalez M-LA, Struman I, et al. Laser-induced choroidal neovascularization model to study age-related macular degeneration in mice. Nat Protoc. (2013) 8:2197–211. doi: 10.1038/nprot.2013.135
104. Lückoff A, Caramoy A, Scholz R, Prinz M, Kalinke U, Langmann T. Interferon-beta signaling in retinal mononuclear phagocytes attenuates pathological neovascularization. EMBO Mol Med. (2016) 8:670–8. doi: 10.15252/emmm.201505994
105. Combadière C, Feumi C, Raoul W, Keller N, Rodéro M, Pézard A, et al. CX3CR1-dependent subretinal microglia cell accumulation is associated with cardinal features of age-related macular degeneration. J Clin Invest. (2007) 117:2920–8. doi: 10.1172/JCI31692
106. Lückoff A, Scholz R, Sennlaub F, Xu H, Langmann T. Comprehensive analysis of mouse retinal mononuclear phagocytes. Nat Protoc. (2017) 12:1136–50. doi: 10.1038/nprot.2017.032
107. Huang H, Parlier R, Shen J, Lutty GA, Vinores SA. VEGF receptor blockade markedly reduces retinal microglia/macrophage infiltration into laser-induced CNV. PLoS ONE. (2013) 8:e71808. doi: 10.1371/journal.pone.0071808
108. RetNetTM—Retinal Information Network. Available online at: https://sph.uth.edu/retnet/ (accessed April 12, 2019).
109. Colella P, Auricchio A. Gene therapy of inherited retinopathies: a long and successful road from viral vectors to patients. Hum Gene Ther. (2012) 23:796–807. doi: 10.1089/hum.2012.123
110. Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. (2006) 368:1795–809. doi: 10.1016/S0140-6736(06)69740-7
111. Keeler CE. The inheritance of a retinal abnormality in white mice. Proc Natl Acad Sci USA. (1924) 10:329–33. doi: 10.1073/pnas.10.7.329
112. Chang B, Hawes NL, Hurd RE, Davisson MT, Nusinowitz S, Heckenlively JR. Retinal degeneration mutants in the mouse. Vision Res. (2002) 42:517–25. doi: 10.1016/S0042-6989(01)00146-8
113. Farber DB, Flannery JG, Bowes-Rickman C. The rd mouse story: seventy years of research on an animal model of inherited retinal degeneration. Prog Retin Eye Res. (1994) 13:31–64. doi: 10.1016/1350-9462(94)90004-3
114. Zeiss CJ, Johnson EA. Proliferation of microglia, but not photoreceptors, in the outer nuclear layer of the rd-1 mouse. Investig Opthalmol Vis Sci. (2004) 45:971–6. doi: 10.1167/iovs.03-0301
115. Zeng H, Zhu X, Zhang C, Yang L-P, Wu L, Tso MOM. Identification of sequential events and factors associated with microglial activation, migration, and cytotoxicity in retinal degeneration in rd Mice. Investig Opthalmol Vis Sci. (2005) 46:2992–9. doi: 10.1167/iovs.05-0118
116. Blank T, Goldmann T, Koch M, Amann L, Schön C, Bonin M, et al. Early microglia activation precedes photoreceptor degeneration in a mouse model of CNGB1-linked retinitis pigmentosa. Front Immunol. (2018) 8:1930. doi: 10.3389/fimmu.2017.01930
117. Guo C, Otani A, Oishi A, Kojima H, Makiyama Y, Nakagawa S, et al. Knockout of ccr2 alleviates photoreceptor cell death in a model of retinitis pigmentosa. Exp Eye Res. (2012) 104:39–47. doi: 10.1016/j.exer.2012.08.013
118. Wang N-K, Lai C-C, Liu C-H, Yeh L-K, Chou CL, Kong J, et al. Origin of fundus hyperautofluorescent spots and their role in retinal degeneration in a mouse model of Goldmann-Favre syndrome. Dis Model Mech. (2013) 6:1113–22. doi: 10.1242/dmm.012112
119. Akhmedov NB, Piriev NI, Chang B, Rapoport AL, Hawes NL, Nishina PM, et al. A deletion in a photoreceptor-specific nuclear receptor mRNA causes retinal degeneration in the rd7 mouse. Proc Natl Acad Sci USA. (2000) 97:5551–6. doi: 10.1073/pnas.97.10.5551
120. Aredo B, Zhang K, Chen X, Wang CX-Z, Li T, Ufret-Vincenty RL. Differences in the distribution, phenotype and gene expression of subretinal microglia/macrophages in C57BL/6N (Crb1 rd8/rd8) versus C57BL6/J (Crb1 wt/wt) mice. J Neuroinflammation. (2015) 12:6. doi: 10.1186/s12974-014-0221-4
121. Chen X, Kezic J, Bemard C, Mcmenamin PG, Med D. Rd8 mutation in the CrbJ gene of COl Jc-eYFP transgenic reporter mice resuits in abnormal numbers of CDJ J c-positive ceils in the retina. J Neuropathol Exp Neurol. (2013) 72:782–90. doi: 10.1097/NEN.0b013e31829e8375
122. Altmann C, Schmidt MHH. The role of microglia in diabetic retinopathy: inflammation, microvasculature defects and neurodegeneration. Int J Mol Sci. (2018) 19:E110. doi: 10.3390/ijms19010110
123. Lechner J, O'Leary OE, Stitt AW. The pathology associated with diabetic retinopathy. Vis Res. (2017) 139:7–14. doi: 10.1016/j.visres.2017.04.003
124. Demircan N, Safran BG, Soylu M, Ozcan AA, Sizmaz S. Determination of vitreous interleukin-1 (IL-1) and tumour necrosis factor (TNF) levels in proliferative diabetic retinopathy. Eye. (2006) 20:1366–9. doi: 10.1038/sj.eye.6702138
125. Koskela UE, Kuusisto SM, Nissinen AE, Savolainen MJ, Liinamaa MJ. High vitreous concentration of IL-6 and IL-8, but not of adhesion molecules in relation to plasma concentrations in proliferative diabetic retinopathy. Ophthalmic Res. (2013) 49:108–14. doi: 10.1159/000342977
126. Muramatsu D, Wakabayashi Y, Usui Y, Okunuki Y, Kezuka T, Goto H. Correlation of complement fragment C5a with inflammatory cytokines in the vitreous of patients with proliferative diabetic retinopathy. Graefe's Arch Clin Exp Ophthalmol. (2013) 251:15–17. doi: 10.1007/s00417-012-2024-6
127. Powell EU, Field R. Diabetic retinopathy and rheumatoid arthritis. Lancet. (1964) 284:17–8. doi: 10.1016/S0140-6736(64)90008-X
128. Boss JD, Singh PK, Pandya HK, Tosi J, Kim C, Tewari A, et al. Assessment of neurotrophins and inflammatory mediators in vitreous of patients with diabetic retinopathy. Investig Opthalmol Vis Sci. (2017) 58:5594–603. doi: 10.1167/iovs.17-21973
129. Abu El Asrar AM, Maimone D, Morse PH, Gregory S, Reder AT. Cytokines in the vitreous of patients with proliferative diabetic retinopathy. Am J Ophthalmol. (1992) 114:731–6. doi: 10.1016/S0002-9394(14)74052-8
130. Tomlinson DR, Gardiner NJ. Glucose neurotoxicity. Nat Rev Neurosci. (2008) 9:36–45. doi: 10.1038/nrn2294
131. Zong H, Ward M, Stitt AW. AGEs, RAGE, and diabetic retinopathy. Curr Diab Rep. (2011) 11:244–52. doi: 10.1007/s11892-011-0198-7
132. Milne R, Brownstein S. Advanced glycation end products and diabetic retinopathy. Amino Acids. (2013) 44:1397–407. doi: 10.1007/s00726-011-1071-3
133. Du Y, Smith MA, Miller CM, Kern TS. Diabetes-induced nitrative stress in the retina, and correction by aminoguanidine. J Neurochem. (2002) 80:771–9. doi: 10.1046/j.0022-3042.2001.00737.x
134. Zeng XX, Ng YK, Ling EA. Neuronal and microglial response in the retina of streptozotocin-induced diabetic rats. Vis Neurosci. (2000) 17:463–71. doi: 10.1017/S0952523800173122
135. Zeng H, Green WR, Tso MOM. Microglial activation in human diabetic retinopathy. Arch Ophthalmol. (2008) 126:227–32. doi: 10.1001/archophthalmol.2007.65
136. Grigsby JG, Cardona SM, Pouw CE, Muniz A, Mendiola AS, Tsin ATC, et al. The role of microglia in diabetic retinopathy. J Ophthalmol. (2014) 2014:705783. doi: 10.1155/2014/705783
137. Kezic JM, Chen X, Rakoczy EP, McMenamin PG. The effects of age and Cx 3 cr1 deficiency on retinal microglia in the Ins2 Akita diabetic mouse. Investig Opthalmol Vis Sci. (2013) 54:854–63. doi: 10.1167/iovs.12-10876
138. Barber AJ, Antonetti DA, Kern TS, Reiter CEN, Soans RS, Krady JK, et al. The Ins2 Akita mouse as a model of early retinal complications in diabetes. Investig Opthalmol Vis Sci. (2005) 46:2210. doi: 10.1167/iovs.04-1340
139. Omri S, Behar-Cohen F, de Kozak Y, Sennlaub F, Verissimo LM, Jonet L, et al. Microglia/macrophages migrate through retinal epithelium barrier by a transcellular route in diabetic retinopathy: role of PKCζ in the Goto Kakizaki rat model. Am J Pathol. (2011) 179:942–53. doi: 10.1016/j.ajpath.2011.04.018
140. Lenzen S. The mechanisms of alloxan- and streptozotocin-induced diabetes. Diabetologia. (2008) 51:216–26. doi: 10.1007/s00125-007-0886-7
141. Gaucher D, Chiappore J-A, Pâques M, Simonutti M, Boitard C, Sahel JA, et al. Microglial changes occur without neural cell death in diabetic retinopathy. Vision Res. (2007) 47:612–23. doi: 10.1016/j.visres.2006.11.017
142. Chen X, Zhou H, Gong Y, Wei S, Zhang M. Early spatiotemporal characterization of microglial activation in the retinas of rats with streptozotocin-induced diabetes. Graefe's Arch Clin Exp Ophthalmol. (2015) 253:519–25. doi: 10.1007/s00417-014-2727-y
143. Jonas JB, Aung T, Bourne RR, Bron AM, Ritch R, Panda-Jonas S. Glaucoma. Lancet. (2017) 390:2183–93. doi: 10.1016/S0140-6736(17)31469-1
144. Quigley HA, Broman AT. The number of people with glaucoma worldwide in 2010 and 2020. Br J Ophthalmol. (2006) 90:262–7. doi: 10.1136/bjo.2005.081224
145. Stevens GA, White RA, Flaxman SR, Price H, Jonas JB, Keeffe J, et al. Global prevalence of vision impairment and blindness: magnitude and temporal trends, 1990–2010. Ophthalmology. (2013) 120:2377–84. doi: 10.1016/j.ophtha.2013.05.025
146. Kass MA, Heuer DK, Higginbotham EJ, Johnson CA, Keltner JL, Miller JP, et al. The ocular hypertension treatment study: a randomized trial determines that topical ocular hypotensive medication delays or prevents the onset of primary open-angle glaucoma. Arch Ophthalmol. (2002) 120:701–13; discussion 829-30. doi: 10.1001/archopht.120.6.701
147. Rudnicka AR, Mt-Isa S, Owen CG, Cook DG, Ashby D. Variations in primary open-angle glaucoma prevalence by age, gender, and race: a bayesian meta-analysis. Investig Opthalmology Vis Sci. (2006) 47:4254–61. doi: 10.1167/iovs.06-0299
148. Leske MC, Wu SY, Honkanen R, Nemesure B, Schachat A, Hyman L, et al. Nine-year incidence of open-angle glaucoma in the barbados eye studies. Ophthalmology. (2007) 114:1058–64. doi: 10.1016/j.ophtha.2006.08.051
149. Bessero A-C, Clarke PG. Neuroprotection for optic nerve disorders. Curr Opin Neurol. (2010) 23:10–15. doi: 10.1097/WCO.0b013e3283344461
150. Baltmr A, Duggan J, Nizari S, Salt TE, Cordeiro MF. Neuroprotection in glaucoma—is there a future role? Exp Eye Res. (2010) 91:554–66. doi: 10.1016/j.exer.2010.08.009
151. Cordeiro MF, Levin LA. Clinical evidence for neuroprotection in glaucoma. Am J Ophthalmol. (2011) 152:715–6. doi: 10.1016/j.ajo.2011.06.015
152. Tezel G. Immune regulation toward immunomodulation for neuroprotection in glaucoma. Curr Opin Pharmacol. (2013) 13:23–31. doi: 10.1016/j.coph.2012.09.013
153. Yan X, Tezel G, Wax MB, Edward DP. Matrix metalloproteinases and tumor necrosis factor α in glaucomatous optic nerve head. Arch Ophthalmol. (2000) 118:666–73. doi: 10.1001/archopht.118.5.666
154. Kuchtey J, Rezaei KA, Jaru-Ampornpan P, Sternberg P, Kuchtey RW, Kuchtey RW. Multiplex cytokine analysis reveals elevated concentration of interleukin-8 in glaucomatous aqueous humor. Invest Ophthalmol Vis Sci. (2010) 51:6441–7. doi: 10.1167/iovs.10-5216
155. Sawada H, Fukuchi T, Tanaka T, Abe H. Tumor necrosis factor-α concentrations in the aqueous humor of patients with Glaucoma. Investig Opthalmology Vis Sci. (2010) 51:903–6. doi: 10.1167/iovs.09-4247
156. Chua J, Vania M, Cheung CMG, Ang M, Chee SP, Yang H, et al. Expression profile of inflammatory cytokines in aqueous from glaucomatous eyes. Mol Vis. (2012) 18:431–8.
157. Zeng H-L, Shi J-M. The role of microglia in the progression of glaucomatous neurodegeneration- a review. Int J Ophthalmol. (2018) 11:143–9. doi: 10.18240/ijo.2018.01.22
158. Yuan L, Neufeld AH. Activated microglia in the human glaucomatous optic nerve head. J Neurosci Res. (2001) 64:523–32. doi: 10.1002/jnr.1104
159. Neufeld AH. Microglia in the optic nerve head and the region of parapapillary chorioretinal atrophy in Glaucoma. Arch Ophthalmol. (1999) 117:1050–6. doi: 10.1001/archopht.117.8.1050
160. Luo C, Yang X, Kain AD, Powell DW, Kuehn MH, Tezel G. Glaucomatous tissue stress and the regulation of immune response through glial toll-like receptor signaling. Investig Opthalmology Vis Sci. (2010) 51:5697–707. doi: 10.1167/iovs.10-5407
161. Ishikawa M, Yoshitomi T, Zorumski CF, Izumi Y. Experimentally induced mammalian models of Glaucoma. Biomed Res Int. (2015) 2015:281214. doi: 10.1155/2015/281214
162. Bosco A, Steele MR, Vetter ML. Early microglia activation in a mouse model of chronic glaucoma. J Comp Neurol. (2011) 519:599–620. doi: 10.1002/cne.22516
163. Inman DM, Horner PJ. Reactive nonproliferative gliosis predominates in a chronic mouse model of glaucoma. Glia. (2007) 55:942–53. doi: 10.1002/glia.20516
164. Tezel G, Yang X, Yang J, Wax MB. Role of tumor necrosis factor receptor-1 in the death of retinal ganglion cells following optic nerve crush injury in mice. Brain Res. (2004) 996:202–12. doi: 10.1016/j.brainres.2003.10.029
165. Yuan L, Neufeld AH. Tumor necrosis factor-alpha: a potentially neurodestructive cytokine produced by glia in the human glaucomatous optic nerve head. Glia. (2000) 32:42–50. doi: 10.1002/1098-1136(200010)32:1<42::AID-GLIA40>3.3.CO;2-V
166. Tezel G, Li LY, Patil RV, Wax MB. TNF-α and TNF-α receptor-1 in the retina of normal and glaucomatous eyes. Invest Ophthalmol Vis Sci. (2001) 42:1787–94. Available online at: https://iovs.arvojournals.org/article.aspx?articleid=2200019
167. Roh M, Zhang Y, Murakami Y, Thanos A, Lee SC, Vavvas DG, et al. Etanercept, a widely used inhibitor of tumor necrosis factor-α (TNF- α), prevents retinal ganglion cell loss in a rat model of glaucoma. PLoS ONE. (2012) 7:e40065. doi: 10.1371/journal.pone.0040065
168. Neufeld AH, Hernandez MR, Gonzalez M. Nitric oxide synthase in the human glaucomatous optic nerve head. Arch Ophthalmol. (1997) 115:497–503. doi: 10.1001/archopht.1997.01100150499009
169. Vidal L, Díaz F, Villena A, Moreno M, Campos JG, Vargas IP de. Nitric oxide synthase in retina and optic nerve head of rat with increased intraocular pressure and effect of timolol. Brain Res Bull. (2006) 70:406–13. doi: 10.1016/j.brainresbull.2006.07.009
170. Cho KJ, Kim JH, Park H-YL, Park CK. Glial cell response and iNOS expression in the optic nerve head and retina of the rat following acute high IOP ischemia–reperfusion. Brain Res. (2011) 1403:67–77. doi: 10.1016/j.brainres.2011.06.005
171. Shibata M, Sugiyama T, Hoshiga M, Hotchi J, Okuno T, Oku H, et al. Changes in optic nerve head blood flow, visual function, and retinal histology in hypercholesterolemic rabbits. Exp Eye Res. (2011) 93:818–24. doi: 10.1016/j.exer.2011.09.014
172. Neufeld AH, Sawada A, Becker B. Inhibition of nitric-oxide synthase 2 by aminoguanidine provides neuroprotection of retinal ganglion cells in a rat model of chronic glaucoma. Proc Natl Acad Sci USA. (1999) 96:9944–8. doi: 10.1073/pnas.96.17.9944
173. Echevarria FD, Formichella CR, Sappington RM. Interleukin-6 deficiency attenuates retinal ganglion cell axonopathy and glaucoma-related vision loss. Front Neurosci. (2017) 11:318. doi: 10.3389/fnins.2017.00318
174. Sappington RM, Chan M, Calkins DJ. Interleukin-6 protects retinal ganglion cells from pressure-induced death. Investig Opthalmol Vis Sci. (2006) 47:2932–42. doi: 10.1167/iovs.05-1407
175. Sappington RM, Calkins DJ. Pressure-induced regulation of IL-6 in retinal glial cells: involvement of the ubiquitin/proteasome pathway and NFκB. Investig Opthalmology Vis Sci. (2006) 47:3860–9. doi: 10.1167/iovs.05-1408
176. Echevarria FD, Rickman AE, Sappington RM. Interleukin-6: a constitutive modulator of glycoprotein 130, neuroinflammatory and cell survival signaling in retina. J Clin Cell Immunol. (2016) 7:439. doi: 10.4172/2155-9899.1000439
177. Hildebrandt H, Dityatev A. Polysialic acid in brain development and synaptic plasticity. Top Curr Chem. (2015) 366:55–96. doi: 10.1007/128_2013_446
178. Schnaar RL, Gerardy-Schahn R, Hildebrandt H. Sialic acids in the brain: gangliosides and polysialic acid in nervous system development, stability, disease, and regeneration. Physiol Rev. (2014) 94:461–518. doi: 10.1152/physrev.00033.2013
179. Rutishauser U. Polysialic acid in the plasticity of the developing and adult vertebrate nervous system. Nat Rev Neurosci. (2008) 9:26–35. doi: 10.1038/nrn2285
180. Angata T, Kerr SC, Greaves DR, Varki NM, Crocker PR, Varki A. Cloning and Characterization of Human Siglec-11. J Biol Chem. (2002) 277:24466–74. doi: 10.1074/jbc.M202833200
181. Wang Y, Neumann H. Alleviation of neurotoxicity by microglial human siglec-11. J Neurosci. (2010) 30:3482–8. doi: 10.1523/JNEUROSCI.3940-09.2010
182. Ulanova M, Asfaha S, Stenton G, Lint A, Gilbertson D, Schreiber A, et al. Involvement of Syk protein tyrosine kinase in LPS-induced responses in macrophages. J Endotoxin Res. (2007) 13:117–25. doi: 10.1177/0968051907079125
183. Gurung P, Fan G, Lukens JR, Vogel P, Tonks NK, Kanneganti T-D. Tyrosine kinase SYK licenses MyD88 adaptor protein to instigate IL-1α-mediated inflammatory disease. Immunity. (2017) 46:635–48. doi: 10.1016/j.immuni.2017.03.014
184. Linnartz B, Neumann H. Microglial activatory (immunoreceptor tyrosine-based activation motif)- and inhibitory (immunoreceptor tyrosine-based inhibition motif)-signaling receptors for recognition of the neuronal glycocalyx. Glia. (2013) 61:37–46. doi: 10.1002/glia.22359
185. Goswami K, Nandakumar DN, Koner BC, Bobby Z, Sen SK. Oxidative changes and desialylation of serum proteins in hyperthyroidism. Clin Chim Acta. (2003) 337:163–8. doi: 10.1016/j.cccn.2003.08.009
186. Moseley R, Waddington R, Embery G. Degradation of glycosaminoglycans by reactive oxygen species derived from stimulated polymorphonuclear leukocytes. Biochim Biophys Acta Mol Basis Dis. (1997) 1362:221–31. doi: 10.1016/S0925-4439(97)00083-5
187. Linnartz-Gerlach B, Schuy C, Shahraz A, Tenner AJ, Neumann H. Sialylation of neurites inhibits complement-mediated macrophage removal in a human macrophage-neuron Co-Culture System. Glia. (2016) 64:35–47. doi: 10.1002/glia.22901
188. Howell GR, Macalinao DG, Sousa GL, Walden M, Soto I, Kneeland SC, et al. Molecular clustering identifies complement and endothelin induction as early events in a mouse model of glaucoma. J Clin Invest. (2011) 121:1429–44. doi: 10.1172/JCI44646
189. Rupprecht R, Papadopoulos V, Rammes G, Baghai TC, Fan J, Akula N, et al. Translocator protein (18 kDa) (TSPO) as a therapeutic target for neurological and psychiatric disorders. Nat Rev Drug Discov. (2010) 9:971–88. doi: 10.1038/nrd3295
190. Karlstetter M, Nothdurfter C, Aslanidis A, Moeller K, Horn F, Scholz R, et al. Translocator protein (18kDa) (TSPO) is expressed in reactive retinal microglia and modulates microglial inflammation and phagocytosis. J Neuroinflammation. (2014) 11:3. doi: 10.1186/1742-2094-11-3
191. Selvaraj V, Stocco DM. The changing landscape in translocator protein (TSPO) function. Trends Endocrinol Metab. (2015) 26:341–8. doi: 10.1016/j.tem.2015.02.007
192. Daugherty DJ, Selvaraj V, Chechneva OV, Liu X-B, Pleasure DE, Deng W. A TSPO ligand is protective in a mouse model of multiple sclerosis. EMBO Mol Med. (2013) 5:891–903. doi: 10.1002/emmm.201202124
193. Scholz R, Caramoy A, Bhuckory M, Rashid K, Chen M, Xu H, et al. Targeting translocator protein (18kDa) (TSPO) dampens pro-inflammatory microglia reactivity in the retina and protects from degeneration. J Neuroinflammation. (2015) 12:201. doi: 10.1186/s12974-015-0422-5
194. Banati RB, Middleton RJ, Chan R, Hatty CR, Wai-Ying Kam W, Quin C, et al. Positron emission tomography and functional characterization of a complete PBR/TSPO knockout. Nat Commun. (2014) 5:5452. doi: 10.1038/ncomms6452
195. Liu G-J, Middleton RJ, Hatty CR, Kam WW-Y, Chan R, Pham T, et al. The 18 kDa translocator protein, microglia and neuroinflammation. Brain Pathol. (2014) 24:631–53. doi: 10.1111/bpa.12196
196. Costa E, Guidotti A. Diazepam binding inhibitor (DBI): a peptide with multiple biological actions. Life Sci. (1991) 49:325–44. doi: 10.1016/0024-3205(91)90440-M
197. Heckenlively JR, Rodriguez JA, Daiger SP. Autosomal dominant sectoral retinitis pigmentosa. Arch Ophthalmol. (1991) 109:84–91. doi: 10.1001/archopht.1991.01080010086038
198. Cruickshanks KJ, Klein R, Klein BEK. Sunlight and age-related macular degeneration. Arch Ophthalmol. (1993) 111:514–8. doi: 10.1001/archopht.1993.01090040106042
199. Cruickshanks KJ, Klein R, Klein BE, Nondahl DM. Sunlight and the 5-year incidence of early age-related maculopathy: the beaver dam eye study. Arch Ophthalmol. (2001) 119:246–50. doi: 10.1001/archopht.119.9.1354
200. Fletcher AE, Bentham GC, Agnew M, Young IS, Augood C, Chakravarthy U, et al. Sunlight exposure, antioxidants, and age-related macular degeneration. Arch Ophthalmol. (2008) 126:1396–403. doi: 10.1001/archopht.126.10.1396
201. Papadopoulos V, Aghazadeh Y, Fan J, Campioli E, Zirkin B, Midzak A. Translocator protein-mediated pharmacology of cholesterol transport and steroidogenesis. Mol Cell Endocrinol. (2015) 408:90–8. doi: 10.1016/j.mce.2015.03.014
202. Jamin N, Neumann J-M, Ostuni MA, Vu TKN, Yao Z-X, Murail S, et al. Characterization of the cholesterol recognition amino acid consensus sequence of the peripheral-type benzodiazepine receptor. Mol Endocrinol. (2005) 19:588–94. doi: 10.1210/me.2004-0308
203. Batarseh A, Barlow KD, Martinez-Arguelles DB, Papadopoulos V. Functional characterization of the human translocator protein (18kDa) gene promoter in human breast cancer cell lines. Biochim Biophys Acta. (2012) 1819:38–56. doi: 10.1016/j.bbagrm.2011.09.001
204. Tu LN, Zhao AH, Stocco DM, Selvaraj V. PK11195 effect on steroidogenesis is not mediated through the translocator protein (TSPO). Endocrinology. (2015) 156:1033–9. doi: 10.1210/en.2014-1707
205. Papadopoulos V, Berkovich A, Krueger KE, Costa E, Guidotti A. Diazepam binding inhibitor and its processing products stimulate mitochondrial steroid biosynthesis via an interaction with mitochondrial benzodiazepine receptors *. Endocrinology. (1991) 129:1481–8. doi: 10.1210/endo-129-3-1481
206. Romeo E, Cavallaro S, Korneyev A, Kozikowski AP, Ma D, Polo A, et al. Stimulation of brain steroidogenesis by 2-aryl-indole-3-acetamide derivatives acting at the mitochondrial diazepam-binding inhibitor receptor complex. J Pharmacol Exp Ther. (1993) 267:462–71.
207. Boujrad N, Hudson JR, Papadopoulos V. Inhibition of hormone-stimulated steroidogenesis in cultured Leydig tumor cells by a cholesterol-linked phosphorothioate oligodeoxynucleotide antisense to diazepam-binding inhibitor. Proc Natl Acad Sci USA. (1993) 90:5728–31. doi: 10.1073/pnas.90.12.5728
208. Owen DR, Fan J, Campioli E, Venugopal S, Midzak A, Daly E, et al. TSPOmutations in rats and a human polymorphism impair the rate of steroid synthesis. Biochem J. (2017) 474:3985–99. doi: 10.1042/BCJ20170648
209. Vasconcelos AR, Cabral-Costa JV, Mazucanti CH, Scavone C, Kawamoto EM. The role of steroid hormones in the modulation of neuroinflammation by dietary interventions. Front Endocrinol. (2016) 7:9. doi: 10.3389/fendo.2016.00009
210. Barnes PJ. How corticosteroids control inflammation: quintiles prize lecture 2005. Br J Pharmacol. (2006) 148:245–54. doi: 10.1038/sj.bjp.0706736
211. Byrne AM, Roche SL, Ruiz-Lopez AM, Jackson ACW, Cotter TG. The synthetic progestin norgestrel acts to increase LIF levels in the rd10 mouse model of retinitis pigmentosa. Mol Vis. (2016) 22:264–74. doi: 10.1016/j.redox.2016.10.002
212. Doonan F, O'Driscoll C, Kenna P, Cotter TG. Enhancing survival of photoreceptor cells in vivo using the synthetic progestin Norgestrel. J Neurochem. (2011) 118:915–27. doi: 10.1111/j.1471-4159.2011.07354.x
213. Roche SL, Wyse-Jackson AC, Ruiz-Lopez AM, Byrne AM, Cotter TG. Fractalkine-CX3CR1 signaling is critical for progesterone-mediated neuroprotection in the retina. Sci Rep. (2017) 7:43067. doi: 10.1038/srep43067
214. Roche SL, Wyse-Jackson AC, Gómez-Vicente V, Lax P, Ruiz-Lopez AM, Byrne AM, et al. Progesterone attenuates microglial-driven retinal degeneration and stimulates protective fractalkine-CX3CR1 signaling. PLoS ONE. (2016) 11:e0165197. doi: 10.1371/journal.pone.0165197
215. Zhu C, Wang S, Wang B, Du F, Hu C, Li H, et al. 17β-Estradiol up-regulates Nrf2 via PI3K/AKT and estrogen receptor signaling pathways to suppress light-induced degeneration in rat retina. Neuroscience. (2015) 304:328–39. doi: 10.1016/j.neuroscience.2015.07.057
216. McNab F, Mayer-Barber K, Sher A, Wack A, O'Garra A. Type I interferons in infectious disease. Nat Rev Immunol. (2015) 15:87–103. doi: 10.1038/nri3787
217. González-Navajas JM, Lee J, David M, Raz E. Immunomodulatory functions of type I interferons. Nat Rev Immunol. (2012) 12:125–35. doi: 10.1038/nri3133
218. Kocur M, Schneider R, Pulm A-K, Bauer J, Kropp S, Gliem M, et al. IFNβ secreted by microglia mediates clearance of myelin debris in CNS autoimmunity. Acta Neuropathol Commun. (2015) 3:20. doi: 10.1186/s40478-015-0192-4
219. Stetson DB, Medzhitov R. Review type I interferons in host defense. Immunity. (2006) 25:373–81. doi: 10.1016/j.immuni.2006.08.007
220. Dendrou CA, Fugger L, Friese MA. Immunopathology of multiple sclerosis. Nat Rev Immunol. (2015) 15:545–58. doi: 10.1038/nri3871
221. Prinz M, Schmidt H, Mildner A, Knobeloch K-P, Hanisch U-K, Raasch J, et al. Distinct and nonredundant in vivo functions of IFNAR on myeloid cells limit autoimmunity in the central nervous system. Immunity. (2008) 28:675–86. doi: 10.1016/j.immuni.2008.03.011
222. Liu J, Copland DA, Horie S, Wu W-K, Chen M, Xu Y, et al. Myeloid cells expressing VEGF and Arginase-1 following uptake of damaged retinal pigment epithelium suggests potential mechanism that drives the onset of choroidal angiogenesis in mice. PLoS ONE. (2013) 8:e72935. doi: 10.1371/journal.pone.0072935
223. Kimoto T, Takahashi K, Tobe T, Fujimoto K, Uyama M, Sone S. Effects of local administration of interferon-beta on proliferation of retinal pigment epithelium in rabbit after laser photocoagulation. Jpn J Ophthalmol. (2002) 46:160–9.
224. Cirino AC, Mathura JR, Jampol LM. of chronic recurrent OF Resolution of activity (choroiditis and choroidal neovascularization) of chronic recurrent punctate inner choroidopathy after treatment with interferon B-1A. Retina. (2006) 26:1091–2. doi: 10.1097/01.iae.0000254891.48272.13
225. Schreiber G. The molecular basis for differential type I interferon signaling. J Biol Chem. (2017) 292:7285–94. doi: 10.1074/jbc.R116.774562
226. Akhtar LN, Qin H, Muldowney MT, Yanagisawa LL, Kutsch O, Clements JE, et al. Suppressor of cytokine signaling 3 inhibits antiviral IFN-beta signaling to enhance HIV-1 replication in macrophages. J Immunol. (2010) 185:2393–404. doi: 10.4049/jimmunol.0903563
227. Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol. (2014) 14:36–49. doi: 10.1038/nri3581
228. Yoshimura A, Naka T, Kubo M. SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol. (2007) 7:454–65. doi: 10.1038/nri2093
229. Krebs DL, Hilton DJ. SOCS Proteins: negative regulators of cytokine signaling. Stem Cells. (2001) 19:378–87. doi: 10.1634/stemcells.19-5-378
230. Chen M, Zhao J, Ali IHA, Marry S, Augustine J, Bhuckory M, et al. Cytokine signaling protein 3 deficiency in myeloid cells promotes retinal degeneration and angiogenesis through arginase-1 up-regulation in experimental autoimmune uveoretinitis. Am J Pathol. (2018) 188:1007–20. doi: 10.1016/j.ajpath.2017.12.021
231. Yu C-R, Mahdi RR, Oh H-M, Amadi-Obi A, Levy-Clarke G, Burton J, et al. Suppressor of cytokine signaling-1 (SOCS1) inhibits lymphocyte recruitment into the retina and protects SOCS1 transgenic rats and mice from ocular inflammation. Invest Ophthalmol Vis Sci. (2011) 52:6978–86. doi: 10.1167/iovs.11-7688
232. Burke JD, Platanias LC, Fish EN. Beta interferon regulation of glucose metabolism is PI3K/Akt dependent and important for antiviral activity against coxsackievirus B3. J Virol. (2014) 88:3485–95. doi: 10.1128/JVI.02649-13
233. Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol. (2005) 5:375–86. doi: 10.1038/nri1604
234. Liu Y, Marin A, Ejlerskov P, Rasmussen LM, Prinz M, Issazadeh-Navikas S. Neuronal IFN-beta–induced PI3K/Akt-FoxA1 signalling is essential for generation of FoxA1 þ T reg cells. Nat Commun. (2017) 8:14709. doi: 10.1038/ncomms14709
235. Dong H, Zhang X, Dai X, Lu S, Gui B, Jin W, et al. Lithium ameliorates lipopolysaccharide-induced microglial activation via inhibition of toll-like receptor 4 expression by activating the PI3K/Akt/FoxO1 pathway. J Neuroinflammation. (2014) 11:140. doi: 10.1186/s12974-014-0140-4
236. Zhu YP, Brown JR, Sag D, Zhang L, Suttles J. Adenosine 5'-monophosphate-activated protein kinase regulates IL-10-mediated anti-inflammatory signaling pathways in macrophages. J Immunol. (2015) 194:584–94. doi: 10.4049/jimmunol.1401024
237. de Oliveira ACP, Candelario-Jalil E, Bhatia HS, Lieb K, Hüll M, Fiebich BL. Regulation of prostaglandin E2 synthase expression in activated primary rat microglia: evidence for uncoupled regulation of mPGES-1 and COX-2. Glia. (2008) 56:844–55. doi: 10.1002/glia.20658
238. de Oliveira AC, Candelario-Jalil E, Langbein J, Wendeburg L, Bhatia HS, Schlachetzki JC, et al. Pharmacological inhibition of Akt and downstream pathways modulates the expression of COX-2 and mPGES-1 in activated microglia. J Neuroinflammation. (2012) 9:2. doi: 10.1186/1742-2094-9-2
239. Jope RS, Cheng Y, Lowell JA, Worthen RJ, Sitbon YH, Beurel E. Stressed and Inflamed, Can GSK3 Be Blamed? Trends Biochem Sci. (2017) 42:180–92. doi: 10.1016/j.tibs.2016.10.009
240. Huang W-C, Lin Y-S, Wang C-Y, Tsai C-C, Tseng H-C, Chen C-L, et al. Glycogen synthase kinase-3 negatively regulates anti-inflammatory interleukin-10 for lipopolysaccharide-induced iNOS/NO biosynthesis and RANTES production in microglial cells. Immunology. (2009) 128:e275–86. doi: 10.1111/j.1365-2567.2008.02959.x
241. Sánchez-Cruz A, Villarejo-Zori B, Marchena M, Zaldivar-Díez J, Palomo V, Gil C, et al. Modulation of GSK-3 provides cellular and functional neuroprotection in the rd10 mouse model of retinitis pigmentosa. Mol Neurodegener. (2018) 13:19. doi: 10.1186/s13024-018-0251-y
242. Garrido-Mesa N, Zarzuelo A, Gálvez J. Minocycline: far beyond an antibiotic. Br J Pharmacol. (2013) 169:337–52. doi: 10.1111/bph.12139
243. Domercq M, Matute C. Neuroprotection by tetracyclines. Trends Pharmacol Sci. (2004) 25:609–12. doi: 10.1016/j.tips.2004.10.001
244. Nikodemova M, Duncan ID, Watters JJ. Minocycline exerts inhibitory effects on multiple mitogen-activated protein kinases and IκBα degradation in a stimulus-specific manner in microglia. J Neurochem. (2006) 96:314–23. doi: 10.1111/j.1471-4159.2005.03520.x
245. Halder SK, Matsunaga H, Ishii KJ, Akira S, Miyake K, Ueda H. Retinal cell type-specific prevention of ischemia-induced damages by LPS-TLR4 signaling through microglia. J Neurochem. (2013) 126:243–60. doi: 10.1111/jnc.12262
246. Kobayashi K, Imagama S, Ohgomori T, Hirano K, Uchimura K, Sakamoto K, et al. Minocycline selectively inhibits M1 polarization of microglia. Cell Death Dis. (2013) 4:e525. doi: 10.1038/cddis.2013.54
247. Peng B, Xiao J, Wang K, So K-F, Tipoe GL, Lin B. Suppression of microglial activation is neuroprotective in a mouse model of human retinitis pigmentosa. J Neurosci. (2014) 34:8139–50. doi: 10.1523/JNEUROSCI.5200-13.2014
248. Krady JK, Basu A, Allen CM, Xu Y, LaNoue KF, Gardner TW, et al. Minocycline reduces proinflammatory cytokine expression, microglial activation, and caspase-3 activation in a rodent model of diabetic retinopathy. Diabetes. (2005) 54:1559–65. doi: 10.2337/diabetes.54.5.1559
249. Wu Y, Chen Y, Wu Q, Jia L, Du X. Minocycline inhibits PARP-1 expression and decreases apoptosis in diabetic retinopathy. Mol Med Rep. (2015) 12:4887–94. doi: 10.3892/mmr.2015.4064
250. Bosco A, Inman DM, Steele MR, Wu G, Soto I, Marsh-Armstrong N, et al. Reduced retina microglial activation and improved optic nerve integrity with minocycline treatment in the DBA/2J mouse model of glaucoma. Investig Opthalmol Vis Sci. (2008) 49:1437–46. doi: 10.1167/iovs.07-1337
251. Baumgartner WA, Baumgartner AM. Rationale for an experimental treatment of retinitis pigmentosa: 140-Month test of hypothesis with one patient. Med Hypotheses. (2013) 81:720–8. doi: 10.1016/j.mehy.2013.07.036
Keywords: retina, microglia, neuroprotection, chronic inflammation, immunomodulation
Citation: Rashid K, Akhtar-Schaefer I and Langmann T (2019) Microglia in Retinal Degeneration. Front. Immunol. 10:1975. doi: 10.3389/fimmu.2019.01975
Received: 27 May 2019; Accepted: 05 August 2019;
Published: 20 August 2019.
Edited by:
Heping Xu, Queen's University Belfast, United KingdomReviewed by:
Wai T. Wong, National Eye Institute (NEI), United StatesPrzemyslaw Sapieha, Université de Montréal, Canada
Copyright © 2019 Rashid, Akhtar-Schaefer and Langmann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thomas Langmann, thomas.langmann@uk-koeln.de