 Daniil Shevyrev
Daniil Shevyrev Valeriy Tereshchenko
Valeriy Tereshchenko- Research Institute for Fundamental and Clinical Immunology (RIFCI), Novosibirsk, Russia
T-regulatory cells (Tregs) represent a unique subpopulation of helper T-cells by maintaining immune equilibrium using various mechanisms. The role of T-cell receptors (TCR) in providing homeostasis and activation of conventional T-cells is well-known; however, for Tregs, this area is understudied. In the last two decades, evidence has accumulated to confirm the importance of the TCR in Treg homeostasis and antigen-specific immune response regulation. In this review, we describe the current view of Treg subset heterogeneity, homeostasis and function in the context of TCR involvement. Recent studies of the TCR repertoire of Tregs, combined with single-cell gene expression analysis, revealed the importance of TCR specificity in shaping Treg phenotype diversity, their functions and homeostatic maintenance in various tissues. We propose that Tregs, like conventional T-helper cells, act to a great extent in an antigen-specific manner, which is provided by a specific distribution of Tregs in niches.
Introduction
As adaptive immunity evolved, there emerged a specialized immune response regulation system. Cartilaginous fish that appeared over 450 million years ago developed a thymus as well as the orthologous genes of key cytokines and transcription factors of the main lymphocyte subpopulations (1); however, whether this class of organisms had a fully developed adaptive immunity and a specific regulation system is debatable. It took some 36 million more years for full-fledged adaptive immunity to emerge, when bony fish appeared (2, 3). A number of aromorphoses, including the secondary immune response, gave bony fish a significant competitive advantage and directed the further evolution of this group of organisms. Despite different subpopulations of effector cells being able to suppress each other (4–7), there emerged a special subpopulation of T cells capable of specific immune response regulation. As such, these lymphocytes were named T regulatory cells, or Tregs. Phylogenesis identifies these lymphocytes in bony fish first, making them the most ancient organisms proven to have basic populations of effector T cells (3). Note that the ontogenetic Treg development in these organisms is linked to the thymus. Further Treg evolution was seemingly associated with the emergence of placental mammals that were the first to feature peripheral induction of Tregs (pTregs) from the effector precursors; cells carrying αβ-TCR and the CD4 co-receptor are hereinafter referred to as Tconv. It is assumed that such pTregs were not only able to control placental immunity but also gained greater involvement in regulating the adaptive immune response along with thymic Tregs (tTregs) (8).
Treg research is of interest when it involves the pathogenesis and therapy of various autoimmune diseases, cancers, and allergies, as well as transplantation studies and tissue engineering. The cells have extremely broad functionality; they ensure tolerance to autoantigens (9) and to the antigens of commensal microflora (10), they limit excess immune response, induce tolerance to food antigens (11), regulate the fetoplacental immunity (12), and play a role in the homeostasis and regeneration of various tissues (13–16). Treg research began more than 40 years ago. As early as the 1970s, Gershon and Kondo experimented on mice with removed thymi and were the first to suggest the existence of suppressor T lymphocytes (17). In the years that followed, great efforts were made to identify and study this population of T cells. Thus, eliminating some subpopulations of T lymphocytes in mice triggered an autoimmune syndrome that affected various organs and tissues, which indirectly proved the existence of a specific T cell population that provides peripheral autotolerance (18). However, studies into suppressor immunity were impeded by a lack of phenotypic Treg markers, the diversity of suppressor mechanisms, and the difficulty of obtaining antigen-specific Treg clones for more subtle cellular and molecular analysis. It was only in the 1990s that the prerequisites for further Treg studies were fulfilled, as there appeared transgenic animals and monoclonal antibodies, and scientists identified the primary suppressive cytokines (IL-10 and TGF-β) as well as their producers (19–21). Nevertheless, defining the Treg phenotype remained an extremely important problem until the early 2000s, i.e., until researchers found that IL-2 and its CD25 receptor were critical to developing and maintaining a Treg pool (22, 23). At the same time, scientists discovered the main transcription factor of these cells, FoxP3, which, as discovered later, enabled Tregs to function and was associated with high CD25 expression (24–26). Thus, the Treg phenotype was defined as CD3+CD4+CD25hiFoxP3+; however, that FoxP3 requires intracellular staining and that CD25 is also a marker of activated Tconv limited the applicability of this phenotype to cell research. In the following years, researchers found a negative correlation of IL-7 receptor (CD127) expression and FoxP3 expression, characterizing Tregs as low CD127 expression cells (27). However, even this factor is not universal because its expression from Tconv may be reduced under certain circumstances, e.g., when affected by IL-7 or other common γ-chain cytokines (28). However, CD3+CD4+CD25hiCD127lo is currently the most common phenotype, especially when the Treg population is isolated by means of FACS or immunomagnetic separation with further evaluation of FoxP3 expression being possible. Further Treg research identified additional phenotypic markers, mainly related to Treg functions.
Treg Heterogeneity
As noted earlier, Tregs may develop ontogenetically in the thymus (tTregs), as well as and peripherally (pTregs) from effector cells. tTregs express FoxP3 constitutively and have a T cell receptor (TCR) of relatively high autoaffinity. These cells are predominant in the bloodstream and in the lymph nodes; they are mainly involved in providing tolerance to autoantigens (9). Peripherally, CD4+-effector cells affected by IL-2 and TGF-β may under certain conditions begin to express FoxP3, thus becoming functional equivalents of tTregs (29). Such pTregs are most common in the peripheral barrier tissue and are mainly involved in preventing local inflammation in the presence of exogenous antigens. It is well-known that Tregs cells and naive CD4+ Tconv cells have non-overlapping TCR repertoires, a small percentage of equal affinity TCRs are found in both CD4+ and Treg cell populations (30). So therefore, the TCR repertories of tTregs cells and pTregs cells have been shown toare be distinct: in the tTreg cell TCR repertoire is biased toward self-recognition, and TCRs expressed in pTregs cells can recognize foreign antigens with high affinity (31). This is has been well-confirmed by analysis of amino acid CDR3 TCR repertoire overlaps, which revealed separate clusterings of Tconv cells and Tregs cells (32). Furthermore, CDR3s containing strongly interacting amino acids are more prominent in the Treg cell TCR pool compared with Tconv cells (33). That is consistent with previously obtained data about the higher TCR affinity of Tregs for self-peptide–MHC complexes? Thus, the TCR specificity spectra of tTregs and pTregs barely overlap. This may be due to the requirement in additional Tregs with specificity to antigens, which are not presented in the thymus by dint of AIRE or Fezf2, such as innocuous environmental antigens. These differences in the tTreg TCR pool from that of Tconv cells is determined during thymic selection based on the strength of the TCR signal (34) and a high TCR Treg affinity to self-antigens, which may enable Treg precursors to compete more efficiently for the limited niche of thymic antigen-presenting cells (33, 35).
It is worth noting that human tTreg- and pTreg-specific markers have not been discovered thus far. The high expression of Helios and Neuropilin-1 in mice suggests a thymic origin (36–38). What distinguishes tTregs and pTregs is the stability of FoxP3 expression in different settings. It has been found that FoxP3 expression by pTregs is transient in nature; in the case of inflammation, pTregs can differentiate into exFoxP3 effector cells that have the phenotype of Th-17 lymphocytes (RORyt+) (39), which are pathogenic for autoimmune disease-affected patients (40, 41). Normally, this transition is observed, for instance, in the intestines, which contain FoxP3+RORyt+ Treg lymphocytes associated with mucosa-associated lymphoid tissue functions (42). The stability of FoxP3 expression greatly depends on the methylation of CpG islets in the locus of the second intron enhancer in the FoxP3 gene, which is also referred to as the conservative non-coding sequence 2 (CNS2) or Major TSDR (Treg-specific demethylated region). CNS2 demethylation stabilizes FoxP3 expression and is characteristic of tTregs (43–45). Attempts are under way to artificially stabilize FoxP3 expression in in vitro induced Tregs for further clinical application (46).
In terms of differentiation, Tregs are subdivided into naive cells (nTregs), central memory cells (cmTregs), effector memory cells (emTregs), and effector Treg (eTreg) lymphocytes (see Figure 1). CCR7 and CD62L molecules enable Treg homing into the secondary lymphoid organs, while CTLA-4 expression reflects suppressive activity of Tregs. Treg lymphocytes function in different tissues and inflammatory sites, which is why their differentiation is associated with the acquisition of corresponding chemokine receptors and adhesion molecules responsible for directed homing. Thus, CCR4 is for migration to the skin, GPR-15 is for migration to the intestines, and CXCR3, LFA-1, VLA-4, CCR2, CCR5, CCR6, CCR8 are for migration to inflammation zones (47–49).
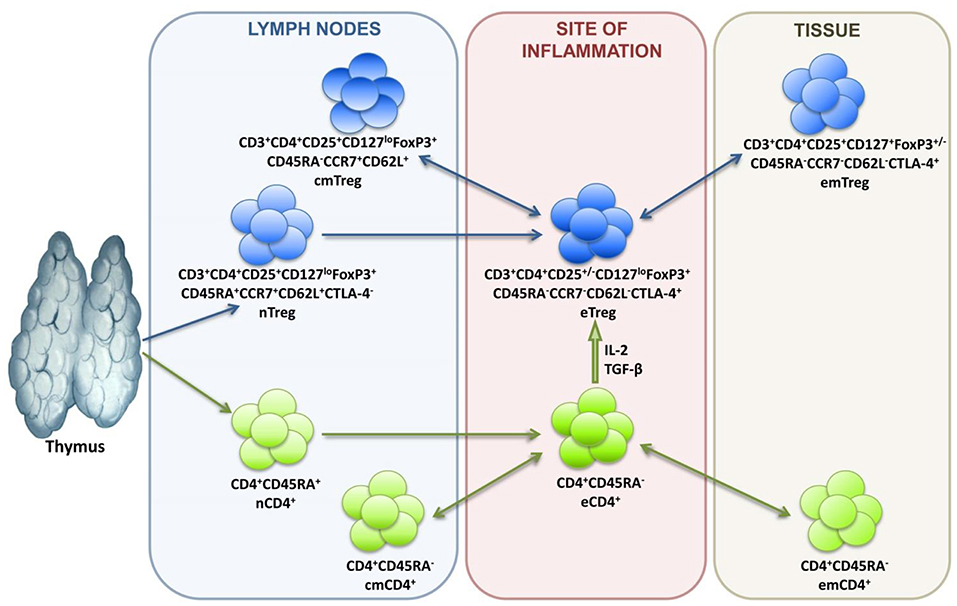
Figure 1. Treg lymphocyte differentiation dynamics.
Transcriptomic analysis of single cells and studying the pathways of Treg differentiation in the transcriptomic space in pseudotime ordering have enabled researchers to study the tissue-specific heterogeneity of the Treg population in detail. They have thus defined the Treg differentiation trajectories in line with their “molecular portraits” and found the percentages of Treg subpopulations in tissues; they have also evaluated the contribution of signaling pathways to maintaining Treg homeostasis (50).
Miragaia et al. assessed the expression of over 30 genes and found that even at their early priming in lymph nodes, Tregs acquire markers of tissue-specific migration. Thus, Tregs that later migrate to the large intestine boost CCR9 and Itga-4 expression (producing integrin-α4), while in mesenteric lymph nodes. Future skin resident Tregs express Cxcr3 and Itgb-1 (producing integrin-β1) genes before migration to the skin, while in shoulder lymph nodes. Thus, the migration properties of T-regulatory cells are already determined in the lymph nodes, although the mechanism for this is unclear (50). This is of particular interest in the context of antigen-specific Treg effects. TCR specificity determines Treg distribution in tissues and lymph nodes; the mechanism behind it is the interaction with dendritic cells in the niche (51–55). Note that after a Treg migrates to a specific tissue, it “matures” or adapts, a process associated with transcriptomic changes (50). At the same time, TCR signal intensity is not related to the Treg activation degree; instead, it determines the phenotype, i.e., Tregs with similar TCR specificity have highly similar transcription profiles (56). In addition, suppressive mechanisms of Tregs have been recently shown to differ if the cells share specificity but differ in TCR affinity. High-affinity receptor cells mostly express TCR-dependent mediators: IL-10, TIGIT, GITR, and CTLA-4; whereas cells having a low-affinity receptor express more Ebi3, which is responsible for IL35-mediated suppressive action. This indicates that affinity determines different functional mechanisms of suppression. In addition to Ebi3, Tregs with low-affinity TCR produce amphiregulin, which is a growth factor that participates in tissue regeneration. Apparently Tregs with low-affinity TCR are more likely to use non-TCR-dependent suppressive mechanisms in the absence of strong TCR signaling in response to humoral inflammatory factors, while high-affinity Tregs preferentially upregulate TCR-dependent regulatory molecules, such as CTLA-4, TIGIT, and IL-10. Nevertheless, both types of cells have suppressive potential and support autotolerance and immune equilibrium (31, 57, 58).
Thus, Treg heterogeneity depends on their origin, differentiation, and migration characteristics, which in turn depend not only on the expression of homing molecules but also on TCR specificity and affinity. Such heterogeneity of the immune response regulating population seems to reflect the diversity of Treg-targeted cells as well as the variety of conditions under which Treg lymphocytes may have to function; in addition, this heterogeneity is associated with the ontogenetic kinship of Tregs and T effectors. Indeed, studies into the Treg/Tconv transcription phenotype identified only a small subset of genes expressed en masse by Tregs that are absent in Tconv. The set is referred to as the Treg signature. Meanwhile, most genes have similar expression profiles in both populations. For instance, the populations overlap in genes whose expression depends on TCR signaling, as well as in genes involved in Treg and Tconv homeostasis in different tissues (50, 56). However, different Treg subpopulations can, in addition to the main signature closely related to the FoxP3 gene, express additional genes responsible for tissue-specific functioning. Research into Treg transcription profiles in a murine spleen identified a common gradient that separates resting and activated Tregs (50), which is largely consistent with the current knowledge of Treg differentiation dynamics (see Figure 1). Unsupervised learning has been employed to identify the clusters of resting nTregs, Tregs in early TCR-dependent activation, and activated Tregs featuring a follicular Treg signature typical of lymph node B zones, various tissues, and sterile inflammation sites (56). Figure 2 presents a simplified diagram of murine Treg transcription heterogeneity in the context of basic signatures and the condition-specific activation of certain genes. It is worth noting that mice and humans have similar Treg transcription profiles (50, 56).

Figure 2. Transcription heterogeneity of the Treg population.
The advancement of single-cell transcriptomics and ML-based multidimensional data clustering opens up great opportunities to study the mechanisms affecting the T cell lifecycle, to analyze the expression of hundreds of genes, and to identify TCR sequences in single cells. This is a great and unprecedented booster for immunology research, one that lays the foundation for potential immunity management.
Treg Suppressive Activity Mechanisms
In recent years, Tregs have been shown to suppress most immune cell populations, including lymphocytes, various types of macrophages, dendritic cells and B cells (59). Interestingly, the manifestation of Treg suppressive activity against a specific T effector population may be associated with the expression of transcription factors typical of this subpopulation. Thus, the expression of T-bet, a Th-1-associated transcription factor, in Tregs is related to the expression of the inhibitory molecule TIGIT, which binds CD155 to dendritic cells to increase the production of IL-10 and reduce that of IL-12 in the dendritic cell, thus inhibiting the activation of T effectors (60, 61). Tregs with the T-bet+TIGIT+ phenotype selectively inhibit the Th1- and Th17-mediated proinflammatory immune response (62, 63). Similarly, the Th2-associated transcription factor IRF-4 enables Treg expression of ICOS and CTLA-4; along with JUNB and RBPJ, it is needed to limit the Th2-mediated immune response (64–66). Meanwhile, the expression of the Th17-typical factor STAT3 in Tregs is closely related to the regulation of the Th17-mediated immune response and enables the expression of IL-10, Ebi3, granzyme-B, and perforin-1 genes (67). It is important that the expression of transcription factors typical of this or that subpopulation of effector T lymphocytes may be associated with unstable FoxP3 expression and loss of Treg suppressive functions. This process appears to be greatly correlated with the cytokine background in a Treg microenvironment and manifests itself in pathology (68). Tregs are believed to acquire the expression of transcription factors typical of effector cells owing to their adaptation to the immune response being polarized toward Th1, Th2, or Th17.
The functional heterogeneity of the Treg population reflects the broad spectrum of suppressive mechanisms Tregs use to control various types of immune responses. Those can be conventionally divided into contact and humoral, antigen-specific or non-specific. Many of these mechanisms are versatile and complementary; however, some are specific to certain types of immune responses.
Antigen-specific suppression is mainly caused by the direct Treg–DC (dendritic cell) interaction enabled by the specific recognition of the antigen (Ag) the DC presents as part of MHC-II by means of Treg TCR. Ultimately, such interaction results in inducing an Ag-specific tolerogenic dendritic cell or rendering the DC unable to present a specific antigen. The mechanisms of such suppression are diverse. They include binding the co-stimulation molecules CD80/86 on the dendritic cell by means of CTLA-4 (69); removing Ag-MHC-II from the DC surface by trans-endocytosis, in which case the DC remains capable of presenting other antigens (70–72); and a CTLA-4-mediated increase in IDO expression in the DC, which lowers the concentration of tryptophan necessary for T effectors to proliferate (73). In general, these mechanisms disrupt Ag presentation, cause T effector anergy, or trigger Ag-specific pTreg induction.
Antigen-non-specific mechanisms include the enzymes CD39/CD73 on the Treg surface, which cause ATP to degrade to adenosine. Increased adenosine concentration in the microenvironment inhibits DC presentation of antigens and suppresses the proliferation of activated T effectors (74). Another non-specific suppression factor is the Treg production of cytokines: TGF-β, IL-10, and IL-35. The range of suppressive effects these cytokines have is extremely broad. They can suppress the activation and proliferation of effector T and B lymphocytes; they can also directly induce pTregs and Bregs (75–78). In addition, TGF-β and IL-10 inhibit antigenic presentation to stimulate the generation of tolerogenic dendritic cells, which in their turn enable pTreg induction (79–83). Note that two pTreg populations have been well-described: Th3 and Tr1, featuring high TGF-β, and IL-10 secretion, respectively. The former has been identified by their role in oral tolerance; the latter by their involvement in preventing autoimmune colitis. Both are generated in chronic inflammation sites as well as in transplanted tissue (84).
Tregs have recently been shown to disrupt the Ca2+ supply to effector lymphocytes, thus disabling the Ca2+-dependent transcription factors NFAT and NF-kB T effectors need in early TCR-dependent activation. This contact suppression mechanism is currently understudied, yet it may play a crucial role in autotolerance (78, 85).
Perforin-granzyme cytolysis is another important contact suppression mechanism characteristic of some subpopulations of activated Tregs. Tregs exhibit perforin-dependent cytotoxicity against a variety of targets, including CD4+, and CD8+ effector T cells (86).
Tumor necrosis factors also are involved in Treg suppressive functions. As Tregs are activated, they acquire TRAIL (TNF-related apoptosis-inducing ligand) expression, while the CD4+ effector cells begin expressing the ligand of this molecule, DR5 (death receptor 5); TRAIL/DR5 interaction induces the apoptosis in effector lymphocytes by activating caspase-8 (87, 88).
Treg lymphocytes simultaneously express the molecule PD-1 and its ligand PD-L1. DC PD-L1 and Treg PD-1 interaction generates a tolerogenic dendritic cell. Treg PD-L1 interacts with PD-1 on the activated effector cells and causes its anergy or even induces the pTreg. In such interaction, the signal is transmitted to the same Treg. The PD-1 transmitted signal is crucial to FoxP3 expression and for maintaining Treg homeostasis. The mechanism is not Treg-specific; it is also important for carcinogenesis and tumor evasion of CD8+ and NK lymphocytes (89).
Because of high IL-2R (CD25) expression, Tregs can reduce IL-2 concentrations in the microenvironment, which will negatively affect the proliferative response of CD8+ cells. The mechanism seems less significant for suppressing the proliferation of CD4+ lymphocytes, which is due to CD4+ and CD8+ differing in their sensitivity to IL-2 (90).
Treg suppressive mechanisms are currently understudied. For example, Tregs expressing HLA-DR manifest early contact suppressive activity associated with high FoxP3 expression. Such HLA-DR+Treg lymphocytes are mature Treg effector cells. At the same time, the antibody blockade of the molecule HLA-DR causes Tregs to lose their suppressive activity in vitro (91). However, the antigen specificity of such suppression remains an unanswered question. Which role MHC-II has to play in this process and how the expression of these molecules emerges on the Treg surface remains to be seen. Some suggest this occurs as part of transendocytosis in Treg-DC interaction, meaning that a Treg is capable of direct antigen-specific contact suppression. However, this conclusion may be premature because this question requires further research (92).
The described mechanisms of Treg-mediated suppression are summarized in Figure 3.

Figure 3. Mechanisms of Treg suppression.
It is worth noting that, in addition to suppressive functions, Tregs regulate tissue repair and regeneration. Tregs interact with innate and adaptive immune cells and regulate their activities after tissue injury. Tregs are involved in tissue-specific repair of the muscle, bone, lung, skin and central nervous system (93). The mechanisms of action may vary from tissue to tissue and include: amphiregulin (a growth factor) production, promotion of proliferation and differentiation of stem cells of different tissues, inhibition of neutrophil extravasation and monocyte activity and also the limitation of osteoclastogenesis (93, 94). In addition, the finding of Rieckmann et al. seems to be an important discovery in the context of TCR Treg specificity. They recently demonstrated that the epitope of myosin heavy chain α is a dominant cardiac antigen triggering CD4+ T cell activation after myocardial infarction in mice. Such Ag-specific T cells selectively accumulated in the myocardium and mediastinal lymph nodes of infarcted mice, acquired a Treg phenotype with a distinct prohealing gene expression profile, and mediated cardioprotection (95). Thus, it seems to be important to investigate new strategies in Treg-mediated tissue regeneration while considering TCR specificity for potential clinical use.
Treg Homeostasis
The common mechanisms of Treg homeostasis are well-known. The main mechanisms are a continuous subliminal signal from TCR, which recognizes the autopeptide in MHC-II (96, 97); co-stimulation signals, in particular those mediated by CD28 in contact with CD80/86 (98, 99); and the effects of humoral factors, primarily IL-2 (100, 101). The combined effects of these factors are observed in the special niches located in the T zones of secondary lymphoid organs, and the effects are DC-mediated. Distribution in niches is tissue-specific and is enabled by TCR affinity to corresponding peptides, as well as by Tregs acquiring characteristic homing molecules as they mature (52–54). Typically, draining lymph nodes function as secondary lymphoid organs. These are key sites for priming the autoreactive T cells, and it seems that they are involved in both inducing and suppressing the tissue-specific immune response (51). This indicates that dLN are the key “arena” of various homeostatic forces, the result of which determines the balance of tolerance and the immune/autoimmune response. This niche theory seemed to be proven by the recent research of Liu et al., who have shown that in secondary lymphoid organs, highly suppressive Tregs are localized in separate clusters and surround autoreactive lymphocytes. These lymphocytes are dominated by activated CD4+ cells that feature a high production of IL-2, which is necessary for Tregs to function. The central place in such clusters is taken by mature DCs that feature high expression of MHC-II and co-stimulatory molecules, mostly of the CD11b+ phenotype. At the same time, Tregs closer to the center express significantly more STAT5 in addition to CTLA-4 and CD73. Further from the center (>100 μm), Tregs have their STAT5, CTLA-4, and CD73 expression drastically reduced, which is due to a lower IL-2 concentration further away from the cluster center. T effectors and their associated increase in IL-2 production trigger compensatory Treg activation, increasing their suppressive activity. In turn, loss of TCR signal or antibody blockade of IL-2 deforms the cluster, which has negative implications for Treg functioning and is accompanied by excessive activation of effector T lymphocytes (102). Thus, autotolerance maintenance is an active process based on subtle feedback regulatory mechanisms implemented in peripheral lymphoid organs at a cluster level involving DCs, Tregs, and effector T lymphocytes (see Figure 4).
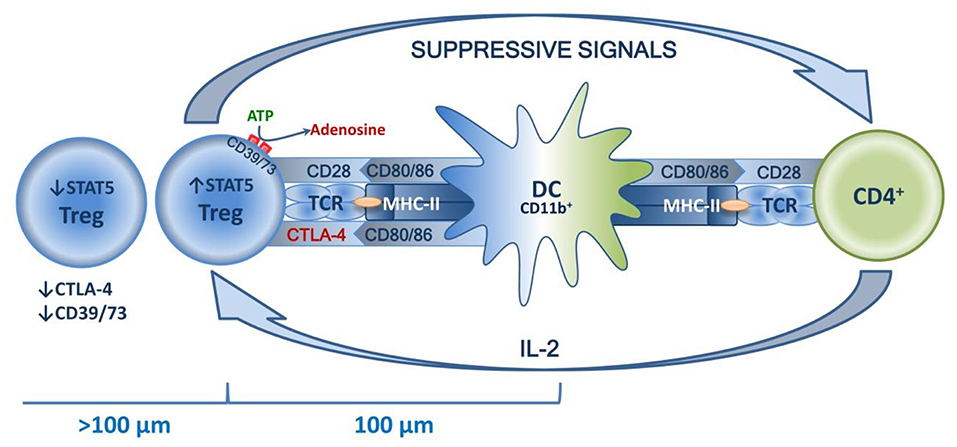
Figure 4. Schematic presentation of a Treg niche.
A number of studies prove the concept presented above. Animal experiments have shown the direct correlation of the peripheral Treg pool with the homeostatic mechanisms above. In transgenic mice, depleting the pool of DCs presenting a specific antigen causes a terminal reduction in the population of Treg clones of specific TCR specificity; increasing this DC pool will dramatically raise the Treg clone population (103, 104). In vivo and in vitro experiments show that disrupting the CD28 and CD80/86 interaction cuts the Treg population and reduces CD25 expression (105–107). In addition, experiments with transgenic mice have revealed a very important pattern: the peripheral Treg pool is a function of the number of effectors capable of IL-2 production (108). Recent research shows that DCs regulate the population and suppressive activity of Tregs by means of the Lkb1-kinase. Knockout of the gene Lkb1 in DCs causes excessive Treg expansion in various organs, deteriorating the antigen-specific immune response. The mechanism behind this phenomenon is associated with increased OX40L expression on DCs, which in turn is caused by activating the nuclear factor NF-kB as Lkb1 activity lowers. The interaction of Treg OX40 and DC OX40L boosts Treg proliferation and suppressive activity. In doing so, researchers discovered that administering LPS to wild mice selectively inhibited Lkb1 expression in DCs and expanded the Tregs while increasing the expression of proinflammatory genes in DCs, which was further confirmed by testing the transcription profile of such DCs. The DC thus remains capable of antigen presentation and priming T effectors. Therefore, Lkb1 may constitute an important factor that DCs use to regulate the immune response. Chen et al. note that the above study calls into question the concept of regulatory DCs; it proves the co-execution of regulatory and inflammatory programs controlled by various signals in the same DC as it is activated and matures (109). More research is necessary to study the mechanisms of controlling Lkb1 expression in DCs, as well as the possible association of changes in the activity of this kinase in various DC types when exposed to different autoimmune diseases or tumors.
Depending on the maturity and tissue distribution, Treg homeostasis may be based on different mechanisms. Just like IL-2, IL-7 is crucial for nTregs circulating between secondary lymphoid organs. This cytokine is produced in lymph nodes by reticuloendothelial cells and enables nTregs to survive and proliferate by increasing the expression of Bcl-2 and Ki-67. As with IL-2, the IL-7 effects are mediated through JAK3/STAT5 and PI3K/Akt signaling pathways (110). It is worth noting that thymopoiesis has quite a low impact on maintaining the nTreg pool in humans, which is confirmed by the minimal effects of age involution and early thymectomy on the nTreg population numbers in adults despite the loss of thymic output and the decrease in the nTreg number at the early stage after thymectomy (110, 111). This is caused by the compensatory peripheral proliferation of naive Treg cells, resulting in the maintenance of the nTreg population. Similar homeostatic maintenance mechanisms are known for cmTreg lymphocytes, as well. Like nTregs, the cells of this weakly proliferating population circulate between secondary lymphoid organs and express anti-apoptotic factors Bcl-2 and Mcl-1 at a high level. IL-2, constitutively produced by Tconv in the lymph node niche, is an important homeostatic factor for these cells (112, 113). Conditions required to maintain Treg memory cell homeostasis have not been studied in such detail because they are tissue-specific, while the emergence of each population invokes multiple mechanisms that vary from location to location. These are long-lived cells emerging from effector Treg lymphocytes after preventing or resolving a primary inflammation; they have a stronger suppressive effect on an immune response caused by secondary contact with a corresponding antigen (114). Experimenting on transgenic mice has shown that differentiation of regulatory memory cells required IL-2 while maintaining a population of these cells required IL-7 but not IL-2. This is reflected by the phenotype of this Treg population because they feature a high expression of IL-2 and IL-7 receptors (CD25+CD127+) (47). Effector Treg lymphocytes are more complicated because they function in different organs and tissues, different inflammation sites, and in the corresponding draining lymph nodes. These cells proliferate actively and are prone to apoptosis because of low Bcl-2 and Mcl-1 expression (112). Their homeostasis is not as dependent on IL-2 and IL-7; rather, it relies on a strong TCR signal that enables the expression of eTreg-specific genes (115). Given that selective Treg effects on different immune response types require different stimuli and are associated with specific transcription changes, drawing a clear border between homeostatic signals and differentiation signals for these cells is difficult.
IL-12 and IFNγ are the main humoral factors typical of the Th1-mediated immune response. However, only the latter is involved in the maturation of Th1-specific Tregs. Binding to IFNγR receptor on Tregs, IFNγ activates STAT1 and triggers T-bet expression, which causes CXCR3 to be expressed and Tregs to migrate to the sites of Th1-mediated inflammation (116). Fluorescent visualization has shown that CXCR3+(T-bet)+Tregs are far closer to T-bet+Th1 and CD8+ lymphocytes than CXCR3−(T-bet)−Tregs, which indicates the lower suppressive activity of CXCR3−(T-bet)−Tregs with respect to the Th1-mediated immune response. Furthermore, analysis into the TCR repertoire has revealed that CXCR3+(T-bet)+Tregs and CXCR3−(T-bet)−Tregs differ in antigenic specificity, which also reflects the functional difference between these subpopulations (62). It is worth noting that T-bet expression is several times lower in Tregs than in Th1 lymphocytes, while the high TCR-dependent expression of FoxP3 suppresses the activation of T-bet-dependent proinflammatory genes in Tregs, thus preventing Tregs from transforming into Th1. Therefore, TCR and IFNγ signals determine the functional maturation and homeostasis of effector Tregs in the context of the Th1-mediated immune response.
eTreg effects on other types of immune responses are less specific and are associated with the TCR-dependent activation of the transcription factor IRF4, which is required for Tregs to function. At the same time, disabling the gene IRF4 in murine Tregs results in an autoimmune syndrome mainly mediated by activating the Th2 type of immune response (64). The expression of this factor is also observed in various populations of effector T cells: Th2, Th9, Th17, Tfh, to which the effector IRF4+Treg can be suppressive (117). IRF4 effects in Tregs are mediated by the transcription factor JunB, which binds IRF4 to target genes in the DNA, thus contributing to the expression of effector Treg molecules such as ICOS and CTLA-4 (66). The expression of ICOS on eTregs is of extreme importance because when interacting with its ligand ICOS-L, this molecule enables Tregs to survive and suppress more efficiently. The mechanism behind ICOS is associated with NFAT activation, which boosts the transcription activity of FoxP3 that, in its turn, enables the expression of the genes IL-4, IL-10, and TGF-β. In addition, the ICOS signal further activates protein kinase B (Akt), which is crucial to Treg survival because it inhibits Treg apoptosis (118). The significance of ICOS in maintaining the homeostasis and functional activity of Tregs has been confirmed by multiple studies. Thus, the lack of expression of this molecule in mice is associated with CNS2 hypermethylation and loss of FoxP3 expression, as well as with a 30% reduction in the Treg pool (119); the antibody blockade of this molecule disrupts Treg functioning (120, 121). Note that the ligand ICOS-L is expressed on the DC; a lack of it may also result in a loss of Treg functionality. Thus, when macroautophagy processes in the DC are disrupted, there accumulates metalloproteinase, which is involved in ICOS-L breakdown. Disrupting the expression of this molecule on DCs negatively affects the suppressive function and stability of Tregs (122). Thus, a strong TCR signal and co-stimulator signals from DCs transmitted from ICOS/ICOS-L are key to the functional activity and homeostasis of the effector eTreg lymphocytes mainly involved in regulating the Th2-mediated immune response.
To act on Th17, eTregs require activating the factor STAT3, which enables selective suppression of the Th17-mediated immune response (see above) (67). STAT3 is activated by various cytokines, including IL-6, and IL-23, which are typical humoral factors of the Th17 immune response. However, STAT3 is only activated in Tregs by means of IL-10, whereas a disrupted expression of IL-10R, as well as disabling the expression of STAT3 in Tregs, accelerates the Th17 response and triggers severe intestinal inflammation (67, 123). IL-10 effects seem to be the most important homeostatic factor that keeps Tregs functional with respect to the Th17-mediated immune response. The effects of this factor are apparently paracrine. It appears that in the case of the Th17-mediated immune response associated with high concentrations of proinflammatory cytokines that negatively affect the stability of FoxP3 expression, the paracrine effects of IL-10 may not suffice to stabilize FoxP3 expression. In that case, Tregs transform into exFoxP3Th17 lymphocytes. This is what frequently accompanies autoimmune diseases such as rheumatoid arthritis.
All of the abovementioned studies prove the paramount role of dendritic cells, co-stimulatory and humoral factors in Treg homeostasis. The relative contribution of these mechanisms to maintaining the homeostasis of different Treg populations may vary from setting to setting. For instance, an immune response, lymphopenia, or inflammation, as well as the emergence of a tumor, may trigger additional contextual mechanisms. Still, they are generally all aimed at enabling the expression of genes of the main Treg signature, as well as that of genes associated with functional adaptation to such conditions; this is indicated by the transcription and functional heterogeneity of Treg populations (50, 56) (Figure 2).
Conclusions
Intensive research in recent decades has described the population of Treg lymphocytes as a separate T cell subpopulation mainly designed to selectively regulate immune response while maintaining autotolerance. Considering selectivity, a basic property of Tregs helps address a contradiction that exists in one of the general autoimmune disease and tumor pathogenesis theories. According to this theory, cancers are triggered by excessive Treg activity while autoimmune processes are due to too low activity. What makes it contradictory is that cancers and autoimmune pathologies may well be concurrent. Given that Tregs are selective to various types of immune responses, and such selectivity depends on antigenic stimuli and the spatial localization of the process, it may be assumed that pathology is caused by the failure of a specific Treg cluster that features a common TCR repertoire of similar antigenic specificity. Ever more data are collected that prove the role TCR plays not only in maintaining the homeostasis, or activation, of Tregs but also in the phenotypic selection because the antigenic specificity of TCR determines the type of immune response Tregs regulate. This is confirmed by the difference in location and transcription profiles of Tregs that differ in TCR specificity (56, 62). This all indicates a clonal organization of the general Treg pool, an organization based on the close antigenic specificity of TCR in the same clone. Indirect evidence of this concept are the positive results obtained in vivo using Tregs with a chimeric antigen receptor (CAR) to treat autoimmune diseases as well as in transplantation. Such CAR-Tregs specifically migrate to target sites and exhibit more pronounced antigen-specific suppressive activity (124), thus filling a gap in a concrete set of Treg clones specific to antigenic determinants with respect to which tolerance is impaired.
A rising Treg population in a tumor positively correlates with disease progression and low survival rates in cancer patients. Research has shown that the effects of Tregs in different tumors are also antigen-specific, which determines the activation and expansion of certain Treg clones in the tumor microenvironment (125). Perhaps novel approaches based on selective suppression of antigen-specific tumor Treg clones will yield better results than using monoclonal antibodies as functional markers characteristic of the general pool of effector Tregs such as PD-1/PD-L1 or CTLA-4.
One additional important piece of evidence of the role of the Ag-specific clonal organization of Treg populations in their functioning was obtained by Bacher's team in 2016. They showed that aeroallergen allergy development linked with imbalance between Ag-specific Treg and Th-2 lymphocytes, which have a specificity of TCR to a narrow set of epitopes. Herewith, such an imbalance was revealed to be associated with the physical entity of these epitopes. These epitopes, which quickly pass into soluble form, predominantly activate Th-2 cells, while particle proteins activate and apparently stimulate proliferation predominantly of Treg cells. This leads to activation of Treg and Th-2 clones with a non-overlapping repertoire of TCR. Thus, a common pool of Treg cells and other Ag-specific Treg clones retain functionality and numbers as in healthy individuals, or have increased functional activity. That demonstrates qualitative and quantitative preservation of a common Treg pool in allergic individuals. This finding highlights necessity to induce Ag-specific Treg responses rather than rely on strategies aimed at activating the existing Treg pool (126).
The negative impact of homeostatic proliferation on the Tconv population leads to changes in the TCR landscape, such as a decrease in the TCR repertoire diversity and oligoclonal expansion (127, 128). Therefore, it seems to be important to investigate the influence of homeostatic proliferation on the Treg pool in the context of clonal organization according to TCR specificity. Because, aside from quantitative maintenance of the nTreg pool, it is important to consider qualitative changes in TCR Treg diversity, which may occur during the homeostatic proliferation of Treg cells with age, and may affect the immune equilibrium in older adults by forming some gaps in the landscape of the naïve Treg TCR repertoire.
Summarizing the data from this review, we conclude that TCR specificity and affinity not only play a key role in thymic selection and maturation of Treg cells but also in determining the further fate of these cells, governing tissue-specific distribution, transcriptomic profile and ultimately determining participation in a particular type of immune response.
In recent years, immunology has seen great advances in its methodologies. New techniques have appeared that enable genomic and transcriptomic analysis, allowing researchers to evaluate the expression of dozens of different proteins in single cells; for example, machine learning-based data mining has become a reality. All of this opens up ample opportunities to research cellular interactions and lays foundations for fundamentally new approaches to the treatment of various diseases by using targeted antigen-specific effects on various components of the immune system to restore the once disturbed equilibrium of the immunity.
Author Contributions
DS contributed to the conception, drafting of the manuscript and design. VT contributed to the conception, revision, and final approval of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Redmond AK, Macqueen DJ, Dooley H. Phylotranscriptomics suggests the jawed vertebrate ancestor could generate diverse helper and regulatory T cell subsets. BMC Evol Biol. (2018) 18:169. doi: 10.1186/s12862-018-1290-2
2. Cao J, Chen Q, Lu M, Hu X, Wang M. Histology and ultrastructure of the thymus during development in tilapia, Oreochromis niloticus. J Anat. (2017) 230:720–33. doi: 10.1111/joa.12597
3. Sugimoto K, Hui PS, Sheng DZ, Nakayama M, Kikuchi K. Zebrafish FOXP3 is required for the maintenance of immune tolerance. Dev. Comp. Immunol. (2017) 73:156–62. doi: 10.1016/j.dci.2017.03.023
4. Gajewski TF, Fitch FW. Anti-proliferative effect of IFN-gamma in immune regulation. I. IFN-gamma inhibits the proliferation of Th2 but not Th1 murine helper T lymphocyte clones. J Immunol. (1988) 140:4245–52.
5. O'Connor WJ, Kamanaka M, Booth CJ, Terrence T, Susumu N, Iwakura Y, et al. A protective function for interleukin 17A in T cell-mediated intestinal inflammation. Nat Immunol. (2009) 10:603–9. doi: 10.1038/ni.1736
6. Wurster AL, Rodgers VL, Satoskar AR. Interleukin 21 is a T helper (Th) cell 2 cytokine that specifically inhibits the differentiation of naive Th cells into interferon gamma-producing Th1 cells. J Exp Med. (2002) 196:969–77. doi: 10.1084/jem.20020620
7. Yssel H, Malefyt WR, Roncarolo MG, Abrams JS, Lahesmaa R, Spits H, et al. IL-10 is produced by subsets of human CD4 + T cell clones and peripheral blood T cells. J Immunol. (1992) 149:2378–84.
8. Samstein RM, Josefowicz SZ, Arvey A, Treuting PM, Rudensky AY. Extrathymic generation of regulatory T cells in placental mammals mitigates maternal-fetal conflict. Cell. (2012) 150:29–38. doi: 10.1016/j.cell.2012.05.031
9. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. (2008) 133:775–87. doi: 10.1016/j.cell.2008.05.009
10. Dembic Z. Beginning of the end of (understanding) the immune response. Scand J Immunol. (2008) 68:381–2. doi: 10.1111/j.1365-3083.2008.02159.x
11. Zhang X, Izikson L, Liu L, Weiner HL. Activation of CD25(+)CD4(+) regulatory T cells by oral antigen administration. J Immunol. (2001) 167:4245–53. doi: 10.4049/jimmunol.167.8.4245
12. Aluvihare VR, Kallikourdis M, Betz AG. Regulatory T cells mediate maternal tolerance to the fetus. Nat Immunol. (2004) 5:266–71. doi: 10.1038/ni1037
13. Eming SA, Wynn TA, Martin P. Inflammation and metabolism in tissue repair and regeneration. Science. (2017) 356:1026–30. doi: 10.1126/science.aam7928
14. Castiglioni A, Corna G, Rigamonti E, Basso V, Vezzoli M, Monno A, et al. FOXP3+ T cells recruited to sites of sterile skeletal muscle injury regulate the fate of satellite cells and guide effective tissue regeneration. PLoS ONE. (2015) 10:e0128094. doi: 10.1371/journal.pone.0128094
15. Ali N, Zirak B, Rodriguez RS, Pauli ML, Truong HA, Lai K, et al. Regulatory T cells in skin facilitate epithelial stem cell differentiation. Cell. (2017) 169:1119–1129. doi: 10.1016/j.cell.2017.05.002
16. Weirather J, Hofmann UD, Beyersdorf N, Ramos GC, Vogel B, Frey A, et al. Foxp3+CD4+T cells improve healing after myocardial infarction by modulating monocyte/macrophage differentiation. Circ Res. (2014) 115:55–67. doi: 10.1161/CIRCRESAHA.115.303895
17. Gershon RK, Kondo K. Cell interactions in the induction of tolerance: the role of thymic lymphocytes. Immunology. (1970) 18:723–37.
18. Sakaguchi S, Fukuma K, Kuribayashi K, Masuda T. Organ-specific autoimmune diseases induced in mice by elimination of T-cell subset. I. Evidence for the active participation of T cells in natural selftolerance: deficit of a T-cell subset as a possible cause of autoimmune disease. J Exp Med. (1985) 161:72–87. doi: 10.1084/jem.161.1.72
19. Kisielow P, Bluthmann H, Staerz UD, Steinmetz M, Boehmer H. Tolerance in T-cell-receptor transgenic mice involves deletion of non-mature CD4+8+ thymocytes. Nature. (1988) 333:742–6. doi: 10.1038/333742a0
20. O'Garra A, Murphy K Role of cytokines in determining T-lymphocyte function. Curr. Opin. Immunol. (1994) 6:458–66. doi: 10.1016/0952-7915(94)90128-7
21. Chen Y, Kuchroo VK, Inobe J, Hafler DA, Weiner HL Regulatory T cell clones induced by oral tolerance: suppression of autoimmune encephalitis. Science. (1994) 265:1237–40. doi: 10.1126/science.7520605
22. Almeida AR, Legrand N, Papiernik M, Freitas AA. Homeostasis of peripheral CD4+ T cells: IL-2R alpha and IL-2 shape a population of regulatory cells that controls CD4+ T cell numbers. J Immunol. (2002) 169:4850–60. doi: 10.4049/jimmunol.169.9.4850
23. Malek TR, Yu A, Vincek V, Scibelli P, Kong L. CD4 regulatory Tcells prevent lethal autoimmunity in IL-2Rbeta-deficient mice. Implications for the non-redundant function of IL-2. Immunity. (2002) 17:167–78. doi: 10.1016/S1074-7613(02)00367-9
24. Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, et al. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet. (2001) 27:68–73. doi: 10.1038/83784
25. Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. (2001) 27:20–1. doi: 10.1038/83713
26. Hori S, Nomura T, Sakaguchi S Control of regulatory T cell development by the transcription factor Foxp3. Science. (2003) 299:1057–61. doi: 10.1126/science.1079490
27. Liu W, Putnam AL, Xu-yu Z, Szot GL, Lee MR, Zhu S, et al. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ Treg cells. J Exp Med. (2006) 203:1701–11. doi: 10.1084/jem.20060772
28. Park JH, Yu Q, Erman B, Appelbaum JS, Montoya-Durango D, Grimes HL, et al., Suppression of IL7Ralpha transcription by IL-7 and other prosurvival cytokines: a novel mechanism for maximizing IL-7-dependent T cell survival. Immunity. (2004) 21:289–302. doi: 10.1016/j.immuni.2004.07.016
29. Zheng SG, Wang J, Wang P, Gray JD, Horwitz DA. IL-2 is essential for TGF-beta to convert naive CD4+CD25- cells to CD25+Foxp3+ regulatory T cells and for expansion of these cells. J Immunol. (2007) 178:2018–27. doi: 10.4049/jimmunol.178.4.2018
30. Pacholczyk R, Ignatowicz H, Kraj P, Ignatowicz L. Origin and T cell receptor diversity of Foxp3+CD4+CD25+ T cells. Immunity. (2006) 25:249–59. doi: 10.1016/j.immuni.2006.05.016
31. Wyss L, Stadinski BD, King CG, Schallenberg S, McCarthy NI, Lee JY, et al. Affinity for self-antigen selects Treg cells with distinct functional properties. Nat Immunol. (2016) 17:1093–101. doi: 10.1038/ni.3522
32. Bolotin DA, Poslavsky S, Davydov AN, Frenkel FE, Fanchi L, Zolotareva OI, et al. Antigen receptor repertoire profiling from RNA-seq data. Nat Biotechnol. (2017) 35:908–11. doi: 10.1038/nbt.3979
33. Izraelson M, Nakonechnaya TO, Moltedo B, Egorov ES, Kasatskaya SA, Putintseva EV, et al. Comparative analysis of murine T-cell receptor repertoires. Immunology. (2018) 153:133–44. doi: 10.1111/imm.12857
34. Moran AE, Holzapfel KL, Xing Y, Cunningham NR, Maltzman JS, Punt J, et al. T cell receptor signal strength in Treg and iNKT cell development demonstrated by a novel fluorescent reporter mouse. J Exp Med. (2011) 208:1279–89. doi: 10.1084/jem.20110308
35. Jordan MS, Boesteanu A, Reed AJ, Petrone AL, Holenbeck AE, Lerman MA, et al. Thymic selection of CD4+CD25+ regulatory T cells induced by an agonist self-peptide. Nat Immunol. (2001) 2:301–6. doi: 10.1038/86302
36. Yadav M, Louvet C, Davini D, Gardner JM, Martinez-Llordella M, Bailey-Bucktrout S, et al., Neuropilin-1 distinguishes natural and inducible regulatory T cells among regulatory T cell subsets in vivo. J Exp Med. (2012) 209:1713–22. doi: 10.1084/jem.20120822
37. Elkord E. Helios should not be cited as a marker of human thymus-derived Tregs. Commentary: Helios(+) and Helios(–) cells coexist within the natural FOXP3(+) T regulatory cell subset in humans. Front Immunol. (2016) 7:276. doi: 10.3389/fimmu.2016.00276
38. Szurek E, Cebula A, Wojciech L, Pietrzak M, Rempala G, Kisielow P, et al. Differences in expression level of Helios and Neuropilin-1 do not distinguish thymus-derived from extrathymically-induced CD4+Foxp3+ regulatory T cells. PLoS ONE. (2015) 10:e0141161. doi: 10.1371/journal.pone.0141161
39. Komatsu N, Okamoto K, Sawa S, Nakashima T, Oh-hora M, Kodama T, et al. Pathogenic conversion of Foxp3+ T-cells into TH17 cells in autoimmune arthritis. Nat Med. (2014) 20:62–70. doi: 10.1038/nm.3432
40. Chen D, Chen Y, Chen H, Hsieh C, Lin C, Lan J. Increasing levels of circulating Th17 cells and interleukin-17 in rheumatoid arthritis patients with an inadequate response to anti-TNF-α therapy. Arthritis Res Ther. (2011) 13:R126. doi: 10.1186/ar3431
41. Hua J, Inomata T, Chen Y, Foulsham W, Stevenson W, Shiang T, et al. Pathological conversion of regulatory T cells is associated with loss of allotolerance. Sci Rep. (2018) 8:7059. doi: 10.1038/s41598-018-25384-x
42. Yang BH, Hagemann S, Mamareli P, Lauer U, Hoffmann U, Beckstette M, et al. Foxp3(+) T cells expressing RORγt represent a stable regulatory T-cell effector lineage with enhanced suppressive capacity during intestinal inflammation. Mucosal Immunol. (2016) 9:444–57. doi: 10.1038/mi.2015.74
43. Feng Y, Arvey A, Chinen T, van der Veeken J, Gasteiger G, Rudensky AY. Control of the inheritance of regulatory T cell identity by a cis element in the Foxp3 locus. Cell. (2014) 158:749–63. doi: 10.1016/j.cell.2014.07.031
44. Kitagawa Y, Ohkura N, Sakaguchi S. Molecular determinants of regulatory T cell development: the essential roles of epigenetic changes. Front Immunol. (2013) 4:106. doi: 10.3389/fimmu.2013.00106
45. Sekiya T, Nakatsukasa H, Lu Q, Yoshimura A. Roles of transcription factors and epigenetic modifications in differentiation and maintenance of regulatory T cells. Microbes Infect. (2016) 18:378–86. doi: 10.1016/j.micinf.2016.02.004
46. Someya K, Nakatsukasa H, Ito M, Kondo T, Tateda KI, Akanuma T, et al. Improvement of Foxp3 stability through CNS2 demethylation by TET enzyme induction and activation. Int Immunol. (2017) 29:365–75. doi: 10.1093/intimm/dxx049
47. Gratz IK, Truong H-A, Yang S-HY, Maurano MM, Lee K, Abbas AK, et al. Memory regulatory T cells require IL-7 and not IL-2 for their maintenance in peripheral tissues. J Immunol. (2013) 190:4483–7. doi: 10.4049/jimmunol.1300212
48. Golubovskaya V, Wu L. Different subsets of T cells, memory, effector functions, and CAR-T. Cancers. (2016) 8:36. doi: 10.3390/cancers8030036
49. Matteucci E, Bartola LD, Giampietro O. Regulatory T cells with effector memory phenotype and glycaemic control in adult type 1 diabetes mellitus. J Diabetes Metab. (2013) S12:003. doi: 10.4172/2155-6156.S12-003
50. Miragaia RJ, Gomes T, Chomka A, Jardine L, Riedel A, Hegazy AN, et al. Single-cell transcriptomics of regulatory T cells reveals trajectories of tissue adaptation. Immunity. (2019) 50:493–504.e7. doi: 10.1016/j.immuni.2019.01.001
51. Scheinecker C, McHugh R, Shevach EM, Germain RN. Constitutive presentation of a natural tissue autoantigen exclusively by dendritic cells in the draining lymph node. J Exp Med. (2002) 196:1079–90. doi: 10.1084/jem.20020991
52. Siegmund K, Feuerer M, Siewert C, Ghani S, Haubold U, Dankof A, et al. Migration matters: regulatory T-cell compartmentalization determines suppressive activity in vivo. Blood. (2005) 106:3097–104. doi: 10.1182/blood-2005-05-1864
53. Dudda JC, Perdue N, Bachtanian E, Campbell DJ. Foxp3+ regulatory T cells maintain immune homeostasis in the skin. J Exp Med. (2008) 205:1559–65. doi: 10.1084/jem.20072594
54. Zhang N, Schröppel B, Lal G, Jakubzick C, Mao X, Chen D, et al. Regulatory T cells sequentially migrate from inflamed tissues to draining lymph nodes to suppress the alloimmune response. Immunity. (2009) 30:458–469. doi: 10.1016/j.immuni.2008.12.022
55. Pohar J, Simon Q, Fillatreau S. Antigen-specificity in the thymic development and peripheral activity of CD4+FOXP3+ T regulatory cells. Front Immunol. (2018) 9:1701. doi: 10.3389/fimmu.2018.01701
56. Zemmour D, Zilionis R, Kiner E, Klein AM, Mathis D, Benoist C. Single-cell gene expression reveals a landscape of regulatory T cell phenotypes shaped by the TCR. Nat Immunol. (2018) 19:291–301. doi: 10.1038/s41590-018-0051-0
57. Sprouse ML, Shevchenko I, Scavuzzo MA, Joseph F, Lee T, Blum S, et al. Cutting edge: low-affinity TCRs support regulatory T cell function in autoimmunity. J Immunol. (2017) 200:909–14. doi: 10.4049/jimmunol.1700156
58. Wei X, Zhang J, Gu Q, Huang M, Zhang W, Guo J, et al. Reciprocal expression of IL-35 and IL-10 defines two distinct effector Treg subsets that are required for maintenance of immune tolerance. Cell Rep. (2017) 27:1853–69. doi: 10.1016/j.celrep.2017.10.090
59. Okeke EB, Uzonna JE. The pivotal role of regulatory T cells in the regulation of innate immune cells. Front Immunol. (2019) 10:680. doi: 10.3389/fimmu.2019.00680
60. Yu X, Harden K, Gonzalez LC, Francesco M, Chiang E, Irving B, et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat Immunol. (2009) 10:48–57. doi: 10.1038/ni.1674
61. Kučan BP, Lenac RT, Cinamon G, Tsukerman P, Mandelboim O, Jonjić S. Targeting PVR (CD155) and its receptors in anti-tumor therapy. Cell Mol Immunol. (2019) 16:40–52. doi: 10.1038/s41423-018-0168-y
62. Levine AG, Mendoza A, Hemmers S, Moltedo B, Niec RE, Schizas M, et al. Stability and function of regulatory T cells expressing the transcription factor T-bet. Nature. (2017) 546:421–5. doi: 10.1038/nature22360
63. Joller N, Lozano E, Burkett PR, Patel B, Xiao S, Zhu C, et al. Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity. (2014) 40:569–81. doi: 10.1016/j.immuni.2014.02.012
64. Zheng Y, Chaudhry A, Kas A, de Roos P, Kim JM, Chu TT, et al. Regulatory T-cell suppressor program co-opts transcription factor IRF4 to control T(H)2 responses. Nature. (2009) 458:351–6. doi: 10.1038/nature07674
65. Delacher M, Schmidl C, Herzig Y, Breloer M, Hartmann W, Brunk F, et al. Rbpj expression in regulatory T cells is critical for restraining TH2 responses. Nat Commun. (2019) 10:1621. doi: 10.1038/s41467-019-09276-w
66. Koizumi SI, Sasaki D, Hsieh TH, Taira N, Arakaki N, Yamasaki S, et al. JunB regulates homeostasis and suppressive functions of effector regulatory T cells. Nat Commun. (2018) 9:5344. doi: 10.1038/s41467-018-07735-4
67. Chaudhry A, Rudra D, Treuting P, Samstein RM, Liang Y, Kas A, et al. CD4+ regulatory T cells control TH17 responses in a Stat3-dependent manner. Science. (2009) 326:986–91. doi: 10.1126/science.1172702
68. Ghoreschi K, Laurence A, Yang XP, Hirahara K, O'Shea JJ. T helper 17 cell heterogeneity and pathogenicity in autoimmune disease. Trends Immunol. (2011) 32:395–401. doi: 10.1016/j.it.2011.06.007
69. Onishi Y, Fehervari Z, Yamaguchi T, Sakaguchi S. Foxp3+ natural regulatory T cells preferentially form aggregates on dendritic cells in vitro and actively inhibit their maturation. Proc Natl Acad Sci USA. (2008) 105:10113–8. doi: 10.1073/pnas.0711106105
70. Akkaya B, Akkaya M, Kamenyeva O, Kabat J, Dorward DW, Holstein AH, et al. Unique interaction dynamics and peptide-MHC class II (pMHC II) transendocytosis lead to antigen-specific T regulatory cell (Treg)-mediated suppression. J Immunol. (2017) 198(Suppl. 1):80.8.
71. Akkaya B, Akkaya M, Oya Y, Souz JA, Holstein AH, Kamenyeva O, et al. Tregs orchestrate antigen specific suppression via stripping cognate peptide-MHCII from the DC surface. J Immunol. (2018) 200(Suppl. 1):47.20.
72. Akkaya B, Oya Y, Akkaya M, Al Souz J, Holstein AH, Kamenyeva O, et al. Regulatory T cells mediate specific suppression by depleting peptide-MHC class II from dendritic cells. Nat Immunol. (2019) 20:218–31. doi: 10.1038/s41590-018-0280-2
73. Fallarino F, Grohmann U, Hwang KW, Orabona C, Vacca C, Bianchi R, et al. Modulation of tryptophan catabolism by regulatory T cells. Nat Immunol. (2003) 4:1206–12. doi: 10.1038/ni1003
74. Ernst PB, Garrison JC, Thompson LF. Much ado about adenosine: adenosine synthesis and function in regulatory T cell biology. J Immunol. (2010) 185:1993–8. doi: 10.4049/jimmunol.1000108
75. Zhu Z, Zhang Y, Ye J, Wang X, Fu X, Yin Y, et al. IL-35 promoted STAT3 phosphorylation and IL-10 production in B cells, but its production was reduced in patients with coronary artery diseases. Hum Immunol. (2018) 79:869–75. doi: 10.1016/j.humimm.2018.10.009
76. Tsuchida Y, Sumitomo S, Ishigaki K, Suzuki A, Kochi Y, Tsuchiya H, et al. TGF-β3 inhibits antibody production by human B cells. PLoS ONE. (2017) 12:e0169646. doi: 10.1371/journal.pone.0169646
77. Dooley A, Quintana M, Cheung M, Sun L, Gupta N. The B cell IL-10 receptor suppresses antibody production. J Immunol. (2018) 200(Suppl. 1):164.9.
78. Schmidt A, Oberle N, Krammer PH. Molecular mechanisms of Treg-mediated T cell suppression. Front Immunol. (2012) 3:51. doi: 10.3389/fimmu.2012.00051
79. Boks MA, Kager-Groenland JR, Haasjes MS, Zwaginga JJ, van Ham SM, Brinke A. IL-10-generated tolerogenic dendritic cells are optimal for functional regulatory T cell induction–a comparative study of human clinical-applicable DC. Clin Immunol. (2012) 142:332–42. doi: 10.1016/j.clim.2011.11.011
80. Strobl H, Knapp W. TGF-beta1 regulation of dendritic cells. Microbes Infect. (1999) 1:1283–90. doi: 10.1016/S1286-4579(99)00256-7
81. Wallet MA, Sen P, Tisch R. Immunoregulation of dendritic cells. Clin Med Res. (2005) 3:166–75. doi: 10.3121/cmr.3.3.166
82. Speck S, Lim J, Shelake S, Matka M, Stoddard J, Farr A, et al. TGF-β signaling initiated in dendritic cells instructs suppressive effects on Th17 differentiation at the site of neuroinflammation. PLoS ONE. (2014) 9:e102390. doi: 10.1371/journal.pone.0102390
83. Thepmalee C, Panya A, Junking M, Chieochansin T, Yenchitsomanus PT. Inhibition of IL-10 and TGF-β receptors on dendritic cells enhances activation of effector T-cells to kill cholangiocarcinoma cells. Hum Vaccin Immunother. (2018) 14:1423–31. doi: 10.1080/21645515.2018.1431598
84. Safinia N, Scotta C, Vaikunthanathan T, Lechler RI, Lombardi G. Regulatory T cells: serious contenders in the promise for immunological tolerance in transplantation. Front Immunol. 6:438. doi: 10.3389/fimmu.2015.00438
85. Schwarz A, Schumacher M, Pfaff D, Schumacher K, Jarius S, Balint B, et al. Fine-tuning of regulatory T cell function: the role of calcium signals and naive regulatory T cells for regulatory T cell deficiency in multiple sclerosis. J Immunol. (2013) 190:4965–70. doi: 10.4049/jimmunol.1203224
86. Grossman WJ, Verbsky JW, Barchet W, Colonna M, Atkinson JP, Ley TJ. Human T regulatory cells can use the perforin pathway to cause autologous target cell death. Immunity. (2004) 21:589–601. doi: 10.1016/j.immuni.2004.09.002
87. Ren X, Ye F, Jiang Z, Chu Y, Xiong S, Wang Y. Involvement of cellular death in TRAIL/DR5-dependent suppression induced by CD4(+)CD25(+) regulatory T cells. Cell Death Differ. (2007) 14:2076–84. doi: 10.1038/sj.cdd.4402220
88. Bodmer JL, Holler N, Reynard S, Vinciguerra P, Schneider P, Juo P, et al. TRAIL receptor-2 signals apoptosis through FADD and caspase-8. Nat Cell Biol. (2000) 2:241–3. doi: 10.1038/35008667
89. Gianchecchi E, Fierabracci A. Inhibitory receptors and pathways of lymphocytes: the role of PD-1 in Treg development and their involvement in autoimmunity onset and cancer progression. Front Immunol. (2018) 9:2374. doi: 10.3389/fimmu.2018.02374
90. Chinen T, Kannan AK, Levine AG, Fan X, Klein U, Zheng Y, et al. An essential role for IL-2 receptor in regulatory T cell function. Nat Immunol. (2016) 17:1322–33. doi: 10.1038/ni.3540
91. Peiser M, Becht A, Wanner R. Antibody blocking of MHC II on human activated regulatory T cells abrogates their suppressive potential. Allergy. (2007) 62:773–80. doi: 10.1111/j.1398-9995.2007.01339.x
92. Baecher-Allan C, Wolf E, Hafler DA. MHC class II expression identifies functionally distinct human regulatory T cells. J Immunol. (2006) 176:4622–31. doi: 10.4049/jimmunol.176.8.4622
93. Li J, Tan J, Martino MM, Lui KO. Regulatory T-cells: potential regulator of tissue repair and regeneration. Front Immunol. (2018) 9:585. doi: 10.3389/fimmu.2018.00585
94. Burzyn D, Kuswanto W, Kolodin D, Shadrach JL, Cerletti M, Jang Y, et al. A special population of regulatory T cells potentiates muscle repair. Cell. (2013) 155:1282–95. doi: 10.1016/j.cell.2013.10.054
95. Rieckmann M, Delgobo M, Gaal C, Büchner L, Steinau P, Reshef D, et al. Myocardial infarction triggers cardioprotective antigen-specific T helper cell responses. J Clin Invest. (2019) 130:4922–36. doi: 10.1172/JCI123859
96. Vahl JC, Drees C, Heger K, Heink S, Fischer JC, Nedjic J, et al. Continuous T cell receptor signals maintain a functional regulatory T cell pool. Immunity. (2014) 41:722–36. doi: 10.1016/j.immuni.2014.10.012
97. Schmidt AM, Lu W, Sindhava VJ, Huang Y, Burkhardt JK, Yang E, et al. Regulatory T cells require TCR signaling for their suppressive function. J Immunol. (2015) 194:4362–70. doi: 10.4049/jimmunol.1402384
98. Golovina TN, Mikheeva T, Suhoski MM, Aqui NA, Tai VC, Shan X, et al. CD28 costimulation is essential for human T regulatory expansion and function. J Immunol. (2008) 181:2855–68. doi: 10.4049/jimmunol.181.4.2855
99. He X, Smeets RL, van Rijssen E, Boots AM, Joosten I, Koenen HJ. Single CD28 stimulation induces stable and polyclonal expansion of human regulatory T cells. Sci Rep. (2017) 7:43003. doi: 10.1038/srep43003
100. Jeon PH, Oh KI. IL2 is required for functional maturation of regulatory T cells. Anim Cells Syst. (2017) 21:1–9. doi: 10.1080/19768354.2016.1272489
101. Fan MY, Low JS, Tanimine N, Finn KK, Priyadharshini B, Germana SK, et al. Differential roles of IL-2 signaling in developing versus mature Tregs. Cell Rep. (2018) 25:1204–13.e4. doi: 10.1016/j.celrep.2018.10.002
102. Liu Z, Gerner MY, Van Panhuys N, Levine AG, Rudensky AY, Germain RN. Immune homeostasis enforced by co-localized effector and regulatory T cells. Nature. (2015) 528:225–30. doi: 10.1038/nature16169
103. Suffner J, Hochweller K, Kühnle MC, Li X, Kroczek RA, Garbi N, et al. Dendritic cells support homeostatic expansion of Foxp3+ regulatory T cells in Foxp3.LuciDTR mice. J Immunol. (2010) 184:1810–20. doi: 10.4049/jimmunol.0902420
104. Darrasse-Jeze G, Deroubaix S, Mouquet H, Victora GD, Eisenreich T, Yao KH, et al. Feedback control of regulatory T cell homeostasis by dendritic cells in vivo. J Exp Med. (2009) 206:1853–62. doi: 10.1084/jem.20090746
105. Bar-On L, Birnberg T, Kim KW, Jung S. Dendritic cell-restricted CD80/86 deficiency results in peripheral regulatory T-cell reduction but is not associated with lymphocyte hyperactivation. Eur J Immunol. (2011) 41:291–8. doi: 10.1002/eji.201041169
106. Tang Q, Henriksen KJ, Boden EK, Tooley AJ, Ye J, Subudhi SK, et al. Cutting edge: CD28 controls peripheral homeostasis of CD4+CD25+ regulatory T cells. J Immunol. (2003) 3348–52. doi: 10.4049/jimmunol.171.7.3348
107. Zheng Y, Manzotti CN, Liu M, Burke F, Mead KI, Sansom DM. CD86 and CD80 differentially modulate the suppressive function of human regulatory T cells. J Immunol. (2004) 172:2778–84. doi: 10.4049/jimmunol.172.5.2778
108. Almeida AR, Zaragoza B, Freitas AA. Indexation as a novel mechanism of lymphocyte homeostasis: the number of CD4+CD25+ regulatory T cells is indexed to the number of IL-2-producing cells. J Immunol. (2006) 177:192–200. doi: 10.4049/jimmunol.177.1.192
109. Chen S, Fang L, Guo W, Zhou Y, Yu G, Li W, et al. Control of Treg cell homeostasis and immune equilibrium by Lkb1 in dendritic cells. Nat Commun. (2018) 9:5298. doi: 10.1038/s41467-018-07545-8
110. Silva SL, Albuquerque AS, Serra-Caetano A, Foxall RB, Pires AR, Matoso P, et al. Human naive regulatory T-cells feature high steady-state turnover and are maintained by IL-7. Oncotarget. (2016) 7:12163–75. doi: 10.18632/oncotarget.7512
111. Schadenberg AW, van den Broek T, Siemelink MA, Algra SO, de Jong PR, Jansen NJ, et al. Differential homeostatic dynamics of human regulatory T-cell subsets following neonatal thymectomy. J Allergy Clin Immunol. (2014) 133:277–80.e1–6. doi: 10.1016/j.jaci.2013.08.030
112. Smigiel KS, Richards E, Srivastava S, Thomas KR, Dudda JC, Klonowski KD, et al. CCR7 provides localized access to IL-2 and defines homeostatically distinct regulatory T cell subsets. J Exp Med. (2014) 211:121–36. doi: 10.1084/jem.20131142
113. Long M, Adler AJ. Cutting edge: paracrine, but not autocrine, IL-2 signaling is sustained during early antiviral CD4 T cell response. J Immunol. (2006) 177:4257–61. doi: 10.4049/jimmunol.177.7.4257
114. Rosenblum MD, Gratz IK, Paw JS, Lee K, Marshak-Rothstein A, Abbas AK. Response to self-antigen imprints regulatory memory in tissues. Nature. (2011) 480:538–42. doi: 10.1038/nature10664
115. Levine AG, Arvey A, Jin W, Rudensky AY. Continuous requirement for the TCR in regulatory T cell function. Nat Immunol. (2014) 15:1070–8. doi: 10.1038/ni.3004
116. Koch MA, Tucker-Heard G, Perdue NR, Killebrew JR, Urdahl KB, Campbell DJ. The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat Immunol. (2009) 10:595–602. doi: 10.1038/ni.1731
117. Huber M, Lohoff M. IRF4 at the crossroads of effector T-cell fate decision. Eur J Immunol. (2014) 44:1886–95. doi: 10.1002/eji.201344279
118. Chen Q, Mo L, Cai X, Wei L, Xie Z, Li H, et al. ICOS signal facilitates Foxp3 transcription to favor suppressive function of regulatory T cells. Int J Med Sci. (2018) 15:666–73. doi: 10.7150/ijms.23940
119. Landuyt AE, Klocke BJ, Colvin TB, Schoeb TR, Maynard CL. Cutting edge: ICOS-deficient regulatory T cells display normal induction of Il10 but readily downregulate expression of Foxp3. J Immunol. (2019) 202:1039–44. doi: 10.4049/jimmunol.1801266
120. Burmeister Y, Lischke T, Dahler AC, Mages HW, Lam KP, Coyle AJ, et al. ICOS controls the pool size of effector-memory and regulatory T cells. J Immunol. (2008) 180:774–82. doi: 10.4049/jimmunol.180.2.774
121. Herman AE, Freeman GJ, Mathis D, Benoist C. CD4+CD25+ T regulatory cells dependent on ICOS promote regulation of effector cells in the prediabetic lesion. J Exp Med. (2004) 199:1479–89. doi: 10.1084/jem.20040179
122. Niven J, Madelon N, Page N, Caruso A, Harlé G, Lemeille S, et al. Macroautophagy in dendritic cells controls the homeostasis and stability of regulatory T cells. Cell Rep. (2019) 28:21–29.e6. doi: 10.1016/j.celrep.2019.05.110
123. Chaudhry A, Samstein RM, Treuting P, Liang Y, Pils MC, Heinrich JM, et al. Interleukin-10 signaling in regulatory T cells is required for suppression of Th17 cell-mediated inflammation. Immunity. (2011) 34:566–78. doi: 10.1016/j.immuni.2011.03.018
124. Zhang Q, Lu W, Liang CL, Chen Y, Liu H, Qiu F, et al. Chimeric antigen receptor (CAR) Treg: a promising approach to inducing immunological tolerance. Front Immunol. (2018) 9:2359. doi: 10.3389/fimmu.2018.02359
125. Ahmadzadeh M, Pasetto A, Jia L, Deniger DC, Stevanovic S, Robbins PF, et al. Tumor-infiltrating human CD4+ regulatory T cells display a distinct TCR repertoire and exhibit tumor and neoantigen reactivity. Sci Immunol. (2019) 4:eaao4310. doi: 10.1126/sciimmunol.aao4310
126. Bacher P, Heinrich F, Stervbo U, Nienen M, Vahldieck M, Iwert C, et al. Regulatory T cell specificity directs tolerance versus allergy against aeroantigens in humans. Cell. (2016) 167:1067–78.e16. doi: 10.1016/j.cell.2016.09.050
127. Egorov ES, Kasatskaya SA, Zubov VN, Izraelson M, Nakonechnaya TO, Staroverov DB, et al. The changing landscape of naive T cell receptor repertoire with human aging. Front Immunol. (2018) 9:1618. doi: 10.3389/fimmu.2018.01618
Keywords: T-regulatory cell, single-cell analysis (SCA), TCR—T cell receptor, antigen-specific activation, immune equilibrium, TCR repertoire
Citation: Shevyrev D and Tereshchenko V (2020) Treg Heterogeneity, Function, and Homeostasis. Front. Immunol. 10:3100. doi: 10.3389/fimmu.2019.03100
Received: 20 September 2019; Accepted: 18 December 2019;
Published: 14 January 2020.
Edited by:
Kjetil Taskén, Oslo University Hospital, NorwayReviewed by:
Olga Britanova, Shemiakin-Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Science, RussiaPaul G. Thomas, St. Jude Children's Research Hospital, United States
Copyright © 2020 Shevyrev and Tereshchenko. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniil Shevyrev, dr.daniil25@mail.ru