 Qihan Zhao1
Qihan Zhao1 Haoran Dai2Xianli Liu2Hanxue Jiang1
Haoran Dai2Xianli Liu2Hanxue Jiang1 Wenbin Liu3
Wenbin Liu3 Zhendong Feng1,4
Zhendong Feng1,4 Na Zhang1
Na Zhang1 Yu Gao1
Yu Gao1 Zhaocheng Dong1,3Xiaoshan Zhou1,3Jieli Du1,3Naiqian Zhang1,3Hongliang Rui5Li Yuan6
Zhaocheng Dong1,3Xiaoshan Zhou1,3Jieli Du1,3Naiqian Zhang1,3Hongliang Rui5Li Yuan6 Baoli Liu1*
Baoli Liu1*- 1Beijing Hospital of Traditional Chinese Medicine Affiliated to Capital Medical University, Beijing, China
- 2Shunyi Branch, Beijing Traditional Chinese Medicine Hospital, Beijing, China
- 3Beijing University of Chinese Medicine, Beijing, China
- 4Beijing Chinese Medicine Hospital Pinggu Hospital, Beijing, China
- 5Beijing Institute of Traditional Chinese Medicine, Beijing Hospital of Traditional Chinese Medicine Affiliated to Capital Medical University, Beijing, China
- 6Department of Nephrology, Affiliated Hospital of Nantong University, Nantong, China
Idiopathic membranous nephropathy (IMN) is an autoimmune disease in which the immune system produces an antibody response to its own antigens due to impaired immune tolerance. Although antibodies are derived from plasma cells differentiated by B cells, the T-B cells also contribute a lot to the immune system. In particular, the subsets of helper T (Th) cells, including the dominant subsets such as Th2, Th17, and follicular helper T (Tfh) cells and the inferior subsets such as regulatory T (Treg) cells, shape the immune imbalance of IMN and promote the incidence and development of autoimmune responses. After reviewing the physiological knowledge of various subpopulations of Th cells and combining the existing studies on Th cells in IMN, the role model of Th cells in IMN was explained in this review. Finally, the existing clinical treatment regimens for IMN were reviewed, and the importance of the therapy for Th cells was highlighted.
Introduction
In 2009, Beck et al. (1) discovered the podocyte autoantigen, i.e., M-type receptor of secretory phospholipase A2 1 (PLA2R1), in the immune deposits of IMN, providing a key evidence of IMN as an autoimmune disease. Later, in addition to PLA2R, more IMN antigens were identified, including thrombospondin type-1 domain-containing 7A (THSD7A), neural epidermal growth factor-like 1 protein (Nell-1), and semaphorin 3B (sema3B), which were all self-components of podocytes (2). In recent years, the incidence of IMN has been increasing year by year, making it the most common primary glomerular disease (3). At present, it is widely accepted that the autoimmune reaction of antibodies and the circulation and combination of target antigens on the cell, formed in situ immune complex deposition in cells and basement membrane space, lead to cell destruction, basement membrane thickening, and glomerular filtration barrier damage, as well as proteinuria and low plasma protein concentration (4).
As a key component of the human adaptive immune system, Helper T (Th) cells play an auxiliary or regulatory role in the immune response by expressing CD4 (5). Before being stimulated by antigens and cytokines, CD4+T cells are in their initial state, namely, naive CD4+ T cells. Upon being stimulated, the naive T cells begin to differentiate into different lineages. The differentiation direction is influenced by T cell receptor (TCR) signaling and specific cytokines in the microenvironment, and the cell fate is determined by major activated transcription factors. At present, 5 subsets of Th cells are relatively well-defined: Th1, Th2, Th17, regulatory T (Treg) cells and follicular helper T (Tfh) cells (6).
Due to their importance to autoimmune response, possible roles of various subsets of Th cells in the induction, immune disorders, and antibody generation of IMN will be discussed, and new clinical therapeutic strategies will be presented.
Understanding Helper T Cells
The subsets of helper T cells are balanced and coordinated with each other, as shown in Figure 1. Th1 and Th2 subsets were the first ones discovered and explained by Mosmann et al. in 1986 (7). When the organism was infected with intracellular pathogens, such as viruses and bacteria, the naive T cells could be induced to differentiate into Th1 cells (8). Such differentiation is mainly promoted by IFN-γ and IL-12, which activate the major transcription factor T-bet through signaling transducer and activator of transcription (STAT)1 and STAT4 signaling, respectively, thus producing more IFN-γ in turn. IFN-γ is the major effector of Th1 cells functions to activate macrophage-mediated cellular immunity (6). IFN-γ also urges T-bet to produce a cascading amplification effect of Th1 cells through autocrine and positive feedback mechanisms (9). In contrast, Th2 cells mainly mediate humoral immune response and assist B cells to produce antibodies. IL-4 activates STAT6 signaling to promote the transformation of naive T cells to Th2 cells, which is regulated by GATA3, the major transcription factor of Th2 cells (10). IL-2 is also important for the formation of Th2 cells by activating STAT5 (11). IL-4 in Th2 cells also plays a similar role to the positive feedback mechanism of IFN-γ in Th1 cells, promoting Th2 cells differentiation (10). Th2 cells can also produce IL-5 and IL-13, etc., and participate in allergic reactions (6). There is an antagonistic relationship between Th1 and Th2 cells. First, when naive T cells receive antigen-presenting signals through TCR, a stronger TCR signal promotes Th1 differentiation, while a weaker TCR signal promotes Th2 differentiation (12). In addition, their major transcription factors T-bet and GATA3 are also inhibiting each other at both gene expression level and protein level (13, 14).
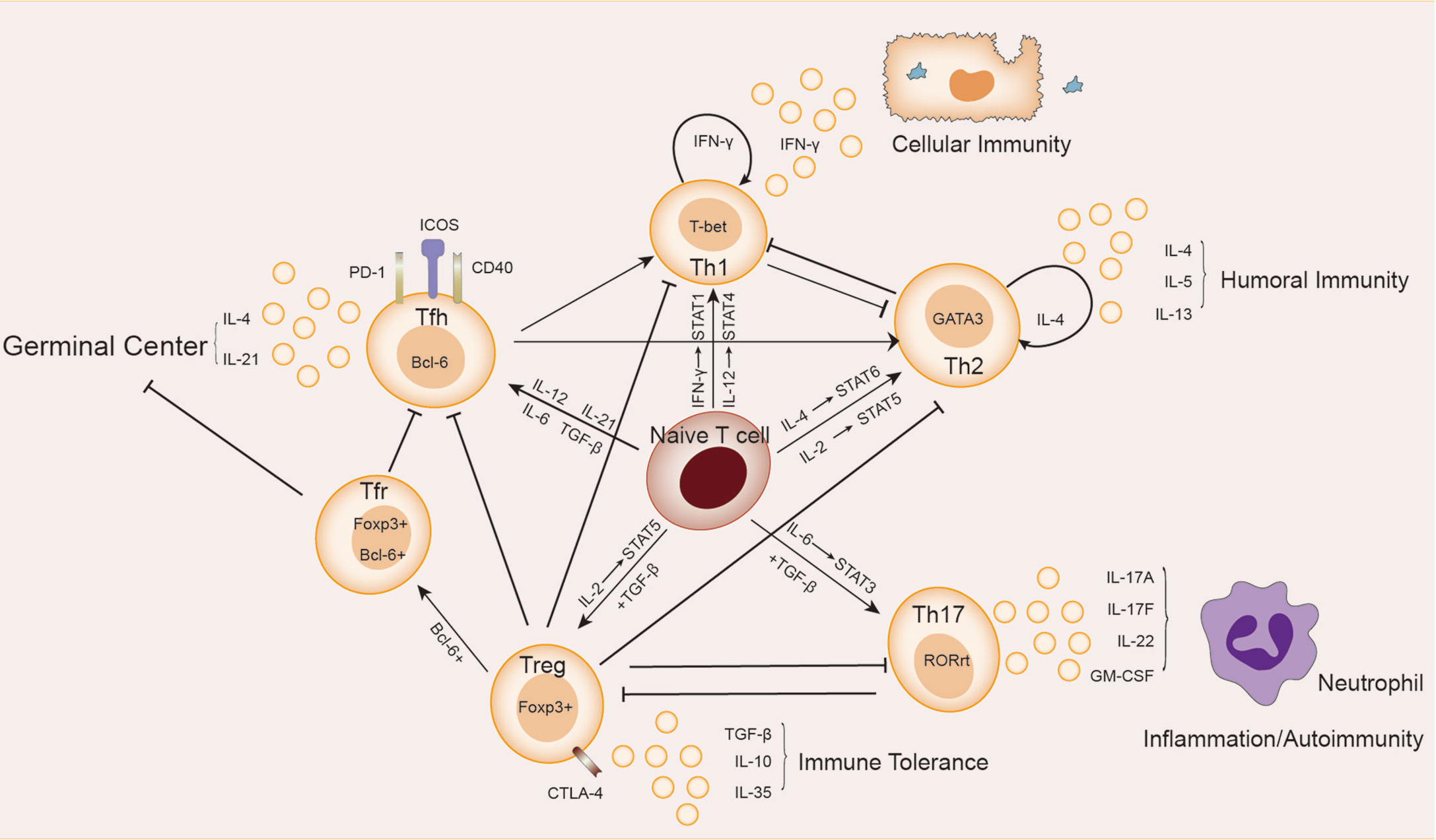
Figure 1 Relationship between Th cells subpopulations. Naive T cells differentiate in different directions under different conditions: IL-12 and IFN-γ activate STAT4 and STAT1 signaling, respectively, inducing the expression of the major transcription factor T-bet, and naive T cells differentiate in the direction of Th1 cells, which secrete cytokines such as IFN-γ and participate in cellular immunity. Th2 cells secrete cytokines such as IL-4, IL-5, and IL-13, which are involved in humoral immunity. In the presence of IL-6 and TGF-β, naive T cells differentiate towards Th17 cells, whose main transcription factor is RORγt. Th17 cells secrete cytokines such as IL-17A, IL-17F, IL-22 and GM-CSF, which are involved in autoimmune diseases or inflammatory responses. Treg cells secrete IL-10, IL-35 and TGF-β to maintain immune tolerance. The naive T cells differentiate towards Tfh cells in response to cytokines such as IL12, IL-21, IL-6 and TGF-β. Treg cells differentiate into Tfr cells in the germinal center. Tfh cells and Tfr cells together participate in the germinal center response.
Approximately 10 years later after the discovery of Th1/Th2 cells, Sakaguchi et al. found a subpopulation (Treg cells) of CD4+T cells expressing the IL-2 receptor α (CD25) in mice that exacted immunosuppressive effects and maintained immune tolerance (15). Treg cells were derived from initial T cells induced by TGF-β alone and mainly regulated by the transcription factor forkhead box P3 (FoxP3) (16). Due to the grouping expression of CD25, Treg cells had a higher affinity with IL-2 than other Th subsets as it helped to achieve optimal inhibition of Treg cells through activation of STAT5 signaling (17). In general, Treg cells were found with high expression of CD4, simultaneous expression of CD25, Foxp3 (cytoplasm), and low expression of CD127 (IL-7 receptor α chain), constituting phenotype for such cells (18). Decrease in the number and/or function of Treg cells has been observed in patients with a variety of autoimmune diseases and mouse models (19). Treg cells have been identified with functional plasticity and different transcriptional characteristics in response to different types of immune responses and environments, thus playing a greater role of immunosuppression. There is also a subset of follicular regulatory T cells (Tfr) located in the germinal center (GC) that, in addition to expressing Foxp3, also express the chemokine CXCR5 and transcription factor Bcl-6, which are also markers of Tfh cells (20). The function of Tfr cells is to inhibit GC reaction and plasma cell differentiation, which is in balance with Tfh cells.
Until 2005, a new subset of Th cells known as Th17 cells which can secrete IL-17 to regulate tissue inflammation was discovered (21, 22). The development of Th17 cells rely on both the induction of TGF-β and the action of the inflammatory factor IL-6. They activate the major transcription factor RORγ-T through the STAT3 signaling pathway, which determines the differentiation of naive T cells to Th17 cells. This induction of IL-6 can also be enhanced in the presence of other cytokines, including IL-1β, TNF-α, IL-23, and IL-21 (23–25). Th17 cells produce IL-17A, IL-17F, IL-22 and granulocyte-macrophage colony-stimulating factor (GM-CSF), recruit inflammatory cells such as neutrophils, and promote inflammation at the infected site (26). An increase in Th17 cells has been observed in a variety of forms of autoimmune diseases, including inflammatory bowel disease (IBD), psoriasis, rheumatoid arthritis (RA), etc. (26), which is contrary to the observed reduction or suppression of Treg cells. There is a balance between Th17 and Treg cells: first, they compete for TGF-β at the site of differentiation; second, both STAT5 and Foxp3 in Treg can inhibit Th17 differentiation, while STAT3 signaling in Th17 can down-regulate Foxp3. All these lead to the differentiation of naive T cells in two different directions under different conditions. It was much believed that the imbalance of Th17/Treg cells was the key to the pathogenesis and therapeutic target of autoimmune diseases (27, 28). Yet, the cause for such imbalance still remains unknown.
Several groups of studies have identified a type of CXCR5+Th cells that have a specific and preferred helper function to B cells in follicles (29–31), known as follicular helper T(Tfh) cells. The main transcription factor of Tfh cells is Bcl-6, which is essential to Tfh formation, assistance to B cells and GC formation (32–34). The expression of Bcl-6 inhibits differentiation of CD4+T cells in directions other than Tfh cells (33), and also hinders the expression of Th1, Th2, Th17 and Treg-related functional receptors (35, 36). In humans, IL-12, IL-21, IL-6, IL-23, and TGF-β synergistically promote Tfh cells, but TGF-β inhibits Tfh cells development in mice (37–40). IL-2 inhibits STAT3 and Bcl-6 by phosphorylating STAT5, and upregulates Blimp-1, thereby inhibiting Tfh cells (41, 42). Tfh cells also secrete IL-21 and express surface molecules programmed cell death protein 1 (PD-1) and recombinant Inducible T cell co-stimulator (ICOS) (43–45), which are critical for regulating the development, migration and function of Tfh cells. Differentiation and development of Tfh cells is mainly accomplished in secondary lymphoid organizations (SLOs). Through the interaction with B cells, Tfh cells gradually migrate from the T cell zone, through the T-B border, to the B cell follicles and germinal center, and finally form GC Tfh cells (46–49), as shown in Figure 2. GC Tfh cells are necessary to maintain GC response and cause three outcomes of B cells: A, differentiation into long-term memory B cells, waiting to be exposed to antigen again; B, differentiated into long-lived plasma cells to continue to produce antibodies; C, re-entry into the dark zone for more proliferation and somatic hypermutation (50–52). Owing to its heterogeneity and plasticity, GC Tfh cells are also able to adapt to different types of immune responses. In addition to secreting IL-21, Tfh cells can also produce IL-4 in response to Th2-mediated antibody response (53).
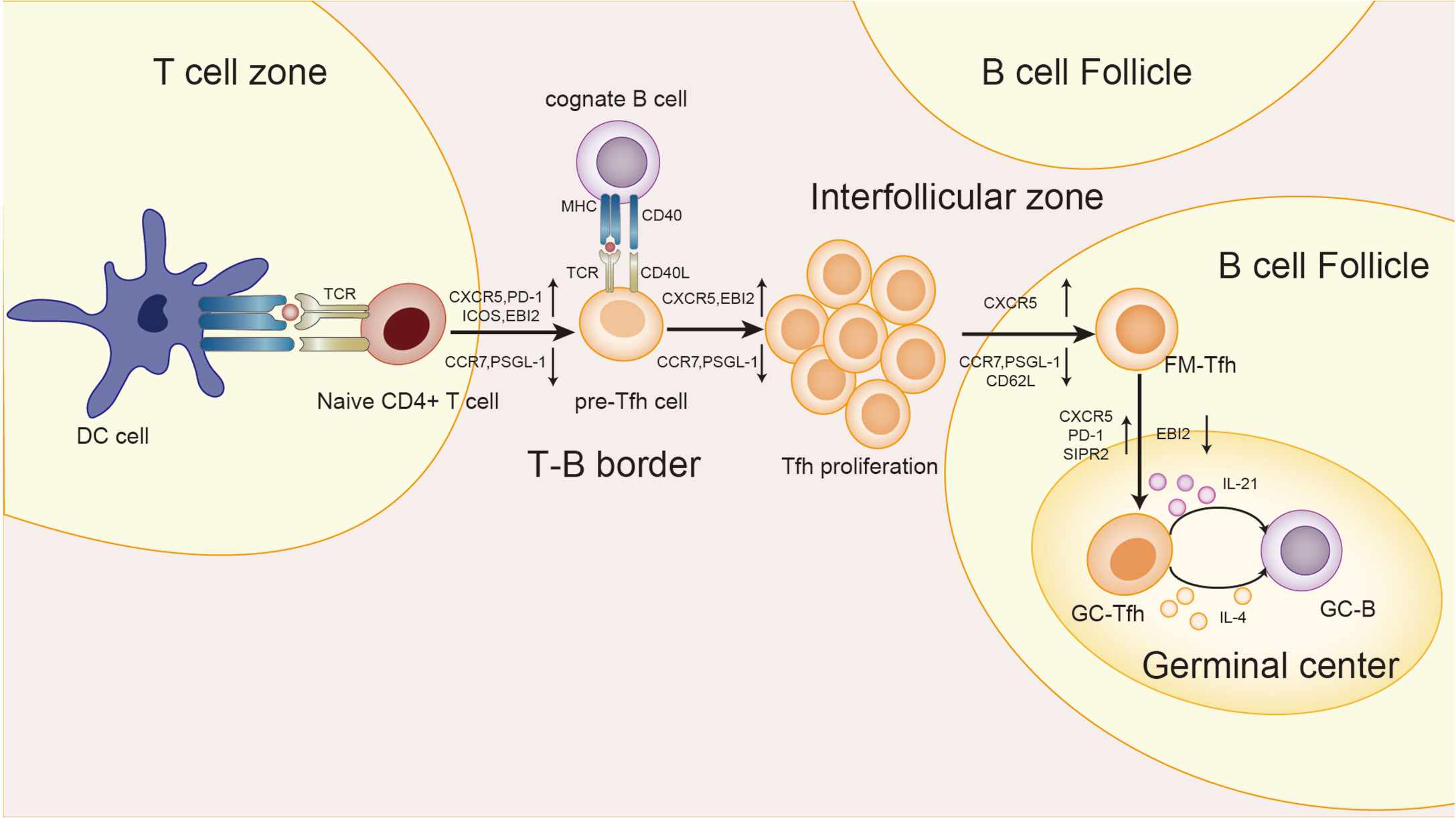
Figure 2 Differentiation and development of Tfh cells. First, in the T cell zone, the naive T cells receive the antigen presentation signal from the DC cells, and Tfh cells begins to differentiate. T cells expressed CXCR5, PD-1, ICOS, and epstein-barr virus-induced gene 2(EBI2), while CCR7 and P-selectionglycoproteinligand-1(PSGL-1) were down-regulated to obtain the pre-Tfh cell phenotype. At the T-B border, cognate B cells interact with T cells to maintain the Tfh cell phenotype. After that, the T-B cell complexes move from the border to the interfollicular zone, where more proliferation takes place. Next, Tfh cells are about to enter the follicle, and the signal from the bystander B cells further upregulates CXCR5 and suppresses CCR7, PSGL-1, and CD62L. Finally, Tfh cells in the follicular fimbria up-regulated CXCR5, PD-1, and sphingosine-1-phosphate receptor 2(S1RP2) surface molecules, down-regulated EBI2, and became GC-Tfh cells. The expression of IL-21 and IL-4 by GC Tfh is essential for the survival, proliferation and differentiation of germinal center B cells.
Since it is difficult to obtain SLOs from patients, attention has been paid to circulating cells with a Tfh phenotype. Some CD4+T cells in the blood with a Tfh-like phenotype (CXCR5+) subpopulation, but without Bcl-6 expression, are referred to as circulating Tfh (cTfh) cells (52). Although the relationship between cTfh cells and true Tfh cells in SLOs is unclear, the frequency of cTfh and its subsets are associated with influenza vaccines, chronic infections, and autoimmune diseases (54–58). Therefore, circulating CXCR5+CD4+T cells are currently considered to be the circulating responders of Tfh cells. According to the different expressions of CXCR3 and CCR6, cTfh can be divided into three subsets expressing different cytokines: A, CXCR3+CCR6-cTfh1, which can secrete IFN-γ; B, CXCR3-CCR6-cTfh2, which can secrete IL-4, IL-5 and IL-13; C, CXCR3-CCR6+cTfh17, which can secrete IL-17A, IL-17F and IL-22 (59). In addition, the activation status of cTfh cells can be distinguished according to the expression of ICOS and PD-1 cTfh2 and cTfh17 can secrete IL-21, which can effectively induce proliferation and differentiation of juvenile B cells and antibody class conversion (59–62).
Helper T Cells in IMN
Th Cells and Induction of IMN
There are many inducing factors of autoimmune diseases, such as the change in autoantigen, the abnormality of immune system, genetic factors, gender and age, etc., as well as their combined forces (63). IMN is usually caused by a single antigen, of which PLA2R accounts for 75%, and 10%-20% of IMN patients have not yet been identified with their antigens (64). Exposure to autoantigen is the major incidence reason, and no direct evidence has been found to reveal this process in IMN. Considering PLA2R as an example, anti-PLA2R antibodies in serum of IMN patients can bind to PLA2R antigen in vitro in a non-reduced state (65), which suggests that the antibody-bound epitopes require PLA2R spatial epitopes and are maintained by disulfide bonds (66). In China, the incidence of IMN is positively correlated with air pollution reflected by PM2.5 (67). We and Paul Brenchley et al. have proposed the hypothesis that lung tissue is stimulated by PM2.5 to cause an inflammatory environment, leading to exposure of PLA2R1 pathogenic epitopes in a strong oxidative microenvironment and then inducing the pathogenesis of IMN (68, 69). Recently, several studies have indeed found enhanced expression of Th17 cells and up-regulation of IL-17 and other cytokines in IMN, suggesting that there is indeed an inflammatory environment in IMN (70–72). Why has PLA2R become the main autoantigen of IMN? This may be related to genetic predisposition. At present, HLA-DQA1 and PLA2R allele risk loci have been found in IMN, which can promote the delivery of antigen epitopes to T cells through major histocompatibility complex (MHC) class II (73, 74), and CD4+T cells receive antigen signals through TCR.
In addition to the exposure of epitopes, the pathogenesis of IMN also involves the breakdown of autoimmune tolerance, including central and peripheral immune tolerance. The production of autoreactive T cells and B cells matters a lot, and the question is how they can escape the numerous tolerance checkpoints. In the process of thymus development of T cells, the V region gene of TCR is rearranged (75). This process may produce TCR against autoantigen, which can be eliminated by negative selection. However, this process may be abnormal in autoimmune diseases, causing abnormalities in the TCR library of T cells arriving at the periphery. Not long ago, Yu Zhang et al. (76) used T-cell receptor repertoire high-throughput sequencing (TCR-HTS) to analyze the TCR β chain repertoire of the circulating T lymphocytes of IMN patients. The result showed that IMN had lower diversity of VJ cassette combination in peripheral blood and a decrease in TCR lineage diversity. A decrease in TCR diversity of peripheral T cells has also been observed in patients with compulsive spondylitis and systemic lupus erythematosus (77, 78). This may explain why autoreactive T cells have escaped central immune tolerance, or why TCR has a shared sequence in patients, increasing the risk of autoimmune diseases (79). Peripheral immune tolerance may also play a key role in autoimmune diseases (80). Treg cells are the key to maintaining peripheral immune tolerance. A large amount of evidence shows that IMN has a reduced proportion of Treg cells in serum and decreased expression of Foxp3 (72), as well as impaired activation and inhibition of Treg cells (81). However, the expression of Treg cells in patients improved by rituximab treatment was significantly up-regulated, and the proportion of Treg cells had a prognostic effect on the treatment of rituximab (82, 83).
Immune response of IMN is dominated by humoral immunity (84), during which differentiation and development of autoreactive B cells are crucial, and Tfh cells play the role of peripheral immune tolerance checkpoint (85, 86). The B cell pool of healthy adults contains a large number of autoreactive B cells, but they have a low affinity and therefore do not cause disease (87, 88). Autoimmune diseases, including IMN, require high affinity with disease-causing antibodies (89, 90), suggesting that these plasma cells that produce these antibodies have undergone affinity maturation and somatic hypermutation (SHM) in GC. In fact, most GC-B cells experience apoptosis, and only a small portion survives and differentiates into memory B cells or plasma cells to leave GC (91–93). GC-B cells can survive and develop only with the assistance of T cells. A competitive model was first proposed, which was positively correlated with B cell receptor (BCR) affinity and antigen presenting ability (52). Professor Carola G. Vinuesa later described this competitive mechanism as positive selection and negative selection of Tfh cells (85, 94). Positive selection meant that Tfh cells provide survival signals to GC-B cells through CD40L, IL-4, IL-21 and other cytokines (94). Negative selection referred to the process Tfh cells transmit death signal to GC-B cells via CD95L. Mice with CD95L deficiency would develop autoimmune diseases (94). B cells without CD40L signal went to apoptosis (95), and the homologous interaction of T-B cells could make B cells enter the dark area again for further division and SHM (85). Studies have shown that the reduction of SHM is associated with the impairment of B cell tolerance, and the increase of cTfh cells and IL-21 in such patients (86). Restrictions on the number and quality (secreting cytokines) of Tfh cells create an environment in which GC B cells must compete for help, making it difficult for some low-affinity B cells, such as autoreactive B cells, to proliferate and differentiate. However, when the amount of Tfh cells increase abnormally, this checkpoint will be damaged, and the loosening of the floodgate will allow some autoreactive B cells to proliferate and differentiate, producing antibodies, and leading to autoimmune disease, which has been confirmed in Sanroque mice (45, 96, 97). In addition, Tfh cells were associated with the occurrence of autoimmune responses in chronic inflammation (98) as well as the process of antigen simulation (99), which has not been further investigated yet.
In fact, no matter the central or peripheral immune tolerance is abnormal or not, the resulting diseases are often multi-antigen pathogenic, such as systemic lupus erythematosus (100). In IMN, although more than one antigen or antibody has been reported (101, 102), the majority of patients are single-antigen pathogenic. Therefore, the abnormalities of Th cells may not be the main cause of the induction of IMN, and the greater significance of such abnormalities lies in the maintenance of the disease state.
Th Cells Involved in the Immune Dysregulation of IMN
The differentiation diversity of Th cells is affected by at least two aspects: on the one hand, the differentiation of naive T cells is affected by cytokine signals in the microenvironment; on the other hand, such differentiation is regulated by TCR downstream signals in the cell. Recently, Mikel Ruterbusch et al. proposed a new differentiation model of CD4+T cells in vivo (103). The studies on Th cell subsets and related cytokines in IMN were reviewed and recorded in Table 1. IMN is identified with obvious Th cells subgroups imbalance, which is mainly reflected in the following aspects:
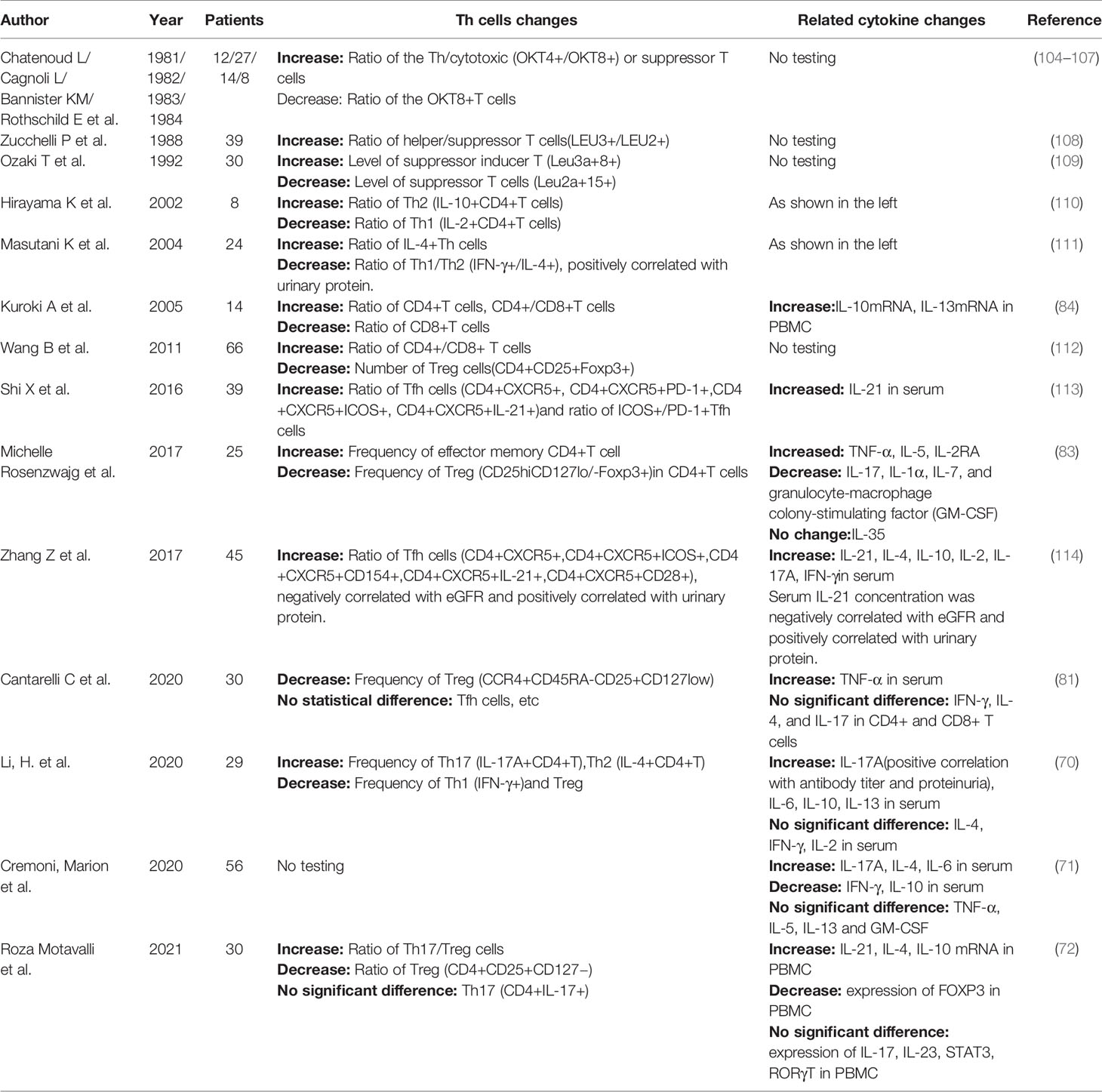
Table 1 Studies of helper T cells in IMN.
First, the CD4+/CD8+T cell ratio increased, and then the Th2/Th1 cell ratio increased, indicating that humoral immunity was dominant in IMN (84). In CD4+T cells, the expression of IL-4 was up-regulated, which was positively correlated with antibody production and disease severity (111). The representative cytokine IFN-γ secreted by Th1 was decreased in IMN (111). Cellular immune-mediated diseases are usually infiltrated by local monocytes and cytotoxic T cells. Although IMN presents as an organ-specific autoimmune disease, there is a local lack of cell infiltration that mediates cellular immunity in the glomerulus, and the generation of proteinuria may be caused by antibody activation of complement that damages the podocytes or antibody affecting podocyte function (115–117). The predictive value of anti-PLA2R antibody titers for clinical prognosis has also been vigorously described (69).
Many studies (112, 115, 118), represented by the rituximab clinical trial conducted by Ronco et al., have shown that IMN reduces Treg cells and destroys immune tolerance, and whether Treg cells can be increased after treatment can predict the therapeutic effect of rituximab. A recent study showed impaired inhibition of Treg cells in IMN (81), which might be attributed to the continuous exposure of antigen and the weakened ability of human immune regulation. TGF-β, IL-35 and IL-10 are the main cytokines secreted by Treg cells that play immunomodulatory roles. Although reductions or no significant changes in TGF-β and IL-35 were observed in IMN, there was an increase in IL-10, and this contradiction could be explained by upregulated regulatory B (Breg) cells in IMN (81). They can also secrete IL-10, but it does not suffice to block the development of the immune response. It was further speculated that the elevated Breg subsets were Br1 cells (119). As mentioned above, there is an antagonistic relationship between Treg and Th17 cells in terms of differentiation, function and other aspects, and imbalance of Th17/Treg has been observed in many autoimmune diseases. Th17 cells in IMN have been a heated topic recently, and studies from different research groups suggested the up-regulation of Th17 and the increase of IL-6 and IL-17A. This indicates that IMN is conducive to Th17 cells differentiation, and also strengthens our confidence that IMN is originated from extrarenal inflammation (115). Increased Th17 cells are also associated with a higher recurrence rate and a higher risk of venous thrombosis (71), which is a concern for clinical treatment.
Autoantibodies are essential to the development and maintenance of IMN, and the production of antibodies requires GC reactions. Tfh cells are professional GC helper B cells, and also serve as the novae of Th cells. In fact, the discovery of Tfh cells has challenged the previous classification of Th cells because their differentiation had been made earlier (103). In addition, it was previously held that the humoral immunity of IMN was driven by IL-4 secreted by Th2 cells, but the present study shows that IL-4 promoting antibody production may come from Tfh cells. Reduction of memory B cells and increase of initial B cells are present in IMN (83), which is consistent with the reports of some other autoimmune diseases (120, 121). An increase in initial B cells, such as Tfh cells, may be associated with the breakdown of tolerance checkpoints (83). The decrease of memory B cells may be caused by the induction of B cells into local tissues by chemokines, or the differentiation into plasma cells to produce antibodies under the action of Tfh cells, or both (85). Two studies from the same group have shown an abnormal increase in Tfh cells in IMN patients, which was correlated with disease severity (113, 114). Earlier studies have also shown that the proportion of CD4+CXCR5+T cells was also up-regulated in the classic model of Heymann nephritis rats (122), a classic animal model of IMN. Nevertheless, there are still many shortcomings, such as discrepancies in the results of studies from the same group. In addition, many questions remain to be explored, such as what causes the abnormal increase in Tfh cells? What is the function of the increased Tfh cells? Are Tfh cells involved in the recurrence of IMN? Research on various lymphocytes in IMN is still insufficient (4), and the role of Tfh cells in the overall immune system of IMN remains to be explored. In addition to the balance between Tfh cells and Treg cells, Tfr cells also form a balance in germinal center. An elevated proportion of circulating Tfh/Tfr cells is found in some autoimmune diseases (123–125), but unfortunately Tfr cells have not been studied in IMN.
We have to point out that although there are many studies on Th cells in IMN, their results are not in good agreement, which is a big obstacle for us to reveal the immunological mechanism of IMN. The underlying reasons may include: A. The included patients are heterogeneous and can be classified by etiological type; B. There are differences in detection methods, especially for cytokines. Measuring cytokine levels after in vitro stimulation may differ a lot compared to direct serological tests, because cytokines can be lost in proteinuria; C. The changes of Th cells and cytokines in IMN are small, suggesting that we should include patients with more active immune responses for observation, such as those with higher autoantibody titers. D. All the above studies are based on non-antigen-specific immunity, so the changes of autoreactive T cells may be attributed to a minority of the total T cells that are neglected. Although PLA2R-specific IgG-producing plasma cells have been identified in IMN (126), the changes of self-reactive T cells, such as PLA2R-specific T cells, are unknown in IMN. IMN is an autoimmune disease with antibody response as its core. In this process, abnormal Th cells provide an immune-promoting environment for autoreactive B cells by secreting cytokine. Therefore, the changes in the population and subpopulation of circulating Th cells can still reflect the immunological and pathological state of IMN, but such changes cannot be completely equivalent to those of antigen-specific Th cells. Further studies shall be conducted to clarify this problem, such as the use of flow cytometric analysis or major histocompatibility complex (MHC) tetramer (IST) staining to detect antigen-specific T cells in IMN. Such a study would be beneficial because Treg and Tfh cells do depend on antigen specificity to a certain extent when acting through cellular contact (127), and the study can also provide a basis for specific immunotolerance therapy in IMN. In rheumatoid arthritis, the degree of CD4+ T cell autoreactivity can determine the mode of immune response and influence the treatment prognosis, which is also enlightening for the study of IMN (128, 129).
The local immunological appearance in the kidney is also of concern. The pathological process of IMN is the binding of circulating antibodies to podocyte antigens and the formation of immune complexes deposited on the basement membrane. This process has been widely recognized. In fact, prior to the discovery of autoantigens on podocytes, it was assumed that the antigens of idiopathic membranous nephropathy were located in the tubules under the influence of Heymann nephritis rats, and CD20+B cell infiltration was observed in the tubulointerstitial area of approximately 50% of patients with membranous nephropathy, of which about 50% were focal infiltration (130). This structure is similar to the ectopic lymphoid structure (ELS) but has not been further described. Data from experimental animal models and patients suggest that Tfh cells or cells with Tfh phenotypic characteristics contribute to the maintenance of the structure and function of ELS (131–133). ELS was associated with interstitial inflammation and poor prognosis in IgA nephropathy (134). In autoimmune diseases, Eels or locally infiltrating clusters of B cells often caused harmful effects (135). For example, local autoantibodies were produced in patients with rheumatoid arthritis (136). A recent study by Kyriaki Kolovou et al. has shown that there is localized B cell infiltration in the kidney in renal diseases characterized by podocyte injury, including membranous nephropathy (137). Huimin Li et al. found infiltrating IL-17+ cells in the renal tubule of IMN patients (138). These findings suggest that T-B cell interaction can play a role in the renal tissue of IMN, especially in the renal tubules, and affect the prognosis of the disease, thus providing a new focus for renal pathological diagnosis.
Th Cells Participating in the Production of IMN Antibodies
In IMN, antibodies against autoantigens are predominantly IgG4, both in renal pathology and in serum, although a small number of other subtypes are also present (139, 140). Why IgG4 is the main pathogenic antibody in membranous nephropathy has always been a problem to be solved. IgG4 is the lowest IgG subtype in the blood of healthy adults, accounting for only 5% (141). Although it has about 90% homology with amino acid sequences of other IgG subtypes, due to changes in individual amino acids, IgG4 is identified with different characteristics, such as Fab arm exchange, weak complement binding force, etc. (142–144). Depending on the environment, IgG4 can play a protective or pathogenic role. In the autoimmune diseases mediated by IgG4, such as IMN and pemphigus, the pathogenic effect of IgG4 is often reflected in blocking the binding of antigen to other proteins and thus affecting its function (90, 144). In IMN, IgG4 combined with THSD7A affects cell adhesion, and thus proteinuria (145), while IgG4 combined with PLA2R may affect IV type collagen fiber adhesion (144, 146), but there are still controversies.
According to V (D) J gene rearrangement, some scholars speculated that IgG4 antibody should be the one with the highest affinity among all IgG subclasses and appear the latest (147). In GC, Tfh cells provide promoting or inhibiting signals to B cells through the competitive mechanism according to their affinity, which is crucial to the production of high-affinity antibodies (148). Among IgG4-mediated autoimmune diseases, including IMN, some other diseases have also demonstrated abnormalities in cTfh cells (83, 84, 110, 113, 114, 149–153), as shown in Table 2. These diseases may share some similarities in pathophysiology. Factors that promote the production of IgG4 mainly include two aspects: long-term exposure of allergens or antigens, and the influence of microenvironment created by cytokines, such as IL-4, IL-13, IL-10, IL-21, etc. (90, 154–156). IMN is an autoimmune disease with long-term exposure to autoantigens, and most of the cytokines involved in IgG4 production are abnormal in IMN (see Table 1). IL-4 or IL-13 combined with IL-10 can promote antibody conversion to the IgG4 category, while IL-4 combined with IL-21 can stimulate plasma cells to produce IgG4 antibodies (154–156). IL-4 is considered to be the hallmark cytokine of Th2 cells, while IL-21 is believed to be the hallmark cytokine of Tfh cells, even though neither of them serves as the sole source (157, 158). Studies have shown that Tfh cells can also express IL-4 and regulate germinal center response, independent of Th2 cells (159). In fact, Tfh cells may express both IL-21 and IL-4 simultaneously, or in sequence (160). These two cytokines all play a key role in the survival and proliferation of B cells, maturation of antibody affinity and class conversion, and the combined effect of IL-21 and IL-4 can promote the production of IgG4 antibody with the support of CD40 co-stimulatory signal (161), which may be related to the regulation of germinal center response. In addition, IL-21 can promote the production of autoantibodies (158).
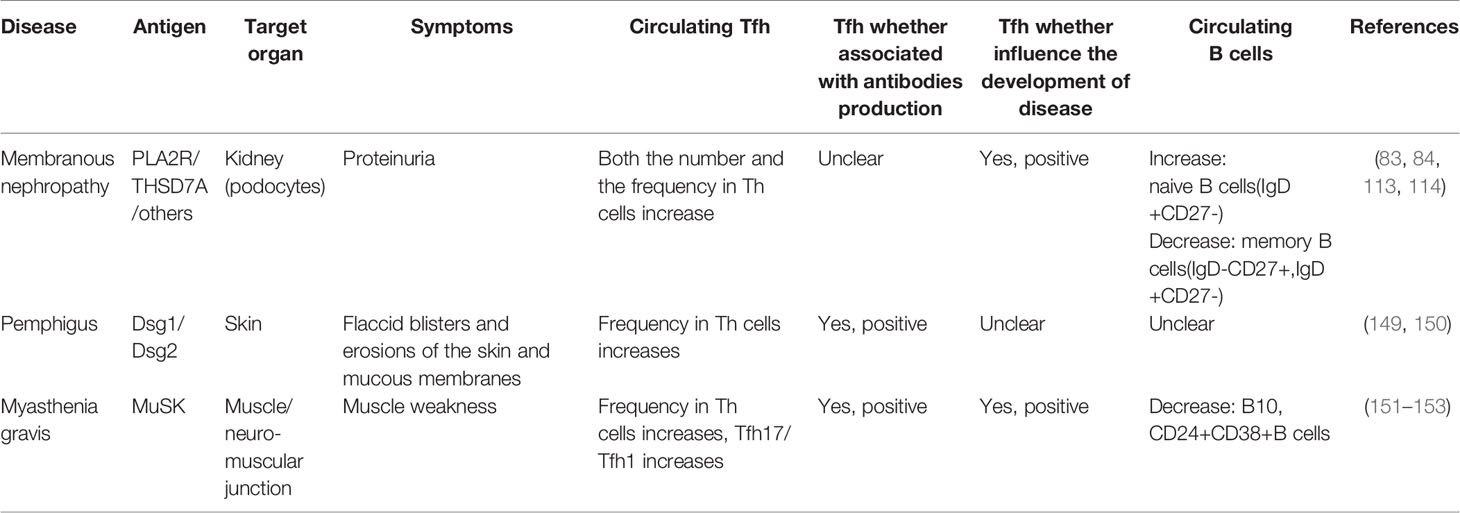
Table 2 Tfh cells in IgG4-mediated autoimmune diseases.
The Role Model of Th Cells in IMN
Based on the above discussions, genetic, immune, and environmental factors may co-participate in the incidence and development of IMN. In the presence of genetic susceptibility and in extrarenal inflammatory environment, autoantigens represented by PLA2R are presented to T cells. The initial cytokine environment pushes the immune response in a Th2-dominated direction. An abnormal increase in Tfh cells enables the proliferation and differentiation of autoreactive B cells, and assists B cells in completing somatic hypermutation in the germinal center, thus promoting the differentiation of B cells into plasma cells to produce IgG4 antibodies. Inflammation up-regulates Th17 cells and affects autoimmune response and inflammation by secreting cytokines such as IL-17. In addition, Th17 cells, Tfh cells and B cells may be partially liable for the damage of the renal tubulointerstitial region in IMN. The number and function of impaired Treg cells could not be maintained under autoimmune tolerance. The autoimmune response of IMN eventually produces antibodies, which bind to the target antigen on the podocytes, resulting in the classical pathological appearance of IMN, as described in Figure 3.
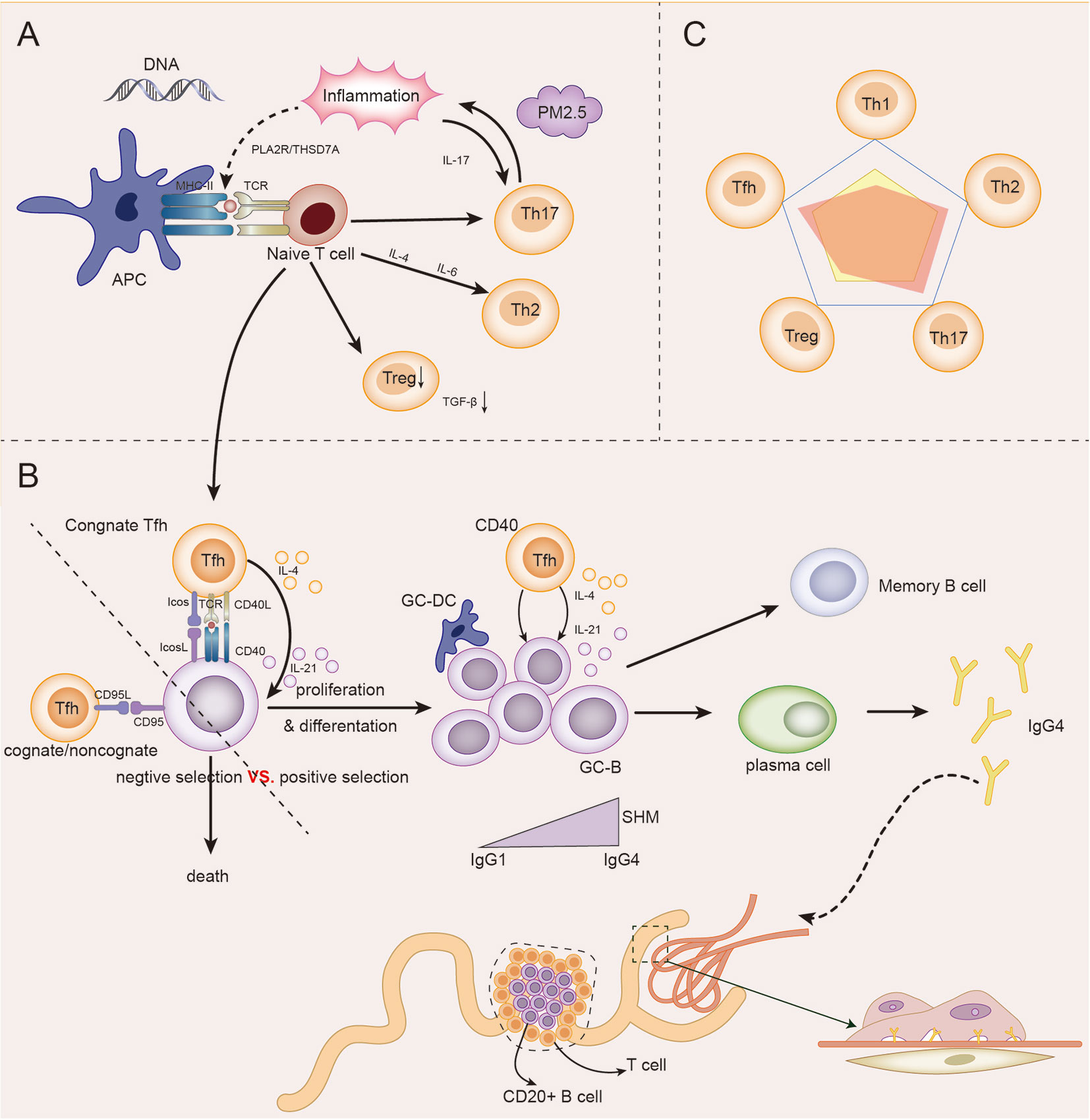
Figure 3 The role model of Th cells in IMN. (A) Under the influence of genetic, inflammatory, and environmental factors (PM2.5), antigen-presenting cells (APCs) present their own antigens to juvenile T cells, and then in the initial microenvironment, the immune response develops towards Th2-dominated direction. Infant T cells differentiate into Th17, which in turn participates in and maintains inflammation and promotes immune response. The differentiation of naive T cells to Treg cells decreased, and the immunosuppressive ability decreased. Naive T cells differentiate into Tfh cells and participate in GC reaction. (B) In germinal centers, homologous Tfh cells transmit survival signals to B cells via CD40L and cytokines (positive selection). Homologous or non-homologous Tfh cells transmit death signals to B cells via CD95L (negative selection). The abnormal increase in Tfh cells, which transmit survival signals, gives autoreactive B cells a chance to proliferate and differentiate. Under the action of IL-4 and IL-21 secreted by GC Tfh cells, GC B cells underwent somatic hypermutation (SHM) and antibody affinity maturation. After GC reaction, some B cells become memory B cells and some plasma cells, and begin to secrete IgG4 antibodies. IgG4 circulates to the glomerulus and binds to podocyte antigens (such as PLA2R) to form immune complexes that lead to the pathological appearance of IMN. In addition, under the influence of some factors, T-B cell infiltration may occur in renal tubules, and even form ectopic lymphatic structure, affecting the prognosis of the disease. (C) The relationship between the five major Th cell subpopulations in IMN was dominated by Th2, Th17, and Tfh cells, while Treg and Th1 cells were impaired.
Treatments for IMN
IMN, as an autoimmune disease, is mainly treated with immunosuppression, which is initiated after a full assessment of the condition, and the patient’s disease status is monitored during the course of treatment. Corticosteroids alone do not work much for the treatment of IMN but are effective when combined with alkylating agents represented by cyclophosphamide (162–164). Cyclophosphamide was originally designed as an antitumor agent and is metabolized by cells to produce phosphoramide mustard (165), which forms cross-links with DNA to achieve cytotoxic effects (166). Cells with high proliferative potential, such as hepatocytes and hematopoietic stem cells, are relatively resistant to cyclophosphamide due to the expression of high levels of aldehyde dehydrogenase (ALDH) (167). Conversely, cyclophosphamide is cytotoxic to mature hematopoietic progenitor cells and almost all lymphocyte subsets (167–169), inducing systemic leukocyte and lymphocyte ablation resulting in rapid suppression of the immune response. However, alkylating agents are associated with a high incidence of adverse events, mainly leukopenia, infection, thrombosis, gonadotoxicity, and increased risk of cancer (170, 171). Calcineurin inhibitors (CNIs) are also widely used in the treatment of IMN, such as tacrolimus and cyclosporine. CNIs can target and block the NFAT signaling pathway, primarily producing an inhibitory effect on T cells, impairing the helper effect of T cells on B cells and thus reducing antibody production. Moreover, some studies have shown that CNIs also have a regulatory effect on the podocyte cytoskeleton (172). The limitations on the clinical use of CNIs lied in their high rate of relapse after drug discontinuation (173) and the association of multiple relapses with progressive renal function (174). Recent studies have shown that the relapse rate after discontinuation of CNIs for IMN can be reduced by the addition of rituximab (175). Rituximab targets the B-cell surface antigen CD20 and cuts the number of B cells other than plasma cells, which can directly reduce antibody titers and induce disease remission (176). In addition to the above drugs, the use of other drugs such as mycophenolate mofetil and belimumab in the treatment of IMN is still being testified.
In addition to remission rates, immunosuppressive therapies for IMN shall also take into account the issues of relapse rates and safety. In terms of the immunological mechanisms of IMN, treatment targeting T or B cells alone may not be comprehensive, and immunosuppressive therapies with multiple targets are yet to be proposed. It has been shown that renal transplant recipient patients treated with a combination of rituximab, tacrolimus, and mycophenolate mofetil are found with Tfh or cTfh cells in the circulation and lymph nodes even when B-cell counts are reduced and GC responses are suppressed (177). Once the B-cell subpopulation recovers after treatment cessation, the residual Tfh may rapidly facilitate B-cell production of auto-reactive antibodies, so the combined or sequential use of rituximab and treatment against Tfh cells may have the potential to reduce relapse rates. Indeed, tacrolimus has a specific inhibitory effect on Tfh cells, which may be due to the greater dependence of Tfh cells on the NFAT signaling pathway (178). In addition, rituximab does not affect increased Th17 cells in IMN, which is associated with relapse and thromboembolism (71). Although many patients can achieve clinical remission with rituximab, maintenance treatment for post-remission immunosuppression, such as targeting Th17 and other Th cells, is also worthy of concern, especially in those patients at high risk of recurrence. It is worth pointing out that the potential therapeutic role of IL-2 in the treatment of autoimmune diseases is gaining increasing attention (179), and recently, a double-blind placebo-controlled trial has demonstrated the efficacy and safety of low-dose IL-2 in the treatment of SLE (180). Different T subpopulations of cells have different affinities with IL-2, with the CD4+FOXP3+ Treg cells subpopulation having a high affinity with IL-2 (181), and Treg cells can be induced to proliferate even at low IL-2, while such dose of IL-2 makes it impossible for other Th cells to proliferate. In addition, IL-2 can inhibit TFH cells responses without relying on Treg cells, which in turn inhibits GC responses and antibodies production (41, 182). By promoting the proliferation of Treg cells and inhibiting the responses of Tfh cells, which are indispensable for the treatment of IMN, IL-2 may have a greater potential in the clinical treatment of IMN.
Conclusion
IMN is a special autoimmune disease mainly caused by autoantibodies. Although antibodies are secreted by plasma cells, T-B cells also contribute a lot in the immune system, and the imbalance of Th1/Th2, Th17/Treg, Tfh/Tfr cells, and other Th cells subsets in IMN jointly shapes the immunological pathological state of IMN. More studies are needed to fully understand the pathological mechanism of IMN. The application of rituximab shifts the scholars’ attention to the study of B cells in IMN, but Th cells are located in the upstream of B cells, and convincing explanation of the changes in B cell subsets hinges on a good understanding of Th cell subsets, which should also be focused on in clinical treatment.
Author Contributions
QZ, HD, XL, and BL contributed to the conception and design of the review study. QZ, HJ, ZWL and ZF wrote the first draft of the manuscript. NZ, YG, ZD, XZ, JD and NQZ wrote sections of the manuscript. HR, LY and BL discussed and revised the content of the review article. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from the National Key Research and Development Project (No. 2019YFC1709402), National Natural Science Foundation of China (No. 81973793 to BL), Capital’s Funds for Health Improvement and Research (No. 2020-2-2234 to BL) and Jiangsu Province TCM science and technology development plan project (No.YB201985).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Beck LH, Bonegio RGB, Lambeau G, Beck DM, Powell DW, Cummins TD, et al. M-Type Phospholipase A2 Receptor as Target Antigen in Idiopathic Membranous Nephropathy. N Engl J Med (2009) 361(1):11–21. doi: 10.1056/NEJMoa0810457
2. Ronco P, Plaisier E, Debiec H. Advances in Membranous Nephropathy. J Clin Med (2021) 10(4):607. doi: 10.3390/jcm10040607
3. Hu R, Quan S, Wang Y, Zhou Y, Zhang Y, Liu L, et al. Spectrum of Biopsy Proven Renal Diseases in Central China: A 10-Year Retrospective Study Based on 34,630 Cases. Sci Rep (2020) 10(1):10994. doi: 10.1038/s41598-020-67910-w
4. Cravedi P, Jarque M, Angeletti A, Fava A, Cantarelli C, Bestard O. Immune-Monitoring Disease Activity in Primary Membranous Nephropathy. Front Med (Lausanne) (2019) 6:241. doi: 10.3389/fmed.2019.00241
5. Saravia J, Chapman NM, Chi H. Helper T Cell Differentiation. Cell Mol Immunol (2019) 16(7):634–43. doi: 10.1038/s41423-019-0220-6
6. Zhu JF. T Helper Cell Differentiation, Heterogeneity, and Plasticity. Csh Perspect Biol (2018) 10(10):a030338. doi: 10.1101/cshperspect.a030338
7. Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two Types of Murine Helper T Cell Clone. I. Definition According to Profiles of Lymphokine Activities and Secreted Proteins. J Immunol (1986) 136(7):2348–57.
8. Espinosa V, Rivera A. Cytokines and the Regulation of Fungus-Specific CD4 T Cell Differentiation. Cytokine (2012) 58(1):100–6. doi: 10.1016/j.cyto.2011.11.005
9. Espinosa V, Rivera A. Cytokines and the Regulation of Fungus-Specific CD4 T Cell Differentiation. Cytokine (2012) 58(1):100–6. doi: 10.1016/j.cyto.2011.11.005
10. Ouyang WJ, Lohning M, Gao ZG, Assenmacher M, Ranganath S, Radbruch A, et al. Stat6-Independent GATA-3 Autoactivation Directs IL-4-Independent Th2 Development and Commitment. Immunity (2000) 12(1):27–37. doi: 10.1016/S1074-7613(00)80156-9
11. Yamane H, Zhu JF, Paul WE. Independent Roles for IL-2 and GATA-3 in Stimulating Naive CD4(+) T Cells to Generate a Th2-Inducing Cytokine Environment. J Exp Med (2005) 202(6):793–804. doi: 10.1084/jem.20051304
12. Morel PA. Differential T-cell Receptor Signals for T Helper Cell Programming. Immunology (2018) 155(1):63–71. doi: 10.1111/imm.12945
13. Zhu J, Jankovic D, Oler AJ, Wei G, Sharma S, Hu G, et al. The Transcription Factor T-bet is Induced by Multiple Pathways and Prevents an Endogenous Th2 Cell Program During Th1 Cell Responses. Immunity (2012) 37(4):660–73. doi: 10.1016/j.immuni.2012.09.007
14. Hwang ES, Szabo SJ, Schwartzberg PL, Glimcher LH. T Helper Cell Fate Specified by Kinase-Mediated Interaction of T-bet With GATA-3. Sci (New York NY) (2005) 307(5708):430–3. doi: 10.1126/science.1103336
15. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic Self-Tolerance Maintained by Activated T Cells Expressing IL-2 Receptor Alpha-Chains (CD25). Breakdown of a Single Mechanism of Self-Tolerance Causes Various Autoimmune Diseases. J Immunol (Baltimore Md 1950) (1995) 155(3):1151–64.
16. Yong Z, Chang L, Mei YX, Yi L. Role and Mechanisms of CD4+CD25+ Regulatory T Cells in the Induction and Maintenance of Transplantation Tolerance. Transpl Immunol (2007) 17(2):120–9. doi: 10.1016/j.trim.2006.09.005
17. Chinen T, Kannan AK, Levine AG, Fan X, Klein U, Zheng Y, et al. An Essential Role for the IL-2 Receptor in Treg Cell Function. Nat Immunol (2016) 17(11):1322–33. doi: 10.1038/ni.3540
18. Santegoets SJAM, Dijkgraaf EM, Battaglia A, Beckhove P, Britten CM, Gallimore A, et al. Monitoring Regulatory T Cells in Clinical Samples: Consensus on an Essential Marker Set and Gating Strategy for Regulatory T Cell Analysis by Flow Cytometry. Cancer Immunol Immunother (2015) 64(10):1271–86. doi: 10.1007/s00262-015-1729-x
19. Dominguez-Villar M, Hafler DA. Regulatory T Cells in Autoimmune Disease. Nat Immunol (2018) 19(7):665–73. doi: 10.1038/s41590-018-0120-4
20. Chung Y, Tanaka S, Chu F, Nurieva RI, Martinez GJ, Rawal S, et al. Follicular Regulatory T Cells Expressing Foxp3 and Bcl-6 Suppress Germinal Center Reactions. Nat Med (2011) 17(8):983–8. doi: 10.1038/nm.2426
21. Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, et al. Interleukin 17-Producing CD4+ Effector T Cells Develop Via a Lineage Distinct From the T Helper Type 1 and 2 Lineages. Nat Immunol (2005) 6(11):1123–32. doi: 10.1038/ni1254
22. Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, et al. A Distinct Lineage of CD4 T Cells Regulates Tissue Inflammation by Producing Interleukin 17. Nat Immunol (2005) 6(11):1133–41. doi: 10.1038/ni1261
23. Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the Context of an Inflammatory Cytokine Milieu Supports De Novo Differentiation of IL-17-Producing T Cells. Immunity (2006) 24(2):179–89. doi: 10.1016/j.immuni.2006.01.001
24. Manel N, Unutmaz D, Littman DR. The Differentiation of Human T(H)-17 Cells Requires Transforming Growth Factor-Beta and Induction of the Nuclear Receptor Rorgammat. Nat Immunol (2008) 9(6):641–9. doi: 10.1038/ni.1610
25. Volpe E, Servant N, Zollinger R, Bogiatzi SI, Hupé P, Barillot E, et al. A Critical Function for Transforming Growth Factor-Beta, Interleukin 23 and Proinflammatory Cytokines in Driving and Modulating Human T(H)-17 Responses. Nat Immunol (2008) 9(6):650–7. doi: 10.1038/ni.1613
26. Lee GR. The Balance of Th17 Versus Treg Cells in Autoimmunity. Int J Mol Sci (2018) 19(3):730. doi: 10.3390/ijms19030730
27. Fasching P, Stradner M, Graninger W, Dejaco C, Fessler J. Therapeutic Potential of Targeting the Th17/Treg Axis in Autoimmune Disorders. Molecules (2017) 22(1):134. doi: 10.3390/molecules22010134
28. Noack M, Miossec P. Th17 and Regulatory T Cell Balance in Autoimmune and Inflammatory Diseases. Autoimmun Rev (2014) 13(6):668–77. doi: 10.1016/j.autrev.2013.12.004
29. Breitfeld D, Ohl L, Kremmer E, Ellwart J, Sallusto F, Lipp M, et al. Follicular B Helper T Cells Express CXC Chemokine Receptor 5, Localize to B Cell Follicles, and Support Immunoglobulin Production. J Exp Med (2000) 192(11):1545–52. doi: 10.1084/jem.192.11.1545
30. Schaerli P, Willimann K, Lang AB, Lipp M, Loetscher P, Moser B. CXC Chemokine Receptor 5 Expression Defines Follicular Homing T Cells With B Cell Helper Function. J Exp Med (2000) 192(11):1553–62. doi: 10.1084/jem.192.11.1553
31. Kim CH, Rott LS, Clark-Lewis I, Campbell DJ, Wu L, Butcher EC. Subspecialization of CXCR5+ T Cells: B Helper Activity is Focused in a Germinal Center-Localized Subset of CXCR5+ T Cells. J Exp Med (2001) 193(12):1373–81. doi: 10.1084/jem.193.12.1373
32. Yu D, Rao S, Tsai LM, Lee SK, He Y, Sutcliffe EL, et al. The Transcriptional Repressor Bcl-6 Directs T Follicular Helper Cell Lineage Commitment. Immunity (2009) 31(3):457–68. doi: 10.1016/j.immuni.2009.07.002
33. Johnston RJ, Poholek AC, DiToro D, Yusuf I, Eto D, Barnett B, et al. Bcl6 and Blimp-1 are Reciprocal and Antagonistic Regulators of T Follicular Helper Cell Differentiation. Science (2009) 325(5943):1006–10. doi: 10.1126/science.1175870
34. Nurieva RI, Chung Y, Martinez GJ, Yang XO, Tanaka S, Matskevitch TD, et al. Bcl6 Mediates the Development of T Follicular Helper Cells. Sci (New York NY) (2009) 325(5943):1001–5. doi: 10.1126/science.1176676
35. Nurieva RI, Chung Y, Martinez GJ, Yang XO, Tanaka S, Matskevitch TD, et al. Bcl6 Mediates the Development of T Follicular Helper Cells. Science (2009) 325(5943):1001–5. doi: 10.1126/science.1176676
36. Crotty S. T Follicular Helper Cell Differentiation, Function, and Roles in Disease. Immunity (2014) 41(4):529–42. doi: 10.1016/j.immuni.2014.10.004
37. Ma CS, Suryani S, Avery DT, Chan A, Nanan R, Santner-Nanan B, et al. Early Commitment of Naïve Human CD4(+) T Cells to the T Follicular Helper (T(FH)) Cell Lineage is Induced by IL-12. Immunol Cell Biol (2009) 87(8):590–600. doi: 10.1038/icb.2009.64
38. Schmitt N, Morita R, Bourdery L, Bentebibel SE, Zurawski SM, Banchereau J, et al. Human Dendritic Cells Induce the Differentiation of Interleukin-21-Producing T Follicular Helper-Like Cells Through Interleukin-12. Immunity (2009) 31(1):158–69. doi: 10.1016/j.immuni.2009.04.016
39. McCarron MJ, Marie JC. TGF-Beta Prevents T Follicular Helper Cell Accumulation and B Cell Autoreactivity. J Clin Invest (2014) 124(10):4375–86. doi: 10.1172/JCI76179
40. Schmitt N, Liu Y, Bentebibel S-E, Munagala I, Bourdery L, Venuprasad K, et al. The Cytokine TGF-β Co-Opts Signaling Via STAT3-STAT4 to Promote the Differentiation of Human TFH Cells. Nat Immunol (2014) 15(9):856–65. doi: 10.1038/ni.2947
41. Ballesteros-Tato A, Leon B, Graf BA, Moquin A, Adams PS, Lund FE, et al. Interleukin-2 Inhibits Germinal Center Formation by Limiting T Follicular Helper Cell Differentiation. Immunity (2012) 36(5):847–56. doi: 10.1016/j.immuni.2012.02.012
42. Johnston RJ, Choi YS, Diamond JA, Yang JA, Crotty S. STAT5 is a Potent Negative Regulator of TFH Cell Differentiation. J Exp Med (2012) 209(2):243–50. doi: 10.1084/jem.20111174
43. Chtanova T, Tangye SG, Newton R, Frank N, Hodge MR, Rolph MS, et al. T Follicular Helper Cells Express a Distinctive Transcriptional Profile, Reflecting Their Role as Non-Th1/Th2 Effector Cells That Provide Help for B Cells. J Immunol (Baltimore Md 1950) (2004) 173(1):68–78. doi: 10.4049/jimmunol.173.1.68
44. Kim CH, Lim HW, Kim JR, Rott L, Hillsamer P, Butcher EC. Unique Gene Expression Program of Human Germinal Center T Helper Cells. Blood (2004) 104(7):1952–60. doi: 10.1182/blood-2004-03-1206
45. Vinuesa CG, Cook MC, Angelucci C, Athanasopoulos V, Rui L, Hill KM, et al. A RING-type Ubiquitin Ligase Family Member Required to Repress Follicular Helper T Cells and Autoimmunity. Nature (2005) 435(7041):452–8. doi: 10.1038/nature03555
46. Kerfoot SM, Yaari G, Patel JR, Johnson KL, Gonzalez DG, Kleinstein SH, et al. Germinal Center B Cell and T Follicular Helper Cell Development Initiates in the Interfollicular Zone. Immunity (2011) 34(6):947–60. doi: 10.1016/j.immuni.2011.03.024
47. Weinstein JS, Bertino SA, Hernandez SG, Poholek AC, Teplitzky TB, Nowyhed HN, et al. B Cells in T Follicular Helper Cell Development and Function: Separable Roles in Delivery of ICOS Ligand and Antigen. J Immunol (2014) 192(7):3166–79. doi: 10.4049/jimmunol.1302617
48. Shulman Z, Gitlin AD, Targ S, Jankovic M, Pasqual G, Nussenzweig MC, et al. T Follicular Helper Cell Dynamics in Germinal Centers. Science (2013) 341(6146):673–7. doi: 10.1126/science.1241680
49. Suan D, Nguyen A, Moran I, Bourne K, Hermes JR, Arshi M, et al. T Follicular Helper Cells Have Distinct Modes of Migration and Molecular Signatures in Naive and Memory Immune Responses. Immunity (2015) 42(4):704–18. doi: 10.1016/j.immuni.2015.03.002
50. MacLennan IC, Toellner KM, Cunningham AF, Serre K, Sze DM, Zuniga E, et al. Extrafollicular Antibody Responses. Immunol Rev (2003) 194:8–18. doi: 10.1034/j.1600-065X.2003.00058.x
51. Kelsoe G. The Germinal Center: A Crucible for Lymphocyte Selection. Semin Immunol (1996) 8(3):179–84. doi: 10.1006/smim.1996.0022
52. Allen CD, Okada T, Cyster JG. Germinal-Center Organization and Cellular Dynamics. Immunity (2007) 27(2):190–202. doi: 10.1016/j.immuni.2007.07.009
53. Weinstein JS, Herman EI, Lainez B, Licona-Limon P, Esplugues E, Flavell R, et al. TFH Cells Progressively Differentiate to Regulate the Germinal Center Response. Nat Immunol (2016) 17(10):1197–205. doi: 10.1038/ni.3554
54. Choi JY, Ho JH, Pasoto SG, Bunin V, Kim ST, Carrasco S, et al. Circulating Follicular Helper-Like T Cells in Systemic Lupus Erythematosus: Association With Disease Activity. Arthritis Rheumatol (2015) 67(4):988–99. doi: 10.1002/art.39020
55. He J, Tsai LM, Leong YA, Hu X, Ma CS, Chevalier N, et al. Circulating Precursor CCR7(lo)PD-1(hi) CXCR5+ CD4+ T Cells Indicate Tfh Cell Activity and Promote Antibody Responses Upon Antigen Reexposure. Immunity (2013) 39(4):770–81. doi: 10.1016/j.immuni.2013.09.007
56. Simpson N, Gatenby PA, Wilson A, Malik S, Fulcher DA, Tangye SG, et al. Expansion of Circulating T Cells Resembling Follicular Helper T Cells is a Fixed Phenotype That Identifies a Subset of Severe Systemic Lupus Erythematosus. Arthritis Rheum (2010) 62(1):234–44. doi: 10.1002/art.25032
57. Liu R, Wu Q, Su D, Che N, Chen H, Geng L, et al. A Regulatory Effect of IL-21 on T Follicular Helper-Like Cell and B Cell in Rheumatoid Arthritis. Arthritis Res Ther (2012) 14(6):R255. doi: 10.1186/ar4100
58. Wang J, Shan Y, Jiang Z, Feng J, Li C, Ma L, et al. High Frequencies of Activated B Cells and T Follicular Helper Cells are Correlated With Disease Activity in Patients With New-Onset Rheumatoid Arthritis. Clin Exp Immunol (2013) 174(2):212–20. doi: 10.1111/cei.12162
59. Morita R, Schmitt N, Bentebibel S-E, Ranganathan R, Bourdery L, Zurawski G, et al. Human Blood CXCR5(+)CD4(+) T Cells are Counterparts of T Follicular Cells and Contain Specific Subsets That Differentially Support Antibody Secretion. Immunity (2011) 34(1):108–21. doi: 10.1016/j.immuni.2010.12.012
60. Locci M, Havenar-Daughton C, Landais E, Wu J, Kroenke MA, Arlehamn CL, et al. Human Circulating PD-1+CXCR3-CXCR5+ Memory Tfh Cells Are Highly Functional and Correlate With Broadly Neutralizing HIV Antibody Responses. Immunity (2013) 39(4):758–69. doi: 10.1016/j.immuni.2013.08.031
61. Boswell KL, Paris R, Boritz E, Ambrozak D, Yamamoto T, Darko S, et al. Loss of Circulating CD4 T Cells With B Cell Helper Function During Chronic HIV Infection. PloS Pathog (2014) 10(1):e1003853. doi: 10.1371/journal.ppat.1003853
62. Bentebibel SE, Lopez S, Obermoser G, Schmitt N, Mueller C, Harrod C, et al. Induction of ICOS+CXCR3+CXCR5+ TH Cells Correlates With Antibody Responses to Influenza Vaccination. Sci Transl Med (2013) 5(176):176ra132. doi: 10.1126/scitranslmed.3005191
63. Adair P, Kim YC, Pratt KP, Scott DW. Avidity of Human T Cell Receptor Engineered CD4(+) T Cells Drives T-Helper Differentiation Fate. Cell Immunol (2016) 299:30–41. doi: 10.1016/j.cellimm.2015.10.003
64. Ronco P, Debiec H. Advances in Membranous Nephropathy: Success Stories of a Long Journey. Clin Exp Pharmacol Physiol (2011) 38(7):460–6. doi: 10.1111/j.1440-1681.2011.05506.x
65. Kao L, Lam V, Waldman M, Glassock RJ, Zhu Q. Identification of the Immunodominant Epitope Region in Phospholipase A2 Receptor-Mediating Autoantibody Binding in Idiopathic Membranous Nephropathy. J Am Soc Nephrol (2015) 26(2):291–301. doi: 10.1681/ASN.2013121315
66. Seitz-Polski B, Dolla G, Payre C, Girard CA, Polidori J, Zorzi K, et al. Epitope Spreading of Autoantibody Response to PLA2R Associates With Poor Prognosis in Membranous Nephropathy. J Am Soc Nephrol (2016) 27(5):1517–33. doi: 10.1681/ASN.2014111061
67. Xu X, Wang G, Chen N, Lu T, Nie S, Xu G, et al. Long-Term Exposure to Air Pollution and Increased Risk of Membranous Nephropathy in China. J Am Soc Nephrol (2016) 27(12):3739–46. doi: 10.1681/ASN.2016010093
68. Liu W, Gao C, Dai H, Zheng Y, Dong Z, Gao Y, et al. Immunological Pathogenesis of Membranous Nephropathy: Focus on PLA2R1 and Its Role. Front Immunol (2019) 10:1809. doi: 10.3389/fimmu.2019.01809
69. van de Logt A-E, Fresquet M, Wetzels JF, Brenchley P. The anti-PLA2R Antibody in Membranous Nephropathy: What We Know and What Remains a Decade After Its Discovery. Kidney Int (2019) 96(6):1292–302. doi: 10.1016/j.kint.2019.07.014
70. Li H, Wu H, Guo Q, Yu H, Xu Y, Yu J, et al. Myeloid-Derived Suppressor Cells Promote the Progression of Primary Membranous Nephropathy by Enhancing Th17 Response. Front Immunol (2020) 11:1777. doi: 10.3389/fimmu.2020.01777
71. Cremoni M, Brglez V, Perez S, Decoupigny F, Zorzi K, Andreani M, et al. Th17-Immune Response in Patients With Membranous Nephropathy Is Associated With Thrombosis and Relapses. Front Immunol (2020) 11:574997. doi: 10.3389/fimmu.2020.574997
72. Motavalli R, Etemadi J, Soltani-Zangbar MS, Ardalan M-R, Kahroba H, Roshangar L, et al. Altered Th17/Treg Ratio as a Possible Mechanism in Pathogenesis of Idiopathic Membranous Nephropathy. Cytokine (2021) 141:155452. doi: 10.1016/j.cyto.2021.155452
73. Xie J, Liu L, Mladkova N, Li Y, Ren H, Wang W, et al. The Genetic Architecture of Membranous Nephropathy and its Potential to Improve Non-Invasive Diagnosis. Nat Commun (2020) 11(1):1600. doi: 10.1038/s41467-020-15383-w
74. Cui Z, Xie LJ, Chen FJ, Pei ZY, Zhang LJ, Qu Z, et al. MHC Class II Risk Alleles and Amino Acid Residues in Idiopathic Membranous Nephropathy. J Am Soc Nephrol (2017) 28(5):1651–64. doi: 10.1681/ASN.2016020114
75. Niemi HJ, Laakso S, Salminen JT, Arstila TP, Tuulasvaara A. A Normal T Cell Receptor Beta CDR3 Length Distribution in Patients With APECED. Cell Immunol (2015) 295(2):99–104. doi: 10.1016/j.cellimm.2015.03.005
76. Zhang Y, Jin Y, Guan Z, Li H, Su Z, Xie C, et al. The Landscape and Prognosis Potential of the T-Cell Repertoire in Membranous Nephropathy. Front Immunol (2020) 11:387. doi: 10.3389/fimmu.2020.00387
77. Cui J-H, Jin Y-B, Lin K-R, Xiao P, Chen X-P, Pan Y-M, et al. Characterization of Peripheral Blood TCR Repertoire in Patients With Ankylosing Spondylitis by High-Throughput Sequencing. Hum Immunol (2018) 79(6):485–90. doi: 10.1016/j.humimm.2018.03.007
78. Sui W, Hou X, Zou G, Che W, Yang M, Zheng C, et al. Composition and Variation Analysis of the TCR β-Chain CDR3 Repertoire in Systemic Lupus Erythematosus Using High-Throughput Sequencing. Mol Immunol (2015) 67(2 Pt B):455–64. doi: 10.1016/j.molimm.2015.07.012
79. Gomez-Tourino I, Kamra Y, Baptista R, Lorenc A, Peakman M. T Cell Receptor Beta-Chains Display Abnormal Shortening and Repertoire Sharing in Type 1 Diabetes. Nat Commun (2017) 8(1):1792. doi: 10.1038/s41467-017-01925-2
80. Pugliese A. Central and Peripheral Autoantigen Presentation in Immune Tolerance. Immunology (2004) 111(2):138–46. doi: 10.1111/j.0019-2805.2003.01804.x
81. Cantarelli C, Jarque M, Angeletti A, Manrique J, Hartzell S, O’Donnell T, et al. A Comprehensive Phenotypic and Functional Immune Analysis Unravels Circulating Anti-Phospholipase A2 Receptor Antibody Secreting Cells in Membranous Nephropathy Patients. Kidney Int Rep (2020) 5(10):1764–76. doi: 10.1016/j.ekir.2020.07.028
82. Ryba-Stanislawowska M, Sakowska J, Zielinski M, Lawrynowicz U, Trzonkowski P. Regulatory T Cells: The Future of Autoimmune Disease Treatment. Expert Rev Clin Immunol (2019) 15(7):777–89. doi: 10.1080/1744666X.2019.1620602
83. Rosenzwajg M, Languille E, Debiec H, Hygino J, Dahan K, Simon T, et al. B- and T-Cell Subpopulations in Patients With Severe Idiopathic Membranous Nephropathy may Predict an Early Response to Rituximab. Kidney Int (2017) 92(1):227–37. doi: 10.1016/j.kint.2017.01.012
84. Kuroki A, Iyoda M, Shibata T, Sugisaki T. Th2 Cytokines Increase and Stimulate B Cells to Produce IgG4 in Idiopathic Membranous Nephropathy. Kidney Int (2005) 68(1):302–10. doi: 10.1111/j.1523-1755.2005.00415.x
85. Vinuesa CG, Sanz I, Cook MC. Dysregulation of Germinal Centres in Autoimmune Disease. Nat Rev Immunol (2009) 9(12):845–57. doi: 10.1038/nri2637
86. Cantaert T, Schickel JN, Bannock JM, Ng YS, Massad C, Delmotte FR, et al. Decreased Somatic Hypermutation Induces an Impaired Peripheral B Cell Tolerance Checkpoint. J Clin Invest (2016) 126(11):4289–302. doi: 10.1172/JCI84645
87. Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, Nussenzweig MC. Predominant Autoantibody Production by Early Human B Cell Precursors. Science (2003) 301(5638):1374–7. doi: 10.1126/science.1086907
88. Primi D, Hammarstrom L, Smith CI, Moller G. Characterization of Self-Reactive B Cells by Polyclonal B-Cell Activators. J Exp Med (1977) 145(1):21–30. doi: 10.1084/jem.145.1.21
89. Ray SK, Putterman C, Diamond B. Pathogenic Autoantibodies are Routinely Generated During the Response to Foreign Antigen: A Paradigm for Autoimmune Disease. Proc Natl Acad Sci USA (1996) 93(5):2019–24. doi: 10.1073/pnas.93.5.2019
90. Huijbers MG, Plomp JJ, van der Maarel SM, Verschuuren JJ. IgG4-mediated Autoimmune Diseases: A Niche of Antibody-Mediated Disorders. Ann N Y Acad Sci (2018) 1413(1):92–103. doi: 10.1111/nyas.13561
91. Hande S, Notidis E, Manser T. Bcl-2 Obstructs Negative Selection of Autoreactive, Hypermutated Antibody V Regions During Memory B Cell Development. Immunity (1998) 8(2):189–98. doi: 10.1016/S1074-7613(00)80471-9
92. Takahashi Y, Cerasoli DM, Dal Porto JM, Shimoda M, Freund R, Fang W, et al. Relaxed Negative Selection in Germinal Centers and Impaired Affinity Maturation in Bcl-Xl Transgenic Mice. J Exp Med (1999) 190(3):399–410. doi: 10.1084/jem.190.3.399
93. Fischer SF, Bouillet P, O’Donnell K, Light A, Tarlinton DM, Strasser A. Proapoptotic BH3-only Protein Bim is Essential for Developmentally Programmed Death of Germinal Center-Derived Memory B Cells and Antibody-Forming Cells. Blood (2007) 110(12):3978–84. doi: 10.1182/blood-2007-05-091306
94. Linterman MA, Vinuesa CG. T Follicular Helper Cells During Immunity and Tolerance. Prog Mol Biol Transl Sci (2010) 92:207–48. doi: 10.1016/S1877-1173(10)92009-7
95. Liu YJ, Joshua DE, Williams GT, Smith CA, Gordon J, MacLennan IC. Mechanism of Antigen-Driven Selection in Germinal Centres. Nature (1989) 342(6252):929–31. doi: 10.1038/342929a0
96. Linterman MA, Rigby RJ, Wong RK, Yu D, Brink R, Cannons JL, et al. Follicular Helper T Cells are Required for Systemic Autoimmunity. J Exp Med (2009) 206(3):561–76. doi: 10.1084/jem.20081886
97. Schwartzberg PL, Mueller KL, Qi H, Cannons JL. SLAM Receptors and SAP Influence Lymphocyte Interactions, Development and Function. Nat Rev Immunol (2009) 9(1):39–46. doi: 10.1038/nri2456
98. Hu R, Kagele DA, Huffaker TB, Runtsch MC, Alexander M, Liu J, et al. miR-155 Promotes T Follicular Helper Cell Accumulation During Chronic, Low-Grade Inflammation. Immunity (2014) 41(4):605–19. doi: 10.1016/j.immuni.2014.09.015
99. Toor D, Sharma N. T Cell Subsets: An Integral Component in Pathogenesis of Rheumatic Heart Disease. Immunol Res (2018) 66(1):18–30. doi: 10.1007/s12026-017-8978-z
100. Cappione A, Anolik JH, Pugh-Bernard A, Barnard J, Dutcher P, Silverman G, et al. Germinal Center Exclusion of Autoreactive B Cells is Defective in Human Systemic Lupus Erythematosus. J Clin Invest (2005) 115(11):3205–16. doi: 10.1172/JCI24179
101. Sethi S, Madden BJ, Debiec H, Charlesworth MC, Gross L, Ravindran A, et al. Exostosin 1/Exostosin 2-Associated Membranous Nephropathy. J Am Soc Nephrol (2019) 30(6):1123–36. doi: 10.1681/ASN.2018080852
102. Larsen CP, Cossey LN, Beck LH. THSD7A Staining of Membranous Glomerulopathy in Clinical Practice Reveals Cases With Dual Autoantibody Positivity. Mod Pathol (2016) 29(4):421–6. doi: 10.1038/modpathol.2016.32
103. Ruterbusch M, Pruner KB, Shehata L, Pepper M. In Vivo CD4(+) T Cell Differentiation and Function: Revisiting the Th1/Th2 Paradigm. Annu Rev Immunol (2020) 38:705–25. doi: 10.1146/annurev-immunol-103019-085803
104. Chatenoud L, Bach MA. Abnormalities of T-cell Subsets in Glomerulonephritis and Systemic Lupus Erythematosus. Kidney Int (1981) 20(2):267–74. doi: 10.1038/ki.1981.130
105. Cagnoli L, Tabacchi P, Pasquali S, Cenci M, Sasdelli M, Zucchelli P. T Cell Subset Alterations in Idiopathic Glomerulonephritis. Clin Exp Immunol (1982) 50(1):70–6.
106. Bannister KM, Drew PA, Clarkson AR, Woodroffe AJ. Immunoregulation in Glomerulonephritis, Henoch–Schonlein Purpura and Lupus Nephritis. Clin Exp Immunol (1983) 53(2):384–90.
107. Rothschild E, Chatenoud L. T Cell Subset Modulation of Immunoglobulin Production in IgA Nephropathy and Membranous Glomerulonephritis. Kidney Int (1984) 25(3):557–64. doi: 10.1038/ki.1984.54
108. Zucchelli P, Ponticelli C, Cagnoli L, Aroldi A, Beltrandi E. Prognostic Value of T Lymphocyte Subset Ratio in Idiopathic Membranous Nephropathy. Am J Nephrol (1988) 8(1):15–20. doi: 10.1159/000167547
109. Ozaki T, Tomino Y, Nakayama S, Koide H. Two-Color Analysis of Lymphocyte Subpopulations in Patients With Nephrotic Syndrome Due to Membranous Nephropathy. Clin Nephrol (1992) 38(2):75–80.
110. Hirayama K, Ebihara I, Yamamoto S, Kai H, Muro K, Yamagata K, et al. Predominance of Type-2 Immune Response in Idiopathic Membranous Nephropathy. Cytoplasmic Cytokine Anal Nephron (2002) 91(2):255–61. doi: 10.1159/000058401
111. Masutani K, Taniguchi M, Nakashima H, Yotsueda H, Kudoh Y, Tsuruya K, et al. Up-Regulated Interleukin-4 Production by Peripheral T-helper Cells in Idiopathic Membranous Nephropathy. Nephrol Dial Transplant (2004) 19(3):580–6. doi: 10.1093/ndt/gfg572
112. Wang B, Zuo K, Wu Y, Huang Q, Qin WS, Zeng CH, et al. Correlation Between B Lymphocyte Abnormality and Disease Activity in Patients With Idiopathic Membranous Nephropathy. J Int Med Res (2011) 39(1):86–95. doi: 10.1177/147323001103900111
113. Shi X, Qu Z, Zhang L, Zhang N, Liu Y, Li M, et al. Increased Ratio of ICOS(+) /PD-1(+) Follicular Helper T Cells Positively Correlates With the Development of Human Idiopathic Membranous Nephropathy. Clin Exp Pharmacol Physiol (2016) 43(4):410–6. doi: 10.1111/1440-1681.12555
114. Zhang Z, Shi Y, Yang K, Crew R, Wang H, Jiang Y. Higher Frequencies of Circulating ICOS(+), Il-21(+) T Follicular Helper Cells and Plasma Cells in Patients With New-Onset Membranous Nephropathy. Autoimmunity (2017) 50(8):458–67. doi: 10.1080/08916934.2017.1385775
115. Liu W, Gao C, Liu Z, Dai H, Feng Z, Dong Z, et al. Idiopathic Membranous Nephropathy: Glomerular Pathological Pattern Caused by Extrarenal Immunity Activity. Front Immunol (2020) 11:1846. doi: 10.3389/fimmu.2020.01846
116. Lateb M, Ouahmi H, Payré C, Brglez V, Zorzi K, Dolla G, et al. Anti-PLA2R1 Antibodies Containing Sera Induce In Vitro Cytotoxicity Mediated by Complement Activation. J Immunol Res (2019) 2019:1324804. doi: 10.1155/2019/1324804
117. Haddad G, Lorenzen JM, Ma H, de Haan N, Seeger H, Zaghrini C, et al. Altered Glycosylation of IgG4 Promotes Lectin Complement Pathway Activation in anti-PLA2R1-associated Membranous Nephropathy. J Clin Invest (2021) 131(5):e140453. doi: 10.1172/JCI140453
118. Fervenza FC, Abraham RS, Erickson SB, Irazabal MV, Eirin A, Specks U, et al. Rituximab Therapy in Idiopathic Membranous Nephropathy: A 2-Year Study. Clin J Am Soc Nephrol (2010) 5(12):2188–98. doi: 10.2215/CJN.05080610
119. Dong Z, Liu Z, Dai H, Liu W, Feng Z, Zhao Q, et al. The Potential Role of Regulatory B Cells in Idiopathic Membranous Nephropathy. J Immunol Res (2020) 2020:7638365. doi: 10.1155/2020/7638365
120. Rodríguez-Bayona B, Ramos-Amaya A, Pérez-Venegas JJ, Rodríguez C, Brieva JA. Decreased Frequency and Activated Phenotype of Blood CD27 IgD IgM B Lymphocytes is a Permanent Abnormality in Systemic Lupus Erythematosus Patients. Arthritis Res Ther (2010) 12(3):R108. doi: 10.1186/ar3042
121. Szabo K, Papp G, Szanto A, Tarr T, Zeher M. A Comprehensive Investigation on the Distribution of Circulating Follicular T Helper Cells and B Cell Subsets in Primary Sjogren’s Syndrome and Systemic Lupus Erythematosus. Clin Exp Immunol (2016) 183(1):76–89. doi: 10.1111/cei.12703
122. Wang YM, Zhang GY, Hu M, Polhill T, Sawyer A, Zhou JJ, et al. CD8+ Regulatory T Cells Induced by T Cell Vaccination Protect Against Autoimmune Nephritis. J Am Soc Nephrol (2012) 23(6):1058–67. doi: 10.1681/ASN.2011090914
123. Zhao S, Ding J, Wang S, Li C, Guo P, Zhang M, et al. Decreased Expression of Circulating Aire and Increased Tfh/Tfr Cells in Myasthenia Gravis Patients. Biosci Rep (2018) 38(6):BSR20180096. doi: 10.1042/BSR20180096
124. Wang X, Yang C, Xu F, Qi L, Wang J, Yang P. Imbalance of Circulating Tfr/Tfh Ratio in Patients With Rheumatoid Arthritis. Clin Exp Med (2019) 19(1):55–64. doi: 10.1007/s10238-018-0530-5
125. Wen Y, Yang B, Lu J, Zhang J, Yang H, Li J. Imbalance of Circulating CD4(+)CXCR5(+)FOXP3(+) Tfr-Like Cells and CD4(+)CXCR5(+)FOXP3(-) Tfh-Like Cells in Myasthenia Gravis. Neurosci Lett (2016) 630:176–82. doi: 10.1016/j.neulet.2016.07.049
126. Cantarelli C, Jarque M, Angeletti A, Manrique J, Hartzell S, O’Donnell T, et al. A Comprehensive Phenotypic and Functional Immune Analysis Unravels Circulating Anti-Phospholipase A2 Receptor Antibody Secreting Cells in Membranous Nephropathy Patients. Kidney Int Rep (2020) 5(10):1764–76. doi: 10.1016/j.ekir.2020.07.028
127. Akkaya B, Oya Y, Akkaya M, Al Souz J, Holstein AH, Kamenyeva O, et al. Regulatory T Cells Mediate Specific Suppression by Depleting peptide-MHC Class II From Dendritic Cells. Nat Immunol (2019) 20(2):218–31. doi: 10.1038/s41590-018-0280-2
128. Perng OA, Aitken M, Rankin AL, Garcia V, Kropf E, Erikson J, et al. The Degree of CD4+ T Cell Autoreactivity Determines Cellular Pathways Underlying Inflammatory Arthritis. J Immunol (2014) 192(7):3043–56. doi: 10.4049/jimmunol.1302528
129. Page A, Fusil F, Cosset FL. Antigen-Specific Tolerance Approach for Rheumatoid Arthritis: Past, Present and Future. Joint Bone Spine (2021) 88(4):105164. doi: 10.1016/j.jbspin.2021.105164
130. Cohen CD, Calvaresi N, Armelloni S, Schmid H, Henger A, Ott U, et al. CD20-Positive Infiltrates in Human Membranous Glomerulonephritis. J Nephrol (2005) 18(3):328–33.
131. Peters A, Pitcher LA, Sullivan JM, Mitsdoerffer M, Acton SE, Franz B, et al. Th17 Cells Induce Ectopic Lymphoid Follicles in Central Nervous System Tissue Inflammation. Immunity (2011) 35(6):986–96. doi: 10.1016/j.immuni.2011.10.015
132. Slight SR, Rangel-Moreno J, Gopal R, Lin Y, Fallert Junecko BA, Mehra S, et al. CXCR5(+) T Helper Cells Mediate Protective Immunity Against Tuberculosis. J Clin Invest (2013) 123(2):712–26. doi: 10.1172/JCI65728
133. Gu-Trantien C, Loi S, Garaud S, Equeter C, Libin M, de Wind A, et al. CD4(+) Follicular Helper T Cell Infiltration Predicts Breast Cancer Survival. J Clin Invest (2013) 123(7):2873–92. doi: 10.1172/JCI67428
134. Pei G, Zeng R, Han M, Liao P, Zhou X, Li Y, et al. Renal Interstitial Infiltration and Tertiary Lymphoid Organ Neogenesis in IgA Nephropathy. Clin J Am Soc Nephrol (2014) 9(2):255–64. doi: 10.2215/CJN.01150113
135. Pitzalis C, Jones GW, Bombardieri M, Jones SA. Ectopic Lymphoid-Like Structures in Infection, Cancer and Autoimmunity. Nat Rev Immunol (2014) 14(7):447–62. doi: 10.1038/nri3700
136. Humby F, Bombardieri M, Manzo A, Kelly S, Blades MC, Kirkham B, et al. Ectopic Lymphoid Structures Support Ongoing Production of Class-Switched Autoantibodies in Rheumatoid Synovium. PloS Med (2009) 6(1):e1. doi: 10.1371/journal.pmed.0060001
137. Kolovou K, Laskari K, Roumelioti M, Tektonidou MG, Panayiotidis P, Boletis JN, et al. B-Cell Oligoclonal Expansions in Renal Tissue of Patients With Immune-Mediated Glomerular Disease. Clin Immunol (Orlando Fla) (2020) 217:108488. doi: 10.1016/j.clim.2020.108488
138. Li H, Wu H, Guo Q, Yu H, Xu Y, Yu J, et al. Myeloid-Derived Suppressor Cells Promote the Progression of Primary Membranous Nephropathy by Enhancing Th17 Response. Front Immunol (2020) 11:1777. doi: 10.3389/fimmu.2020.01777
139. Filippone EJ. Idiopathic Membranous Nephropathy and IgG4: An Interesting Relationship. Clin Nephrol (2014) 82(1):7–15. doi: 10.5414/CN107768
140. Huang CC, Lehman A, Albawardi A, Satoskar A, Brodsky S, Nadasdy G, et al. Igg Subclass Staining in Renal Biopsies With Membranous Glomerulonephritis Indicates Subclass Switch During Disease Progression. Modern Pathol: an Off J U States Can Acad Pathol Inc (2013) 26(6):799–805. doi: 10.1038/modpathol.2012.237
141. Aucouturier P, Danon F, Daveau M, Guillou B, Sabbah A, Besson J, et al. Measurement of Serum IgG4 Levels by a Competitive Immunoenzymatic Assay With Monoclonal Antibodies. J Immunol Methods (1984) 74(1):151–62. doi: 10.1016/0022-1759(84)90376-4
142. van der Neut Kolfschoten M, Schuurman J, Losen M, Bleeker WK, Martinez-Martinez P, Vermeulen E, et al. Anti-Inflammatory Activity of Human IgG4 Antibodies by Dynamic Fab Arm Exchange. Science (2007) 317(5844):1554–7. doi: 10.1126/science.1144603
143. van der Zee JS, van Swieten P, Aalberse RC. Inhibition of Complement Activation by IgG4 Antibodies. Clin Exp Immunol (1986) 64(2):415–22.
144. Koneczny I. A New Classification System for IgG4 Autoantibodies. Front Immunol (2018) 9:97. doi: 10.3389/fimmu.2018.00097
145. Tomas NM, Hoxha E, Reinicke AT, Fester L, Helmchen U, Gerth J, et al. Autoantibodies Against Thrombospondin Type 1 Domain-Containing 7A Induce Membranous Nephropathy. J Clin Invest (2016) 126(7):2519–32. doi: 10.1172/JCI85265
146. Jurgensen HJ, Johansson K, Madsen DH, Porse A, Melander MC, Sorensen KR, et al. Complex Determinants in Specific Members of the Mannose Receptor Family Govern Collagen Endocytosis. J Biol Chem (2014) 289(11):7935–47. doi: 10.1074/jbc.M113.512780
147. Collins AM, Jackson KJ. A Temporal Model of Human IgE and IgG Antibody Function. Front Immunol (2013) 4:235. doi: 10.3389/fimmu.2013.00235
148. Vinuesa CG, Sanz I, Cook MC. Dysregulation of Germinal Centres in Autoimmune Disease. Nat Rev Immunol (2009) 9(12):845–57. doi: 10.1038/nri2637
149. Hennerici T, Pollmann R, Schmidt T, Seipelt M, Tackenberg B, Mobs C, et al. Increased Frequency of T Follicular Helper Cells and Elevated Interleukin-27 Plasma Levels in Patients With Pemphigus. PloS One (2016) 11(2):e0148919. doi: 10.1371/journal.pone.0148919
150. Kim AR, Han D, Choi JY, Seok J, Kim S-E, Seo S-H, et al. Targeting Inducible Costimulator Expressed on CXCR5PD-1 T Cells Suppresses the Progression of Pemphigus Vulgaris. J Allergy Clin Immunol (2020) 146(5):1070–9. doi: 10.1016/j.jaci.2020.03.036
151. Zhang C-J, Gong Y, Zhu W, Qi Y, Yang C-S, Fu Y, et al. Augmentation of Circulating Follicular Helper T Cells and Their Impact on Autoreactive B Cells in Myasthenia Gravis. J Immunol (Baltimore Md 1950) (2016) 197(7):2610–7. doi: 10.4049/jimmunol.1500725
152. Yi JS, Guptill JT, Stathopoulos P, Nowak RJ, O’Connor KC. B Cells in the Pathophysiology of Myasthenia Gravis. Muscle Nerve (2018) 57(2):172–84. doi: 10.1002/mus.25973
153. Li Y, Guptill JT, Russo MA, Howard JF, Massey JM, Juel VC, et al. Imbalance in T Follicular Helper Cells Producing IL-17 Promotes Pro-Inflammatory Responses in MuSK Antibody Positive Myasthenia Gravis. J Neuroimmunol (2020) 345:577279. doi: 10.1016/j.jneuroim.2020.577279
154. Aalberse RC, van der Gaag R, van Leeuwen J. Serologic Aspects of IgG4 Antibodies. I. Prolonged Immunization Results in an IgG4-restricted Response. J Immunol (1983) 130(2):722–6.
155. Gascan H, Gauchat JF, Roncarolo MG, Yssel H, Spits H, de Vries JE. Human B Cell Clones can be Induced to Proliferate and to Switch to IgE and IgG4 Synthesis by Interleukin 4 and a Signal Provided by Activated CD4+ T Cell Clones. J Exp Med (1991) 173(3):747–50. doi: 10.1084/jem.173.3.747
156. Wood N, Bourque K, Donaldson DD, Collins M, Vercelli D, Goldman SJ, et al. Il-21 Effects on Human IgE Production in Response to IL-4 or IL-13. Cell Immunol (2004) 231(1-2):133–45. doi: 10.1016/j.cellimm.2005.01.001
157. Lundgren M, Persson U, Larsson P, Magnusson C, Smith CI, Hammarstrom L, et al. Interleukin 4 Induces Synthesis of IgE and IgG4 in Human B Cells. Eur J Immunol (1989) 19(7):1311–5. doi: 10.1002/eji.1830190724
158. Ettinger R, Kuchen S, Lipsky PE. The Role of IL-21 in Regulating B-Cell Function in Health and Disease. Immunol Rev (2008) 223:60–86. doi: 10.1111/j.1600-065X.2008.00631.x
159. Crotty S. Follicular Helper CD4 T Cells (TFH). Annu Rev Immunol (2011) 29:621–63. doi: 10.1146/annurev-immunol-031210-101400
160. Belanger S, Crotty S. Dances With Cytokines, Featuring TFH Cells, IL-21, IL-4 and B Cells. Nat Immunol (2016) 17(10):1135–6. doi: 10.1038/ni.3561
161. Unger PA, Lighaam LC, Vermeulen E, Kruithof S, Makuch M, Culver EL, et al. Divergent Chemokine Receptor Expression and the Consequence for Human Igg4 B Cell Responses. Eur J Immunol (2020) 50(8):1113–25. doi: 10.1002/eji.201948454
162. Hofstra JM, Wetzels JF. Alkylating Agents in Membranous Nephropathy: Efficacy Proven Beyond Doubt. Nephrol Dial Transplant (2010) 25(6):1760–6. doi: 10.1093/ndt/gfq017
163. Maas R, Hofstra JM, Wetzels JF. An Overview of Immunosuppressive Therapy in Idiopathic Membranous Nephropathy. Minerva Med (2012) 103(4):253–66.
164. van de Logt AE, Dahan K, Rousseau A, van der Molen R, Debiec H, Ronco P, et al. Immunological Remission in PLA2R-antibody-associated Membranous Nephropathy: Cyclophosphamide Versus Rituximab. Kidney Int (2018) 93(4):1016–7. doi: 10.1016/j.kint.2017.12.019
165. Boddy AV, Yule SM. Metabolism and Pharmacokinetics of Oxazaphosphorines. Clin Pharmacokinet (2000) 38(4):291–304. doi: 10.2165/00003088-200038040-00001
166. Emadi A, Jones RJ, Brodsky RA. Cyclophosphamide and Cancer: Golden Anniversary. Nat Rev Clin Oncol (2009) 6(11):638–47. doi: 10.1038/nrclinonc.2009.146
167. Jones RJ, Barber JP, Vala MS, Collector MI, Kaufmann SH, Ludeman SM, et al. Assessment of Aldehyde Dehydrogenase in Viable Cells. Blood (1995) 85(10):2742–6. doi: 10.1182/blood.V85.10.2742.bloodjournal85102742
168. Gordon MY, Goldman JM, Gordon-Smith EC. 4-Hydroperoxycyclophosphamide Inhibits Proliferation by Human Granulocyte-Macrophage Colony-Forming Cells (GM-CFC) But Spares More Primitive Progenitor Cells. Leuk Res (1985) 9(8):1017–21. doi: 10.1016/0145-2126(85)90072-4
169. Ozer H, Cowens JW, Colvin M, Nussbaum-Blumenson A, Sheedy D. In Vitro Effects of 4-Hydroperoxycyclophosphamide on Human Immunoregulatory T Subset Function. I. Selective Effects on Lymphocyte Function in T-B Cell Collaboration. J Exp Med (1982) 155(1):276–90. doi: 10.1084/jem.155.1.276
170. van den Brand JAJG, van Dijk PR, Hofstra JM, Wetzels JFM. Long-Term Outcomes in Idiopathic Membranous Nephropathy Using a Restrictive Treatment Strategy. J Am Soc Nephrol JASN (2014) 25(1):150–8. doi: 10.1681/ASN.2013020185
171. van den Brand JA, van Dijk PR, Hofstra JM, Wetzels JF. Cancer Risk After Cyclophosphamide Treatment in Idiopathic Membranous Nephropathy. Clin J Am Soc Nephrol (2014) 9(6):1066–73. doi: 10.2215/CJN.08880813
172. Faul C, Donnelly M, Merscher-Gomez S, Chang YH, Franz S, Delfgaauw J, et al. The Actin Cytoskeleton of Kidney Podocytes is a Direct Target of the Antiproteinuric Effect of Cyclosporine A. Nat Med (2008) 14(9):931–8. doi: 10.1038/nm.1857
173. Fervenza FC, Appel GB, Barbour SJ, Rovin BH, Lafayette RA, Aslam N, et al. Rituximab or Cyclosporine in the Treatment of Membranous Nephropathy. N Engl J Med (2019) 381(1):36–46. doi: 10.1056/NEJMoa1814427
174. Kalliakmani P, Koutroulia E, Sotsiou F, Vlachojannis JG, Goumenos DS. Benefit and Cost From the Long-Term Use of Cyclosporine-A in Idiopathic Membranous Nephropathy. Nephrol (Carlton) (2010) 15(8):762–7. doi: 10.1111/j.1440-1797.2010.01301.x
175. Fernández-Juárez G, Rojas-Rivera J, A-Evd L, Justino J, Sevillano A, Caravaca-Fontán F, et al. The STARMEN Trial Indicates That Alternating Treatment With Corticosteroids and Cyclophosphamide Is Superior to Sequential Treatment With Tacrolimus and Rituximab in Primary Membranous Nephropathy. Kidney Int (2021) 99(4):986–98. doi: 10.1016/j.kint.2020.10.014
176. Dahan K, Debiec H, Plaisier E, Cachanado M, Rousseau A, Wakselman L, et al. Rituximab for Severe Membranous Nephropathy: A 6-Month Trial With Extended Follow-Up. J Am Soc Nephrol (2017) 28(1):348–58. doi: 10.1681/ASN.2016040449
177. Wallin EF, Jolly EC, Suchánek O, Bradley JA, Espéli M, Jayne DRW, et al. Human T-follicular Helper and T-Follicular Regulatory Cell Maintenance Is Independent of Germinal Centers. Blood (2014) 124(17):2666–74. doi: 10.1182/blood-2014-07-585976
178. Wallin EF, Hill DL, Linterman MA, Wood KJ. The Calcineurin Inhibitor Tacrolimus Specifically Suppresses Human T Follicular Helper Cells. Front Immunol (2018) 9:1184. doi: 10.3389/fimmu.2018.01184
179. Ballesteros-Tato A. Beyond Regulatory T Cells: The Potential Role for IL-2 to Deplete T-follicular Helper Cells And Treat Autoimmune Diseases. Immunotherapy (2014) 6(11):1207–20. doi: 10.2217/imt.14.83
180. He J, Zhang R, Shao M, Zhao X, Miao M, Chen J, et al. Efficacy and Safety of Low-Dose IL-2 in the Treatment of Systemic Lupus Erythematosus: A Randomised, Double-Blind, Placebo-Controlled Trial. Ann Rheum Dis (2020) 79(1):141–9. doi: 10.1136/annrheumdis-2019-215396
181. Ballesteros-Tato A. Beyond Regulatory T Cells: The Potential Role for IL-2 to Deplete T-Follicular Helper Cells and Treat Autoimmune Diseases. Immunotherapy (2014) 6(11):1207–20. doi: 10.2217/imt.14.83
Keywords: idiopathic membranous nephropathy (IMN), helper T cells (Th cells), autoimmune, antibodies, germinal center (GC)
Citation: Zhao Q, Dai H, Liu X, Jiang H, Liu W, Feng Z, Zhang N, Gao Y, Dong Z, Zhou X, Du J, Zhang N, Rui H, Yuan L and Liu B (2021) Helper T Cells in Idiopathic Membranous Nephropathy. Front. Immunol. 12:665629. doi: 10.3389/fimmu.2021.665629
Received: 08 February 2021; Accepted: 06 May 2021;
Published: 20 May 2021.
Edited by:
Barbara Seitz-Polski, University of Nice Sophia Antipolis, FranceReviewed by:
Paolo Cravedi, Icahn School of Medicine at Mount Sinai, United StatesUlf Panzer, Medical School Hamburg, Germany
Copyright © 2021 Zhao, Dai, Liu, Jiang, Liu, Feng, Zhang, Gao, Dong, Zhou, Du, Zhang, Rui, Yuan and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Baoli Liu, liubaoli@bjzhongyi.com