 Espen Basmo Ellingsen
Espen Basmo Ellingsen Sara M. Mangsbo
Sara M. Mangsbo Eivind Hovig
Eivind Hovig Gustav Gaudernack
Gustav Gaudernack- 1Department of Tumor Biology, Institute for Cancer Research, The Norwegian Radium Hospital, Oslo, Norway
- 2Faculty of Medicine, University of Oslo, Oslo, Norway
- 3Research and Development, Ultimovacs ASA, Oslo, Norway
- 4Research and Development, Ultimovacs AB, Uppsala, Sweden
- 5Department of Pharmaceutical Biosciences, Science for Life Laboratory, Uppsala University, Uppsala, Sweden
- 6Centre for Bioinformatics, Department of Informatics, University of Oslo, Oslo, Norway
Telomerase-based therapeutic cancer vaccines (TCVs) have been under clinical investigation for the past two decades. Despite past failures, TCVs have gained renewed enthusiasm for their potential to improve the efficacy of checkpoint inhibition. Telomerase stands as an attractive target for TCVs due to its almost universal presence in cancer and its essential function promoting tumor growth. Herein, we review tumor telomerase biology that may affect the efficacy of therapeutic vaccination and provide insights on optimal vaccine design and treatment combinations. Tumor types possessing mechanisms of increased telomerase expression combined with an immune permissive tumor microenvironment are expected to increase the therapeutic potential of telomerase-targeting cancer vaccines. Regardless, rational treatment combinations, such as checkpoint inhibitors, are likely necessary to bring out the true clinical potential of TCVs.
Introduction
Preventive vaccines against infectious agents have been one of the major advancements in medical history. However, the transfer of this technology to the treatment of cancer has for several reasons proven to be a difficult task. Multiple therapeutic cancer vaccines (TCVs) have been evaluated in clinical trials since the 1990s, often inducing vaccine-specific immune responses, but rarely translating to clinical efficacy (1, 2). Nevertheless, the recent advances in immunotherapy have rekindled the interest in TCVs, exemplified by an increase from 612 to 855 cancer vaccine candidates in the overall immuno-oncology drug pipeline from 2017 to 2020 (3).
The advent of checkpoint inhibitors (CPIs) has provided significant improvement in survival outcomes for patients with various cancer types, most notably in malignant melanoma (4, 5). CPIs disrupt intrinsic and tumor-induced suppressor mechanisms restricting a spontaneous anti-tumor immune response, as evidenced by associations between tumor PD-L1 expression, tumor-infiltrating lymphocytes (TILs), tumor mutational burden (TMB), neoantigen load, and response to therapy (6–9). Although many patients experience remarkable durable clinical responses to CPIs, most patients eventually progress. A lack of response to CPIs is believed to be caused by either an insufficient spontaneously primed immune response against tumor antigens, an immunosuppressive tumor microenvironment (TME), or a combination of the two (10). These insights into the balance between the immune system and the tumor suggest TCVs as a logical next step to improve clinical outcomes by strengthening the immune response against the tumor and possibly overcoming TME immunosuppression. Moreover, there are good reasons to believe that the immune checkpoint molecules contributed to the earlier failure of TCVs in the clinic (11). Combining CPIs with TCVs may allow unchecked expansion and function of vaccine-induced T cells both in tumor-draining lymph nodes and in the tumor, thereby achieving superior anti-tumor immune responses, balancing the immune system in favor of tumor control.
An essential property of a TCV is the antigen it targets. Tumor-associated antigens (TAAs) are antigens that are over-expressed by the tumor and preferably have a restricted expression pattern in healthy tissues. Several TAAs have been the target of TCVs and can be divided into three broad categories: 1) germline antigens (e.g., MAGE and NY-ESO-1) (12), 2) cell lineage antigens (e.g., gp100, MART-1, PSA/PAP/PMSA) (2, 13–16), and 3) differentially expressed antigens (e.g., telomerase and Her2) (17, 18). A challenge with targeting endogenous antigens is the possibility of central tolerance, where self-reactive high-affinity T cell clones are deleted through negative selection in the thymus and are thus absent or present in low numbers with low affinity in the T cell repertoire (19). Another challenge is the risk for on-target off-tumor autoimmunity, where vaccination induces an immune response to healthy tissues expressing the antigen. One TCV strategy to circumvent these challenges is to vaccinate against predicted tumor neoantigens. Tumor-specific somatic mutations may give rise to aberrant peptides presented as T cell targetable neoantigens on the cancer cell surface in the context of an MHC molecule (20, 21). Though neoantigens are seemingly attractive targets for vaccination, subclonal expression of neoantigens due to substantial intratumoral heterogeneity may provide resistance and escape mechanisms for the tumor (9). Furthermore, such personalized TCVs require comprehensive logistics and subsequent delay in the onset of treatment.
In this review, we will discuss cancer vaccines targeting the differentially expressed TAA telomerase reverse transcriptase (hTERT), offering an “off-the-shelf” alternative to personalized TCVs. Telomerase is almost ubiquitously expressed in cancer, and TCVs targeting hTERT is thus an attractive approach to achieve T cell infiltration and epitope-spreading. We will focus on tumor biological considerations, immunologically rational indications and treatment combinations to optimize for clinical efficacy from therapeutic vaccination against telomerase. Furthermore, we will provide insights into possible causes of failure in previous clinical trials and an update on ongoing studies.
Telomerase as a Target for Therapeutic Vaccination
An Almost Universal Cancer Antigen
Telomeres and Telomerase
The 3’ ends of the chromosomes consist of a repeating sequence of nucleotides, TTAGGG, termed telomeres. The telomeres serve to protect the chromosomal ends from inducing DNA damage responses, which would otherwise be activated upon breaks in the double-stranded DNA. Since DNA polymerases cannot replicate the DNA ends, the telomeres are progressively shortened with approximately 50 bps with each cell division (22). This progressive shortening of the telomeres ultimately leads to telomere crisis and chromosomal instability and, consequently, a limited number of cell divisions that can occur before the cell enters senescence or apoptosis (23). This replicative cellular senescence phenomenon led to the identification of the Hayflick Limit, defined as the maximum number of cell divisions that can occur in a somatic cell (24). The reverse transcriptase enzyme component of the telomerase complex (hTERT) can be activated in specific cell types, leading to replication of the telomeric DNA and thereby increasing the proliferative potential of the cell (23).
In somatic cells, telomerase is restricted to certain rapidly proliferating tissues, such as the intestinal epithelium, premenopausal endometrium, the testis, and tissues containing a high population of activated lymphocytes, such as secondary lymphoid organs (25). Telomerase is also expressed by stem cells, and their telomerase activity is closely related to the proliferation rate, explaining the relatively low activity in adult stem cells compared to embryonic or cancer cells (26).
Telomerase in Cancer
Telomerase has been extensively studied in cancer, and telomerase activity has been documented in >90% of all cancers (27, 28). Telomerase activation is a major cell immortalization mechanism and is implicated as an essential step in carcinogenesis (29). Through telomerase activation, cancer cells acquire the ability of unlimited proliferation. Telomerase activity is also linked to epithelial-to-mesenchymal transition and cancer stemness, providing cancer cells with metastatic potential (30, 31). Telomerase is expressed in most tumor types across all stages of development and is thus an attractive target for therapeutic vaccination (Figure 1). To restrict possibilities of resistance mutations to develop, epitopes within the essential hTERT component of the telomerase complex are commonly used as antigens for TCVs, as loss of hTERT would abolish tumor growth. Moreover, due to its ubiquitous expression, hTERT serves as a cancer antigen being independent of clonal diversity within a tumor. Tumor telomerase activity is considered a negative prognostic factor for several cancers (33–37), while spontaneous anti-hTERT CD4+ immune responses have been identified as a positive prognostic factor in non-small cell lung cancer (NSCLC) (38), substantiating both the natural immunogenicity of hTERT and its relevance as a target for TCVs.
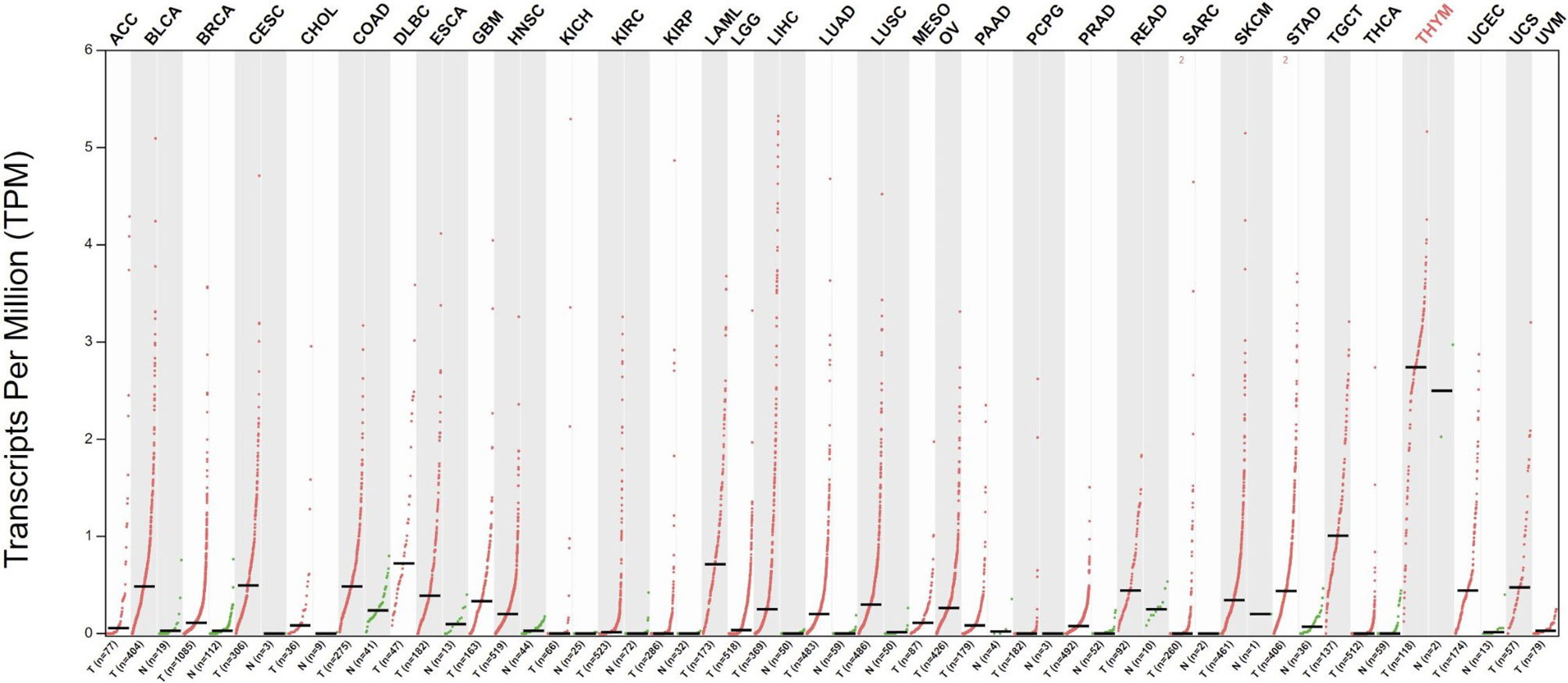
Figure 1 Differential expression of hTERT in tumors vs. adjacent normal tissues. Red dots indicate primary tumor, and green adjacent healthy tissue. Data gathered from The Cancer Genome Atlas through GEPIA (32). Q-value cut-off was set to 0.01. ACC, Adrenocortical carcinoma; BLCA, Bladder Urothelial Carcinoma; BRCA, Breast invasive carcinoma; CESC, Cervical squamous cell carcinoma and endocervical adenocarcinoma; CHOL, Cholangiocarcinoma; COAD, Colon adenocarcinoma; DLBC, Lymphoid Neoplasm Diffuse Large B-cell Lymphoma; ESCA, Esophageal carcinoma; Glioblastoma multiforme GBM, Head and Neck squamous cell carcinoma HNSC, KICH, Kidney Chromophobe; KIRC, Kidney renal clear cell carcinoma; KIRP, Kidney renal papillary cell carcinoma; LAML, Acute Myeloid Leukemia; LGG, Brain Lower Grade Glioma; LIHC, Liver hepatocellular carcinoma; LUAD, Lung adenocarcinoma; LUSC, MESO, Lung squamous cell carcinoma; Mesothelioma; OV, Ovarian serous cystadenocarcinoma; PAAD, Pancreatic adenocarcinoma; PCPG, Pheochromocytoma and Paraganglioma; PRAD, Prostate adenocarcinoma; READ, Rectum adenocarcinoma; SARC, Sarcoma; SKCM, Skin Cutaneous Melanoma; STAD, Stomach adenocarcinoma; TGCT, Testicular Germ Cell Tumors; THCA, Thyroid carcinoma; THYM, Thymoma; UCEC, Uterine Corpus Endometrial Carcinoma; UCS, Uterine Carcinosarcoma; UVM, Uveal Melanoma.
Telomerase Vaccination
TCVs aim to induce T cells that target a tumor antigen leading to improved anti-tumor immune responses and, ultimately, cancer cell death. As recently reviewed, telomerase vaccination has been evaluated across 34 clinical trials spanning almost two decades (39). However, there have been no positive late-phase studies, and as such, there is an obvious need for improvement, either in vaccine design (including sequence choice, formulation, and delivery), selection of indications, or treatment combination strategies. Optimal hTERT targeting TCVs should be designed to effectively induce the appropriate immune response phenotype, which can be further augmented through rational therapeutic combination strategies.
The Phenotype of the Induced Immune Response
CD8+ cytotoxic T lymphocytes were long considered the most potent anti-tumor effectors in the adaptive immune system, but lately, the focus has shifted towards the importance of CD4+ T helper lymphocytes as an opportunity to achieve tumor recognition and T cell infiltration in an immunosuppressive TME. CD8+ T cells have the ability to directly kill cancer cells expressing their cognate antigen in the context of an HLA class I molecule. For this interaction to occur, the antigen must be processed internally by the cancer cell in a multi-step process to be loaded onto HLA class I molecules (40). CD4+ T cells are, on the other hand, activated through interaction with their antigen in the context of an HLA class II molecule, typically expressed by antigen-presenting cells (APCs), but also upregulated on cancer cells by IFN-γ stimulation and thus frequently expressed by immunogenic tumors (41). Activated CD4+ T cells orchestrate an immune response through the release of pro-inflammatory cytokines. As reviewed elsewhere (42–44), critical features of CD4+ T helper 1 (Th1) cells in anti-tumor immunity include induction of effective antigen presentation by APCs, augmentation of CD8+ T cell responses, T cell homing to the tumor (45), direct and indirect tumor cell killing (46–48), and formation of memory T cells (49, 50). The multifaceted functions of Th1 cells may thus support virtually all steps in the cancer immunity cycle (51) (Figure 2). As hTERT is expressed throughout the tumorigenesis, this CD4+ Th1 response may stay activated and relevant regardless of the tumor’s rapidly evolving genetic makeup, providing the immune system with an opportunity to mount an individually tailored immune response to relevant target antigens. This concept of an “in vivo personalized vaccine” stands as an alternative strategy to “ex vivo personalized vaccines” where the selection of tumor targets is based on predicted HLA class I binding neoantigens.
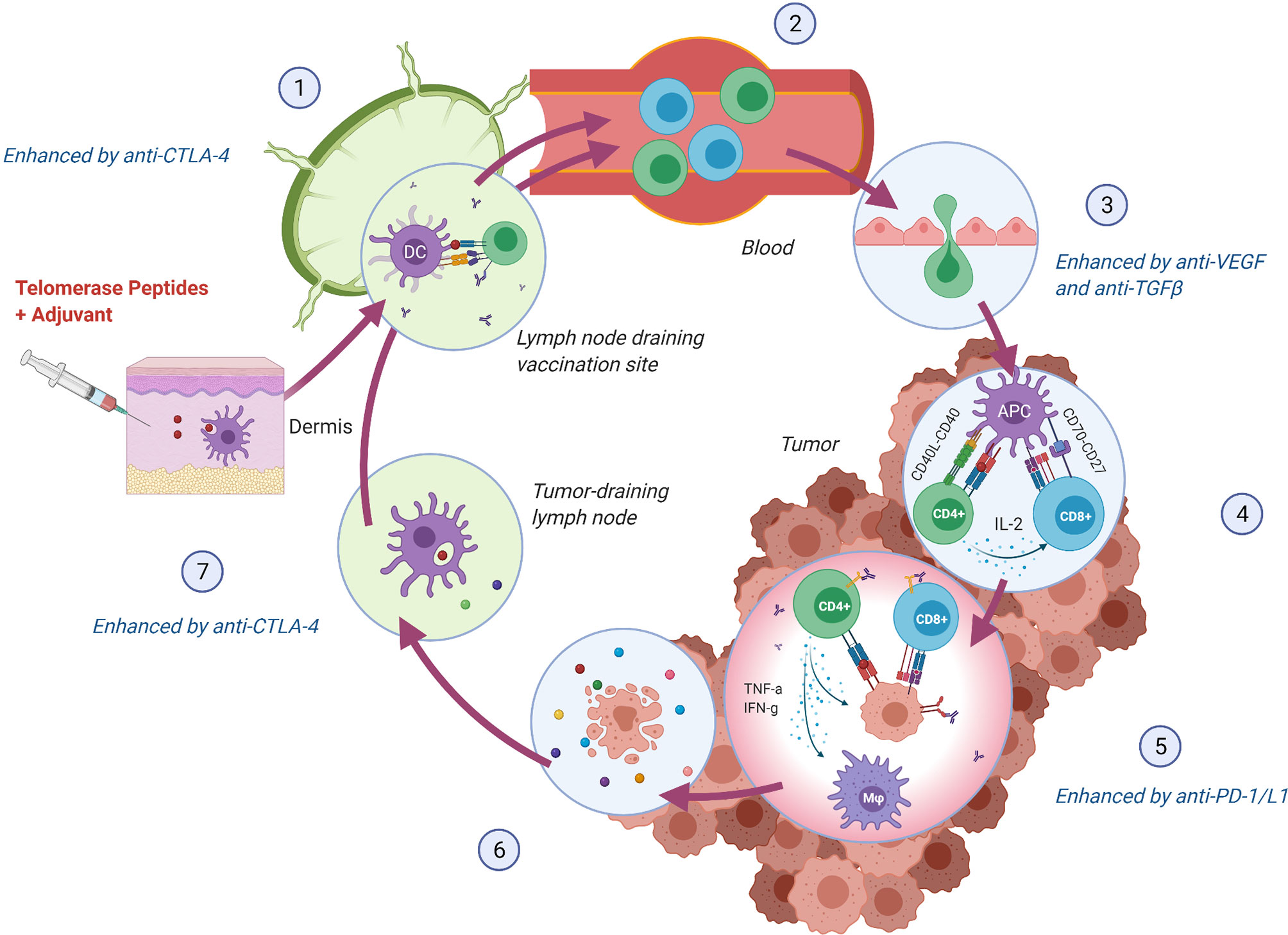
Figure 2 hTERT specific CD4+ Th1 cells support crucial steps in the Cancer Immunity Cycle (51). (1) Vaccine peptides are presented to naïve T cells by DCs in the lymph node draining the vaccination site. Anti-CTLA-4 monoclonal antibody (mAb) may lead to increased expansion of vaccine-induced T cells. (2) hTERT-specific T cells enter circulation and (3) infiltrate the tumor. Normalization of the tumor vasculature through inhibition of VEGF may facilitate an increased influx of T cells. (4) T cells recognize hTERT on local antigen-presenting cells in the context of an MHC class II molecule and directly stimulate local CD8+ T cells through IL-2 secretion and indirectly through co-stimulation of DCs (CD40L-CD40 interaction), leading to enhanced cross-presentation 44. (5) MHC class II expressing tumor cells can be directly killed through cytokine secretion or indirectly through activation of CD8+ cells and macrophages (M φ) (44, 48). Anti-PD-1/L1 mAb may provide increased effector activity of vaccine-induced T cells in the tumor by blocking regulatory signals on T cells (PD-1) or tumor cells (PD-L1). (6) Lysed tumor cells release hTERT or mutated peptides, which in turn are (7) phagocytosed by DCs and presented to T cells providing either intra- or intermolecular epitope spreading and broadening of the anti-tumor immune response (52). Anti-CTLA-4 mAb may, in turn, support further priming and expansion of anti-tumor T cells. Figure created with BioRender.com.
A caveat of the CD4+ immune response is the different subtypes that are considered good or bad with respect to anti-tumor immunity. The Th1 phenotype is typically considered ideal, and T(reg) (CD4+, FOXP3+) is considered immunosuppressive (44). The roles of other phenotypes, such as Th17 and Th2, in cancer immunity, are not as well established (53). The differentiation into the Th subsets relies on the priming environment (54), and as such, the phenotype may be affected by vaccine design, vaccine administration route, and use of an adjuvant. Furthermore, Hansen et al. found that samples from the CTN-2000 trial, where patients received hTERT vaccination as monotherapy, displayed a more Th1-polarized phenotype than samples from the CTN-2006 trial evaluating the same vaccine, GV1001, as maintenance treatment after chemoradiotherapy (55). This indicates that disease stage and previous therapies may also affect the phenotype of vaccine-induced T cells and thus anti-tumor efficacy.
Telomerase-Based TCV Platforms
The most frequently utilized vaccination platform for telomerase-based TCVs is peptide vaccines (23/34 clinical trials). Peptide vaccines aim to elicit an adaptive immune response by in vivo uptake of the peptides by APCs at the vaccination site and subsequent presentation of embedded epitopes to naïve T cells leading to their expansion. Peptides are probably the preferred platform owing to their relatively long shelf-life, simple synthesis and administration route, requiring only intradermal or subcutaneous injection along with a vaccine adjuvant. The skin serves as an ideal administration route, as it contains a dense population of various dendritic cell subsets (56). The first TCVs developed commonly consisted of short peptides (up to 10 amino acids) as they can be loaded directly onto HLA class I molecules and induce CD8+ immune responses (57). Recently, however, synthetic long peptides (SLPs) have been in focus since they have the potential to provide cross-presentation by APCs leading to both class I and II presentation, and hence CD8+ and CD4+ immune responses, respectively (58, 59). The use of epitope dense SLPs also allows enrollment of patients independently of their HLA types (such as with GV1001 and UV1), whereas many short peptide vaccines have been tailored to fit single HLA class I molecules, thus limiting inclusion to patients harboring this HLA type (39) (Table 1).
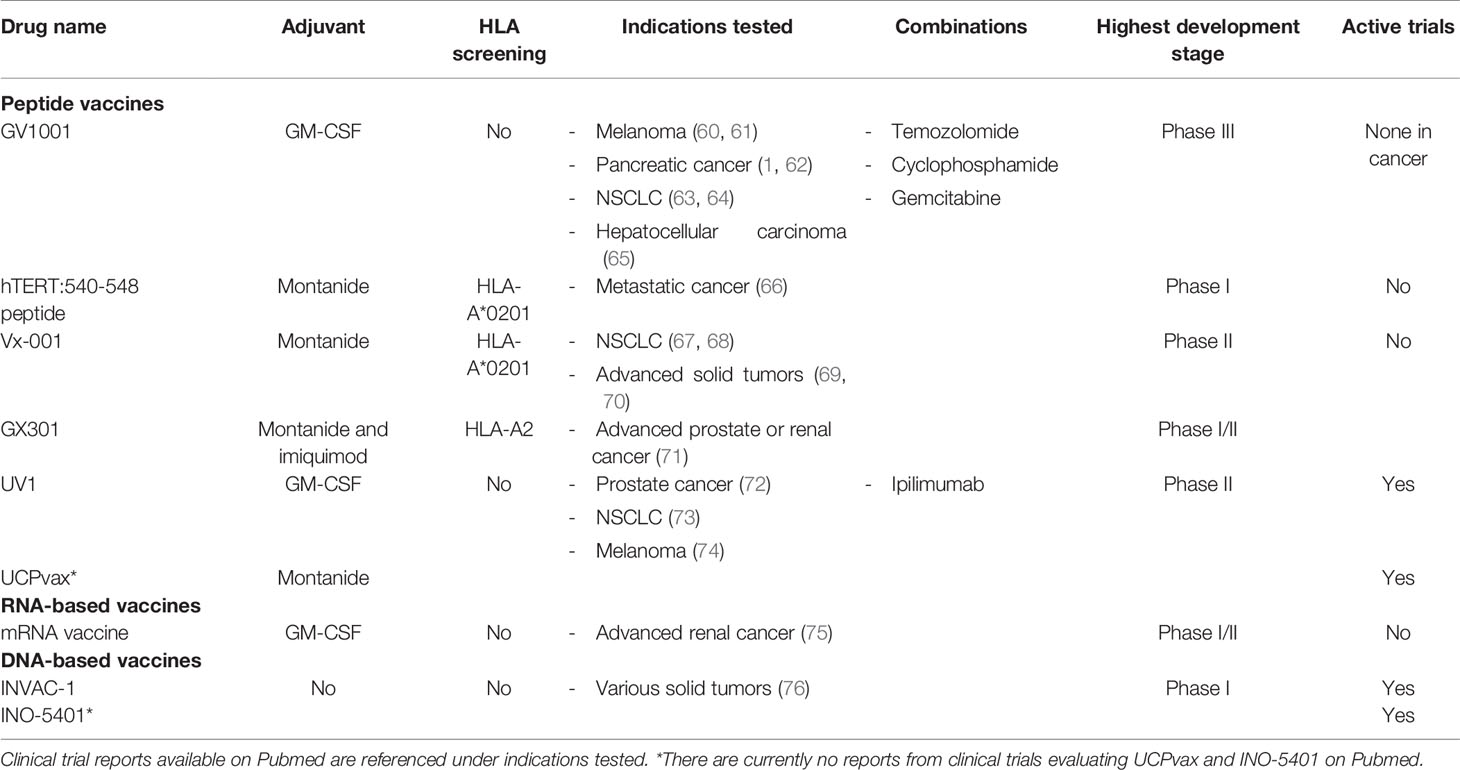
Table 1 hTERT TCV candidates evaluated in clinical trials covering various indications over the past two decades (autologous cell-based therapies are not included).
Other vaccine platforms include autologous dendritic cell (DC) vaccines, utilized in 10/34 clinical trials, ensuring in vitro antigen loading in DCs for presentation to T cells in vivo. This platform necessitates complex logistics and competence, including patient leukapheresis and subsequent DC cell culture and antigen pulsing, after which the treated DCs are transfused back to the patient (77). An mRNA vaccine has also emerged in the hTERT TCV pipeline, coding for several TAAs (MUC1, CEA, Her-2/neu, telomerase, survivin, and MAGE-A1), showing induction of CD4+ and CD8+ immune responses in a phase I/II trial (75). The last platform evaluated in clinical trials is DNA vaccines. Two hTERT-based vaccines utilize this platform, INVAC-1 and INO-5401. INVAC-1 consists of a plasmid DNA that encodes a modified inactive version of hTERT and has been shown to induce CD4+ and CD8+ immune responses in a phase I study (76). The advantage of a DNA or RNA-based vaccine is the incorporation of the whole gene. Still, the possible lack of adjuvant capability of DNA vaccines, in general, remains the biggest bottleneck for future clinical efficacy (78, 79). Results from studies with INO-5401 are not yet published in peer-reviewed journals. Dillard et al. recently described a novel telomerase immunotherapy platform, hTERT-targeting T cell receptor (TCR) therapy, showing both in vitro and in vivo anti-tumor efficacy in an animal model (80).
Vaccine Adjuvants
Peptide vaccines require an adjuvant to elicit an immune response, as naked peptides by themselves are poorly immunogenic. There are primarily two adjuvants currently employed in telomerase TCV trials. Granulocyte-macrophage colony stimulating factor (GM-CSF) is the most frequent, being utilized in 12/34 clinical trials, and incomplete Freund’s adjuvant (IFA)(Montanide ISA-51) being the second most common (11/34). Recombinant human GM-CSF acts to recruit APCs to the vaccination site and initiate differentiation and activation (81). Importantly, GM-CSF is water-soluble as opposed to IFA, which is a water-in-oil emulsion. As described by Hailemichael et al., IFA creates a depot effect at the vaccination site, which may sequester antigen-specific T cells and thus prevent migration of T cells to the tumor site (82), possibly explaining the failure of the gp100 vaccine (with IFA adjuvant) in the landmark study of ipilimumab and gp100 combination vs. ipilimumab alone or gp100 alone (2). Thus, GM-CSF or other water-soluble adjuvants may be the most suitable vaccine adjuvant for peptide cancer vaccines, but likely requires repeated administrations to compensate for the lack of depot effect. GM-CSF can also display dual roles as a proinflammatory signaling moiety in the immune system, impacting immunological responses depending on the cell type expanded in response to GM-CSF stimulation (83). As a drug, the dose of GM-CSF and possibly the formulation of GM-CSF may also impact the pharmacodynamic response. Nevertheless, hTERT-targeting TCVs with both GM-CSF and IFA adjuvants have been shown to induce immune responses in blood with no apparent differences in frequency.
Recently, compounds that conjugate an adjuvant directly with the vaccine peptides have garnered interest and hold promise due to their efficiency in providing targeted adjuvant effect (84, 85). Such compounds have, however, not been evaluated with telomerase peptides.
Monitoring of Immune Responses in Clinical Trials
Inherent to the potential clinical benefit of a therapeutic cancer vaccine is its ability to induce immune responses, and several clinical trials with telomerase vaccines have shown a positive correlation between vaccine-induced immune responses and survival (60, 62, 63). The rate of immune responders has varied across the clinical trials, ranging from zero percent in a trial in hepatocellular carcinoma (65) to 78 percent in a trial in melanoma (60) with the most frequently evaluated hTERT TCV, GV1001. The discrepancy observed across trials may have several causes other than suboptimal vaccine design. The fraction of vaccine-specific T cells in circulation is likely low since T cells primarily reside in lymphoid tissues (or the tumor) and are thus not captured by drawing <100 ml of peripheral blood (86, 87). Thus, ideal methodologies to monitor immune responses have yet to be developed. Regardless, the relatively high immune response rates observed across different hTERT trials suggest that central tolerance is not imposed on hTERT-specific T cells and that vaccination may induce high-affinity T cell responses. Furthermore, longitudinal immune monitoring with the proliferation assay has demonstrated persisting immune responses detectable up to 7 years after initial vaccination and serve as evidence of induction of immune memory with lasting proliferative potential (60, 63). Interestingly, late peaks in immune responses have coincided with clinical events, suggesting a natural boosting of the immune response by recurring tumors (74).
Safety
The most reported side effect with telomerase-targeting cancer vaccines is injection site reactions, including local erythema and pruritus, and flu-like symptoms such as fever, muscle and joint pain, and fatigue. Generally, telomerase vaccination appears to be well tolerated, with only a few cases of serious adverse events reported (39). As an endogenous self-antigen, an immune response against hTERT is associated with a theoretical risk for off-tumor on-target toxicity. However, due to the physiologic function of hTERT, its expression pattern is limited to specific sites of highly proliferating cells and stem cells such as bone marrow, testis, embryo, and placenta (25, 26). These tissues are described as specific immune-privileged sites due to their local tolerogenic environment (88–93). Immune tolerance mechanisms are put in place at these tissues to protect from immunological insults, which would potentially be deleterious given the critical physiologic functions of these cells. These factors are likely also preventing autoimmunity resulting from off-tumor on-target reactions from vaccine-induced T cells, further substantiated by the lack of immune-related adverse events observed in hTERT-expressing tissues in clinical trials with various telomerase vaccines. To explore the theoretical risk of inducing immune responses against stem cells, bone marrow histological examinations have been performed in a subset of clinical trials with telomerase peptides or DC vaccines, showing no discernable changes after vaccination (64, 94, 95). Although the reports from conducted clinical trials indicate a tolerable safety profile, novel compounds utilizing different vaccine platforms and treatment combinations may elicit more potent immune responses, and consequently, increase the risk for off-tumor effects. Checkpoint inhibitor combinations are especially relevant in this context, as their physiologic function is to limit autoimmunity, and their inhibition may thus lower the threshold for such off-tumor reactions. A clinical trial of an hTERT vaccine in combination with the checkpoint inhibitor ipilimumab did not report a change in side effect panorama in treated patients (n=12) (74). However, larger studies are needed to conclude on the safety of combining checkpoint inhibitors and telomerase vaccines.
Considerations for Optimizing the Therapeutic Potential of Telomerase-Targeting Vaccines
Considering that most tumors rely on hTERT, telomerase appears to be an almost universal cancer antigen, and hTERT targeting TCVs are therefore potentially broadly applicable, reflected in the extensive set of indications tested (see Table 1 below) (39). Still, to optimize the therapeutic potential of hTERT-vaccination, the selection of indications should be made based on tumor hTERT expression and factors that may restrict the intratumoral activity of T cells.
Telomerase Activation and Expression
Tumor hTERT Promoter Mutations
Mechanisms of hTERT gene activation in cancer have been widely studied, and somatic mutations in hTERT are the most well described. Somatic mutations in the coding region of hTERT appear to be rare, but mutations in the promoter region are common (appx. 19% of tumors) (96). Furthermore, two recurrent, mutually exclusive, hTERT promoter mutations (C228T and C250T, collectively referred to as hTERTp) have been found in 71% of melanomas and confer a 2-4-fold increase in hTERT promoter transcriptional activity (97). These two mutations have the same functional consequence, creating an hTERT promoter binding site (GGAA) for ETS (E-twenty-six) transcription factors. These mutations have also been significantly associated with BRAF mutations in melanoma (96–98). BRAF mutations lead to elevated ETS transcription factors, which may bind to the hTERT promoter binding site resulting from the promoter mutations, thereby working in concert to increase hTERT activity (99).
As reported by others (36, 97, 98), hTERTp appears to be more frequent in metastatic than in primary melanoma tumors. These findings suggest that hTERTp is either a late event or confer a survival advantage for the cancer clone, leading to its higher frequency in advanced disease. In contrast, in another study of 58 matching primary and metastatic melanoma lesions, hTERTp appeared more frequently in primary tumors and were exclusively observed in primary or metastatic lesions in 17% and 7% of cases, respectively (37). Additionally, a clonal distribution of hTERT promoter mutations has been reported in early-stage hepatocellular carcinoma (100). These discordant observations indicate variations in the phylogeny of promoter mutations across tumors and possibly indicate different roles of hTERTp in the tumorigenesis of the individual tumor. The heterogeneity of hTERTp could indicate an intratumoral heterogeneity of hTERT expression. In line with this concept, there are variations in the hTERT expression levels both within and across cancer cell lines (101). This observation may be due to cell-level variation in the transcription of hTERT occurring at different phases of the cell cycle (102), giving rise to variations at a specific moment in time and a continuous fluctuation with longitudinal observations. However, the regulation of hTERT expression is convoluted and a contended area of research (103). Notably, these promoter mutations were not found in benign lesions, kidney cancers, pheochromocytomas, or gastrointestinal stromal tumors (96, 104).
Copy Number Alterations
Another possible mechanism of hTERT activation is copy number amplification (CNA) of the hTERT gene. hTERT CNA is over-represented in melanomas (105) and also described with a relatively high frequency (30%) in lung cancer, breast cancer, cervical carcinomas, and neuroblastoma (106). Another study of 2,210 solid tumors found that CNA at chromosome 5p, where the hTERT gene is located, was the eighth most common chromosomal gain (13.2%) (107). Furthermore, hTERT CNA is associated with increased hTERT expression among cancer cell lines and primary solid tumors (106).
Epigenetic Changes Affecting Telomerase Expression
Genetic, as well as epigenetic changes, can affect telomerase activity. Epigenetic changes are not linked to changes in the nucleotide sequence, but are still preserved with cell division, impacting gene activity and expression. Commonly, the impact on gene regulation via epigenetic changes is controlled by specific methylation patterns or histone modifications (acetylation), often at non-coding sites such as the promoter region. In the case of telomerase, there are two different regions in the promoter that affect expression, one of which is a non-methylated region in the proximal region of the promoter, which, in its non-methylated state, is associated with active telomerase expression. However, it has also been discovered that there is a region called the TERT Hypermethylated Oncological Region (THOR) (108–112). The THOR is unusual in its behavior, as it is repressing expression in its unmethylated state, and as such, it is related to hTERT activation and cancer progression upon hypermethylation. Interestingly, THOR hypermethylation is more common in cancers known to have a low frequency of hTERTp (prostate, lung, colon, and breast cancer), indicating THOR hypermethylation as an alternative hTERT activation mechanism for these tumor types. Mutations in the promoter region can also affect the hypermethylation pattern (113) and possibly give rise to synergistic effects between the genetic and epigenetic changes when it comes to hTERT expression.
Alternative Lengthening of Telomeres
Some tumors utilize another mechanism of telomere maintenance than hTERT, termed alternative lengthening of telomeres (ALT), documented in <5% of all cancers (114). This mechanism is important as tumors that harbor the ALT phenotype would likely not benefit from hTERT vaccination. ALT appears to be more regularly employed in non-epithelial cancers such as sarcomas and brain tumors, and mutations in the telomere binding proteins ATRX and DAXX have been described to induce the ALT phenotype (115). hTERT promoter and ATRX mutations are mutually exclusive, indicating that hTERT activation and ALT do not co-occur in the same tumor (104).
Although these findings indicate hTERT and ALT being two distinct telomere maintenance pathways, the transfection of hTERT into an ALT-utilizing cell line has shown that these two mechanisms can run in parallel. However, when a telomerase-positive cell line is fused with an ALT-utilizing cell line, ALT is repressed, indicating that a factor other than hTERT represses ALT in non-transfected models (116). The possibility of ALT activation as a mechanism of resistance from hTERT inhibition has been evaluated in cancer cell line models, and they did indeed show that cells surviving the telomere crisis after telomerase inhibition could elongate the telomeres in an ALT-like manner. However, resistant, telomerase-negative cancer cells were significantly less invasive and tumorigenic (117, 118). These findings are in line with the described tumorigenic contribution of hTERT besides telomere elongation (119).
Although the direct inhibition of hTERT may lead to resistance through activation of the ALT pathway, the immune pressure imposed on tumors through vaccination likely avoids this type of resistance mechanism. CD4+ T cells are activated upon interaction with its cognate antigen on HLA class II expressing cells, such as APCs. APCs scavenge the TME and phagocytose remnants of dying cancer cells. They present this content to CD4+ T cells, which in turn release inflammatory cytokines and stimulate other immune cells (120). Thus, this indirect and dynamic approach to enhance anti-tumor immune responses likely circumvents tumor resistance by mechanisms typically seen with direct inhibition, such as tyrosine kinase inhibitors. More plausible resistance mechanisms are likely similar to those of acquired resistance to CPIs, characterized by evasion from the immune system through disruption of shared pathways of immune activation (e.g., tumor loss of HLA, B2M, and IFN-γ signaling) (121).
Immune Permissive Tumor Microenvironment
The anti-tumor effect of vaccine-induced T cells likely relies on their potential to home to the tumor or tumor-draining lymph node. Multiple mechanisms are exploited by the tumor to restrict the infiltrative potential of T cells (122) and could thus limit the anti-tumor efficacy of TCVs. Tumors can be characterized into three broad categories based on the presence of tumor-infiltrating lymphocytes (TILs) and immunosuppressive factors in the TME, or the so-called immunophenotype (123). These categories include the inflamed, excluded, and desert phenotypes. Inflamed tumors have infiltration and activation of immune cells, marked by elevated PD-L1 expression and IFN-γ signaling, and they typically respond well to CPI therapy. Immune excluded tumors have an abundance of immunosuppressive factors such as TGF-β and MDSCs. Both the inflamed and excluded phenotypes can harbor TILs, but at a much lower level in the excluded phenotype. The immune desert phenotype has few TILs and is characterized by elevated WNT/β-catenin signaling and fatty acid metabolism.
PD-L1 expression and IFN-γ signaling provide evidence of infiltration and activation of T cells and indicate an immune permissive TME. However, a lack of these features may not accurately determine the T cells’ infiltrative potential in these tumors, as evidenced by responses to CPIs in TMB high and PD-L1 low tumors. Thus, no accurate predictive marker exists to determine an immune permissive TME, and variations between the inflamed, excluded, and desert immunophenotypes exist both within and across tumors, making the selection of exclusively permissive tumor types difficult (124). Lastly, the tumor consists of a dynamic environment that may be affected by TCV-induced T cells overcoming immunosuppression via intratumoral activation of CD4 T cells and secretion of pro-inflammatory cytokines. However, tumor types where CPIs have demonstrated clinical responses may be the best and easiest guide for selecting immune permissive tumors.
Immunologically Rational Treatment Combinations
Although telomerase vaccines have proven to induce immune responses in blood, the T cell population expanded through vaccination is likely constrained by intrinsic and tumor-induced regulatory mechanisms, such as the checkpoint molecules CTLA-4 and PD-1/L1, respectively, and a varying degree of immunosuppression within the TME (depending on immunophenotype). Previous late-phase TCV trials may have failed due to the lack of appropriate treatment combinations addressing these regulatory mechanisms or the immunosuppressive milieu of the tumor. Therefore, it is necessary to leverage the recent advancements in immunotherapy and combine TCVs with checkpoint inhibitors or other therapeutic molecules modulating the TME in favor of T cell expansion, infiltration, and effector function.
The CTLA-4 checkpoint primarily acts to regulate the expansion of activated T cells by competitive inhibition of the binding of CD28 on the T cells with B7 ligands on the APCs, thereby disrupting co-stimulation of primed T cells (125). In the TCV setting, the systemic administration of an anti-CTLA-4 monoclonal antibody may provide not only enhanced expansion of spontaneously primed T cells in the tumor-draining lymph node, but also vaccine-induced T cells in the lymph node draining the vaccination site. Thus, the combination of anti-CTLA-4 and a TCV may allow increased expansion of vaccine-induced T cells after priming, addressing a central challenge when targeting TAAs. Only one completed clinical trial has combined the anti-CTLA-4 monoclonal antibody ipilimumab with a telomerase TCV (NCT02275416). Results from this trial evaluating UV1 combined with ipilimumab in advanced melanoma were presented at ASCO-SITC 2020 and showed early induction of immune responses in 10/11 (91%) of the evaluable patients (74).
The PD-1 immune checkpoint is upregulated on T cells upon activation, and its ligand PD-L1/L2 is upregulated on tumor cells in response to inflammatory cytokines (IFN-γ). This axis thus serves to restrict the effector capacity of T cells within the tumor by promoting T cell anergy and exhaustion (125). By blocking this interaction, the vaccine-induced T cells may achieve greater effector activity and tumor cell killing. The anti-tumor synergy of vaccination and dual checkpoint blockade has previously been demonstrated in animal models (126–128).
There are at least nine ongoing clinical trials investigating hTERT vaccines with an anti-PD-1/L1 monoclonal antibody (Table 2). The telomerase peptide vaccine UV1 is combined with pembrolizumab (anti-PD-1) in a phase I clinical trial (NCT03538314) and with nivolumab (anti-PD-1) and ipilimumab in two randomized phase II clinical trials in malignant melanoma (NCT04382664) and mesothelioma (NCT04300244), respectively. Two more studies that have yet to begin patient recruitment are investigating UV1 in combination with durvalumab (anti-PD-L1) and olaparib (PARP inhibitor) in relapsed ovarian cancer and with pembrolizumab in head and neck cancer. The peptide vaccine UCPVax is combined with nivolumab in a randomized phase II clinical trial (NCT04263051) and with atezolizumab (anti-PD-L1) in HPV+ cancers (NCT03946358). INO-5401, a DNA vaccine targeting Wilms tumor gene-1 (WT1), prostate-specific membrane antigen (PSMA), and hTERT, is evaluated in two phase I/IIa studies, in combination with atezolizumab in a urothelial carcinoma study (NCT03502785) and in combination with cemiplimab (anti-PD-L1) in newly diagnosed glioblastoma (NCT03491683), respectively. Although the combination with anti-CTLA-4 and anti-PD-1/L1 may be superior in expanding vaccine-induced T cells, this combination also poses a severe toxicity profile. Therefore, adding a TCV must not significantly exacerbate toxicity to achieve a feasible risk/benefit profile for the combination treatment.
Treatment with inhibitors of vascular endothelial growth factor (VEGF) can normalize the vasculature to allow increased infiltration of T cells, synergizing with adoptive cell transfer (129), and conceivably also TCVs (130). Furthermore, the immunosuppressive cytokine TGF-β is involved in modulating the immune excluded TME and can drive the differentiation of primed CD4+ T cells to the immunosuppressive T(reg) subtype (131–133). Recently, TGF- β has been identified to selectively suppress CD4+ Th2 cells (134). Targeting these suppressive TME factors, or so-called cancer environment immunotherapy, may be a novel approach that synergizes with TCVs (135–137).
Telomerase-Based TCVs in Clinical Development
Several anti-telomerase vaccine candidates have been evaluated in clinical trials during the last two decades (Table 1). Opportunities for scientifically rational treatment combinations (e.g., checkpoint inhibitors) have provided renewed interest in TCVs targeting telomerase with 13 ongoing trials (Table 2). Novel vaccine platforms have also emerged in the hTERT TCV pipeline to include DNA and RNA-based vaccines. TCVs that are either in active development or have been evaluated in more than one trial are described in more detail below.
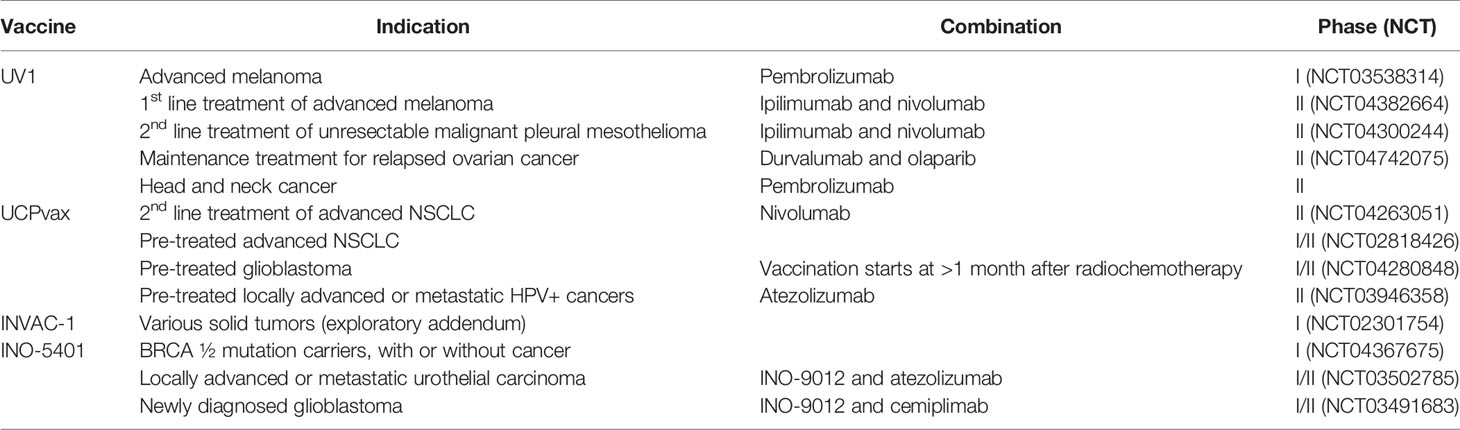
Table 2 Several hTERT targeting TCVs are currently evaluated in active clinical trials and are often combined with different checkpoint inhibitors.
GV1001
The only hTERT-based vaccine with market approval is the telomerase vaccine is GV1001, a 16-mer peptide covering the active site of hTERT. GV1001 was also one of the first telomerase-based TCVs evaluated in the clinic and has been assessed in 7 clinical trials, covering pancreatic cancer, melanoma, NSCLC, and hepatocellular carcinoma. GV1001 vaccination encompasses intradermal injection of 0.56 mg of vaccine peptide and 75μg of GM-CSF as an adjuvant. The treatment schedule consists of 3 vaccinations during week 1, and one vaccination on weeks 2, 3, 4, and 6, and monthly vaccinations thereafter.
GV1001 was evaluated in the only phase III trial with hTERT-targeting TCVs to date, assessing chemotherapy with or without GV1001 in patients with locally advanced or metastatic pancreatic cancer (1). The study did not meet its primary endpoint of improved overall survival with the addition of GV1001 to chemotherapy. There are several possible causes for the failure to meet the primary endpoint in this trial. First, the immune response rate was substantially lower than expected, at 38%. This low rate essentially reduces the population eligible for a clinical effect of vaccination by almost 2/3. Second, as previously argued, immunologically rational treatment combinations are likely necessary to bring out the true clinical potential of TCVs, and even though chemotherapy may induce immunogenic cancer cell death, it is uncertain which effect it has on a vaccine’s ability to induce appropriate T cells responses. Third, pancreatic cancer is known to be desmoplastic with a high concentration of cancer-associated fibroblasts inducing an immunosuppressive TME, limiting the potential for immunotherapy (138), as supported by a lack of efficacy of anti-PD-L1 in pancreatic cancer (139). hTERT expression in pancreatic cancer is also relatively low compared to other cancer types (Figure 1). Although the study failed to achieve an OS benefit in the intention-to-treat population, retrospective subgroup analysis showed that eotaxin levels predicted the benefit of the addition of GV1001 to chemotherapy (140). Based on these data, GV1001 received conditional market approval in Korea for patients with locally advanced or metastatic pancreatic cancer and an elevated serum eotaxin level. Eotaxins function as chemoattractants for immune cells and could thus enhance infiltration of T cells to the tumor. However, the mechanistic link and its predictive value need confirmation in future studies.
UV1
In the CTN-2000 trial, which evaluated GV1001 in NSCLC (n=26) (63), patients who mounted a vaccine-specific immune response had an improved overall survival (OS) compared to non-immune responders (median 19 months vs. 3.5 months; P < 0.001). Inderberg et al. subsequently characterized the immune response in long vs. short-term survivors to elucidate immunological mechanisms characteristic for patients with a clinical benefit (52). They evaluated immune responses in patients using a library of overlapping peptides, 24 15-mers and one 30-mer, covering the active site of hTERT. Long-term surviving immune responders demonstrated so-called intramolecular epitope spreading, i.e., induction of de novo immune responses against other, structurally unrelated, epitopes within the hTERT molecule. Moreover, immune responses against specific hTERT peptides were correlated with survival benefit and were not detected in short-term survivors. Resultantly, three highly immunogenic epitope dense peptides with broad HLA-coverage associated with improved survival were selected for a next-generation telomerase vaccine, UV1. These peptides were relatively long (two 15-mers and one 30-mer), requiring intracellular processing by the APCs, allowing individual selection of epitopes matching patient-specific HLA-alleles, thereby ensuring wide population coverage and CD8+ and CD4+ immune responses. The UV1 vaccine is administered intradermally at 300 μg of peptides with 75 μg GM-CSF as an adjuvant. The treatment schedule consists of 3 vaccinations during week 1 and up to 5 booster vaccinations thereafter.
INVAC-1
INVAC-1 is a DNA-based vaccine containing a modified and enzymatically inert hTERT gene variant that can be administered by intradermal injection and give rise to hTERT protein exposure in vaccinated individuals. The vaccine has been modified to improve protein degradation through a ubiquitin sequence introduction. In a recent publication presenting the phase I safety readout, the vaccine was found safe with no dose-limiting toxicity with vaccine-induced T cell expansion established (76).
VX-001
VX-001 is a peptide-based telomerase vaccine containing two 9 amino-acid peptides (one wild-type sequence and one mutated/optimized sequence) that aim to expand cytotoxic hTERT specific T cells in an HLA-A2 selected population. The peptides are formulated in Montanide and at low non-physiological pH. In a phase 2 study, the first and second vaccination was performed using the mutated/optimized sequence, and the third vaccination and onwards (four additional vaccinations) were performed using the wild type sequence. In patients with a tumor under control, vaccination was continued every 3 months. Despite the failure of meeting the primary endpoint in the non-selected cohort, there was a significantly improved overall survival in immune responders, and this was also established in immune responders with an otherwise unfavorable prognosis based on elevated LDH and γGT (141). This may indicate that the vaccine provided an effect and that the improved survival in immune responders was not only a result of a selection of patients that would survive longer regardless of treatment.
Future Perspectives
As the tumor develops, so do its mechanisms for evading the immune system, necessitating strategic treatment combinations to overcome tumor intrinsic or extrinsic immune escape mechanisms in the advanced disease setting. To fully harvest the potential of synergy by combinatorial treatment strategies, there is a need to understand the kinetics of each component to align for optimal efficacy. Traditionally, treatments are given simultaneously to impose the greatest pressure on the tumor. However, with immunotherapeutic approaches, there could be additional gains achieved by appropriate timing, as demonstrated for sequential administration of a tumor cell vaccine and anti-CTLA mAb (142) and concurrent administration of anti-PD-1 and vaccination (143). A further understanding of how to best combine various immunotherapies will be essential in future testing. To date, most hTERT-targeting TCVs have been evaluated in advanced disease and in heavily pre-treated patients. As hTERT is a relevant antigen along the cancer disease continuum, these TCVs could be employed in earlier disease settings, perhaps with reduced reliance on treatment combinations. Indeed, there are studies showing promising results of monotherapy TCVs in patients with low tumor burden (144–147).
No clinical trials have yet incorporated relevant target biomarkers, such as tumor hTERT expression. Such biomarkers could potentially allow a narrower selection of patients eligible for effect from vaccination and thus improve on clinical efficacy. Another interesting approach is the application of hTERT-targeting TCVs in TMB-low tumors, where CPIs show limited effect and other TCV strategies such as neoantigen vaccines are less relevant. Biomarkers of possible resistance should also be considered for future studies. Such predictive biomarkers can include tumor loss of function mutations in HLA, B2M, and IFN-γ signaling.
Conclusion
Although telomerase vaccines have been under investigation for almost two decades, recent studies elucidating the mechanisms behind the lack of effect from CPIs provide renewed enthusiasm for TCVs, in general, as a means to improve clinical outcomes. Telomerase as a TCV target has apparent advantages due to its universal presence and essential function in almost all cancer types, providing spatiotemporal relevance to the induced immune response and limiting possible escape mechanisms for the tumor.
TCVs should robustly elicit both the CD4+ and CD8+ compartments of the adaptive immune system for optimal intratumoral activity of the induced immune response. Higher tumor expression of telomerase is likely to confer a heightened anti-tumor immune response in vaccinated patients. Several factors are involved in regulating telomerase expression, with hTERT promoter mutations being the most well described. Furthermore, BRAF mutations have been shown to act synergistically with hTERT promoter mutations to increase telomerase activity. Considering the high frequency of hTERT promoter mutations, BRAF mutations, and copy number amplification of the hTERT gene in melanoma, patients with this cancer are more likely to achieve benefit from vaccination.
Immunologically rational combinations, such as anti-CTLA-4 and anti-PD-1/L1, are likely necessary to bring out the true clinical potential of hTERT-targeting TCVs. There are already several phase II randomized controlled trials evaluating hTERT targeting TCVs in combination with CPIs with anticipated read-outs. The tumor type targeted should be assessed for its microenvironment as multiple factors, such as TGF-β, are to a varying degree contributing to local immunosuppression across tumor types. Although highly immunosuppressive tumors are likely to be more challenging to target, novel compounds addressing these tumor environmental factors are emerging and could possibly provide synergistic effects with vaccination.
Development of better methodologies to evaluate immune responses in patients is needed and should provide a more comprehensive quantification of the induced immune responses and insights into the optimal phenotype of the T cells. Additional translational studies on the intratumoral activity of the induced T cells would strengthen the rationale for further development of hTERT targeting TCVs.
Author Contributions
EBE and SM have reviewed the field. EH and GG have provided guidance on relevant considerations with respect to tumor biology and anti-tumor immunity. All authors contributed to the article and approved the submitted version.
Funding
The Norwegian Research Councils grant number is 298864 and Ultimovacs ASA fund the Ph.D. project for the corresponding author. The funder was not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication. The University of Oslo funds the article processing fee.
Conflict of Interest
EBE and GG are employees of Ultimovacs ASA. SM and GG are shareholders in Ultimovacs ASA. SM is a founder, shareholder, and board member of Immuneed AB, Vivologica AB, and Strike pharma AB and an employee of Ultimovacs AB. GG is an inventor of a UV1 vaccine patent.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Middleton G, Silcocks P, Cox T, Valle J, Wadsley J, Propper D, et al. Gemcitabine and Capecitabine With or Without Telomerase Peptide Vaccine GV1001 in Patients With Locally Advanced or Metastatic Pancreatic Cancer (TeloVac): An Open-Label, Randomised, Phase 3 Trial. Lancet Oncol (2014) 15(8):829–40. doi: 10.1016/s1470-2045(14)70236-0
2. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved Survival With Ipilimumab in Patients With Metastatic Melanoma. New Engl J Med (2010) 363(8):711–23. doi: 10.1056/NEJMoa1003466
3. Upadhaya S, Hubbard-Lucey VM, Yu JX. Immuno-Oncology Drug Development Forges on Despite COVID-19. Nat Rev Drug Discov (2020) 19(11):751–2. doi: 10.1038/d41573-020-00166-1
4. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Rutkowski P, Lao CD, et al. Five-Year Survival With Combined Nivolumab and Ipilimumab in Advanced Melanoma. N Engl J Med (2019) 381(16):1535–46. doi: 10.1056/NEJMoa1910836
5. Robert C, Ribas A, Schachter J, Arance A, Grob J-J, Mortier L, et al. Pembrolizumab Versus Ipilimumab in Advanced Melanoma (KEYNOTE-006): Post-Hoc 5-Year Results From an Open-Label, Multicentre, Randomised, Controlled, Phase 3 Study. Lancet Oncol (2019) 20(9):1239–51. doi: 10.1016/s1470-2045(19)30388-2
6. Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 Pathway Blockade for Cancer Therapy: Mechanisms, Response Biomarkers, and Combinations. Sci Trans Med (2016) 8(328):328rv4–rv4. doi: 10.1126/scitranslmed.aad7118
7. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJM, Robert L, et al. PD-1 Blockade Induces Responses by Inhibiting Adaptive Immune Resistance. Nature (2014) 515(7528):568–71. doi: 10.1038/nature13954
8. Kim JY, Kronbichler A, Eisenhut M, Hong SH, van der Vliet HJ, Kang J, et al. Tumor Mutational Burden and Efficacy of Immune Checkpoint Inhibitors: A Systematic Review and Meta-Analysis. Cancers (Basel) (2019) 11(11):1798 doi: 10.3390/cancers11111798
9. McGranahan N, Furness AJS, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, et al. Clonal Neoantigens Elicit T Cell Immunoreactivity and Sensitivity to Immune Checkpoint Blockade. Science (2016) 351(6280):1463–9. doi: 10.1126/science.aaf1490
10. Jenkins RW, Barbie DA, Flaherty KT. Mechanisms of Resistance to Immune Checkpoint Inhibitors. Br J Cancer (2018) 118(1):9–16. doi: 10.1038/bjc.2017.434
11. Melero I, Gaudernack G, Gerritsen W, Huber C, Parmiani G, Scholl S, et al. Therapeutic Vaccines for Cancer: An Overview of Clinical Trials. Nat Rev Clin Oncol (2014) 11(9):509–24. doi: 10.1038/nrclinonc.2014.111
12. Gjerstorff MF, Burns J, Ditzel HJ. Cancer-Germline Antigen Vaccines and Epigenetic Enhancers: Future Strategies for Cancer Treatment. Expert Opin Biol Ther (2010) 10(7):1061–75. doi: 10.1517/14712598.2010.485188
13. Tarhini AA, Leng S, Moschos SJ, Yin Y, Sander C, Lin Y, et al. Safety and Immunogenicity of Vaccination With MART-1 (26–35, 27L), Gp100 (209–217, 210M), and Tyrosinase (368–376, 370D) in Adjuvant With PF-3512676 and GM-CSF in Metastatic Melanoma. J Immunother Cancer (2012) 35(4):359–66. doi: 10.1097/CJI.0b013e31825481fe
14. Kubler H, Scheel B, Gnad-Vogt U, Miller K, Schultze-Seemann W, Vom Dorp F, et al. Self-Adjuvanted Mrna Vaccination in Advanced Prostate Cancer Patients: A First-in-Man Phase I/IIa Study. J Immunother Cancer (2015) 3:26. doi: 10.1186/s40425-015-0068-y
15. Wargowski E, Johnson LE, Eickhoff JC, Delmastro L, Staab MJ, Liu G, et al. Prime-Boost Vaccination Targeting Prostatic Acid Phosphatase (PAP) in Patients With Metastatic Castration-Resistant Prostate Cancer (Mcrpc) Using Sipuleucel-T and a DNA Vaccine. J Immunother Cancer (2018) 6(1):21. doi: 10.1186/s40425-018-0333-y
16. Hammerstrom AE, Cauley DH, Atkinson BJ, Sharma P. Cancer Immunotherapy: Sipuleucel-T and Beyond. Pharmacotherapy: J Hum Pharmacol Drug Ther (2011) 31(8):813–28. doi: 10.1592/phco.31.8.813
17. Dosset M, Castro A, Carter H, Zanetti M. Telomerase and CD4 T Cell Immunity in Cancer. Cancers (Basel) (2020) 12(6):1687. doi: 10.3390/cancers12061687
18. Brown TA, Mittendorf EA, Hale DF, Myers JW, Peace KM, Jackson DO, et al. Prospective, Randomized, Single-Blinded, Multi-Center Phase II Trial of Two HER2 Peptide Vaccines, GP2 and AE37, in Breast Cancer Patients to Prevent Recurrence. Breast Cancer Res Treat (2020) 181(2):391–401. doi: 10.1007/s10549-020-05638-x
19. Zheng X, Gao JX, Zhang H, Geiger TL, Liu Y, Zheng P. Clonal Deletion of Simian Virus 40 Large T Antigen-Specific T Cells in the Transgenic Adenocarcinoma of Mouse Prostate Mice: An Important Role for Clonal Deletion in Shaping the Repertoire of T Cells Specific for Antigens Overexpressed in Solid Tumors. J Immunol (2002) 169(9):4761–9. doi: 10.4049/jimmunol.169.9.4761
20. Sahin U, Derhovanessian E, Miller M, Kloke BP, Simon P, Lower M, et al. Personalized RNA Mutanome Vaccines Mobilize Poly-Specific Therapeutic Immunity Against Cancer. Nature (2017) 547(7662):222–6. doi: 10.1038/nature23003
21. Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, et al. An Immunogenic Personal Neoantigen Vaccine for Patients With Melanoma. Nature (2017) 547(7662):217–21. doi: 10.1038/nature22991
22. Maciejowski J, de Lange T. Telomeres in Cancer: Tumour Suppression and Genome Instability. Nat Rev Mol Cell Biol (2017) 18(3):175–86. doi: 10.1038/nrm.2016.171
23. Leão R, Apolónio JD, Lee D, Figueiredo A, Tabori U, Castelo-Branco P. Mechanisms of Human Telomerase Reverse Transcriptase (hTERT) Regulation: Clinical Impacts in Cancer. J Biomed Sci (2018) 25(1):22. doi: 10.1186/s12929-018-0422-8
24. Hayflick L, Moorhead PS. The Serial Cultivation of Human Diploid Cell Strains. Exp Cell Res (1961) 25:585–621. doi: 10.1016/0014-4827(61)90192-6
25. Hiyama E, Hiyama K, Yokoyama T, Shay JW. Immunohistochemical Detection of Telomerase (hTERT) Protein in Human Cancer Tissues and a Subset of Cells in Normal Tissues. Neoplasia (2001) 3(1):17–26. doi: 10.1038/sj.neo.7900134
26. Hiyama E, Hiyama K. Telomere and Telomerase in Stem Cells. Br J Cancer (2007) 96(7):1020–4. doi: 10.1038/sj.bjc.6603671
27. Shay JW, Bacchetti S. A Survey of Telomerase in Human Cancer. Eur J Cancer (1997) 33(5):787–91. doi: 10.1016/S0959-8049(97)00062-2
28. Kim N, Piatyszek M, Prowse K, Harley C, West M, Ho P, et al. Specific Association of Human Telomerase Activity With Immortal Cells and Cancer. Science (1994) 266(5193):2011–5. doi: 10.1126/science.7605428
29. Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
30. Liu Z, Li Q, Li K, Chen L, Li W, Hou M, et al. Telomerase Reverse Transcriptase Promotes Epithelial-Mesenchymal Transition and Stem Cell-Like Traits in Cancer Cells. Oncogene (2013) 32(36):4203–13. doi: 10.1038/onc.2012.441
31. Hannen R, Bartsch JW. Essential Roles of Telomerase Reverse Transcriptase Htert in Cancer Stemness and Metastasis. FEBS Lett (2018) 592(12):2023–31. doi: 10.1002/1873-3468.13084
32. Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: A Web Server for Cancer and Normal Gene Expression Profiling and Interactive Analyses. Nucleic Acids Res (2017) 45(W1):W98–W102. doi: 10.1093/nar/gkx247
33. Taga S, Osaki T, Ohgami A, Imoto H, Yasumoto K. Prognostic Impact of Telomerase Activity in non-Small Cell Lung Cancers. Ann Surg (1999) 230(5):715–20. doi: 10.1097/00000658-199911000-00015
34. Bertorelle R, Briarava M, Rampazzo E, Biasini L, Agostini M, Maretto I, et al. Telomerase is an Independent Prognostic Marker of Overall Survival in Patients With Colorectal Cancer. Br J Cancer (2013) 108(2):278–84. doi: 10.1038/bjc.2012.602
35. Clark GM, Osborne CK, Levitt D, Wu F, Kim NW. Telomerase Activity and Survival of Patients With Node-Positive Breast Cancer. JNCI: J Natl Cancer Institute (1997) 89(24):1874–81. doi: 10.1093/jnci/89.24.1874
36. Juratli TA, Thiede C, Koerner MVA, Tummala SS, Daubner D, Shankar GM, et al. Intratumoral Heterogeneity and TERT Promoter Mutations in Progressive/Higher-Grade Meningiomas. Oncotarget (2017) 8(65):109228–37. doi: 10.18632/oncotarget.22650
37. Hugdahl E, Kalvenes MB, Mannelqvist M, Ladstein RG, Akslen LA. Prognostic Impact and Concordance of TERT Promoter Mutation and Protein Expression in Matched Primary and Metastatic Cutaneous Melanoma. Br J Cancer (2018) 118(1):98–105. doi: 10.1038/bjc.2017.384
38. Laheurte C, Dosset M, Vernerey D, Boullerot L, Gaugler B, Gravelin E, et al. Distinct Prognostic Value of Circulating Anti-Telomerase CD4(+) Th1 Immunity and Exhausted PD-1(+)/TIM-3(+) T Cells in Lung Cancer. Br J Cancer (2019) 121(5):405–16. doi: 10.1038/s41416-019-0531-5
39. Negrini S, De Palma R, Filaci G. Anti-Cancer Immunotherapies Targeting Telomerase. Cancers (2020) 12(8):2260. doi: 10.3390/cancers12082260
40. Lee MY, Jeon JW, Sievers C, Allen CT. Antigen Processing and Presentation in Cancer Immunotherapy. J Immunotherapy Cancer (2020) 8(2):e001111. doi: 10.1136/jitc-2020-001111
41. Seliger B, Kloor M, Ferrone S. HLA Class II Antigen-Processing Pathway in Tumors: Molecular Defects and Clinical Relevance. OncoImmunology (2017) 6(2):e1171447. doi: 10.1080/2162402X.2016.1171447
42. Melssen M, Slingluff CL Jr. Vaccines Targeting Helper T Cells for Cancer Immunotherapy. Curr Opin Immunol (2017) 47:85–92. doi: 10.1016/j.coi.2017.07.004
43. Borst J, Ahrends T, Babala N, Melief CJM, Kastenmuller W. CD4(+) T Cell Help in Cancer Immunology and Immunotherapy. Nat Rev Immunol (2018) 18(10):635–47. doi: 10.1038/s41577-018-0044-0
44. Tay RE, Richardson EK, Toh HC. Revisiting the Role of CD4(+) T Cells in Cancer Immunotherapy-New Insights Into Old Paradigms. Cancer Gene Ther (2020) 28(1):5–17. doi: 10.1038/s41417-020-0183-x
45. Wong SB, Bos R, Sherman LA. Tumor-Specific CD4+ T Cells Render the Tumor Environment Permissive for Infiltration by Low-Avidity CD8+ T Cells. J Immunol (2008) 180(5):3122–31. doi: 10.4049/jimmunol.180.5.3122
46. Tran E, Turcotte S, Gros A, Robbins PF, Lu Y-C, Dudley ME, et al. Cancer Immunotherapy Based on Mutation-Specific CD4+ T Cells in a Patient With Epithelial Cancer. Science (2014) 344(6184):641–5. doi: 10.1126/science.1251102
47. Oh DY, Kwek SS, Raju SS, Li T, McCarthy E, Chow E, et al. Intratumoral CD4(+) T Cells Mediate Anti-Tumor Cytotoxicity in Human Bladder Cancer. Cell (2020) 181(7):1612–25 e13. doi: 10.1016/j.cell.2020.05.017
48. Haabeth OA, Tveita AA, Fauskanger M, Schjesvold F, Lorvik KB, Hofgaard PO, et al. How do CD4(+) T Cells Detect and Eliminate Tumor Cells That Either Lack or Express MHC Class II Molecules? Front Immunol (2014) 5:174. doi: 10.3389/fimmu.2014.00174
49. Janssen EM, Droin NM, Lemmens EE, Pinkoski MJ, Bensinger SJ, Ehst BD, et al. CD4 T-Cell Help Controls CD8 T-Cell Memory via TRAIL-mediated activation-induced cell death. Nature (2005) 434(7029):88–93. doi: 10.1038/nature03337
50. Laidlaw BJ, Craft JE, Kaech SM. The Multifaceted Role of CD4(+) T Cells in CD8(+) T Cell Memory. Nat Rev Immunol (2016) 16(2):102–11. doi: 10.1038/nri.2015.10
51. Chen DS, Mellman I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity (2013) 39(1):1–10. doi: 10.1016/j.immuni.2013.07.012
52. Inderberg-Suso EM, Trachsel S, Lislerud K, Rasmussen AM, Gaudernack G. Widespread CD4+ T-Cell Reactivity to Novel hTERT Epitopes Following Vaccination of Cancer Patients With a Single hTERT Peptide GV1001. Oncoimmunology (2012) 1(5):670–86. doi: 10.4161/onci.20426
53. Dobrzanski M. Expanding Roles for CD4 T Cells and Their Subpopulations in Tumor Immunity and Therapy. Front Oncol (2013) 3(63):63. doi: 10.3389/fonc.2013.00063
54. Geginat J, Paroni M, Maglie S, Alfen JS, Kastirr I, Gruarin P, et al. Plasticity of Human CD4 T Cell Subsets. Front Immunol (2014) 5(630):630. doi: 10.3389/fimmu.2014.00630
55. Hansen GL, Gaudernack G, Brunsvig PF, Cvancarova M, Kyte JA. Immunological Factors Influencing Clinical Outcome in Lung Cancer Patients After Telomerase Peptide Vaccination. Cancer Immunol Immunother (2015) 64(12):1609–21. doi: 10.1007/s00262-015-1766-5
56. Clausen BE, Stoitzner P. Functional Specialization of Skin Dendritic Cell Subsets in Regulating T Cell Responses. Front Immunol (2015) 6(534):534. doi: 10.3389/fimmu.2015.00534
57. Maeurer MJ, Storkus WJ, Kirkwood JM, Lotze MT. New Treatment Options for Patients With Melanoma: Review of Melanoma-Derived T-Cell Epitope-Based Peptide Vaccines. Melanoma Res (1996) 6(1):11–24. doi: 10.1097/00008390-199602000-00003
58. Rosalia RA, Quakkelaar ED, Redeker A, Khan S, Camps M, Drijfhout JW, et al. Dendritic Cells Process Synthetic Long Peptides Better Than Whole Protein, Improving Antigen Presentation and T-Cell Activation. Eur J Immunol (2013) 43(10):2554–65. doi: 10.1002/eji.201343324
59. Zhang H, Hong H, Li D, Ma S, Di Y, Stoten A, et al. Comparing Pooled Peptides With Intact Protein for Accessing Cross-Presentation Pathways for Protective CD8+ and CD4+ T Cells. J Biol Chem (2009) 284(14):9184–91. doi: 10.1074/jbc.M809456200
60. Kyte JA, Gaudernack G, Dueland S, Trachsel S, Julsrud L, Aamdal S. Telomerase Peptide Vaccination Combined With Temozolomide: A Clinical Trial in Stage IV Melanoma Patients. Clin Cancer Res (2011) 17(13):4568–80. doi: 10.1158/1078-0432.CCR-11-0184
61. Hunger RE, Kernland Lang K, Markowski CJ, Trachsel S, Møller M, Eriksen JA, et al. Vaccination of Patients With Cutaneous Melanoma With Telomerase-Specific Peptides. Cancer Immunol Immunotherapy (2011) 60(11):1553. doi: 10.1007/s00262-011-1061-z
62. Bernhardt SL, Gjertsen MK, Trachsel S, Moller M, Eriksen JA, Meo M, et al. Telomerase Peptide Vaccination of Patients With non-Resectable Pancreatic Cancer: A Dose Escalating Phase I/II Study. Br J Cancer (2006) 95(11):1474–82. doi: 10.1038/sj.bjc.6603437
63. Brunsvig PF, Kyte JA, Kersten C, Sundstrom S, Moller M, Nyakas M, et al. Telomerase Peptide Vaccination in NSCLC: A Phase II Trial in Stage III Patients Vaccinated After Chemoradiotherapy and an 8-Year Update on a Phase I/II Trial. Clin Cancer Res (2011) 17(21):6847–57. doi: 10.1158/1078-0432.CCR-11-1385
64. Brunsvig PF, Aamdal S, Gjertsen MK, Kvalheim G, Markowski-Grimsrud CJ, Sve I, et al. Telomerase Peptide Vaccination: A Phase I/II Study in Patients With non-Small Cell Lung Cancer. Cancer Immunol Immunother (2006) 55(12):1553–64. doi: 10.1007/s00262-006-0145-7
65. Greten TF, Forner A, Korangy F, N’Kontchou G, Barget N, Ayuso C, et al. A Phase II Open Label Trial Evaluating Safety and Efficacy of a Telomerase Peptide Vaccination in Patients With Advanced Hepatocellular Carcinoma. BMC Cancer (2010) 10:209–. doi: 10.1186/1471-2407-10-209
66. Parkhurst MR, Riley JP, Igarashi T, Li Y, Robbins PF, Rosenberg SA. Immunization of Patients With the hTERT:540-548 Peptide Induces Peptide-Reactive T Lymphocytes That do Not Recognize Tumors Endogenously Expressing Telomerase. Clin Cancer Res (2004) 10(14):4688–98. doi: 10.1158/1078-0432.Ccr-04-0325
67. Bolonaki I, Kotsakis A, Papadimitraki E, Aggouraki D, Konsolakis G, Vagia A, et al. Vaccination of Patients With Advanced non–Small-Cell Lung Cancer With an Optimized Cryptic Human Telomerase Reverse Transcriptase Peptide. J Clin Oncol (2007) 25(19):2727–34. doi: 10.1200/JCO.2006.10.3465
68. Kotsakis A, Papadimitraki E, Vetsika EK, Aggouraki D, Dermitzaki EK, Hatzidaki D, et al. A Phase II Trial Evaluating the Clinical and Immunologic Response of HLA-A2(+) non-Small Cell Lung Cancer Patients Vaccinated With an hTERT Cryptic Peptide. Lung Cancer (Amsterdam Netherlands) (2014) 86(1):59–66. doi: 10.1016/j.lungcan.2014.07.018
69. Mavroudis D, Bolonakis I, Cornet S, Myllaki G, Kanellou P, Kotsakis A, et al. A Phase I Study of the Optimized Cryptic Peptide TERT 572Y in Patients With Advanced Malignancies. Oncology (2006) 70(4):306–14. doi: 10.1159/000096252
70. Kotsakis A, Vetsika EK, Christou S, Hatzidaki D, Vardakis N, Aggouraki D, et al. Clinical Outcome of Patients With Various Advanced Cancer Types Vaccinated With an Optimized Cryptic Human Telomerase Reverse Transcriptase (TERT) Peptide: Results of an Expanded Phase II Study. Ann Oncol (2012) 23(2):442–9. doi: 10.1093/annonc/mdr396
71. Fenoglio D, Traverso P, Parodi A, Tomasello L, Negrini S, Kalli F, et al. A Multi-Peptide, Dual-Adjuvant Telomerase Vaccine (GX301) is Highly Immunogenic in Patients With Prostate and Renal Cancer. Cancer Immunol Immunotherapy (2013) 62(6):1041–52. doi: 10.1007/s00262-013-1415-9
72. Lilleby W, Gaudernack G, Brunsvig PF, Vlatkovic L, Schulz M, Mills K, et al. Phase I/IIa Clinical Trial of a Novel hTERT Peptide Vaccine in Men With Metastatic Hormone-Naive Prostate Cancer. Cancer Immunol Immunother (2017) 66(7):891–901. doi: 10.1007/s00262-017-1994-y
73. Brunsvig PF, Guren TK, Nyakas M, Steinfeldt-Reisse CH, Rasch W, Kyte JA, et al. Long-Term Outcomes of a Phase I Study With UV1, a Second Generation Telomerase Based Vaccine, in Patients With Advanced non-Small Cell Lung Cancer. Front Immunol (2020) 11(3088):572172. doi: 10.3389/fimmu.2020.572172
74. Ellingsen EB, Aamdal E, Inderberg EM, Rasch W, Brunsvig P, Aamdal S, et al. A Phase I/IIa Clinical Trial Investigating the Therapeutic Cancer Vaccine UV1 in Combination With Ipilimumab in Patients With Malignant Melanoma: Four-Year Survival Update. J Clin Oncol (2020) 38(5_suppl):62. doi: 10.1200/JCO.2020.38.5_suppl.62
75. Rittig SM, Haentschel M, Weimer KJ, Heine A, Muller MR, Brugger W, et al. Intradermal Vaccinations With RNA Coding for TAA Generate CD8+ and CD4+ Immune Responses and Induce Clinical Benefit in Vaccinated Patients. Mol Ther (2011) 19(5):990–9. doi: 10.1038/mt.2010.289
76. Teixeira L, Medioni J, Garibal J, Adotevi O, Doucet L, Durey M-AD, et al. A First-in-Human Phase I Study of INVAC-1, an Optimized Human Telomerase DNA Vaccine in Patients With Advanced Solid Tumors. Clin Cancer Res (2020) 26(3):588–97. doi: 10.1158/1078-0432.Ccr-19-1614
77. Mantia-Smaldone GM. Chu CS. A Review of Dendritic Cell Therapy for Cancer: Progress and Challenges. BioDrugs (2013) 27(5):453–68. doi: 10.1007/s40259-013-0030-9
78. Suschak JJ, Williams JA, Schmaljohn CS. Advancements in DNA Vaccine Vectors, non-Mechanical Delivery Methods, and Molecular Adjuvants to Increase Immunogenicity. Hum Vaccines Immunotherapeutics (2017) 13(12):2837–48. doi: 10.1080/21645515.2017.1330236
79. Jorritsma SHT, Gowans EJ, Grubor-Bauk B, Wijesundara DK. Delivery Methods to Increase Cellular Uptake and Immunogenicity of DNA Vaccines. Vaccine (2016) 34(46):5488–94. doi: 10.1016/j.vaccine.2016.09.062
80. Dillard P, Koksal H, Maggadottir SM, Winge-Main A, Pollmann S, Menard M, et al. Targeting Telomerase With an HLA Class II-Restricted TCR for Cancer Immunotherapy. Mol Ther (2020) 29(3):1199–213. doi: 10.1016/j.ymthe.2020.11.019
81. Shi Y, Liu CH, Roberts AI, Das J, Xu G, Ren G, et al. Granulocyte-Macrophage Colony-Stimulating Factor (GM-CSF) and T-Cell Responses: What We do and Don’t Know. Cell Res (2006) 16(2):126–33. doi: 10.1038/sj.cr.7310017
82. Hailemichael Y, Woods A, Fu T, He Q, Nielsen MC, Hasan F, et al. Cancer Vaccine Formulation Dictates Synergy With CTLA-4 and PD-L1 Checkpoint Blockade Therapy. J Clin Invest (2018) 128(4):1338–54. doi: 10.1172/JCI93303
83. Bhattacharya P, Budnick I, Singh M, Thiruppathi M, Alharshawi K, Elshabrawy H, et al. Dual Role of GM-CSF as a Pro-Inflammatory and a Regulatory Cytokine: Implications for Immune Therapy. J Interferon Cytokine Res (2015) 35(8):585–99. doi: 10.1089/jir.2014.0149
84. Baharom F, Ramirez-Valdez RA, Tobin KKS, Yamane H, Dutertre CA, Khalilnezhad A, et al. Intravenous Nanoparticle Vaccination Generates Stem-Like TCF1(+) Neoantigen-Specific CD8(+) T Cells. Nat Immunol (2020) 22(1):41–52. doi: 10.1038/s41590-020-00810-3
85. Zom GG, Willems MMJHP, Khan S, van der Sluis TC, Kleinovink JW, Camps MGM, et al. Novel TLR2-Binding Adjuvant Induces Enhanced T Cell Responses and Tumor Eradication. J ImmunoTherapy Cancer (2018) 6(1):146. doi: 10.1186/s40425-018-0455-2
86. Kumar BV, Connors TJ, Farber DL. Human T Cell Development, Localization, and Function Throughout Life. Immunity (2018) 48(2):202–13. doi: 10.1016/j.immuni.2018.01.007
87. Buggert M, Vella LA, Nguyen S, Wu VH, Chen Z, Sekine T, et al. The Identity of Human Tissue-Emigrant CD8(+) T Cells. Cell (2020) 183(7):1946–61.e15. doi: 10.1016/j.cell.2020.11.019
88. Fujisaki J, Wu J, Carlson AL, Silberstein L, Putheti P, Larocca R, et al. In Vivo Imaging of Treg Cells Providing Immune Privilege to the Haematopoietic Stem-Cell Niche. Nature (2011) 474(7350):216–9. doi: 10.1038/nature10160
89. Zhao S, Zhu W, Xue S, Han D. Testicular Defense Systems: Immune Privilege and Innate Immunity. Cell Mol Immunol (2014) 11(5):428–37. doi: 10.1038/cmi.2014.38
90. Nancy P, Erlebacher A. T Cell Behavior at the Maternal-Fetal Interface. Int J Dev Biol (2014) 58(2-4):189–98. doi: 10.1387/ijdb.140054ae
91. Erlebacher A. Mechanisms of T Cell Tolerance Towards the Allogeneic Fetus. Nat Rev Immunol (2013) 13(1):23–33. doi: 10.1038/nri3361
92. Niederkorn JY. See No Evil, Hear No Evil, do No Evil: The Lessons of Immune Privilege. Nat Immunol (2006) 7(4):354–9. doi: 10.1038/ni1328
93. Fournel S, Aguerre-Girr M, Huc X, Lenfant F, Alam A, Toubert A, et al. Cutting Edge: Soluble HLA-G1 Triggers CD95/CD95 Ligand-Mediated Apoptosis in Activated CD8+ Cells by Interacting With CD8. J Immunol (2000) 164(12):6100–4. doi: 10.4049/jimmunol.164.12.6100
94. Danet-Desnoyers GA, Luongo JL, Bonnet DA, Domchek SM, Vonderheide RH. Telomerase Vaccination has No Detectable Effect on SCID-Repopulating and Colony-Forming Activities in the Bone Marrow of Cancer Patients. Exp Hematol (2005) 33(11):1275–80. doi: 10.1016/j.exphem.2005.07.011
95. Vonderheide RH, Domchek SM, Schultze JL, George DJ, Hoar KM, Chen D-Y, et al. Vaccination of Cancer Patients Against Telomerase Induces Functional Antitumor CD8 T Lymphocytes. Clin Cancer Res (2004) 10(3):828–39. doi: 10.1158/1078-0432.Ccr-0620-3
96. Vinagre J, Almeida A, Populo H, Batista R, Lyra J, Pinto V, et al. Frequency of TERT Promoter Mutations in Human Cancers. Nat Commun (2013) 4:2185. doi: 10.1038/ncomms3185
97. Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin L, Garraway LA. Highly Recurrent TERT Promoter Mutations in Human Melanoma. Science (2013) 339(6122):957–9. doi: 10.1126/science.1229259
98. Horn S, Figl A, Rachakonda PS, Fischer C, Sucker A, Gast A, et al. TERT Promoter Mutations in Familial and Sporadic Melanoma. Science (2013) 339(6122):959–61. doi: 10.1126/science.1230062
99. Gabler L, Lotsch D, Kirchhofer D, van Schoonhoven S, Schmidt HM, Mayr L, et al. TERT Expression is Susceptible to BRAF and ETS-Factor Inhibition in BRAF(V600E)/TERT Promoter Double-Mutated Glioma. Acta Neuropathol Commun (2019) 7(1):128. doi: 10.1186/s40478-019-0775-6
100. Losic B, Craig AJ, Villacorta-Martin C, Martins-Filho SN, Akers N, Chen X, et al. Intratumoral Heterogeneity and Clonal Evolution in Liver Cancer. Nat Commun (2020) 11(1):291. doi: 10.1038/s41467-019-14050-z
101. Rowland TJ, Dumbovic G, Hass EP, Rinn JL, Cech TR. Single-Cell Imaging Reveals Unexpected Heterogeneity of Telomerase Reverse Transcriptase Expression Across Human Cancer Cell Lines. Proc Natl Acad Sci USA (2019) 116(37):18488–97. doi: 10.1073/pnas.1908275116
102. Murofushi Y, Nagano S, Kamizono J, Takahashi T, Fujiwara H, Komiya S, et al. Cell Cycle-Specific Changes in Htert Promoter Activity in Normal and Cancerous Cells in Adenoviral Gene Therapy: A Promising Implication of Telomerase-Dependent Targeted Cancer Gene Therapy. Int J Oncol (2006) 29(3):681–8. doi: 10.3892/ijo.29.3.681
103. Kyo S, Inoue M. Complex Regulatory Mechanisms of Telomerase Activity in Normal and Cancer Cells: How can We Apply Them for Cancer Therapy? Oncogene (2002) 21(4):688–97. doi: 10.1038/sj.onc.1205163
104. Killela PJ, Reitman ZJ, Jiao Y, Bettegowda C, Agrawal N, Diaz LA Jr., et al. TERT Promoter Mutations Occur Frequently in Gliomas and a Subset of Tumors Derived From Cells With Low Rates of Self-Renewal. Proc Natl Acad Sci USA (2013) 110(15):6021–6. doi: 10.1073/pnas.1303607110
105. Pirker C, Holzmann K, Spiegl-Kreinecker S, Elbling L, Thallinger C, Pehamberger H, et al. Chromosomal Imbalances in Primary and Metastatic Melanomas: Over-Representation of Essential Telomerase Genes. Melanoma Res (2003) 13:483–92. doi: 10.1097/01.cmr.0000056264.56735.73
106. Zhang A, Zheng C, Lindvall C, Hou M, Ekedahl J, Lewensohn R, et al. Frequent Amplification of the Telomerase Reverse Transcriptase Gene in Human Tumors. Cancer Res (2000) 60(22):6230–5.
107. Gaspar TB, Sa A, Lopes JM, Sobrinho-Simoes M, Soares P, Vinagre J. Telomere Maintenance Mechanisms in Cancer. Genes (Basel) (2018) 9(5):241. doi: 10.3390/genes9050241
108. Guilleret I, Yan P, Grange F, Braunschweig R, Bosman FT, Benhattar J. Hypermethylation of the Human Telomerase Catalytic Subunit (hTERT) Gene Correlates With Telomerase Activity. Int J Cancer (2002) 101(4):335–41. doi: 10.1002/ijc.10593
109. Castelo-Branco P, Choufani S, Mack S, Gallagher D, Zhang C, Lipman T, et al. Methylation of the TERT Promoter and Risk Stratification of Childhood Brain Tumours: An Integrative Genomic and Molecular Study. Lancet Oncol (2013) 14(6):534–42. doi: 10.1016/s1470-2045(13)70110-4
110. Castelo-Branco P, Leão R, Lipman T, Campbell B, Lee D, Price A, et al. A Cancer Specific Hypermethylation Signature of the TERT Promoter Predicts Biochemical Relapse in Prostate Cancer: A Retrospective Cohort Study. Oncotarget (2016) 7(36):57726–336. doi: 10.18632/oncotarget.10639
111. Lee DD, Leão R, Komosa M, Gallo M, Zhang CH, Lipman T, et al. DNA Hypermethylation Within TERT Promoter Upregulates TERT Expression in Cancer. J Clin Invest (2019) 129(1):223–9. doi: 10.1172/JCI121303
112. Zinn RL, Pruitt K, Eguchi S, Baylin SB, Herman JG. Htert is Expressed in Cancer Cell Lines Despite Promoter DNA Methylation by Preservation of Unmethylated DNA and Active Chromatin Around the Transcription Start Site. Cancer Res (2007) 67(1):194–201. doi: 10.1158/0008-5472.Can-06-3396
113. Leão R, Lee D, Figueiredo A, Hermanns T, Wild P, Komosa M, et al. Combined Genetic and Epigenetic Alterations of the TERT Promoter Affect Clinical and Biological Behavior of Bladder Cancer. Int J Cancer (2019) 144(7):1676–84. doi: 10.1002/ijc.31935
114. Heaphy CM, Subhawong AP, Hong SM, Goggins MG, Montgomery EA, Gabrielson E, et al. Prevalence of the Alternative Lengthening of Telomeres Telomere Maintenance Mechanism in Human Cancer Subtypes. Am J Pathol (2011) 179(4):1608–15. doi: 10.1016/j.ajpath.2011.06.018
115. Heaphy CMW, de Wilde RF, Jiao Y, Klein AP, Edil BH, Shi C, et al. Altered Telomeres in Tumors With ATRX and DAXX Mutations. Science (2011) 333:425. doi: 10.1126/science.1207313
116. Perrem K, Colgin LM, Neumann AA, Yeager TR, Reddel RR. Coexistence of Alternative Lengthening of Telomeres and Telomerase in hTERT-Transfected GM847 Cells. Mol Cell Biol (2001) 21(12):3862–75. doi: 10.1128/MCB.21.12.3862-3875.2001
117. Bechter OE, Zou Y, Walker W, Wright WE, Shay JW. Telomeric Recombination in Mismatch Repair Deficient Human Colon Cancer Cells After Telomerase Inhibition. Cancer Res (2004) 64(10):3444–51. doi: 10.1158/0008-5472.Can-04-0323
118. Chen W, Xiao BK, Liu JP, Chen SM, Tao ZZ. Alternative Lengthening of Telomeres in hTERT-Inhibited Laryngeal Cancer Cells. Cancer Sci (2010) 101(8):1769–76. doi: 10.1111/j.1349-7006.2010.01611.x
119. Stewart SA, Hahn WC, O’Connor BF, Banner EN, Lundberg AS, Modha P, et al. Telomerase Contributes to Tumorigenesis by a Telomere Length-Independent Mechanism. Proc Natl Acad Sci (2002) 99(20):12606–11. doi: 10.1073/pnas.182407599
120. Bandola-Simon J, Roche PA. Dysfunction of Antigen Processing and Presentation by Dendritic Cells in Cancer. Mol Immunol (2019) 113:31–7. doi: 10.1016/j.molimm.2018.03.025
121. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell (2017) 168(4):707–23. doi: 10.1016/j.cell.2017.01.017
122. Lanitis E, Dangaj D, Irving M, Coukos G. Mechanisms Regulating T-Cell Infiltration and Activity in Solid Tumors. Ann Oncol (2017) 28:xii18–32. doi: 10.1093/annonc/mdx238
123. Hegde PS, Karanikas V, Evers S. The Where, the When, and the How of Immune Monitoring for Cancer Immunotherapies in the Era of Checkpoint Inhibition. Clin Cancer Res (2016) 22(8):1865–74. doi: 10.1158/1078-0432.CCR-15-1507
124. Hegde PS, Chen DS. Top 10 Challenges in Cancer Immunotherapy. Immunity (2020) 52(1):17–35. doi: 10.1016/j.immuni.2019.12.011
125. Wei SC, Duffy CR, Allison JP. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discovery (2018) 8(9):1069–86. doi: 10.1158/2159-8290.CD-18-0367
126. Duraiswamy J, Kaluza KM, Freeman GJ, Coukos G. Dual Blockade of PD-1 and CTLA-4 Combined With Tumor Vaccine Effectively Restores T-Cell Rejection Function in Tumors. Cancer Res (2013) 73(12):3591–603. doi: 10.1158/0008-5472.Can-12-4100
127. Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 Combination Blockade Expands Infiltrating T Cells and Reduces Regulatory T and Myeloid Cells Within B16 Melanoma Tumors. Proc Natl Acad Sci (2010) 107(9):4275–80. doi: 10.1073/pnas.0915174107
128. Duperret EK, Wise MC, Trautz A, Villarreal DO, Ferraro B, Walters J, et al. Synergy of Immune Checkpoint Blockade With a Novel Synthetic Consensus DNA Vaccine Targeting TERT. Mol Ther (2018) 26(2):435–45. doi: 10.1016/j.ymthe.2017.11.010
129. Shrimali RK, Yu Z, Theoret MR, Chinnasamy D, Restifo NP, Rosenberg SA. Antiangiogenic Agents can Increase Lymphocyte Infiltration Into Tumor and Enhance the Effectiveness of Adoptive Immunotherapy of Cancer. Cancer Res (2010) 70(15):6171–80. doi: 10.1158/0008-5472.CAN-10-0153
130. Mougel A, Terme M, Tanchot C. Therapeutic Cancer Vaccine and Combinations With Antiangiogenic Therapies and Immune Checkpoint Blockade. Front Immunol (2019) 10(467):467. doi: 10.3389/fimmu.2019.00467
131. Chen W, Jin W, Hardegen N, K-j L, Li L, Marinos N, et al. Conversion of Peripheral CD4+CD25– Naive T Cells to CD4+CD25+ Regulatory T Cells by TGF-β Induction of Transcription Factor Foxp3. J Exp Med (2003) 198(12):1875–86. doi: 10.1084/jem.20030152
132. Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. TGFβ Attenuates Tumour Response to PD-L1 Blockade by Contributing to Exclusion of T Cells. Nature (2018) 554(7693):544–8. doi: 10.1038/nature25501
133. Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, et al. TGFβ Drives Immune Evasion in Genetically Reconstituted Colon Cancer Metastasis. Nature (2018) 554(7693):538–43. doi: 10.1038/nature25492
134. Liu M, Kuo F, Capistrano KJ, Kang D, Nixon BG, Shi W, et al. TGF-Beta Suppresses Type 2 Immunity to Cancer. Nature (2020) 587(7832):115–20. doi: 10.1038/s41586-020-2836-1
135. Bonaventura P, Shekarian T, Alcazer V, Valladeau-Guilemond J, Valsesia-Wittmann S, Amigorena S, et al. Cold Tumors: A Therapeutic Challenge for Immunotherapy. Front Immunol (2019) 10:168. doi: 10.3389/fimmu.2019.00168
136. Li S, Liu M, Do MH, Chou C, Stamatiades EG, Nixon BG, et al. Cancer Immunotherapy via Targeted TGF-Beta Signalling Blockade in TH Cells. Nature (2020) 587(7832):121–5. doi: 10.1038/s41586-020-2850-3
137. Terabe M, Robertson FC, Clark K, De Ravin E, Bloom A, Venzon DJ, et al. Blockade of Only TGF-β 1 and 2 is Sufficient to Enhance the Efficacy of Vaccine and PD-1 Checkpoint Blockade Immunotherapy. OncoImmunology (2017) 6(5):e1308616. doi: 10.1080/2162402X.2017.1308616
138. Ho WJ, Jaffee EM, Zheng L. The Tumour Microenvironment in Pancreatic Cancer - Clinical Challenges and Opportunities. Nat Rev Clin Oncol (2020) 17(9):527–40. doi: 10.1038/s41571-020-0363-5
139. Brahmer JR, Tykodi SS, Chow LQM, Hwu W-J, Topalian SL, Hwu P, et al. Safety and Activity of Anti–PD-L1 Antibody in Patients With Advanced Cancer. New Engl J Med (2012) 366(26):2455–65. doi: 10.1056/NEJMoa1200694
140. Neoptolemos JP, Greenhalf W, Cox TF, Costello E, Shaw V, Valle JW, et al. Predictive Cytokine Biomarkers for Survival in Patients With Advanced Pancreatic Cancer Randomized to Sequential Chemoimmunotherapy Comprising Gemcitabine and Capecitabine (GemCap) Followed by the Telomerase Vaccine GV1001 Compared to Concurrent Chemoimmunotherapy in the Telovac Phase III Trial. J Clin Oncol (2014) 32(15_suppl):4121. doi: 10.1200/jco.2014.32.15_suppl.4121
141. Gridelli C, Ciuleanu T, Domine M, Szczesna A, Bover I, Cobo M, et al. Clinical Activity of a Htert (vx-001) Cancer Vaccine as Post-Chemotherapy Maintenance Immunotherapy in Patients With Stage IV non-Small Cell Lung Cancer: Final Results of a Randomised Phase 2 Clinical Trial. Br J Cancer (2020) 122(10):1461–6. doi: 10.1038/s41416-020-0785-y
142. Wada S, Jackson CM, Yoshimura K, Yen H-R, Getnet D, Harris TJ, et al. Sequencing CTLA-4 Blockade With Cell-Based Immunotherapy for Prostate Cancer. J Trans Med (2013) 11(1):89. doi: 10.1186/1479-5876-11-89
143. Verma V, Shrimali RK, Ahmad S, Dai W, Wang H, Lu S, et al. PD-1 Blockade in Subprimed CD8 Cells Induces Dysfunctional PD-1+CD38hi Cells and Anti-PD-1 Resistance. Nat Immunol (2019) 20(9):1231–43. doi: 10.1038/s41590-019-0441-y
144. Trimble CL, Morrow MP, Kraynyak KA, Shen X, Dallas M, Yan J, et al. Safety, Efficacy, and Immunogenicity of VGX-3100, a Therapeutic Synthetic DNA Vaccine Targeting Human Papillomavirus 16 and 18 E6 and E7 Proteins for Cervical Intraepithelial Neoplasia 2/3: A Randomised, Double-Blind, Placebo-Controlled Phase 2b Trial. Lancet (2015) 386(10008):2078–88. doi: 10.1016/S0140-6736(15)00239-1
145. Choi YJ, Hur SY, Kim T-J, Hong SR, Lee JK, Cho C-H, et al. Prospective, Randomized, Multicenter, Open-Label Study of GX-188E, an HPV DNA Vaccine, in Patients With Cervical Intraepithelial Neoplasia 3. Clin Cancer Res (2020) 26(7):1616. doi: 10.1158/1078-0432.CCR-19-1513
146. Kenter GG, Welters MJP, Valentijn ARPM, Lowik MJG, Berends-van der Meer DMA, Vloon APG, et al. Vaccination Against HPV-16 Oncoproteins for Vulvar Intraepithelial Neoplasia. New Engl J Med (2009) 361(19):1838–47. doi: 10.1056/NEJMoa0810097
147. van Poelgeest MIE, Welters MJP, Vermeij R, Stynenbosch LFM, Loof NM, Berends-van der Meer DMA, et al. Vaccination Against Oncoproteins of HPV16 for Noninvasive Vulvar/Vaginal Lesions: Lesion Clearance is Related to the Strength of the T-Cell Response. Clin Cancer Res (2016) 22(10):2342. doi: 10.1158/1078-0432.CCR-15-2594
Keywords: cancer, telomerase, immunotherapy, cancer vaccine, hTERT, melanoma, immune response, immuno-oncology
Citation: Ellingsen EB, Mangsbo SM, Hovig E and Gaudernack G (2021) Telomerase as a Target for Therapeutic Cancer Vaccines and Considerations for Optimizing Their Clinical Potential. Front. Immunol. 12:682492. doi: 10.3389/fimmu.2021.682492
Received: 18 March 2021; Accepted: 21 June 2021;
Published: 05 July 2021.
Edited by:
Lu Si, Peking University Cancer Hospital, ChinaReviewed by:
A. J. Robert McGray, University at Buffalo, United StatesAlan L. Epstein, University of Southern California, United States
Copyright © 2021 Ellingsen, Mangsbo, Hovig and Gaudernack. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Espen Basmo Ellingsen, espen.ellingsen@ultimovacs.com