Update on molecular pathology of cutaneous melanocytic lesions: What is new in diagnosis and molecular testing for treatment?
 Adriana C. H. van Engen-van Grunsven1
Heidi Kusters-Vandevelde2
Patricia J. T. A. Groenen1
Adriana C. H. van Engen-van Grunsven1
Heidi Kusters-Vandevelde2
Patricia J. T. A. Groenen1  Willeke A. M. Blokx1*
Willeke A. M. Blokx1*- 1Department of Pathology, Radboud University Medical Center, Nijmegen, Netherlands
- 2Department of Pathology, Canisius Wilhelmina Hospital, Nijmegen, Netherlands
In this article, we give an update on recent findings regarding molecular pathology in cutaneous melanocytic tumors. The focus lies on use of genetics in the diagnosis of distinct subtypes of spitzoid tumors that are often characterized by specific phenotypic–genotypic alterations that can frequently be recognized by adequate histological examination. Typical illustrating cases are given in order to increase recognition of these lesions in daily dermatopathology practice. New molecular findings in the pathogenesis of congenital melanocytic tumors and neurocutaneous melanosis are reviewed. In addition, use of mutation analysis in the differential diagnosis of melanoma metastasis is discussed. Finally, application of mutation analysis in targeted therapy in advanced melanoma with advantages of new techniques such as next generation sequencing is described.
Introduction
During the past 20 years, there has been a rapid development of molecular techniques increasing the possibilities of genetic testing in all kinds of tumors including melanocytic tumors. This has led to a rapid gain in our knowledge on the development of melanocytic tumors, which can help in diagnosis, prognosis, and treatment of melanocytic tumors. In 2010, we wrote a review on the molecular pathology of these tumors (1). Since developments in the field are fast, we will address in this paper relevant new findings from the recent years within the field of molecular pathology in melanocytic lesions.
The first part has a focus on new findings with respect to diagnosis and pathogenesis. The focus will be on new molecular findings in spitzoid tumors, in congenital melanocytic nevi (CMN) and neurocutaneous melanocytosis, and the use of molecular tests in the (differential) diagnosis of melanoma.
The second part will will be devoted to application of molecular pathology in the treatment of melanoma, and will briefly address new technological developments such as next generation sequencing (NGS) techniques in this setting.
Part 1: Molecular Pathology in the Diagnosis and Pathogenesis of Melanocytic Tumors
Early events in the development of melanocytic tumors are often hotspot mutations in genes involved in the MAPK pathway, which is one of the most important pathways involved in melanocytic tumor development (Figure 1). Important oncogenes in this pathway are BRAF (7q34), NRAS (1p13), HRAS (11p15), GNAQ (9p21), GNA11 (19p13), and KIT (4q12) (2–5). Mutations in these genes are mostly mutually exclusive, by themselves do not cause malignant progression, stay present with malignant progression, and activate the MAPK pathway. Different subtypes of benign and malignant melanocytic tumors are characterized by different mutations in these genes of the MAPK pathway.
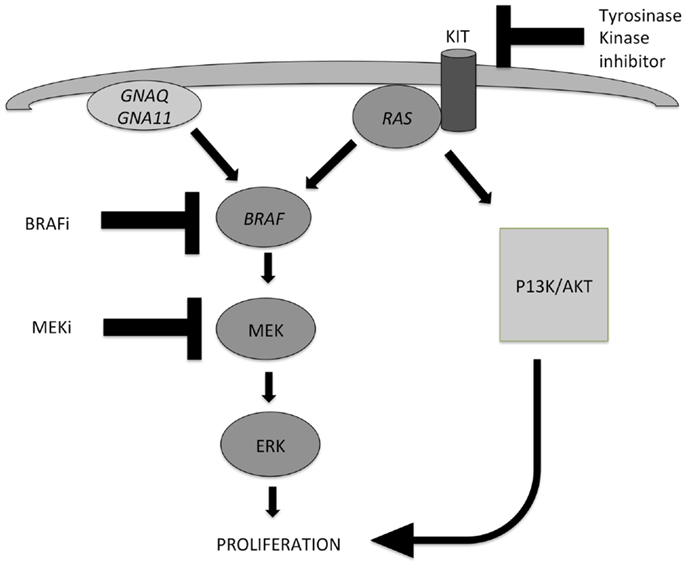
Figure 1. Two important pathways involved in the development of melanocytic tumors and melanoma: the MAPK pathway and the AKT/PI3K-pathway. Activation of both routes leads to proliferation. NRAS plays an important role in both pathways. Mutations in GNAQ, GNA11, and BRAF lead to activation of the MAPK pathway only, while mutations in KIT and NRAS can activate both pathways. Different inhibitors can be applied in targeted therapy of advanced melanoma patients, and their points of action are indicated also. MEK inhibitors can be effective in case of several different gene mutations in the MAPK pathway, because they exert their effect in the distal part of the pathway.
In common nevi for instance, BRAF and NRAS mutations are present in 60–87.5% (6, 7) and 20%, respectively. In large congenital nevi upto 80%, NRAS mutations are reported (7, 8). In blue nevi, mainly GNAQ (83%) and GNA11 (7%) mutations are found (9), and in Spitz nevi, HRAS mutations are reported in 20–29% (7, 10).
Especially, in Spitz tumors, several new data indicate that these tumors are genetically more diverse than was previously thought. We will discuss these new findings below in part 1, together with new insights in the pathogenesis of CMN and the rare disease of neurocutaneous melanocytosis. We will also address the role of molecular pathology in the differential diagnosis of (metastatic) melanoma.
The distinct mutations in different melanoma types will be discussed later in part 2 (see also Table 1).
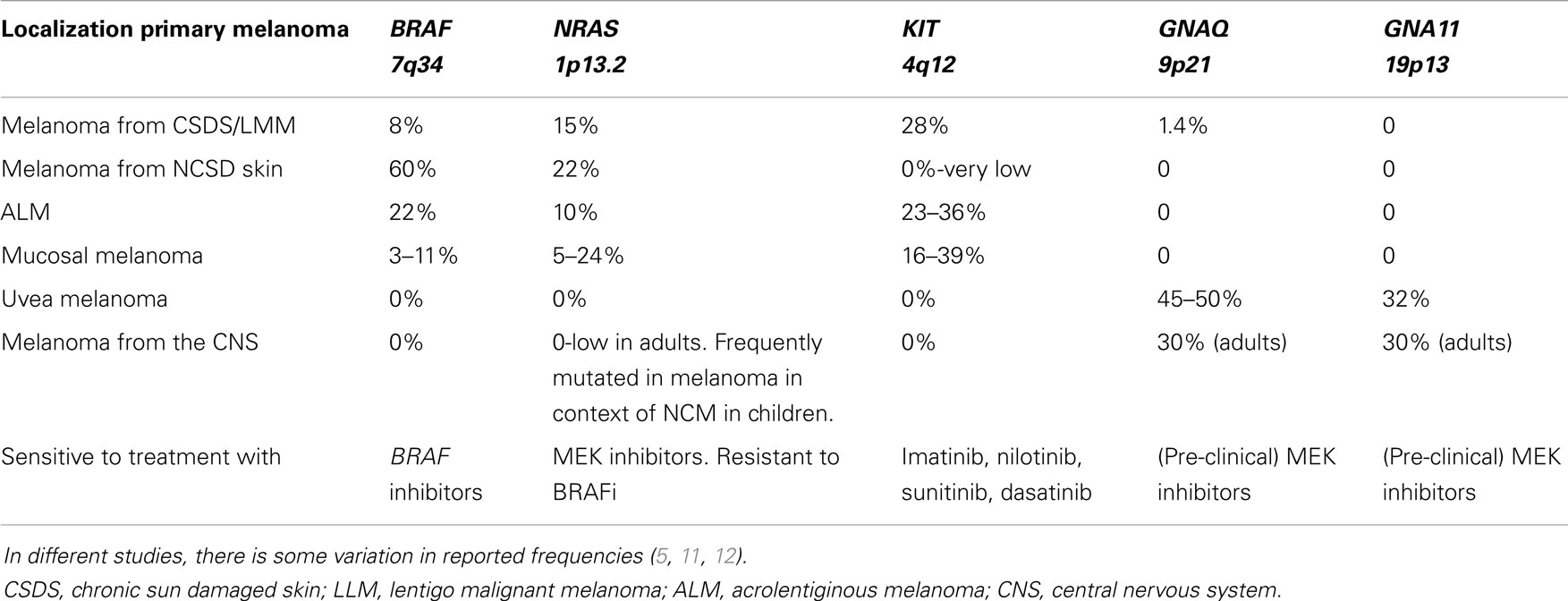
Table 1. Overview of frequencies of gene mutations in different melanoma subtypes derived from different locations.
What is New in Spitzoid Melanocytic Tumors?
At present, roughly three subgroups of spitzoid melanocytic tumors can be identified based on distinct genetic alterations. The first one is the group of the HRAS-mutated spitzoid tumors (13). The second group is the one of the BAP1-mutated “spitzoid lesions” (14, 15), and the third group consists of spitzoid tumors with kinase fusion (16).
The first two groups seem to be characterized mostly by a typical phenotype that can be recognized or at least suspected upon histological evaluation.
Most of the HRAS-mutated spitzoid tumors are typically wedge shaped, dermal-based lesions, with an infiltrative margin, consisting of mostly spindle-shaped cells, and showing marked desmoplasia (13, 17). This group is relevant to discriminate because of the favorable prognosis and to prevent melanoma overdiagnosis.
Several studies have reported the presence of HRAS mutations in spitzoid tumors with benign behavior, and absence in clear-cut spitzoid melanomas (10, 17, 18). There is only one recent paper mentioning the occurrence of HRAS mutations in upto 10% (2/20 cases examined) of primary cutaneous melanomas (19). In this paper, no histology of the lesions is shown or described; therefore, whether these lesions were spitzoid or not remains unclear, and no follow-up data of the patients are included to confirm the proposed diagnosis of melanoma by the authors. Furthermore, this paper also gives mutation frequencies of BRAF (25%) and NRAS (10%) that are quite different from most studies in the melanoma field.
In 2010, we described a series of 24 HRAS-mutated spitzoid melanocytic tumors (13). In 7/24 (29%) of these lesions, the initial diagnosis or important differential diagnosis had been melanoma based upon histological examination alone. These were mainly cases in adults that displayed rather frequent or deep mitotic activity. In five cases, more than 2 mitoses/1 mm2 or deep mitoses were present. In this series with a mean and median follow-up of 10.5 years, no recurrences or metastases occurred. An example of a HRAS-mutated spitzoid lesion is depicted in Figure 2.
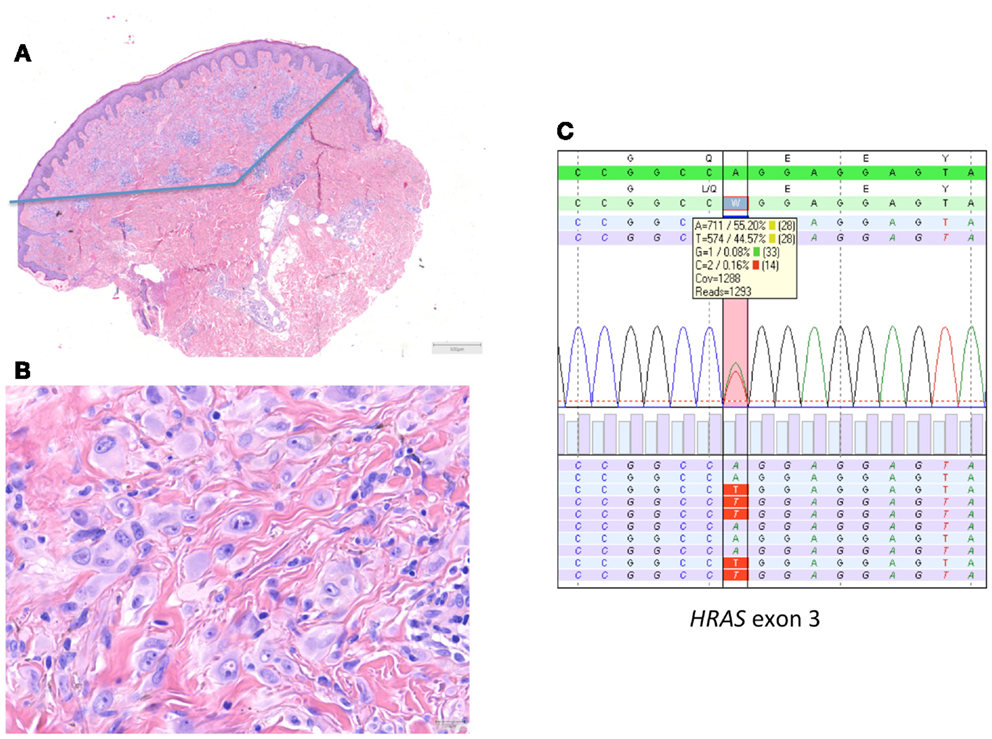
Figure 2. HRAS-mutated spitzoid tumor in a female patient aged 47 years. The lesion was located on the right lower arm and measured 0.8 cm in diameter. The lesion was diagnosed elsewhere as STUMP (Spitzoid tumor of uncertain malignant potential). (A,B) The lesion was symmetrical, wedge shaped, mainly dermal located, with epithelioid spitzoid-looking cells in an eosinophilic hyalinized stroma [(A) HE 25×, (B) HE 400×]. There were 2 mitoses present per 1 mm2. Deep mitoses were absent. (C) Sequence plot of the mutation HRAS c.182A >T(p.(Gln61Leu)) with 44% of mutant alleles. The genomic sequence of the gene investigated is marked in green bars, and the protein sequence information is on top of the gene sequence. The combined (forward and reverse) gene sequence information is highlighted in light green, and the expected protein sequence is placed on top. The forward sequence information is indicated as light blue bars and reverse sequence information as purple bars. The square box shows the number and percentage of the different nucleotides at the variant position; the red vertical bar indicates the HRAS hotspot mutation position. The sequence plots are generated in SeqNext (from JSI Medical Systems GmbH). Note that the HRAS RefSeq is NM_005343.2.
The second group, which we preferentially call MBAITs (melanocytic BAP1-associated intradermal tumors, but are also called Wiesner tumor or BAPoma) are often polypoid, dermal based, consisting of large epitheloid “spitzoid-looking” cells, that can have a small common nevus component at the margin (especially in BAP1-germline mutated lesions), and in one-third of cases, there are prominent tumor-infiltrating lymphocytes (TILs).
These BAP1-mutated melanocytic lesions were first described by Wiesner et al. They described two families, one in Australia and one in Germany, in which a total of 16 individuals were affected by atypical cutaneous melanocytic tumors, in association with cutaneous and uvea melanomas (14). The affected family members were found to have a BAP1-germline mutation. Subsequently, it was found that these spitzoid-looking MBAITs, besides a BAP1 mutation, also contained a BRAF mutation. Later, these lesions were also described in a sporadic setting in so-called atypical spitzoid tumors (ASTs), without having an underlying BAP1-germline mutation (15, 20). In 2012, Wiesner et al. described a series of 32 ASTs (20). Nine cases (28%) showed BAP 1 protein expression loss while BRAF protein expression was present. In 8/9 cases (89%), a BRAFV600E mutation was found, and in 5/9 cases (55%), a somatic BAP1 mutation was present. No HRAS mutations were found. Histology was comparable to the BAP1-germline mutation associated MBAITs, demonstrating a dermal-based lesion, plump epithelioid cells, giant cells, with in 1/3 cases prominent TILs, and absence of prominent fibrosis. These cases were not described to contain a small nevoid component in contrast to the germline BAP1-associated cases.
Yeh et al. recently described genomic loss, determined with array CGH, of >1 Mbp of chromosome 3 in a region containing the BAP1 locus in a series of 29 cases out of 436 ambiguous melanocytic tumors (6.7%) (21): 22 cases showed partial loss of chromosome 3, while 7 cases demonstrated monosomy of chromosome 3. In 11 cases BAP1 mutation analysis was performed with in 10 cases a loss of function mutation of BAP1, and in the remaining single case with wild-type BAP1, BAP1 protein expression was lost on the immunohistochemical level. In the cases with loss of 1 copy of BAP1, the BAP1 protein expression was always lost. So, immunohistochemistry for BAP1 protein seems to correlate well with the genetic findings. Reported follow-up in their series was good, although short (median 17 months), with no recurrences, and in one patient a negative sentinel lymph node. Morphology of 17 lesions with biallelic loss of BAP1 that looked spitzoid was as described previously by others, with TILs being present in 50% of cases, and 31% having a small nevoid component at the margin. In these cases, a junctional component was present composed of the common nevus component. In one case without a common nevus component, a junctional spitzoid component was present. In 12/17 cases, a BRAFV600E mutation was present, 4 cases were wildtype BRAF, and 1 case showed an NRASQ61R mutation. The latter is rarely reported at present, but we have also encountered such a case ourselves recently (paper submitted). In our patient, no underlying BAP1 mutation was found. The other reported NRAS-mutated case (21) in literature was not tested for a germline mutation in BAP1. Most of the MBAIT cases reported thus far do not seem to be tested for the presence of NRAS mutations, so the number of NRAS-mutated cases may be larger, and the genetic make-up of these BAP1-associated melanocytic lesions could be broader than currently thought.
The fact that the combined MBAIT lesions show only BAP1 loss in the epithelioid component suggests that they probably develop from a common nevus (that is mostly BRAF and seldom NRAS mutated) (7).
MBAITs probably have a low risk of developing into melanoma, but at present, data about behavior are insufficient to draw definite conclusions.
In the two largest series thus far by Pouryazdanparast et al. (22) and Yeh et al. (21), reporting 28 and 29 cases, respectively, follow-up was favorable without recurrences. Follow-up was relatively short with a mean of 21 months and median of 17 months, respectively. Pouryazdanparast performed FISH [using probes targeting chromosome 6p25 (RREB1), chromosome 6q23 (MYB), chromosome 11q13 (CCND1), and the centromeric portion of chromosome 6 (CEP 6)] on these lesions, which was negative in all cases.
The difference in outcome between the uveal lesions and the skin lesions with a BAP1 mutation may be related to the presence of different oncogenic driver mutations in uveal lesions, which harbor GNAQ or GNA11 mutations (23) instead of BRAF or NRAS mutations.
The suggested progression-promoting effect of mutated BAP1 is in line with the tumor suppressive function of intact BAP1 as a deubiquitylase required for efficient assembly of the homologous recombination (HR) factors BRCA1 and RAD51 after DNA double-strand breaks (DSBs) (24, 25). BAP1 is recruited to DNA damage sites together with ASXL1 and deubiquitilates Ub-H2AK119 at sites of DNA damage (24, 25). In this way, it promotes error-free repair of these lesions. Defective HR and increased sensitivity to radiation due to BAP1 deficiency may, therefore, lead to genomic instability, a hallmark of cancer (24, 25). Moreover, BAP1 prevents proteasomal degradation of the conserved epigenetic regulator host cell factor-1 (HCF-1) and, consistent with this, Dey et al. showed that BAP1 KO splenocytes contained far less HCF-1 than their wild-type counterparts (26). It is thought that BAP1 regulates gene expression via stabilization of HCF-1 (26). These two examined functions of BAP1 could explain tumor progression due to altered BAP1 expression.
The most important reason for recognition of MBAITs at present is that they can be a marker of an underlying BAP1-associated germline mutation/cancer syndrome. Individuals with a BAP1-germline mutation have an increased risk to develop cutaneous and ocular melanoma and mesothelioma, apart from the risk to develop multiple MBAITs (27).
Management of MBAIT lesions should consist of complete removal of the lesion and advise for genetic counseling to exclude a potential underlying cancer syndrome.
Typical BRAF-mutated MBAIT cases from our own practice are shown in Figures 3 and 4 (Courtesy Dr. R. Kornegoor, Department of Pathology, Gelre Hospitals, Apeldoorn, The Netherlands).
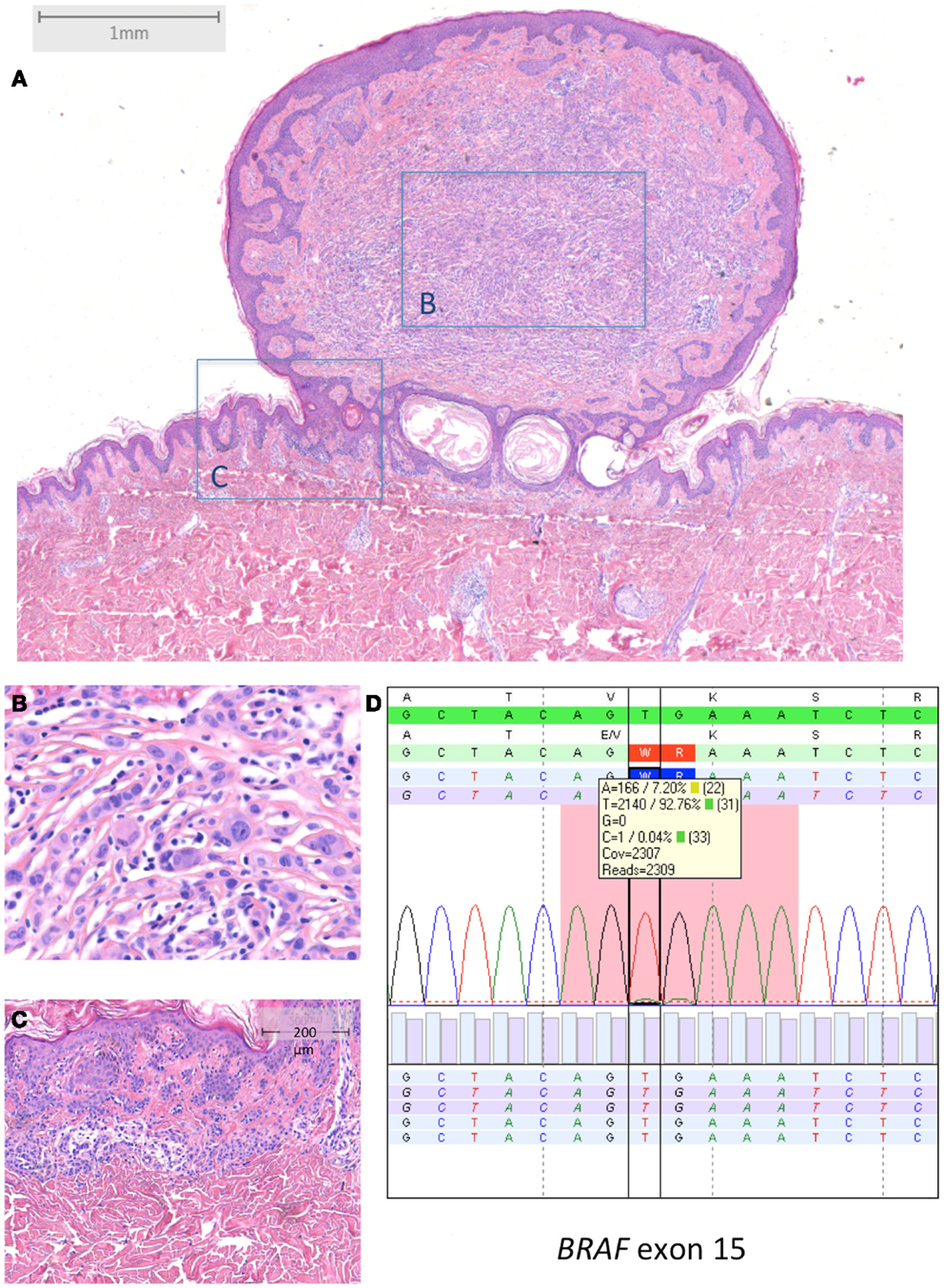
Figure 3. BRAF-mutated MBAIT in a 31-year-old female located on the lower back (Courtesy Dr. R. Kornegoor, Gelre Hospitals, Apeldoorn, The Netherlands). (A) Overview (HE 25×), showing a polypoid lesion above the skin surface. The central part (B) (HE 400×) consists of large epithelioid cells with intermingled lymphocytes. The margin (C) (HE 100×) shows a common compound nevus. (D) Molecular analysis showed a BRAFV600E mutation. In (D), sequence plot of the mutation BRAF c.1799_1800TG >AA (p.(Val600Glu) alias p.V600E) with 7% mutant allel (very low load). Note that the BRAF RefSeq is NM_004333.4. The sequence plots are visualized as described under Figure 2. Immunohistochemistry demonstrated positive BRAF protein expression, and loss of BAP1 protein expression in the large epithelioid cells (not shown).
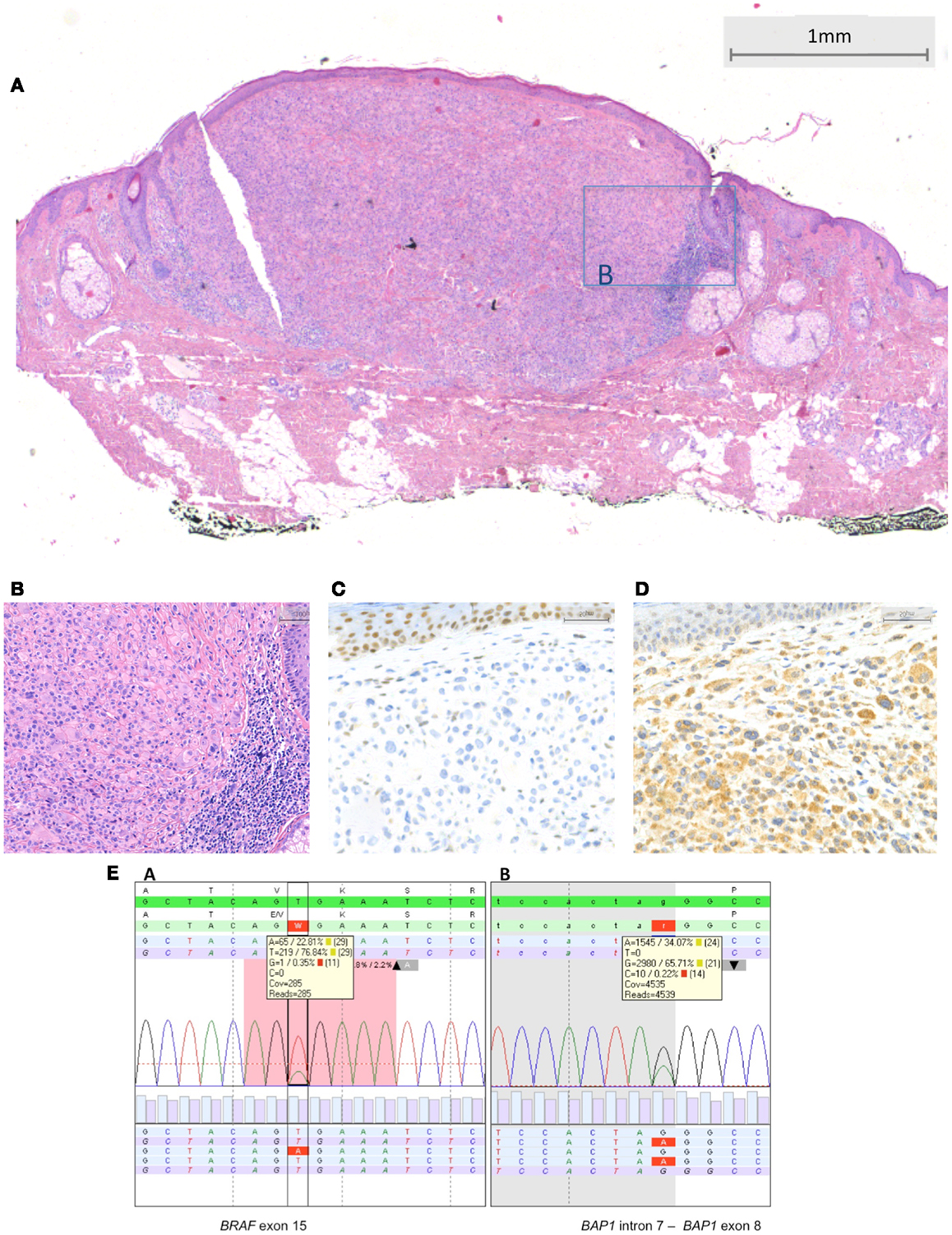
Figure 4. BRAF-mutated MBAIT lesion located on the posterior side of the ear in a 55-year-old female (Courtesy Dr. R. Kornegoor, Gelre Hospitals, Apeldoorn, The Netherlands). In (A), an overview showing a nodular dermal-based lesion with lymphocytes at the lateral margin. In this case, no common nevus component was present (HE, 25×). (B) A higher magnification demonstrates the large epithelioid aspect of the cells (HE, 200×). In (C), absence of BAP1 protein expression is seen in the large epithelioid melanocytes. Nuclei in the keratinocytes in the epidermis stain positive, serving as a positive internal control (400×). In (D), BRAF protein is expressed by the large epithelioid melanocytes (400×). (E) Sequence plots of the mutation BRAF c.1799T >A (p.(Val600Glu) alias p.V600E) with 23% mutant alleles and of the BAP1 (RefSeq NM_004656.2) c.581-1G >A splice site mutation with 34% mutant alleles. The sequence plots are visualized as described under Figure 2. In the BAP1 plot, the intron–exon boundary is represented as gray-white parts.
In case of suspicion of a MBAIT lesion, we recommend at least a BRAF and BAP1 immunostaining, but preferentially BRAF, NRAS, HRAS, and BAP1 mutation analysis is performed. In case a laboratory cannot perform these, we recommend consultation because of the important implications of a correct diagnosis. Mutation analysis of the BAP1 gene is difficult, since it is a complex gene and mutations can be present along all of the 17 exons of the gene. A low tumor percentage due to the small size of a lesion or small size of the spitzoid component, or the presence of a lot of TILs, can all hamper BAP1 mutation analysis in this setting.
The third group of spitzoid lesions, those with kinase fusions has only recently been described. Wiesner et al. described the presence of alterations in ROS1, NTRK1, ALK, BRAF, and RET in, respectively, 17%, 16%, 10%, 5%, and 3% of spitzoid tumors. These alterations were present along the entire spectrum of the spitzoid tumors (55% in Spitz nevi, 56% in AST, and 39% in spitzoid melanoma), and these alterations therefore seem early events, and are not useful (yet) for differentiating benign from malignant spitzoid lesions. No clear distinct phenotypes were described at this time.
Recently, Busam et al. described 17 cases of Spitz tumors with ALK fusion, including 5 Spitz nevi and 12 AST (28). Clinically, these lesions were often polypoid. Histology showed a compound, mostly dermal located lesions with a plexiform growth, and consisting of fusiform melanocytes. In only 2 cases, the lesional cells contained pigment.
All cases showed ALK protein expression. ALK FISH was positive in all cases (using a commercially available break-apart probe, Abbott Molecular, Des Plaines, IL): in 11/17 cases, the fusion partner was tropomyosin 3 (TPM-3), in 6/17 cases, the fusion partner was dynactin 1 (DCTN-1). FISH for copy number alterations did not meet the criteria for melanoma diagnosis in any case. Array CGH revealed no chromosomal gains or losses. In 2 cases, a sentinel node procedure was performed and in both cases showed small nests in the subcapsular sinus. Both patients were alive and well after <1 year and after 4 years of follow-up. At present, the follow-up time and number of cases is too limited to draw definite conclusions about the prognosis of this group of Spitz tumors with ALK fusions.
An ALK-positive Spitz tumor is presented in Figure 5 (Courtesy Prof. Dr. J. van den Oord, KU Leuven, Belgium).
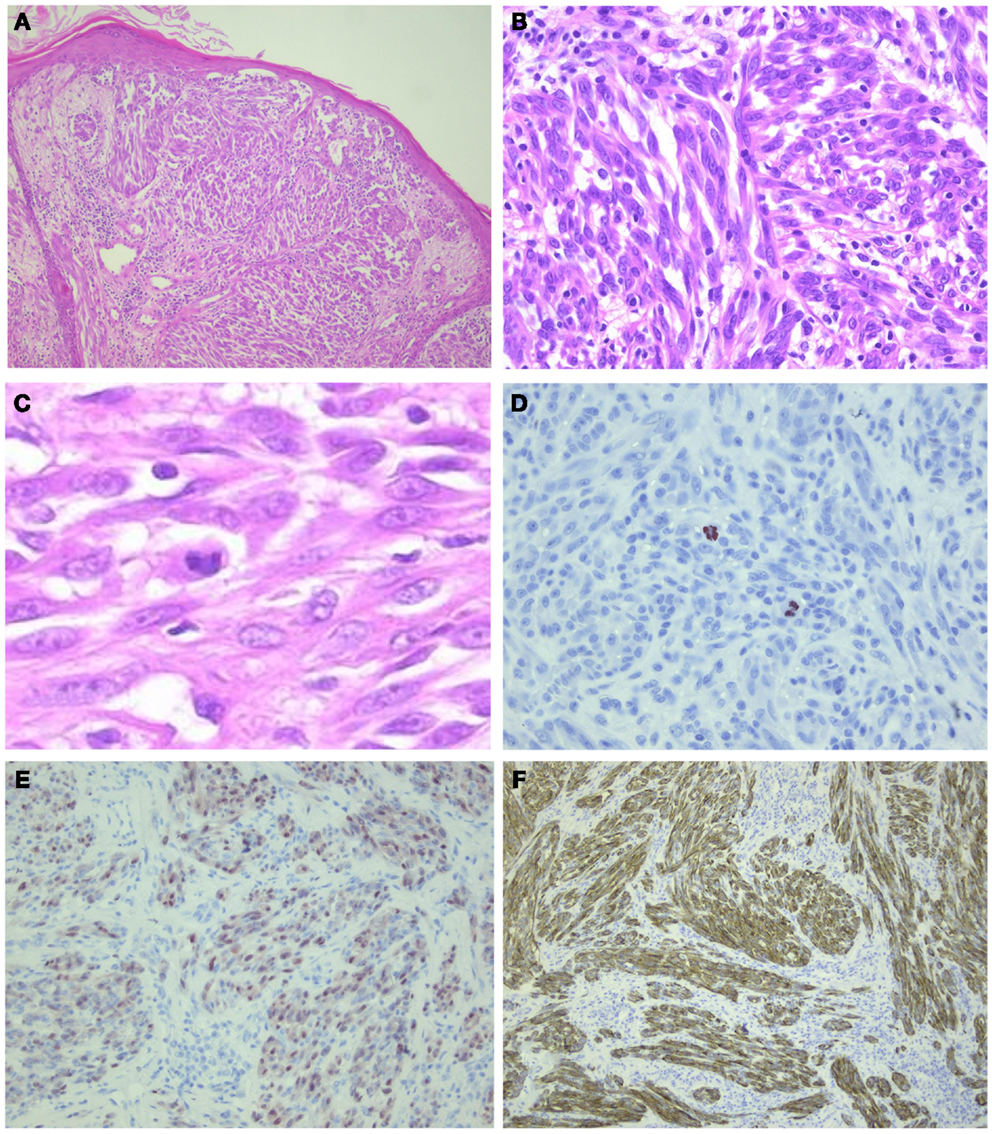
Figure 5. ALK-positive spitzoid tumor diagnosed as spitzoid tumor of uncertain malignant potential (STUMP). This lesion was seen in consultation elsewhere (Courtesy Prof. Dr. J. van den Oord, KU Leuven, Belgium). The lesion was clinically polypoid and presented on the arm of a 4-year-old girl. There were 11 mitoses/1 mm2 present. Follow-up data are not available yet. In (A), HE overview (25×); in (B,C), larger magnification (HE 200–400×, respectively) showing spindle-shaped cells and some lymphocytes intermingled; (D) PHH3 staining demonstrating mitoses (400×); (E) mitf (250×); and (F) ALK (250×).
The anaplastic lymphoma kinase (ALK) gene, located on chromosome 2p23, is a receptor tyrosinase kinase protein and is capable of causing diverse tumor types of different lineages through a variety of molecular mechanisms (29). The most common mechanism of ALK activation is a genomic rearrangement involving the ALK locus, with a breakpoint in the 3′ end of the ALK gene, with the other breakpoint involving a diverse group of genes leading to formation of a fusion oncogene that encodes for a fusion oncoprotein that is able to self-associate (29). Another way of ALK activation is by gain-of-function point mutations, but these are less frequent than ALK rearrangements. Also, ALK amplifications have been described in several tumor types, leading to the presence of multiple copies of the wild-type full-length ALK gene. But the way this contributes to tumor development is poorly understood (29).
As mentioned above, in spitzoid tumors at present, only ALK rearrangements have been tested and described, and present in upto 19% of cases (16), with TPM-3 and DCTN-1 as fusion partners (16, 28). ALK rearrangements leading to TPM3-ALK fusion have also been reported in anaplastic large cell lymphoma and papillary thyroid carcinoma, and result in a constitutive tyrosinase kinase activity, in this way causing tumor development (29).
Relevance of diagnosis of Spitz tumors with kinase fusions at the moment is mainly for treatment in malignant lesions (e.g., ALK, RET, ROS 1 alterations can be targeted with kinase inhibitors, such as crizotinib, cabozantinib, and vandetanib).
Table 2 summarizes the characteristic features of the three above-described subtypes of spitzoid melanocytic lesions, including the most common histological findings, and genetic and prognostic features, with clinical relevance of recognition of these subtypes.
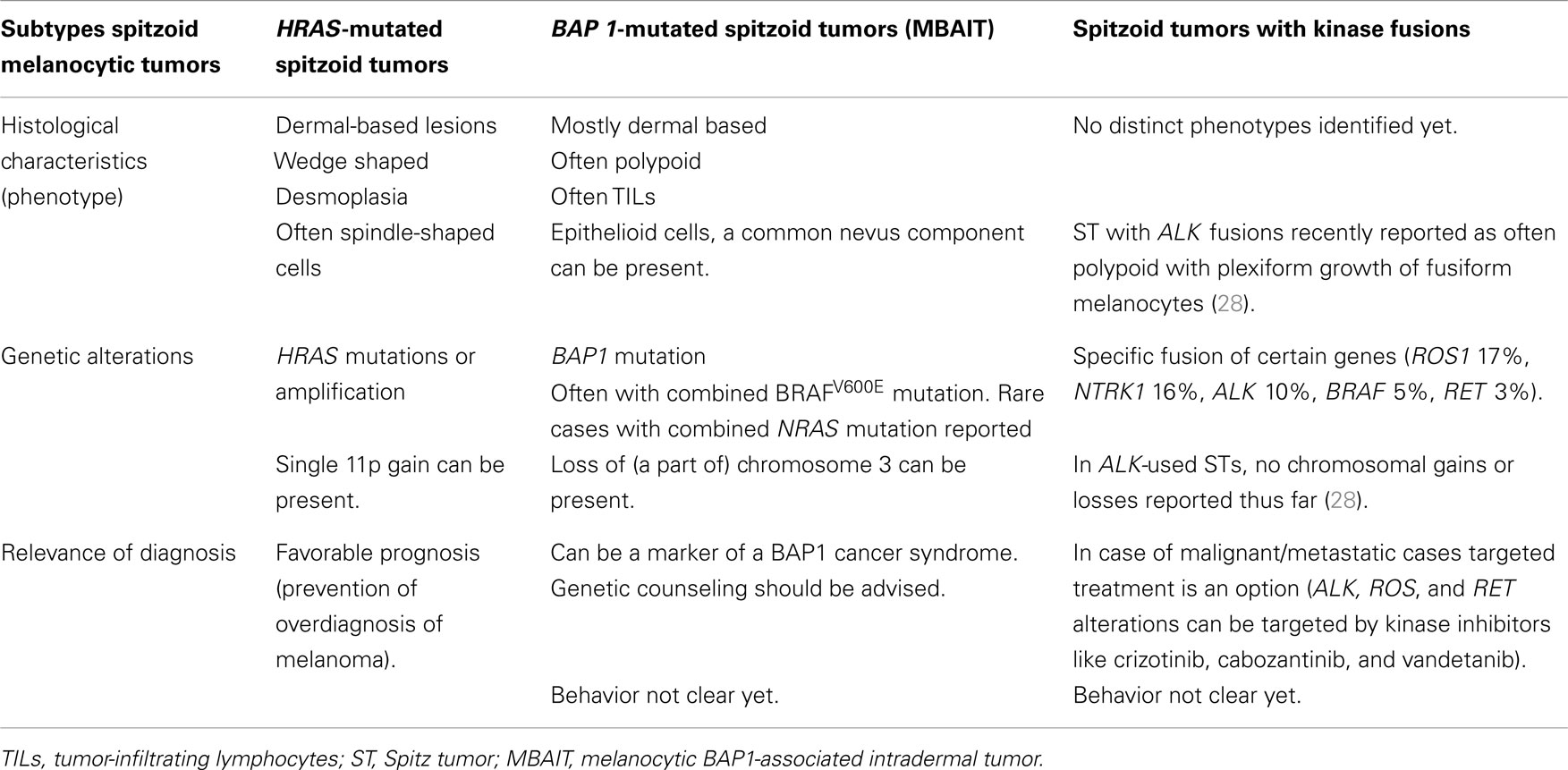
Table 2. Subtypes of spitzoid melanocytic tumors with their phenotypical and genotypical characteristics, and relevance of diagnosis.
Molecular Background of Congenital Melanocytic Nevi (CMN) and Neurocutaneous Melanosis (NCM)
Congenital melanocytic nevi are pigmented moles present at birth or shortly thereafter (30). They vary in size from small to very large or “giant” and hundreds of CMN can be present in one patient (31). CMN are considered to be a sporadic event with the exception of a few familial cases (32, 33). Especially giant CMN ( >40 cm in size) are associated with an increased risk for cutaneous melanoma (up to 10–15% life-time risk) (34).
Both NRAS and BRAF mutations have been detected in CMN in a mutually exclusive pattern, and there is a genotype–phenotype correlation between the size of CMN and type of mutation. Especially, large and giant CMN harbor somatic NRAS mutations ( >75%) in contrast to medium-sized CMN that are less frequently NRAS mutated and especially small CMN and acquired melanocytic nevi that frequently carry BRAFV600E mutations (up to 80% in acquired nevi) (35). Activating BRAFV600E mutations in large CMN are rare (approximately 15%) (6). A few cases of large CMN were shown to harbor a chromosomal translocation involving BRAF resulting in gain-of-function, and this could represent an alternative mechanism of BRAF activation in BRAF- or NRAS-wild-type CMN (36).
Recently, using several highly sensitive techniques, Charbel et al. showed that large and giant CMN contain NRASQ61 mutations (Q61R or Q61K) in up to 94.7% of cases (37). In addition, using whole-exome sequencing, they found no other coding mutations in five large/giant CMN implying that at present NRAS mutations are the sole recurrent mutation in these lesions. Somatic NRAS mutations, therefore, seem to be sufficient to drive melanocytic proliferations in utero. In addition, identical NRASQ61 mutations were recently demonstrated in multiple CMN samples from the same patient (38). This finding suggests that multiple CMN are clonal proliferations caused by a single, postzygotic NRAS mutation in a neuro-ectodermal precursor cell rather than de novo proliferations arising from different mutations.
Kinsler et al. recently proposed the term “CMN syndrome” as they observed that patients with CMN often have characteristic facial features (such as wide or prominent forehead and apparent hypertelorism) (39). The osteocartilageneous structures of the face are neuro-ectodermal in origin and can be affected by mutations in components of the RAS/RAF/MEK/ERK pathway in patients with germline RASopathies who have characteristic facial features (40). Kinsler et al. hypothesized that the occurrence of a postzygotic, somatic NRAS mutation in early neuro-ectodermal precursors might be responsible for the characteristic facial features in patients with CMN as well (38). In addition, as they observed a high prevalence of red hair in families of children with CMN, a germline predisposition for the development of the “CMN syndrome” was hypothesized. Indeed, they showed that certain germline allele variants of the melanocortin-1-receptor (MC1R), known to be responsible for the red hair/fair skin/freckling phenotype, are associated with the presence of CMN (as well as with more extensive CMN) (41).
Congenital melanocytic nevi can be associated with a spectrum of neurological abnormalities, including malformations (for instance, Dandy–Walker malformation), communicating hydrocephalus, arachnoid cysts, CNS tumors (for instance, astrocytoma and choroid plexus papilloma) and also melanotic abnormalities (38). The latter is called neurocutaneous melanosis (NCM) and refers to the presence of large of multiple CMN in association with melanin depositions in the brain parenchym (visible on T1-weighted MRI) or melanocytic tumors like leptomeningeal melanocytosis or melanoma (42). Leptomeningeal melanocytosis consists of a diffuse proliferation of histological benign appearing melanocytes in the leptomeninges, without CNS invasion, and carries a poor prognosis once symptomatic (42).
Recently, it was shown that postzygotic, somatic NRAS mutations contribute to the pathogenesis of NCM. Identical, somatic NRASQ61 mutations were detected in multiple CMN as well as in the CNS melanocytic tumor in the same NCM patients (43). Kinsler et al. also detected a somatic NRASQ61 mutation in three non-melanocytic CNS tumors occurring in patients with CMN (including choroid plexus papilloma, neurocristic hamartoma, and meningioma) (38). The presence of identical, somatic NRASQ61 mutations in both CMN and in CNS melanocytic (and non-melanocytic) neoplasms in the same patients suggests that these mutations occur in the developing neuro-ectoderm early during embryogenesis. In fact, this pathogenetic mechanism fits the spectrum of mosaic RASopathies that are characterized by postzygotic mutations resulting in the presence of at least two genetically distinct cell populations in the same organism (44). For instance, postzygotic, somatic RAS mutations (HRAS, KRAS) were recently shown to be present in distinct lesions of the keratinocytic epidermal nevus syndrome, a mosaic RASopathy characterized by the presence of epidermal nevi in association with extra-cutaneous abnormalities (CNS, ocular, skeletal, cardiovascular, and genitourinary system) (44). The phenotype in the spectrum of mosaic RASopathies is most likely determined by type of mutation, the timing of the mutation, and affected cell type (44, 45).
A mouse model has demonstrated a role for postzygotic, early embryonic NRAS mutations in the pathogenesis of NCM. By using the Cre-loxP technology, Pedersen et al. showed that expression of oncogenic NRASG12D in melanocytes early during embryogenesis resulted in a mouse phenotype strongly resembling NCM in human beings (43) As NRAS mutations occur in benign lesions such as CMN, they are in itself insufficient for malignant transformation of melanocytes, and better insight in the genetic aberrations eventually leading to melanoma is needed. NRAS mutations are a therapeutic target for treatment with MEK inhibitors. In a Phase II trial, treatment with MEK162 was shown to have effect in some patients with advanced NRAS-mutated melanoma and a Phase III trial is ongoing (46). For patients with NRAS-mutated CNS melanocytic tumors, treatment with MEK162 might be of benefit as well (47).
Molecular Diagnostics in the Differential Diagnosis of Melanoma (Metastasis)
Melanomas are known for their wide range of cytomorphologic features and architectural patterns and may mimic various non-melanocytic tumors [reviewed by Banerjee et al. (48, 49)]. In most cases, a diagnosis can be rendered by careful examination of the histomorphology and by sometimes adding immunohistochemical stains. In some cases, however, especially in recurrences and metastases, melanomas can show an aberrant immunophenotype with loss of lineage-specific markers. In part of these cases, molecular analysis of genes in the MAPK pathway and CDKN2A (7) mutation analysis can be useful in the differential diagnosis. In addition, molecular analysis can be of help in determining the site of the primary melanoma in case of metastasis with an unknown primary since melanomas from different locations have different mutation types (see Table 1). Furthermore, molecular analysis can help to discriminate between a melanoma metastasis and a second primary melanoma.
One of the lesions that can be hard to distinguish from melanoma is clear cell sarcoma (CCS) as they share histopathological features and cannot be distinguished by immunohistochemistry (50). Classically, CCS is a deep soft tissue tumor associated with tendons and aponeuroses (51), but it can also present as a cutaneous lesion (52), and then has to be differentiated from metastatic or primary nodular melanoma. While most cutaneous melanomas harbor a BRAF or NRAS mutation (see Table 1), CCS in approximately 75% have a t(12;22)(a13;q12) or less commonly a t(2;22)(q34;q12) translocation leading to the EWSR1/ATF1 or EWSR1/CREB1 fusion transcripts (53, 54). Yang et al. (55) performed BRAF and NRAS mutation analysis, as well as FISH analysis for the EWST1/ATF1 fusion gene in 31 melanoma cases and 16 CCSs. They found the translocation in 78.6% of the CCSs but in none of the melanomas, whereas BRAF and NRAS mutations were present in, respectively, 51.6 and 12.9% of the melanomas and not in any of the CCSs. Hantschke et al. (52) described 12 cases of cutaneous CCS in which FISH analysis for the t(12;22)(a13;q12) translocation contributed in the differential diagnosis with melanoma. This implicates that this type of analysis can be of great aid in the differential diagnosis between CCS and melanoma.
Recently, we described two cases in which mutation analysis lead to the correct diagnosis of (dedifferentiated) metastatic melanoma (56). Both patients had a history of melanoma and presented several years after their primary diagnosis with a lesion histologically and immunohistochemically different from the primary melanoma and mimicking a solitary fibrous tumor (SFT). Using BRAF and NRAS mutation analysis, it could be proven that both lesions were melanoma metastases instead of SFT. Many other soft tissue tumors can mimick melanoma or melanoma metastasis, such as dermatofibrosarcoma protuberans, malignant peripheral nerve sheath tumors (MPNST) (57), and sarcomas (48, 58–60).
Although CDKN2A mutation frequency in sporadic melanomas as well as consistency in melanoma metastasis are reported to be low [reported CDKN2A mutation frequency in upto 25% of primary melanomas, 0–14% in melanoma metasases, with a 31% consistency being reported between a primary and the metastasis (61, 62)], CDKN2A mutation analysis can be an alternative way to confirm a diagnosis of metastatic melanoma by showing a clonal relationship between a primary melanoma and a metastasis [after exclusion of a CDKN2A germline mutation (63)], as is illustrated by a recent case from our own practice. A 36-year-old woman with a history of a superficial spreading melanoma of the back 16 years earlier, presented with a large lung mass, which was thought to be a non-small cell lesion, probably squamous cell carcinoma based on fine needle aspiration (EUS-FNA); melanoma markers HMB45 and MART were negative, while the squamous cell marker – p63 – was weakly positive. Evaluation of the pneumonectomy specimen, however, led to a differential diagnosis with metastatic melanoma, since immunohistochemical stainings on the complete tumor now available for histological evaluation showed in part features consistent with melanocytic differentiation. Vimentin, tyrosinase, and mitf were weakly positive; however, S100 and SOX10 were both negative. Therefore, additional BRAF and CDKN2A mutation analysis was performed. Identical mutations in both genes were present in the primary cutaneous melanoma and the lung mass – in absence of a germline CDKN2A mutation – confirming that the lung mass was a late metastasis of the cutaneous melanoma. This case is illustrated in Figure 6.
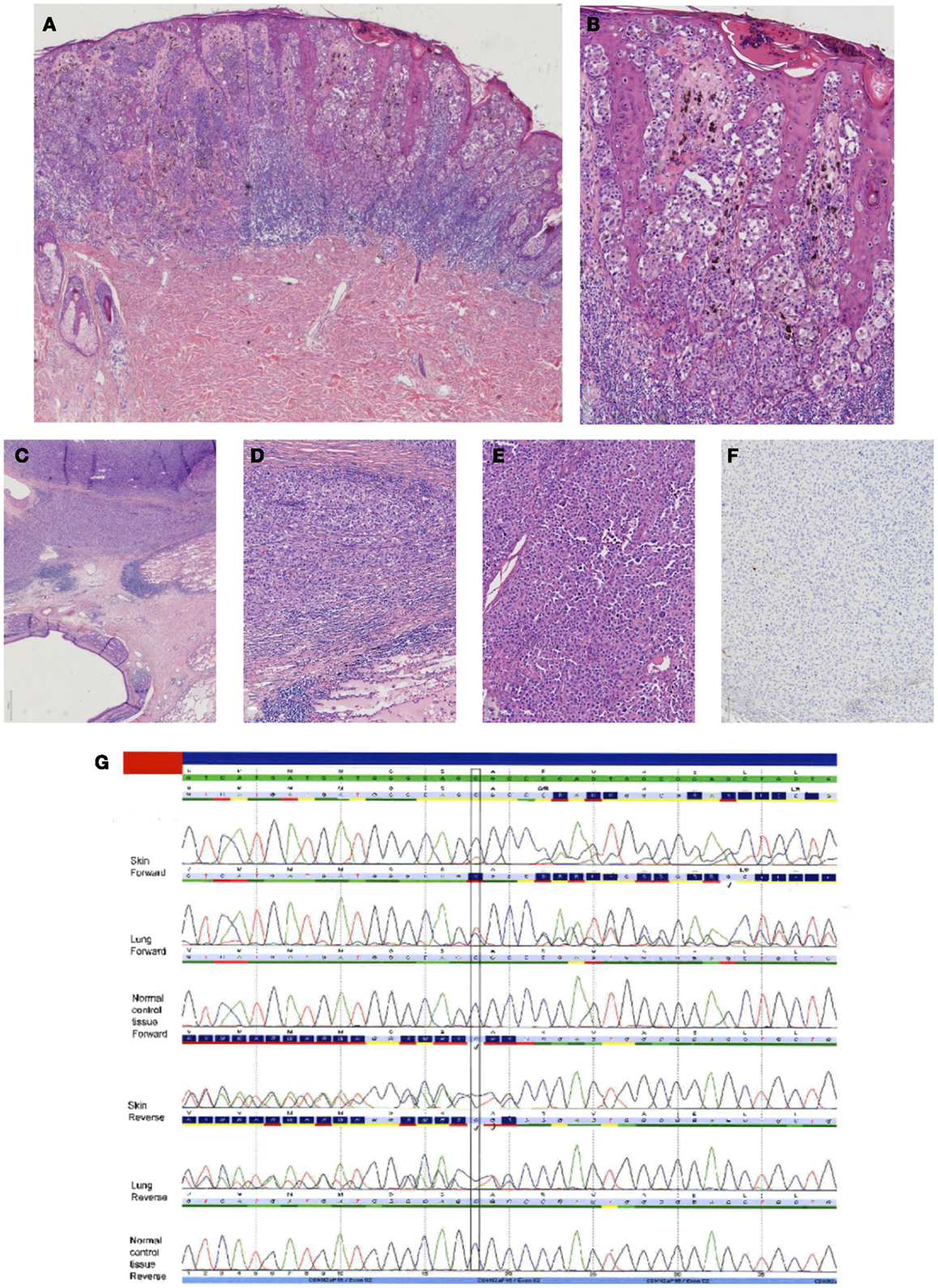
Figure 6. Case of a superficial spreading melanoma of the back with lung metastasis 16 years later in a female patient. In (A), an overview of the primary cutaneous melanoma showing centrally an asymmetrical melanocytic lesion (HE, 25×). In (B), a more detailed picture showing clear atypia with loss of maturation and ascension in the epidermis (HE, 100×). In (C–E), the lung lesion at different magnifications [HE C 25×, (D,E) 100×] showing a nodular proliferation of atypical partly plasmacytoid cells resembling the primary cutaneous melanoma. Immunohistochemical S100 stain, however, was negative [(F) S100 immunostaining, 100×]. Internal and external controls were positive. (G) Sequence plots demonstrated an identical c.168_170delinsTG(p.(Arg58fs)) in the CDKN2A gene in both the skin melanoma and the lung melanoma, which was absent in normal tissue of the patient. This confirmed that the lung tumor was a metastasis of the previous melanoma. The sequence plots are visualized as described under Figure 2.
In a patient with a history of melanoma, it can occasionally be difficult to differentiate between a second primary cutaneous melanoma and a melanoma metastasis based on histomorphology, especially when there is epidermotropism (64). If the first melanoma was not a cutaneous melanoma, the problem can often be solved by mutation analysis since different types of melanomas have mutations in different genes of the MAPK pathway as mentioned in Table 1. If the primary melanoma was a cutaneous melanoma, mutation analysis of NRAS and BRAF can be of help, but since these mutations are hotspot mutations, it is of limited use. Usage is additionally hampered by frequent occurrence of heterogeneity in melanoma with respect to NRAS and BRAF status, leading to a substantial discordance in mutation status in these genes in a primary compared to the metastases (61, 65).
CDKN2A mutation analysis can sometimes be helpful as we have published before (66), and is illustrated by the case above, to differentiate between a new primary and a melanoma metastasis. The advantage of using CDKN2A mutations for clonality is that CDKN2A mutations are unique mutations instead of hot spot mutations. Although CDKN2A mutation frequency in sporadic melanomas is low, CDKN2A mutation analysis to our opinion is worth trying, as there is a substantial difference in the prognosis between a second primary melanoma and a melanoma metastasis.
Part 2: Molecular Pathology in Melanoma Treatment
Metastatic melanoma treatment got a great impulse after the discovery that the (hot spot) mutations in genes involved in the MAPK pathway in melanoma, as well as in GNAQ and GNA11, which proved targetable on the protein level by specific inhibitors. GNAQ and GNA11 mutations are mainly present in uveal and primary brain melanomas, and combinations of MEK inhibitors with either PI3K or mTOR inhibitors have shown efficacy in GNAQ- and GNA11-mutant melanomas (67, 68). A simplified scheme of the most important pathways involved in melanoma pathogenesis, and points of action of specific inhibitors are depicted in Figure 1.
Melanomas from distinct locations have been shown to contain different mutation types and frequencies (5, 11, 12). An overview of the most frequent mutations in the distinct melanoma subtypes and their frequencies, as well as the different therapeutic options, are depicted in Table 1.
The most frequently tested genes at present in melanoma are BRAF and NRAS, because these mutations are most prevalent in melanoma, especially in those arising from the skin. Testing for targeted treatment in melanoma should be guided by the localization of the primary tumor. Nowadays, this issue becomes less important because several molecular labs use platforms that detect various regions from multiple genes within one test.
In case of molecular testing for targeted therapy in metastatic melanoma, there are several important issues.
Molecular testing for therapy requires a close collaboration between clinician, pathologist, and molecular biologist. Testing for treatment should only be performed when targeted therapy is considered as a therapeutic option. This can be judged best by the clinician. In most cases, testing is only indicated in inoperable stage 3 or stage 4 melanoma patients.
Tests need to be performed in an accredited molecular laboratory that guarantees that the laboratory techniques and processes are performed standardized and yield high-quality results, which imply that only validated tests are used.
The role of the pathologist is to confirm the diagnosis of melanoma metastasis, to check representativity of the tumor tissue to be examined, to indicate the area for macrodissection (if needed) by the technician for DNA extraction, to indicate possible tumor heterogeneity, and to estimate the neoplastic cell percentage within the tested sample.
The knowledge and the expertise of trained molecular biologists are used to come to optimal test results and adequate interpretation and reporting of molecular test results. The molecular report should contain information on the specific tissue block tested, the percentage of neoplastic cells, the type of molecular test used, and the sensitivity of the test, the type of genes, and exons or mutations thereof which are tested, and an accurate description of the mutation present according to the Human Genome Variation Society nomenclature. The molecular biologist, together with the pathologist, is responsible for proper integration and interpretation of the molecular results in the pathology report.
The molecular test is preferentially performed on a recent metastasis. First, reason for this is confirmation of the diagnosis of melanoma metastasis. Second, there is considerable tumor heterogeneity with a reported discrepancy by Colombino et al. of 7.5–29% in the BRAF and NRAS mutation status of the metastatic melanoma when compared to the primary (61, 65). Concordance seems dependent on the location of the metastases and is highest for visceral (92.5%) and nodal metastases (91%), but relatively low for brain (79%) and skin metastases (71%). Especially, in the latter, testing should be performed on the metastasis.
Saint-Jean et al. (69) also reported discordant BRAF results. They performed multiple tests in a subgroup of 74 patients: in 10/74 (13.5%) of these patients, BRAF status was discordant between distinct samples of a patient. In two patients, the discordance was present between the primary and a metastasis. In six patients, the discordance was present between two distinct metastases. The authors state that without repeated testing, five patients would unjustly have been excluded from treatment with the BRAF inhibitor, vemurafenib. They advocate to test other samples in case no BRAF mutation has been detected. We can agree with this especially in selected cases, when the primary location is likely to be associated with a BRAF mutation and when no mutation in other targetable genes (like NRAS or KIT) have been identified.
A molecular test should be able to detect all relevant and targetable mutations in a gene. The most frequently tested gene for melanoma treatment at present is BRAF.
The most frequent BRAF mutation is a mononucleotide point mutation in codon 600 (CTG) of exon 15, c.1799T >A (p.(Val600Glu)), in which the valine (V) of codon 600 is replaced by glutamine (E). This mutation is, therefore, known as the BRAFV600E mutation and is present in about 70–75% of all BRAF-mutated melanomas (11, 70, 71). In a recent study in which 1112 primary and metastatic melanomas from different locations were analyzed for BRAF mutations (774 skin melanomas, 111 acral melanomas, 26 mucosal melanomas, 23 uveal melanomas, 1 leptomeningeal melanoma, and 177 metastases), 44.9% of the cases harbored a BRAF mutation: in 75.4% of the cases, mutations were BRAFV600E either deriving from the c.1799T >A or from a c.1799 1800delinsAA mutation. Of the remaining BRAFnon-V600E cases (24.6%), the most frequently seen mutation was the BRAFV600K (17.2%); BRAFV600R or BRAFV600D mutated cases were found in low percentages (4.6%). BRAF exon 11 mutations were also observed in a low percentage (0.4%).
There are several studies that have reported that all melanomas with BRAF codon 600 mutations are sensitive to BRAF inhibitors, such as vemurafenib (Zelboraf®, Roche Molecular Systems Inc.) and dabrafenib (GSK2118436) (72, 73). This implicates that a BRAF test needs to be able to detect not only the BRAFc.1799T >A(p.(Val600Glu)/BRAFV600E mutation but the other codon 600 mutations as well.
A test must be able to detect BRAF mutations that have been reported to be insensitive to BRAF-inhibitor treatment, like the kinase-dead mutation BRAFD594 (74, 75).
The molecular test for therapy should be performed within a short turnaround time since mostly this kind of targeted therapy will be given in rapidly progressive metastatic melanoma patients. A turnaround time of 5 working days is feasible within our hands.
At present, we perform NGS-based mutation testing using Ion Torrent Personal Genome Machine (IT-PGM) from Life Technologies for analysis of gene-panels for diagnostic purposes.
At the moment, there is a tendency toward testing with NGS methods for targeted treatment in diverse cancer types. The sensitivity of NGS is higher than Sanger sequencing (detection of 2–10% versus 15–25% allele frequency). Moreover, the amount of DNA that is needed for the analysis of gene panels is very low, only 10 ng for all amplicons for instance when using the IT-PGM from Life Technologies versus 10 ng needed per amplicon for Sanger sequencing.
The turnaround time and costs of NGS methods can be competitive with respect to low throughput technologies in centers that have sufficient numbers of samples.
For diagnostic requests in melanoma in our department, a custom-designed gene panel for amplicon-sequencing of BRAF (NM_004333.4) exon 15, NRAS (NM_002524.4) exon 2 and 3, HRAS (NM_005343.2) exon 2 and 3, AKT1 (NM_005163.2) exon 3, GNAQ (NM_002072.2) exon 4 and 5, GNA11 (NM_002067.2) exon 4 and 5, KIT NM_000222.2) exon 8, 9, 11, 13, and 14, and PDGFRA (NM_006206.4) exon 12, 14, and 18 is performed.
The use of small, dedicated gene panels and efficient loading of the chips for IT-PGM also significantly reduce costs per case.
The major benefit of (targeted) NGS is that it uncovers all kinds of mutations in selected genomic regions instead of only mutations at predefined positions.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Blokx WA, van Dijk MC, Ruiter DJ. Molecular cytogenetics of cutaneous melanocytic lesions – diagnostic, prognostic and therapeutic aspects. Histopathology (2010) 56:121–32. doi: 10.1111/j.1365-2559.2009.03452.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Curtin JA, Fridlyand J, Kageshita T, Patel HN, Busam KJ, Kutzner H, et al. Distinct sets of genetic alterations in melanoma. N Engl J Med (2005) 353:2135–47. doi:10.1056/NEJMoa050092
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Solus JF, Kraft S. Ras, Raf, and MAP kinase in melanoma. Adv Anat Pathol (2013) 20:217–26. doi:10.1097/PAP.0b013e3182976c94
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Takata M, Murata H, Saida T. Molecular pathogenesis of malignant melanoma: a different perspective from the studies of melanocytic nevus and acral melanoma. Pigment Cell Melanoma Res (2010) 23:64–71. doi:10.1111/j.1755-148X.2009.00645.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Whiteman DC, Pavan WJ, Bastian BC. The melanomas: a synthesis of epidemiological, clinical, histopathological, genetic, and biological aspects, supporting distinct subtypes, causal pathways, and cells of origin. Pigment Cell Melanoma Res (2011) 24:879–97. doi:10.1111/j.1755-148X.2011.00880.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Ichii-Nakato N, Takata M, Takayanagi S, Takashima S, Lin J, Murata H, et al. High frequency of BRAFV600E mutation in acquired nevi and small congenital nevi, but low frequency of mutation in medium-sized congenital nevi. J Invest Dermatol (2006) 126:2111–8. doi:10.1038/sj.jid.5700366
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Bastian BC. The molecular pathology of melanoma: an integrated taxonomy of melanocytic neoplasia. Annu Rev Pathol (2014) 9:239–71. doi:10.1146/annurev-pathol-012513-104658
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Bauer J, Curtin JA, Pinkel D, Bastian BC. Congenital melanocytic nevi frequently harbor NRAS mutations but no BRAF mutations. J Invest Dermatol (2007) 127:179–82. doi:10.1038/sj.jid.5700490
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Van Raamsdonk CD, Bezrookove V, Green G, Bauer J, Gaugler L, O’Brien JM, et al. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature (2009) 457:599–602. doi:10.1038/nature07586
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. van Dijk MC, Bernsen MR, Ruiter DJ. Analysis of mutations in B-RAF, N-RAS, and H-RAS genes in the differential diagnosis of Spitz nevus and spitzoid melanoma. Am J Surg Pathol (2005) 29:1145–51. doi:10.1097/01.pas.0000157749.18591.9e
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Greaves WO, Verma S, Patel KP, Davies MA, Barkoh BA, Galbincea JM, et al. Frequency and spectrum of BRAF mutations in a retrospective, single-institution study of 1112 cases of melanoma. J Mol Diagn (2013) 15:220–6. doi:10.1016/j.jmoldx.2012.10.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Lovly CM, Dahlman KB, Fohn LE, Su Z, Dias-Santagata D, Hicks DJ, et al. Routine multiplex mutational profiling of melanomas enables enrollment in genotype-driven therapeutic trials. PLoS One (2012) 7:e35309. doi:10.1371/journal.pone.0035309
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. van Engen-van Grunsven AC, van Dijk MC, Ruiter DJ, Klaasen A, Mooi WJ, Blokx WA. HRAS-mutated Spitz tumors: a subtype of Spitz tumors with distinct features. Am J Surg Pathol (2010) 34:1436–41. doi:10.1097/PAS.0b013e3181f0a749
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Wiesner T, Obenauf AC, Murali R, Fried I, Griewank KG, Ulz P, et al. Germline mutations in BAP1 predispose to melanocytic tumors. Nat Genet (2011) 43:1018–21. doi:10.1038/ng.910
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Busam KJ, Wanna M, Wiesner T. Multiple epithelioid Spitz nevi or tumors with loss of BAP1 expression: a clue to a hereditary tumor syndrome. JAMA Dermatol (2013) 149:335–9. doi:10.1001/jamadermatol.2013.1529
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Wiesner T, He J, Yelensky R, Esteve-Puig R, Botton T, Yeh I, et al. Kinase fusions are frequent in Spitz tumours and spitzoid melanomas. Nat Commun (2014) 5:3116. doi:10.1038/ncomms4116
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Bastian BC, LeBoit PE, Pinkel D. Mutations and copy number increase of HRAS in Spitz nevi with distinctive histopathological features. Am J Pathol (2000) 157:967–72. doi:10.1016/S0002-9440(10)64609-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Da Forno PD, Pringle JH, Fletcher A, Bamford M, Su L, Potter L, et al. BRAF, NRAS and HRAS mutations in spitzoid tumours and their possible pathogenetic significance. Br J Dermatol (2009) 161:364–72. doi:10.1111/j.1365-2133.2009.09181.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Richetta AG, Silvestri V, Giancristoforo S, Rizzolo P, D’Epiro S, Graziano V, et al. Mutational profiling in melanocytic tumors: multiple somatic mutations and clinical implications. Oncology (2014) 86:104–8. doi:10.1159/000356878
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Wiesner T, Murali R, Fried I, Cerroni L, Busam K, Kutzner H, et al. A distinct subset of atypical Spitz tumors is characterized by BRAF mutation and loss of BAP1 expression. Am J Surg Pathol (2012) 36:818–30. doi:10.1097/PAS.0b013e3182498be5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Yeh I, Mully TW, Wiesner T, Vemula SS, Mirza SA, Sparatta AJ, et al. Ambiguous melanocytic tumors with loss of 3p21. Am J Surg Pathol (2014) 38(8):1088–95. doi:10.1097/PAS.0000000000000209
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Pouryazdanparast P, Haghighat Z, Beilfuss BA, Guitart J, Gerami P. Melanocytic nevi with an atypical epithelioid cell component: clinical, histopathologic, and fluorescence in situ hybridization findings. Am J Surg Pathol (2011) 35:1405–12. doi:10.1097/PAS.0b013e31822678d2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Field MG, Harbour JW. Recent developments in prognostic and predictive testing in uveal melanoma. Curr Opin Ophthalmol (2014) 25:234–9. doi:10.1097/ICU.0000000000000051
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Yu H, Pak H, Hammond-Martel I, Ghram M, Rodrigue A, Daou S, et al. Tumor suppressor and deubiquitinase BAP1 promotes DNA double-strand break repair. Proc Natl Acad Sci U S A (2014) 111:285–90. doi:10.1073/pnas.1309085110
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Ismail IH, Davidson R, Gagne JP, Xu ZZ, Poirier G, Hendzel MJ. Germ-line mutations in BAP1 impair its function in DNA double-strand break repair. Cancer Res (2014) 74(16):4282–94. doi:10.1158/0008-5472.CAN-13-3109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Dey A, Seshasayee D, Noubade R, French DM, Liu J, Chaurushiya MS, et al. Loss of the tumor suppressor BAP1 causes myeloid transformation. Science (2012) 337:1541–6. doi:10.1126/science.1221711
27. Carbone M, Ferris LK, Baumann F, Napolitano A, Lum CA, Flores EG, et al. BAP1 cancer syndrome: malignant mesothelioma, uveal and cutaneous melanoma, and MBAITs. J Transl Med (2012) 10:179. doi:10.1186/1479-5876-10-179
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Busam KJ, Kutzner H, Cerroni L, Wiesner T. Clinical and pathologic findings of Spitz nevi and atypical spitz tumors with ALK fusions. Am J Surg Pathol (2014) 38:925–33. doi:10.1097/PAS.0000000000000187
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Marino-Enriquez A, Dal Cin P. ALK as a paradigm of oncogenic promiscuity: different mechanisms of activation and different fusion partners drive tumors of different lineages. Cancer Genet (2013) 206:357–73. doi:10.1016/j.cancergen.2013.07.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Krengel S. Nevogenesis – new thoughts regarding a classical problem. Am J Dermatopathol (2005) 27:456–65. doi:10.1097/01.dad.0000175532.27368.3f
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Ruiz-Maldonado R. Measuring congenital melanocytic nevi. Pediatr Dermatol (2004) 21:178–9. doi:10.1111/j.0736-8046.2004.21222.x
32. Danarti R, Konig A, Happle R. Large congenital melanocytic nevi may reflect paradominant inheritance implying allelic loss. Eur J Dermatol (2003) 13:430–2.
33. de Wijn RS, Zaal LH, Hennekam RC, van der Horst CM. Familial clustering of giant congenital melanocytic nevi. J Plast Reconstr Aesthet Surg (2010) 63:906–13. doi:10.1016/j.bjps.2009.02.090
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Krengel S, Hauschild A, Schafer T. Melanoma risk in congenital melanocytic naevi: a systematic review. Br J Dermatol (2006) 155:1–8. doi:10.1111/j.1365-2133.2006.07218.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Dessars B, De Raeve LE, Morandini R, Lefort A, El Housni H, Ghanem GE, et al. Genotypic and gene expression studies in congenital melanocytic nevi: insight into initial steps of melanotumorigenesis. J Invest Dermatol (2009) 129:139–47. doi:10.1038/jid.2008.203
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Dessars B, De Raeve LE, El Housni H, Debouck CJ, Sidon PJ, Morandini R, et al. Chromosomal translocations as a mechanism of BRAF activation in two cases of large congenital melanocytic nevi. J Invest Dermatol (2007) 127:1468–70. doi:10.1038/sj.jid.5700725
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Charbel C, Fontaine RH, Malouf GG, Picard A, Kadlub N, El-Murr N, et al. NRAS mutation is the sole recurrent somatic mutation in large congenital melanocytic nevi. J Invest Dermatol (2014) 134:1067–74. doi:10.1038/jid.2013.429
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Kinsler VA, Thomas AC, Ishida M, Bulstrode NW, Loughlin S, Hing S, et al. Multiple congenital melanocytic nevi and neurocutaneous melanosis are caused by postzygotic mutations in codon 61 of NRAS. J Invest Dermatol (2013) 133(9):2229–36. doi:10.1038/jid.2013.70
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Kinsler V, Shaw AC, Merks JH, Hennekam RC. The face in congenital melanocytic nevus syndrome. Am J Med Genet A (2012) 158A:1014–9. doi:10.1002/ajmg.a.34217
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Zenker M. Clinical manifestations of mutations in RAS and related intracellular signal transduction factors. Curr Opin Pediatr (2011) 23:443–51. doi:10.1097/MOP.0b013e32834881dd
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Kinsler VA, Abu-Amero S, Budd P, Jackson IJ, Ring SM, Northstone K, et al. Germline melanocortin-1-receptor genotype is associated with severity of cutaneous phenotype in congenital melanocytic nevi: a role for MC1R in human fetal development. J Invest Dermatol (2012) 132:2026–32. doi:10.1038/jid.2012.95
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Kadonaga JN, Frieden IJ. Neurocutaneous melanosis: definition and review of the literature. J Am Acad Dermatol (1991) 24:747–55. doi:10.1016/0190-9622(91)70115-I
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Pedersen M, Kusters-Vandevelde HV, Viros A, Groenen PJ, Sanchez-Laorden B, Gilhuis JH, et al. Primary melanoma of the CNS in children is driven by congenital expression of oncogenic NRAS in melanocytes. Cancer Discov (2013) 3:458–69. doi:10.1158/2159-8290.CD-12-0464
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Hafner C, Groesser L. Mosaic RASopathies. Cell Cycle (2013) 12:43–50. doi:10.4161/cc.23108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Avitan-Hersh E, Tatur S, Indelman M, Gepstein V, Shreter R, Hershkovitz D, et al. Postzygotic HRAS mutation causing both keratinocytic epidermal nevus and thymoma and associated with bone dysplasia and hypophosphatemia due to elevated FGF23. J Clin Endocrinol Metab (2014) 99:E132–6. doi:10.1210/jc.2013-2813
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Ascierto PA, Schadendorf D, Berking C, Agarwala SS, van Herpen CM, Queirolo P, et al. MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: a non-randomised, open-label phase 2 study. Lancet Oncol (2013) 14:249–56. doi:10.1016/S1470-2045(13)70024-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Kusters-Vandevelde HV, Willemsen AE, Groenen PJ, Kusters B, Lammens M, Wesseling P, et al. Experimental treatment of NRAS-mutated neurocutaneous melanocytosis with MEK162, a MEK-inhibitor. Acta Neuropathol Commun (2014) 2:41. doi:10.1186/2051-5960-2-41
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Banerjee SS, Harris M. Morphological and immunophenotypic variations in malignant melanoma. Histopathology (2000) 36:387–402. doi:10.1046/j.1365-2559.2000.00894.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Banerjee SS, Eyden B. Divergent differentiation in malignant melanomas: a review. Histopathology (2008) 52:119–29.
50. Bearman RM, Noe J, Kempson RL. Clear cell sarcoma with melanin pigment. Cancer (1975) 36:977–84. doi:10.1002/1097-0142(197509)36:3<977::AID-CNCR2820360321>3.0.CO;2-D
51. Enzinger FM. Clear-cell sarcoma of tendons and aponeuroses. An analysis of 21 cases. Cancer. (1965) 18:1163–74.
52. Hantschke M, Mentzel T, Rutten A, Palmedo G, Calonje E, Lazar AJ, et al. Cutaneous clear cell sarcoma: a clinicopathologic, immunohistochemical, and molecular analysis of 12 cases emphasizing its distinction from dermal melanoma. Am J Surg Pathol (2010) 34:216–22. doi:10.1097/PAS.0b013e3181c7d8b2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Mrozek K, Karakousis CP, Perez-Mesa C, Bloomfield CD. Translocation t(12;22)(q13;q12.2-12.3) in a clear cell sarcoma of tendons and aponeuroses. Genes Chromosomes Cancer (1993) 6:249–52. doi:10.1002/gcc.2870060412
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Antonescu CR, Nafa K, Segal NH, Dal Cin P, Ladanyi M. EWS-CREB1: a recurrent variant fusion in clear cell sarcoma – association with gastrointestinal location and absence of melanocytic differentiation. Clin Cancer Res (2006) 12:5356–62. doi:10.1158/1078-0432.CCR-05-2811
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Yang L, Chen Y, Cui T, Knosel T, Zhang Q, Geier C, et al. Identification of biomarkers to distinguish clear cell sarcoma from malignant melanoma. Hum Pathol (2012) 43:1463–70. doi:10.1016/j.humpath.2011.10.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Bekers EM, van Engen-van Grunsven AC, Groenen PJ, Westdorp H, Koornstra RH, Bonenkamp JJ, et al. Metastatic melanoma mimicking solitary fibrous tumor: report of two cases. Virchows Arch (2014) 464:247–51. doi:10.1007/s00428-014-1542-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. King R, Busam K, Rosai J. Metastatic malignant melanoma resembling malignant peripheral nerve sheath tumor: report of 16 cases. Am J Surg Pathol (1999) 23:1499–505. doi:10.1097/00000478-199912000-00007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Lodding P, Kindblom LG, Angervall L. Metastases of malignant melanoma simulating soft tissue sarcoma. A clinico-pathological, light- and electron microscopic and immunohistochemical study of 21 cases. Virchows Arch A Pathol Anat Histopathol (1990) 417:377–88. doi:10.1007/BF01606026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Zelger BG, Steiner H, Wambacher B, Zelger B. Malignant melanomas simulating various types of soft tissue tumors. Dermatol Surg (1997) 23:1047–54. doi:10.1111/j.1524-4725.1997.tb00446.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Magro CM, Crowson AN, Mihm MC. Unusual variants of malignant melanoma. Mod Pathol (2006) 19(Suppl 2):S41–70. doi:10.1038/modpathol.3800516
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Colombino M, Capone M, Lissia A, Cossu A, Rubino C, De Giorgi V, et al. BRAF/NRAS mutation frequencies among primary tumors and metastases in patients with melanoma. J Clin Oncol (2012) 30:2522–9. doi:10.1200/JCO.2011.41.2452
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Piccinin S, Doglioni C, Maestro R, Vukosavljevic T, Gasparotto D, D’Orazi C, et al. p16/CDKN2 and CDK4 gene mutations in sporadic melanoma development and progression. Int J Cancer (1997) 74:26–30. doi:10.1002/(SICI)1097-0215(19970220)74:1<26::AID-IJC5>3.0.CO;2-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Monzon J, Liu L, Brill H, Goldstein AM, Tucker MA, From L, et al. CDKN2A mutations in multiple primary melanomas. N Engl J Med (1998) 338:879–87. doi:10.1056/NEJM199803263381305
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Colombino M, Lissia A, Capone M, De Giorgi V, Massi D, Stanganelli I, et al. Heterogeneous distribution of BRAF/NRAS mutations among Italian patients with advanced melanoma. J Transl Med (2013) 11:202. doi:10.1186/1479-5876-11-202
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Blokx WA, Lesterhuis WJ, Andriessen MP, Verdijk MA, Punt CJ, Ligtenberg MJ. CDKN2A (INK4A-ARF) mutation analysis to distinguish cutaneous melanoma metastasis from a second primary melanoma. Am J Surg Pathol (2007) 31:637–41. doi:10.1097/PAS.0b013e318030718d
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Khalili JS, Yu X, Wang J, Hayes BC, Davies MA, Lizee G, et al. Combination small molecule MEK and PI3K inhibition enhances uveal melanoma cell death in a mutant GNAQ- and GNA11-dependent manner. Clin Cancer Res (2012) 18:4345–55. doi:10.1158/1078-0432.CCR-11-3227
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Ho AL, Musi E, Ambrosini G, Nair JS, Deraje Vasudeva S, de Stanchina E, et al. Impact of combined mTOR and MEK inhibition in uveal melanoma is driven by tumor genotype. PLoS One (2012) 7:e40439. doi:10.1371/journal.pone.0040439
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Saint-Jean M, Quereux G, Nguyen JM, Peuvrel L, Brocard A, Vallee A, et al. Is a single BRAF wild-type test sufficient to exclude melanoma patients from vemurafenib therapy? J Invest Dermatol (2014) 134:1468–70. doi:10.1038/jid.2013.378
70. Rubinstein JC, Sznol M, Pavlick AC, Ariyan S, Cheng E, Bacchiocchi A, et al. Incidence of the V600K mutation among melanoma patients with BRAF mutations, and potential therapeutic response to the specific BRAF inhibitor PLX4032. J Transl Med (2010) 8:67. doi:10.1186/1479-5876-8-67
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Amanuel B, Grieu F, Kular J, Millward M, Iacopetta B. Incidence of BRAF p.Val600Glu and p.Val600Lys mutations in a consecutive series of 183 metastatic melanoma patients from a high incidence region. Pathology (2012) 44:357–9. doi:10.1097/PAT.0b013e3283532565
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Klein O, Clements A, Menzies AM, O’Toole S, Kefford RF, Long GV. BRAF inhibitor activity in V600R metastatic melanoma. Eur J Cancer (2012) 49(5):1073–9. doi:10.1016/j.ejca.2012.11.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Menzies AM, Long GV, Murali R. Dabrafenib and its potential for the treatment of metastatic melanoma. Drug Des Devel Ther (2012) 6:391–405. doi:10.2147/DDDT.S38998
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Heidorn SJ, Milagre C, Whittaker S, Nourry A, Niculescu-Duvas I, Dhomen N, et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell (2010) 140:209–21. doi:10.1016/j.cell.2009.12.040
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Stones CJ, Kim JE, Joseph WR, Leung E, Marshall ES, Finlay GJ, et al. Comparison of responses of human melanoma cell lines to MEK and BRAF inhibitors. Front Genet (2013) 4:66. doi:10.3389/fgene.2013.00066
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: review, Spitz, congenital nevus, melanoma, molecular pathology, targeted therapy
Citation: van Engen-van Grunsven ACH, Kusters-Vandevelde H, Groenen PJTA and Blokx WAM (2014) Update on molecular pathology of cutaneous melanocytic lesions: What is new in diagnosis and molecular testing for treatment? Front. Med. 1:39. doi: 10.3389/fmed.2014.00039
Received: 30 September 2014; Accepted: 26 August 2014;
Published online: 31 October 2014.
Edited by:
Robert Léon Baran, Nail Disease Center, FranceReviewed by:
Koen Quint, Leiden University Medical Centre, NetherlandsEdith Duhard-Brohan, Société française de Dermatologie, France
Copyright: © 2014 van Engen-van Grunsven, Kusters-Vandevelde, Groenen and Blokx. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Willeke A. M. Blokx, Department of Pathology, Radboud University Medical Center, P.O. Box 9101, Nijmegen 6500 HB, Netherlands e-mail: willeke.blokx@radboudumc.nl