Epidermolysis Bullosa Acquisita: The 2019 Update
 Hiroshi Koga1
Hiroshi Koga1  Catherine Prost-Squarcioni2
Catherine Prost-Squarcioni2  Hiroaki Iwata3
Hiroaki Iwata3  Marcel F. Jonkman4
Marcel F. Jonkman4  Ralf J. Ludwig5*
Ralf J. Ludwig5*  Katja Bieber5
Katja Bieber5- 1Department of Dermatology, Kurume University School of Medicine, and Kurume University Institute of Cutaneous Cell Biology, Fukuoka, Japan
- 2Department of Dermatology, APHP, Avicenne Hospital, Referral Center for Autoimmune Bullous Diseases, Bobigny, France
- 3Department of Dermatology, Hokkaido University Graduate School of Medicine, Sapporo, Japan
- 4Department of Dermatology, Center for Blistering Diseases, University Medical Center Groningen, University of Groningen, Groningen, Netherlands
- 5Lübeck Institute of Experimental Dermatology, University of Lübeck, Lübeck, Germany
Epidermolysis bullosa acquisita (EBA) is an orphan autoimmune disease. Patients with EBA suffer from chronic inflammation as well as blistering and scarring of the skin and mucous membranes. Current treatment options rely on non-specific immunosuppression, which in many cases, does not lead to a remission of treatment. Hence, novel treatment options are urgently needed for the care of EBA patients. During the past decade, decisive clinical observations, and frequent use of pre-clinical model systems have tremendously increased our understanding of EBA pathogenesis. Herein, we review all of the aspects of EBA, starting with a detailed description of epidemiology, clinical presentation, diagnosis, and current treatment options. Of note, pattern analysis via direct immunofluorescence microscopy of a perilesional skin lesion and novel serological test systems have significantly facilitated diagnosis of the disease. Next, a state-of the art review of the current understanding of EBA pathogenesis, emerging treatments and future perspectives is provided. Based on pre-clinical model systems, cytokines and kinases are among the most promising therapeutic targets, whereas high doses of IgG (IVIG) and the anti-CD20 antibody rituximab are among the most promising “established” EBA therapeutics. We also aim to raise awareness of EBA, as well as initiate basic and clinical research in this field, to further improve the already improved but still unsatisfactory conditions for those diagnosed with this condition.
Epidemiology
The incidence of most autoimmune blistering diseases is increasing. Although the incidence of epidermolysis bullosa acquisita (EBA) is not known in detail, it is estimated to be rare. The most common autoimmune subepidermal blistering disease, bullous pemphigoid (BP), is reported to have an annual estimated incidence between 2.4 and 21.7 per million (1, 2). By contrast, the estimated incidence of EBA is reported to be <0.5 per million (3–7). In South Korea, the incidence and prevalence of EBA is estimated to be higher than that of previous reports (8), but the exact epidemiologic data have not been surveyed. In Germany, the EBA prevalence has recently been determined to be 2.8 cases per million (9). This ethnical difference may be due to the reported association of EBA with the human leukocyte antigen (HLA) class II (10–12). EBA occurs at any age; the onset age in previous case reports exhibit a wide range from 1 to 94 years old (13–15). Two onset age peaks are reported; the second and seventh decades (9).
Clinical Presentation
Several clinical EBA manifestations have been described: (i) the classical/mechano-bullous form and (ii) the non-classical/non-mechano-bullous forms (16). The latter includes BP-like EBA that meets the clinical criteria of both EBA and BP, mucous membrane (MM)-EBA that is clinically defined by predominant mucous membrane lesions, IgA-EBA that is defined by the IgA class of immune deposits, and Brunsting-Perry-like EBA (Figure 1). Few patients may have a MM-IgA-EBA. The relative frequencies of these different clinical forms of EBA reported in the few series in the literature (17–21) depend on the morphological and/or serological diagnostic means available to the authors (Table 1). The two most common presentations of EBA are the classical/mechano-bullous and the BP-like forms.
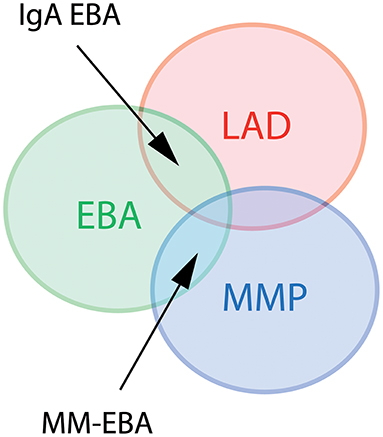
Figure 1. Diagram of different clinical forms of EBA. LAD, linear IgA disease. MMP, mucous membrane pemphigoid; EBA, epidermolysis bullosa acquisita; MM-EBA, mucous membrane EBA.
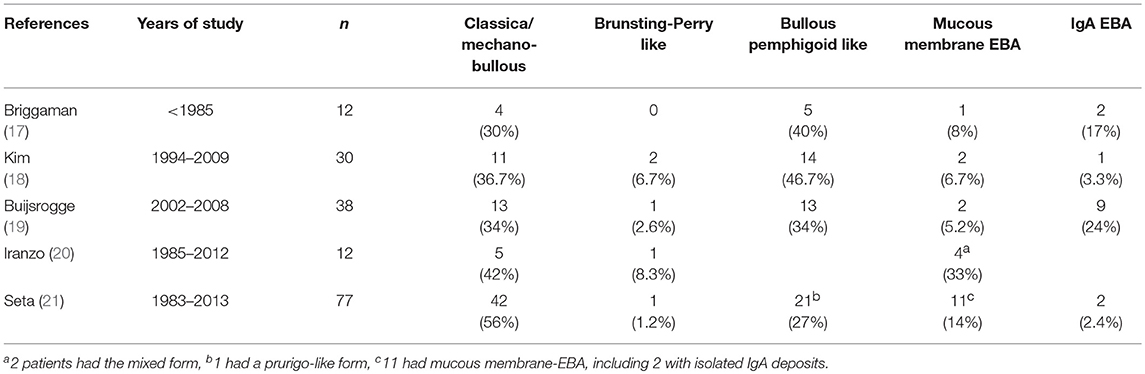
Table 1. Clinical variants of EBA in the series of the literature.
It should be recognized that in an individual EBA patient, clinical presentation may change over time. Notably, patients may switch from a BP-like form to a classical/mechanobullous form or when mucous membrane lesions appear secondarily from a BP-like form to a MM-EBA (22).
Regardless of the clinical form, patients present with cutaneous-mucous fragility, which is easily suspected when the lesions are on trauma-prone areas. Bullous lesions or erosions that are linear or with angular contours can also provide evidence of this fragility (Figure 2). Questioning the patient can confirm that bullous lesions appear immediately or a few hours after a trauma which can be minimal. This fragility can be quantified by applying an analogical visual scale. Because in EBA the subepidermal cleavage is deep on the dermal side of the basement membrane zone (BMZ), the cutaneous blisters can persist for a long time, collapsing and becoming flaccid before their rupture; they can also be haemorrhagic.
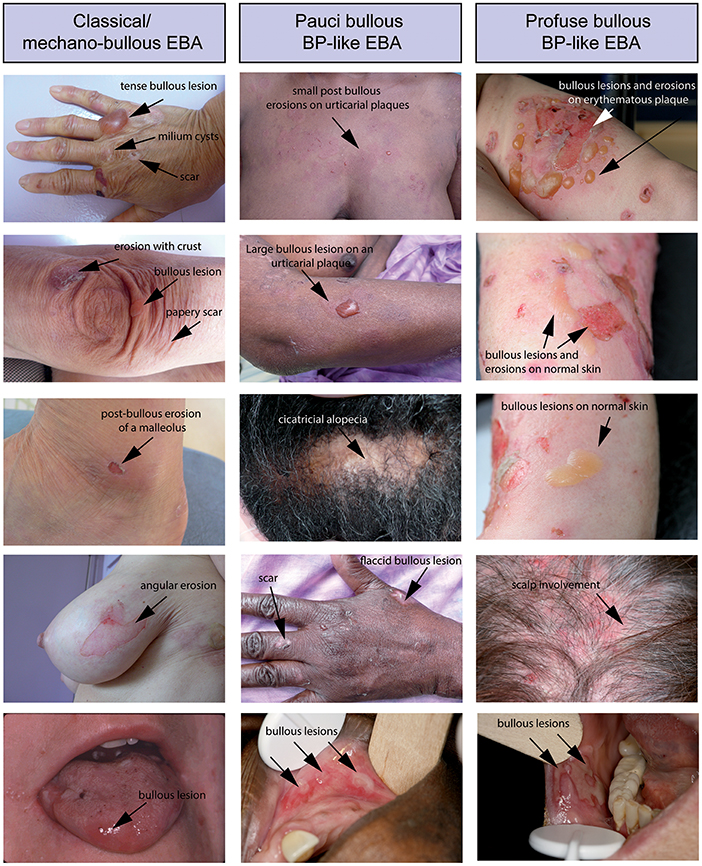
Figure 2. Different clinical forms of EBA. All patients were documented at the first visit in a center for auto-immune bullous disease. (Left) Patient with a classical/mechanobullous form of EBA: lesions are preferably localized to the extensor skin surfaces and trauma-prone sites, i.e., dorsal hands knees elbows and ankles. Tense or flaccid bullous lesions are surrounded by non-inflamed skin; erosions are covered or not by crusts; one erosion with angular contours had been induced by adhesive plaster; old lesions have healed with milium formation and/or are atrophic papery scar. (Middle) Patient with a BP-like form of EBA, with little blistering: urticarial plaques with small or large bullous lesions as in BP, but location of lesions on extensor areas of limbs, hands and scalp, and scars and extensor areas of the face (not shown) and limbs (atypical for BP). (Right) Patient with a BP-like form of EBA, with extensive blistering: bullous lesions and erosions on erythematous skin in flexural areas of limbs (tight and arm) as in BP but also bullous lesions and erosions on normal skin and involvement of extensor area of the limbs and scalp, atypical for a BP involvement of the scalp (not shown) and both flexural and extensor areas of limbs extremities with bullous lesions and erosions on erythematous but also normal skin. The tongue and the lips are the most frequent sites of mucosal lesions in all EBA variants. Other mucosal lesions (not shown) are possible regardless of the variant of EBA The involvement of nasal and buccal mucous membrane are visible in all EBA variants.
Classical/Mechano-Bullous EBA
Two cases of an adult-onset, acquired blistering disease resembling patients with hereditary dystrophic epidermolysis bullosa were reported by Elliott (23). Other similar cases were described in early decades of the twentieth century (24, 25). The first to actually coin the term “epidermolysis bullosa acquisita” was probably Hundley and Smith (26). A landmark paper by Roenigk and colleagues was published in 1971, who described three new cases of EBA, reviewed the world literature and proposed the first diagnostic criteria for classical/mechano-bullous EBA (27).
The criteria of Roenigk were modified once immunological tests for EBA diagnosis had been developed. Advanced clinical criteria of the classical/mechanobullous form have been published in 2017 (16). Current clinical diagnostic criteria for the classical/mechanobullous form are skin fragility, blisters, or erosions on non-inflamed or scarred skin, scarring and milium formation, preferably located at trauma-prone sites and the extensor skin surface (dorsal hands, elbows, knees, Achilles tendon, feet) with possible nail dystrophy and scarring alopecia (Figure 2). Fibrosis of the hands and fingers leading to a mitten-like deformity may occur in patients with severe disease reminiscent of hereditary dystrophic epidermolysis bullosa. Mucosal involvement may also occur, but it is not predominant.
BP-like EBA
BP-like form of EBA was first described by Gammon et al. (28). This occurred shortly after the demonstration by Nieboer et al. (29) and Yaoita et al. (30) that EBA autoantibodies are deposited in the anchor fibrils zone, which allowed for a definite diagnosis by immunoelectron microscopy (IEM), even though the clinic was (based on the criteria by Roenigk) atypical. Indeed, patients with a BP-like form of EBA have generally profuse skin lesions suggestive of a BP in some areas and an EBA in others (17, 31, 32). The patients have pruritus, tense bullae and erosions on inflamed erythematous or urticarial skin as well as trauma-induced bullous lesions surrounded by normal skin (Figure 2). The lesions are on the trunk and folds but also on limb extensor areas and distal extremities. The face can be affected. Mucosal involvement is also possible, but it is not predominant. Finally, lesions heal most often leaving atrophic scars and milia cysts as in the mechanobullous form.
MM-EBA
The high frequency of mucosal lesions in EBA, in particular tongue and lip involvement, was highlighted by Dahl (33). Currently, MM-EBA cases are defined as EBA that mainly affects mucous membranes with a squamous epithelium (16, 33–35), such as the mucous membrane of the mouth, pharynx, esophagus, epiglottis, conjunctiva, genitalia, anus, and respiratory tract in malpighian metaplasia, especially the trachea and bronchi. Only one of these sites can be involved and remain so for a long time before a second localization appears in the case of inadvertent discontinuation of the treatment, a decrease in dose or no treatment (36). These cases are frequently misdiagnosed.
As on the skin, bullous lesions of mucous membranes rupture late in MM-EBA. Thus, intact blisters are frequently seen on mucous membranes in comparison to mucous membrane pemphigoid (MMP), in which they are rare (Figure 2). The erosions on the mucous membranes are similar in MM-EBA and classical MMPs, except in the esophagus where they can be linear, caused by mucous membrane fragility and the passage of the fibroscope (37). The cicatricial lesions (atrophic scars, synechiae, and stenosis) are identical in MM-EBA and MMP. The cicatricial lesions have mild consequences in the mouth, genitals and anus, but cause severe impairment in the esophagus, larynx, trachea, bronchi, and conjunctiva, which dictate more aggressive treatment and multidisciplinary management.
Esophageal stenosis, usually as a web located at the upper esophagus, causes the most severe damage to the esophagus. Esophageal stenosis causes dysphagia, weight loss and, at worst, malnutrition and/or false routes and pulmonary infection (34, 36–50).
Severe lesions at the nose and throat are perforation of the nasal septum and/or stenosis of nostrils, choanal, pharynx, and larynx (39, 51–53). Involvement of the trachea and bronchi may also rarely occur (51, 54). Scarring of the larynx or trachea are potentially life-threatening because this may lead to asphyxiation if tracheostomy is not performed. In general, mucous membrane lesions in EBA patients are, however, asymptomatic in 30% of cases (51).
Few case reports and small series of ocular involvement in EBA have been reported (13, 33, 34, 55–64). Patients presented the involvement of at least two other sites. Interestingly, in MM-EBA, immune deposits of IgA are present in half of the cases and the only Ig class in a third. Patients displayed a fibrosing conjunctivitis that might worsen and eventually cause blindness.
The frequency of esophageal, nasal and throat, and conjunctival involvement was, respectively, 6, 11, and 25% of 39 EBA cases in a French series (65). Bladder involvement has also been reported in one case (66).
In addition to MM-EBA, the involvement of mucous membranes in “skin-predominant” EBA is common (34). Hence, after the diagnosis of EBA, an interdisciplinary approach is needed for both diagnosis and treatment.
IgA-EBA
Currently, IgA-EBA is defined as an EBA that presents with linear IgA deposits at the BMZ. IgA-EBA may resemble linear IgA bullous disease with erythematous cutaneous arciform lesions and a few scars and milium cysts, in particular in children. However, it IgA-EBA may also develop into a more severe clinical manifestation, especially with scarring at mucosal sites, as reported in 30% of the 82 cases in the Vodegel literature review, including 4% with severe ocular involvement (16, 19, 67, 68).
Brunsting-Perry Type EBA
Patients with Brunsting-Perry type EBA present only cutaneous lesions, without erythematous or urticarial plaques, which predominate in the head and neck and heal leaving very atrophic scars. Review of seven of the eight cases reported in the literature has recently been published by Asfour et al. (69–76).
Non-Inflammatory vs. Inflammatory Forms of EBA
The definition of inflammatory and non-inflammatory forms of EBA varies with the authors in the literature (17–21): (i) for most of them but one, the non-inflammatory form of EBA overlays the classical/non-mechano-bullous form; (ii) Buijsrogge et al. included the Brunsting-Perry like type in the mechano-bullous phenotype (19); (iii) for Briggaman et al. (17) who were the first to describe the inflammatory form of EBA, and for Kim et al. (18), the inflammatory form of EBA is synonymous to the BP-like form; (iv) for Buijsrogge et al. (19); and Iranzo et al. (20), all the patients who have not a mechano-bullous phenotype form, have an inflammatory phenotype and (v), for Seta et al. (21), inflammatory lesions are characteristic of the BP-like form of EBA but can also be seen in some patients with MM-EBA or IgA EBA.
The authors of the consensus conference (16) agreed on the following: (i) BP-like EBA are usually inflammatory forms of EBA, (ii) MM-EBA may be inflammatory forms of EBA, (iii) IgA-EBA may be inflammatory forms of EBA, (iv) Brunsting-Perry-like EBA are usually non-inflammatory forms of EBA, and (v) Brunsting-Perry-like EBA are not classical/mechano-bullous forms of EBA.
Quality of Life in EBA and Associated Disorders
In general, EBA has a significant impact on the quality of life, which is now to be measured by generalized scores and the “autoimmune bullous disease quality of life” (ABQOL) and “treatment-based autoimmune bullous disease quality of life” (TABQOL) scores created specifically for autoimmune bullous diseases (77, 78).
Many systemic diseases have been reported to be associated with EBA, such as amyloidosis, thyroiditis, multiple endocrinopathy syndrome, rheumatoid arthritis, pulmonary fibrosis, chronic lymphocytic leukemia, thymoma, and diabetes [review in Gupta et al. (22)]. Most of these reports are, however, anecdotal. The only unarguable association of EBA with other diseases is with chronic inflammatory bowel diseases, in particular Crohn's disease, which has been reported to be present in 25% of EBA patients (65, 79). In B-cell lymphomas, presence of circulating and tissue-bound auto-antibodies to type VII collagen (COL7) has also been described in association with a frequency of 6% in 100 EBA cases, but the patients did not have clinical features suggestive of EBA (80). Furthermore, EBA associated with systemic lupus erythematosus, but not fulfilling the criteria of bullous erythematosus systemic lupus, are described (81, 82).
Diagnosis
If clinically suspected, the minimal diagnostic criteria for EBA diagnosis are the detection of linear immunoglobulin- or C3-deposits along the dermal-epidermal junction in a perilesional skin biopsy with detection of a u-serrated pattern of Ig-binding.
Routine histopathology from a lesional skin (or mucous membrane) biopsy does not allow to distinguish EBA from other subepidermal AIBD. It shows: (i) initially, papillary oedema and vacuolar alteration along the dermo-epidermal junction and at a later stage, a subepidermal or subepithelial cleavage, (ii) a great variability in the magnitude and/or quality of the inflammatory infiltrate, (iii) milia cysts and fibrosis in older lesions (Figure 3).
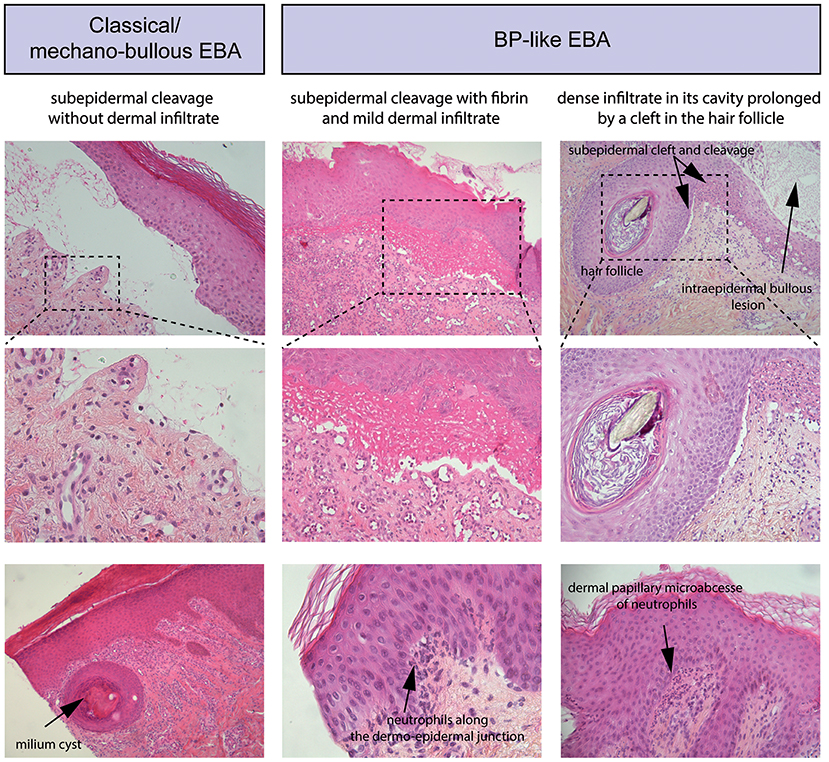
Figure 3. Standard histology. (Left) Histological study of lesional skin biopsies in a patient with a classical/mechanobullous form of EBA, subepidermal cleavage without dermal infiltrate; bottom, milium cyst in the dermis. (Middle) Patient with a BP-like form of EBA, subepidermal cleavage with fibrin in the blister cavity and mild dermal infiltrate. Bottom, Neutrophils along the dermo-epidermal junction (Right) Patient with a BP-like form of EBA, one intraepidermal bullous lesion and one subepidermal bullous lesion with dense infiltrate in its cavity prolonged by a cleft in the hair follicle; bottom, a dermal papillary microabcesse of neutrophils.
Definite diagnosis can be performed by either of the following methods: (i) serration pattern analysis of linear immunoglobulin deposits in the perilesional skin biopsy, (ii) fluorescent overlay antigen mapping (FOAM), (iii) immunoelectron microscopy, and/or (iv) detection of circulating antibodies against COL7.
If possible and needed, more than one of the above may be used to diagnose or exclude EBA. In addition, serology, i.e., detection of circulating anti-COL7 antibodies, should be performed, and, if positive, can serve as a biomarker of disease severity (83).
Diagnosis of EBA can be made by indirect immunofluorescence microscopy (IIF) using 1 M NaCl-split skin (SSS) as a substrate (84). Here, binding of antibodies to the dermal site (floor) of the blister is observed. By immunoblot analysis, binding to the 290-kDa antigen by the patient IgG is detected. The newly developed COL7 ELISA has a sensitivity of 45%. Combining SSS and ELISA reaches a sensitivity of 50%. Thus, half of the patients with EBA are sero-negative (85), and thus a negative serological finding does not exclude EBA as a differential diagnosis.
In serological negative cases, direct immunofluorescence (DIF) on sodium chloride-separated skin biopsy might reveal the diagnosis. Specifically, the diagnosis can be made using DIF serration pattern analysis, which shows distinct, EBA-specific, linear u-serrated immune-depositions at the BMZ (86). DIF serration pattern analysis by n-vs.-u may consider require expertise, which can be studied online: “n-vs.-u UMCG” (https://www.umcg.nl/NL/UMCG/Afdelingen/dermatologie/Wetenschappelijk_Onderzoek/NversusU/Paginas/default.aspx.
Direct Immunodetection of Perilesional Skin Biopsies
All pemphigoid diseases are characterized by a linear deposition of immunoglobulins and/or complement along the epidermal basement membrane zone (Figure 4A). These antibodies are directed against various hemidesmosomal proteins: (i) type XVII collagen (BP180) in BP, MMP, pemphigoid gestationis, lichen planus pemphigoides, and LAD, (ii) BP230 in BP, (iii) laminin-332 in anti-laminin-332 pemphigoid, (iv) integrin β4 in ocular MMP, and (v) p200 in anti-p200 pemphigoid. Moreover, in EBA and bullous systemic lupus erythematosus (SLE), antibodies against COL7, present in the sublamina densa, also give rise to a linear deposition pattern (87).
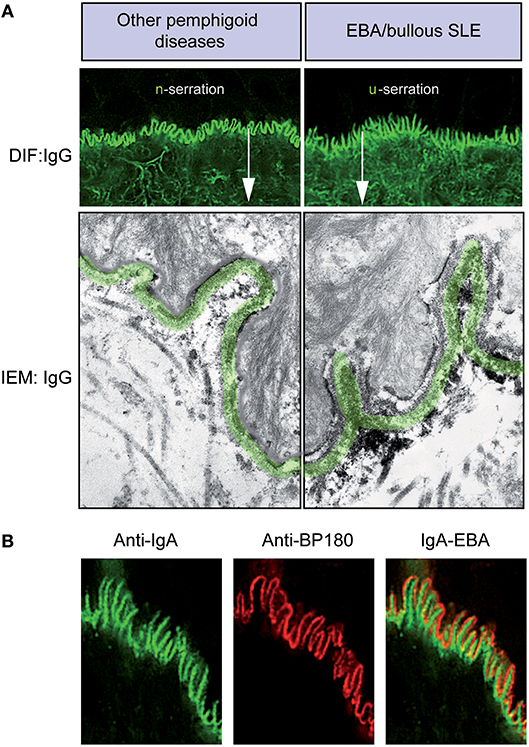
Figure 4. Diagnosis of EBA by direct immunofluorescence. (A) All pemphigoid variants are characterized by linear deposition of immunoglobulins and/or complement along the epidermal basement membrane zone. Serration pattern can be separated in an n-serrated pattern (blistering diseases with binding above the lamina densa with antibodies against hemidesmosomal components, e.g., BP, pemphigoid gestationis, mucous membrane pemphigoid, anti-p200 pemphigoid, and anti-laminin 332 pemphigoid, exept MM-EBA) and a u-serrated pattern (sublamina densa binding diseases caused by autoantibodies against COL7, e.g., EBA and bullous SLE). (B) In some cases, it is not possible to determine the serration pattern, especially in mucosal biopsies. In these cases, the level of the deposition of antibodies can be determined by fluorescent overlay antigen mapping (FOAM). FOAM is a technique that is based on the possibility of visualizing a targeted antigen relative to a topographic marker. For instance, using red staining, it is used for BP180 as a topographic reference marker, and green staining is used for IgA deposits. In the case of IgA-EBA, separate patterns of IgA deposits (green) and BP180 (red) can be observed with red staining on the dermal side.
If in suspected pemphigoid disease, a linear Ig- and/or C3- deposition is observed, it is important to determine the targeted autoantigen. In most variants of BP and in EBA, the deposits consist of IgG and complement. Mixed IgG/IgA depositions are usually encountered, especially in mucosal dominant pemphigoid. In some patients, IgA is the only present Ig-subtype, leading to a diagnosis of LAD or IgA EBA (67, 88, 89). However, in mucosal dominant pemphigoid with mixed IgA/IgG depositions, the IgG component may be faint, which sometimes result in a misdiagnosis of LAD. In very few patients, linear IgM deposition may be present in addition to IgG and C3. Even less cases have been described with linear IgM deposition only (90, 91).
Bullous SLE is characterized by antibodies against COL7 in patients fulfilling the diagnostic criteria for SLE. In bullous SLE, in addition to, or superimposed on a linear IgG deposition, a biopsy might show a lupus band, which is characterized by granular deposition of immunoglobulins and complement, in coincidence with epidermal anti-nuclear antibodies.
In most pemphigoid patients, a linear-serrated pattern can be discerned in direct IF microscopy of a peri-lesional skin biopsy. This serration pattern can be divided into an n-serrated and a u-serrated pattern (Figure 4A). The identification of these particular patterns allows to differentiate between (i) sublamina densa binding diseases caused by autoantibodies against COL7, e.g., EBA and bullous SLE and (ii) blistering diseases with binding above the lamina densa with antibodies against hemidesmosomal components, e.g., BP, pemphigoid gestationis, MMP, anti-p200 pemphigoid, and anti-laminin 332 pemphigoid. The u- and n-serrated patterns form based on the molecular architecture of the dermal-epidermal junction (DEJ): Specifically, if autoantibodies against COL7 are present, the immunodeposits are located between the rootlets of the basal keratinocytes, leading to the EBA-associated u-serrated pattern (Figure 4A). If, however, immune-depositions are located above the lamina densa, they trail the plasma membrane in the basal cell rootlets, resulting in the n-serrated pattern (Figure 4A). If it is not possible to determine the serration pattern, which occurs in few cases, it is recommended wise to cut thinner sections or obtain an additional peri-lesional skin biopsy.
However, if diagnosis of EBA cannot be established based on the serration pattern, the level of antibody deposition can be determined by fluorescent overlay antigen mapping (FOAM), which is based on the visualization of a targeted antigen relative to a known topographic marker. For instance, in Figure 4B, red staining is used for BP180 as a topographic reference, whereas green staining corresponds to IgA deposits. In the case of IgA-EBA, separate patterns of IgA deposits (green) and BP180 (red) can be seen with red staining on the dermal side (89). FOAM can be performed using a standard immunofluorescence microscope. However, confocal microscopy usually yields better results.
Direct immunoelectron microscopy provides a more detailed location of the deposition site, i.e., the lamina densa and/or sublamina densa at the DEJ (29). This location is distinct from that in other AIBDs (29, 30, 92).
Indirect Immunofluorescence on Salt-Split Skin
Diagnosis of EBA can be made by indirect immunofluorescence (IIF) using 1 M NaCl-split skin (SSS) as a substrate. Here, antibody binding of to the dermal site (floor) of the blister is observed. By immunoblot analysis binding to the 290-kDa antigen of patient immunoglobulin is detected (84). Using SSS as a substrate, IIF shows IgG binding on the dermal side of the split. IgG binding can therefore be easily distinguished from BP and, in some cases, MMP, in which the immunoglobulins bind on the epidermal side of the split.
COL7-Specific Serological Analyses by ELISA
For the serological diagnosis of EBA, three different assays are available: (i) an enzyme-linked immunosorbent assay (ELISA) that uses the non-collagenous (NC)1 and NC2 domains of COL7 (15, 93–95), (ii) an ELISA that is based on the NC1 domain alone (83), and (iii) an indirect IF test employing the NC1 domain (83). If they can be detected, serum levels anti-COL7 IgG correlate with disease activity (96). However, no correlation has been detected between the antibody specificity and the clinical phenotype (97).
Therapy
First, the treatment of EBA remains challenging because no randomized control trials have been documented due to its rare prevalence (98, 99). Similar to other AIBDs, systemic corticosteroids are widely accepted as a first choice in the treatment of EBA. Initial doses range from 0.5 to 2.0 mg/kg/day (87). With systemic corticosteroids, steroid-sparing agents, including colchicine, diaminodiphenyl sulfone (DDS, dapsone), methotrexate (MTX), azathioprine (AZA), cyclosporine (CSA), mycophenolate mofetil (MMF), and cyclophosphamide (CPA), have been reported in treatment of EBA. Other therapeutic options, including high-dose intravenous Immunoglobulin (IVIG), rituximab (RTX), plasmapheresis and immunoadsorption (IA), and extracorporeal photochemotherapy (ECP) also have been reported (98, 100).
A retrospective analysis included 30 cases from the EBA cohort study regarding treatment and outcome (18). In this cohort study, the median time to remission was 9 months, and complete remission (CR) was observed in 33, 33, and 45% at 1 year, 3 years, and 6 years follow-up, respectively. Relapse sometimes occurs while receiving therapy, while a rate of relapse has not been reported in cohort studies.
Very recently, we collected information that included the treatment of EBA cases who met current diagnostic criteria published between 1971 and 2016 (101). Among all the reported treatments, we found IVIG and RTX to be associated with CR. Based on this retrospective analysis and previous reports in other autoimmune bullous diseases as a reference, each treatment is shortly summarized below.
Colchicine
In EBA, colchicine is usually used at 1–2 mg/day. Of note, colchicine monotherapy (2 mg/day) has been reported (20, 102). By some experts, colchicine is considered a first line EBA treatment, especially for mild cases due to the relatively minor side effects compared with other therapeutic choices (103–105). There is currently no controlled study reporting the efficacy of this treatment, and no experimental data are available. Hence, colchicine is a triable treatment for mild cases, and it should be considered if other immunosuppressant treatments are ineffective.
Diaminodiphenyl Sulfone (DDS, Known as Dapsone)
Based on expert opinion, DDS is considered a safe and relatively effective first line treatment (18). Although DDS monotherapy (100 mg/day) has been reported with CR in single IgA-EBA cases (67), 25–150 mg/day of DDS is usually used as an adjuvant therapy with systemic corticosteroids. Adverse effects, haemolysis, methemoglobinemia, agranulocytosis, peripheral neuropathy, and psychosis might be observed (106). A rare adverse effect called “dapsone syndrome” should be considered. Some experts recommend that this therapy can be effective (105, 107). Similar to BP (108), DDS appears to have a corticosteroid-sparing effect in EBA.
Methotrexate (MTX)
Usually, MTX is used in combination with systemic corticosteroid with/without other immunosuppressants at 20–25 mg weekly. A retrospective review for MTX in the treatment of pemphigus and pemphigoid has reported the use of 5–50 mg weekly of MTX. Adverse effects, including nausea, anemia, and infection sometimes led to the discontinuation of treatment (109). In EBA, no case-series study has focused on the efficacy of MTX. Based on the efficacy in BP (109), MTX may be a viable treatment for EBA as a corticosteroid-sparing agent.
Azathioprine (AZA)
In EBA, AZA was the most frequently used immunosuppressant as an adjuvant therapy (101). Two types of adverse effects have been reported; non-dose-related adverse effects, including pancreatitis, fever, rash, malaise, nausea, diarrhea, and hepatitis, and dose-related adverse effects, including leucopoenia and some forms of hepatitis (110). Like MTX, no case-series study has focused on the efficacy of AZA in EBA. Based on the efficacy in BP (108), AZA may not be a beneficial treatment for EBA, and further analysis is needed.
Cyclosporine (CSA)
CSA is mainly used as an adjuvant therapy (101). Renal dysfunction, hypertension, headache, tremor, paraesthesia, hypertrichosis, and hyperlipidaemia may be observed as adverse effects, and most persistent renal dysfunction is related to prolonged therapy or doses >5 mg/kg/day (111).
Mycophenolate Mofetil (MMF)
In our recent, retrospective analysis (101), 1–3 g/day of MMF was used in combination with systemic corticosteroids. A randomized clinical trial of methylprednisolone plus-AZA vs. -MMF therapy in pemphigus showed a slightly lower frequency of adverse effects, including hypertension, hyperglycaemia, and infection in the MMF-treated group, although it was not a significant difference (112). The results from the randomized control study in pemphigus suggested that 2 g/day offered a better risk–benefit profile than 3 g/day (113). In a case-series study, EBA cases were successfully treated with MMF as a steroid-sparing agent (114). Hence, MMF could be a steroid-sparing agent in EBA.
Cyclophosphamide (CPA)
There has been no case-series study focused on the efficacy of CPA in EBA, and relativity fewer reports have examined treatment with CPA compared with other immunosuppressants. Therefore, CPA appears to be a therapeutic option when other immunosuppressants cannot control the disease, but limited data is available (101).
High-dose Intravenous Immunoglobulin (IVIG)
In our recent, retrospective analysis (101), 31 EBA cases were treated with IVIG, among which 24 cases providing information on the regimen of IVIG and outcome are summarized in Table 2. Ahmed et al. reported 10 cases treated with 2 g/kg/cycle (divided into 3 consecutive days) of IVIG in EBA with severe disease and non-responsive to conventional therapies (118). After 16–22 cycles of IVIG therapy, clinical remission was observed from 29 to 123 (mean 53.9) months without any other therapies. The main adverse effect was headache, which increased the intervals of each infusion in two cases, although no severe adverse effects were observed. These two cases are also well summarized in previous reports of IVIG treatment in EBA. In most cases, 2 g/kg/cycle for 3 days or 400 mg/kg/day for 5 sequential days were used with clinical improvement. In experimental mouse studies of EBA and BP, beneficial effects of IVIG have been demonstrated (128–131). Recently, a randomized double-blind trial of IVIG was reported in BP showing a lower disease activity score in the IVIG-treated group compared with placebo (132). Therefore, IVIG is an effective treatment in severe or intractable cases of EBA.
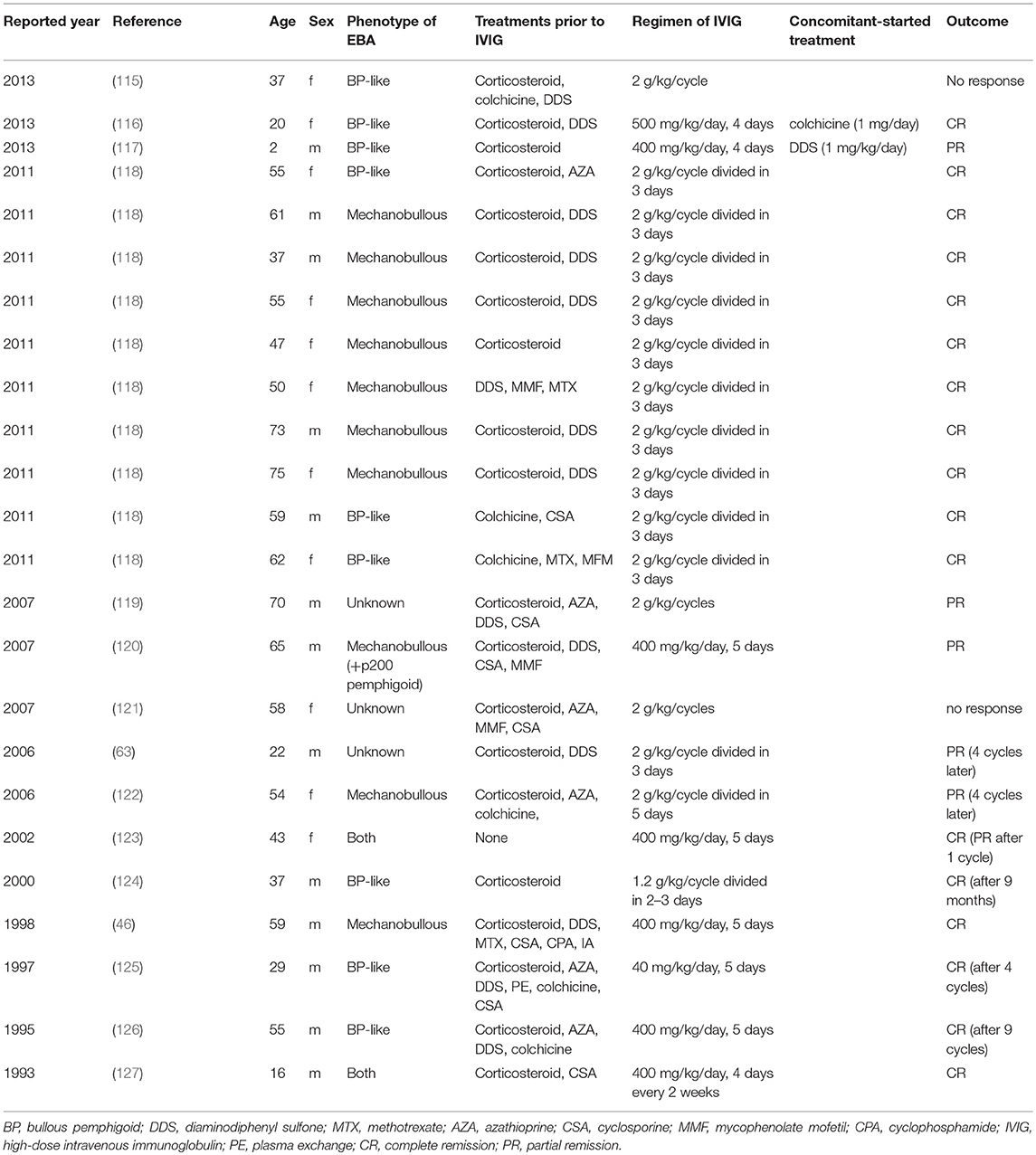
Table 2. Reports of IVIG treatment in EBA.
Rituximab (RTX)
Rituximab, a humanized anti-CD20 monoclonal antibody, was used in 10 cases with a protocol of 375 mg/m2 weekly for 4 weeks (in most cases) and was associated with CR in our recent, retrospective analysis (101), which is summarized in Table 3. In pemphigus, another regimen, 1,000 mg every 2 weeks on day 0 and day 14 twice and 500 mg at 12 and 18 months, showed good efficacy in an open-label randomized trial (140). A recent study reported 4 EBA patients treated with 1,000 mg of RTX every 2 weeks twice, similar to the regimen in pemphigus with PR and CR outcomes in each case and no response in 2 cases (141). In EBA animal models, depletion of B cells at the induction of experimental disease showed that B cells, in addition to developing into plasma cells, serve as important antigen-presenting cells. Specifically, if anti-CD20 treatment was applied at the time of immunization, development of antigen-specific CD4+ T cells was significantly hampered in immunization-induced EBA (142). Hence, RTX seems to be a promising treatment option for EBA. Further controlled clinical studies are required to determine which regimen of RTX is most effective in EBA.
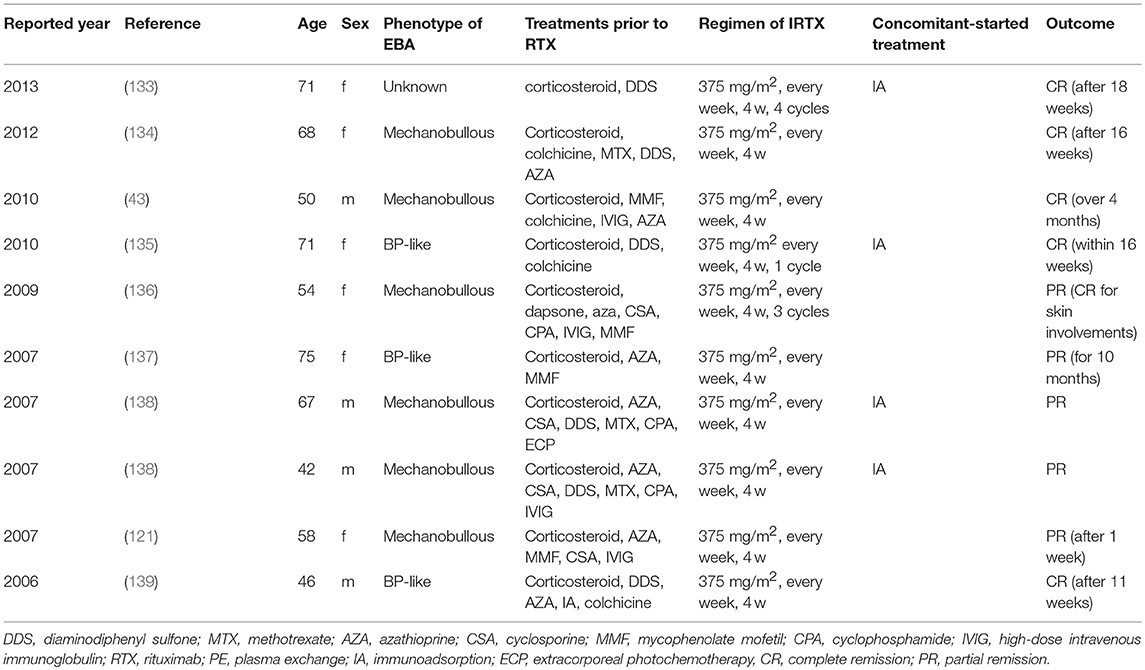
Table 3. Reports of RTX in EBA.
Plasmapheresis and Immunoadsorption (IA)
Although plasmapheresis has been used for the treatment of pemphigus and pemphigoid, including EBA, it makes the shift to IA because of its advantages compared with plasmapheresis: (i) selective removal of immunoglobulin from the circulation; (ii) no requirement for the substitution of plasma components, such as human albumin or fresh frozen plasma; (iii) two to three times more processing capacity per treatment session than plasmapheresis; and (iv) fewer side-effects, such as infections and allergic reactions (90). Interestingly, there are several reports of combination therapy with IA and RTX in EBA that might provide an effective treatment protocol in EBA (133, 135, 138). Kolesnik et al. (133) and Kubisch et al. (135) reported that each patient was treated with IA for 3 consecutive days followed by IA every week and 375 mg/m2 of RTX on the day after IA for 4 weeks, leading CR after 18 months and CR within 16 weeks, respectively. Niedermeier et al. (138) reported 2 intractable cases treated with 2 cycles of IA for 4 consecutive days at 4-week intervals followed by RTX (375 mg/m2, every week for 4 w), leading to PR. Interestingly, antigen-specific immunoadsorption, i.e., where only autoantibodies specific for the respective autoantigen are removed from the circulation, are in pre-clinical development (143, 144).
Extracorporeal Photochemotherapy (ECP)
ECP has been reported in the treatment of Sezary syndrome, mycosis fungoides, and autoimmune bullous diseases (145). There are several reports of the use of ECP in refractory EBA with outcomes of CR and PR in 3 cases, respectively, and no response in 1 case (61, 100, 146, 147). The mode of action of ECP in the treatment of EBA is still unknown, although one report has shown a decrease in circulating antibody detected by immunofluorescence and an increase in suction blister time (100). Despite the low number of published EBA patients, due to the reported success rates, ECP should be considered a therapeutic option in patients with refractory EBA.
Others
Daclizumab, a humanized monoclonal antibody against the a-subunit of the high-affinity interleukin-2 receptor also known as the Tac antigen or CD25, was reported in the treatment of EBA (148), in which only one of 3 cases showed clinical improvement. Sulfasalazine was used in a patient with EBA associated with Crohn's disease, resulting in no improvement of the skin lesion (149). The usefulness of doxycycline has been reported in BP (150). Doxycycline and another tetracycline, minocycline, were found in the literature on EBA cases, although its usefulness remains unclear in EBA treatment (151–153).
Prognosis
EBA is a chronic disease characterized by exacerbations and remissions over the course of months to years. Although data on the prognostic factors in EBA are lacking in the literature, the experts admit that the prognosis of EBA depends on its severity at the time of diagnosis and propose treatment accordingly.
Analogous to BP (154, 155) and MMP (35), an EBA is considered severe if the patient has 10 or more cutaneous bullous lesions and/or 3 or more instances of mucosal sites and/or conjunctival, laryngo-tracheal or esophageal involvement. Otherwise, the EBA is classified as moderate or minimal. The MMP-DAI (disease activity index) score (156) can be used to quantify the extent of the disease, but the cut-offs between the severe, moderate, and minimal forms of EBA have not been established to date.
The goal of the treatment is to obtain control of the disease followed by CR, i.e., the absence of active lesions (erythema, urticaria, bullous lesions, and erosions) without worsening of the cicatricial lesions, which are irreversible.
A CR off treatment of EBA is not possible since a long-term maintenance treatment is recommended. CR under minimal treatment may occur after months to years in mild or moderate forms (unpublished data), but minimal skin fragility without bullous lesions can persist for several months to years. The milium cysts may eventually disappear.
The prognosis has been reserved in severe forms, as evidenced by the publication of numerous case reports in therapeutic failure. Indeed, in some patients, the disease may progress quickly with periods of severe exacerbation and rapid scarring. The cicatricial lesions (synechiae, stenosis, joint contractures) may engage the functional prognosis and be life-threatening. In a retrospective study of 30 patients with EBA, all of whom were initially treated with a combination of methylprednisolone, dapsone, and colchicine (six who did not respond were subsequently treated with other immunosuppressants), 8 of 24 patients (33 percent) achieved complete remission and 5 of 24 (21 percent) achieved partial remission within 1 year (18). The prognosis of these severe forms could improve because of recent publications demonstrating the therapeutic success of intravenous immunoglobulins and rituximab (see above). The overall prognosis and response to treatment may be more favorable in children than in adults (157, 158).
Taken together, these findings underline the need for an early diagnosis, multidisciplinary care by experienced practitioner and prompt implementation of appropriate treatment to improve the prognosis of EBA.
Pathogenesis
COL7 as the Autoantigen in EBA
Nearly a century after the first description of EBA, the carboxyl terminus of COL7 was identified as the autoantigen in EBA. Since that time, it has been shown that most patients develop autoantibodies that bind to epitopes located within the NC1 domain of COL7 (159–162), whereas antibody reactivity to either the collagenous domain (163) or the NC2 domain (164) is detected in a very small minority of patients. No correlation was detected between antibody specificity and clinical phenotype (159). In a recent multicentre study with 95 EBA patients, NC1/NC2 ELISA showed a higher sensitivity (97.9%) than NC1 ELISA (89.5%), supporting a considerable number of patients with antibodies against NC2 (95).
Interestingly, the humoral autoimmune response toward COL7 encompasses almost all IgG subclasses. Most commonly, COL7 autoantibodies are IgG, but in ~10% of EBA patients, IgA autoantibodies against COL7 are detected. Few cases of IgE- and IgM-COL7-reactive immunoglobulins have been described (101). The nature and/or cause of this broad immunoglobulin isotype reactivity against COL7 is, however, unknown.
Genetic and Environmental Factors Contribute to Tolerance Loss in EBA
As with most autoimmune diseases, the exact cause of the disease is unknown. With regard to EBA, the data indicate a certain genetic predisposition as well as a contribution of environmental factors to EBA pathogenesis. Due to the small number of EBA cases, it is difficult to study the influence of certain environmental factors or infections. EBA susceptibility is associated with genes in and outside the major histocompatibility complex (MHC) locus. Specifically, an association with the MHC locus (HLA-DR2) has been documented in humans in two independent studies (10, 11). The association with the MHC locus is also supported by animal studies, where an association of susceptibility to immunization-induced EBA is linked to the H2s locus (165). Evidence for the involvement of genes outside the MHC locus arises from one case of coincident EBA in members of a family provided further support for the genetic control of EBA (166). The contribution of genes outside the MHC locus is again underscored by corresponding observations in experimental EBA (165, 167, 168). First, when C57Bl6/J mice are immunized with COL7, they develop autoantibodies but no clinical disease. When mice on the same genetic background lack expression of the inhibitory Fc gamma receptor (FcγR) IIB, they also develop clinically overt blistering (165, 168). Similarly, mice carrying the EBA-associated H2s allele develop severe clinical disease when on the B6 genetic background but only moderate disease when on the C57BL/10 background (165, 169). To pinpoint the mutations associated with EBA susceptibility, mice of an advanced, autoimmune-prone intercross line were immunized with COL7. Herein, one third of the mice developed clinical disease, while the remaining mice remained phenotypically healthy (170, 171) Classical quantitative trait loci mapping identified several genes outside the MHC that were associated with either the onset or severity of clinical disease (170). However, the number of genes is still too large to pinpoint the association with clinical disease to single genes, yet in a nutshell, it provides evidence for a genetic basis of EBA susceptibility.
In addition to genetic factors, animal models of EBA clearly indicated an influence of resident microbial communities in disease pathogenesis (172, 173) (Figure 5). By the use of outbred mice in immunization-induced EBA, it could be shown that Firmicutes were the most abundant (54%), followed by Proteobacteria (21%), Actinobacteria (12%), and Bacteroidetes (6%), which is similar to previous studies in the skin. At the genus level, Staphylococcus (36%), Corynebacterium (9%), and Ralstonia (8%) were most abundant (172). Ellebrecht et al. used the same model to show skin community changes before and after immunization. Among SJL/J mice that were immunized with COL7, only 80% of the mice developed disease, whereas the others remained healthy. Interestingly, the specific antibody concentrations and binding of antibodies to the DEJ were unaffected. By contrast, immunized mice that did not develop clinical phenotypes showed a greater alpha diversity, compared to mice that developed EBA symptoms after immunization (173).
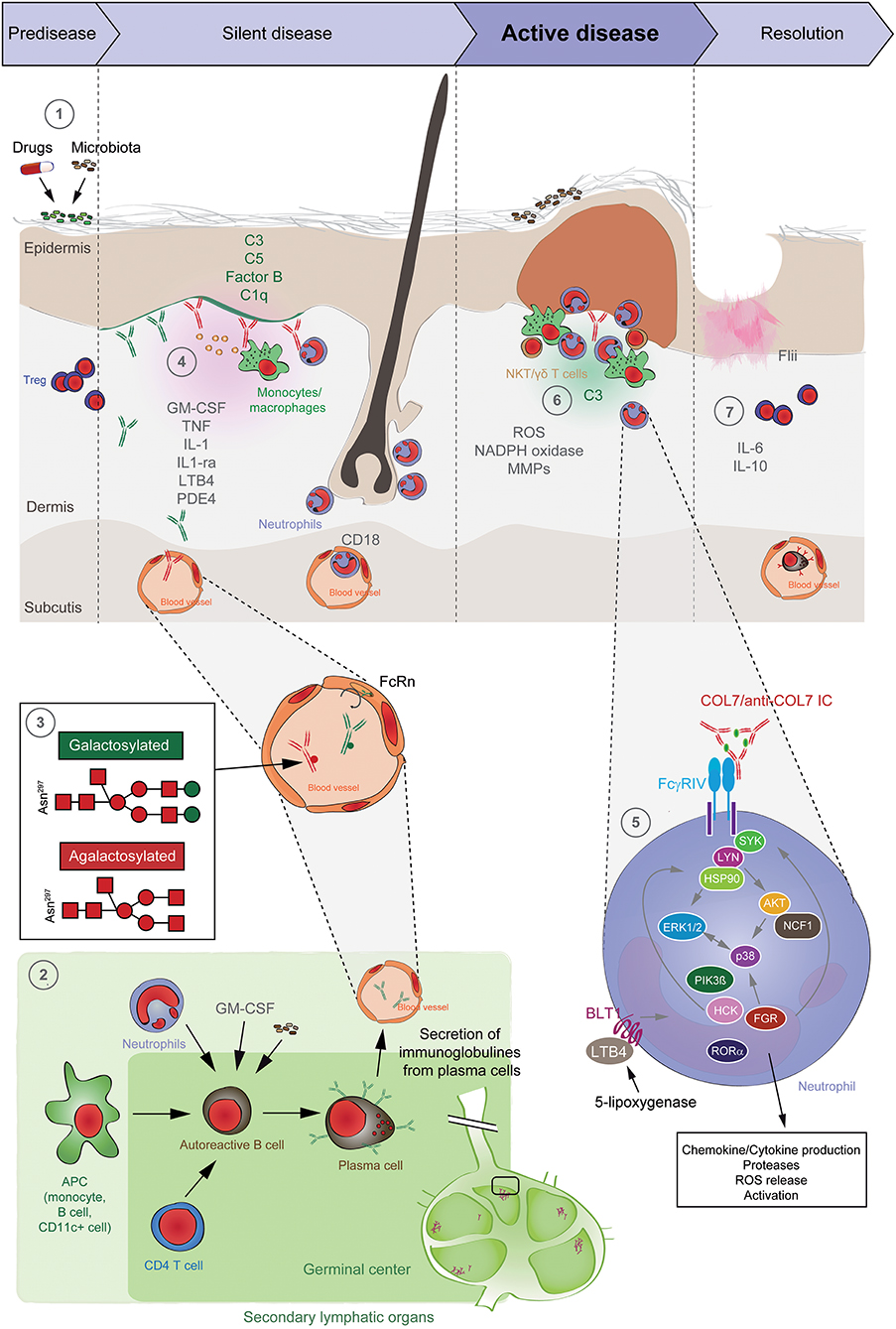
Figure 5. Pathogenesis of EBA. (1) Genetic factors and the skin microbiome promote a tolerance loss. (2) This phenomenon is mediated by the interaction of APCs with autoreactive B and T cells, leading to clonal expansion and differentiation into plasma cells. Autoantibodies against COL7 are released into the blood circulation and effector organs. (3) During inflammation, galactosylation of antibodies may differ. High galactosylation of IgG is crucial for these anti-inflammatory properties, whereas low galactosylation is pro-inflammatory. (4) Binding of autoantibodies to DEJ in the skin induces complement deposition, pro-inflammatory cytokine and mediator release and subsequently leukocyte extravasation. (5) Immune complexes bind in a Fc-dependent manner to neutrophils and induce a signaling cascade leading to activation, including the (6) release of ROS and matrix metalloproteases. In addition to neutrophils, other cell types are involved in split formation, as shown for monocytes/macrophages, NKT and γδ T cells. By contrast, Treg cells have an inhibitory effect on EBA progression. (7) Resolution of autoantibody-induced tissue injury. Treg, regulatory T cell; NKT, natural killer cell; C, complement; GM-CSF, granulocyte-macrophage colony-stimulating factor; IL, interleukin; LTB4, leukotriene B4; PDE4, phosphodiesterase 4; ROS, reactive oxygen species; NADPH, nicotinamide adenine dinucleotide phosphate; MMPs, matrix metalloproteinases; APC, antigen-presenting cell; CD, cluster of differentiation; SYK, spleen tyrosine kinase; Lyn, tyrosine-Protein Kinase Lyn; HSP, heat shock protein; AKT, protein kinase B; NCF1, neutrophil cytosolic factor 1; ERK, extracellular signal-regulated kinase; PI3K, phosphatidylinositol-4,5-bisphosphate 3-kinase; HCK, tyrosine-protein kinase HCK; FGR, tyrosine-protein kinase FGR; RORα, retinoid-related orphan receptor-alpha; BLT1, leukotriene B4 receptor 1; LTB4, leukotriene B4.
The Afferent Phase: CD4 T Cell-Dependent Production of Auto-Antibodies Against COL7
EBA is characterized and causes autoantibodies directed against COL7. Unfortunately, little human data are available for this relevant and complex phase of pathogenesis, which includes the interaction of various immune cells, such as antigen-presenting cells, autoreactive B cells, T cells and neutrophils, and subsequently leads to antibody production. Regarding human data, COL7-specific T cells (isolated from the blood) can be detected EBA patients (174, 175), but most data concerning the interaction of these cell in the afferent phase has been derived from animal models (167).
In the immunization-induced EBA model, T-cell-deficient mice do not develop COL7-specific antibodies and, consequently, clinical disease development, indicating that autoantibody production in this model is T cell-dependent. Furthermore, disease susceptibility could be restored in T cell-deficient SJL/J mice by T cells transfer from COL7-immunized wild-type mice (176). To delineate T-cell subsets involved in the generation of anti-COL7 antibodies in this model of immunization-induced EBA, CD4+ and CD8+ T cells were depleted for 2 weeks, starting at immunization. Depletion of CD4+ T cells led to a delay of both autoantibody production and the clinical disease onset. By contrast, CD8+ T cells depletion at the same time period did not impact production of COL7-specific autoantibodies or clinical disease (142). Therefore, in experimental EBA, CD4+ T are required for induction of autoantibody production. Since only few and specific inbred mouse strains developed clinically overt disease after COL7 immunization, the autoantibody response of clinically healthy vs. diseased mice after immunization was contrasted. Here, complement-fixing antibodies were linked to clinical EBA manifestation (169). Furthermore, by determination of the IgG isotype of the autoantibodies, a Th1 polarization of the immune response was noted. In addition, an increase in the Interferon (IFN)-γ/Interleukin (IL)-4 ratio in the draining lymph nodes of EBA-susceptible mice compared with EBA-resistant strains was observed (169). Regarding the involvement of neutrophils in autoantibody production, GM-CSF-deficient mice generated less COL7-specific autoantibodies, which was paralleled by reduced neutrophil numbers in peripheral lymph nodes, draining the immunized site. The same effect was observed in neutrophil-depleted wild-type mice (177).
To further address which antigen-presenting cells are required the formation of antigen-specific CD4+ T cells, B cells were depleted in mice and subsequently immunized with COL7. In the absence of B cells, the antigen-specific CD4+ T-cell response was completely abolished. Furthermore, depletion of dendritic cells and macrophages had similar effects. Hence, the development of COL7-specific CD4+ T cells requires the presence of APC—specifically B cells, dendritic cells, and macrophages (142) (Figure 5). More interestingly, the absence of Treg cells in scurfy mice cells led to blistering via the formation of pathogenic autoantibodies, demonstrating a critical involvement of these cell types in the afferent phase of EBA (178, 179).
In contrast to T cells, autoreactive B cells are almost exclusively found in the peripheral lymph nodes in the immunization-induced EBA mouse model (180, 181), which may be due to missing expression of homing-associated CXCR3 and CXCR4 chemokine receptors. In immunization-induced EBA, COL7-specific plasma cells have a half-life of ~7 weeks. This resembles an intermediate between short- and long-living plasma cells (180). A similar intermediate plasma cell type is most likely also present in patients because autoantibody titers in patients with autoimmune bullous diseases slowly decline over a period of 8–12 weeks after B cell-depleting rituximab treatment (182).
An important molecular requirement for autoantibody production in experimental EBA is heat-shock protein 90 (Hsp90). Specifically, pharmacological HSP90 inhibition suppressed autoantibody production in immunization-induced EBA. In the same model, HSP90 blockade impaired the onset of clinical disease manifestation when injected prior to immunization with COL7. Furthermore, clinical disease progression was ameliorated when the compounds were applied in therapeutic experimental settings. Interestingly, B cell development was unaffected by the HSP90 inhibition, while T-cell proliferation was impaired. Overall, this identified T cells as targets of HSP90 inhibition in experimental EBA (174).
Circulation and Pathogenicity of Autoantibodies Against COL7
Targeting the Half-Life of Anti-COL7 Autoantibodies
After (auto)-antibodies are present in the circulation, their half-life is controlled by the neonatal Fc receptor (FcRn). FcRn is constructed as a heterodimer, consisting of an alpha-chain and a beta-2-microglobulin light chain (183). Among other functions, FcRn protects IgG from catabolism (184). Inhibition of the FcRn leads to the enhanced clearance of IgG, including autoantibodies. In antibody transfer-induced animal models of EBA (167), disease induction in mice is completely blocked (128, 185). However, this protection can be overridden by the transfer of large amounts of antibodies (185). Similarly, treatment of experimental EBA or other pemphigoid diseases with high doses of IgG (IVIG), which by saturation also inhibits the FcRn, reduces circulating autoantibody titers and leads to disease improvement (128, 129, 186, 187), although FcRn inhibition is most likely not the only mode of action of IVIG in EBA (131, 188, 189).
Interestingly, two clinical trials are currently being conducted to evaluate the safety and efficacy of FcRn inhibitory treatments in patients with other autoimmune skin blistering diseases (NCT03334058) (190). Hence, this may become a treatment option for EBA patients in the not too distant future (167).
Targeting the Pathogenicity of Anti-COL7 Autoantibodies
In addition to its half-life, the pathogenicity of an antibody can also affect disease progression. In general, IgG antibodies have one conserved N-glycosylation site in each of their constant heavy chain regions. These Fc glycans have a major impact on their structure as well as their effector functions. Non-galactosylated (agalactosylated; G0) IgG antibodies have long been thought to have pro-inflammatory effector functions in autoimmune patients with rheumatoid arthritis. In contrast, sialylated IgGs mediate anti-inflammatory effects. Recent evidence also suggests that pro-inflammatory immune responses, including autoimmune reactions, mainly induce antigen-specific G0 IgGs, whereas tolerance leads to the generation of immunosuppressive, galactosylated, and sialylated IgGs. Under normal conditions, differentially glycosylated IgGs clearly mediate their pro- or anti-inflammatory effector functions as immune complexes in an antigen-specific manner (191) (Figure 5). In agreement with these findings, the use of EndoS, an endoglycosidase derived from Streptococcus pyogenes that selectively hydrolyses the N-linked glycan of native IgG, impaired split formation at the DEJ in skin cryosections. In EBA mouse models, EndoS abrogated clinical disease induction in mice (192, 193). This observation raises the possibility that the glycosylation status of IgG can also affect the onset, severity and progression of disease.
The Efferent Phase of EBA: Attraction and Activation of Leukocytes Leads to Blister Formation
Based on the current understanding of EBA pathogenesis (Figure 5), the effector phase of EBA, i.e., autoantibody-induced inflammation and blistering, can be divided into (i) autoantibody binding to COL7, (ii) complement activation and the formation of a pro-inflammatory milieu, (iii) leukocyte extravasation, (iv) activation by Fcγ receptors, and (v) tissue damage. Mechanisms leading to non-inflammatory blistering in EBA are, in contrast, poorly understood. With the increased understanding of pathomechanisms of epitope-dependent pathogenicity-associated (194), non-inflammatory BP, new insights into mechanobullous EBA can be expected. However, due to a lack of data on non-inflammatory mechanisms of blistering in EBA, the following text relates to inflammatory EBA.
Autoantibody-induced tissue injury in EBA is initiated by (i) the deposition of autoantibodies at the DEJ. Apart from the skin anti-COL7 antibodies bind to the esophagus, stomach, small intestine, and colon because of the autoantigen expression at these sites (195, 196). Yet, not all isotypes of anti-COL7 have the potential to induce dermal-epidermal separation: Ex vivo, only human IgG1 and IgG3, but not IgG2 and IgG4, are capable to cause blistering (197). Furthermore, immune complexes containing IgA1 or IgA2 COL7 autoantibodies activate neutrophils and also induce subepidermal blistering when in cryosections of human skin. Of note, and in contrast to IgG1 autoantibodies, neither IgA1 nor IgA2 leads to complement deposition at the dermal-epidermal junction. Because complement activation has traditionally been thought a prerequisite for blister induction, this may be compensated by so far unknown soluble factors and/or by a stronger activation of neutrophil granulocytes when engaging IgA immune complexes (198).
Thereafter, (ii) a pro-inflammatory milieu is generated in the skin, which includes activation of the complement system (199). The complement system consists of circulating proteins that, upon activation, initiate a highly controlled cascade that is an integral part of the innate humoral immune response (200). C5-deficient mice (168) are either completely or (168) partially (201) protected from induction of experimental EBA by antibody transfer.
Dissecting the specific role of each complement activation pathway (classical, lectin, and alternative pathway) showed the following: MBL deficient mice showed a similar EBA phenotype to the wild-type controls. C1q-deficient mice showed weak and partial protection, while factor-B-deficient mice showed clinically relevant protection from EBA induction by antibody transfer (199). This identified the alternative pathway of the complement system as a main driver of skin blistering and inflammation in antibody transfer-induced EBA (202). Downstream of C5, C5ar1-deficient mice are significantly protected from experimental EBA, whereas C6-deficient mice developed widespread blistering disease, excluding the membrane attack complex as a pharmacological target for EBA. In line, pharmacological blockade of C5, factor B, or C5aR1 led to a significant improvement of the blistering phenotype in antibody transfer-induced EBA (202, 203).
In addition to the complement system, the lipid mediator leukotriene B4 (LTB4) is a potent granulocyte chemoattractant (204, 205) and activator (206) and is abundant in the blister fluids of bullous pemphigoid patients, but its pathogenic significance for pemphigoid diseases had been unknown until recently. LTB4 is biosynthesized from arachidonic acid through sequential enzymatic conversion by 5-lipoxygenase and LTA4-hydrolase. The 5-lipoxygenase is most abundant in neutrophils, and it is activated upon cell stimulation by, for example, immune complexes or the complement fragment C5a (207). Mice deficient in 5-lipoxygenase, a key enzyme in LTB4 biosynthesis, or BLT1-deficient mice are completely resistant to the induction of experimental EBA by antibody transfer (208). In addition to complement and lipid mediators, several cytokines have been identified to modulate the effector phase of EBA. Cytokines that are differentially regulated in experimental EBA (209) or cytokines associated with those functional data (210) are summarized in Table 4 (171, 224).

Table 4. Mediators of the EBA effector phase.
The pro-inflammatory milieu induces the (iii) attraction of different leukocyte populations (Table 5). Unfortunately, the composition of these cells has not been investigated thus far in EBA patients, but it is known from BP patients that the infiltrate includes cells such as lymphocytes, histiocytes, eosinophils, neutrophils, and mast cells (228, 229). Subsequent mechanistic studies using the antibody transfer-induced model have uncovered neutrophils as the major culprits responsible for blister formation (208). The recruitment of neutrophils into the skin is mediated by CD18- and ICAM-1 (216, 230–232). In addition, CD18 crucially regulates neutrophil adhesion as an indispensable step leading to tissue damage (233).

Table 5. Cell lineage in the effector phase.
Concerning the functional role of T cells, it was recently demonstrated that T cells can enhance neutrophil recruitment into the site of inflammation by modulating the expression of the cell surface integrin CD18 on neutrophils. Interestingly, this effect was neither mediated by CD4 nor CD8 cells, but rather γδT and NKT cells (225). Interestingly, blockade of Treg led to a dramatic worsening of the clinical disease manifestation in antibody transfer-induced EBA (224).
In addition to granulocytes and lymphocytes, macrophages/monocytes may be involved in the blister formation in EBA. However, the blockade of these cells is technically difficult and does not effectively impair disease progression (142). Nevertheless, more studies are needed because macrophages/monocytes are able to produce high amounts of reactive oxygen species (ROS) after immune complex stimulation and induce ex vivo split formation in human skin sections (214). Furthermore, the application of high concentrations of anti-COL7 IgG has been shown to induce mast cell activation, but mast cell deficient mice develop experimental EBA just like wild type animals, indicating that mast cells do not contribute to the immune-mediated tissue injury (234–236). Concerning the role of additional cell types in inflammation, a possible role of plasma cells (227) has been discussed, but further studies are needed to unravel the cellular orchestration responsible for the lesional sites.
After extravasation from the blood into the skin, (iv) myeloid effector cells bind to the skin-bound immune complexes in a FcγR-dependent fashion (Table 6). In EBA the full IgG molecule of the autoantibodies, but not their corresponding F(ab)2 fragments, are pathogenic. Specifically, only the full anti-COL7 IgG elicits dermal-epidermal separation when, together with PMN, incubated on cryosections of human skin (248). Likewise, and unlike the full IgG, F(ab′)2 fragments of anti-COL7 IgG do not induce clinical EBA manifestation when injected into mice (168). The central role of these Fc-FcγR interactions for mediating skin inflammation and subepidermal blistering in experimental EBA is further supported by the complete protection of mice toward EBA induction when injected with chicken anti-mouse COL7 IgY, which is known not bind to murine complement and Fc receptors (249). In addition, the therapeutic effects observed when blocking these interactions, i.e., using soluble CD32/SM101 (231), highlights the key role of Fc-FcγR interactions in EBA pathogenesis. Furthermore, IgG glycosylation has been shown to have preventive and therapeutic effects in mouse models of chronic inflammatory diseases, including EBA (191). Further studies eluted on the differential contribution of the different FcγR (250). In mice, three different activating FcγR and one inhibitory FcγR are described: FcγRI, FcγRIII, and FcγRIV are activating FcγR, all with specific binding avidities toward IgG. The FcγRIIB is the only inhibitory FcγR (250). Of note, an increased expression of FcγRIV has been demonstrated in the skin of mice with experimental EBA (181). Subsequent functional studies identified the FcγRIV as the key mediator of tissue injury in EBA. By contrast, blockade of FcγRI, FcγRIII, or both receptors in combination had no effect on the induction of experimental EBA by antibody transfer. In FcγRIIB deficient mice enhanced blistering was observed in antibody transfer-induced EBA, as well as BP (181, 251), indicating a protective role of this FcRγ in experimental EBA. In human ex vivo models of BP, FcγRIIA, and FcγRIIIB contributed to the autoantibody-induced tissue damage (252). Once the neutrophils are bound to the immune complexes, a multifaceted signaling cascade is initiated (Table 6). This involves activation of the retinoid-related orphan receptor (ROR)α (230), heat shock protein (HSP)90 (241), phosphodiesterase 4 (240), phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K) β and δ (238, 239), p38, AKT, ERK1/2 (244), the spleen tyrosine kinase SYK (171, 245), and src kinases (247), as well as CARD9 (246)—which have been reviewed in detail elsewhere (253). The exact temporal and spatial order of these signaling events is currently unknown. Ultimately, the signaling cascade leads to the activation of myeloid effector cells, specifically release of ROS and proteases, both of which are required for subepidermal blistering in EBA (216, 225).

Table 6. Receptors and signaling in the efferent phase of EBA.
Recently the contribution of cytokines in EBA pathogenesis has been eluted both on the morphological, as well as the functional level (254). Because biologics targeting cytokines are already in clinical use—although, with few exceptions such as rituximab and omalizumab, for other indications (255)—we here highlight their contribution for EBA pathogenesis. Several cytokines have been shown to contribute to blister formation in experimental EBA (Table 4). IL-6 acts as a pro-inflammatory cytokine in various autoimmune diseases (256, 257). Hence, IL-6 has emerged as a potential target for the treatment of autoimmune diseases, and IL-6-targeting therapies are licensed for rheumatoid arthritis and juvenile idiopathic arthritis (256). In EBA patients and in experimental EBA models, serum IL-6 levels are elevated. Unexpectedly, mice lacking IL-6 expression show an increased clinical phenotype in experimental EBA compared with controls. Furthermore, treatment with recombinant IL-6 leads to a dose-dependent reduction of clinical disease manifestation in EBA induced by antibody transfer. Thus, in experimental EBA, IL-6 has a profound anti-inflammatory activity (209). At the molecular level, the protective effect of IL-6 is mediated by the IL-6-dependent release of IL-1ra (213). In line, IL-1R–deficient mice or wild-type mice treated with either the IL-1R antagonist Anakinra or a IL-1β function blocking antibody are also partially protected from the EBA-inducing effects of anti-COL7 IgG. Mechanistically, IL-1β increases ICAM-1 expression on endothelial cells, suggesting that IL-1β supports recruitment of inflammatory cells into the skin. Interestingly, these effects of IL-1 were independent of caspase-1 because of caspase-1/11–deficient mice showed a similar phenotype to wild type control animals when injected with anti-COL7 IgG (213).
Additionally, the evaluation of cytokines affecting neutrophil functions, such as IL-8 (CXCL1 and CXCL2 in the mouse) and GM-CSF in experimental EBA, showed an increased expression. To evaluate if the increased expression is of functional relevance these cytokines were inhibited by either antibodies of mice lacking expression of the respective cytokine(s). Treatment with allosteric CXCR1 and 2 inhibitors (DF2156A) impaired the induction of skin blistering in antibody transfer-induced EBA.
In a therapeutic setting, the administration of DF2156A improves the clinical manifestation of EBA after disease onset in immunization-induced EBA (212). For the evaluation of GM-CSF, anti-COL7 IgG was injected into GM-CSF-deficient mice or wild type mice treated with a function-blocking GM-CSF antibody. The induction of experimental EBA was impaired if the function of GM-CSF was blocked in comparison to appropriate controls. In vitro studies have demonstrated the requirement of GM-CSF for neutrophil recruitment from bone marrow into the blood and from the blood into the skin. Furthermore, GM-CSF preactivates neutrophils, leading to an enhancement of immune complex-induced neutrophil activation. In a therapeutic setting, the blockade of GM-CSF in mice with already established immunization-induced EBA has demonstrated beneficial therapeutic effects (177, 213). In addition to these cytokines, increased expression of TNF has been observed in experimental EBA, and prophylactic blockade of TNF and therapeutic use of etanercept in the immunization-induced EBA model impair the induction and progression of experimental EBA (214). By contrast, increased expression of CCL3/MIP1α is a mandatory cytokine for disease development (221).
The Resolution Phase of EBA
For most autoimmune bullous diseases, the location of blisters differs over time, indicating a frequent, but unfortunately overall insufficient, healing process in the skin (Figure 5). Flightless I (Flii), an actin remodeling protein, has been shown to modulate the resolution of skin blistering in experimental models of EBA. In vivo, the induction of EBA leads to increased cutaneous Flii expression, resulting in impaired Claudin-1 and Claudin-4 tight junction protein expression, as well as a delay in the recovery from blistering (218, 258). Overexpression of Flii produces severe blistering post-induction of EBA, while decreased Flii reduces blister severity, elevates integrin expression, and improves COL7 production. In addition, topically applied Flii neutralizing antibodies improve the healing of blistered skin in murine EBA (218–220, 259).
An interesting experiment could show a protective effect of IL-10-positive plasma cells toward the neutrophil-dependent inflammation in EBA. After the start of skin inflammation, plasmacytosis is induced by injection of goat anti-mouse IgD serum and provides protection from skin inflammation and neutrophil infiltration for at least another 3 weeks. Suppression of EBA skin inflammation is abrogated by the co-injection of a neutralizing IL-10 receptor antibody. Despite its anti-inflammatory effect, plasmacytosis neither reduces the numbers of autoreactive COL7-specific plasma cells nor the autoantibodies that trigger the disease (227). These observations point toward a significant contribution of pathways that are involved in the resolution of cutaneous involvement. Therefore, EBA may manifest not only when many pro-inflammatory stimuli are present but also when the balance of pro-inflammatory, anti-inflammatory, and resolving pathways are unbalanced, driving the cells toward pro-inflammatory mechanisms.
CD11b is an integrin family member that pairs with CD18 to form the CR3 heterodimer. CD11b is expressed on the surface of many leukocytes, including monocytes, neutrophils, natural killer cells, granulocytes, and macrophages. Unexpectedly, in the antibody transfer-induced model of EBA, CD11b-deficient mice develop more severe disease symptoms than wild-type mice in the late phase of the disease. Furthermore, compared to wild-type controls, CD11b-deficient mice express increased levels of circulating IFN-γ and IL-4, suggesting an anti-inflammatory role for CD11b in the resolution phase of experimental autoimmune diseases, such as EBA (237).
Emerging Treatments
The current (unsatisfactory) treatment options for EBA have been outlined above. An increasing understanding of EBA pathogenesis indicates new potential therapeutic targets that interfere in different phases of disease progression, including the generation of autoantibodies, maintaining autoantibodies in the circulation, and autoantibody-induced tissue injury.
Due to the lack of clinical studies, we categorized the emerging treatments into three categories:
Treatments Described in Case Reports
Several case reports are known for the treatment of EBA using ECP (145) and Daclizumab (148). Furthermore, anti-C1s mAb, a mouse monoclonal IgG2a antibody, inhibits the activation of C1s, which is a serine protease of the classical complement system. TNT003 completely blocks complement classical pathway activation by the reduction of C4a and C5a production induced by incubation of the sera from patients with BP on cryosections of human skin (260). The disease induction of experimental EBA is also complement-dependent. This finding suggests that TNT003 might work even in EBA. A “humanized version” of TNT003, TNT009 is currently evaluated in a phase I clinical trial in BP patients (NCT02502903) and data reported on congresses indicates a favorable safety, as well as efficacy in some patients. Sulfasalazine was used in a patient with EBA associated with Crohn's disease, resulting in no improvement of the skin lesion (149). The utility of doxycycline has been reported in BP (150). Doxycycline and another tetracycline, minocycline, have been described in the literature in EBA cases, although their usefulness remains unclear (151–153).
Treatments Applied in Therapeutic Settings in Experimental EBA
Due to the low incidence, clinical studies with EBA patients are not performed. Therefore, animal models are indispensable for investigating the pathogenesis of EBA and other autoimmune blistering diseases and to test new target substances for treatment; these models include the transfer of autoantibodies to experimental animals, adoptive transfer of autoantibody-producing B cells to immune-deficient mice and construction of transgenic mice that produce autoantibodies (167). Although animal models have many benefits, as described above, both the advantages and disadvantages of each model should be considered. Although the models duplicate the clinical, histopathological, ultrastructural, and immunological features of the human disease, these systems are mostly completely murine, and it is possible that findings in mice will not translate to humans.
The most important mouse model to test treatments applied in therapeutic settings is the immunization-induced EBA model (167, 168). Immunization with parts of the NC1 domain of COL7 with an adjuvant induces anti-COL7 IgG production in mice. The susceptibility of experimental EBA is closely associated with the H2s haplotype in mice (165). During a time period of 4–6 weeks, the mice develop antibodies against mCOL7 that bind to the DEJ and induce neutrophil activation and blistering. Using the immunization-induced EBA model, the pathomechanisms of blistering and potential therapeutic options have been studied, and more importantly, this model is used to test potential new drug candidates and therapeutics (167). Successfully applied drugs are summarized in Table 7.
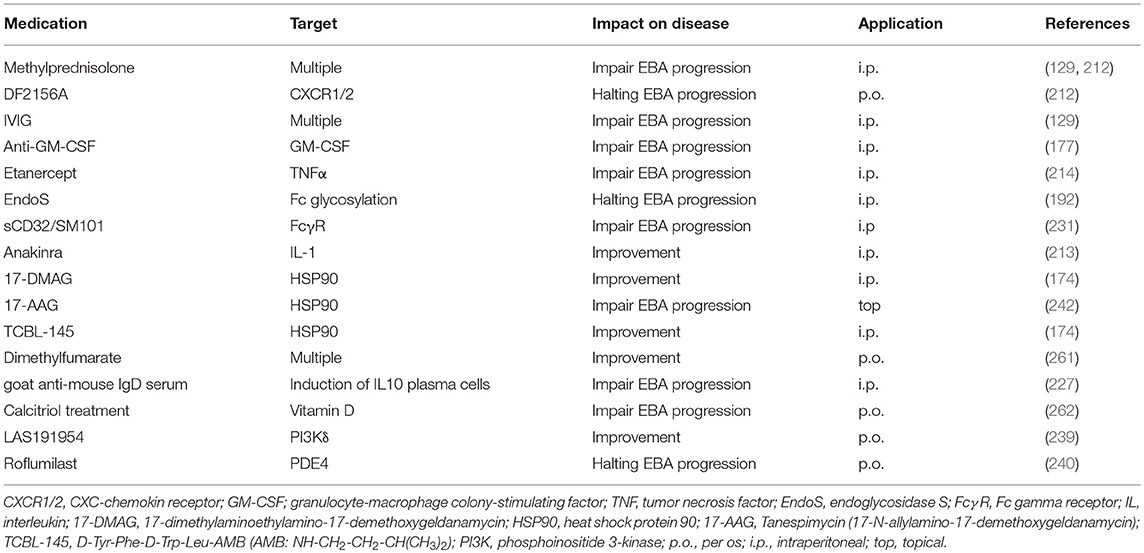
Table 7. Experimental treatments in pre-clinical immunization-induced EBA.
Treatments Applied in Preventive Settings in Experimental EBA
Beneath the validation of potential drug candidates in the therapeutic setting in immunization-induced EBA, some authors have also investigated several drugs in antibody transfer-induced EBA. In this model, rabbit sera or whole rabbit IgG immunized with COL7 NC1 domain are injected into mice. These mice develop clinical and histological subepidermal blistering within a few days. The antibody transfer models are used to investigate the cellular and molecular basis of blistering and to investigate preventive effects of several therapeutic options. The use of antibody transfer models enables examination of the effector phase of EBA. Testing novel drugs or investigating the pathogenesis of EBA is straightforwardly done because clinical symptoms are visible days after starting the experiment. However, similar to most animal models, the situation in human patients is only partially reflected—mostly, because the signaling cascade and the interaction of different cell types vary among human and mice. Additionally, induced disease may last less than a few days after the transfer of antibodies. Furthermore, the loss of tolerance and the generation of autoantibodies cannot be studied in models based on antibody transfer. Therefore, the investigation of potential drug candidates should always be performed in addition to immunization-induced EBA in a therapeutic setting.
Complement
Complement activation has been described as a prerequisite for blister formation. In the antibody-transfer model, blister induction is completely dependent on complement C5 because C5-deficient mice or mice injected with F(ab)2 fragments of the immune IgGs are devoid of blisters. This finding indicates that the complement system can be targeted in animal models. Targeting therapies for complement system factor B, C5, and C5R ameliorate disease severity in antibody-transfer models (199, 201, 203, 211, 260). Interestingly, in antibody transfer-induced BP, C5ar1(−/−) mice are protected from disease development, whereas the extent of skin lesions is increased in C5ar2(−/−) animals.
SYK
SYK is a non-receptor cytoplasmic enzyme that is mainly expressed in hematopoietic cells. SYK regulates cellular responses to extracellular antigens or antigen-immunoglobulin complexes. In EBA pathogenesis, myeloid SYK is a central player in driving inflammation in prototypical autoantibody-transfer models. The SYK inhibitor (BAY61-3606) protects mice from inflammation in antibody-transfer EBA (171, 245).
Cell-Derived Nanoparticles (CDNPs)
CDNPs are intercellular protein complexes, such as annexin A1, annexin A5, actin, 14-3-3ε, 14-3-3ζ, galectin-3, and heat-shock proteins 27 and 70. CDNPs play a therapeutic role against viral infections, cancer and in the experimental EBA model. Mice that receive anti-COL7 IgG develop skin lesions. Treatment with CDNPs significantly reduces the affected skin lesions and increases IL-4 expression (263).
LTB4
LTB4 is a potent chemoattractant and activator of myeloid cells, particularly of neutrophils. The 5-lipoxygenase–deficient and BLT1-deficient mice exhibit neither any clinical nor histological signs of disease in the antibody-transfer model. The 5-lipoxygenase inhibitor (zileuton) targeting the LTB4/BLT1 pathway ameliorates disease severity by ~50% in antibody-transfer models (208).
Flii I
Flii is a member of the gelsolin family of actin remodeling proteins in regulating cell adhesion and intracellular signaling. Flii expression is increased in the epidermis during development and during epidermal stratification to maintain barrier functions, such as tight junctions. Topical application of Flii-neutralizing antibody significantly reduces the clinical severity of blistering and histological separations in antibody-transfer EBA mice. In addition, the barrier function measured by transepidermal water loss (TEWL) is significantly decreased in Flii-neutralizing antibody-treated mice (218, 219).
IL-6 (Protective)
IL-6 is a proinflammatory cytokine and plays a role during the transition from innate to acquired immunity. In the initial immune response to pathogen, IL-6 attracts neutrophils to the affected tissue. In addition, IL-6 plays a crucial role in B- and T-cell differentiation. Elevated IL-6 concentrations are observed in many inflammatory diseases and often correlate with disease activity. IL-6 expression is increased in the skin of EBA patients. Interestingly, the EBA severity is enhanced in IL-6-deficient mice in an experimental model. Furthermore, treatment with recombinant IL-6 dose-dependently impairs the induction of experimental EBA (209).
Galactosylated Immunoglobulins
Antibody-transfer EBA model activation is FcgR-dependent in blister formation. Immune-complexes induce the classical complement pathway via Fc fragment. The mouse IgG1 subclass preferentially binds to inhibitory FcgRIIB and suppresses the inflammatory response. Glycosylation of Asn297 in the IgG Fc fragment plays an important role in complement activation. Highly galactosylated immune-complex treatment reduces the development of cutaneous lesions in an antibody-transfer model (202).
CARD9
Caspase recruitment domain-containing protein 9 (CARD9) is an intracellular adapter protein that is expressed in myeloid-lineage cells. CARD9 plays a critical role in host defense against pathogens in both mice and humans. Card9 deficient mice are significantly protected against skin blistering in antibody-transfer EBA. In this setting, CARD9 is required for development of the inflammatory response. These pieces of evidence suggest a therapeutic target in EBA (246).
Signal Transduction by Scr Kinases
Src family kinases, such as Hck, Fgr, and Lyn, play roles in malignant transformation and tumor progression, and therefore they can be targets of cancer therapy. In addition, Src family kinases are present in many types of immune cells. It is known that Src family kinases have a role in integrin signal transduction in neutrophils and macrophages. Hck, Fgr, and Lyn-deficient mice are completely protected in antibody-transfer EBA (247).
T Cells
Tregs are of importance in modulating host responses to tumors and infections and in inhibiting the development of autoimmunity and allergies. T cell-deficient mice are protected from induction of skin lesions in antibody-transfer EBA. In particular, specific depletion of Tregs increases disease progression in antibody transfer-induced EBA. In a similar experimental setting, NKT-deficient mice and γδT cell-deficient mice are protected against the induction of experimental EBA (224, 225).
Signal Transduction by p38, MAPK AKT
In the antibody-transfer model, neutrophils are crucial for inducing clinical disease. Neutrophil activation is mediated by the phosphorylation of ERK1/2, p38 MAPK, and Akt. Methylprednisolone inhibits the phosphorylation of Akt, ERK1/2, and p38 MAPKs in neutrophils. Chemical inhibitors of Akt (Akt inhibitor VIII), ERK1/2 (U0126), and p38 MAPK (SB203580) statistically suppress ex-vivo dermal-epidermal separation. In addition, ERK1/2 (U0126), and p38 MAPK (SB203580) demonstrate an ~10% reduction of disease severity compared with the control (244).
RORα
RORα is a steroid nuclear hormone receptor and a transcription factor. Experimental EBA shows strain-dependent disease severity. To elucidate the strain-dependency, RORα is found to be a risk gene for the antibody-transfer model. The RORα agonist SR3335 impairs blister formation in both types of antibody-transfer EBA (230).
Signal Transduction by PI3Kβ
PI3Ks play major roles in the signaling pathways that link cell surface receptors to the control of cell function. There are four defined isoforms of PI3Ks. In particular, PI3Kβ is widely expressed and blocks arterial thrombus formation. PI3Kβ-deficient mice are protected from the development of blisters in the antibody-transfer EBA model.
Others
This antibody-transfer mouse model can be used to validate or, at least as important exclude novel therapeutic targets in EBA. For example, TREM1, MIP1a, CD11b, caspase 1/11, and S100, which are all increasingly expressed in experimental EBA, have no impact on disease manifestation (213, 221–223, 237).
Conclusions
Since the first description of EBA over 100 years ago, our understanding of this serious disease has dramatically improved. The knowledge of the cellular and molecular mechanisms led to the development of new therapies, some of which have been successfully tested in preclinical models and in concept studies in bullous pemphigoid patients. In the future, new drug targets will evolve to improve the treatment and living standards of people with EBA.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by grants from the Excellence Cluster Inflammation at Interfaces (EXC306/2), the University of Lübeck, the Research Training Group Modulation of Autoimmunity (GRK 1727), and Clinical Research Pemphigoid Diseases (KFO 303).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
17-AAG, tanespimycin; 17-DMAG, 17-imethylaminoethylamino- 17-demethoxygeldanamycin; ABQOL, autoimmune bullous disease quality of life; AIBD, autoimmune blistering disease; AKT, protein kinase B; APC, antigen-presenting cell; AZA, azathioprine; BLT, leukotriene B4 receptor; BMZ, basement membrane zone; BP, bullous pemphigoid; C, complement factor; CARD9, caspase recruitment domain-containing protein 9; CD, cluster of differentiation; CDNP, cell-derived nanoparticles; COL, collagen; CPA, cyclophosphamide; CR, complete remission; CSA, cyclosporine; CXCR, CXC-chemokin receptor; DDS, diaminodiphenyl sulfone; DEJ, dermal-epidermal junction; DIF, direct immunofluorescence; DMF, dimethylfumarate; EBA, epidermolysis bullosa acquisita; ECP, extracorporeal photochemotherapy; ELISA, enzyme-linked immunosorbent assay; EndoS, endoglycosidase S; ERK, extracellular signal-regulated kinase; FcgR, Fc gamma receptor; FcRn, neonatal Fc receptor; Flii, flightless I; FOAM, fluorescent overlay antigen mapping; G0, agalactosylated antibodies; GM-CSF, granulocyte-macrophage colony-stimulating factor; HLA, human leukocyte antigen; Hsp, heat-shock protein; i.p., intraperitoneal; IA, immunoadsorption; IC, immune complexes; IEM, immunoelectron microscopy; IFN, interferon; Ig, immunoglobulin; IIF, indirect immunofluorescence microscopy; IL, interleukin; IVIG, high-dose intravenous immunoglobulin; JAK2, janus kinase 2; LAD, linear IgA bullous disease; LTB4, leukotriene B4; MHC, major histocompatibility complex; MIP1a, macrophage inflammatory protein1a; MM-EBA, mucous membrane EBA; MMF, mycophenolate mofetil; MMP, mucous membrane pemphigoid; MMPs, matrix metalloproteases; MTX, methotrexate; NADPH, nicotinamide adenine dinucleotide phosphate; NC, non-collagenous; NCF1, neutrophil cytosolic factor 1; NKT, natural killer T cells; p.o., per os; PI3K, phosphatidylinositol-4,5-bisphosphate 3-kinase; PR, partial remission; RORa, retinoid-related orphan receptor-alpha; ROS, reactive oxygen species; RTX, rituximab; SLE, systemic lupus erythematodes; SSS, NaCl-split skin; SYK, Spleen tyrosine kinase; TABQOL, treatment-based autoimmune bullous disease quality of life; Th, T-helper; TNF-α, tumor necrosis factor α; Treg, regulatory T cells; Trem1, triggering receptor expressed on myeloid cells-1.
References
1. Kridin K. Subepidermal autoimmune bullous diseases: overview, epidemiology, and associations. Immunol Res. (2018) 66:6–17. doi: 10.1007/s12026-017-8975-2
2. Kridin K, Ludwig RJ. The growing incidence of bullous pemphigoid: overview and potential explanations. Front Med. (2018) 5:220. doi: 10.3389/fmed.2018.00220
3. Bertram F, Brocker EB, Zillikens D, Schmidt E. Prospective analysis of the incidence of autoimmune bullous disorders in Lower Franconia, Germany. J Dtsch Dermatol Ges. (2009) 7:434–40. doi: 10.1111/j.1610-0387.2008.06976.x
4. Wong SN, Chua SH. Spectrum of subepidermal immunobullous disorders seen at the National Skin Center, Singapore: a 2-year review. Br J Dermatol. (2002) 147:476–80. doi: 10.1046/j.1365-2133.2002.04919.x
5. Nanda A, Dvorak R, Al-Saeed K, Al-Sabah H, Alsaleh QA. Spectrum of autoimmune bullous diseases in Kuwait. Int J Dermatol. (2004) 43:876–81. doi: 10.1111/j.1365-4632.2004.02292.x
6. Bernard P, Vaillant L, Labeille B, Bedane C, Arbeille B, Denoeux JP, et al. Incidence and distribution of subepidermal autoimmune bullous skin diseases in three French regions. Bullous Diseases French Study Group. Arch Dermatol. (1995) 131:48–52. doi: 10.1001/archderm.1995.01690130050009
7. Zillikens D, Wever S, Roth A, Weidenthaler-Barth B, Hashimoto T, Brocker EB. Incidence of autoimmune subepidermal blistering dermatoses in a region of central Germany. Arch Dermatol. (1995) 131:957–8. doi: 10.1001/archderm.131.8.957
8. Lee CW. Prevalences of subacute cutaneous lupus erythematosus and Epidermolysis bullosa acquisita among Korean/Oriental populations. Dermatology (1998) 197:187.
9. Hubner F, Recke A, Zillikens D, Linder R, Schmidt E. Prevalence and age distribution of pemphigus and pemphigoid diseases in Germany. J Invest Dermatol. (2016) 136:2495–98. doi: 10.1016/j.jid.2016.07.013
10. Gammon WR, Heise ER, Burke WA, Fine JD, Woodley DT, Briggaman RA. Increased frequency of HLA-DR2 in patients with autoantibodies to epidermolysis bullosa acquisita antigen: evidence that the expression of autoimmunity to type VII collagen is HLA class II allele associated. J Invest Dermatol. (1988) 91:228–32. doi: 10.1111/1523-1747.ep12470317
11. Zumelzu C, Le Roux-Villet C, Loiseau P, Busson M, Heller M, Aucouturier F, et al. Black patients of African descent and HLA-DRB1*15:03 frequency overrepresented in epidermolysis bullosa acquisita. J Invest Dermatol. (2011) 131:2386–93. doi: 10.1038/jid.2011.231
12. Lee CW, Kim SC, Han H. Distribution of HLA class II alleles in Korean patients with epidermolysis bullosa acquisita. Dermatol (1996) 193:328–9.
13. Caux F, Kirtschig G, Lemarchand-Venencie F, Venencie PY, Hoang-Xuan T, Robin H, et al. IgA-epidermolysis bullosa acquisita in a child resulting in blindness. Br J Dermatol. (1997) 137:270–5. doi: 10.1046/j.1365-2133.1997.18191915.x
14. Trigo-Guzman FX, Conti A, Aoki V, Maruta CW, Santi CG, Resende Silva CM, et al. Epidermolysis bullosa acquisita in childhood. J Dermatol. (2003) 30:226–9. doi: 10.1111/j.1346-8138.2003.tb00376.x
15. Marzano AV, Cozzani E, Fanoni D, De Pita O, Vassallo C, Berti E, et al. Diagnosis and disease severity assessment of epidermolysis bullosa acquisita by ELISA for anti-type VII collagen autoantibodies: an Italian multicentre study. Br J Dermatol. (2013) 168:80–4. doi: 10.1111/bjd.12011
16. Prost-Squarcioni C, Caux F, Schmidt E, Jonkman MF, Vassileva S, Kim SC, et al. International Bullous Diseases Group: consensus on diagnostic criteria for epidermolysis bullosa acquisita. Br J Dermatol. (2017) 179:30–41. doi: 10.1111/bjd.16138
17. Briggaman RA, Gammon WR, Woodley DT. Epidermolysis bullosa acquisita of the immunopathological type (dermolytic pemphigoid). J Invest Dermatol. (1985) 85:79s−84s. doi: 10.1111/1523-1747.ep12275505
18. Kim JH, Kim YH, Kim SC. Epidermolysis bullosa acquisita: a retrospective clinical analysis of 30 cases. Acta Derm Venereol. (2011) 91:307–12. doi: 10.2340/00015555-1065
19. Buijsrogge JJ, Diercks GF, Pas HH, Jonkman MF. The many faces of epidermolysis bullosa acquisita after serration pattern analysis by direct immunofluorescence microscopy. Br J Dermatol. (2011) 165:92–8. doi: 10.1111/j.1365-2133.2011.10346.x
20. Iranzo P, Herrero-Gonzalez JE, Mascaro-Galy JM, Suarez-Fernandez R, Espana A. Epidermolysis bullosa acquisita: a retrospective analysis of 12 patients evaluated in four tertiary hospitals in Spain. Br J Dermatol. (2014) 171:1022–30. doi: 10.1111/bjd.13144
21. Seta V, Aucouturier F, Bonnefoy J, Le Roux-Villet C, Pendaries V, Alexandre M, et al. Comparison of 3 type VII collagen (C7) assays for serologic diagnosis of epidermolysis bullosa acquisita (EBA). J Am Acad Dermatol. (2016) 74:1166–72. doi: 10.1016/j.jaad.2016.01.005
22. Gupta R, Woodley DT, Chen M. Epidermolysis bullosa acquisita. Clin Dermatol. (2012) 30:60–9. doi: 10.1016/j.clindermatol.2011.03.011
24. Kablitz R. Ein Beitrag Zur Frage der Epidermolysis bullosa (heriditaria et acquisita). Dissertation, Rostock (1904).
25. Wise F, Lautman MF. Epidermolysis bullosa beginning in adult life. the acquired for of the disease, with the report of a case and review of the literature. J Cutan Dis. (1915) 33:44.
26. Hundley JL, Smith DC. Epidermolysis bullosa acquisita. South Med J. (1941) 34:364. doi: 10.1097/00007611-194104000-00004
27. Roenigk HH Jr, Ryan JG, Bergfeld WF. Epidermolysis bullosa acquisita. Report of three cases and review of all published cases. Arch Dermatol. (1971) 103:1–10. doi: 10.1001/archderm.1971.04000130003001
28. Gammon WR, Briggaman RA, Wheeler CE Jr. Epidermolysis bullosa acquisita presenting as an inflammatory bullous disease. J Am Acad Dermatol. (1982) 7:382–7. doi: 10.1016/S0190-9622(82)80319-8
29. Nieboer C, Boorsma DM, Woerdeman MJ, Kalsbeek GL. Epidermolysis bullosa acquisita. Immunofluorescence, electron microscopic and immunoelectron microscopic studies in four patients. Br J Dermatol. (1980) 102:383–92. doi: 10.1111/j.1365-2133.1980.tb06550.x
30. Yaoita H, Briggaman RA, Lawley TJ, Provost TT, Katz SI. Epidermolysis bullosa acquisita: ultrastructural and immunological studies. J Invest Dermatol. (1981) 76:288–92. doi: 10.1111/1523-1747.ep12526124
32. Woodley DT, Briggaman RA, Gammon WT. Review and update of epidermolysis bullosa acquisita. Semin Dermatol. (1988) 7:111–22.
33. Dahl MG. Epidermolysis bullosa acquisita–a sign of cicatricial pemphigoid? Br J Dermatol. (1979) 101:475–84. doi: 10.1111/j.1365-2133.1979.tb00030.x
34. Luke MC, Darling TN, Hsu R, Summers RM, Smith JA, Solomon BI, et al. Mucosal morbidity in patients with epidermolysis bullosa acquisita. Arch Dermatol. (1999) 135:954–9. doi: 10.1001/archderm.135.8.954
35. Chan LS, Ahmed AR, Anhalt GJ, Bernauer W, Cooper KD, Elder MJ, et al. The first international consensus on mucous membrane pemphigoid: definition, diagnostic criteria, pathogenic factors, medical treatment, and prognostic indicators. Arch Dermatol. (2002) 138:370–9. doi: 10.1001/archderm.138.3.370
36. Schattenkirchner S, Lemann M, Prost C, Caux F, Guigui B, Cadot M, et al. Localized epidermolysis bullosa acquisita of the esophagus in a patient with Crohn's disease. Am J Gastroenterol. (1996) 91:1657–9.
37. Zehou O, Raynaud JJ, Le Roux-Villet C, Alexandre M, Airinei G, Pascal F, et al. Oesophageal involvement in 26 consecutive patients with mucous membrane pemphigoid. Br J Dermatol. (2017) 177:1074–85. doi: 10.1111/bjd.15592
38. Schattenkirchner S, Eming S, Hunzelmann N, Krieg T, Smola H. Treatment of epidermolysis bullosa acquisita with mycophenolate mofetil and autologous keratinocyte grafting. Br J Dermatol. (1999) 141:932–3. doi: 10.1046/j.1365-2133.1999.03176.x
39. Delgado L, Aoki V, Santi C, Gabbi T, Sotto M, Maruta C. Clinical and immunopathological evaluation of epidermolysis bullosa acquisita. Clin Exp Dermatol. (2011) 36:12–8. doi: 10.1111/j.1365-2230.2010.03845.x
40. Tokuda Y, Amagai M, Yaoita H, Kawachi S, Ito T, Matsuyama I, et al. A case of an inflammatory variant of epidermolysis bullosa acquisita: chronic bullous dermatosis associated with nonscarring mucosal blisters and circulating IgG anti-type-VII-collagen antibody. Dermatol (1998) 197:58–61.
41. Miyagawa S, Iida T, Hachisuka H, Yamashina Y, Shirai T. Epidermolysis bullosa acquisita with oesophageal stenosis. Br J Dermatol. (1992) 127:172–6. doi: 10.1111/j.1365-2133.1992.tb08052.x
42. Taniuchi K, Inaoki M, Nishimura Y, Mori T, Takehara K. Nonscarring inflammatory epidermolysis bullosa acquisita with esophageal involvement and linear IgG deposits. J Am Acad Dermatol. (1997) 36:320–2. doi: 10.1016/S0190-9622(97)80408-2
43. Meissner C, Hoefeld-Fegeler M, Vetter R, Bellutti M, Vorobyev A, Gollnick H, et al. Severe acral contractures and nail loss in a patient with mechano-bullous Epidermolysis bullosa acquisita. Eur J Dermatol. (2010) 20:543–4. doi: 10.1684/ejd.2010.1002
44. Moura EG, Couto-Junior DS, Alvarado-Escobar H, da Costa-Martins B, Sallum RA, Artifon EL, et al. Epidermolysis bullosa acquisita complicated by esophageal stenosis. endoscopic treatment with thermoplastic dilators and intralesional steroid injection. Rev Gastroenterol Mex. (2011) 76:279–85.
45. Stewart MI, Woodley DT, Briggaman RA. Epidermolysis bullosa acquisita and associated symptomatic esophageal webs. Arch Dermatol. (1991) 127:373–7. doi: 10.1001/archderm.1991.01680030093013
46. Harman KE, Whittam LR, Wakelin SH, Black MM. Severe, refractory epidermolysis bullosa acquisita complicated by an oesophageal stricture responding to intravenous immune globulin. Br J Dermatol. (1998) 139:1126–7. doi: 10.1046/j.1365-2133.1998.2576m.x
47. Chua S, Dodd H, Saeed IT, Chakravarty K. Dysphagia in a patient with lupus and review of the literature. Lupus (2002) 11:322–4. doi: 10.1191/0961203302lu195cr
48. Shipman AR, Agero AL, Cook I, Scolyer RA, Craig P, Pas HH, et al. Epidermolysis bullosa acquisita requiring multiple oesophageal dilatations. Clin Exp Dermatol. (2008) 33:787–9. doi: 10.1111/j.1365-2230.2008.02875.x
49. Tu J, Kumarasinghe PW. Epidermolysis bullosa acquisita with moderately severe Dysphagia due to esophageal strictures. Indian J Dermatol. (2011) 56:224–7. doi: 10.4103/0019-5154.80428
50. Ishii N, Furumura M, Hamada T, Mori O, Ohzono A, Ueda A, et al. Oesophageal involvement in epidermolysis bullosa acquisita. Br J Dermatol. (2015) 172:288–90. doi: 10.1111/bjd.13224
51. Alexandre M, Brette MD, Pascal F, Tsianakas P, Fraitag S, Doan S, et al. A prospective study of upper aerodigestive tract manifestations of mucous membrane pemphigoid. Medicine (2006) 85:239–52. doi: 10.1097/01.md.0000231954.08350.52
52. Kiniwa Y, Ashida A, Ohashi A, Kitoh R, Fukuda S, Hashimoto T, et al. A case of epidermolysis bullosa acquisita associated with laryngeal stenosis. Acta Derm Venereol. (2012) 92:93–4. doi: 10.2340/00015555-1163
53. Benton EC, Bhogal B, Oakley R, Groves RW. Beware the blistering patient with dysphonia. Clin Exp Dermatol. (2013) 38:691–2. doi: 10.1111/ced.12009
54. Poirier E, Soued I, Alexandre M, Boussoura S, Lamberto C, Uzunhan Y, et al. Pemphigoide des muqueuses avec sténose laryngée ou trachéale. In: Annual Meeting of the Société Française de Dermatologie. Paris, December 2014. (2014) 141 (Hs12):S262.
55. Lang PG Jr, Tapert MJ. Severe ocular involvement in a patient with epidermolysis bullosa acquisita. J Am Acad Dermatol. (1987) 16:439–43. doi: 10.1016/S0190-9622(87)70057-7
56. Zierhut M, Thiel HJ, Weidle EG, Steuhl KP, Sonnichsen K, Schaumburg-Lever G. Ocular involvement in epidermolysis bullosa acquisita. Arch Ophthalmol. (1989) 107:398–401. doi: 10.1001/archopht.1989.01070010408035
57. Zambruno G, Manca V, Kanitakis J, Cozzani E, Nicolas JF, Giannetti A. Linear IgA bullous dermatosis with autoantibodies to a 290 kd antigen of anchoring fibrils. J Am Acad Dermatol. (1994) 31:884–8. doi: 10.1016/S0190-9622(94)70252-7
58. Aclimandos WA. Corneal perforation as a complication of epidermolysis bullosa acquisita. Eye (1995) 9(Pt 5):633–6. doi: 10.1038/eye.1995.153
59. Hoang-Xuan T, Robin H, Heller M, Caux F, Prost C. Epidermolysis bullosa acquisita diagnosed by direct immunoelectron microscopy of the conjunctiva. Ophthalmology (1997) 104:1414–20. doi: 10.1016/S0161-6420(97)30122-5
60. Bauer JW, Schaeppi H, Metze D, Muss W, Pohla-Gubo G, Hametner R, et al. Ocular involvement in IgA-epidermolysis bullosa acquisita. Br J Dermatol. (1999) 141:887–92. doi: 10.1046/j.1365-2133.1999.03163.x
61. Camara A, Becherel PA, Bussel A, Lagrange S, Chosidow O, Joly P, et al. Resistant acquired bullous epidermolysis with severe ocular involvement: the success of extracorporeal photochemotherapy. Ann Dermatol Venereol. (1999) 126:612–5.
62. Dantas PE, Nishiwaki-Dantas MC, Seguim MH, Cursino JW. Bilateral corneal involvement in epidermolysis bullosa acquisita. Cornea (2001) 20:664–7. doi: 10.1097/00003226-200108000-00022
63. Letko E, Bhol K, Anzaar F, Perez VL, Ahmed AR, Foster CS. Chronic cicatrizing conjunctivitis in a patient with epidermolysis bullosa acquisita. Arch Ophthalmol. (2006) 124:1615–8. doi: 10.1001/archopht.124.11.1615
64. Cox NH, Bearn MA, Herold J, Ainsworth G, Liu C. Blindness due to the IgA variant of epidermolysis bullosa acquisita, and treatment with osteo-odonto-keratoprosthesis. Br J Dermatol. (2007) 156:775–7. doi: 10.1111/j.1365-2133.2006.07739.x
65. Le Roux-Villet C, Prost-Squarcioni C. Epidermolysis bullosa acquisita: clinical, histological and immunologicalanalysis of 39 cases. Ann Dermatol Venereol. (2002) 129(Suppl. 1):S71–2.
66. Lee CW. Epidermolysis bullosa acquisita associated with vesicular cystitis. Br J Dermatol. (1988) 119:101–5. doi: 10.1111/j.1365-2133.1988.tb07109.x
67. Vodegel RM, de Jong MC, Pas HH, Jonkman MF. IgA-mediated epidermolysis bullosa acquisita: two cases and review of the literature. J Am Acad Dermatol. (2002) 47:919–25. doi: 10.1067/mjd.2002.125079
68. Gottlieb J, Ingen-Housz-Oro S, Alexandre M, Grootenboer-Mignot S, Aucouturier F, Sbidian E, et al. Idiopathic linear IgA bullous dermatosis: prognostic factors based on a case series of 72 adults. Br J Dermatol. (2017) 177:212–22. doi: 10.1111/bjd.15244
69. Lee CW, Jun KM. Epidermolysis bullosa acquisita presenting with localized facial blisters. Clin Exp Dermatol. (1992) 17:363–5. doi: 10.1111/j.1365-2230.1992.tb00234.x
70. Joly P, Ruto F, Thomine E, Delpech A, Balguerie X, Tron F, et al. Brunsting-Perry cicatricial bullous pemphigoid: a clinical variant of localized acquired epidermolysis bullosa? J Am Acad Dermatol. (1993) 28:89–92. doi: 10.1016/0190-9622(93)70016-M
71. Kurzhals G, Stolz W, Maciejewski W, Karpati S, Meurer M, Breit R. Localized cicatricial pemphigoid of the Brunsting-Perry type with transition into disseminated cicatricial pemphigoid. report of a case proved by preembedding immunogold electron microscopy. Arch Dermatol. (1995) 131:580–5. doi: 10.1001/archderm.1995.01690170082012
72. Choi GS, Lee ES, Kim SC, Lee S. Epidermolysis bullosa acquisita localized to the face. J Dermatol. (1998) 25:19–22. doi: 10.1111/j.1346-8138.1998.tb02339.x
73. Woodley DT. Epidermolysis bullosa acquisita. In: Freedberg IM, Eisen AZ, Wolff K, editors. Dermatology in General Medicine. New York, NY: Mc Graw Hill (2003). p. 609–16.
74. Tanaka N, Dainichi T, Ohyama B, Yasumoto S, Oono T, Iwatsuki K, et al. A case of epidermolysis bullosa acquisita with clinical features of Brunsting-Perry pemphigoid showing an excellent response to colchicine. J Am Acad Dermatol. (2009) 61:715–9. doi: 10.1016/j.jaad.2008.12.020
75. Minato H, Ishii N, Fukuda S, Wakasa T, Wakasa K, Sogame R, et al. Heterogeneity of Brunsting-Perry type pemphigoid: a case showing blister formation at the lamina lucida, immune deposition beneath the lamina densa and autoantibodies against the 290-kD polypeptide along the lamina densa. J Dermatol. (2011) 38:887–92. doi: 10.1111/j.1346-8138.2010.01172.x
76. Asfour L, Chong H, Mee J, Groves R, Singh M. Epidermolysis bullosa acquisita (brunsting-perry pemphigoid variant) localized to the face and diagnosed with antigen identification using skin deficient in type VII collagen. Am J Dermatopathol. (2017) 39:e90–6. doi: 10.1097/DAD.0000000000000829
77. Sebaratnam DF, Hanna AM, Chee SN, Frew JW, Venugopal SS, Daniel BS, et al. Development of a quality-of-life instrument for autoimmune bullous disease: the Autoimmune Bullous Disease Quality of Life questionnaire. JAMA Dermatol. (2013) 149:1186–91. doi: 10.1001/jamadermatol.2013.4972
78. Tjokrowidjaja A, Daniel BS, Frew JW, Sebaratnam DF, Hanna AM, Chee S, et al. The development and validation of the treatment of autoimmune bullous disease quality of life questionnaire, a tool to measure the quality of life impacts of treatments used in patients with autoimmune blistering disease. Br J Dermatol. (2013) 169:1000–6. doi: 10.1111/bjd.12623
79. Chen M, O'Toole EA, Sanghavi J, Mahmud N, Kelleher D, Weir D, et al. The epidermolysis bullosa acquisita antigen (type VII collagen) is present in human colon and patients with crohn's disease have autoantibodies to type VII collagen. J Invest Dermatol. (2002) 118:1059–64. doi: 10.1046/j.1523-1747.2002.01772.x
80. Aractingi S, Bachmeyer C, Prost C, Caux F, Flageul B, Fermand JP. Subepidermal autoimmune bullous skin diseases associated with B-cell lymphoproliferative disorders. Medicine (1999) 78:228–35. doi: 10.1097/00005792-199907000-00003
81. Camisa C, Sharma HM. Vesiculobullous systemic lupus erythematosus. report of two cases and a review of the literature. J Am Acad Dermatol. (1983) 9:924–33. doi: 10.1016/S0190-9622(83)70210-0
82. Gammon WR, Briggaman RA. Bullous SLE: a phenotypically distinctive but immunologically heterogeneous bullous disorder. J Invest Dermatol. (1993) 100:28S−34S. doi: 10.1111/1523-1747.ep12355210
83. Komorowski L, Muller R, Vorobyev A, Probst C, Recke A, Jonkman MF, et al. Sensitive and specific assays for routine serological diagnosis of epidermolysis bullosa acquisita. J Am Acad Dermatol. (2013) 68:e89–95. doi: 10.1016/j.jaad.2011.12.032
84. Gammon WR, Kowalewski C, Chorzelski TP, Kumar V, Briggaman RA, Beutner EH. Direct immunofluorescence studies of sodium chloride-separated skin in the differential diagnosis of bullous pemphigoid and epidermolysis bullosa acquisita. J Am Acad Dermatol. (1990) 22:664–70. doi: 10.1016/0190-9622(90)70094-X
85. Terra JB, Jonkman MF, Diercks GF, Pas HH. Low sensitivity of type VII collagen enzyme-linked immunosorbent assay in epidermolysis bullosa acquisita: serration pattern analysis on skin biopsy is required for diagnosis. Br J Dermatol. (2013) 169:164–7. doi: 10.1111/bjd.12300
86. Meijer JM, Atefi I, Diercks GFH, Vorobyev A, Zuiderveen J, Meijer HJ, et al. Serration pattern analysis for differentiating epidermolysis bullosa acquisita from other pemphigoid diseases. J Am Acad Dermatol. (2018) 78:754–59 e6. doi: 10.1016/j.jaad.2017.11.029
87. Schmidt E, Zillikens D. Pemphigoid diseases. Lancet (2013) 381:320–32. doi: 10.1016/S0140-6736(12)61140-4
88. Wilson BD, Beutner EH, Kumar V, Chorzelski TP, Jablonska S. Linear IgA bullous dermatosis. an immunologically defined disease. Int J Dermatol. (1985) 24:569–74. doi: 10.1111/j.1365-4362.1985.tb05575.x
89. Vodegel RM, de Jong MC, Meijer HJ, Weytingh MB, Pas HH, Jonkman MF. Enhanced diagnostic immunofluorescence using biopsies transported in saline. BMC Dermatol. (2004) 4:10. doi: 10.1186/1471-5945-4-10
90. Schmidt E, Klinker E, Opitz A, Herzog S, Sitaru C, Goebeler M, et al. Protein A immunoadsorption: a novel and effective adjuvant treatment of severe pemphigus. Br J Dermatol. (2003) 148:1222–9. doi: 10.1046/j.1365-2133.2003.05302.x
91. Suchniak JM, Diaz LA, Lin MS, Fairley JA. IgM-mediated epidermolysis bullosa acquisita. Arch Dermatol. (2002) 138:1385–6. doi: 10.1001/archderm.138.10.1385
92. Caux F. Diagnosis and clinical features of epidermolysis bullosa acquisita. Dermatol Clin. (2011) 29:485–91. doi: 10.1016/j.det.2011.03.017
93. Saleh MA, Ishii K, Kim YJ, Murakami A, Ishii N, Hashimoto T, et al. Development of NC1 and NC2 domains of type VII collagen ELISA for the diagnosis and analysis of the time course of epidermolysis bullosa acquisita patients. J Dermatol Sci. (2011) 62:169–75. doi: 10.1016/j.jdermsci.2011.03.003
94. Marzano AV, Cozzani E, Biasin M, Russo I, Alaibac M. The use of Biochip immunofluorescence microscopy for the serological diagnosis of epidermolysis bullosa acquisita. Arch Dermatol Res. (2016) 308:273–6. doi: 10.1007/s00403-016-1632-0
95. Schmidt T, Hoch M, Lotfi Jad SS, Solimani F, Di Zenzo G, Marzano AV, et al. Serological diagnostics in the detection of IgG autoantibodies against human collagen VII in epidermolysis bullosa acquisita: a multicentre analysis. Br J Dermatol. (2017) 177:1683–92. doi: 10.1111/bjd.15800
96. Kim JH, Kim YH, Kim S, Noh EB, Kim SE, Vorobyev A, et al. Serum levels of anti-type VII collagen antibodies detected by enzyme-linked immunosorbent assay in patients with epidermolysis bullosa acquisita are correlated with the severity of skin lesions. J Eur Acad Dermatol Venereol. (2013) 27:e224–30. doi: 10.1111/j.1468-3083.2012.04617.x
97. Vorobyev A, Ludwig RJ, Schmidt E. Clinical features and diagnosis of epidermolysis bullosa acquisita. Expert Rev Clin Immunol. (2017) 13:157–69. doi: 10.1080/1744666X.2016.1221343
98. Witte M, Koga H, Hashimoto T, Ludwig RJ, Bieber K. Discovering potential drug-targets for personalized treatment of autoimmune disorders - what we learn from epidermolysis bullosa acquisita. Expert Opin Ther Targets (2016) 20:985–98. doi: 10.1517/14728222.2016.1148686
99. Ludwig RJ. Clinical Presentation, Pathogenesis, Diagnosis, and Treatment of Epidermolysis Bullosa Acquisita. ISRN Dermatol. (2013) 2013:812029. doi: 10.1155/2013/812029
100. Gordon KB, Chan LS, Woodley DT. Treatment of refractory epidermolysis bullosa acquisita with extracorporeal photochemotherapy. Br J Dermatol. (1997) 136:415–20. doi: 10.1111/j.1365-2133.1997.tb14957.x
101. Iwata H, Vorobyev A, Koga H, Recke A, Zillikens D, Prost-Squarcioni C, et al. Meta-analysis of the clinical and immunopathological characteristics and treatment outcomes in epidermolysis bullosa acquisita patients. Orphanet J Rare Dis. (2018) 13:153. doi: 10.1186/s13023-018-0896-1
102. Megahed M, Scharffetter-Kochanek K. Epidermolysis bullosa acquisita–successful treatment with colchicine. Arch Dermatol Res. (1994) 286:35–46. doi: 10.1007/BF00375841
103. Dasgeb B, Kornreich D, McGuinn K, Okon L, Brownell I, Sackett DL. Colchicine: an ancient drug with novel applications. Br J Dermatol. (2018) 178:350–6. doi: 10.1111/bjd.15896
104. Cunningham BB, Kirchmann TT, Woodley D. Colchicine for epidermolysis bullosa acquisita. J Am Acad Dermatol. (1996) 34:781–4. doi: 10.1016/S0190-9622(96)90013-4
105. Ishii N, Hamada T, Dainichi T, Karashima T, Nakama T, Yasumoto S, et al. Epidermolysis bullosa acquisita: what's new? J Dermatol. (2010) 37:220–30. doi: 10.1111/j.1346-8138.2009.00799.x
106. Zhu YI, Stiller MJ. Dapsone and sulfones in dermatology: overview and update. J Am Acad Dermatol. (2001) 45:420–34. doi: 10.1067/mjd.2001.114733
107. Hashimoto T, Ishii N, Ohata C, Furumura M. Pathogenesis of epidermolysis bullosa acquisita, an autoimmune subepidermal bullous disease. J Pathol. (2012) 228:1–7. doi: 10.1002/path.4062
108. Sticherling M, Franke A, Aberer E, Glaser R, Hertl M, Pfeiffer C, et al. An open, multicentre, randomized clinical study in patients with bullous pemphigoid comparing methylprednisolone and azathioprine with methylprednisolone and dapsone. Br J Dermatol. (2017) 177:1299–305. doi: 10.1111/bjd.15649
109. Gurcan HM, Ahmed AR. Analysis of current data on the use of methotrexate in the treatment of pemphigus and pemphigoid. Br J Dermatol. (2009) 161:723–31. doi: 10.1111/j.1365-2133.2009.09246.x
110. Fraser AG, Orchard TR, Jewell DP. The efficacy of azathioprine for the treatment of inflammatory bowel disease: a 30 year review. Gut (2002) 50:485–9. doi: 10.1136/gut.50.4.485
111. Ryan C, Amor KT, Menter A. The use of cyclosporine in dermatology: part II. J Am Acad Dermatol. (2010) 63:949–72. doi: 10.1016/j.jaad.2010.02.062
112. Beissert S, Werfel T, Frieling U, Bohm M, Sticherling M, Stadler R, et al. A comparison of oral methylprednisolone plus azathioprine or mycophenolate mofetil for the treatment of pemphigus. Arch Dermatol. (2006) 142:1447–54. doi: 10.1001/archderm.142.11.1447
113. Beissert S, Mimouni D, Kanwar AJ, Solomons N, Kalia V, Anhalt GJ. Treating pemphigus vulgaris with prednisone and mycophenolate mofetil: a multicenter, randomized, placebo-controlled trial. J Invest Dermatol. (2010) 130:2041–8. doi: 10.1038/jid.2010.91
114. Sami N. Mycophenolate mofetil (MMF) in the treatment of epidermolysis bullosa acquisita (EBA) long-term follow-up. JAAD Case Rep. (2015) 1:321–3. doi: 10.1016/j.jdcr.2015.07.007
115. Barreiro-Capurro A, Mascaro-Galy JM, Iranzo P. Retrospective study of the clinical, histologic, and immunologic features of epidermolysis bullosa acquisita in 9 patients. Actas Dermosifiliogr. (2013) 104:904–14. doi: 10.1016/j.ad.2013.05.005
116. Gual A, Guilabert A, Iranzo P, Flores G, Diaz LA, Mascaro JM Jr. IgG autoantibody subclass analysis as a tool to differentiate epidermolysis bullosa acquisita with overlapping features of bullous systemic lupus erythematosus. J Am Acad Dermatol. (2013) 69:e34–6. doi: 10.1016/j.jaad.2013.01.025
117. Baican A, Chiriac G, Torio-Padron N, Sitaru C. Childhood epidermolysis bullosa acquisita associated with severe dental alterations: a case presentation. J Dermatol. (2013) 40:410–1. doi: 10.1111/1346-8138.12107
118. Ahmed AR, Gurcan HM. Treatment of epidermolysis bullosa acquisita with intravenous immunoglobulin in patients non-responsive to conventional therapy: clinical outcome and post-treatment long-term follow-up. J Eur Acad Dermatol Venereol. (2012) 26:1074–83. doi: 10.1111/j.1468-3083.2011.04205.x
119. Busch JO, Sticherling M. Epidermolysis bullosa acquisita and neuroendocrine pancreatic cancer - coincidence or patho-genetic relationship? J Dtsch Dermatol Ges. (2007) 5:916–8. doi: 10.1111/j.1610-0387.2007.06338.x
120. Pastar Z, Rados J, Lipozencic J, Dobric I, Marinovic B, Ishii N, et al. Case of concurrent epidermolysis bullosa acquisita and anti-p200 pemphigoid–how to treat it? Int J Dermatol. (2007) 46:295–8. doi: 10.1111/j.1365-4632.2006.02969.x
121. Crichlow SM, Mortimer NJ, Harman KE. A successful therapeutic trial of rituximab in the treatment of a patient with recalcitrant, high-titre epidermolysis bullosa acquisita. Br J Dermatol. (2007) 156:194–6. doi: 10.1111/j.1365-2133.2006.07596.x
122. Campos M, Silvente C, Lecona M, Suarez R, Lazaro P. Epidermolysis bullosa acquisita: diagnosis by fluorescence overlay antigen mapping and clinical response to high-dose intravenous immunoglobulin. Clin Exp Dermatol. (2006) 31:71–3. doi: 10.1111/j.1365-2230.2005.01989.x
123. Gourgiotou K, Exadaktylou D, Aroni K, Rallis E, Nicolaidou E, Paraskevakou H, et al. Epidermolysis bullosa acquisita: treatment with intravenous immunoglobulins. J Eur Acad Dermatol Venereol. (2002) 16:77–80. doi: 10.1046/j.1468-3083.2002.00386.x
124. Jappe U, Zillikens D, Bonnekoh B, Gollnick H. Epidermolysis bullosa acquisita with ultraviolet radiationsensitivity. Br J Dermatol. (2000) 142:517–20. doi: 10.1046/j.1365-2133.2000.03368.x
125. Kofler H, Wambacher-Gasser B, Topar G, Weinlich G, Schuler G, Hintner H, et al. Intravenous immunoglobulin treatment in therapy-resistant epidermolysis bullosa acquisita. J Am Acad Dermatol. (1997) 36:331–5. doi: 10.1016/S0190-9622(97)80411-2
126. Mohr C, Sunderkotter C, Hildebrand A, Biel K, Rutter A, Rutter GH, et al. Successful treatment of epidermolysis bullosa acquisita using intravenous immunoglobulins. Br J Dermatol. (1995) 132:824–6. doi: 10.1111/j.1365-2133.1995.tb00735.x
127. Meier F, Sonnichsen K, Schaumburg-Lever G, Dopfer R, Rassner G. Epidermolysis bullosa acquisita: efficacy of high-dose intravenous immunoglobulins. J Am Acad Dermatol. (1993) 29:334–7. doi: 10.1016/0190-9622(93)70189-Z
128. Li N, Zhao M, Hilario-Vargas J, Prisayanh P, Warren S, Diaz LA, et al. Complete FcRn dependence for intravenous Ig therapy in autoimmune skin blistering diseases. J Clin Invest. (2005) 115:3440–50. doi: 10.1172/JCI24394
129. Hirose M, Tiburzy B, Ishii N, Pipi E, Wende S, Rentz E, et al. Effects of intravenous immunoglobulins on mice with experimental epidermolysis bullosa acquisita. J Invest Dermatol. (2015) 135:768–75. doi: 10.1038/jid.2014.453
130. Sasaoka T, Ujiie H, Nishie W, Iwata H, Ishikawa M, Higashino H, et al. Intravenous IgG reduces pathogenic autoantibodies, serum IL-6 levels, and disease severity in experimental bullous pemphigoid models. J Invest Dermatol. (2018) 138:1260–67. doi: 10.1016/j.jid.2018.01.005
131. Kamaguchi M, Iwata H, Mori Y, Toyonaga E, Ujiie H, Kitagawa Y, et al. Anti-idiotypic antibodies against BP-IgG prevent type XVII collagen depletion. Front Immunol. (2017) 8:1669. doi: 10.3389/fimmu.2017.01669
132. Amagai M, Ikeda S, Hashimoto T, Mizuashi M, Fujisawa A, Ihn H, et al. A randomized double-blind trial of intravenous immunoglobulin for bullous pemphigoid. J Dermatol Sci. (2017) 85:77–84. doi: 10.1016/j.jdermsci.2016.11.003
133. Kolesnik M, Becker E, Reinhold D, Ambach A, Heim MU, Gollnick H, et al. Treatment of severe autoimmune blistering skin diseases with combination of protein A immunoadsorption and rituximab: a protocol without initial high dose or pulse steroid medication. J Eur Acad Dermatol Venereol. (2014) 28:771–80. doi: 10.1111/jdv.12175
134. Kim JH, Lee SE, Kim SC. Successful treatment of epidermolysis bullosa acquisita with rituximab therapy. J Dermatol. (2012) 39:477–9. doi: 10.1111/j.1346-8138.2011.01360.x
135. Kubisch I, Diessenbacher P, Schmidt E, Gollnick H, Leverkus M. Premonitory epidermolysis bullosa acquisita mimicking eyelid dermatitis: successful treatment with rituximab and protein A immunoapheresis. Am J Clin Dermatol. (2010) 11:289–93. doi: 10.2165/11533210-000000000-00000
136. Saha M, Cutler T, Bhogal B, Black MM, Groves RW. Refractory epidermolysis bullosa acquisita: successful treatment with rituximab. Clin Exp Dermatol. (2009) 34:e979–80. doi: 10.1111/j.1365-2230.2009.03608.x
137. Wallet-Faber N, Franck N, Batteux F, Mateus C, Gilbert D, Carlotti A, et al. Epidermolysis bullosa acquisita following bullous pemphigoid, successfully treated with the anti-CD20 monoclonal antibody rituximab. Dermatol (2007) 215:252–5. doi: 10.1159/000106585
138. Niedermeier A, Eming R, Pfutze M, Neumann CR, Happel C, Reich K, et al. Clinical response of severe mechanobullous epidermolysis bullosa acquisita to combined treatment with immunoadsorption and rituximab (anti-CD20 monoclonal antibodies). Arch Dermatol. (2007) 143:192–8. doi: 10.1001/archderm.143.2.192
139. Schmidt E, Benoit S, Brocker EB, Zillikens D, Goebeler M. Successful adjuvant treatment of recalcitrant epidermolysis bullosa acquisita with anti-CD20 antibody rituximab. Arch Dermatol. (2006) 142:147–50. doi: 10.1001/archderm.142.2.147
140. Joly P, Maho-Vaillant M, Prost-Squarcioni C, Hebert V, Houivet E, Calbo S, et al. First-line rituximab combined with short-term prednisone versus prednisone alone for the treatment of pemphigus (Ritux 3): a prospective, multicentre, parallel-group, open-label randomised trial. Lancet (2017) 389:2031–40. doi: 10.1016/S0140-6736(17)30070-3
141. Lamberts A, Euverman HI, Terra JB, Jonkman MF, Horvath B. Effectiveness and safety of rituximab in recalcitrant pemphigoid diseases. Front Immunol. (2018) 9:248. doi: 10.3389/fimmu.2018.00248
142. Iwata H, Bieber K, Tiburzy B, Chrobok N, Kalies K, Shimizu A, et al. B cells, dendritic cells, and macrophages are required to induce an autoreactive CD4 helper T cell response in experimental epidermolysis bullosa acquisita. J Immunol. (2013) 191:2978–88. doi: 10.4049/jimmunol.1300310
143. Langenhan J, Dworschak J, Saschenbrecker S, Komorowski L, Schlumberger W, Stocker W, et al. Specific immunoadsorption of pathogenic autoantibodies in pemphigus requires the entire ectodomains of desmogleins. Exp Dermatol. (2014) 23:253–9. doi: 10.1111/exd.12355
144. Mersmann M, Dworschak J, Ebermann K, Komorowski L, Schlumberger W, Stocker W, et al. Immunoadsorber for specific apheresis of autoantibodies in the treatment of bullous pemphigoid. Arch Dermatol Res. (2016) 308:31–8. doi: 10.1007/s00403-015-1606-7
145. Woodley DT, Remington J, Chen M. Autoimmunity to type VII collagen: epidermolysis bullosa acquisita. Clin Rev Allergy Immunol. (2007) 33:78–84. doi: 10.1007/s12016-007-0027-6
146. Baroudjian B, Le Roux-Villet C, Brechignac S, Alexandre M, Caux F, Prost-Squarcioni C, et al. Long-term efficacy of extracorporeal photochemotherapy in a patient with refractory epidermolysis bullosa acquisita. Eur J Dermatol. (2012) 22:795–7. doi: 10.1684/ejd.2012.1840
147. Miller JL, Stricklin GP, Fine JD, King LE, Arzubiaga MC, Ellis DL. Remission of severe epidermolysis bullosa acquisita induced by extracorporeal photochemotherapy. Br J Dermatol. (1995) 133:467–71. doi: 10.1111/j.1365-2133.1995.tb02680.x
148. Egan CA, Brown M, White JD, Yancey KB. Treatment of epidermolysis bullosa acquisita with the humanized anti-Tac mAb daclizumab. Clin Immunol. (2001) 101:146–51. doi: 10.1006/clim.2001.5113
149. Labeille B, Gineston JL, Denoeux JP, Capron JP. Epidermolysis bullosa acquisita and Crohn's disease. a case report with immunological and electron microscopic studies. Arch Intern Med. (1988) 148:1457–9. doi: 10.1001/archinte.1988.00380060221040
150. Williams HC, Wojnarowska F, Kirtschig G, Mason J, Godec TR, Schmidt E, et al. Doxycycline versus prednisolone as an initial treatment strategy for bullous pemphigoid: a pragmatic, non-inferiority, randomised controlled trial. Lancet (2017) 389:1630–8. doi: 10.1016/S0140-6736(17)30560-3
151. Cavailhes A, Balme B, Gilbert D, Skowron F. Successful use of combined corticosteroids and rituximab in the treatment of recalcitrant epidermolysis bullosa acquisita. Ann Dermatol Venereol. (2009) 136:795–9. doi: 10.1016/j.annder.2009.02.007
152. Kawase K, Oshitani Y, Mizutani Y, Shu E, Fujine E, Seishima M. Inflammatory epidermolysis bullosa acquisita effectively treated with minocycline. Acta Derm Venereol. (2014) 94:6145–6. doi: 10.2340/00015555-1804
153. Nagano T, Tani M, Hiramatsu Y, Kohriyama K, Ishihara K, Nei M, et al. A case of epidermolysis bullosa acquisita with bleeding tendency due to factor VIII inhibitor (acquired haemophilia). Br J Dermatol. (2004) 151:716–7. doi: 10.1111/j.1365-2133.2004.06150.x
154. Joly P, Roujeau JC, Benichou J, Delaporte E, D'Incan M, Dreno B, et al. A comparison of two regimens of topical corticosteroids in the treatment of patients with bullous pemphigoid: a multicenter randomized study. J Invest Dermatol. (2009) 129:1681–7. doi: 10.1038/jid.2008.412
155. Murrell DF, Daniel BS, Joly P, Borradori L, Amagai M, Hashimoto T, et al. Definitions and outcome measures for bullous pemphigoid: recommendations by an international panel of experts. J Am Acad Dermatol. (2012) 66:479–85. doi: 10.1016/j.jaad.2011.06.032
156. Murrell DF, Marinovic B, Caux F, Prost C, Ahmed R, Wozniak K, et al. Definitions and outcome measures for mucous membrane pemphigoid: recommendations of an international panel of experts. J Am Acad Dermatol. (2015) 72:168–74. doi: 10.1016/j.jaad.2014.08.024
157. Callot-Mellot C, Bodemer C, Caux F, Bourgault-Villada I, Fraitag S, Goudie G, et al. Epidermolysis bullosa acquisita in childhood. Arch Dermatol. (1997) 133:1122–6. doi: 10.1001/archderm.1997.03890450070008
158. Edwards S, Wakelin SH, Wojnarowska F, Marsden RA, Kirtschig G, Bhogal B, et al. Bullous pemphigoid and epidermolysis bullosa acquisita: presentation, prognosis, and immunopathology in 11 children. Pediatr Dermatol. (1998) 15:184–90. doi: 10.1111/j.1525-1470.1998.tb01311.x
159. Vorobyev A, Ujiie H, Recke A, Buijsrogge JJ, Jonkman MF, Pas HH, et al. Autoantibodies to multiple epitopes on the non-collagenous-1 domain of type VII collagen induce blisters. J Invest Dermatol. (2015) 135:1565–73. doi: 10.1038/jid.2015.51
160. Lapiere JC, Woodley DT, Parente MG, Iwasaki T, Wynn KC, Christiano AM, et al. Epitope mapping of type VII collagen. identification of discrete peptide sequences recognized by sera from patients with acquired epidermolysis bullosa. J Clin Invest. (1993) 92:1831–9. doi: 10.1172/JCI116774
161. Gammon WR, Murrell DF, Jenison MW, Padilla KM, Prisayanh PS, Jones DA, et al. Autoantibodies to type VII collagen recognize epitopes in a fibronectin-like region of the noncollagenous (NC1) domain. Journal Invest Dermatol. (1993) 100:618–22. doi: 10.1111/1523-1747.ep12472291
162. Chen M, Doostan A, Bandyopadhyay P, Remington J, Wang X, Hou Y, et al. The cartilage matrix protein subdomain of type VII collagen is pathogenic for epidermolysis bullosa acquisita. Am J Pathol. (2007) 170:2009–18. doi: 10.2353/ajpath.2007.061212
163. Ishii N, Yoshida M, Ishida-Yamamoto A, Fritsch A, Elfert S, Bruckner-Tuderman L, et al. Some epidermolysis bullosa acquisita sera react with epitopes within the triple-helical collagenous domain as indicated by immunoelectron microscopy. Br J Dermatol. (2009) 160:1090–3. doi: 10.1111/j.1365-2133.2008.08952.x
164. Ishii N, Yoshida M, Hisamatsu Y, Ishida-Yamamoto A, Nakane H, Iizuka H, et al. Epidermolysis bullosa acquisita sera react with distinct epitopes on the NC1 and NC2 domains of type VII collagen: study using immunoblotting of domain-specific recombinant proteins and postembedding immunoelectron microscopy. Br J Dermatol. (2004) 150:843–51. doi: 10.1111/j.1365-2133.2004.05933.x
165. Ludwig RJ, Recke A, Bieber K, Muller S, Marques Ade C, Banczyk D, et al. Generation of antibodies of distinct subclasses and specificity is linked to H2s in an active mouse model of epidermolysis bullosa acquisita. J Invest Dermatol. (2011) 131:167–76. doi: 10.1038/jid.2010.248
166. Noe MH, Chen M, Woodley DT, Fairley JA. Familial epidermolysis bullosa acquisita. Dermatol Online J. (2008) 14:2. Available online at: https://escholarship.org/uc/item/14b7543k
167. Bieber K, Koga H, Nishie W. In vitro and in vivo models to investigate the pathomechanisms and novel treatments for pemphigoid diseases. Exp Dermatol. (2017) 26:1163–70. doi: 10.1111/exd.13415
168. Sitaru C, Chiriac MT, Mihai S, Buning J, Gebert A, Ishiko A, et al. Induction of complement-fixing autoantibodies against type VII collagen results in subepidermal blistering in mice. J Immunol. (2006) 177:3461–8. doi: 10.4049/jimmunol.177.5.3461
169. Hammers CM, Bieber K, Kalies K, Banczyk D, Ellebrecht CT, Ibrahim SM, et al. Complement-fixing anti-type VII collagen antibodies are induced in Th1-polarized lymph nodes of epidermolysis bullosa acquisita-susceptible mice. J Immunol. (2011) 187:5043–50. doi: 10.4049/jimmunol.1100796
170. Ludwig RJ, Muller S, Marques A, Recke A, Schmidt E, Zillikens D, et al. Identification of quantitative trait loci in experimental epidermolysis bullosa acquisita. J Invest Dermatol. (2012) 132:1409–15. doi: 10.1038/jid.2011.466
171. Samavedam UK, Mitschker N, Kasprick A, Bieber K, Schmidt E, Laskay T, et al. Whole-genome expression profiling in skin reveals SYK as a key regulator of inflammation in experimental epidermolysis bullosa acquisita. Front Immunol. (2018) 9:249. doi: 10.3389/fimmu.2018.00249
172. Srinivas G, Moller S, Wang J, Kunzel S, Zillikens D, Baines JF, et al. Genome-wide mapping of gene-microbiota interactions in susceptibility to autoimmune skin blistering. Nat Commun. (2013) 4:2462. doi: 10.1038/ncomms3462
173. Ellebrecht CT, Srinivas G, Bieber K, Banczyk D, Kalies K, Kunzel S, et al. Skin microbiota-associated inflammation precedes autoantibody induced tissue damage in experimental epidermolysis bullosa acquisita. J Autoimmun. (2016) 68:14–22. doi: 10.1016/j.jaut.2015.08.007
174. Kasperkiewicz M, Müller R, Manz R, Magens M, Hammers CM, Somlai C, et al. Heat-shock protein 90 inhibition in autoimmunity to type VII collagen: evidence that nonmalignant plasma cells are not therapeutic targets. Blood (2011) 117:6135–42. doi: 10.1182/blood-2010-10-314609
175. Bieber K, Ernst AL, Tukaj S, Holtsche MM, Schmidt E, Zillikens D, et al. Analysis of serum markers of cellular immune activation in patients with bullous pemphigoid. Exp Dermatol. (2017) 26:1248–52. doi: 10.1111/exd.13382
176. Sitaru AG, Sesarman A, Mihai S, Chiriac MT, Zillikens D, Hultman P, et al. T cells are required for the production of blister-inducing autoantibodies in experimental epidermolysis bullosa acquisita. J Immunol. (2010) 184:1596–603. doi: 10.4049/jimmunol.0901412
177. Samavedam UK, Iwata H, Muller S, Schulze FS, Recke A, Schmidt E, et al. GM-CSF modulates autoantibody production and skin blistering in experimental epidermolysis bullosa acquisita. J Immunol. (2014) 192:559–71. doi: 10.4049/jimmunol.1301556
178. Haeberle S, Wei X, Bieber K, Goletz S, Ludwig RJ, Schmidt E, et al. Regulatory T-cell deficiency leads to pathogenic bullous pemphigoid antigen 230 autoantibody and autoimmune bullous disease. J Allergy Clin Immunol. (2018) S0091-6749:30614-6. doi: 10.1016/j.jaci.2018.04.006
179. Muramatsu K, Ujiie H, Kobayashi I, Nishie W, Izumi K, Ito T, et al. Regulatory T-cell dysfunction induces autoantibodies to bullous pemphigoid antigens in mice and human subjects. J Allergy Clin Immunol. (2018) S0091-6749:30615-8. doi: 10.1016/j.jaci.2018.03.014
180. Tiburzy B, Szyska M, Iwata H, Chrobok N, Kulkarni U, Hirose M, et al. Persistent autoantibody-production by intermediates between short-and long-lived plasma cells in inflamed lymph nodes of experimental epidermolysis bullosa acquisita. PLoS ONE (2013) 8:e83631. doi: 10.1371/journal.pone.0083631
181. Kasperkiewicz M, Nimmerjahn F, Wende S, Hirose M, Iwata H, Jonkman MF, et al. Genetic identification and functional validation of FcgammaRIV as key molecule in autoantibody-induced tissue injury. J Pathol. (2012) 228:8–19. doi: 10.1002/path.4023
182. Nagel A, Hertl M, Eming R. B-cell-directed therapy for inflammatory skin diseases. J Invest Dermatol. (2009) 129:289–301. doi: 10.1038/jid.2008.192
183. Martin WL, West AP Jr, Gan L, Bjorkman PJ. Crystal structure at 2.8 A of an FcRn/heterodimeric Fc complex: mechanism of pH-dependent binding. Mol Cell (2001) 7:867–77. doi: 10.1016/S1097-2765(01)00230-1
184. Kuo TT, Baker K, Yoshida M, Qiao S-W, Aveson VG, Lencer WI, et al. Neonatal Fc receptor: from immunity to therapeutics. J Clin Immunol. (2010) 30:777–89. doi: 10.1007/s10875-010-9468-4
185. Sesarman A, Sitaru AG, Olaru F, Zillikens D, Sitaru C. Neonatal Fc receptor deficiency protects from tissue injury in experimental epidermolysis bullosa acquisita. J Mol Med. (2008) 86:951–9. doi: 10.1007/s00109-008-0366-7
186. Liu Z, Roopenian DC, Zhou X, Christianson GJ, Diaz LA, Sedmak DD, et al. Beta2-microglobulin-deficient mice are resistant to bullous pemphigoid. J Exp Med. (1997) 186:777–83. doi: 10.1084/jem.186.5.777
187. Rath T, Baker K, Pyzik M, Blumberg RS. Regulation of immune responses by the neonatal fc receptor and its therapeutic implications. Front Immunol. (2014) 5:664. doi: 10.3389/fimmu.2014.00664
188. Schwab I, Mihai S, Seeling M, Kasperkiewicz M, Ludwig RJ, Nimmerjahn F. Broad requirement for terminal sialic acid residues and FcgammaRIIB for the preventive and therapeutic activity of intravenous immunoglobulins in vivo. Eur J Immunol. (2014) 44:1444–53. doi: 10.1002/eji.201344230
189. Ishii N, Hashimoto T, Zillikens D, Ludwig RJ. High-dose intravenous immunoglobulin (IVIG) therapy in autoimmune skin blistering diseases. Clin Rev Allergy Immunol. (2010) 38:186–95. doi: 10.1007/s12016-009-8153-y
190. Werth VP, Culton D, Blumberg L, Humphries J, Blumberg R, Hall R. 538 FcRn blockade with SYNT001 for the treatment of pemphigus. J Invest Dermatol. (2018) 138:S92. doi: 10.1016/j.jid.2018.03.546
191. Collin M, Ehlers M. The carbohydrate switch between pathogenic and immunosuppressive antigen-specific antibodies. Exp Dermatol. (2013) 22:511–4. doi: 10.1111/exd.12171
192. Hirose M, Vafia K, Kalies K, Groth S, Westermann J, Zillikens D, et al. Enzymatic autoantibody glycan hydrolysis alleviates autoimmunity against type VII collagen. J Autoimmun. (2012) 39:304–14. doi: 10.1016/j.jaut.2012.04.002
193. Mihai S, Albert H, Ludwig RJ, Iwata H, Bjorck L, Collin M, et al. In vivo enzymatic modulation of IgG antibodies prevents immune complex-dependent skin injury. Exp Dermatol. (2017) 26:691–6. doi: 10.1111/exd.13163
194. Wada M, Nishie W, Ujiie H, Izumi K, Iwata H, Natsuga K, et al. Epitope-dependent pathogenicity of antibodies targeting a major bullous pemphigoid autoantigen collagen XVII/BP180. J Invest Dermatol. (2016) 136:938–46. doi: 10.1016/j.jid.2015.11.030
195. Ishii N, Recke A, Mihai S, Hirose M, Hashimoto T, Zillikens D, et al. Autoantibody-induced intestinal inflammation and weight loss in experimental epidermolysis bullosa acquisita. J Pathol. (2011) 224:234–44. doi: 10.1002/path.2857
196. Paller AS, Queen LL, Woodley DT, Lane AT, Gammon WR, Briggaman RA. Organ-specific, phylogenetic, and ontogenetic distribution of the epidermolysis bullosa acquisita antigen. J Invest Dermatol. (1986) 86:376–9. doi: 10.1111/1523-1747.ep12285623
197. Recke A, Sitaru C, Vidarsson G, Evensen M, Chiriac MT, Ludwig RJ, et al. Pathogenicity of IgG subclass autoantibodies to type VII collagen: induction of dermal-epidermal separation. J Autoimmun. (2010) 34:435–44. doi: 10.1016/j.jaut.2009.11.003
198. Recke A, Trog LM, Pas HH, Vorobyev A, Abadpour A, Jonkman MF, et al. Recombinant human IgA1 and IgA2 autoantibodies to type VII collagen induce subepidermal blistering ex vivo. J Immunol. (2014) 193:1600–8. doi: 10.4049/jimmunol.1400160
199. Mihai S, Chiriac MT, Takahashi K, Thurman JM, Holers VM, Zillikens D, et al. The alternative pathway of complement activation is critical for blister induction in experimental epidermolysis bullosa acquisita. J Immunol. (2007) 178:6514–21. doi: 10.4049/jimmunol.178.10.6514
200. Klos A, Tenner AJ, Johswich K-O, Ager RR, Reis ES, Köhl J. The role of the anaphylatoxins in health and disease. Mol Immunol. (2009) 46:2753–66. doi: 10.1016/j.molimm.2009.04.027
201. Iwata H, Witte M, Samavedam UK, Gupta Y, Shimizu A, Ishiko A, et al. Radiosensitive hematopoietic cells determine the extent of skin inflammation in experimental epidermolysis bullosa acquisita. J Immunol. (2015) 195:1945–54. doi: 10.4049/jimmunol.1501003
202. Karsten CM, Pandey MK, Figge J, Kilchenstein R, Taylor PR, Rosas M, et al. Anti-inflammatory activity of IgG1 mediated by Fc galactosylation and association of FcγRIIB and dectin-1. Nat Med. (2012) 18:1401–06. doi: 10.1038/nm.2862
203. Mihai S, Hirose M, Wang Y, Thurman JM, Holers VM, Morgan BP, et al. Specific inhibition of complement activation significantly ameliorates autoimmune blistering disease in mice. Front Immunol. (2018) 9:535. doi: 10.3389/fimmu.2018.00535
204. Afonso PV, Janka-Junttila M, Lee YJ, McCann CP, Oliver CM, Aamer KA, et al. LTB4 is a signal-relay molecule during neutrophil chemotaxis. Dev Cell. (2012) 22:1079–91. doi: 10.1016/j.devcel.2012.02.003
205. Lammermann T, Afonso PV, Angermann BR, Wang JM, Kastenmuller W, Parent CA, et al. Neutrophil swarms require LTB4 and integrins at sites of cell death in vivo. Nature (2013) 498:371–5. doi: 10.1038/nature12175
206. Peters-Golden M, Henderson WR Jr. Leukotrienes. N Engl J Med. (2007) 357:1841–54. doi: 10.1056/NEJMra071371
207. Sadik CD, Kim ND, Iwakura Y, Luster AD. Neutrophils orchestrate their own recruitment in murine arthritis through C5aR and FcgammaR signaling. Proc Natl Acad Sci USA. (2012) 109:E3177–85. doi: 10.1073/pnas.1213797109
208. Sezin T, Krajewski M, Wutkowski A, Mousavi S, Chakievska L, Bieber K, et al. The leukotriene B4 and its receptor BLT1 act as critical drivers of neutrophil recruitment in murine bullous pemphigoid-like epidermolysis bullosa acquisita. J Invest Dermatol. (2017) 137:1104–13. doi: 10.1016/j.jid.2016.12.021
209. Samavedam UK, Kalies K, Scheller J, Sadeghi H, Gupta Y, Jonkman MF, et al. Recombinant IL-6 treatment protects mice from organ specific autoimmune disease by IL-6 classical signalling-dependent IL-1ra induction. J Autoimmun. (2013) 40:74–85. doi: 10.1016/j.jaut.2012.08.002
210. Ludwig RJ, Vanhoorelbeke K, Leypoldt F, Kaya Z, Bieber K, McLachlan SM, et al. Mechanisms of autoantibody-induced pathology. Front Immunol. (2017) 8:603. doi: 10.3389/fimmu.2017.00603
211. Sitaru C, Mihai S, Otto C, Chiriac MT, Hausser I, Dotterweich B, et al. Induction of dermal-epidermal separation in mice by passive transfer of antibodies specific to type VII collagen. J Clin Invest. (2005) 115:870–8. doi: 10.1172/JCI200521386
212. Hirose MBL, Zimmer D, Götz J, Westermann J, Allegretti M, Moriconi A, et al. The allosteric CXCR1/2 inhibitor DF2156A improves experimental epidermolysis bullosa acquisita. J Genet Syndr Gene Ther. (2013) 2013:9. doi: 10.4172/2157-7412.S3-005
213. Sadeghi H, Lockmann A, Hund AC, Samavedam UK, Pipi E, Vafia K, et al. Caspase-1-independent IL-1 release mediates blister formation in autoantibody-induced tissue injury through modulation of endothelial adhesion molecules. J Immunol. (2015) 194:3656–63. doi: 10.4049/jimmunol.1402688
214. Hirose M, Kasprick A, Beltsiou F, Dieckhoff Schulze K, Schulze FS, Samavedam UK, et al. Reduced skin blistering in experimental epidermolysis bullosa acquisita after anti-TNF treatment. Mol Med. (2016) 22:918–26. doi: 10.2119/molmed.2015.00206
215. Wannick M, Yu X, Iwakura Y, Ludwig R, Petersen F, Hölscher C. The role of IL-17A in the pathogenesis of Epidermolysis bullosa acquisita. In: Experimental Dermatology Conference abstract in Inflammatory Skin Disease Summit - The Translational Revolution. Vienna (2014).
216. Chiriac MT, Roesler J, Sindrilaru A, Scharffetter-Kochanek K, Zillikens D, Sitaru C. NADPH oxidase is required for neutrophil-dependent autoantibody-induced tissue damage. J Pathol. (2007) 212:56–65. doi: 10.1002/path.2157
217. Shimanovich I, Mihai S, Oostingh GJ, Ilenchuk TT, Brocker EB, Opdenakker G, et al. Granulocyte-derived elastase and gelatinase B are required for dermal-epidermal separation induced by autoantibodies from patients with epidermolysis bullosa acquisita and bullous pemphigoid. J Pathol. (2004) 204:519–27. doi: 10.1002/path.1674
218. Kopecki Z, Yang GN, Arkell RM, Jackson JE, Melville E, Iwata H, et al. Flightless I over-expression impairs skin barrier development, function and recovery following skin blistering. J Pathol. (2014) 232:541–52. doi: 10.1002/path.4323
219. Kopecki Z, Ruzehaji N, Turner C, Iwata H, Ludwig RJ, Zillikens D, et al. Topically applied flightless I neutralizing antibodies improve healing of blistered skin in a murine model of epidermolysis bullosa acquisita. J Invest Dermatol. (2013) 133:1008–16. doi: 10.1038/jid.2012.457
220. Kopecki Z, O'Neill GM, Arkell RM, Cowin AJ. Regulation of focal adhesions by flightless i involves inhibition of paxillin phosphorylation via a Rac1-dependent pathway. J Invest Dermatol. (2011) 131:1450–9. doi: 10.1038/jid.2011.69
221. Kasperkiewicz M, Kalies K, Pagel R, Bieber K, Zillikens D, Ludwig RJ. CCL3/MIP1alpha represents a biomarker but not a mandatory cytokine for disease development in experimental epidermolysis bullosa acquisita. J Dermatol Sci. (2017) 88:248–50. doi: 10.1016/j.jdermsci.2017.06.019
222. Akbarzadeh R, Yu X, Vogl T, Ludwig RJ, Schmidt E, Zillikens D, et al. Myeloid-related proteins-8 and−14 are expressed but dispensable in the pathogenesis of experimental epidermolysis bullosa acquisita and bullous pemphigoid. J Dermatol Sci. (2016) 81:165–72. doi: 10.1016/j.jdermsci.2015.12.001
223. Drager S, Kalies K, Sidronio TB, Witte M, Ludwig RJ, Bieber K. Increased TREM-1 expression in inflamed skin has no functional impact on the pathogenesis of cutaneous disorders. J Dermatol Sci. (2017) 88:152–5. doi: 10.1016/j.jdermsci.2017.05.016
224. Bieber K, Sun S, Witte M, Kasprick A, Beltsiou F, Behnen M, et al. Regulatory T cells suppress inflammation and blistering in pemphigoid diseases. Front Immunol. (2017) 8:1628. doi: 10.3389/fimmu.2017.01628
225. Bieber K, Witte M, Sun S, Hundt JE, Kalies K, Drager S, et al. T cells mediate autoantibody-induced cutaneous inflammation and blistering in epidermolysis bullosa acquisita. Sci Rep. (2016) 6:38357. doi: 10.1038/srep38357
226. Schmidt E, Ambach A, Bastian B, Brocker EB, Zillikens D. Elevated levels of interleukin-8 in blister fluid of bullous pemphigoid compared with suction blisters of healthy control subjects. J Am Acad Dermatol. (1996) 34:310–2. doi: 10.1016/S0190-9622(96)80146-0
227. Kulkarni U, Karsten CM, Kohler T, Hammerschmidt S, Bommert K, Tiburzy B, et al. IL-10 mediates plasmacytosis-associated immunodeficiency by inhibiting complement-mediated neutrophil migration. J Allergy Clin Immunol. (2016) 137:1487–97 e6. doi: 10.1016/j.jaci.2015.10.018
228. Hussein MR, Ali FM, Omar AE. Immunohistological analysis of immune cells in blistering skin lesions. J Clin Pathol. (2007) 60:62–71. doi: 10.1136/jcp.2006.037010
229. Ambach A, Zillikens D, Klingert B, Hartmann AA, Burg G. Immune phenotyping of mononuclear infiltrate in bullous pemphigoid. Hautarzt (1992) 43:81–5.
230. Sadeghi H, Gupta Y, Moller S, Samavedam UK, Behnen M, Kasprick A, et al. The retinoid-related orphan receptor alpha is essential for the end-stage effector phase of experimental epidermolysis bullosa acquisita. J Pathol. (2015) 237:111–22. doi: 10.1002/path.4556
231. Iwata H, Pipi E, Mockel N, Sondermann P, Vorobyev A, van Beek N, et al. Recombinant soluble CD32 suppresses disease progression in experimental epidermolysis bullosa acquisita. J Invest Dermatol. (2015) 135:916–9. doi: 10.1038/jid.2014.451
232. Engineer L, Dow EC, Braverman IM, Ahmed AR. Epidermolysis bullosa acquisita and multiple myeloma. J Am Acad Dermatol. (2002) 47:943–6. doi: 10.1067/mjd.2002.113682
233. Yu X, Akbarzadeh R, Pieper M, Scholzen T, Gehrig S, Schultz C, et al. Neutrophil adhesion is a prerequisite for antibody-mediated proteolytic tissue damage in experimental models of epidermolysis bullosa acquisita. J Invest Dermatol. (2018) 138:1990–8. doi: 10.1016/j.jid.2018.03.1499
234. Kasprick A, Yu X, Scholten J, Hartmann K, Pas HH, Zillikens D, et al. Conditional depletion of mast cells has no impact on the severity of experimental epidermolysis bullosa acquisita. Eur J Immunol. (2015) 45:1462–70. doi: 10.1002/eji.201444769
235. Yu X, Kasprick A, Hartmann K, Petersen F. The role of mast cells in autoimmune bullous dermatoses. Front Immunol. (2018) 9:386. doi: 10.3389/fimmu.2018.00386
236. Fang H, Zhang Y, Li N, Wang G, Liu Z. The autoimmune skin disease bullous pemphigoid: the role of mast cells in autoantibody-induced tissue injury. Front Immunol. (2018) 9:407. doi: 10.3389/fimmu.2018.00407
237. Deng F, Chen Y, Zheng J, Huang Q, Cao X, Zillikens D, et al. CD11b-deficient mice exhibit an increased severity in the late phase of antibody transfer-induced experimental epidermolysis bullosa acquisita. Exp Dermatol. (2017) 26:1175–8. doi: 10.1111/exd.13434
238. Kulkarni S, Sitaru C, Jakus Z, Anderson KE, Damoulakis G, Davidson K, et al. PI3Kbeta plays a critical role in neutrophil activation by immune complexes. Sci Signal. (2011) 4:ra23. doi: 10.1126/scisignal.2001617
239. Koga H, Kasprick A, Lopez R, Auli M, Pont M, Godessart N, et al. Therapeutic effect of a novel PI3Kδ inhibitor in experimental epidermolysis bullosa acquisita. Front Immunol. (2018) 9:1558. doi: 10.3389/fimmu.2018.01558
240. Koga H, Recke A, Vidarsson G, Pas HH, Jonkman MF, Hashimoto T, et al. PDE4 inhibition as potential treatment of epidermolysis bullosa acquisita. J Invest Dermatol. (2016) 136:2211–20. doi: 10.1016/j.jid.2016.06.619
241. Tukaj S, Hellberg L, Ueck C, Hansel M, Samavedam U, Zillikens D, et al. Heat shock protein 90 is required for ex vivo neutrophil-driven autoantibody-induced tissue damage in experimental epidermolysis bullosa acquisita. Exp Dermatol. (2015) 24:471–3. doi: 10.1111/exd.12680
242. Tukaj S, Bieber K, Kleszczynski K, Witte M, Cames R, Kalies K, et al. Topically applied Hsp90 blocker 17AAG inhibits autoantibody-mediated blister-inducing cutaneous inflammation. J Invest Dermatol. (2017) 137:341–9. doi: 10.1016/j.jid.2016.08.032
243. Samavedam UKS, Scheuber J, Seavey MM, Koga H, Witte M, Schulze FS, et al. Therapeutic efficacy of a novel selective JAK2 inhibitor (CEP-33779) in organ-specific, autoantibody-induced tissue injury. J Invest Dermatol. (2014) 2014:S16.
244. Hellberg L, Samavedam UK, Holdorf K, Hansel M, Recke A, Beckmann T, et al. Methylprednisolone blocks autoantibody-induced tissue damage in experimental models of bullous pemphigoid and epidermolysis bullosa acquisita through inhibition of neutrophil activation. J Invest Dermatol. (2013) 133:2390–9. doi: 10.1038/jid.2013.91
245. Nemeth T, Virtic O, Sitaru C, Mocsai A. The Syk tyrosine kinase is required for skin inflammation in an in vivo mouse model of epidermolysis bullosa acquisita. J Invest Dermatol. (2017) 137:2131–9. doi: 10.1016/j.jid.2017.05.017
246. Nemeth T, Futosi K, Sitaru C, Ruland J, Mocsai A. Neutrophil-specific deletion of the CARD9 gene expression regulator suppresses autoantibody-induced inflammation in vivo. Nat Commun. (2016) 7:11004. doi: 10.1038/ncomms11004
247. Kovacs M, Nemeth T, Jakus Z, Sitaru C, Simon E, Futosi K, et al. The Src family kinases Hck, Fgr, and Lyn are critical for the generation of the in vivo inflammatory environment without a direct role in leukocyte recruitment. J Exp Med. (2014) 211:1993–2011. doi: 10.1084/jem.20132496
248. Sitaru C, Kromminga A, Hashimoto T, Brocker EB, Zillikens D. Autoantibodies to type VII collagen mediate Fcgamma-dependent neutrophil activation and induce dermal-epidermal separation in cryosections of human skin. Am J Pathol. (2002) 161:301–11. doi: 10.1016/S0002-9440(10)64182-X
249. Sesarman A, Mihai S, Chiriac MT, Olaru F, Sitaru AG, Thurman JM, et al. Binding of avian IgY to type VII collagen does not activate complement and leucocytes and fails to induce subepidermal blistering in mice. Br J Dermatol. (2008) 158:463–71. doi: 10.1111/j.1365-2133.2007.08388.x
250. Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol. (2008) 8:34–47. doi: 10.1038/nri2206
251. Schulze FS, Beckmann T, Nimmerjahn F, Ishiko A, Collin M, Kohl J, et al. Fcgamma receptors III and IV mediate tissue destruction in a novel adult mouse model of bullous pemphigoid. Am J Pathol. (2014) 184:2185–96. doi: 10.1016/j.ajpath.2014.05.007
252. Yu X, Holdorf K, Kasper B, Zillikens D, Ludwig RJ, Petersen F. FcgammaRIIA and FcgammaRIIIB are required for autoantibody-induced tissue damage in experimental human models of bullous pemphigoid. J Invest Dermatol. (2010) 130:2841–4. doi: 10.1038/jid.2010.230
253. Ludwig RJ. Signalling and targeted therapy of inflammatory cells in epidermolysis bullosa acquisita. Exp Dermatol. (2017) 26:1179–86. doi: 10.1111/exd.13335
254. Ludwig RJ, Schmidt E. Cytokines in autoimmune bullous skin diseases. Epiphenomena or contribution to pathogenesis? G Ital Dermatol Venereol. (2009) 144:339–49.
255. Reichert JM. Marketed therapeutic antibodies compendium. MAbs (2012) 4:413–5. doi: 10.4161/mabs.19931
256. Tanaka T, Narazaki M, Kishimoto T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb Perspect Biol. (2014) 6:a016295. doi: 10.1101/cshperspect.a016295
257. Luo Y, Zheng SG. Hall of fame among pro-inflammatory cytokines: interleukin-6 gene and its transcriptional regulation mechanisms. Front Immunol. (2016) 7:604. doi: 10.3389/fimmu.2016.00604
258. Kopecki Z, Arkell RM, Strudwick XL, Hirose M, Ludwig RJ, Kern JS, et al. Overexpression of the Flii gene increases dermal-epidermal blistering in an autoimmune ColVII mouse model of epidermolysis bullosa acquisita. J Pathol. (2011) 225:401–13. doi: 10.1002/path.2973
259. Kopecki Z, Ludwig RJ, Cowin AJ. Cytoskeletal regulation of inflammation and its impact on skin blistering disease epidermolysis bullosa acquisita. Int J Mol Sci. (2016) 17:1116. doi: 10.3390/ijms17071116
260. Kasprick A, Holtsche MM, Rose EL, Hussain S, Schmidt E, Petersen F, et al. The anti-C1s antibody TNT003 prevents complement activation in the skin induced by bullous pemphigoid autoantibodies. J Invest Dermatol. (2018) 138:458–61. doi: 10.1016/j.jid.2017.08.030
261. Muller S, Behnen M, Bieber K, Moller S, Hellberg L, Witte M, et al. Dimethylfumarate impairs neutrophil functions. J Invest Dermatol. (2015) 136:117–26. doi: 10.1038/JID.2015.361
262. Tukaj S, Bieber K, Witte M, Ghorbanalipoor S, Schmidt E, Zillikens D, et al. Calcitriol treatment ameliorates inflammation and blistering in mouse models of epidermolysis bullosa acquisita. J Invest Dermatol. (2018) 138:301–9. doi: 10.1016/j.jid.2017.09.009
Keywords: epidermolysis bullosa acquisita, animal models, diagnosis, treatment, pathogenesis
Citation: Koga H, Prost-Squarcioni C, Iwata H, Jonkman MF, Ludwig RJ and Bieber K (2019) Epidermolysis Bullosa Acquisita: The 2019 Update. Front. Med. 5:362. doi: 10.3389/fmed.2018.00362
Received: 15 August 2018; Accepted: 19 December 2018;
Published: 10 January 2019.
Edited by:
Robert Gniadecki, University of Alberta, CanadaReviewed by:
Marian Dmochowski, Poznan University of Medical Sciences, PolandTakashi Hashimoto, Osaka University, Japan
Copyright © 2019 Koga, Prost-Squarcioni, Iwata, Jonkman, Ludwig and Bieber. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ralf J. Ludwig, ralf.ludwig@uksh.de