Eosinophilic Granulomatosis With Polyangiitis: Dissecting the Pathophysiology
 Filippo Fagni
Filippo Fagni Federica Bello
Federica Bello Giacomo Emmi
Giacomo Emmi- Department of Experimental and Clinical Medicine, University of Firenze, Firenze, Italy
Eosinophilic Granulomatosis with Polyangiitis (EGPA) is a rare multisystemic disease classified both amongst hypereosinophilic disorders and ANCA-associated vasculitis. Vessel inflammation and eosinophilic proliferation are the hallmarks of the disease and main effectors of organ damage. Two distinct disease phenotypes have classically been described according to ANCA-status: the ANCA-negative subset with eosinophil-driven manifestation and the ANCA-positive one with vasculitic manifestations. An analogous dichotomization has also been backed by histological findings and a distinct genetic background. EGPA is typically consider a Th2-mediated disease and blood and tissue eosinophilia represent the cornerstone of diagnosis. Besides, ANCA are known for inducing endothelial injury and vascular inflammation by activating the circulating neutrophils. Thus, the pathogenesis of EGPA seems to be mediated by two coexisting mechanisms. However, the verbatim application of this strict dualism cannot always be translated into routine clinical practice. In the present review we describe the current knowledge on the eosinophilic and ANCA-mediated aspects of EGPA pathogenesis. Finally, we review the rationale of the currently proposed EGPA dichotomy and future research perspectives.
Introduction
Eosinophilic Granulomatosis with Polyangiitis (EGPA) is a rare disease characterized by granulomatous and eosinophil rich inflammation and systemic necrotizing vasculitis affecting small-to-medium sized vessels. EGPA occurs in patients with asthma and peripheral and tissue eosinophilia, and ~30% of the patients present antineutrophil cytoplasm antibodies (ANCA) mainly specific for myeloperoxidase (MPO) (1). The disease is unique in its genre as it combines asthmatic manifestations with hypereosinophilic disorders and ANCA-associated vasculitis (AAV) features. Therefore, a full comprehension of its pathophysiology still lies beyond our reach.
An increasing amount of evidence indicates that EGPA's clinical phenotypes tends to segregate according to ANCA-status, as the major eosinophil-driven complications are most frequently found in the ANCA-negative subset of EGPA, namely lung infiltrates, myocardiopathy, and gastrointestinal manifestations. In contrast, MPO-ANCA-positive patients present a more “vasculitic phenotype,” which comprises palpable purpura, peripheral neuropathy, rapidly progressive glomerulonephritis and, rarely alveolar hemorrhage (2, 3) (Table 1). An analogous dichotomy is also supported by histological findings, as biopsy-proven vasculitis is found more frequently in ANCA-positive patients than in ANCA-negative ones, whereas eosinophilic infiltrates and granulomas are found with a similar frequency in the two groups (24).
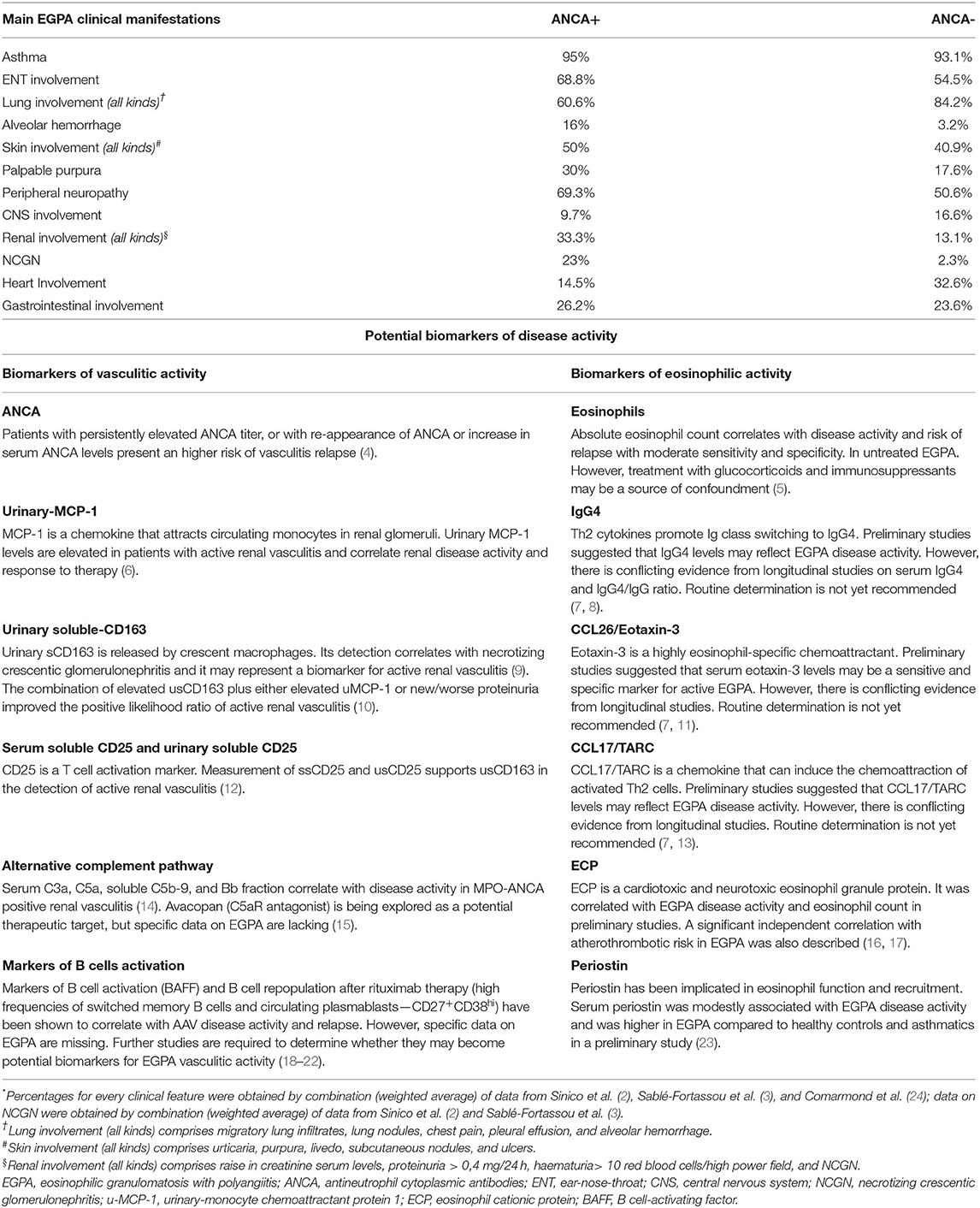
Table 1. Prevalence of EGPA clinical characteristics according to ANCA-status and biomarkers of vasculitic and eosinophilic activity*.
The dualism between ANCA-positive and ANCA-negative EGPA is also supported by genetic background. A recent genome wide association study (GWAS) found differential association of genetic variants between the two serological subsets. MPO/ANCA-positive EGPA has a significant association with HLA class II DQ haplotype, which is shared with the other MPO-AAV (i.e., microscopic polyangiitis, MPA), while ANCA-negativity is associated with GP33 and IL5/IRF1 loci, indicating a possible mucosal/barrier dysfunction origin (25).
The pathogenesis of the disease also results from the complex interaction among innate and adaptive immunity, including eosinophils, neutrophils, T-helper lymphocytes, and B lymphocytes (26).
Based on these premises, the present review will focus on untangling the interactions between the two main pathogenic processes in EGPA (i.e., eosinophilic vs. vasculitic). The validity of the current two-faced model of the disease will also be examined.
EGPA as an Eosinophilic Disorder
Eosinophils are granulocyte innate immune cells that have classically been described in allergy, host defense against parasites, myelo- and lympho-proliferative disorders, and in autoimmune diseases. Particularly, blood and tissue eosinophilia represent the diagnostic cornerstone of EGPA, making it the prototype of eosinophilic vasculitis (5). From a pathophysiological point of view, EGPA shares intrinsic mechanisms with allergy and anti-helminthic response (27–30). It is characterized by the en masse polarization of T helper lymphocytes toward a Th2 phenotype, the upregulation of eosinophil-selective eotaxin chemokines (particularly eotaxin-3), and an increased secretion of eosinophilotropic cytokines [i.e., interleukin (IL)-4, IL-5, IL-9, IL-13, and IL-25] (28, 31). So-called “allergic granulomas,” consisting of palisading giant cells surrounding a core of necrotizing eosinophils, are also a distinctive histopathological feature of EGPA and are a sign of chronic eosinophilic inflammation which have also been described within persistent helminthic infections (29). Eosinophil-mediated organ damage is a shared feature of both EGPA and hypereosinophilic syndrome (HES), and clinical aspects overlap considerably (32). From a pathogenic standpoint however, a myeloid or lymphoid clonal origin can be detected in nearly half of HES (32, 33), and sensitivity to imatinib has been reported in a number of FIP1L1-PDGFRA (F/P)-negative HES patients bearing other novel fusion genes (34–36). Nonetheless, imatinib also anecdotally showed efficacy in F/P-unmutated EGPA, while a F/P-positive EGPA patient was reported, suggesting possible shared pathogenic mechanisms with HES (37, 38).
Eosinophils' Cytotoxicity
Eosinophils exhibit a wide spectrum of cytotoxicity, that is mediated by an array of enzymes stored in cytoplasmic granules, each of which associated with distinct type of clinically observable organ damage (39) (Figure 1A). Cardiac involvement is the major cause of mortality and morbidity in EGPA and has been widely associated to eosinophilia (40–42). In vitro evidence suggests that cardiotoxicity is mainly mediated by eosinophilic cationic protein (ECP) by altering the membrane sodium permeability of cardiomyocytes and inhibiting mitochondrial respiration (41). ECP also mediates fibrogenesis by inducing the release of fibrogenic cytokines transforming growth factor β (TGF- β), IL-1α, and IL-1β (43). Consistently, the presence of eosinophilic infiltrates and granule proteins has been widely documented in fibrotic tissues, including endomyocardial biopsy specimens of patients with EGPA (44, 45). The neurotoxic properties of eosinophils are clinically evident in the form of axonal neuropathy, a frequent finding in EGPA. Histologically, this relates to the presence of infiltrating eosinophils in the endoneurium and epineural vessels of EGPA patients (46). Fiber damage is probably due to the activity of eosinophilic neurotoxin (ENT) and ECP, which have been found to induce it in vivo (47). Interestingly, ENT acts as activating factor for myeloid dendritic cells by triggering the Toll-like receptor 2 (TLR2)–MyD88 signaling pathway, which is associated with the secretion of Th2 type interleukins (48). Thus, ENT might have similar properties to an endogenous alarmin, alerting the immune system for preferential enhancement of antigen-specific Th2 response (48). Airway remodeling, subepithelial fibrosis, and ciliated cells destruction have been linked to the activity of major basic protein (MBP) in vivo (49). MBP is an abundant granule protein that can induce histamine release from basophiles, superoxide generation by alveolar macrophages, and fibrogenesis through TGF-β signaling (39, 50). Consistently, toxic-range concentrations of MBP were found in sputum and pleural fluid from asthmatics (51). An emerging aspect in eosinophils pathophysiology is their ability to induce a prothrombotic microenvironment on the endothelium. This clinically relates to an increased risk of arterial and venous thrombosis, which can be observed in EGPA (52, 53). Eosinophils can autonomously generate thrombin and induce tissue factor exposure on endothelial cells. This leads to increase platelet adhesion to the vascular wall and thrombus growth (54). Intravascular eosinophil activation also induces the formation of eosinophil extracellular traps, which can be found in human thrombi and could have a potential role in injury-related thrombosis (55). Prothrombotic alterations are also linked to ECP- and MBP-mediated interference with the coagulation cascade and to aberrant eosinophil-derived reactive oxygen species (ROS) production (56–58). Eosinophil NADPH-oxidase works in concert with eosinophil peroxidase (EPO) to generate high levels of ROS from H2O2, which in turn interact with endotheliocytes' cellular signaling to upregulate genes for adhesion molecules, tissue factor, and vasoactive substances (58, 59). Most importantly, eosinophil-derived ROS promotes lipoperoxidation, thereby contributing to atheromatous plaque formation and destabilization (55).
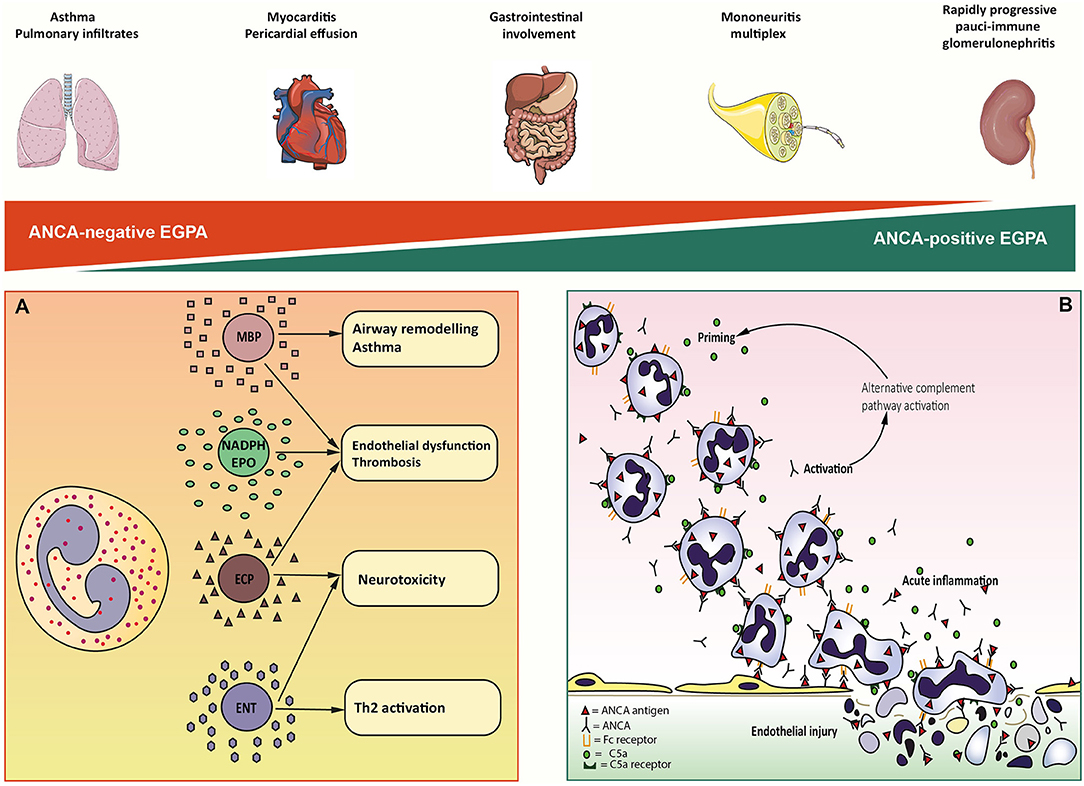
Figure 1. Overview of eosinophil-mediated and ANCA-mediated mechanisms of EGPA immunopathogenesis. On top, a schematic representation of the spectral distribution of clinical features in MPO-ANCA-negative (red triangle) and MPO-ANCA-positive EGPA (green triangle). At the extreme ends of the spectrum, lung involvement and rapidly progressive pauci-immune glomerulonephritis come as the result of predominantly eosinophilic and vasculitic mechanisms of organ damage, respectively. In contrast, the other clinical manifestations of EGPA result from the combination of the two processes. (A) Eosinophil-mediated pathogenesis of EGPA. Eosinophil granule proteins are highly cationic compounds that act synergically to mediate the cell's cytotoxicity. Mayor Basic Protein (MBP) is associated with airway remodeling and asthma, fibrogenesis, and procoagulant activity. NADPH-oxidase (NADPH) and Eosinophil Peroxidase (EPO) generate high quantities of reactive oxygen species that contribute to endothelial dysfunction and thrombosis. Eosinophil Cationic Proteins (ECP) is implied in cardiac toxicity, procoagulant activity, and nerve fibers degeneration. Eosinophil Neurotoxin (ENT) has marked neurotoxic potential in vivo. (B) Putative sequence of neutrophil-mediated endothelial injury. Circulating neutrophils get primed for ANCA activation by inflammatory cytokines and C5a complement factor. Priming induces the exposition on neutrophils cell-surface of ANCA antigens. Circulating ANCA bind to ANCA-antigens through F(ab)2 fragment and activate neutrophils by interaction with Fc receptor. Activated neutrophils release cytotoxic enzymes and factors that activate the alternative complement pathway, producing C5a that further enhances neutrophils priming. ANCA-activated neutrophils marginate and penetrate the vessel wall, where they undergo respiratory burst, degranulation, NETosis, and necrosis causing endothelial damage.
Genetic Background of Eosinophilia
Our knowledge of eosinophils' biology allowed their identification as the sole perpetrators of non-vasculitic clinical manifestations in EGPA. However, our current understanding of the primitive pathological alterations underlying the triggers and drivers of eosinophilic inflammation in EGPA is still incomplete (25).
A predisposition based on immunogenetic factors is known. EGPA is associated to HLA alleles DRB1*04 and *07 and with HLA-DRB4, suggesting a strong link with CD4+ T-lymphocyte activation (60, 61). Furthermore, functionally relevant variations of the IL-10 gene promoter were associated with EGPA in general (62), whereas IRF1/IL5 and GPA33 genes variants were associated with MPO-ANCA-negative EGPA (25). Interestingly, IL-10 and IRF1/IL5 both relate to eosinophilic inflammation. IL-10 is pivotal for the activation of the Th2pathway, while IRF1/IL5 can interact with the regulatory regions of IL-4 and IL-5 (25, 62). Analyzed IRF1/IL5 variants were associated with an increased risk to develop EGPA, higher eosinophils, and severe asthma (25). Intriguingly, GPA33 encodes a surface glycoprotein that contributes to intestinal and bronchial mucosal function, hinting at a role of barrier dysfunction and innate immunity in disease development (63).
Adaptive and Innate Immune Response and Eosinophils
Ultimately, a dysfunctional communication between innate and adaptive type 2 immunity seems to be at the root of eosinophilia in EGPA. EGPA is generally considered as a Th2-response-mediated disease due to the high eosinophil activity and the characteristically elevated serum levels of Th2cytokines (7, 28). Amongst them, IL-5 has the most relevant impact on the differentiation, proliferation, and survival of eosinophils and proved to be a promising therapeutic target (64). Indeed, the surface expression of the IL-5 receptor (CD125/CD131) is a key terminal step in eosinophil haematopoiesis and circulating IL-5 levels regulate the mobilization of eosinophils from the bone marrow (65). At a tissue level, the Th2 differentiation marker CD294 is abundantly present in biopsies from EGPA patients (13), and a Th2-dominant transcriptomic profile (STAT3, STAT6, GATA3, IL10, and IL4) was also described in bronchoalveolar lavage (66). Tissue and circulating eosinophils in EGPA also secrete IL-25, a potent eosinophilotropic cytokine that induces their own proliferation and Th2-response, thereby maintaining a vicious cycle (67). Eosinophils is EGPA are also impaired qualitatively. This is revealed by an increase in surface-expressed eosinophil activation CD69 and CD25, and by evidence of dysfunctional apoptotic pathways (68, 69). EGPA is associated with variants of the apoptosis-controlling BCL2L11 and MORRBID genes (25), the latter of which is dysregulated in hypereosinophilic syndromes (70). Furthermore, several proapoptotic genes (BCL2L13, CASP2, and CARD4) were found underexpressed in the eosinophils of EGPA patients (69), and high circulating levels of soluble CD95 (an inhibitor of Fas-mediated apoptosis) were also described (71). Finally, although their role has yet to be fully characterized, it must be mentioned that IL-5-producing innate lymphoid cells type 2 (ILC2) were also found elevated in EGPA and their blood concentration correlates with eosinophil count and disease activity (72). ILC2s are strategically embedded in peripheral tissues and orchestrate the crosstalk between epithelial cells and the immune system. Their activity has been linked to the initiation of type-2 immune responses via IL-5 and IL-13 production, eosinophils and mast cells recruitment, and M2 macrophage polarization (73). Although their real contribution to EGPA still needs to be investigated, their privileged position as an interface between innate immunity and the adaptive Th2 response could be promising for future research developments (74, 75).
EGPA as a Vasculitic Disorder
Animal and Human Models of ANCA-Mediated Vascular Inflammation
ANCA prevalence in EGPA varies from 30 to 40%. Of the EGPA patients that test positive for ANCA, 90-to 100% have MPO-ANCA specificity. Although the pathogenic role of anti-MPO ANCA has not been overtly demonstrated in EGPA, it is presumed that similar mechanisms to the ones known in MPA occur. Several animal models have demonstrated the direct noxious role of ANCA toward endothelial cells and their key interaction with neutrophils in vasculitis pathogenesis as the cause of necrotizing-crescentic glomerulonephritis (NCGN) and pulmonary hemorrhage (76). The pathogenic potential of MPO-ANCA has been documented either by injection of anti-MPO IgG in mice (77), by injection of splenocytes containing anti-MPO positive B-cells in Rag2−/− mice (which lack B and T cell responses) (77) or by transplanting bone marrow that contain MPO-positive myeloid cells in irradiated MPO−/− mice previously immunized with MPO antigen (78).
However, the relationship between ANCA and EGPA manifestations appears more complex as autoantibody titer does not always correlate with disease severity and ANCA can persist in remission phases or re-appear without clear disease activity (79, 80). Furthermore, a percentage of patients with vasculitic manifestations test negative for ANCA (81, 82), and conversely low-titer non-pathogenic ANCA have been described in healthy individuals (83, 84). These elements suggest that, despite ANCA being directly pathogenic, not all ANCA appear effectively involved. Indeed, more than 20 MPO epitopes have been identified. Antibodies to MPO specific for disease active phases were proven to be strong ROS inducers from neutrophils, whereas antibodies in healthy individuals evoked a poor neutrophilic response (85).
Recently anti-lactoferrin antibodies have been described in EGPA patients, but not in GPA and MPA, and have been directly correlated to disease activity (86). However, anti-lactoferrin antibodies can also be found in other several autoimmune diseases. Thus, whether these autoantibodies are effectively pathogenic or represent just an epiphenomenon is still unknown. Nevertheless, their detection may suggest the presence of ANCA directed toward alternative epitopes, which we are not able to identify yet.
Studies on mouse models also showed that disease manifestations could be limited by modification of ANCA IgG glycosylation on the Fc fragment (87), affecting FcγR-antibody interaction or through inhibition of p38 mitogen-activated protein kinase (MAPK), which is thought to prime and activate neutrophils (88). The scenario proposed for ANCA-mediated vascular injury starts with neutrophil priming by circulating inflammatory cytokines (Figure 1B). Once primed, neutrophils expose a small amount of ANCA antigens (normally sequestered in the cytoplasmic granules) on cell surface. ANCA F(ab)2 fragment binds to surface antigens, while the Fc fragment interacts with FcγRIIa and FcγRIIIb, triggering the respiratory burst (89). Activated neutrophils penetrate the vessel wall and release ROS and toxic enzymes causing the necrosis of endothelial cells and adjacent matrix. In addition, monocytes can be similarly activated by ANCA. Activated monocytes contribute to further neutrophil recruitment and activation and to granulomatous lesions formation. Several studies have also suggested the participation of alternative complement pathway in ANCA-induced inflammation (90, 91). Activated neutrophils release factors which activate the alternative complement pathway, resulting in the generation of C5a fragment, which in turn attracts neutrophils at the site of inflammation and primes the incoming neutrophils for ANCA activation (92).
ANCA and Adaptive Immune Responses
Adaptive immune responses also appear to be involved. A significant contribution comes from T cells, which can be found in NCGN biopsy specimens and in EGPA granulomatous lesions. It has been shown that activated neutrophils do not only cause endothelial injury but also deposit MPO-antigen in glomeruli and in the basal membrane. Anti-MPO CD4+ T cells recognize the planted glomerular MPO and amplify the immune-mediated damage (93, 94).
Increased frequencies of Th17 lymphocytes in peripheral blood samples from EGPA patients are reported during relapse of vasculitic manifestations (95). It has been proposed that MPO-ANCA activated neutrophils, through IL-6, IL-17, and IL-23 production create a permissive environment for T cells to differentiate toward a Th17 phenotype (96, 97). This generates an amplification loop in which Th17 lymphocytes promote neutrophils recruitment and activation (98).
Origin of Pathogenic-ANCA
If it is almost ascertained that ANCA play a direct role in the pathogenesis of vasculitic manifestations, the initial events leading to tolerance breakdown and to autoantibodies production remain still enigmatic. Since GWAS studies have linked MPO production with specific HLA haplotypes, it is possible to theorize that in patients with a genetically determined predisposition in antigen recognition, ANCA are produced as an initially appropriate immune response, that would lately transform into an aberrant autoimmune process. The proposed antigens include microbial peptides (99, 100), drugs (namely hydralazine, minocycline, propylthiouracil, and levamisole-adulterated cocaine) (101) or endogenously produced antisense transcripts of MPO or PR3 genes (102, 103). However, such a process has never been proved neither for MPO or PR3 ANCA.
Another factor that may influence ANCA production is epigenetic modification of MPO expression, which appear disrupted in AAV patients, potentially contributing to overexpression of ANCA antigens on neutrophil surface (104). Central loss of tolerance toward MPO was proposed to be involved in ANCA production. MPO is expressed in an AIRE-dependent manner in the thymus and its expression is involved in the central deletion of potentially autoreactive anti-MPO CD4+ T cells (105). Moreover, a defective activity both in regulatory T cells (Tregs) and regulatory B cells (Breg) have been reported in EGPA (106–108). From a functional point of view, it appears that the depletion of Tregs and Bregs, which normally would suppress immune responses, could facilitate the production of ANCA from effector B cells. The role of Bregs and autoreactive B cells is also suggested by the efficacy of rituximab (anti-CD20 antibody) (109, 110). In patients treated with rituximab, the peripheral Breg restoration rate correlates with a more effective remission of the vasculitic process (111). Finally, recent studies have focused on the possible role of a mechanism known as NETosis in ANCA production. Neutrophil extracellular traps (NETs) are a framework of chromatin fibers and antimicrobial proteins, including MPO that are released from dying neutrophils as a defense mechanism against microbes. Even in the absence of infective stimuli, the formation of NETs is enhanced in AAV patients compared to healthy controls. NETs are thought to facilitates ANCA developing by presenting antigens to the adaptive immune system (112, 113).
Intriguingly, ANCA have been detected in sputum samples of EGPA patients, irrespective to their serum ANCA-status. Despite unknown specific targets, sputum autoantibodies induced both neutrophil and eosinophil extracellular traps in vitro, suggesting their possible pathogenicity (114). It is tempting to speculate that sputum-ANCA may preceded the development of serum-ANCA positivity in a subset of EGPA patients.
Despite the presented evidence, the role of ANCA in EGPA pathogenesis is still under query. ANCA-positive EGPA patients suffer more, albeit not excursively, from vasculitis symptoms, and contrarily, not all patients which developed vasculitic manifestations display ANCA positivity.
Furthermore, the clinical differences of EGPA with other MPO-positive vasculitis contribute to the puzzling picture. Indeed, in ANCA-positive EGPA the prevalence of renal involvement varies from 27 to 51% (2, 3, 24), while in MPA, NCGN involves almost all patients (70 to 90–100%) (115, 116). A similar gap may be found also for alveolar hemorrhage, whose prevalence varies from 2 to 16% in ANCA-positive EGPA patients (24, 117, 118) against 12–30% in MPA patients (116, 119). The reasons for this attenuated vasculitic phenotype in MPO-positive EGPA compared with MPA remains undercover.
What is Missing in EGPA Dichotomy?
Progressive improvements in our clinical and pathophysiological understanding of EGPA have reflected into significant advances in the early diagnosis and treatment of the disease. However, the processes through which molecular- and cellular-scale disease mechanisms translate into macroscopic clinical changes still needs to be thoroughly elucidated.
While the pathophysiology of EGPA could be dichotomised as either “eosinophil-driven” or “ANCA-driven,” the same principle cannot be applied clinically. EGPA has been classically described to evolve through a prodromic allergic phase characterized by asthma and rhinosinusitis, an eosinophilic phase with blood and tissue eosinophilia, and a vasculitic phase with organ involvement secondary to small-vessel vasculitis. However, these phases partially overlap and may not appear in such a defined order.Besides, clinical features typical of vasculitis, such as glomerulonephritis or neuropathy, can be observed in both ANCA-negative and ANCA-positive EGPA regardless of the disease phase (81). Moreover, some clinical manifestations including cardiomyopathy and neuropathy could come as a result of the overlapping influence of eosinophilic infiltration and vasculitis (120, 121). These phases and the varied pathological findings suggest that the pathophysiology of the disorder might evolve over time. We could therefore speculate that EGPA may represent a spectrum of disease phenotypes that cannot be perfectly encapsulated by a single serological or clinical descriptor (Figure 1). For instance, a clear phenotypic subdivision is not commonly observed in the clinical practice. Cluster analysis performed on the basis of clinical features supports this observation, as it failed in demonstrating an explicit dichotomy and an intermediate phenotype with frequent renal, gastrointestinal, and cardiovascular involvement emerged (122).
Furthermore, the two subgroups share clinical, pathological and genetic features. Indeed, patients display severe asthmatic manifestations, and a percentage has a history of allergy, independently from ANCA-status. Eosinophilia and granulomatous lesions are found with analogous frequencies in both subgroups and several genetic variants associated with asthma and elevated eosinophil levels are shared (25). We can therefore speculate that EGPA is primarily caused by intrinsic eosinophil dysfunction, upon which a group of genetically predisposed patients, which present HLA-DQ variant, develop anti-MPO autoantibodies in response to an unidentified stimulus.
Additionally, the clinical value of ANCA-positivity should not be overestimated. Since MPO targeted epitopes have never been characterized, it is tempting to speculate that alternative MPO epitopes, other than MPA ones, develop in ANCA-positive EGPA, thus contributing to a mitigated vasculitic phenotype.
Therefore, stronger efforts should be made to better characterize the mechanisms underlying EGPA pathogenesis. Further studies are required in order to molecularly characterize clinical phenotypes by taking into account complex -omics data (e.g., genomics, epigenomics, transcriptomics, proteomics, and metabolomics). Shifting the subject of EGPA research from clinical phenotype to molecular endotype would possibly allow to identify valuable biomarkers and therapeutic targets that could improve diagnostic precision and therapeutic outcomes.
Author Contributions
FF and FB equally contributed to the conception and design of the work and drafted the article. GE critically revised the article. All authors approved the final version of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Vaglio A, Buzio C, Zwerina J. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss): state of the art. Allergy. (2013) 68:261–73. doi: 10.1111/all.12088
2. Sinico RA, Di Toma L, Maggiore U, Bottero P, Radice A, Tosoni C, et al. Prevalence and clinical significance of antineutrophil cytoplasmic antibodies in Churg-Strauss syndrome. Arthritis Rheum. (2005) 52:2926–35. doi: 10.1002/art.21250
3. Sablé-Fourtassou R, Cohen P, Mahr A, Pagnoux C, Mouthon L, Jayne D, et al. Antineutrophil cytoplasmic antibodies and the Churg-Strauss syndrome. Ann Intern Med. (2005) 143:632–8. doi: 10.7326/0003-4819-143-9-200511010-00006
4. Kitching AR, Anders HJ, Basu N, Brouwer E, Gordon J, Jayne DR, et al. ANCA-associated vasculitis. Nat Rev Dis Prim. (2020) 6:71. doi: 10.1038/s41572-020-0204-y
5. Khoury P, Grayson PC, Klion AD. Eosinophils in vasculitis: characteristics and roles in pathogenesis. Nat Rev Rheumatol. (2014) 10:474–83. doi: 10.1038/nrrheum.2014.98
6. Tam FW, Sanders JS, George A, Hammad T, Miller C, Dougan T, et al. Urinary monocyte chemoattractant protein-1 (MCP-1) is a marker of active renal vasculitis. Nephrol Dial Transpl. (2004) 19:2761–8. doi: 10.1093/ndt/gfh487
7. Dejaco C, Oppl B, Monach P, Cuthbertson D, Carette S, Hoffman G, et al. Serum biomarkers in patients with relapsing eosinophilic granulomatosis with polyangiitis (Churg-Strauss). PLoS ONE. (2015) 10:e0121737. doi: 10.1371/journal.pone.0121737
8. Vaglio A, Strehl JD, Manger B, Maritati F, Alberici F, Beyer C, et al. IgG4 immune response in Churg-Strauss syndrome. Ann Rheum Dis. (2012) 71:390–3. doi: 10.1136/ard.2011.155382
9. Moran SM, Monach PA, Zgaga L, Cuthbertson D, Carette S, Khalidi NA, et al. Urinary soluble CD163 and monocyte chemoattractant protein-1 in the identification of subtle renal flare in anti-neutrophil cytoplasmic antibody-associated vasculitis. Nephrol Dial Transpl. (2020) 35:283–91. doi: 10.1093/ndt/gfy300
10. O'Reilly VP, Wong L, Kennedy C, Elliot LA, O'Meachair S, Coughlan AM, et al. Urinary soluble CD163 in active renal vasculitis. J Am Soc Nephrol. (2016) 27:2906–16. doi: 10.1681/ASN.2015050511
11. Zwerina J, Bach C, Martorana D, Jatzwauk M, Hegasy G, Moosig F, et al. Eotaxin-3 in Churg-Strauss syndrome: a clinical and immunogenetic study. Rheumatology. (2011) 50:1823–7. doi: 10.1093/rheumatology/keq445
12. Dekkema GJ, Abdulahad WH, Bijma T, Moran SM, Ryan L, Little MA, et al. Urinary and serum soluble CD25 complements urinary soluble CD163 to detect active renal anti-neutrophil cytoplasmic autoantibody-associated vasculitis: a cohort study. Nephrol Dial Transpl. (2019) 34:234–42. doi: 10.1093/ndt/gfy018
13. Dallos T, Heiland GR, Strehl J, Karonitsch T, Gross WL, Moosig F, et al. CCL17/thymus and activation-related chemokine in Churg-Strauss syndrome. Arthritis Rheum. (2010) 62:3496–503. doi: 10.1002/art.27678
14. Gou SJ, Yuan J, Chen M, Yu F, Zhao MH. Circulating complement activation in patients with anti-neutrophil cytoplasmic antibody-associated vasculitis. Kidney Int. (2013) 83:129–37. doi: 10.1038/ki.2012.313
15. Jayne DRW, Bruchfeld AN, Harper L, Schaier M, Venning MC, Hamilton P, et al. Randomized trial of C5a receptor inhibitor avacopan in ANCA-associated vasculitis. J Am Soc Nephrol. (2017) 28:2756–67. doi: 10.1681/ASN.2016111179
16. Niccoli G, Ferrante G, Cosentino N, Conte M, Belloni F, Marino M, et al. Eosinophil cationic protein: a new biomarker of coronary atherosclerosis. Atherosclerosis. (2010) 211:606–11. doi: 10.1016/j.atherosclerosis.2010.02.038
17. Guilpain P, Auclair JF, Tamby MC, Servettaz A, Mahr A, Weill B, et al. Serum eosinophil cationic protein: a marker of disease activity in Churg-Strauss syndrome. Ann N Y Acad Sci. (2007) 1107:392–9. doi: 10.1196/annals.1381.041
18. Venhoff N, Niessen L, Kreuzaler M, Rolink AG, Hassler F, Rizzi M, et al. Reconstitution of the peripheral B lymphocyte compartment in patients with ANCA-associated vasculitides treated with rituximab for relapsing or refractory disease. Autoimmunity. (2014) 47:401–8. doi: 10.3109/08916934.2014.914174
19. Schneeweis C, Rafalowicz M, Feist E, Buttgereit F, Rudolph PE, Burmester GR, et al. Increased levels of BLyS and sVCAM-1 in anti-neutrophil cytoplasmatic antibody (ANCA)-associated vasculitides (AAV). Clin Exp Rheumatol. (2010) 28(1Suppl.57):62–6.
20. McClure M, Gopaluni S, Jayne D, Jones R. B cell therapy in ANCA-associated vasculitis: current and emerging treatment options. Nat Rev Rheumatol. (2018) 14:580–91. doi: 10.1038/s41584-018-0065-x
21. Jayne D, Blockmans D, Luqmani R, Moiseev S, Ji B, Green Y, et al. Efficacy and safety of belimumab and azathioprine for maintenance of remission in antineutrophil cytoplasmic antibody-associated vasculitis: a randomized controlled study. Arthritis Rheumatol. (2019) 71:952–63. doi: 10.1002/art.40802
22. von Borstel A, Land J, Abdulahad WH, Rutgers A, Stegeman CA, Diepstra A, et al. CD27+CD38hi B cell frequency during remission predicts relapsing disease in granulomatosis with polyangiitis patients. Front Immunol. (2019) 10:2221. doi: 10.3389/fimmu.2019.02221
23. Rhee RL, Holweg CTJ, Wong K, Cuthbertson D, Carette S, Khalidi NA, et al. Serum periostin as a biomarker in eosinophilic granulomatosis with polyangiitis. PLoS ONE. (2018) 13:e0205768. doi: 10.1371/journal.pone.0205768
24. Comarmond C, Pagnoux C, Khellaf M, Cordier JF, Hamidou M, Viallard JF, et al. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss): clinical characteristics and long-term followup of the 383 patients enrolled in the French Vasculitis Study Group cohort. Arthritis Rheum. (2013) 65:270–81. doi: 10.1002/art.37721
25. Lyons PA, Peters JE, Alberici F, Liley J, Coulson RMR, Astle W, et al. Genome-wide association study of eosinophilic granulomatosis with polyangiitis reveals genomic loci stratified by ANCA status. Nat Commun. (2019) 10:5120. doi: 10.1038/s41467-019-12515-9
26. Vaglio A, Moosig F, Zwerina J. Churg-strauss syndrome: update on pathophysiology and treatment. Curr Opin Rheumatol. (2012) 24:24–30. doi: 10.1097/BOR.0b013e32834d85ce
27. Akuthota P, Weller PF. Eosinophils and disease pathogenesis. Semin Hematol. (2012) 49:113–9. doi: 10.1053/j.seminhematol.2012.01.005
28. Kiene M, Csernok E, Müller A, Metzler C, Trabandt A, Gross WL. Elevated interleukin-4 and interleukin-13 production by T cell lines from patients with Churg-Strauss syndrome. Arthritis Rheum. (2001) 44:469–73. doi: 10.1002/1529-0131(200102)44:2<469::AID-ANR66>3.0.CO;2-0
29. Swartz JM, Dyer KD, Cheever AW, Ramalingam T, Pesnicak L, Domachowske JB, et al. Schistosoma mansoni infection in eosinophil lineage-ablated mice. Blood. (2006) 108:2420–7. doi: 10.1182/blood-2006-04-015933
30. de Carli ML, Miyazawa M, Nonogaki S, Shirata NK, Oliveira DT, Pereira AAC, et al. M2 macrophages and inflammatory cells in oral lesions of chronic paracoccidioidomycosis. J Oral Pathol Med. (2016) 45:141–7. doi: 10.1111/jop.12333
31. Pagnoux C, Nair P, Xi Y, Khalidi NA, Carette S, Cuthbertson D. Serum cytokine and chemokine levels in patients with eosinophilic granulomatosis with polyangiitis, hypereosinophilic syndrome, or eosinophilic asthma. Clin Exp Rheumatol. (2019) 37:40.
32. Kahn JE, Groh M, Lefèvre G. (A critical appraisal of) classification of hypereosinophilic disorders. Front Med. (2017) 4:216. doi: 10.3389/fmed.2017.00216
33. Cools J, DeAngelo DJ, Gotlib J, Stover EH, Legare RD, Cortes J, et al. A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N Engl J Med. (2003) 348:1201–14. doi: 10.1056/NEJMoa025217
34. Walz C, Curtis C, Schnittger S, Schultheis B, Metzgeroth G, Schoch C, et al. Transient response to imatinib in a chronic eosinophilic leukemia associated with ins(9;4)(q33;q12q25) and aCDK5RAP2-PDGFRA fusion gene. Genes Chromosom Cancer. (2006) 45:950–6. doi: 10.1002/gcc.20359
35. Curtis CE, Grand FH, Musto P, Clark A, Murphy J, Perla G, et al. Two novel imatinib-responsive PDGFRA fusion genes in chronic eosinophilic leukemia. Br J Haematol. (2007) 138:77–81. doi: 10.1111/j.1365-2141.2007.06628.x
36. Score J, Curtis C, Waghorn K, Stalder M, Jotterand M, Grand FH, et al. Identification of a novel imatinib responsive KIF5B-PDGFRA fusion gene following screening for PDGFRA overexpression in patients with hypereosinophilia. Leukemia. (2006) 20:827–32. doi: 10.1038/sj.leu.2404154
37. Emmi G, Silvestri E, Marconi R, Carrai V, Fanelli T, Zucchini P, et al. First report of FIP1L1-PDGFRα -positive eosinophilic granulomatosis with polyangiitis: figure 1. Rheumatology. (2015) 54:1751–3. doi: 10.1093/rheumatology/kev242
38. Triggianese P, D'Antonio A, Conigliaro P, Buccisano F, Fonti GL, Chimenti MS, et al. A new focus on thyrosine kinases inhibitors in eosinophilic granulomatosis with polyangiitis. Clin Exp Rheumatol. (2020).
39. Acharya KR, Ackerman SJ. Eosinophil granule proteins: form and function. J Biol Chem. (2014) 289:17406–15. doi: 10.1074/jbc.R113.546218
40. Cereda AF, Pedrotti P, De Capitani L, Giannattasio C, Roghi A. Comprehensive evaluation of cardiac involvement in eosinophilic granulomatosis with polyangiitis (EGPA) with cardiac magnetic resonance. Eur J Intern Med. (2017) 39:51–6. doi: 10.1016/j.ejim.2016.09.014
41. Spry CJF, Tai PC. The cardiotoxicity of eosinophils. Postgrad Med J. (1983) 59:147–53. doi: 10.1136/pgmj.59.689.147
42. Zampieri M, Emmi G, Beltrami M, Fumagalli C, Urban ML, Dei L-L, et al. Cardiac involvement in eosinophilic granulomatosis with polyangiitis (formerly Churg-Strauss syndrome): Prospective evaluation at a tertiary referral centre. Eur J Intern. Med. (2020). doi: 10.1016/j.ejim.2020.12.008
43. Zagai U, Dadfar E, Lundahl J, Venge P, Sköld CM. Eosinophil cationic protein stimulates TGF-β1 release by human lung fibroblasts in vitro. Inflammation. (2007) 30:153–60. doi: 10.1007/s10753-007-9032-4
44. Noguchi H, Kephart GM, Colby TV, Gleich GJ. Tissue eosinophilia and eosinophil degranulation in syndromes associated with fibrosis. Am J Pathol. (1992) 140:521–8.
45. Arima M, Kanoh T. Eosinophilic myocarditis associated with dense deposits of eosinophil cationic protein (ECP) in endomyocardium with high serum ECP. Heart. (1999) 81:669–75. doi: 10.1136/hrt.81.6.669
46. Nishi R, Koike H, Ohyama K, Fukami Y, Ikeda S, Kawagashira Y, et al. Differential clinicopathologic features of EGPA-associated neuropathy with and without ANCA. Neurology. (2020) 94:E1726–37. doi: 10.1212/WNL.0000000000009309
47. Fredens K, Dahl R, Venge P. The Gordon phenomenon induced by the eosinophil cationic protein and eosinophil protein X. J Allergy Clin Immunol. (1982) 70:361–6. doi: 10.1016/0091-6749(82)90025-2
48. Yang D, Chen Q, Shao BS, Zhang P, Kurosaka K, Caspi RR, et al. Eosinophil-derived neurotoxin acts as an alarmin to activate the TLR2-MyD88 signal pathway in dendritic cells and enhances Th2 immune responses. J Exp Med. (2008) 205:79–90. doi: 10.1084/jem.20062027
49. Frigas E, Gleich GJ. The eosinophil and the pathophysiology of asthma. J Allergy Clin Immunol. (1986) 77:527–37. doi: 10.1016/0091-6749(86)90341-6
50. Soragni A, Yousefi S, Stoeckle C, Soriaga AB, Sawaya MR, Kozlowski E, et al. Toxicity of eosinophil MBP is repressed by intracellular crystallization and promoted by extracellular aggregation. Mol Cell. (2015) 57:1011–21. doi: 10.1016/j.molcel.2015.01.026
51. Frigas E, Loegering DA, Solley GO. Elevated levels of the eosinophil granule major basic protein in the sputum of patients with bronchial asthma. Mayo Clin Proc. (1981) 56:345–53.
52. Allenbach Y, Seror R, Pagnoux C, Teixeira L, Guilpain P, Guillevin L, et al. High frequency of venous thromboembolic events in Churg-Strauss syndrome, Wegener's granulomatosis and microscopic polyangiitis but not polyarteritis nodosa: a systematic retrospective study on 1,130 patients. Ann Rheum Dis. (2009) 68:564–7. doi: 10.1136/ard.2008.099051
53. Bettiol A, Sinico RA, Schiavon F, Monti S, Bozzolo EP, Franceschini F, et al. Risk of acute arterial and venous thromboembolic events in Eosinophilic Granulomatosis with Polyangiitis (Churg-Strauss syndrome). Eur Respir J. (2021) 0:2004158. doi: 10.1183/13993003.04158-2020
54. Marx C, Novotny J, Salbeck D, Zellner KR, Nicolai L, Pekayvaz K, et al. Eosinophil-platelet interactions promote atherosclerosis and stabilize thrombosis with eosinophil extracellular traps. Blood. (2019) 134:1859–72. doi: 10.1182/blood.2019000518
55. Uderhardt S, Ackermann JA, Fillep T, Hammond VJ, Willeit J, Santer P, et al. Enzymatic lipid oxidation by eosinophils propagates coagulation, hemostasis, and thrombotic disease. J Exp Med. (2017) 214:2121–38. doi: 10.1084/jem.20161070
56. Mukai HY, Ninomiya H, Ohtani K, Nagasawa T, Abe T. Major basic protein binding to thrombomodulin potentially contributes to the thrombosis in patients with eosinophilia. Br J Haematol. (1995) 90:892–9. doi: 10.1111/j.1365-2141.1995.tb05211.x
57. Slungaard A, Vercellotti GM, Tran T, Gleich GJ, Key NS. Eosinophil cationic granule proteins impair thrombomodulin function: a potential mechanism for thromboembolism in hypereosinophilic heart disease. J Clin Invest. (1993) 91:1721–30. doi: 10.1172/JCI116382
58. Wang JG, Mahmud SA, Thompson JA, Geng JG, Key NS, Slungaard A. The principal eosinophil peroxidase product, HOSCN, is a uniquely potent phagocyte oxidant inducer of endothelial cell tissue factor activity: a potential mechanism for thrombosis in eosinophilic inflammatory states. Blood. (2006) 107:558–65. doi: 10.1182/blood-2005-05-2152
59. Henderson JP, Byun J, Mueller DM, Heinecke JW. The eosinophil peroxidase-hydrogen peroxide-bromide system of human eosinophils generates 5-bromouracil, a mutagenic thymine analog. Biochemistry. (2001) 40:2052–9. doi: 10.1021/bi002015f
60. Wieczorek S, Hellmich B, Gross WL, Epplen JT. Associations of Churg-Strauss syndrome with the HLA-DRB1 locus, and relationship to the genetics of antineutrophil cytoplasmic antibody-associated vasculitides: comment on the article by Vaglio et al. Arthritis Rheum. (2008) 58:329–30. doi: 10.1002/art.23209
61. Vaglio A, Martorana D, Maggiore U, Grasselli C, Zanetti A, Pesci A, et al. HLA-DRB4 as a genetic risk factor for Churg-Strauss syndrome. Arthritis Rheum. (2007) 56:3159–66. doi: 10.1002/art.22834
62. Wieczorek S, Hellmich B, Arning L, Moosig F, Lamprecht P, Gross WL, et al. Functionally relevant variations of the interleukin-10 gene associated with antineutrophil cytoplasmic antibody-negative Churg-Strauss syndrome, but not with Wegener's granulomatosis. Arthritis Rheum. (2008) 58:1839–48. doi: 10.1002/art.23496
63. Williams BB, Tebbutt NC, Buchert M, Putoczki TL, Doggett K, Bao S, et al. Glycoprotein A33 deficiency: a new mouse model of impaired intestinal epithelial barrier function and inflammatory disease. DMM Dis Model Mech. (2015) 8:805–15. doi: 10.1242/dmm.019935
64. Wechsler ME, Akuthota P, Jayne D, Khoury P, Klion A, Langford CA, et al. Mepolizumab or placebo for eosinophilic granulomatosis with polyangiitis. N Engl J Med. (2017) 376:1921–32. doi: 10.1056/NEJMoa1702079
65. Kouro T, Takatsu K. IL-5- and eosinophil-mediated inflammation: from discovery to therapy. Int Immunol. (2009) 21:1303–9. doi: 10.1093/intimm/dxp102
66. Jakiela B, Szczeklik W, Plutecka H, Sokolowska B, Mastalerz L, Sanak M, et al. Increased production of IL-5 and dominant Th2-type response in airways of Churg-Strauss syndrome patients. Rheumatol. (2012) 51:1887–93. doi: 10.1093/rheumatology/kes171
67. Terrier B, Bièche I, Maisonobe T, Laurendeau I, Rosenzwajg M, Kahn JE, et al. Interleukin-25: a cytokine linking eosinophils and adaptive immunity in Churg-Strauss syndrome. Blood. (2010) 116:4523–31. doi: 10.1182/blood-2010-02-267542
68. Tsukadaira A. Eosinophil active cytokines and surface analysis of eosinophils in churg-strauss syndrome. Allergy Asthma Proc. (1999) 20:39–44. doi: 10.2500/108854199778681486
69. Jakiela B, Szczeklik W, Sokolowska B, Mastalerz L, Sanak M, Plutecka H, et al. Intrinsic pathway of apoptosis in peripheral blood eosinophils of Churg-Strauss syndrome. Rheumatology. (2009) 48:1202–7. doi: 10.1093/rheumatology/kep209
70. Kotzin JJ, Spencer SP, McCright SJ, Kumar DBU, Collet MA, Mowel WK, et al. The long non-coding RNA Morrbid regulates Bim and short-lived myeloid cell lifespan. Nature. (2016) 537:239–43. doi: 10.1038/nature19346
71. Müschen M, Warskulat U, Perniok A, Even J, Moers C, Kismet B, et al. Involvement of soluble CD95 in Churg-Strauss syndrome. Am J Pathol. (1999) 155:915–25. doi: 10.1016/S0002-9440(10)65191-7
72. Tsurikisawa N, Oshikata C, Watanabe M, Tsuburai T, Kaneko T, Saito H. Innate immune response reflects disease activity in eosinophilic granulomatosis with polyangiitis. Clin Exp Allergy. (2018) 48:1305–16. doi: 10.1111/cea.13209
73. Karta MR, Broide DH, Doherty TA. Insights into group 2 innate lymphoid cells in human airway disease. Curr Allergy Asthma Rep. (2016) 16:1–11. doi: 10.1007/s11882-015-0581-6
74. Herbert DR, Douglas B, Zullo K. Group 2 innate lymphoid cells (ILC2): type 2 immunity and helminth immunity. Int J Mol Sci. (2019) 20:92276. doi: 10.3390/ijms20092276
75. Dion J, London J, Chaigne B, Dumoitier N, Dunogué B, Cohen P, et al. Analysis of innate and adaptive immune responses in eosinophilic granulomatosis with polyangiitis (Churg-Strauss). Arthritis Rheumatol. (2016) 68:2448–9.
76. Shochet L, Holdsworth S, Kitching AR. Animal models of ANCA associated vasculitis. Front Immunol. (2020) 11:525. doi: 10.3389/fimmu.2020.00525
77. Xiao H, Heeringa P, Hu P, Liu Z, Zhao M, Aratani Y, et al. Antineutrophil cytoplasmic autoantibodies specific for myeloperoxidase cause glomerulonephritis and vasculitis in mice. J Clin Invest. (2002) 110:955–63. doi: 10.1172/JCI0215918
78. Schreiber A, Xiao H, Falk RJ, Jennette JC. Bone marrow-derived cells are sufficient and necessary targets to mediate glomerulonephritis and vasculitis induced by anti-myeloperoxidase antibodies. J Am Soc Nephrol. (2006) 17:3355–64. doi: 10.1681/ASN.2006070718
79. Tomasson G, Grayson PC, Mahr AD, Lavalley M, Merkel PA. Value of ANCA measurements during remission to predict a relapse of ANCA-associated vasculitis–a meta-analysis. Rheumatology. (2012) 51:100–9. doi: 10.1093/rheumatology/ker280
80. Moiseev S, Bossuyt X, Arimura Y, Blockmans D, Csernok E, Damoiseaux J, et al. International consensus on ANCA testing in eosinophilic granulomatosis with polyangiitis. Am J Respir Crit Care Med. (2020) 202:1–41. doi: 10.1164/rccm.202005-1628SO
81. Cottin V, Bel E, Bottero P, Dalhoff K, Humbert M, Lazor R, et al. Revisiting the systemic vasculitis in eosinophilic granulomatosis with polyangiitis (Churg-Strauss): a study of 157 patients by the Groupe d'Etudes et de Recherche sur les Maladies Orphelines Pulmonaires and the European Respiratory Society Taskforce on e. Autoimmun Rev. (2017) 16:1–9. doi: 10.1016/j.autrev.2016.09.018
82. Eisenberger U, Fakhouri F, Vanhille P, Beaufils H, Mahr A, Guillevin L, et al. ANCA-negative pauci-immune renal vasculitis: histology and outcome. Nephrol Dial Transplant. (2005) 20:1392–9. doi: 10.1093/ndt/gfh830
83. Cui Z, Zhao M, Segelmark M, Hellmark T. Natural autoantibodies to myeloperoxidase, proteinase 3, and the glomerular basement membrane are present in normal individuals. Kidney Int. (2010) 78:590–7. doi: 10.1038/ki.2010.198
84. Xu P-C, Cui Z, Chen M, Hellmark T, Zhao M-H. Comparison of characteristics of natural autoantibodies against myeloperoxidase and anti-myeloperoxidase autoantibodies from patients with microscopic polyangiitis. Rheumatology. (2011) 50:1236–43. doi: 10.1093/rheumatology/ker085
85. Roth AJ, Ooi JD, Hess JJ, Van Timmeren MM, Berg EA, Poulton CE, et al. Epitope specificity determines pathogenicity and detectability in anca-associated vasculitis. J Clin Invest. (2013) 123:1773–83. doi: 10.1172/JCI65292
86. Shida H, Nakazawa D, Tateyama Y, Miyoshi A, Kusunoki Y, Hattanda F, et al. The presence of anti-lactoferrin antibodies in a subgroup of eosinophilic granulomatosis with polyangiitis patients and their possible contribution to enhancement of neutrophil extracellular trap formation. Front Immunol. (2016) 7:636. doi: 10.3389/fimmu.2016.00636
87. van Timmeren MM, van der Veen BS, Stegeman CA, Petersen AH, Hellmark T, Collin M, et al. IgG glycan hydrolysis attenuates ANCA-mediated glomerulonephritis. J Am Soc Nephrol. (2010) 21:1103–14. doi: 10.1681/ASN.2009090984
88. van der Veen BS, Chen M, Müller R, van Timmeren MM, Petersen AH, Lee PA, et al. Effects of p38 mitogen-activated protein kinase inhibition on anti-neutrophil cytoplasmic autoantibody pathogenicity in vitro and in vivo. Ann Rheum Dis. (2011) 70:356–65. doi: 10.1136/ard.2010.129106
89. Kettritz R. How anti-neutrophil cytoplasmic autoantibodies activate neutrophils. Clin ExpImmunol. (2012) 169:220. doi: 10.1111/j.1365-2249.2012.04615.x
90. Xiao H, Dairaghi DJ, Powers JP, Ertl LS, Baumgart T, Wang Y, et al. C5a receptor (CD88) blockade protects against MPO-ANCA GN. J Am Soc Nephrol. (2014) 25:225–31. doi: 10.1681/ASN.2013020143
91. Xing G, Chen M, Liu G, Heeringa P, Zhang J, Zheng X, et al. Complement activation is involved in renal damage in human antineutrophil cytoplasmic autoantibody associated pauci-immune vasculitis. J Clin Immunol. (2009) 29:282–91. doi: 10.1007/s10875-008-9268-2
92. Jennette JC, Falk RJ. Pathogenesis of antineutrophil cytoplasmic autoantibody-mediated disease. Nat Rev Rheumatol. (2014) 10:463–73. doi: 10.1038/nrrheum.2014.103
93. Ruth A-J, Kitching AR, Kwan RYQ, Odobasic D, Ooi JDK, Timoshanko JR, et al. Anti-neutrophil cytoplasmic antibodies and effector CD4+ cells play non-redundant roles in anti-myeloperoxidase crescentic glomerulonephritis. J Am Soc Nephrol. (2006) 17:1940–9. doi: 10.1681/ASN.2006020108
94. Gan P-Y, Holdsworth SR, Kitching AR, Ooi JD. Myeloperoxidase (MPO)-specific CD4+ T cells contribute to MPO-anti-neutrophil cytoplasmic antibody (ANCA) associated glomerulonephritis. Cell Immunol. (2013) 282:21–7. doi: 10.1016/j.cellimm.2013.04.007
95. Saito H, Tsurikisawa N, Tsuburai T, Oshikata C, Akiyama K. Cytokine production profile of CD4+ T cells from patients with active Churg-Strauss syndrome tends toward Th17. Int Arch Allergy Immunol. (2009) 149(Suppl.):61–5. doi: 10.1159/000210656
96. Gan P-Y, Steinmetz OM, Tan DSY, O'Sullivan KM, Ooi JD, Iwakura Y, et al. Th17 cells promote autoimmune anti-myeloperoxidase glomerulonephritis. J Am Soc Nephrol. (2010) 21:925–31. doi: 10.1681/ASN.2009070763
97. Nogueira E, Hamour S, Sawant D, Henderson S, Mansfield N, Chavele K-M, et al. Serum IL-17 and IL-23 levels and autoantigen-specific Th17 cells are elevated in patients with ANCA-associated vasculitis. Nephrol Dial Transplant. (2010) 25:2209–17. doi: 10.1093/ndt/gfp783
98. Hoshino A, Nagao T, Nagi-Miura N, Ohno N, Yasuhara M, Yamamoto K, et al. MPO-ANCA induces IL-17 production by activated neutrophils in vitro via classical complement pathway-dependent manner. J Autoimmun. (2008) 31:79–89. doi: 10.1016/j.jaut.2008.03.006
99. Popa ER, Stegeman CA, Kallenberg CGM, Tervaert JWC. Staphylococcus aureus and Wegener's granulomatosis. Arthritis Res. (2002) 4:77–9. doi: 10.1186/ar392
100. Davies DJ, Moran JE, Niall JF, Ryan GB. Segmental necrotising glomerulonephritis with antineutrophil antibody: possible arbovirus etiology? Br Med J. 285:606.6297657. doi: 10.1136/bmj.285.6342.606
101. Pendergraft WF, Niles JL. Trojan horses. Curr Opin Rheumatol. (2014) 26:42–9. doi: 10.1097/BOR.0000000000000014
102. Pendergraft WF, Preston GA, Shah RR, Tropsha A, Carter CW, Jennette JC, et al. Autoimmunity is triggered by cPR-3(105-201), a protein complementary to human autoantigen proteinase-3. Nat Med. (2004) 10:72–9. doi: 10.1038/nm968
103. Preston GA, Pendergraft WF, Falk RJ. New insights that link microbes with the generation of antineutrophil cytoplasmic autoantibodies: the theory of autoantigen complementarity. Curr Opin Nephrol Hypertens. (2005) 14:217–22. doi: 10.1097/01.mnh.0000165886.93427.b1
104. Ciavatta DJ, Yang J, Preston GA, Badhwar AK, Xiao H, Hewins P, et al. Epigenetic basis for aberrant upregulation of autoantigen genes in humans with ANCA vasculitis. J Clin Invest. (2010) 120:3209–19. doi: 10.1172/JCI40034
105. Tan DSY, Gan PY, O'Sullivan KM, Hammett MV, Summers SA, Ooi JD, et al. Thymic deletion and regulatory T cells prevent antimyeloperoxidase GN. J Am Soc Nephrol. (2013) 24:573–85. doi: 10.1681/ASN.2012090898
106. Tsurikisawa N, Saito H, Oshikata C, Tsuburai T, Akiyama K. Decreases in the numbers of peripheral blood regulatory T cells, and increases in the levels of memory and activated B cells, in patients with active eosinophilic granulomatosis and polyangiitis. J Clin Immunol. (2013) 33:965–76. doi: 10.1007/s10875-013-9898-x
107. Saito H, Tsurikisawa N, Tsuburai T, Akiyama K. Involvement of regulatory T cells in the pathogenesis of Churg-Strauss syndrome. Int Arch Allergy Immunol. (2008) 146(Suppl.):73–6. doi: 10.1159/000126065
108. Wilde B, Thewissen M, Damoiseaux J, Knippenberg S, Hilhorst M, van Paassen P, et al. Regulatory B cells in ANCA-associated vasculitis. Ann Rheum Dis. (2013) 72:1416–9. doi: 10.1136/annrheumdis-2012-202986
109. Mohammad AJ, Hot A, Arndt F, Moosig F, Guerry M-J, Amudala N, et al. Rituximab for the treatment of eosinophilic granulomatosis with polyangiitis (Churg-Strauss). Ann Rheum Dis. (2016) 75:396–401. doi: 10.1136/annrheumdis-2014-206095
110. Emmi G, Rossi GM, Urban ML, Silvestri E, Prisco D, Goldoni M, et al. Scheduled rituximab maintenance reduces relapse rate in eosinophilic granulomatosis with polyangiitis. Ann Rheum Dis. (2017) 77:952–4. doi: 10.1136/annrheumdis-2017-211897
111. Bunch DO, McGregor JG, Khandoobhai NB, Aybar LT, Burkart ME, Hu Y, et al. Decreased CD5+ B cells in active ANCA vasculitis and relapse after rituximab. Clin J Am Soc Nephrol. (2013) 8:382–91. doi: 10.2215/CJN.03950412
112. Kessenbrock K, Krumbholz M, Schönermarck U, Back W, Gross WL, Werb Z, et al. Netting neutrophils in autoimmune small-vessel vasculitis. Nat Med. (2009) 15:623–5. doi: 10.1038/nm.1959
113. Söderberg D, Segelmark M. Neutrophil extracellular traps in ANCA-associated vasculitis. Front Immunol. (2016) 7:256. doi: 10.3389/fimmu.2016.00256
114. Mukherjee M, Thomas SR, Radford K, Dvorkin-Gheva A, Davydchenko S, Kjarsgaard M, et al. Sputum antineutrophil cytoplasmic antibodies in serum antineutrophil cytoplasmic antibody-negative eosinophilic granulomatosis with polyangiitis. Am J Respir Crit Care Med. (2019) 199:158–70. doi: 10.1164/rccm.201804-0809OC
115. Guillevin L, Durand-Gasselin B, Cevallos R, Gayraud M, Lhote F, Callard P, et al. Microscopic polyangiitis: clinical and laboratory findings in eighty-five patients. Arthritis Rheum. (1999) 42:421–30. doi: 10.1002/1529-0131(199904)42:3<421::AID-ANR5>3.0.CO;2-6
116. Villiger PM, Guillevin L. Microscopic polyangiitis: clinical presentation. Autoimmun Rev. (2010) 9:812–9. doi: 10.1016/j.autrev.2010.07.009
117. Durel C-A, Berthiller J, Caboni S, Jayne D, Ninet J, Hot A. Long-term followup of a multicenter cohort of 101 patients with eosinophilic granulomatosis with polyangiitis (Churg-Strauss). Arthritis Care Res. (2016) 68:374–87. doi: 10.1002/acr.22686
118. Cottin V, Bel E, Bottero P, Dalhoff K, Humbert M, Lazor R, et al. Respiratory manifestations of eosinophilic granulomatosis with polyangiitis (Churg-Strauss). Eur Respir J. (2016) 48:1429–41. doi: 10.1183/13993003.00097-2016
119. Lhote F, Cohen P, Généreau T, Gayraud M, Guillevin L. Microscopic polyangiitis: clinical aspects and treatment. Ann Med Interne. (1996) 147:165–77.8796093
120. Groh M, Masciocco G, Kirchner E, Kristen A, Pellegrini C, Varnous S, et al. Heart transplantation in patients with eosinophilic granulomatosis with polyangiitis (Churg-Strauss syndrome). J Heart Lung Transplant. (2014) 33:842–50. doi: 10.1016/j.healun.2014.02.023
121. Oka N, Kawasaki T, Matsui M, Shigematsu K, Unuma T, Sugiyama H. Two subtypes of Churg-Strauss syndrome with neuropathy: the roles of eosinophils and ANCA. Mod Rheumatol. (2011) 21:290–5. doi: 10.3109/s10165-010-0400-9
Keywords: Eosinophilic Granulomatosis with Polyangiitis, Churg-Strauss syndrome, eosinophils, hypereosinophilic syndromes, ANCA-associated vasculitis, neutrophils, myeloperoxidase, EGPA classification
Citation: Fagni F, Bello F and Emmi G (2021) Eosinophilic Granulomatosis With Polyangiitis: Dissecting the Pathophysiology. Front. Med. 8:627776. doi: 10.3389/fmed.2021.627776
Received: 10 November 2020; Accepted: 18 January 2021;
Published: 24 February 2021.
Edited by:
Augusto Vaglio, University of Parma, ItalyReviewed by:
Sergey Brodsky, Ohio State University Hospital, United StatesBeom Jin Lim, Yonsei University College of Medicine, South Korea
Copyright © 2021 Fagni, Bello and Emmi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giacomo Emmi, giacomo.emmi@unifi.it
†These authors have contributed equally to this work and share first authorship