NAD+ Homeostasis in Diabetic Kidney Disease
 Jing Xu
Jing Xu Munehiro Kitada
Munehiro Kitada Daisuke Koya
Daisuke Koya- 1Department of Diabetology and Endocrinology, Kanazawa Medical University, Uchinada, Japan
- 2Department of Endocrinology and Metabolism, The Affiliated Hospital of Guizhou Medical University, Guiyang, China
- 3Division of Anticipatory Molecular Food Science and Technology, Medical Research Institute, Kanazawa Medical University, Uchinada, Japan
The redox reaction and energy metabolism status in mitochondria is involved in the pathogenesis of metabolic related disorder in kidney including diabetic kidney disease (DKD). Nicotinamide adenine dinucleotide (NAD+) is a cofactor for redox reactions and energy metabolism in mitochondria. NAD+ can be synthesized from four precursors through three pathways. The accumulation of NAD+ may ameliorate oxidative stress, inflammation and improve mitochondrial biosynthesis via supplementation of precursors and intermediates of NAD+ and activation of sirtuins activity. Conversely, the depletion of NAD+ via NAD+ consuming enzymes including Poly (ADP-ribose) polymerases (PARPs), cADPR synthases may contribute to oxidative stress, inflammation, impaired mitochondrial biosynthesis, which leads to the pathogenesis of DKD. Therefore, homeostasis of NAD+ may be a potential target for the prevention and treatment of kidney diseases including DKD. In this review, we focus on the regulation of the metabolic balance of NAD+ on the pathogenesis of kidney diseases, especially DKD, highlight benefits of the potential interventions targeting NAD+-boosting in the treatment of these diseases.
Introduction
The kidney is rich in mitochondria where generates the majority of adenosine triphosphate (ATP) required for various cellular metabolic activities (1). Nicotinamide adenine dinucleotide (NAD+) is considered as a critical cofactor and intermediary for turning fuel into energy through redox reaction (2). During the reduction reactions, which participate in glycolysis, fatty acid oxidation, and tricarboxylic acid (TCA) cycle, NAD+ serves as a hydrogen ion acceptor to generate its reduced form, NADH. Correspondingly, NADH participates in mitochondrial ATP generation by removing hydrogen ions through oxidation reactions (2, 3). NAD+ is also a cosubstrate for enzymes involved in non-redox reactions such as the sirtuins family, poly (ADP-ribose) polymerases (PARPs), and the cyclic ADP-ribose (cADPR) synthases, such as CD38 and CD157, to participate in the regulation of multiple cellular process, especially in energy metabolism (4, 5). Decreased cellular NAD+ concentrations and NAD+ /NADH ratio are closely related to the pathogenesis of multiple age-related and metabolic diseases, including diabetic kidney disease (DKD) (6). Thus, boosting NAD+ levels may be a potential therapeutic strategy for preventing the pathogenesis of DKD. In this review, we present the biogenesis of NAD+ including its synthesis, degradation, and other regulatory signaling particularly mitochondrial quality control in the development and progression of DKD, highlight the role of NAD+ supplementation and potential therapy targeting increasing NAD+ on the treatment of DKD.
Biology of NAD+
NAD+ Anabolism
NAD+ can be synthesized from four precursors including nicotinamide (NAM), tryptophan, nicotinic acid (a form of niacin, also known as vitamin B3), and nicotinamide riboside (NR), obtained from daily diet (including milk, meats, nuts, et al.), through three pathways (2, 5) (Figure 1). In mammals, the main synthetic pathway for NAD+ is salvage pathway (7, 8). In salvage pathway, the precursors including NAM, nicotinic acid and NR are converted into an intermediate called nicotinamide mononucleotide (NMN) through a rate-limiting enzyme, nicotinamide phosphoribosyltransferase (NAMPT). The intermediate NMN can be converted into NAD+ via nicotinamide mononucleotide adenylyltransferase (NMNAT). NAD+ generated by this pathway is consumed by multiple enzymes including sirtuins, PARPs, cADPR synthases, to generate NAM, which can be reused in salvage pathway again (2, 5). In Preiss-Handler pathway, nicotinic acid obtained from the daily diet can be converted into nicotinate mononucleotide (NAMN) and nicotinic acid adenine dinucleotide (NAAD) via some key enzymes such as nicotinic acid phosphoribosyltransferase (NAPRT) and NMN transferase (NMNAT), respectively, then further generate NAD+ (5). In kidney, except salvage pathway, de novo pathway is another main source of NAD+ (9). In de novo pathway, tryptophan can be converted into NAMN via quinolinate phosphoribosyltransferase (QPRT), and then into NAD+ via the Preiss-Handler pathway (5). The regulation of rate-limiting enzymes in these pathways and the supplementation of precursors and intermediates may be potential treatments for metabolic related disorder including diabetes and obesity (7, 10, 11).
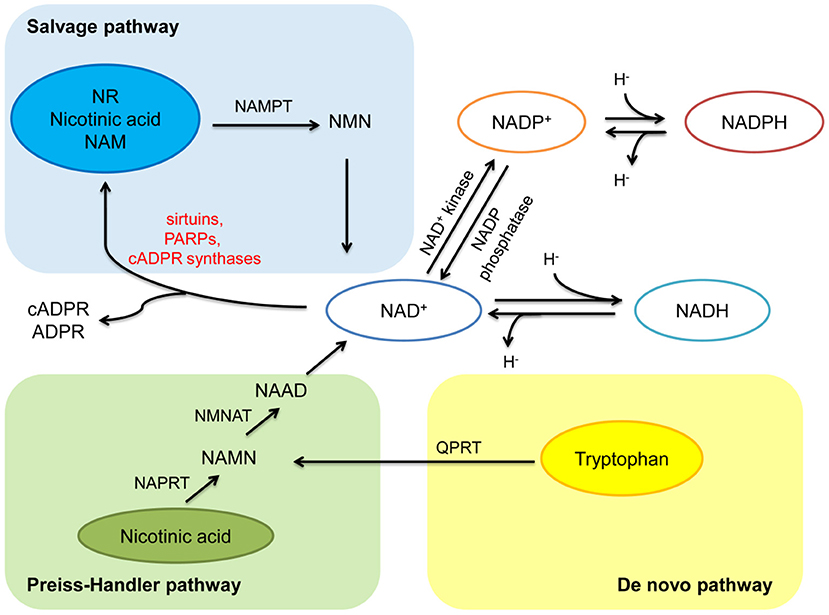
Figure 1. Anabolism and catabolism of NAD+. NAD+ is synthesized from four precursors including nicotinamide (NAM), tryptophan, nicotinic acid, and nicotinamide riboside (NR) through three pathways (salvage pathway, de novo pathway, and Preiss-Handler pathway). In salvage pathway, the precursors are converted into an intermediate called nicotinamide mononucleotide (NMN) through nicotinamide phosphoribosyltransferase (NAMPT). Then NMN is converted into NAD+ via nicotinamide mononucleotide adenylyltransferase (NMNAT). NAD+ generated by this pathway is consumed by multiple enzymes including sirtuins, PARPs, and cADPR synthases, to generate NAM, and reused in salvage pathway. In Preiss-Handler pathway, nicotinic acid is converted into nicotinate mononucleotide (NAMN) through nicotinic acid phosphoribosyltransferase (NAPRT), then NAMN is converted into nicotinic acid adenine dinucleotide (NAAD) through NMN transferase (NMNAT), further generate NAD+. In de novo pathway, tryptophan is converted into NAMN via quinolinate phosphoribosyltransferase (QPRT), and then into NAD+ via the Preiss-Handler pathway. In redox reactions, NAD+ can be phosphorylated to NADP+ by NAD+ kinase. NADP+ can be dephosphorylated to NAD+ by NADP phosphatase. Both the oxidized forms (NAD+ and NADP+) serve as hydride acceptors to generate their reduced forms (NADH and NADPH).
NAD+ Catabolism
The catabolism of NAD+ mainly involves three types of enzymes, including sirtuins, PARPs, and cADPR synthases (5) (Figure 1). These NAD+-degrading enzymes are involved in a variety of metabolic pathways and play key roles in the pathogenesis of aging-related diseases.
Sirtuins (SIRT1-7), a highly conserved homologous family from bacteria to mammals, is recognized as antiaging molecules (12). Among them, SIRT1, 6, 7 are mainly expressed in the nucleus, SIRT2 is mainly expressed in the cytoplasm, and SIRT3, 4, 5 are highly expressed in mitochondria (13). Deacetylation dependent on the consuming of NAD+ is the most important function of sirtuins, which involved in multiple metabolic regulation (14). Both calorie restriction (CR) (15, 16) and AMP-activated kinase (AMPK) (4) can induce SIRT1 activity by sensing elevated NAD+ concentration. SIRT1 deacetylates histones, including H4, H3, and H1 (17) and other transcription factors to regulate the posttranslational modifications of target genes and the expression of downstream proteins. SIRT1 deacetylates nuclear factor-κB (NF-κB) to suppress inflammatory signaling pathway (18), deacetylates signal transducer and activator of transcription 3 (STAT3) to protect from apoptosis (19), deacetylates microtubule-associated protein 1A/1B-light chain 3 (LC3) to activate autophagy (20), and deacetylates peroxisome proliferator-activated receptor gamma coactivator 1α (PGC-1α) (21, 22), Mfn (mitofusin)1 and 2 (23, 24), further participate in the regulation of mitochondrial biogenesis, oxidative stress and fusion process, respectively. SIRT2 deacetylates the forkhead box O (FOXO) 3a to suppress oxidative stress (25), deacetylates PGC-1α to regulate fatty acid oxidation (26). In erythrocytes, in response to increased oxidative stress, SIRT2 deacetylates glucose-6-phosphate dehydrogenase (G6PD), a key enzyme involved in pentose phosphate pathway, to increase the production of nicotinamide adenine dinucleotide phosphate (NADPH) (27). SIRT3 can also be activated by CR and AMPK through increased NAD+ levels (28, 29). SIRT3 deacetylates superoxide dismutase 2 (SOD2) and isocitrate dehydrogenase 2 (IDH2) to alleviate oxidative stress (30), deacetylates optic atrophy 1 (OPA1) to regulate mitochondrial fusion (31), deacetylates PGC-1α to improve mitochondrial biogenesis (32). Different from SIRT1-3, which predominately function as deacetylases, SIRT4 functions as ADP-ribose via NAD+ utilization and is responsible for cellular insulin secretion (33). SIRT4 suppresses insulin secretion via inhibiting glutamate dehydrogenase (GDH) activity through ADP-ribosylation (33) and SIRT4 knockdown increases fatty acid oxidation in liver and muscle cells (34). Although SIRT5 is also a NAD+-dependent deacetylase (35), it predominantly presents desuccinylated effect on IDH2, which suppresses cellular oxidative stress (36). SIRT6 deacetylates histone H3K9 and H3K56 to restore high glucose-impaired mitochondrial dysfunction, suppresses apoptosis and inflammation in potocytes (37). SIRT7 is less studied, previous study showed it suppresses the nuclear export of NF-κB p65 via deacetylating Ras-related nuclear antigen in the nucleus (38). These evidences combine sirtuins with mitochondrial biosynthesis, oxidative stress, apoptosis, and inflammation, suggesting a crucial role of cellular NAD+ levels regulated by sirtuins in maintaining cellular homeostasis.
PARP is a family containing at least 17 enzymes. Among them, PARP-1 is the most the most widely studied enzyme which can catalyze the synthesis of ADP-ribose on target proteins in response to DNA damage and genotoxic stress via NAD+ consumption (39, 40). High glucose induced DNA damage may contribute to excessive activation of PARP (41). Subsequent studies have also confirmed that PARP-1 is closely related to the activation of oxidative stress and inflammation (42). Selective PARP-1 inhibitor suppresses oxidative stress, inflammation via activating SIRT1/ PGC-1α signaling in diabetic mice (43) (Figure 3A and Table 1).
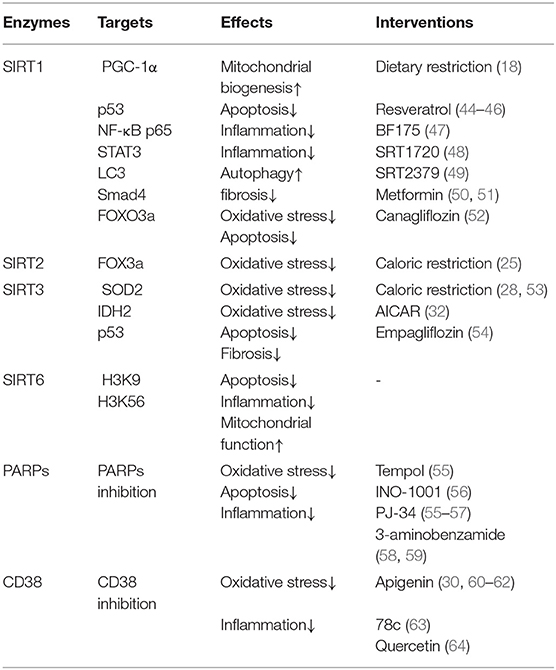
Table 1. Deacetylated targets of NAD+-consuming enzymes in DKD.
The cADPR synthases CD38 and its homolog CD157 can be induced by inflammatory cytokines, which is associated with aging-related decrease of NAD+ levels (65). CD38 catalytic activity via degrading NAD+ mainly generates NAM and cADPR through salvage pathway. One molecule of cADPR generation via CD38 for every 100 molecules of NAD+ hydrolyzed (66). CD38 also degrades NAD+ precursor NR and intermediate NMN (67). CD38 and CD157 also serve as Ca2+-transporting second messengers to stimulate Ca2+ release, which participates in the regulation of cardio muscle, renal vasoconstriction (68, 69), and insulin secretion in pancreatic β cells (70). CD38 inhibition in pancreatic β cells suppresses insulin signaling (71, 72). CD38 knockout mice show decreased cADPR and increased NAD+ concentrations, which may protect from inflammation, apoptosis, oxidative stress, and high-fat diet induced obesity (30, 73, 74) (Figure 3B and Table 1).
NADP+ and NADPH
In redox reactions, NAD+ can also be phosphorylated to NADP+ by NAD+ kinase. In contrast, NADP+ can be dephosphorylated to NAD+ by NADP phosphatase. Both The oxidized forms (NAD+ and NADP+) serve as hydride acceptors to generate their reduced forms (NADH and NADPH) (5, 75). These processes involve in the synthesis and consumption of NAD+ form a redox reaction cycle (Figure 1). Mitochondria are the main organelle that produces ROS in kidney. NADPH has antioxidant effects in mitochondrial biogenesis, while NADPH oxidases transfer electrons from NADPH and interact with oxygen to form superoxide to aggravate the production of ROS in mitochondria, which is also a main source of ROS in kidney (76) (Figure 2).
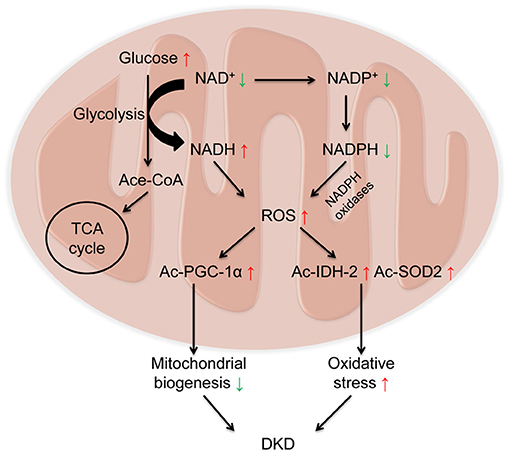
Figure 2. Decreased NAD+ and increased Reactive oxygen species (ROS) in diabetic kidney disease (DKD). Under diabetic condition, glucose is metabolized to acetylated-CoA, which enters the tricarboxylic acid cycle (TCA) cycle via consuming NAD+. On one hand, NAD+ receives hydrogen to be reduced into NADH, leading to the overload of NADH and the elevated levels of ROS. On the other hand, NADPH oxidases transfer electrons from NADPH and interact with oxygen to form superoxide to aggravate the production of ROS. Overloaded ROS in mitochondria results in the acetylation of peroxisome proliferator-activated receptor gamma coactivator 1α (PGC-1α) to impair mitochondrial biogenesis, and acetylation of isocitrate dehydrogenase 2 (IDH2) and superoxide dismutase 2 (SOD2) to aggravate oxidative stress, ultimately leading to the progression of DKD.
Regulation of NAD+ Levels in the Pathogenesis of DKD
The pathogenesis of DKD involves multiple mechanisms, including mitochondrial dysfunction, oxidative stress, and inflammation (77). Imbalance of NAD+ and NADH is a hallmark of diabetes and its chronic complications (78). Under diabetic condition, with the activation of glycolytic pathway, glucose is metabolized into acetylated-CoA, which enters the TCA cycle. During this procedure, NAD+ obtain hydrogen to become NADH via the catalysis of glyceraldehyde 3-phosphate dehydrogenase and pyruvate dehydrogenase, leading to the overload of NADH and the elevated production of reactive oxygen species (ROS), which further results in oxidative stress (79, 80). The decline in NAD+ levels also results in the acetylation of many proteins involved in oxidative stress and mitochondrial biogenesis, such as IDH2, SOD2(30), and PGC-1α (44), which ultimately leads to the progression of DKD (Figure 2). Therefore, as a key organelle for cellular redox reactions, mitochondrial function is essential for the maintenance of NAD+ levels. In this part, we mainly focus on the effects of sirtuins (especially SIRT1-6), PARPs, and cADPR synthases (particularly CD38) in the pathogenesis of DKD (Table 1).
Nuclear Sirtuins
SIRT1 deacetylates a variety of proteins involved in mitochondrial biogenesis, oxidative stress, inflammation apoptosis and autophagy via utilizing cellular NAD+ (81, 82). CR is a crucial activator of SIRT1 activity via increasing intracellular NAD+ levels. AMPK also enhances SIRT1 activity by increasing NAD+ to regulate the deacetylation of SIRT1 targets proteins (4, 83). In turn, SIRT1 can deacetylate liver kinase B1 (LKB1), a classic AMPK activating kinase to regulate the activity of AMPK (84). SIRT1 have confirmed its beneficial effects via NAD+ dependent deacetylation on amelioration of mitochondrial biogenesis, suppression of oxidative stress, fibrosis, inflammation, and apoptosis in kidney (82) (Table 1). Although mainly expressed in the nucleus, SIRT1 has been implicated in mitochondrial functions. SIRT1 deacetylates PGC-1α to increase mitochondrial biogenesis and mitochondrial fatty acid oxidation (21, 22) and attenuate high glucose-induced mitochondrial oxidative stress in potocytes (44, 45). Moreover, SIRT1 is related to autophagy and mitochondrial autophagy (mitophagy). Previous studies demonstrated that SIRT1 knockout mouse embryonic fibroblasts could not activate autophagy under starvation (85) and SIRT1 deacetylates nuclear LC3 to initiate autophagy (20). Increasing SIRT1 activity induced by elevated NAD+/NADH ratio results in increased mitochondrial membrane potential, LC3-II and proteins that regulate mitochondrial fusion and fission (86). SIRT1 reduced FOXO3a acetylation, preventing podocytes from oxidative stress and apoptosis (87). With the depletion of cellular NAD+ level in diabetic kidney, the activity of SIRT1 was suppressed, leading to acetylation of p53 and consequently cell death via apoptosis (82, 88). Our previous study and other research also demonstrated diabetic animal models showed decreased SIRT1 levels and increased acetylation of NF-κB p65 and STAT3, accompanying with elevated inflammation-related genes, leading to the injury and apoptosis of potocytes and proximal tubule (18, 19). SIRT1 also suppresses advanced glycation end-product (AGE)-induced diabetic renal fibrosis through antioxidative effects (89) and reduces epithelial-to-mesenchymal transition (EMT) to ameliorate injury-induced kidney fibrosis via deacetylating mothers against decapentaplegic homolog 4 (Smad4) in tubular epithelial cells (90). Besides, SIRT1 alleviates kidney fibrosis via suppresses transforming growth factor β (TGF-β) pathway in mesangial cells, proximal tubular cells and endothelial cells (89, 91–93).
The NAD+-dependent deacetylated effects of SIRT6 exert renoprotective effects in diabetic rodent models and high-glucose treated cells (Table 1). Previous studies reported that the expression of SIRT6 is decreased under diabetic condition (94, 95). SIRT6 inhibits high glucose-induced mitochondrial dysfunction and apoptosis in potocytes via deacetylating histone H3K9 and H3K56 (95). Proximal tubule-specific SIRT6 knockout mice exhibit enhanced fibrogenic extracellular matrix remodeling in kidney under high glucose condition (96). Podocyte-specific knockout SIRT6 mice showed renal injury and proteinuria under diabetic condition. SIRT6 inhibits Notch 1 and 4 via deacetylating histone H3K9, to suppress inflammation, apoptosis in potocytes (37) and overexpression of SIRT6 in macrophages protected podocytes from high glucose-induced renal injury (97).
Cytoplasmic and Mitochondrial Sirtuins
Studies between SIRT2 and DKD are limited. One research showed that SIRT2 is highly expressed in kidney and can deacetylate FOXO3a to decrease cellular ROS in the kidney of caloric-restricted mice (25) (Table 1).
Mitochondrial SIRT3 is a NAD+-dependent deacetylase, which predominantly exerts antioxidant activities on preventing aging-related diseases (98, 99). SIRT3 activity is decreased in diabetic patients and rodent animal models (30, 100, 101). SIRT3 deficiency presents impaired insulin secretion, renal fibrosis, elevated acetylation of mitochondrial proteins and increased mitochondrial oxidative stress (30, 102). Rat glomerular mesangial cells exposed to high glucose showed decreased NAD+/NADH ratio and SIRT3 activity, leading to oxidative stress and mesangial hypertrophy (101). Our previous studies observed decreased intracellular NAD+/NADH ratio and SIRT3 activity in diabetic rats, resulting in the activation of acetylated-SOD2 and acetylated-IDH2 in kidney mitochondria and tubular cells, then ultimately increasing ROS levels to aggravate oxidative stress (30, 103). Another study indicated overexpression of SIRT3 can ameliorated high glucose induced oxidative stress and apoptosis via Protein kinase B (Akt)/FOXO signaling in human renal tubular epithelial cells (104). In addition, SIRT3 deficiency also has a pathogenic effect on acute kidney injury (AKI). On one hand, SIRT3 deficiency increased dynamin related protein 1 (Drp1) and decreased OPA1 and PGC-1α, leading to a shift of mitochondria from fusion to fission, which exacerbates cisplatin-AKI and stress (31, 32). On the other hand, SIRT3 inhibition acetylated SOD2 and p53, leading to oxidative stress and apoptosis in ischemia/reperfusion -induced AKI (105) (Table 1).
SIRT4 functions as NAD+-dependent ADP ribosyltransferase to participate in the regulation of insulin secretion in β cells (106). SIRT4 ADP-ribosylates and suppresses GDH, a key enzyme in glutamine metabolism and ATP production, while suppression of SIRT4 activates glucose-induced insulin secretion (33, 106). SIRT4 knockout mice exert elevated basal and stimulated insulin secretion via GDH activation, resulting in glucose intolerance and insulin resistance (107). The effects of SIRT4 in renal function are limited. SIRT4 overexpression suppresses high glucose- induced overproduction of ROS and inflammatory cytokine including TNFα, IL-6, IL-1β, which protects podocytes from oxidative stress and inflammation (108).
SIRT5 is another mitochondrial NAD+-dependent deacetylase, involved in the regulation of mitochondrial quality control. Other post-translational modifications of SIRT5 include demalonylation and desuccinylation to participate in glycolysis, oxidative stress, and fatty acid oxidation (36, 109–112). In mouse liver, SIRT5 expression is suppressed by AMPK and activated by PGC-1α. SIRT5 overexpression in HepG2 cells increased ATP synthesis and oxygen consumption (113). SIRT5 knockout mice showed activated malonylation (114) and glutarylation (115) in multiple organs including kidney. SIRT5 induced the acetylation of NF-κB p65 and its downstream inflammatory cytokines, such as IL-6, TNFα, and monocyte chemoattractant protein 1 (MCP-1) (116). The role of SIRT5 in DKD is limited. However, some studies indicated that SIRT5 protects from AKI via increasing Nrf2 to suppress apoptosis (117) and regulating fatty acid oxidation to improve mitochondrial function in proximal tubule (118).
PARPs
Diabetic rodent models present hyper-activation of PARP and increased consumption of NAD+ in renal cortex. The activation of PARP is closely related to elevated levels of endothelin-1 (ET-1), a potent vasoconstrictor, and ET receptors in kidney (58, 59, 119, 120). PRAP activation is also responsible for apoptosis (55, 121, 122), inflammation and fibrosis (123), which leads to the progression of DKD (Figure 3A and Table 1). Moreover, PARP activation exacerbates oxidative stress via consuming NAD+ (55, 59). PARP-1 deficiency in diabetic mice ameliorates high glucose-induced kidney hypertrophy, mesangial expansion, collagen deposition, and urinary albumin (120).
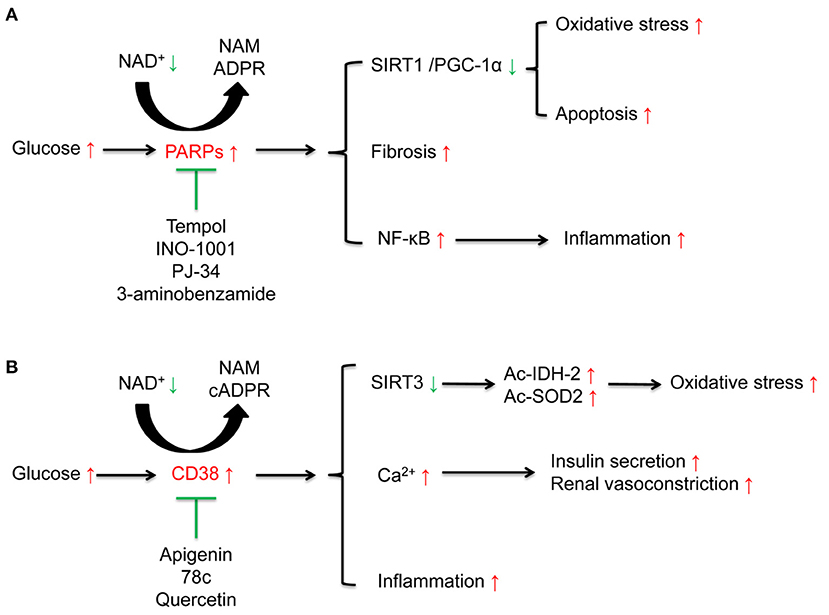
Figure 3. Effects of poly (ADP-ribose) polymerases (PARPs), and the cyclic ADP-ribose (cADPR) synthases CD38 in diabetic kidney disease (DKD). (A) High glucose activates NAD+ consuming enzyme PARPs. The activated PARPs can induce oxidative stress and apoptosis by inhibiting the sirtuins 1(SIRT1)/peroxisome proliferator-activated receptor gamma coactivator 1α (PGC-1α) signaling, accelerate renal fibrosis, and enhance inflammation by activating nuclear factor-κB (NF-κB). PARP inhibitors such as tempol, INO-1001, PJ-34, 3-aminobenzamide, can ameliorate these alterations induced by high glucose. (B) High glucose activates NAD+ consuming enzyme CD38. On one hand, CD38 induces the acetylation of isocitrate dehydrogenase 2 (IDH2) and superoxide dismutase 2 (SOD2) to aggravate oxidative stress via suppression of SIRT3 activity and is closely related to inflammation. On the other hand, CD38 is responsible for insulin secretion and renal vasoconstriction via regulating Ca2+ release. CD38 inhibitors including apigenin, 78c, and quercetin can suppress these alterations induced by high glucose.
CD38
CD38 contributes to cellular NAD+ degradation and is involved in the regulation of cellular glucose metabolism and insulin secretion (124). Compared to the wild type mice, CD38 knockout mice present higher NAD+ levels in kidney (73). The functions of CD38 in kidney partially link to nuclear and mitochondrial sirtuins, especially SIRT1 and SIRT3. CD38 inhibition protects from high-fat diet-induced obesity via NAD+-dependent SIRT1/PGC-1α signaling (74) and attenuates renal vasoconstriction caused by angiotensin II, ET-1, and norepinephrine (69). Our previous studies demonstrated that high glucose induced-CD38 is responsible for the decreased NAD+/NADH ratio and SIRT3 activity inhibition, further results in oxidative stress characterized as elevated acetylated-SOD2 and acetylated-IDH2 in renal tubular cells (30, 103) (Figure 3B and Table 1).
Potential Interventions Targeting on NAD+ in the Treatment of DKD
Based on the effect of maintaining intracellular NAD+ stability, interventions targeting the activation of the NAD+ synthesis pathway and the inhibition of the metabolic pathway have become potential therapeutic directions.
Supplementation of NAD+ Synthesis
NAD+ supplementary therapy can be derived from supplementation of NAD or precursors and intermediates of NAD+. Rat glomerular mesangial cells incubated with NAD suppresses high glucose induced mesangial hypertrophy via SIRT1 and SIRT3 mediated-AMPK/mTOR pathway (101). NR supplementation increases NAD+ and induces the activity of SIRT1 and SIRT3, which protects from oxidative stress, improves insulin sensitivity (11) and ameliorates hepatic inflammation via suppressing NLRP3 inflammasome in T2DM mice (125). Administration of exogenous NMN, a key intermediate of NAD+ synthesis, significantly induces NAD+ levels, resulting in the improvement of impaired glucose tolerance, enhancement of insulin sensitivity and suppression of inflammation characterized as decreased acetylated NF-κB p65 in liver of high fat diet- and age-induced diabetic mice (10). NMN treatment also suppresses inflammatory cytokines including TNFα, IL-1β, restores impaired β cell function in islet of fructose-induced diabetic mice. In kidney, NMN treatment alleviates inflammatory and fibrosis in glomerular mesangial cells (126) and STZ-induced diabetic rats via inhibiting endogenous NAMPT (127). A recent study also demonstrated that short-term NMN administration (for 2 weeks) increases NAD+ levels, SIRT1 expression and NAD+ salvage pathway in kidney, ameliorating urinary albumin excretion, mesangium expansion, and foot process effacement in db/db mice (128). Besides, supplementation of NMN increased NAD+ level and protected mice from cisplatin-induced AKI via activating SIRT1 and suppressing the c-Jun N-terminal kinase (JNK) signaling (129).
However, since NAD+ synthesis is a complex process involving multiple enzymes, signaling, and metabolites, benefits of supplementation of exogenous NAD+ intermediates are still controversial. Previous study showed that NAMPT, a limiting enzyme of NAD+ synthesis, is increased in streptozotocin (STZ)-induced diabetic rats, which may be an adaptive, protective response to high glucose-induced inflammation, while exogenous NAMPT may induce inflammation in tubular cells (127, 130). Endogenous NAMPT induces inflammatory and fibrosis in glomerular mesangial cells and STZ-induced diabetic rats through activating NF-κB p65 and suppressing SIRT1 (127). Besides, some studies also showed that exogenous NR administration had no benefits in young healthy animals or humans (131, 132). The results of clinical trials of NAD+ supplementation may not reverse AKI. Supplementation of NAM could not relieve inflammation, renal dysfunction and kidney injury in animals and patients of AKI (133). Another randomized, double-blind, placebo-control study demonstrated that although combination of NR supplementation and pterostillbene, a sirtuins activator, can increase NAD+ levels after 48 h treatment, there was no benefit on renal function including creatinine and estimated glomerular filtration rate in patients with AKI (134). More researches are needed to identify whether supplementation of certain intermediates in NAD+ synthesis can benefit by increasing the intracellular NAD+ concentration.
Activation of Sirtuins
Caloric/dietary restriction is an effective way to activate sirtuins and protects the progression of DKD. Dietary restriction ameliorated kidney inflammation in diabetic mice via activating SIRT1 to inhibit acetylated-NF-κB (18). Calorie restriction reduces renal oxidative stress and inflammation by SIRT2 (25) and attenuates palmitate-induced ROS production and inflammation in proximal tubular cells via SIRT3-mediated deacetylation (28, 53).
Multiple sirtuin activating compounds have been identified to protect from the progression of DKD via suppression mitochondrial oxidative stress, apoptosis, and inflammation (12). SIRT1 agonists, resveratrol (44–46), BF175 (47), ameliorated mitochondrial oxidative stress and apoptosis in podocytes of diabetic mice via regulating SIRT1/PGC-1α and SIRT1/p53 signaling. Resveratrol also ameliorated high glucose-induced mitochondrial dysfunction via activating SIRT1/Nrf-antioxidant response element (ARE) pathway (135). SRT1720 attenuated renal fibrosis and oxidative stress (48). SRT2379 inhibited LPS-stimulated JNK and IκB kinase (IKK) inflammatory pathways in macrophages (49). AMPK agonist, AICAR reduced cisplatin-induced AKI and improved renal function via the activating SIRT3 deacetylation effect and further activating mitochondrial fusion process and mitopahgy (32). In addition to these compounds under development, some anti-diabetic drugs which have been widely used clinically can also activate sirtuins. Metformin, the first-line medication for T2DM treatment, is an AMPK and sirtuin agonist. Previous studies have confirmed that metformin reduced oxidative stress, enhanced autophagy, and ameliorated insulin resistance by activating AMPK/ SIRT1 /FOXO1 signaling in rat mesangial cells and potocytes, further protected against the pathogenesis of DKD (50, 51). SGLT2 inhibitors are effective anti-diabetic drugs which present renoprotective effects. Previous studies showed SGLT2 inhibitors canagliflozin reversed high glucose-induced SIRT1 suppression (52, 136) in human renal tubular cells and db/db mice to protect against DKD. Our research indicated that SGLT2 inhibitor empagliflozin restored high glucose-suppressed SIRT3, which in part suppressed EMT and kidney fibrosis (54) (Figure 4 and Table 1).
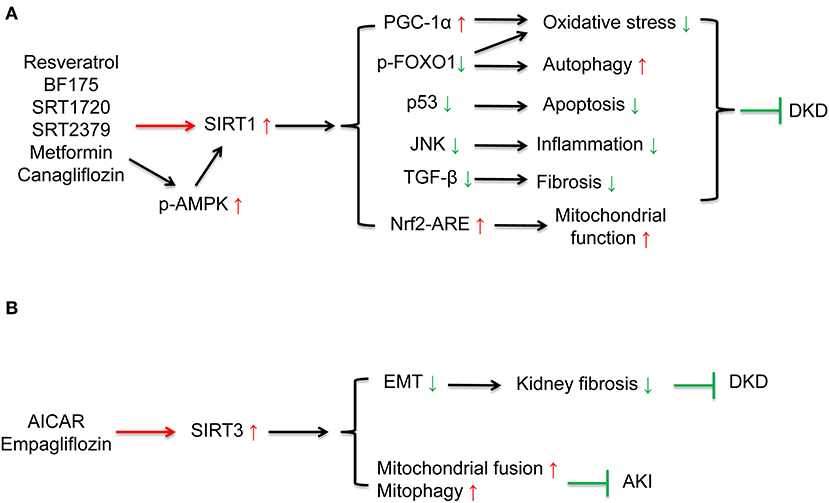
Figure 4. Drug interventions on sirtuins activation in the treatment of kidney diseases. (A) SIRT1 activation can ameliorate oxidative stress, apoptosis, fibrosis, inflammation, restore autophagy, and mitochondrial function. SIRT1 agonists, resveratrol, BF175, SRT1720, SRT2379, and some anti-diabetic drugs including metformin and canagliflozin can activate SIRT1 to protect against DKD. (B) AICAR can increase mitochondrial fusion and mitophagy via activating SIRT3 to protect against DKD. SGLT2 inhibitor empagliflozin can ameliorate kidney fibrosis via restoring high glucose-suppressed SIRT3 to protect against DKD.
Inhibition of PARPs
Given the role of PARPs in high glucose-induced oxidative stress, apoptosis and inflammation, inhibitors targeting PARP may be a potential targets for the treatment of DKD. Tempol reduces podocytes apoptosis via suppressing PARP signaling in STZ-induced diabetic rats (55). PARP inhibitors, INO-1001 and PJ-34, suppressed high glucose induced ROS levels and nuclear NF-κB in potocytes and db/db mice and potocytes apoptosis in STZ-induced diabetic rats (55, 56). Another PARP inhibitor, 3-aminobenzamide, inhibited high glucose induced oxidative stress and ET-1 expression in diabetic rodent models (58, 59). The effect of PARP inhibitor is also related to SIRT1 activation. One study demonstrated that PARP inhibitor PJ-34 interacted with SIRT1 to suppress the accumulation of renal extracellular matrix via activating AMPK/ PGC-1α signaling in db/db mice (57) (Figure 3A and Table 1).
Inhibition of CD38
Suppression NAD+-degrading enzyme such as CD38 is an effective way to increasing endogenous NAD+ level and restores impaired mitochondrial functions (63, 67). CD38 inhibitor, apigenin, increased NAD+ to decrease acetylation of p53 and NF-κB p65 (60). Based on these studies, our research demonstrated that apigenin suppressed high glucose-induced acetylation of SOD2 and IDH2 to ameliorate mitochondrial oxidative stress, via increasing NAD+/NADH ratio and SIRT3 activity in renal tubular cells of diabetic rats (30). Apigenin also ameliorated renal inflammation via inhibiting mitogen-activated protein kinase (MAPK) pathway in STZ-induced diabetic rats (61) and increased expression of NF-E2-related factor 2 (Nrf2) to protective from high glucose-induced oxidative stress, injury and inflammation in human renal tubular epithelial cells (62). For other CD38 inhibitors, 78c increased NAD+ to activate sirtuins, and AMPK, further improved glucose tolerance, muscle function, exercise capacity, and cardiac function in aged mouse model (63). Quercetin inhibited LPS-induced inflammation in macrophages via suppressing NF-κB signaling activation to relieve kidney inflammation and protect from AKI (64) (Figure 3B and Table 1).
Conclusions
The regulation of intracellular NAD+ levels has become a crucial direction for exploring the potential mechanisms of multiple age-related metabolic diseases, including DKD. The regulation of NAD+ levels mainly involves two aspects, synthesis and catabolism. Many studies have shown that increasing the precursors and intermediates of NAD+ can benefit via increasing intracellular NAD+ levels, but some studies also have shown that exogenous supplementation or activation of some certain NAD+ synthesis key enzymes (such as NAMPT) may play a negative role in metabolic pathways. Further studies are still needed to confirm the therapeutic effects of supplementing precursors and intermediates on DKD. The results of studies on NAD+ catabolism are relatively certain. On the one hand, activation of the sirtuins family (especially SIRT1, 2, 3, 6) of NAD+-dependent deacetylases can inhibit mitochondrial oxidative stress, improve mitochondrial biogenesis, alleviate inflammation and reduce apoptosis, thereby preventing the progression of DKD. On the other hand, by inhibiting NAD+-consuming enzymes such as PARPs and cADPR synthetase (especially CD38), also ameliorate mitochondrial oxidative stress, inflammation and apoptosis in DKD. There have been a variety of drugs targeting the NAD+-consuming enzymes, such as activators of the sirtuins family and inhibitors of PARPs and cADPR, which is benefit for the treatment of DKD in cell and animal models. The regulation of NAD+ catabolism may be a potential target for the treatment of DKD.
Author Contributions
JX contributed to drafting and writing the article. MK and DK contributed to the discussion of the review. MK is responsible for the integrity of the content. All authors revised the manuscript critically for important intellectual content and approved the final version to be published.
Funding
This study was supported in part by the Japan China Sasakawa Medical Fellowship to JX. The funders had no role in the interpretation or writing of the manuscript.
Conflict of Interest
Boehringer Ingelheim, Mitsubishi Tanabe Pharma, Taisho Pharmaceutical Co. and Ono Pharmaceutical Co. contributed to establishing the Division of Anticipatory Molecular Food Science and Technology.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Forbes JM. Mitochondria-power players in kidney function? Trends Endocrinol Metab. (2016) 27:441–2. doi: 10.1016/j.tem.2016.05.002
2. Ralto KM, Rhee EP, Parikh SM. NAD(+) homeostasis in renal health and disease. Nat Rev Nephrol. (2020) 16:99–111. doi: 10.1038/s41581-019-0216-6
3. Hershberger KA, Martin AS, Hirschey MD. Role of NAD(+) and mitochondrial sirtuins in cardiac and renal diseases. Nat Rev Nephrol. (2017) 13:213–25. doi: 10.1038/nrneph.2017.5
4. Canto C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, et al. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. (2009) 458:1056–60. doi: 10.1038/nature07813
5. Verdin E. NAD? in aging, metabolism, and neurodegeneration. Science. (2015) 350:1208–13. doi: 10.1126/science.aac4854
6. Tilton RG, Baier LD, Harlow JE, Smith SR, Ostrow E, Williamson JR. Diabetes-induced glomerular dysfunction: links to a more reduced cytosolic ratio of NADH/NAD+. Kidney Int. (1992) 41:778–88. doi: 10.1038/ki.1992.121
7. Revollo JR, Grimm AA, Imai S. The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J Biol Chem. (2004) 279:50754–63. doi: 10.1074/jbc.M408388200
8. Evans J, Wang TC, Heyes MP, Markey SP. LC/MS analysis of NAD biosynthesis using stable isotope pyridine precursors. Anal Biochem. (2002) 306:197–203. doi: 10.1006/abio.2002.5715
9. Liu L, Su X, Quinn WJ, III, Hui S, Krukenberg K, Frederick DW, et al. Quantitative analysis of NAD synthesis-breakdown fluxes. Cell Metab. (2018) 27:1067–80. doi: 10.1016/j.cmet.2018.03.018
10. Yoshino J, Mills KF, Yoon MJ, Imai S. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. (2011) 14:528–36. doi: 10.1016/j.cmet.2011.08.014
11. Canto C, Houtkooper RH, Pirinen E, Youn DY, Oosterveer MH, Cen Y, et al. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. (2012) 15:838–47. doi: 10.1016/j.cmet.2012.04.022
12. Wood JG, Rogina B, Lavu S, Howitz K, Helfand SL, Tatar M, et al. Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature. (2004) 430:686–9. doi: 10.1038/nature02789
13. Morigi M, Perico L, Benigni A. Sirtuins in renal health and disease. J Am Soc Nephrol. (2018) 29:1799–809. doi: 10.1681/ASN.2017111218
14. Guarente L, Franklin H. Epstein lecture: sirtuins, aging, and medicine. N Engl J Med. (2011) 364:2235–44. doi: 10.1056/NEJMra1100831
15. Kume S, Uzu T, Horiike K, Chin-Kanasaki M, Isshiki K, Araki S, et al. Calorie restriction enhances cell adaptation to hypoxia through Sirt1-dependent mitochondrial autophagy in mouse aged kidney. J Clin Invest. (2010) 120:1043–55. doi: 10.1172/JCI41376
16. Chen D, Bruno J, Easlon E, Lin SJ, Cheng HL, Alt FW, et al. Tissue-specific regulation of SIRT1 by calorie restriction. Genes Dev. (2008) 22:1753–7. doi: 10.1101/gad.1650608
17. Vaquero A, Scher M, Lee D, Erdjument-Bromage H, Tempst P, Reinberg D. Human SirT1 interacts with histone H1 and promotes formation of facultative heterochromatin. Mol Cell. (2004) 16:93–105. doi: 10.1016/j.molcel.2004.08.031
18. Kitada M, Takeda A, Nagai T, Ito H, Kanasaki K, Koya D. Dietary restriction ameliorates diabetic nephropathy through anti-inflammatory effects and regulation of the autophagy via restoration of Sirt1 in diabetic Wistar fatty (fa/fa) rats: a model of type 2 diabetes. Exp Diabetes Res. (2011) 2011:908185. doi: 10.1155/2011/908185
19. Liu R, Zhong Y, Li X, Chen H, Jim B, Zhou MM, et al. The role of transcription factor acetylation in diabetic kidney disease. Diabetes. (2014) 63:2240–53. doi: 10.2337/db13-1810
20. Huang R, Xu Y, Wan W, Shou X, Qian J, You Z, et al. Deacetylation of nuclear LC3 drives autophagy initiation under starvation. Mol Cell. (2015) 57:456–66. doi: 10.1016/j.molcel.2014.12.013
21. Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. (2005) 434:113–8. doi: 10.1038/nature03354
22. Li L, Pan R, Li R, Niemann B, Aurich AC, Chen Y, et al. Mitochondrial biogenesis and peroxisome proliferator-activated receptor-gamma coactivator-1alpha (PGC-1alpha) deacetylation by physical activity: intact adipocytokine signaling is required. Diabetes. (2011) 60:157–67. doi: 10.2337/db10-0331
23. Oanh NTK, Park YY, Cho H. Mitochondria elongation is mediated through SIRT1-mediated MFN1 stabilization. Cell Signal. (2017) 38:67–75. doi: 10.1016/j.cellsig.2017.06.019
24. Biel TG, Lee S, Flores-Toro JA, Dean JW, Go KL, Lee MH, et al. Sirtuin 1 suppresses mitochondrial dysfunction of ischemic mouse livers in a mitofusin 2-dependent manner. Cell Death Differ. (2016) 23:279–90. doi: 10.1038/cdd.2015.96
25. Wang F, Nguyen M, Qin FX, Tong Q. SIRT2 deacetylates FOXO3a in response to oxidative stress and caloric restriction. Aging Cell. (2007) 6:505–14. doi: 10.1111/j.1474-9726.2007.00304.x
26. Krishnan J, Danzer C, Simka T, Ukropec J, Walter KM, Kumpf S, et al. Dietary obesity-associated Hif1alpha activation in adipocytes restricts fatty acid oxidation and energy expenditure via suppression of the Sirt2-NAD+ system. Genes Dev. (2012) 26:259–70. doi: 10.1101/gad.180406.111
27. Wang YP, Zhou LS, Zhao YZ, Wang SW, Chen LL, Liu LX, et al. Regulation of G6PD acetylation by SIRT2 and KAT9 modulates NADPH homeostasis and cell survival during oxidative stress. Embo J. (2014) 33:1304–20. doi: 10.1002/embj.201387224
28. Qiu X, Brown K, Hirschey MD, Verdin E, Chen D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. (2010) 12:662–7. doi: 10.1016/j.cmet.2010.11.015
29. Zhang B, Pan Y, Xu L, Tang D, Dorfman RG, Zhou Q, et al. Berberine promotes glucose uptake and inhibits gluconeogenesis by inhibiting deacetylase SIRT3. Endocrine. (2018) 62:576–87. doi: 10.1007/s12020-018-1689-y
30. Ogura Y, Kitada M, Xu J, Monno I, Koya D. CD38 inhibition by apigenin ameliorates mitochondrial oxidative stress through restoration of the intracellular NAD(+)/NADH ratio and Sirt3 activity in renal tubular cells in diabetic rats. Aging. (2020) 12:11325–36. doi: 10.18632/aging.103410
31. Samant SA, Hong Z, Pillai VB, Sundaresan NR, Wolfgeher D, Archer SL, et al. SIRT3 deacetylates and activates OPA1 to regulate mitochondrial dynamics during stress. Mol Cell Biol. (2014) 5:807–19. doi: 10.1128/MCB.01483-13
32. Morigi M, Perico L, Rota C, Longaretti L, Conti S, Rottoli D, et al. Sirtuin 3-dependent mitochondrial dynamic improvements protect against acute kidney injury. J Clin Invest. (2015) 125:715–26. doi: 10.1172/JCI77632
33. Haigis MC, Mostoslavsky R, Haigis KM, Fahie K, Christodoulou DC, Murphy AJ, et al. SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell. (2006) 126:941–54. doi: 10.1016/j.cell.2006.06.057
34. Nasrin N, Wu X, Fortier E, Feng Y, Bare OC, Chen S, et al. SIRT4 regulates fatty acid oxidation and mitochondrial gene expression in liver and muscle cells. J Biol Chem. (2010) 285:31995–2002. doi: 10.1074/jbc.M110.124164
35. Nakagawa T, Lomb DJ, Haigis MC, Guarente L. SIRT5 Deacetylates carbamoyl phosphate synthetase 1 and regulates the urea cycle. Cell. (2009) 137:560–70. doi: 10.1016/j.cell.2009.02.026
36. Zhou L, Wang F, Sun R, Chen X, Zhang M, Xu Q, et al. SIRT5 promotes IDH2 desuccinylation and G6PD deglutarylation to enhance cellular antioxidant defense. EMBO Rep. (2016) 17:811–22. doi: 10.15252/embr.201541643
37. Liu M, Liang K, Zhen J, Zhou M, Wang X, Wang Z, et al. Sirt6 deficiency exacerbates podocyte injury and proteinuria through targeting Notch signaling. Nat Commun. (2017) 8:413. doi: 10.1038/s41467-017-00498-4
38. Sobuz SU, Sato Y, Yoshizawa T, Karim F, Ono K, Sawa T, et al. SIRT7 regulates the nuclear export of NF-kappaB p65 by deacetylating Ran. Biochim Biophys Acta Mol Cell Res. (2019) 1866:1355–67. doi: 10.1016/j.bbamcr.2019.05.001
39. Kraus WL. PARPs and ADP-ribosylation: 50 years … and counting. Mol Cell. (2015) 58:902–10. doi: 10.1016/j.molcel.2015.06.006
40. Murata MM, Kong X, Moncada E, Chen Y, Imamura H, Wang P, et al. NAD+ consumption by PARP1 in response to DNA damage triggers metabolic shift critical for damaged cell survival. Mol Biol Cell. (2019) 30:2584–97. doi: 10.1091/mbc.E18-10-0650
41. Fan C, Ma Q, Xu M, Qiao Y, Zhang Y, Li P, et al. Ginsenoside Rb1 attenuates high glucose-induced oxidative injury via the NAD-PARP-SIRT axis in rat retinal capillary endothelial cells. Int J Mol Sci. (2019) 20:4936. doi: 10.3390/ijms20194936
42. Brady PN, Goel A, Johnson MA. Poly(ADP-Ribose) polymerases in host-pathogen interactions, inflammation, and immunity. Microbiol Mol Biol Rev. (2019) 83:e00038-18. doi: 10.1128/MMBR.00038-18
43. Waldman M, Nudelman V, Shainberg A, Abraham NG, Kornwoski R, Aravot D, et al. PARP-1 inhibition protects the diabetic heart through activation of SIRT1-PGC-1α axis. Exp Cell Res. (2018) 373:112–8. doi: 10.1016/j.yexcr.2018.10.003
44. Zhang T, Chi Y, Kang Y, Lu H, Niu H, Liu W, et al. Resveratrol ameliorates podocyte damage in diabetic mice via SIRT1/PGC-1alpha mediated attenuation of mitochondrial oxidative stress. J Cell Physiol. (2019) 234:5033–43. doi: 10.1002/jcp.27306
45. Zhang T, Chi Y, Ren Y, Du C, Shi Y, Li Y. Resveratrol reduces oxidative stress and apoptosis in podocytes via Sir2-related enzymes, Sirtuins1 (SIRT1)/peroxisome proliferator-activated receptor gamma co-activator 1alpha (PGC-1alpha) axis. Med Sci Monitor. (2019) 25:1220–31. doi: 10.12659/MSM.911714
46. Wang XL, Wu LY, Zhao L, Sun LN, Liu HY, Liu G, et al. SIRT1 activator ameliorates the renal tubular injury induced by hyperglycemia in vivo and in vitro via inhibiting apoptosis. Biomed Pharmacother. (2016) 83:41–50. doi: 10.1016/j.biopha.2016.06.009
47. Hong Q, Zhang L, Das B, Li Z, Liu B, Cai G, et al. Increased podocyte Sirtuin-1 function attenuates diabetic kidney injury. Kidney Int. (2018) 93:1330–43. doi: 10.1016/j.kint.2017.12.008
48. Ren Y, Du C, Shi Y, Wei J, Wu H, Cui H. The Sirt1 activator, SRT1720, attenuates renal fibrosis by inhibiting CTGF and oxidative stress. Int J Mol Med. (2017) 39:1317–24. doi: 10.3892/ijmm.2017.2931
49. Yoshizaki T, Schenk S, Imamura T, Babendure JL, Sonoda N, Bae EJ, et al. SIRT1 inhibits inflammatory pathways in macrophages and modulates insulin sensitivity. Am J Physiol Endocrinol Metab. (2010) 298:E419–28. doi: 10.1152/ajpendo.00417.2009
50. Rogacka D, Audzeyenka I, Rychłowski M, Rachubik P, Szrejder M, Angielski S, et al. Metformin overcomes high glucose-induced insulin resistance of podocytes by pleiotropic effects on SIRT1 and AMPK. Biochim Biophys Acta Mol Basis Dis. (2018) 1864:115–25. doi: 10.1016/j.bbadis.2017.10.014
51. Ren H, Shao Y, Wu C, Ma X, Lv C, Wang Q. Metformin alleviates oxidative stress and enhances autophagy in diabetic kidney disease via AMPK/SIRT1-FoxO1 pathway. Mol Cell Endocrinol. (2020) 500:110628. doi: 10.1016/j.mce.2019.110628
52. Umino H, Hasegawa K, Minakuchi H, Muraoka H, Kawaguchi T, Kanda T, et al. High basolateral glucose increases sodium-glucose cotransporter 2 and reduces sirtuin-1 in renal tubules through glucose transporter-2 detection. Sci Rep. (2018) 8:6791. doi: 10.1038/s41598-018-25054-y
53. Koyama T, Kume S, Koya D, Araki S, Isshiki K, Chin-Kanasaki M, et al. SIRT3 attenuates palmitate-induced ROS production and inflammation in proximal tubular cells. Free Radic Biol Med. (2011) 51:1258–67. doi: 10.1016/j.freeradbiomed.2011.05.028
54. Li J, Liu H, Takagi S, Nitta K, Kitada M, Srivastava SP, et al. Renal protective effects of empagliflozin via inhibition of EMT and aberrant glycolysis in proximal tubules. JCI insight. (2020) 5:129034. doi: 10.1172/jci.insight.129034
55. Peixoto EB, Papadimitriou A, Lopes de Faria JM, Lopes de Faria JB. Tempol reduces podocyte apoptosis via PARP signaling pathway in experimental diabetes mellitus. Nephron Exp Nephrol. (2012) 120:e81–90. doi: 10.1159/000337364
56. Szabó C, Biser A, Benko R, Böttinger E, Suszták K. Poly(ADP-ribose) polymerase inhibitors ameliorate nephropathy of type 2 diabetic Leprdb/db mice. Diabetes. (2006) 55:3004–12. doi: 10.2337/db06-0147
57. Zhu H, Fang Z, Chen J, Yang Y, Gan J, Luo L, et al. PARP-1 and SIRT-1 are interacted in diabetic nephropathy by activating AMPK/PGC-1α signaling pathway. Diabetes Metab Syndr Obes. (2021) 14:355–66. doi: 10.2147/DMSO.S291314
58. Chiu J, Xu BY, Chen S, Feng B, Chakrabarti S. Oxidative stress-induced, poly(ADP-ribose) polymerase-dependent upregulation of ET-1 expression in chronic diabetic complications. Can J Physiol Pharmacol. (2008) 86:365–72. doi: 10.1139/Y08-033
59. Xu B, Chiu J, Feng B, Chen S, Chakrabarti S. PARP activation and the alteration of vasoactive factors and extracellular matrix protein in retina and kidney in diabetes. Diabetes Metab Res Rev. (2008) 24:404–12. doi: 10.1002/dmrr.842
60. Escande C, Nin V, Price NL, Capellini V, Gomes AP, Barbosa MT, et al. Flavonoid apigenin is an inhibitor of the NAD+ ase CD38: implications for cellular NAD+ metabolism, protein acetylation, and treatment of metabolic syndrome. Diabetes. (2013) 62:1084–93. doi: 10.2337/db12-1139
61. Malik S, Suchal K, Khan SI, Bhatia J, Kishore K, Dinda AK, et al. Apigenin ameliorates streptozotocin-induced diabetic nephropathy in rats via MAPK-NF-κB-TNF-α and TGF-β1-MAPK-fibronectin pathways. Am J Physiol Renal Physiol. (2017) 313:F414–22. doi: 10.1152/ajprenal.00393.2016
62. Zhang J, Zhao X, Zhu H, Wang J, Ma J, Gu M. Apigenin protects against renal tubular epithelial cell injury and oxidative stress by high glucose via regulation of NF-E2-related factor 2 (Nrf2) Pathway. Med Sci Monitor. (2019) 25:5280–8. doi: 10.12659/MSM.915038
63. Tarrago MG, Chini CCS, Kanamori KS, Warner GM, Caride A, de Oliveira GC, et al. A potent and specific CD38 inhibitor ameliorates age-related metabolic dysfunction by reversing tissue NAD(+) decline. Cell Metab. (2018) 27:1081–95. doi: 10.1016/j.cmet.2018.03.016
64. Shu B, Feng Y, Gui Y, Lu Q, Wei W, Xue X, et al. Blockade of CD38 diminishes lipopolysaccharide-induced macrophage classical activation and acute kidney injury involving NF-κB signaling suppression. Cell Signal. (2018) 42:249–58. doi: 10.1016/j.cellsig.2017.10.014
65. Chini EN, Chini CCS, Espindola Netto JM, de Oliveira GC, van Schooten W. The pharmacology of CD38/NADase: an emerging target in cancer and diseases of aging. Trends Pharmacol Sci. (2018) 39:424–36. doi: 10.1016/j.tips.2018.02.001
66. Aksoy P, White TA, Thompson M, Chini EN. Regulation of intracellular levels of NAD: a novel role for CD38. Biochem Biophys Res Commun. (2006) 345:1386–92. doi: 10.1016/j.bbrc.2006.05.042
67. Camacho-Pereira J, Tarrago MG, Chini CCS, Nin V, Escande C, Warner GM, et al. CD38 Dictates age-related NAD decline and mitochondrial dysfunction through an SIRT3-dependent mechanism. Cell Metab. (2016) 23:1127–39. doi: 10.1016/j.cmet.2016.05.006
68. Mericskay M. Nicotinamide adenine dinucleotide homeostasis and signalling in heart disease: pathophysiological implications and therapeutic potential. Arch Cardiovasc Dis. (2016) 109:207–15. doi: 10.1016/j.acvd.2015.10.004
69. Thai TL, Arendshorst WJ. Mice lacking the ADP ribosyl cyclase CD38 exhibit attenuated renal vasoconstriction to angiotensin II, endothelin-1, and norepinephrine. Am J Physiol Renal Physiol. (2009) 297:F169–76. doi: 10.1152/ajprenal.00079.2009
70. Johnson JD, Ford EL, Bernal-Mizrachi E, Kusser KL, Luciani DS, Han Z, et al. Suppressed insulin signaling and increased apoptosis in CD38-null islets. Diabetes. (2006) 55:2737–46. doi: 10.2337/db05-1455
71. Takasawa S, Okamoto H. Pancreatic beta-cell death, regeneration and insulin secretion: roles of poly(ADP-ribose) polymerase and cyclic ADP-ribose. Int J Exp Diabetes Res. (2002) 3:79–96. doi: 10.1080/15604280214485
72. Antonelli A, Baj G, Marchetti P, Fallahi P, Surico N, Pupilli C, et al. Human anti-CD38 autoantibodies raise intracellular calcium and stimulate insulin release in human pancreatic islets. Diabetes. (2001) 50:985–91. doi: 10.2337/diabetes.50.5.985
73. Young GS, Choleris E, Lund FE, Kirkland JB. Decreased cADPR and increased NAD+ in the Cd38-/- mouse. Biochem Biophys Res Commun. (2006) 346:188–92. doi: 10.1016/j.bbrc.2006.05.100
74. Barbosa MT, Soares SM, Novak CM, Sinclair D, Levine JA, Aksoy P, et al. The enzyme CD38 (a NAD glycohydrolase, EC 3.2.2.5) is necessary for the development of diet-induced obesity. Faseb J. (2007) 21:3629–39. doi: 10.1096/fj.07-8290com
75. Kawai S, Murata K. Structure and function of NAD kinase and NADP phosphatase: key enzymes that regulate the intracellular balance of NAD(H) and NADP(H). Biosci Biotechnol Biochem. (2008) 72:919–30. doi: 10.1271/bbb.70738
76. Irazabal MV, Torres VE. Reactive oxygen species and redox signaling in chronic kidney disease. Cells. (2020) 9:1342. doi: 10.3390/cells9061342
77. Xu J, Kitada M, Koya D. The impact of mitochondrial quality control by Sirtuins on the treatment of type 2 diabetes and diabetic kidney disease. Biochim Biophys Acta Mol Basis Dis. (2020) 1866:165756. doi: 10.1016/j.bbadis.2020.165756
78. Cantó C, Menzies KJ, Auwerx J. NAD(+) metabolism and the control of energy homeostasis: a balancing act between mitochondria and the nucleus. Cell Metab. (2015) 22:31–53. doi: 10.1016/j.cmet.2015.05.023
79. Fan L, Cacicedo JM, Ido Y. Impaired nicotinamide adenine dinucleotide (NAD(+)) metabolism in diabetes and diabetic tissues: implications for nicotinamide-related compound treatment. J Diabetes Investig. (2020) 11:1403–19. doi: 10.1111/jdi.13303
80. Luo X, Li R, Yan LJ. Roles of pyruvate, NADH, and mitochondrial complex i in redox balance and imbalance in β cell function and dysfunction. J Diabetes Res. (2015) 2015:512618. doi: 10.1155/2015/512618
81. Kitada M, Kume S, Takeda-Watanabe A, Kanasaki K, Koya D. Sirtuins and renal diseases: relationship with aging and diabetic nephropathy. Clin Sci (Lond). (2013) 124:153–64. doi: 10.1042/CS20120190
82. Kong L, Wu H, Zhou W, Luo M, Tan Y, Miao L, et al. Sirtuin 1: a target for kidney diseases. Mol Med (Cambridge, Mass). (2015) 21:87–97. doi: 10.2119/molmed.2014.00211
83. Tang BL. Sirt1 and the mitochondria. Mol Cells. (2016) 39:87–95. doi: 10.14348/molcells.2016.2318
84. Lan F, Cacicedo JM, Ruderman N, Ido Y. SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. Possible role in AMP-activated protein kinase activation. J Biol Chem. (2008) 283:27628–35. doi: 10.1074/jbc.M805711200
85. Lee IH, Cao L, Mostoslavsky R, Lombard DB, Liu J, Bruns NE, et al. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc Natl Acad Sci USA. (2008) 105:3374–9. doi: 10.1073/pnas.0712145105
86. Jang SY, Kang HT, Hwang ES. Nicotinamide-induced mitophagy: event mediated by high NAD+/NADH ratio and SIRT1 protein activation. J Biol Chem. (2012) 287:19304–14. doi: 10.1074/jbc.M112.363747
87. Hasegawa K, Wakino S, Yoshioka K, Tatematsu S, Hara Y, Minakuchi H, et al. Sirt1 protects against oxidative stress-induced renal tubular cell apoptosis by the bidirectional regulation of catalase expression. Biochem Biophys Res Commun. (2008) 372:51–6. doi: 10.1016/j.bbrc.2008.04.176
88. Braidy N, Guillemin GJ, Mansour H, Chan-Ling T, Poljak A, Grant R. Age related changes in NAD+ metabolism oxidative stress and Sirt1 activity in wistar rats. PLoS ONE. (2011) 6:e19194. doi: 10.1371/journal.pone.0019194
89. Huang K, Huang J, Xie X, Wang S, Chen C, Shen X, et al. Sirt1 resists advanced glycation end products-induced expressions of fibronectin and TGF-beta1 by activating the Nrf2/ARE pathway in glomerular mesangial cells. Free Radic Biol Med. (2013) 65:528–40. doi: 10.1016/j.freeradbiomed.2013.07.029
90. Simic P, Williams EO, Bell EL, Gong JJ, Bonkowski M, Guarente L. SIRT1 suppresses the epithelial-to-mesenchymal transition in cancer metastasis and organ fibrosis. Cell Rep. (2013) 3:1175–86. doi: 10.1016/j.celrep.2013.03.019
91. Huang XZ, Wen D, Zhang M, Xie Q, Ma L, Guan Y, et al. Sirt1 activation ameliorates renal fibrosis by inhibiting the TGF-β/Smad3 pathway. J Cell Biochem. (2014) 115:996–1005. doi: 10.1002/jcb.24748
92. Mortuza R, Feng B, Chakrabarti S. SIRT1 reduction causes renal and retinal injury in diabetes through endothelin 1 and transforming growth factor β1. J Cell Mol Med. (2015) 19:1857–67. doi: 10.1111/jcmm.12557
93. Sun Z, Ma Y, Chen F, Wang S, Chen B, Shi J. miR-133b and miR-199b knockdown attenuate TGF-β1-induced epithelial to mesenchymal transition and renal fibrosis by targeting SIRT1 in diabetic nephropathy. Eur J Pharmacol. (2018) 837:96–104. doi: 10.1016/j.ejphar.2018.08.022
94. Caliskan Z, Mutlu T, Guven M, Tuncdemir M, Niyazioglu M, Hacioglu Y, et al. SIRT6 expression and oxidative DNA damage in individuals with prediabetes and type 2 diabetes mellitus. Gene. (2018) 642:542–8. doi: 10.1016/j.gene.2017.11.071
95. Fan Y, Yang Q, Yang Y, Gao Z, Ma Y, Zhang L, et al. Sirt6 suppresses high glucose-induced mitochondrial dysfunction and apoptosis in podocytes through AMPK activation. Int J Biol Sci. (2019) 15:701–13. doi: 10.7150/ijbs.29323
96. Muraoka H, Hasegawa K, Sakamaki Y, Minakuchi H, Kawaguchi T, Yasuda I, et al. Role of Nampt-Sirt6 axis in renal proximal tubules in extracellular matrix deposition in diabetic nephropathy. Cell Rep. (2019) 27:199–212. doi: 10.1016/j.celrep.2019.03.024
97. Ji L, Chen Y, Wang H, Zhang W, He L, Wu J, et al. Overexpression of Sirt6 promotes M2 macrophage transformation, alleviating renal injury in diabetic nephropathy. Int J Oncol. (2019) 55:103–15. doi: 10.3892/ijo.2019.4800
98. Perico L, Morigi M, Benigni A. Mitochondrial sirtuin 3 and renal diseases. Nephron. (2016) 134:14–9. doi: 10.1159/000444370
99. Kitada M, Xu J, Ogura Y, Monno I, Koya D. Manganese superoxide dismutase dysfunction and the pathogenesis of kidney disease. Front Physiol. (2020) 11:755. doi: 10.3389/fphys.2020.00755
100. Caton PW, Richardson SJ, Kieswich J, Bugliani M, Holland ML, Marchetti P, et al. Sirtuin 3 regulates mouse pancreatic beta cell function and is suppressed in pancreatic islets isolated from human type 2 diabetic patients. Diabetologia. (2013) 56:1068–77. doi: 10.1007/s00125-013-2851-y
101. Zhuo L, Fu B, Bai X, Zhang B, Wu L, Cui J, et al. NAD blocks high glucose induced mesangial hypertrophy via activation of the sirtuins-AMPK-mTOR pathway. Cell Physiol Biochem. (2011) 27:681–90. doi: 10.1159/000330077
102. De Marchi U, Galindo AN, Thevenet J, Hermant A, Bermont F, Lassueur S, et al. Mitochondrial lysine deacetylation promotes energy metabolism and calcium signaling in insulin-secreting cells. Faseb J. (2019) 33:4660–74. doi: 10.1096/fj.201801424R
103. Ogura Y, Kitada M, Monno I, Kanasaki K, Watanabe A, Koya D. Renal mitochondrial oxidative stress is enhanced by the reduction of Sirt3 activity, in Zucker diabetic fatty rats. Redox Rep. (2018) 23:153–9. doi: 10.1080/13510002.2018.1487174
104. Jiao X, Li Y, Zhang T, Liu M, Chi Y. Role of Sirtuin3 in high glucose-induced apoptosis in renal tubular epithelial cells. Biochem Biophys Res Commun. (2016) 480:387–93. doi: 10.1016/j.bbrc.2016.10.060
105. Ouyang J, Zeng Z, Fang H, Li F, Zhang X, Tan W. SIRT3 inactivation promotes acute kidney injury through elevated acetylation of SOD2 and p53. J Surg Res. (2019) 233:221–30. doi: 10.1016/j.jss.2018.07.019
106. Ahuja N, Schwer B, Carobbio S, Waltregny D, North BJ, Castronovo V, et al. Regulation of insulin secretion by SIRT4, a mitochondrial ADP-ribosyltransferase. J Biol Chem. (2007) 282:33583–92. doi: 10.1074/jbc.M705488200
107. Anderson KA, Huynh FK, Fisher-Wellman K, Stuart JD, Peterson BS, Douros JD, et al. SIRT4 is a lysine deacylase that controls leucine metabolism and insulin secretion. Cell Metab. (2017) 25:838–55. doi: 10.1016/j.cmet.2017.03.003
108. Shi JX, Wang QJ, Li H, Huang Q. SIRT4 overexpression protects against diabetic nephropathy by inhibiting podocyte apoptosis. Exp Ther Med. (2017) 13:342–8. doi: 10.3892/etm.2016.3938
109. Du J, Zhou Y, Su X, Yu JJ, Khan S, Jiang H, et al. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science. (2011) 334:806–9. doi: 10.1126/science.1207861
110. Nakamura Y, Ogura M, Ogura K, Tanaka D, Inagaki N. SIRT5 deacetylates and activates urate oxidase in liver mitochondria of mice. FEBS Lett. (2012) 586:4076–81. doi: 10.1016/j.febslet.2012.10.009
111. de Moura MB, Uppala R, Zhang Y, Van Houten B, Goetzman ES. Overexpression of mitochondrial sirtuins alters glycolysis and mitochondrial function in HEK293 cells. PLoS ONE. (2014) 9:e106028. doi: 10.1371/journal.pone.0106028
112. Rardin MJ, He W, Nishida Y, Newman JC, Carrico C, Danielson SR, et al. SIRT5 regulates the mitochondrial lysine succinylome and metabolic networks. Cell Metab. (2013) 18:920–33. doi: 10.1016/j.cmet.2013.11.013
113. Buler M, Aatsinki SM, Izzi V, Uusimaa J, Hakkola J. SIRT5 is under the control of PGC-1alpha and AMPK and is involved in regulation of mitochondrial energy metabolism. Faseb J. (2014) 28:3225–37. doi: 10.1096/fj.13-245241
114. Nishida Y, Rardin MJ, Carrico C, He W, Sahu AK, Gut P, et al. SIRT5 regulates both cytosolic and mitochondrial protein malonylation with glycolysis as a major target. Mol Cell. (2015) 59:321–32. doi: 10.1016/j.molcel.2015.05.022
115. Tan M, Peng C, Anderson KA, Chhoy P, Xie Z, Dai L, et al. Lysine glutarylation is a protein posttranslational modification regulated by SIRT5. Cell Metab. (2014) 19:605–17. doi: 10.1016/j.cmet.2014.03.014
116. Park J, Chen Y, Tishkoff DX, Peng C, Tan M, Dai L, et al. SIRT5-mediated lysine desuccinylation impacts diverse metabolic pathways. Mol Cell. (2013) 50:919–30. doi: 10.1016/j.molcel.2013.06.001
117. Li W, Yang Y, Li Y, Zhao Y, Jiang H. Sirt5 attenuates cisplatin-induced acute kidney injury through regulation of Nrf2/HO-1 and Bcl-2. BioMed Res Int. (2019) 2019:4745132. doi: 10.1155/2019/4745132
118. Chiba T, Peasley KD, Cargill KR, Maringer KV, Bharathi SS, Mukherjee E, et al. Sirtuin 5 regulates proximal tubule fatty acid oxidation to protect against AKI. J Am Soc Nephrol. (2019) 30:2384–98. doi: 10.1681/ASN.2019020163
119. Minchenko AG, Stevens MJ, White L, Abatan OI, Komjáti K, Pacher P, et al. Diabetes-induced overexpression of endothelin-1 and endothelin receptors in the rat renal cortex is mediated via poly(ADP-ribose) polymerase activation. Faseb J. (2003) 17:1514–6. doi: 10.1096/fj.03-0013fje
120. Shevalye H, Maksimchyk Y, Watcho P, Obrosova IG. Poly(ADP-ribose) polymerase-1 (PARP-1) gene deficiency alleviates diabetic kidney disease. Biochim Biophys Acta. (2010) 1802:1020–7. doi: 10.1016/j.bbadis.2010.07.004
121. Shen H, Fang K, Guo H, Wang G. high glucose-induced apoptosis in human kidney cells was alleviated by miR-15b-5p mimics. Biol Pharm Bull. (2019) 42:758–63. doi: 10.1248/bpb.b18-00951
122. Katsoulieris EN, Drossopoulou GI, Kotsopoulou ES, Vlahakos DV, Lianos EA, Tsilibary EC. High glucose impairs insulin signaling in the glomerulus: an in vitro and ex vivo approach. PLoS ONE. (2016) 11:e0158873. doi: 10.1371/journal.pone.0158873
123. Zakaria EM, El-Maraghy NN, Ahmed AF, Ali AA, El-Bassossy HM. PARP inhibition ameliorates nephropathy in an animal model of type 2 diabetes: focus on oxidative stress, inflammation, and fibrosis. Naunyn-Schmiedeberg's Arch Pharmacol. (2017) 390:621–31. doi: 10.1007/s00210-017-1360-9
124. Okamoto H. The CD38-cyclic ADP-ribose signaling system in insulin secretion. Mol Cell Biochem. (1999) 193:115–8. doi: 10.1007/978-1-4419-8740-2_17
125. Lee HJ, Hong YS, Jun W, Yang SJ. Nicotinamide riboside ameliorates hepatic metaflammation by modulating NLRP3 inflammasome in a rodent model of type 2 diabetes. J Med Food. (2015) 18:1207–13. doi: 10.1089/jmf.2015.3439
126. Caton PW, Kieswich J, Yaqoob MM, Holness MJ, Sugden MC. Nicotinamide mononucleotide protects against pro-inflammatory cytokine-mediated impairment of mouse islet function. Diabetologia. (2011) 54:3083–92. doi: 10.1007/s00125-011-2288-0
127. Chen Y, Liang Y, Hu T, Wei R, Cai C, Wang P, et al. Endogenous Nampt upregulation is associated with diabetic nephropathy inflammatory-fibrosis through the NF-κB p65 and Sirt1 pathway; NMN alleviates diabetic nephropathy inflammatory-fibrosis by inhibiting endogenous Nampt. Exp Ther Med. (2017) 14:4181–93. doi: 10.3892/etm.2017.5098
128. Yasuda I, Hasegawa K, Sakamaki Y, Muraoka H, Kawaguchi T, Kusahana E, et al. Pre-emptive short-term nicotinamide mononucleotide treatment in a mouse model of diabetic nephropathy. J Am Soc Nephrol. (2021) 32:1355–70. doi: 10.1681/ASN.2020081188
129. Guan Y, Wang SR, Huang XZ, Xie QH, Xu YY, Shang D, et al. Nicotinamide mononucleotide, an NAD(+) precursor, rescues age-associated susceptibility to AKI in a sirtuin 1-dependent manner. J Am Soc Nephrol. (2017) 28:2337–52. doi: 10.1681/ASN.2016040385
130. Benito-Martin A, Ucero AC, Izquierdo MC, Santamaria B, Picatoste B, Carrasco S, et al. Endogenous NAMPT dampens chemokine expression and apoptotic responses in stressed tubular cells. Biochim Biophys Acta. (2014) 1842:293–303. doi: 10.1016/j.bbadis.2013.11.022
131. Kourtzidis IA, Dolopikou CF, Tsiftsis AN, Margaritelis NV, Theodorou AA, Zervos IA, et al. Nicotinamide riboside supplementation dysregulates redox and energy metabolism in rats: Implications for exercise performance. Exp Physiol. (2018) 103:1357–66. doi: 10.1113/EP086964
132. Kourtzidis IA, Stoupas AT, Gioris IS, Veskoukis AS, Margaritelis NV, Tsantarliotou M, et al. The NAD(+) precursor nicotinamide riboside decreases exercise performance in rats. J Int Soc Sports Nutr. (2016) 13:32. doi: 10.1186/s12970-016-0143-x
133. Piedrafita A, Balayssac S, Mayeur N, Gazut S, Grossac J, Buleon M, et al. The tryptophan pathway and nicotinamide supplementation in ischaemic acute kidney injury. Clin Kidney J. (2021) 1–7. doi: 10.1093/ckj/sfab050
134. Simic P, Vela Parada XF, Parikh SM, Dellinger R, Guarente LP, Rhee EP. Nicotinamide riboside with pterostilbene (NRPT) increases NAD(+) in patients with acute kidney injury (AKI): a randomized, double-blind, placebo-controlled, stepwise safety study of escalating doses of NRPT in patients with AKI. BMC Nephrol. (2020) 21:342. doi: 10.1186/s12882-020-02006-1
135. Zhang Q, Deng Q, Zhang J, Ke J, Zhu Y, Wen RW, et al. Activation of the Nrf2-are pathway ameliorates hyperglycemia-mediated mitochondrial dysfunction in podocytes partly through Sirt1. Cell Physiol Biochem. (2018) 48:1–15. doi: 10.1159/000491658
Keywords: nicotinamide adenine dinucleotide, diabetic kidney disease, oxidative stress, sirtuins, PARPs, CD38
Citation: Xu J, Kitada M and Koya D (2021) NAD+ Homeostasis in Diabetic Kidney Disease. Front. Med. 8:703076. doi: 10.3389/fmed.2021.703076
Received: 30 April 2021; Accepted: 29 June 2021;
Published: 21 July 2021.
Edited by:
Katalin Susztak, University of Pennsylvania, United StatesReviewed by:
Ashish Verma, Brigham and Women's Hospital and Harvard Medical School, United StatesRanjan Das, Rush University Medical Center, United States
Copyright © 2021 Xu, Kitada and Koya. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Munehiro Kitada, kitta@kanazawa-med.ac.jp