miR-7 and miR-153 protect neurons against MPP+-induced cell death via upregulation of mTOR pathway
 Apostolia Fragkouli
Apostolia Fragkouli Epaminondas Doxakis
Epaminondas Doxakis- Lab of Molecular and Cellular Neuroscience, Center for Basic Research, Biomedical Research Foundation of the Academy of Athens, Athens, Greece
Differential expression of microRNAs (miRs) in the brain of patients with neurodegenerative diseases suggests that they may have key regulatory roles in the development of these disorders. Two such miRs, miR-7, and miR-153 have recently been shown to target α-synuclein, a protein critically involved in the pathological process of Parkinson's disease. By using a well-established in culture Parkinson's disease model that of neurotoxin 1-Methyl-4-Phenyl-Pyridinium (MPP+), we examined whether miR-7 and miR-153 display neuroprotective properties. Herein, we demonstrate that treatment of cortical neurons with MPP+ induced a dose-dependent cell death with apoptotic characteristics. This was reflected in altered intracellular signaling characterized by increased levels of activated kinases p38MAPK and ERK1/2 and reduced levels of activated AKT, p70S6K, and SAPK/JNK. Overexpression of miR-7 or miR-153 by adenoviral transduction protected cortical neurons from MPP+-induced toxicity, restored neuronal viability and anti-apoptotic BCL-2 protein levels while attenuated activation of caspase-3. Moreover, both miR-7 and miR-153 interfered with MPP+-induced alterations in intracellular signaling pathways in a partially overlapping manner; specifically, they preserved activation of mTOR and SAPK/JNK signaling pathways in the MPP+-treated neurons, while miR-153 also attenuated MPP+-induced activation of p38MAPK. No major effects were observed in the rest of signaling cascades or proteins investigated. Furthermore, the neuroprotective effect of miR-7 and miR-153 was alleviated when MPP+ was co-administered with rapamycin. Taken together, our results suggest that miR-7 and miR-153 protect neurons from cell death by interfering with the MPP+-induced downregulation of mTOR signaling.
Introduction
Parkinson's disease (PD) is a heterogeneous neurodegenerative disorder that affects 1% of the population aged over 65. It perturbs both dopaminergic (substantia nigra pars compacta) and non-dopaminergic (locus coeruleous, raphe nuclei, nucleus basalis of Meynert, hypothalamus, pedunculopontine nucleus) neuronal systems. Our current understanding of the disease points toward a variety of genetic, cellular, and environmental factors that independently or in combination cause progressive neurodegeneration. These factors lead to oxidative stress, abnormal protein degradation, autophagy, reduced protein synthesis, and altered signal transduction that combined induce neuronal death. Which of these mechanisms is more important to PD pathogenesis and progression remains unknown (Obeso et al., 2010). So far, epidemiological data and therapeutic studies using neuroprotective substances such as caffeine, nicotine, ginsenosides, flavonoids, vitamins, and growth factors have pointed out that drugs directed against a single molecular target are likely to be ineffective in treating the disease while agents with multiple pharmacological targets appear more suitable. Consistently, treatments with generic neuroprotective factors and various combinations of approved drugs are now vigorously explored (reviewed in Seidl and Potashkin, 2011; Mythri et al., 2012; Rodnitzky, 2012; Santos, 2012; Kordower and Bjorklund, 2013).
microRNAs (miRs) are a class of highly conserved small, about 22 nucleotides in length, non-coding endogenous RNA molecules that act to inhibit protein expression by partially hybridizing to complementary sequences, in mainly the 3′ UTR, of target RNA transcripts (reviewed in Doxakis, 2013). Each miR is estimated to regulate multiple target mRNAs, and the combinatorial action of miRs is expected to regulate the expression of hundreds of mRNAs. They display a wide variety of expression patterns and many are differentially expressed during development or disease (reviewed in Wienholds and Plasterk, 2005). With respect to PD, it has been shown that two miRs, miR-34, and miR-133, are significantly reduced in affected brain regions relative to controls (reviewed in Mouradian, 2012). Moreover, we and others have reported that two additional miRs, miR-7, and miR-153, target α-synuclein, a protein critically involved in both familial and sporadic pathological processes of PD (Junn et al., 2009; Doxakis, 2010). Importantly, miR-7 and miR-153 are neuron-enriched and show highest levels of expression in murine midbrain (Doxakis, 2010). In addition, miR-7 levels are down-regulated in the midbrain of mice intraperitoneally injected with the PD neurotoxin, MPTP (Junn et al., 2009) while miR-153 has been shown to regulate amyloid β precursor protein (APP) expression and its levels are significantly reduced in Alzheimer's disease brains (Liang et al., 2007; Long et al., 2012). Finally, both miR-7 and miR-153 have been known to modulate intracellular signaling by targeting upstream components of the AKT pathway (Kefas et al., 2008; Fang et al., 2012; Song et al., 2012; Sanchez et al., 2013; Wang et al., 2013; Wu et al., 2013).
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) is a neurotoxin that was discovered accidentally in exposed humans. Young drug addicts developed an idiopathic parkinsonian syndrome after intravenous self-administration of a synthetic heroin with this contaminant (Davis et al., 1979; Langston et al., 1983). Significantly, most of the biochemical, neuropathological, and clinical characteristics observed, corresponded to the cardinal symptoms of human PD with the exemption of the formation of Lewy bodies (Langston et al., 1983; Ballard et al., 1985). At the molecular level, MPTP is transformed into its toxic derivative 1-methyl-4-phenylpyridinium ion (MPP+) by the enzyme MAO-B in astrocytes (Langston et al., 1984; Nicklas et al., 1985). Today, MPTP and MPP+ represent the most relevant and frequently used parkinsonian toxins for animal and in culture PD models, respectively. A number of studies have, thus far, indicated that inhibition of complex I of the mitochondria electron transport chain, elevation of oxidative stress, activation of pro-apoptotic ERK-1/2 and p38 MAPK and suppression of pro-survival AKT and mTOR signaling pathways contribute to MPP+-induced cell death (Mizuno et al., 1987; Deguil et al., 2007; Karunakaran et al., 2008; Cui et al., 2011).
Based on the above, our current study was undertaken to evaluate the ability of miR-7 and miR-153 to prevent MPP+-induced toxicity in neurons and delineate the underlying mechanism. Our results demonstrate that miR-7 and miR-153 could protect cortical neurons against MPP+-induced death by preserving the activation of the downstream master integrating signaling pathway of mTOR. We argue that these findings may have important therapeutic preclinical applications for PD.
Materials and Methods
Ethics Statement
All rodent tissues were obtained in accordance with European Union (2003/65/CE) guidelines regarding the use of laboratory animals. Experimental protocols were approved by the Institutional Animal Care and Use Committee of BRFAA and the Veterinary Services of Attica prefecture (K/2134).
Antibodies
The rabbit polyclonal antibodies phospho-S6 ribosomal protein (Ser240/244 CST#2215), phospho-eEF2k (Ser366, CST#3691), phospho-p70 S6 kinase (Thr389, CST#9234), phospho-AKT (Ser473, CST#9271), phospho-ERK1/2 (Thr202/Tyr204, CST#9101), phospho-p38 (Thr180/Tyr182, CST#4511), phospho-SAPK/JNK (Thr183/Tyr185, CST#4668), phospho-GSK3β (Ser9, CST#), phospho-Mapkapk2 (Thr334, CST#3007), S6 ribosomal protein (CST#2217), p70 S6 kinase (CST#9202), AKT (CST#9272), ERK1/2 (CST#9102), p38 (CST#9212), SAPK/JNK (CST#92588), and cleaved caspase-3 (Asp175, CST#9664) were purchased from Cell Signaling Technologies (Beverly, MA, USA). The mouse monoclonal IgG antibodies against BAX (sc-493) and BCL-2 (sc-7382) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The anti-GAPDH (GT239) monoclonal antibody was purchased from Genetex (Irvine, CA, USA). The mouse (CST#7076) and rabbit (CST#7074) HRP-conjugated secondary antibodies were from Cell Signaling Technologies.
Generation of DNA Constructs
The construction of pcDNA6.2-GW/EmGFP- scramble/pri-miR-7/pri-miR-153 and pri-miR-7/153 plasmids has been described previously (Doxakis, 2010). The entry plasmids pENTR/EmGFP- scramble/pri-miR-7/pri-miR-153 and pri-miR-7/153 were constructed by inserting the EmGFP-pri-miR cassettes from the pcDNA6.2-GW/EmGFP-pri-miR plasmids into the XhoI/NotI sites of the pENTR Gateway plasmid (Life Technologies, Carlsbad, CA, USA). Using LR clonase II enzyme (Life Technologies) the EmGFP-pri-miR cassettes were, subsequently, transferred by LR recombination from the pENTR plasmid into the pAd5 destination adenoviral vector (Life Technologies). All pAd5/EmGFP-pri-miR vectors were verified by sequencing before use.
Adenoviral Production
pAd5/EmGFP-pri-miR vectors were digested with the PacI enzyme, to lineralize DNA, before transfecting into HEK293A producer cell line in 12-well plates by using Lipofectamine 2000 according to the manufacturer's instructions (Life Technologies). Two days later, cells were trypsinized and transferred onto 10 cm dishes. Culture media were replaced with fresh every 2–3 days until visible regions of cytopathic effect were observed (typically 5–8 days post-transfection). Adenovirus-containing cells and media were harvested when approximately 50% of cells were detached from dish. Crude lysates were prepared by 3 freeze/thaw cycles followed by centrifugation at 3000 rpm for 15 min. To amplify viral stock, 1% of crude adenoviral stocks were used to infect freshly-plated HEK293A cells. Infections were allowed to proceed until 80–90% of the cells have rounded up and were floating (typically 2 days later). High-titer viral stocks were, once again, prepared by 3 freeze/thaw cycles followed by centrifugation at 3000 rpm for 15 min. Adenoviral titers were determined by standard viral plaque assays. Titers were approximately 5 × 108 infectious units per ml.
Neuron Culture and Transduction
Dissociated, embryonic day 16 murine cortical neurons (>95% pure, 9 × 105 cells/ml), were grown in Neurobasal/DMEM 1:1 medium (Life Technologies) with 0.5 × B-27 supplement (Life Technologies), 5% heat-inactivated horse serum and Glutamax (Life Technologies) in poly-L-lysine (SIGMA, St-Louis, USA) coated culture plates in the absence of trophic factors (Doxakis et al., 2004). Neurons were transduced by adenoviruses at multiplicity of infection (MOI) 40 at 6–7 days after plating and lysed 48 h post-transduction.
Pharmacological Treatments
MPP+ (SIGMA) and rapamycin (SIGMA) were dissolved at the stock concentration of 100 mM in distilled water and tissue culture grade dimethylsulfoxide (DMSO, Applichem, Darmstadt, Germany), respectively. All experiments were initiated at day 7 or 8 after plating when all of the neurons had developed extensive neurite outgrowths. In the case of transduced neurons all pharmacological treatments were performed 24 h post-transduction. In MPP+ experiments, neurons were treated with 5–50 μM MPP+ for 24 h before analysis while in mTOR experiments, neurons were treated with 20 or 50 nM rapamycin for either 1 h (for signal transduction analysis) or 24 h (for cell viability analysis). When neuronal cultures were co-treated with MPP+ and rapamycin, the later compound was added 1 h earlier.
Methyl Thiazol Tetrazolium (MTT) Assay
MTT assay, a measure of mitochondrial dehydrogenase activity in live cells, was performed in neurons cultured in 96-well poly-L-lysine coated plates. Once the different treatments have been completed, 10 μl of MTT (Applichem) solution in PBS (5 mg/ml) was added to each well and the plate was placed back to incubator for a further 1.5 h. The MTT formazan precipitants formed by live cells were, subsequently, dissolved in 150 μl DMSO and the absorbance was measured at 570 nm by an ELISA microplate reader (ELx800, Bio-Tek Instruments, Winooski, VT, USA).
Immunoblotting
Immunoblotting was used to assay the protein levels of various intracellular signaling components in 8–9 days old cultures of cortical neurons transduced and/or treated with pharmacological compounds. Neurons were harvested in a lysis buffer containing 25 mM Tris pH 7.5, 150 mM NaCl, 1 mM EDTA, 1%Triton X-100, phosphatase (PhosSTOP®, Roche Applied Sciences, Penzberg, Bavaria, Germany) and protease (Complete®, Roche Applied Sciences) inhibitor cocktails. Cellular protein content was determined by the Bradford assay (Biorad, Hercules, CA, USA). Equal amounts of cell extracts were supplemented with 6x SDS sample buffer (375 mM Tris pH6.8, 10% SDS, 50% glycerol, 10% β-mercaptoethanol, 0.03% bromophenol blue), boiled for 5 min and subjected to SDS-PAGE under reducing conditions on 10 or 12% polyacrylamide gels, depending on the molecular mass of the proteins under examination. After electrophoresis, the resolved proteins were transferred to Protran® nitrocellulose membrane (Whatman, Kent, UK) by electroblotting. Subsequently membranes were saturated for 1 h at room temperature in 5% non-fat milk/0.1% Tween-20 in TBS and incubated at overnight at 4°C in 5% non-fat milk/TBS containing the primary antibody. All primary antibodies were used at 1:1000 dilution as recommended by vendors. The following day, membranes were washed in TBS, incubated for 1 h at room temperature in 5% non-fat milk/TBS containing the appropriate HRP-conjugated secondary antibody, washed in TBS and finally developed using the Western Lighting Plus ECL reagents (PerkinElmer, Waltham, MA, USA) according to the manufacturer's instructions. To ensure equal loading, following film exposure membranes were washed in 0.1% Tween-20 in TBS (TBST), incubated for 30 min at 50°C in stripping buffer (2% SDS, 0.8% mercaptoethanol, 62.5 mM Tris-HCl pH6.8), extensively washed in TBST and after saturation reprobed with the appropriate primary antibodies. Each sample was tested in duplicate and samples obtained from three or four independent experiments were used for analysis. Densitometric analysis of immunoblotting images was performed using the image analysis software Image J, NIH USA.
Statistical Analysis
Mean values were derived from three to five independent experiments performed in duplicate. The effect of treatment on the different parameters examined was assessed using One-Way ANOVA with treatment as independent factor. Bonferroni post-hoc analysis was performed where applicable. Significance was defined as p < 0.05. All statistical analyses were performed using the SPSS software (Release 10.0.1, SPSS, Chicago, IL, USA).
Results
MPP+ Treatment Induced Apoptosis in Cortical Neurons in a Concentration-Dependent Manner, Accompanied by Alterations in All Major Intracellular Signaling Cascades
In order to study the mechanism of MPP+-induced cell death in our experimental system, 7 days old primary cultures of cortical neurons were treated with various concentrations of MPP+ for 24 h. Neuronal viability was, initially, monitored using the MTT assay. As shown in Figure 1A, a 24-h treatment with MPP+ induced cytotoxicity in a concentration-dependent manner; loss of viability extended from 7 to 67% with MPP+ concentrations ranging between 5 and 50 μM. Statistical analysis revealed a significant effect of MPP+ treatment [F(5, 35) = 45.662, P < 0.001 and post-hoc] at 10, 20, 30, and 50 μM, but not 5 μM, of MPP+.
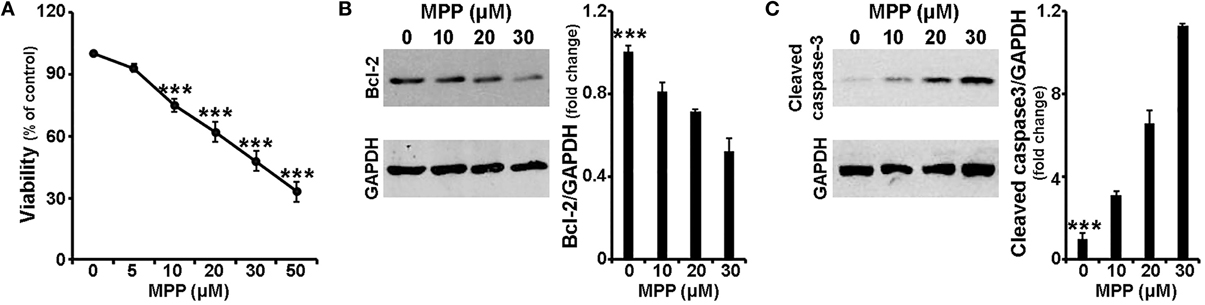
Figure 1. MPP+ induced neurotoxicity in cortical neurons. Seven-day primary cortical neurons were treated with various concentrations of MPP+ for 24 h. (A) Cell viability following dose-dependent treatments was assayed by measuring MTT reduction by live neurons. Note that a significant reduction in neuronal viability was observed upon treatment with 10, 20, 30, and 50 μM, but not 5 μM, of MPP+. (B,C) Equal amounts of total protein from lysates of cortical neurons cultured for 24 h in the presence of 10, 20, and 30 μM MPP+ were analyzed on 12% SDS-PAGE and immunoblotted with antibodies specific for BCL-2 (B) and cleaved caspase-3 (C). To ensure equal loading, membranes were re-probed against GAPDH. Note that compared to untreated controls, primary cortical neurons treated for 24 h with MPP+ displayed a dose-dependent decrease of BCL-2 protein levels, as well as a dose-dependent increase of cleaved caspase-3 protein levels. Quantification of the results in (B,C) was performed by scanning densitometry. Bars in all the presented graphs depict mean ± s.e.m. ***P ≤ 0.001.
Subsequently, the protein levels of the apoptotic-related factors BCL-2, BAX and cleaved caspase-3 were assayed by immunoblot analysis. Consistent with the loss in cell viability, levels of BCL-2, a major pro-survival protein, were significantly reduced by 20, 30, and 48% in cortical neurons treated for 24 h with 10, 20, and 30 μM of MPP+, respectively [F(3, 11) = 23.699, P < 0.001 and post-hoc; Figure 1B]. In contrast, a dose-dependent increase was observed in protein levels of cleaved caspase-3, an important effector caspase. More specifically, compared to untreated controls, primary cortical neurons treated for 24 h with 10, 20 and 30 μM of MPP+ displayed a significant 3.2-, 6.5-, and 11.3-fold increase of cleaved caspase-3 levels, respectively [F(3, 11) = 52.150, P < 0.001 and post-hoc; Figure 1C]. In our experimental system, the levels of BAX, a major pro-apoptotic factor, were not significantly altered (data not shown). Collectively, these data indicate that MPP+ induced a dose-dependent neuronal death that displayed apoptotic features.
It is well established that cell apoptosis and survival are regulated by intracellular signaling cascades; thus, activation by phosphorylation of the major signaling effectors was next examined in the same experimental system. As shown in Figures 2A,B, and consistent with previous studies (Junyent et al., 2010; Cui et al., 2011; Hashimoto et al., 2012), the levels of phosphorylated AKT, a major pro-survival kinase, were significantly reduced by ~30% in cortical neurons treated with 20 and 30 μM of MPP+ [F(3, 15) = 10.932, P = 0.001 and post-hoc], whereas levels of phosphorylated p38 MAPK, a major pro-apoptotic kinase, were significantly increased at all MPP+ concentrations applied [F(3, 15) = 5.996, P = 0.01 and post-hoc]. Similar results were observed in the levels of phosphorylated GSK-3β and MAPKAPK-2, downstream effectors of AKT and p38 MAPK, respectively (data not shown). Moreover, cortical neurons treated for 24 h with 20 or 30 μM, but not 10 μM, of MPP+ displayed significant alterations in the levels of phosphorylated ERK1/2, a dubious MAPK, as well as of phosphorylated stress-induced kinases SAPK/JNK; levels of phosphorylated ERK1/2 were up-regulated by 110 and 170% [F(3, 11) = 11.806, P < 0.01 and post-hoc; Figure 2C] and those of phosphorylated SAPK/JNK were down-regulated by 38 and 60% [F(3, 15) = 33.699, P < 0.001 and post-hoc; Figure 2D] at 20 and 30 μM of MPP+, respectively. Finally, as shown in Figure 2E, in our experimental system MPP+ treatment also significantly reduced the phosphorylation status of the mTOR effector p70S6 kinase, even when applied at 10 μM; this reduction extended from 27% at 10 μM to 72% at 30 μM of MPP+ [F(3, 15) = 20.701, P < 0.001 and post-hoc]. Similar reductions were also observed in the levels of activated S6RP [F(3, 15) = 60.445, P < 0.001 and post-hoc; Figure 2F] and of activated eEF2K [F(3, 11) = 18.310, P = 0.001 and post-hoc; data not shown], two p70S6K substrates which mediate translation and cell growth (reviewed in Laplante and Sabatini, 2013). It is therefore evident that in cortical neurons, a 24-h treatment with MPP+ leads to changes in the phosphorylation status of all major signaling kinases, in a concentration-dependent manner.
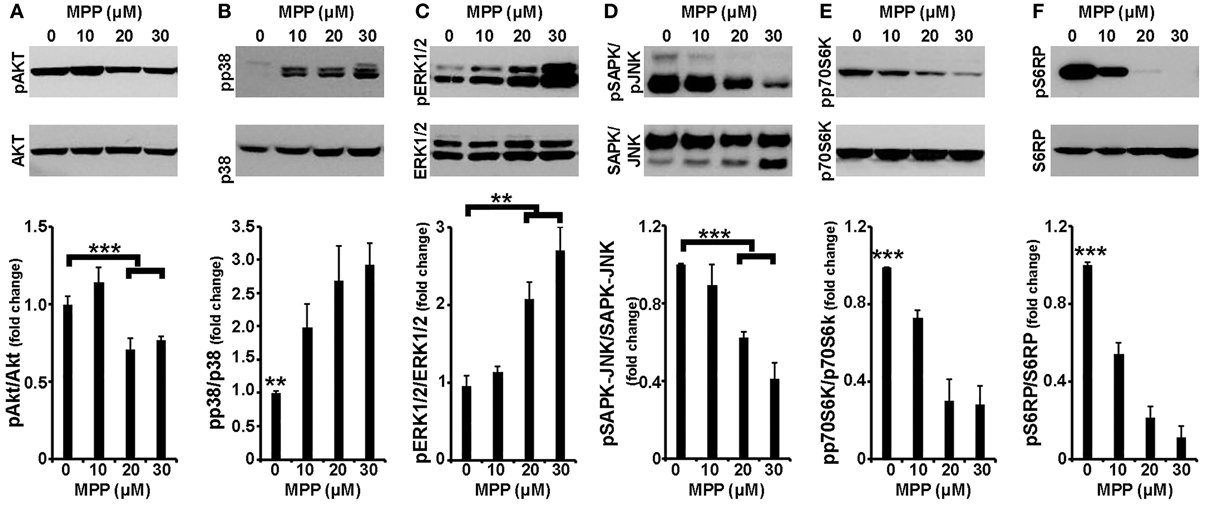
Figure 2. Cortical neurons treated with MPP+ displayed alterations in all major intracellular signaling cascades. Seven-day primary cortical neurons were treated with 10, 20, and 30 μM MPP+ for 24 h. Equal amounts of total protein from lysates of cortical neurons were analyzed on 10% SDS-PAGE and immunoblotted with antibodies specific for phosphorylated forms of AKT (A), p38 MAPK (B), ERK1/2 (C), JNK/SAPK (D), p70S6K (E), as well as for the phosphorylated forms of p70S6K substrate S6RP (F). To ensure equal loading membranes were re-probed against AKT, p38 MAPK, ERK1/2, JNK/SAPK, p70S6K, and S6RP, respectively. Quantification of the results was performed by scanning densitometry. Bars in the graph depict mean ± s.e.m. Note that MPP+-treatment changed the phosphorylation status of all the major signaling kinases examined in a concentration dependent manner. **P ≤ 0.01, ***P ≤ 0.001.
Over-Expression of miR-7 and/or miR-153 in Cortical Neurons Attenuated MPP+-Induced Neurotoxicity
It has been shown that miR-7 and miR-153 target α-synuclein, a protein critically involved in PD pathogenesis (Junn et al., 2009; Doxakis, 2010) and most importantly that miR-7 levels are down-regulated in the midbrain of mice intraperitoneally injected with the PD neurotoxin, MPTP (Junn et al., 2009). Therefore, in order to evaluate possible neuroprotective effects of miR-7 and/or miR-153 against MPP+ insult, 6- to 7-day old primary cortical neurons were transduced with adenoviral particles expressing scramble miR, miR-7, miR-153, or both of these two miRs, miR-7/153. It should be noted that irrespective of the adenoviral particles used, overall adenoviral infection of primary cortical neurons affected cell viability, whereas adenoviral over-expression of miR-7 and/or miR-153 had no effect on neuronal viability compared to adenoviral expression of scramble miR (Supplemental Figure 1). Thus, in order to avoid any confounding effects due to the infection per se, all subsequent comparisons were performed between primary neurons transduced with adenoviral particles expressing a scramble miR and primary neurons transduced with adenoviral particles expressing the miR(s) of interest. Twenty four hours post-transduction, cortical neurons were exposed to MPP+ concentrations ranging between 5 and 50 μM and left in culture for additional 24 h. Neuronal viability was then monitored by the MTT assay. As shown in Figure 3A and similar to uninfected neuronal cultures (see Supplemental Figure 2) in the scramble miR transduced cultures loss of viability extended from 10 to 62% with MPP+ concentrations ranging between 5 and 50 μM. Statistical analysis revealed a significant effect of MPP+ treatment [F(5, 29) = 48.968, P < 0.001 and post-hoc] at 10, 20, 30, and 50 μM, but not 5 μM, of MPP+. In contrast, neuronal viability in miR-7 or miR-153 transduced cultures was not impaired when treated with 5, 10, or 20 μM of MPP+ and it was only reduced at the higher concentrations applied i.e., 30 and 50 μM [miR-7: F(5, 29) = 32.948, P < 0.001 and post-hoc; miR-153: F(5, 29) = 24.816, P < 0.001 and post-hoc; Figure 3A]. Interestingly, in neuronal cultures transduced with adenoviral particles expressing both miR-7/153 neuronal viability was only impaired upon 24-h treatment with 50 μM MPP+ [F(5, 29) = 11.803, P < 0.001 and post-hoc; Figure 3A], Nevertheless, even at the highest concentration applied, compared to scramble miR transduced control cultures, neuronal cultures transduced with both miR-7/153 displayed an approximately 2-fold increase in neuronal viability upon 24-h treatment with 50 μM MPP+ [F(3, 19) = 11.444, P < 0.001 and post-hoc; Figure 3A].
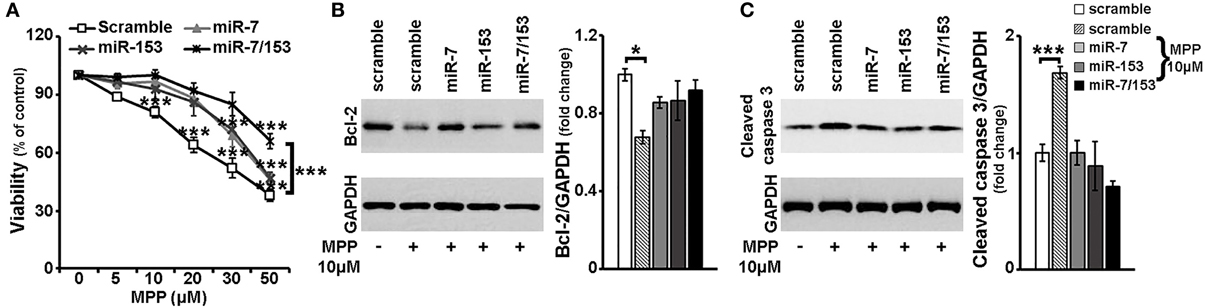
Figure 3. Neuroprotective effects of miR-7 and miR-153 against MPP+-toxicity. Six to seven days old primary cortical neurons were transduced with adenoviral particles expressing scramble miR, miR-7, miR-153, or both miR-7/153. After 24 h, transduced neurons were exposed for additional 24 h to various concentrations of MPP+. (A) Neuronal viability following MPP+-treatment was monitored by the MTT assay. Note that over-expression of miR-7 and/or miR-153 attenuated MPP+-induced cell death. (B,C) Equal amounts of total protein from lysates of transduced cortical neurons cultured for 24 h in the presence of 10 μM MPP+ were analyzed on 12% SDS-PAGE and immunoblotted with antibodies specific for BCL-2 (B) and cleaved caspase-3 (C). To ensure equal loading membranes were re-probed against GAPDH. Quantification of the results was performed by scanning densitometry. Note that compared to scramble-transduced untreated controls, only scramble-transduced cortical neurons displayed significant changes in BCL-2 and cleaved caspase-3 protein levels. Bars in all the depicted graphs correspond to mean ± s.e.m. *P ≤ 0.05, ***P ≤ 0.001.
Finally, as depicted in Figures 3B,C, compared to scramble miR transduced untreated controls, only scramble-transduced cortical neurons displayed a significant decrease in the levels of anti-apoptotic BCL-2 [F(4, 14) = 4.567, P < 0.05 and post-hoc], accompanied by a significant increase of cleaved caspase-3 levels [F(4, 14) = 10.438, P = 0.001 and post-hoc], upon 24-h treatment with 10 μM of MPP+. No such changes were observed among scramble-transduced untreated controls and MPP+-treated neuronal cultures transduced with miR-7, miR-153, or miR-7/153 adenoviruses, suggesting that over-expression of miR-7 and miR-153 in cortical neurons attenuated both the MPP+-induced down-regulation of pro-survival BCL-2 protein and activation of the pro-apoptotic caspase-3.
miR-7 and miR-153 Activated p70s6K Signaling Cascade in Primary Cortical Neurons and Attenuated the Effects of Rapamycin on mTOR Signaling and Cell Viability
Given a number of studies that show that miR-7 and miR-153 modulate intracellular signaling (Kefas et al., 2008; Fang et al., 2012; Song et al., 2012; Sanchez et al., 2013; Wang et al., 2013, Wu et al., 2013) in non-neuronal cells, we next investigated whether these miRs affect the activation of the major intracellular signaling cascades in neurons. Therefore, 6–7 days old primary cortical neurons were transduced with adenoviral particles expressing scramble miR, miR-7, or miR-153, and the activation of signaling kinases was assessed 48 h later by immunoblotting. As depicted in Figure 4A, overexpression of miR-7 or miR-153 in cortical neurons did not affect the phosphorylation status of pro-survival kinase AKT or of the pro-apoptotic p38 MAPK and similar results were also observed in the levels of phosphorylated GSK-3β and MAPKAPK-2, downstream effectors of AKT and p38 MAPK, respectively (data not shown). Finally, no significant change was either observed in the levels of phosphorylated ERK1/2 or those of activated stress-induced kinases SAPK/JNK (Figure 4A).
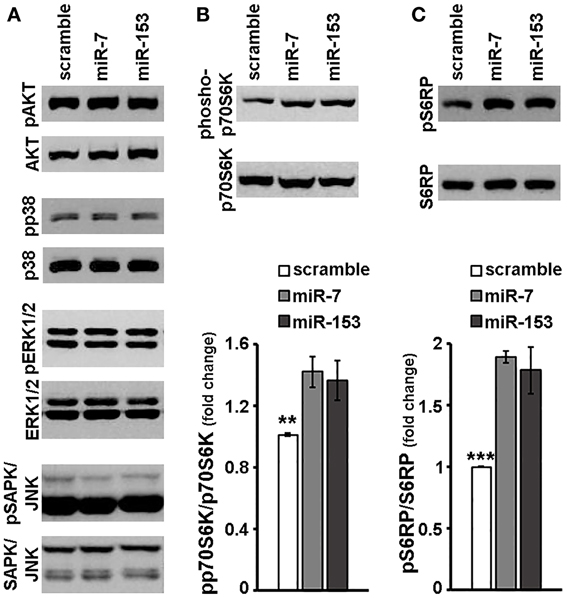
Figure 4. miR-7 and miR-153 activated p70S6K signaling cascade in cortical neurons. Six to seven days old primary cortical neurons were transduced with adenoviral particles expressing scramble miR, miR-7, or miR-153 and were lysed 48 h post-transduction. (A) Equal amounts of total protein from lysates of cortical neurons were analyzed on 10% SDS-PAGE and immunoblotted with antibodies specific for phosphorylated forms of AKT, p38 MAPK, ERK1/2 SAPK/JNK. To ensure equal loading membranes were re-probed against AKT, p38 MAPK, ERK1/2, and SAPK/JNK, respectively. (B,C) Equal amounts of total protein from lysates of cortical neurons were analyzed on 10% SDS-PAGE and immunoblotted with antibodies specific for phosphorylated forms of p70S6K (B) as well as for the phosphorylated forms of p70S6K substrate, S6RP (C). To ensure equal loading, membranes were re-probed against p70S6K and S6RP, respectively. Quantification of the results was performed by scanning densitometry. Bars in the graph depict mean ± s.e.m. Note that compared to scramble-transduced controls, primary cortical neurons transduced with miR-7 or miR-153 expressing adenoviruses displayed a significant increase only in the levels of phosphorylated forms of p70S6K and S6RP. **P ≤ 0.01, ***P ≤ 0.001.
Markedly, in the same experimental system, both miR-7 and miR-153 appeared to induce p70S6 kinase signaling downstream of the mTOR signaling cascade. More specifically, levels of phosphorylated p70S6K were significantly up-regulated by 45 and 52% in cortical neurons transduced with adenoviral particles expressing miR-7 and miR-153, respectively [ANOVA: F(2, 14) = 8.056, P < 0.01 and post-hoc; Figure 4B]. Consistent with the above, phosphorylation of p70S6K substrates S6RP and eEF2K was also significantly increased; levels of phosphorylated S6RP were up-regulated by 89 and 83% [ANOVA: F(2, 11) = 20.084, P < 0.001 and post-hoc; Figure 4C], whereas levels of phosphorylated eEF2K were increased by 36 and 62% [ANOVA: F(2, 11) = 7.475, P < 0.05 and post-hoc; data not shown] upon overexpression of miR-7 and miR-153, respectively. The latter mTOR downstream activation by miR-7 or miR-153 is unlikely to be attributed to an unspecific scramble miR effect, since transduction with adenoviral particles expressing scramble miR appeared to have no effect on the phosphorylation of p70S6K and of its substrate S6RP compared to transduced empty control neurons (Supplemental Figure 3). Taken together the latter observations suggest that miR-7 and miR-153 may activate the mTOR signaling cascade.
To further explore the latter hypothesis, 6–7 days old primary cortical neurons were again transduced with adenoviral particles expressing scramble miR, miR-7, or miR-153 and left in culture for additional 48 h. One hour before harvest, cultures were supplemented with 20 nM rapamycin, a potent mTORC1 (and mTORC2 at higher doses and long-term treatment) inhibitor (Sarbassov et al., 2006; Rosner and Hengstschlager, 2008; Chen et al., 2010) and levels of phosphorylated mTORC1 effector p70S6K and its phosphorylated substrates S6RP and eEF2K were determined. As shown in Figure 5A, irrespective of the adenovirus used, 1-h rapamycin treatment resulted to a significant decrease in the levels of phosphorylated p70S6K [ANOVA: F(3, 11) = 36.857, P < 0.001 and post-hoc]; nevertheless, primary cortical neurons transduced with miR-7 or miR-153 sustained phosphorylated p70S6K levels at more than 2-fold higher than scramble miR-transduced controls (post-hoc, P < 0.01). Consistently, overexpression of miR-7 and miR-153 attenuated the effect of rapamycin on the phosphorylation of S6RP [ANOVA: F(3, 11) = 71.638, P < 0.001 and post-hoc; Figure 5B], and of eEF2K [ANOVA: F(3, 11) = 65.623, P < 0.001 and post-hoc; Figure 5B], More specifically, compared to scramble-transduced rapamycin-treated controls, primary cortical neurons transduced with miR-7 or miR-153 and treated with rapamycin displayed significantly increased levels of phosphorylated S6RP (4.7- and 5.4-fold increase in the case of miR-7, miR-153 overexpression, respectively; post-hoc, P < 0.001), as well as of phosphorylated eEF2K (by 90% for miR-7 and 65% for miR-153, post-hoc, P < 0.001). It appears, therefore, that miR-7 and miR-153 may act as “activators” of mTOR signaling pathway.
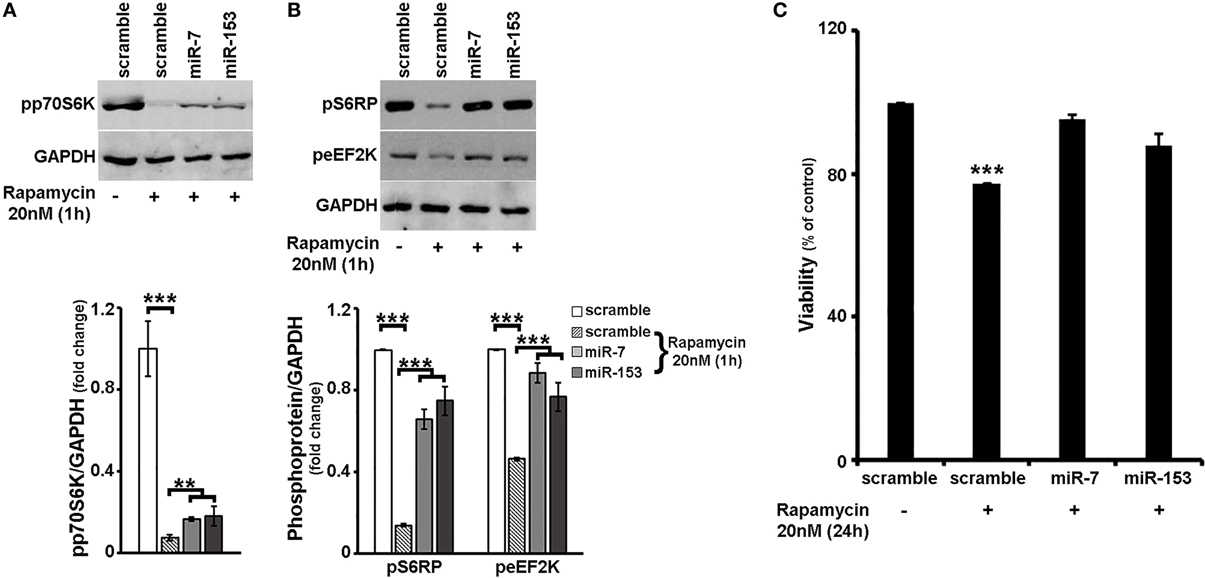
Figure 5. Overexpression of miR-7 or miR-153 attenuated the effects of rapamycin in cortical neurons. (A,B) Six to seven days old primary cortical neurons were transduced with adenoviral particles expressing scramble miR, miR-7, or miR-153 and left in culture for additional 48 h. One hour before harvest, cultures were supplemented with 20 nM rapamycin. Equal amounts of total protein from lysates of cortical neurons were analyzed on 10% SDS-PAGE and immunoblotted with antibodies specific for phosphorylated forms of p70S6K, S6RP, and eEF2K. To ensure equal loading, membranes were re-probed against GAPDH. Quantification of the results was performed by scanning densitometry. Note that overexpression of miR-7, as well as that of miR-153 attenuated the effect of rapamycin on the phosphorylation of all proteins examined. (C) Six to seven days old primary cortical neurons were transduced with the same adenoviral particles and after 24 h were exposed to 20 nM of rapamycin for an additional of 24 h. Neuronal viability following 24-h treatment with rapamycin was monitored by the MTT assay. Note that overexpression of miR-7 and/or miR-153 attenuated rapamycin-induced cell death. Bars in all the presented graphs depict mean ± s.e.m. **P ≤ 0.01, ***P ≤ 0.001.
Given that mTOR signaling pathway is a downstream regulator of neuronal survival (see also Supplemental Figure 4), we next wanted to examine whether overexpression of miR-7 and/or miR-153 is able to interfere with the effect of mTOR signaling inhibition on cell survival. In order to address this, 6–7 days old primary cortical neurons were transduced with adenoviral particles expressing scramble miR, miR-7, or miR-153, supplemented with 20 nM rapamycin 24 h post-transduction, and left in culture for an additional 24 h. Neuronal viability was monitored using the MTT assay. As depicted in Figure 5C, a 24-h treatment with rapamycin reduced significantly the viability of scramble-transduced neuronal cultures to 77% [ANOVA: F(3, 11) = 32.804, P < 0.001 and post-hoc], a reduction that was comparable to the one observed in untransduced cortical neuronal cultures (Supplemental Figure 4). In contrast, viability of miR-7-, or miR-153- transduced neuronal cultures was not significantly impaired, providing further support that these two miRs sustain mTOR signaling in neurons.
Overexpression of miR-7 and/or miR-153 in Cortical Neurons Attenuated MPP+-Induced Neurotoxicity Via Upregulation of mTOR Pathway
Our results so far suggest that miR-7 and miR-153 are able to induce rapamycin-sensitive mTOR downstream signaling, which appeared significantly impaired in cortical neurons upon MPP+ treatment. Therefore, in order to evaluate whether miR-7 and/or miR-153 exert their neuroprotective effect through upregulation of mTOR signaling pathway, 6- to 7- day old primary cortical neurons were again transduced with adenoviral particles expressing scramble miR, miR-7, or miR-153, as well as with an adenoviral construct expressing both of these two miRs. Twenty four hours post-transduction, cortical neurons were exposed to 10 μM of MPP+ and the activation of p70S6K and its substrates was assessed 24 h later by immunoblotting. As shown in Figures 6A,B, compared to scramble-transduced untreated controls, 24-h treatment with 10 μM of MPP+ induced a significant decrease in the levels of phosphorylated p70S6K [F(4, 14) = 5.072, P < 0.05 and post-hoc] and of its phosphorylated substrate S6RP [F(4, 14) = 5.241, P < 0.05 and post-hoc] in only the scramble-transduced cortical neurons; overexpression of miR-7 or miR-153 attenuated the MPP+-induced reduction in the activation of p70S6K and its downstream targets, while overexpression of both miRs restored their phosphorylation status to that of scramble-transduced untreated controls. To further explore the possibility that sustained mTOR downstream signaling activation underlies the neuroprotective effects of miR-7 and miR-153, 20 nM of rapamycin was co-administered with 10 μM MPP+ in these neuronal cultures. As shown in Figure 6C, in scramble miR-transduced cultures all treatments lead to a significant reduction in neuronal viability [F(3, 11) = 25.244, P < 0.001 and post-hoc]. In contrast, in miR-7, miR-153, or miR-7/153 -transduced cultures, neuronal viability was significantly impaired only when MPP+ was co-administered with rapamycin [miR-7: F(3, 11) = 41.253, P < 0.001 and post-hoc; miR-153: F(3, 11) = 14.061, P = 0.001 and post-hoc, miR-7/153: F(3, 11) = 26.653, P < 0.001 see Figure 6C]. Taken together the above results suggest that miR-7 and/or miR-153 induced activation of mTOR pathway largely mediates their neuroprotective effect against MPP+ toxicity in cortical neurons.
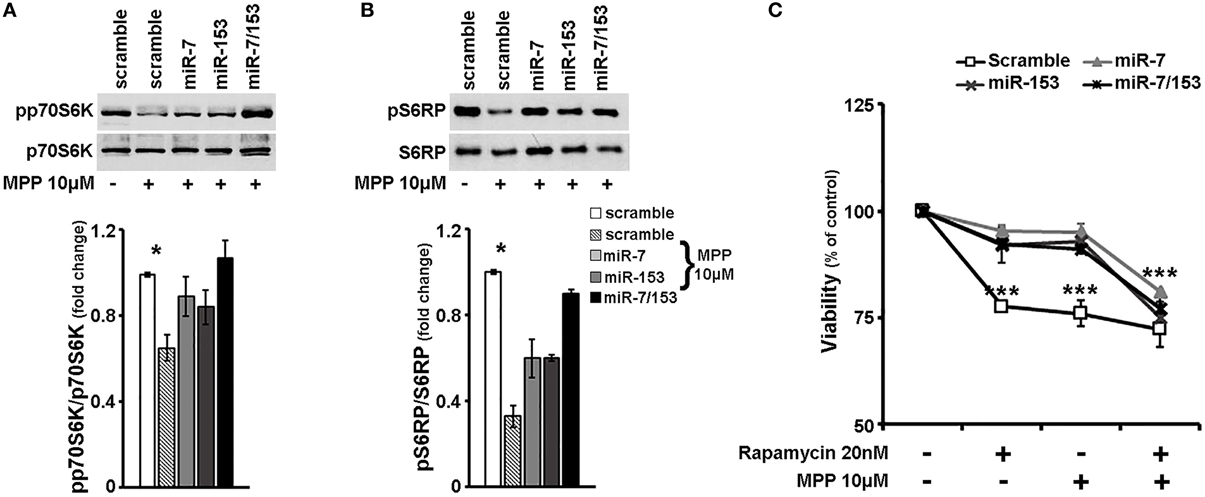
Figure 6. miR-7 and/or miR-153 attenuated MPP+-induced neurotoxicity via upregulation of rapamycin sensitive mTOR pathway. (A,B) Six to seven days old primary cortical neurons were transduced with adenoviral particles expressing scramble miR, miR-7, miR-153, or both miR-7/153. After 24 h, transduced neurons were exposed for additional 24 h to 10 μM of MPP+. Equal amounts of total protein from lysates of transduced cortical neurons cultured for 24 h in the presence of 10 μM MPP+ were analyzed on 10% SDS-PAGE and immunoblotted with antibodies specific for phosphorylated forms of p70S6K (A) and S6RP (B). To ensure equal loading membranes were re-probed against p70S6K and S6RP, respectively. Note that overexpression of miR-7 and/or miR-153 attenuated the effect of MPP+ on the phosphorylation of all proteins examined. Quantification of the results was performed by scanning densitometry. (C) Six to seven days old cultures of primary cortical neurons were transduced as described above, and 24 h later were exposed to 20 nM of rapamycin, 10 μM of MPP+, or both. Neuronal viability was monitored 24 h post-exposure by the MTT assay. Note that in the presence of rapamycin, overexpression of miR-7 and/or miR-153 failed to protect cortical neurons against MPP+-induced cell death. Bars in all presented graphs depict mean ± s.e.m. *P < 0.05, ***P ≤ 0.001.
Finally, in order to explore whether miR-7 and/or miR-153 interfere with MPP+-induced changes in other than mTOR intracellular signaling cascades, the phosphorylation status of other major signaling effectors was examined in cortical neurons transduced with scramble miR, miR-7, miR-153, or miR-7/153 adenoviruses and treated for 24 h with 10 μM MPP+. As shown in Figure 7A, in MPP+-treated cortical neurons overexpression of both miR-7/153, but not that of miR-7 or miR-153 alone, attenuated the MPP+-induced reduction in the levels of phosphorylated AKT [F(4, 24) = 12.314, P = 0.01 and post-hoc]. Interestingly, miR-153, but not miR-7 or miR-7/153, attenuated the MPP+-induced activation of pro-apoptotic p38 MAPK [F(4, 24) = 12.978, P = 0.01 and post-hoc; Figure 7B]. Finally, overexpression of miR-7 and/or miR-153 had no significant effect on the phosphorylation status of ERK1/2 (Figure 7C), but resulted in a significant increase of phosphorylated SAPK/JNK levels to above control levels [F(4, 24) = 15.894, P < 0.001 and post-hoc; Figure 7D]. It, therefore, appears that miR-7 and/or miR-153 alter the intracellular response of cortical neurons to MPP+ insult and thus interfere with MPP+-induced neurotoxicity.
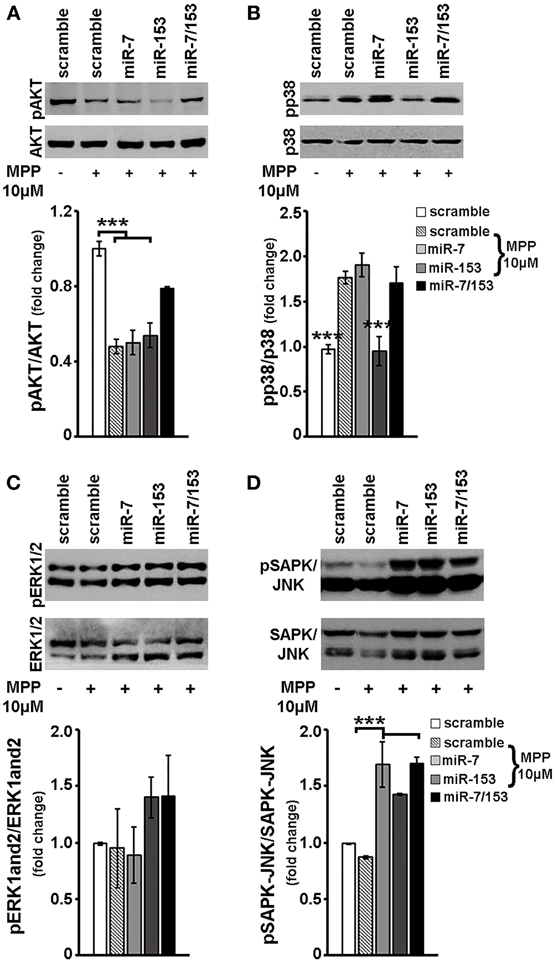
Figure 7. Effect of miR-7 and/or mir-153 overexpression on AKT, p38, ERK-1/2, and SAPK/JNK signaling in MPP+-treated cortical neurons. Six to seven days old primary cortical neurons were transduced with adenoviral particles expressing scramble miR, miR-7, miR-153, or both miR-7/153. After 24 h, transduced neurons were exposed for additional 24 h to 10 μM of MPP+. Equal amounts of total protein from lysates of transduced cortical neurons cultured for 24 h in the presence of 10 μM MPP+ were analyzed on 10% SDS-PAGE and immunoblotted with antibodies specific for phosphorylated forms of AKT (A), p38 MAPK (B), ERK1/2 (C), and SAPK/JNK (D). To ensure equal loading membranes were re-probed against AKT, p38 MAPK, ERK1/2, and SAPK/JNK, respectively. Quantification of the results was performed by scanning densitometry. Bars in all the presented graphs depict mean ± s.e.m. Note that miR-7, miR-153, or miR-7/153 altered in an opposite way MPP+-induced changes in intracellular signaling cascades. ***P < 0.001.
Discussion
The mechanisms underlying chronic neurodegeneration in PD remain obscure. An emerging hypothesis is that neuronal systems deteriorate and eventually degenerate due to failure of intrinsic cellular pathways that mediate neuronal homeostasis. This failure maybe due to lack of external neurotrophic support or to mutations in intrinsic factors such as the PARK genes that modify intracellular signaling (reviewed in Wang et al., 2012). Thus, far, a great number of studies have indicated that neurotrophic factors or herbal extracts protect neurons from PD insults by enhancing pro-survival and/or decreasing pro-apoptotic signaling pathways (Nakaso et al., 2008; Wang et al., 2010; Cui et al., 2011; Zhang et al., 2011; Bao et al., 2012; Hashimoto et al., 2012). In addition, manipulation of specific intracellular signaling cascades by either overexpressing or inhibiting signaling protein kinases has revealed that they modulate most PD neurotoxin effects (Malagelada et al., 2006; Zhu et al., 2007, 2012; Nakaso et al., 2008; Cui et al., 2011; Bao et al., 2012; Piao et al., 2012). Most importantly, these findings phenocopy data from the analysis of human postmortem PD brains that show decreased phosphorylation of pro-survival and enhanced activation of pro-apoptotic pathways (Zhu et al., 2002, 2003; Malagelada et al., 2006; Timmons et al., 2009; Reinhardt et al., 2013).
Previous work from our group has shown that mir-7 and mir-153 significantly regulate the expression of α-synuclein, a protein encoded by the gene SNCA that belongs to the PARK gene family (Doxakis, 2010). A-synuclein plays a seminal role in neurodegeneration and has been shown, among others, to affect signaling by modulating neurotrophin BDNF expression and AKT activity (Yuan et al., 2010; Chung et al., 2011). Based on the intrinsic property of miRs to regulate the expression of multiple proteins and possibly the activation of signaling cascades, in the present study we wished to investigate if miR-7 and miR-153 protect neurons exposed to PD insults via altering intracellular signaling. Thus, we evaluated whether overexpression of mir-7 and/or mir-153 could prevent MPP+-induced toxicity in cortical neurons. Cortical neurons were selected because they are directly affected in PD by showing progressive pathology (Trojanowski et al., 1998; Braak et al., 2006) and they can be isolated in great numbers relative free of glial cells. MPP+, on the other hand, is a widely used neurotoxin that reproduces the neuronal dysfunction of PD both in vivo and in different cell systems in vitro. MPP+ enters cells through the dopamine re-uptake system, present in dopaminergic neurons; however, at higher concentrations it can enter all cell types by passive diffusion (Reinhard et al., 1990) and/or by the extraneuronal monoamine transporter (Russ et al., 1996). The mechanism of MPP+ toxicity in cells is rather ubiquitous and involves inhibition of the mitochondrial respiratory chain, elevation of oxidative stress and alteration of intracellular signaling. The vulnerability of neurons to MPP+ is modified by microglia numbers in the vicinity of neurons, neurotrophic support, glutathione, or superoxide dismutase content (antioxidant capacity), the content of redox active molecules or elements (such as dopamine or iron), the ratio of anti-apoptotic vs. pro-apoptotic BCL-2 family proteins and basal levels of phosphorylated signaling kinases (Lawson et al., 1990; Kim et al., 2000; Zigmond et al., 2002; Wu et al., 2003; Zecca et al., 2004; Willis et al., 2007). Noteworthy, modulation of intracellular signaling pathways has been shown to mediate most of the MPP+ effects in neurons indicating that signaling cascades are downstream of MPP+ targets and/or can reverse pro-apoptotic effects (Nakaso et al., 2008; Wang et al., 2010; Cui et al., 2011; Bao et al., 2012; Hashimoto et al., 2012; Piao et al., 2012).
Based on the above, we initially characterized the molecular mechanisms underlying MPP+-induced neuronal death in cortical neurons since most studies have been carried out in dividing neuroblastoma cells and/or were limited to two or three signaling pathways. Hence, the levels of apoptosis-related BCL-2 family members and the major signaling pathways, AKT, ERK-1/2, p38, SAPK/JNK, and mTOR were determined. Consistent with previous studies, we found that MPP+-induced neurotoxicity displayed apoptotic characteristics, as documented by the reduced levels of BCL-2 and the increased levels of cleaved caspase-3, and was accompanied by enhanced activities of the pro-apoptotic p38 and ERK-1/2 MAPK signaling pathways as well as by reduced activation of pro-survival AKT and p70S6K kinases (Deguil et al., 2007; Junyent et al., 2010; Cui et al., 2011; Bao et al., 2012; Hashimoto et al., 2012; Rodriguez-Blanco et al., 2012). Finally, contrary to most other findings (Wang et al., 2010; Zhang et al., 2011; Hashimoto et al., 2012; Rodriguez-Blanco et al., 2012), and with the exception of a single study (Sun and Chang, 2003), activation of the SAPK/JNK kinase was suppressed in a dose-dependent manner by MPP+ treatment of cortical neurons.
Subsequently, the effect of miR-7 and miR-153 overexpression in neurons was determined. We found that miR overexpression did not, overall, alter neuronal viability or the activity of AKT, ERK-1/2, p38, and SAPK/JNK signaling pathways. However, a significant upregulation of mTORC1 downstream signaling was observed by the overexpression of both miRs, as evident by the increased levels of phosphorylated p70S6K and its downstream targets S6RP and eEF2K. mTOR complexes (mTORC1/2) serve as central regulators of cell metabolism, growth and survival by integrating intracellular (energy status, oxygen, and amino acids) and extracellular signals (growth factors) (Wu et al., 2004; Takei et al., 2009; reviewed in Swiech et al., 2008; Laplante and Sabatini, 2013). Mutant mTOR embryos lack telencephalon and die by midgestation, an effect that is phenocopied by the mTOR inhibitor, rapamycin, validating the importance of this pathway in brain development (Hentges et al., 2001). In cultured neurons, mTORC1, the best studied mTOR complex, has been shown to regulate soma size, dendrite axonal growth, dendrite development, and regeneration (Campbell and Holt, 2001; Jaworski et al., 2005; Kumar et al., 2005; Tavazoie et al., 2005; Verma et al., 2005; Li et al., 2008; Park et al., 2008). Our finding that the activation of mTOR downstream effectors was significantly increased in cortical neurons over-expressing miR-7 or miR-153, suggests that these two miRs may act as “activators” of mTOR signaling pathway. The latter hypothesis is further supported by our observations showing that overexpression of miR-7 or miR-153 in primary neurons is able to attenuate the effects of rapamycin on both the activation of mTOR downstream effectors and neuronal viability.
Probing the effect of miR-7 and miR-153 overexpression in MPP+-treated neurons, we revealed that they could, either alone or together, significantly protect neurons from cell death. We reasoned that this was due to enhanced mTOR signaling as this was the only pathway that was upregulated by the overexpression of both miRs. Consistent with the latter hypothesis, overexpression of miR-7 and/or miR-153 attenuated the MPP+-induced reduction on the activation of p70S6K and its downstream targets, whereas treatment of transduced neurons with rapamycin abolished the pro-survival effects of miR-7 and miR-153 upon MPP+ exposure.
To further explore the modulation of intracellular signaling by miR-7 and miR-153 overexpression in MPP+-treated neurons, the activation of the remaining pathways was also determined. It should be noted that compared to untransduced cortical neurons, transduced primary cortical neurons used in the present study appeared less resistant to MPP+-treatment and therefore displayed a more robust intracellular response to the same MPP+ concentration i.e., 10 μM; this is likely to be attributed to the adenoviral transduction, given that it comprises an additional, to that of MPP+, insult for the cortical neurons. Taking the latter observation into account, herein we found that the activity of AKT which is known to activate mTORC1 by alleviating the inhibition induced by TSC2 and PRAS40 proteins (Dan et al., 2002; Inoki et al., 2002; Manning et al., 2002; Vander Haar et al., 2007; Zhu et al., 2007), was not restored by either miR-7 or miR-153; however, overexpressing both miR-7 and miR-153 significantly relieved the suppression of AKT activation by MPP+, likely by having overlapping or additive effects on their targets. In addition, the finding that AKT was activated at Ser473, known to be mediated by mTORC2 complex (Sarbassov et al., 2005), may indicate that mTORC2 signaling is also contributing to the survival of neurons transduced by both miRs. p38 is a stress kinase that has been linked to neuro-inflammation and MPP+-mediated apoptosis (Karunakaran et al., 2008; Thomas et al., 2008b). It should be noted that miR-153, but not miR-7, significantly prevented the activation of p38 by MPP+ which may have partly contributed to its pro-survival effects in cortical neurons. Overexpressing miR-7 and miR-153 together alleviated the negative effect of miR-153 on p38 phosphorylation indicating that miR-7 targets may block mir-153 responses on p38 signaling pathway activation. The role of ERK-1/2 activation in neuronal survival is context-specific; some reports show positive or negative input on survival after induction by growth factors, glutamate, or okadaic acid (Runden et al., 1998; Bonni et al., 1999; Satoh et al., 2000; Stanciu et al., 2000; Cui et al., 2011) while others implicate it in MPP+- and 6-hydroxydopamine- induced mitophagy/autophagy and cell death (Zhu et al., 2007, 2012). In the present study, neither miR-7 nor miR-153 overexpression significantly changed ERK-1/2 phosphorylation in the MPP+-treated neurons. SAPK/JNK is a kinase with an indispensable role in microtubule stability in neurons. It stimulates dendrite formation, axodendritic length, axonal regeneration, mediates fast axonal transport, and contributes to the regulation of synaptic plasticity (Bjorkblom et al., 2005; Chen et al., 2005; Zhu et al., 2005; Tararuk et al., 2006; Thomas et al., 2008a; Morfini et al., 2009; Barnat et al., 2010; Podkowa et al., 2010). At the same time it has been linked to stress-induced apoptosis in different pathological conditions as a result of its inhibition of autophagy and the induction of pro-apoptotic BCL-2 family members (Jia et al., 2006; Hubner et al., 2008; Xu et al., 2011). In our cell culture system, miR-7 and miR-153 overexpression significantly lifted SAPK/JNK activation in the MPP+-treated neurons. Overexpression of both miRs together did not further induce SAPK/JNK activation indicating that they modulate a similar target group. Additional experiments will be required to determine if the effect of miR-7 and miR-153 overexpression on SAPK/JNK phosphorylation partly negates their neuroprotective responses via mTOR signaling, and/or maintains the axodendritic growth of neurons which is impaired by MPP+-induced microtubule dysfunction (Cartelli et al., 2010) and/or negates MPP+-induced ERK-1/2-mediated enhanced autophagy in neurons.
Taken together, our data suggest that miR-7 and miR-153 protect neurons against MPP+-induced toxicity via upregulation of mTOR downstream targets. In addition, we show that miR-7 and miR-153 modulate the signaling pathways of SAPK/JNK and p38 in MPP+-treated cells, however, their effect on neuronal viability maybe less important. Given also our previous study showing that miR-7 and miR-153 regulate α-synuclein expression, it appears these two miRs may prove good therapeutic candidates for the treatment of PD. Evidence from successful medical interventions based on miRs has already been shown in cornerstone studies to lower plasma cholesterol levels in rodents and primates (Krutzfeldt et al., 2005; Elmen et al., 2008a,b). Currently, a large number of miRs are studied in preclinical and clinical settings by biotechnology companies (Lindow and Kauppinen, 2012). In future, it will be important to characterize the effect of miR-7 and miR-153 on neurite outgrowth and synaptogenesis and test if they can support neurons treated with other PD neurotoxins.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Paulos Alexakos for excellent veterinary assistance. Epaminondas Doxakis has received funding from the Greek General Secretariat for Research and Technology (Grant IDs 09Syn-12-876 and 12RUS-11-65).
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fncel.2014.00182/abstract
Supplemental Figure 1 | Overexpression of miR-7 or miR-153 has no effect on neuronal viability. (A) Six to seven days old primary cortical neurons were left untreated or were transduced with adenoviral particles expressing scramble miR, miR-7, miR-153, or both miR-7153 and neuronal viability was assayed by MTT reduction 48 h later. It should be noted that overall adenoviral infection of primary cortical neurons affected cell viability [F(4, 19) = 2.979, *P = 0.05 and post-hoc]; nevertheless, no significant difference was observed among transduced primary neurons, i.e., those transduced with adenoviral particles expressing a scramble miR or the miR (s) of interest. (B) Six to seven days old primary cortical neurons were left untreated or were transduced with the same adenoviral particles and lysed 48 h later. Equal amounts of total protein from lysates of cortical neurons were analyzed on 12% SDS-PAGE and immunoblotted with antibodies specific for BCL-2 and cleaved caspase-3. To ensure equal loading membranes were re-probed against GAPDH. Compared to scramble-transduced controls, primary cortical neurons transduced with miR-7, miR-153, or miR-7/153 displayed no significant change on either BCL-2 or cleaved caspase-3 protein levels; it should be noted that a statistically insignificant increase in the levels of cleaved caspase-3 was observed among transduced and untransduced primary neurons.
Supplemental Figure 2 | MMP+ treatment induced neurotoxicity and alterations in major signaling cascades in both untransduced and scramble miR transduced cortical neurons in a similar manner. (A) Six to seven days old primary cortical neurons were left untreated or were transduced with adenoviral particles expressing scramble miR. After 24 h, neuronal cultures were exposed for additional 24 h to various concentrations of MPP+. Cell viability following dose-dependent treatments was assayed by measuring MTT reduction by live neurons. Note that similar significant reductions in neuronal viability was observed in both untransduced and scramble miR transduced neurons upon treatment with 10, 20, 30, and 50 μM, but not 5 μM, of MPP+. (B,C) Six to seven days old primary cortical neurons were left untreated or were transduced with adenoviral particles expressing scramble miR. After 24 h, neuronal cultures were exposed for additional 24 h to 10 μM of MPP+. Equal amounts of total protein from lysates of cortical neurons were analyzed on 10% SDS-PAGE and immunoblotted with antibodies specific for phosphorylated forms of AKT, p38 MAPK, p70S6K, SAPK/JNK, and ERK1/2. To ensure equal loading membranes were re-probed against AKT, p38 MAPK, p70S6K, SAPK/JNK, and ERK1/2, respectively. Note that in contrast to untransduced neurons, scramble miR transduced neurons displayed significant reduction of phosphorylated AKT levels even at 10 μM of MPP+. No difference was observed in the phosphorylation status of the other major signaling kinases examined among untransduced and scramble miR transduced neurons upon treatment with 10 μM of MPP+. ***P < 0.001.
Supplemental Figure 3 | Adenoviral overexpression of miR-7 and miR-153 in cortical neurons, but not of scramble miR, activated p70S6K signaling cascade. Six to seven days old primary cortical neurons were transduced with empty adenoviral particles or adenoviral particles expressing scramble miR, miR-7, or miR-153 and were lysed 48 later. Equal amounts of total protein from lysates of untransduced and transduced cortical neurons were analyzed on 10% SDS-PAGE and immunoblotted with antibodies specific for phosphorylated forms of p70S6K (A) and S6RP (B). To ensure equal loading, membranes were re-probed against GAPDH. Note that no difference was observed in the levels of phosphorylated p70S6K and S6RP between transduced empty control and scramble miR transduced cortical neurons.
Supplemental Figure 4 | Rapamycin induced neurotoxicity in cortical neurons. Seven-day primary cortical neurons were treated with various concentrations of rapamycin and MPP+ for 24 h. Cell viability following dose-dependent treatments was assayed by measuring MTT reduction by live neurons. Note that a significant ~20% reduction in neuronal viability was observed upon treatment with rapamycin at concentrations 20–50 nM [F(4, 14) = 20.852, P < 0.001]. No synergistic effect was observed among rapamycin and MPP+. ***P < 0.001.
References
Ballard, P. A., Tetrud, J. W., and Langston, J. W. (1985). Permanent human parkinsonism due to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP): seven cases. Neurology 35, 949–956. doi: 10.1212/WNL.35.7.949
Bao, X. Q., Kong, X. C., Qian, C., and Zhang, D. (2012). FLZ protects dopaminergic neuron through activating protein kinase B/mammalian target of rapamycin pathway and inhibiting RTP801 expression in Parkinson's disease models. Neuroscience 202, 396–404. doi: 10.1016/j.neuroscience.2011.11.036
Barnat, M., Enslen, H., Propst, F., Davis, R. J., Soares, S., and Nothias, F. (2010). Distinct roles of c-Jun N-terminal kinase isoforms in neurite initiation and elongation during axonal regeneration. J. Neurosci. 30, 7804–7816. doi: 10.1523/JNEUROSCI.0372-10.2010
Bjorkblom, B., Ostman, N., Hongisto, V., Komarovski, V., Filen, J. J., Nyman, T. A., et al. (2005). Constitutively active cytoplasmic c-Jun N-terminal kinase 1 is a dominant regulator of dendritic architecture: role of microtubule-associated protein 2 as an effector. J. Neurosci. 25, 6350–6361. doi: 10.1523/JNEUROSCI.1517-05.2005
Bonni, A., Brunet, A., West, A. E., Datta, S. R., Takasu, M. A., and Greenberg, M. E. (1999). Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science 286, 1358–1362. doi: 10.1126/science.286.5443.1358
Braak, H., Rub, U., Schultz, C., and Del Tredici, K. (2006). Vulnerability of cortical neurons to Alzheimer's and Parkinson's diseases. J. Alzheimers Dis. 9, 35–44.
Campbell, D. S., and Holt, C. E. (2001). Chemotropic responses of retinal growth cones mediated by rapid local protein synthesis and degradation. Neuron 32, 1013–1026. doi: 10.1016/S0896-6273(01)00551-7
Cartelli, D., Ronchi, C., Maggioni, M. G., Rodighiero, S., Giavini, E., and Cappelletti, G. (2010). Microtubule dysfunction precedes transport impairment and mitochondria damage in MPP+-induced neurodegeneration. J. Neurochem. 115, 247–258. doi: 10.1111/j.1471-4159.2010.06924.x
Chen, J. T., Lu, D. H., Chia, C. P., Ruan, D. Y., Sabapathy, K., and Xiao, Z. C. (2005). Impaired long-term potentiation in c-Jun N-terminal kinase 2-deficient mice. J. Neurochem. 93, 463–473. doi: 10.1111/j.1471-4159.2005.03037.x
Chen, X. G., Liu, F., Song, X. F., Wang, Z. H., Dong, Z. Q., Hu, Z. Q., et al. (2010). Rapamycin regulates Akt and ERK phosphorylation through mTORC1 and mTORC2 signaling pathways. Mol. Carcinog. 49, 603–610. doi: 10.1002/mc.20628
Chung, J. Y., Lee, S. J., Lee, S. H., Jung, Y. S., Ha, N. C., Seol, W., et al. (2011). Direct interaction of alpha-synuclein and AKT regulates IGF-1 signaling: implication of Parkinson disease. Neurosignals 19, 86–96. doi: 10.1159/000325028
Cui, W., Li, W., Han, R., Mak, S., Zhang, H., Hu, S., et al. (2011). PI3-K/Akt and ERK pathways activated by VEGF play opposite roles in MPP+-induced neuronal apoptosis. Neurochem. Int. 59, 945–953. doi: 10.1016/j.neuint.2011.07.005
Dan, H. C., Sun, M., Yang, L., Feldman, R. I., Sui, X. M., Ou, C. C., et al. (2002). Phosphatidylinositol 3-kinase/Akt pathway regulates tuberous sclerosis tumor suppressor complex by phosphorylation of tuberin. J. Biol. Chem. 277, 35364–35370. doi: 10.1074/jbc.M205838200
Davis, G. C., Williams, A. C., Markey, S. P., Ebert, M. H., Caine, E. D., Reichert, C. M., et al. (1979). Chronic Parkinsonism secondary to intravenous injection of meperidine analogues. Psychiatry Res. 1, 249–254. doi: 10.1016/0165-1781(79)90006-4
Deguil, J., Jailloux, D., Page, G., Fauconneau, B., Houeto, J. L., Philippe, M., et al. (2007). Neuroprotective effects of pituitary adenylate cyclase-activating polypeptide (PACAP) in MPP+-induced alteration of translational control in Neuro-2a neuroblastoma cells. J. Neurosci. Res. 85, 2017–2025. doi: 10.1002/jnr.21318
Doxakis, E. (2010). Post-transcriptional regulation of alpha-synuclein expression by mir-7 and mir-153. J. Biol. Chem. 285, 12726–12734. doi: 10.1074/jbc.M109.086827
Doxakis, E. (2013). Principles of miRNA-Target Regulation in Metazoan Models. Int. J. Mol. Sci. 14, 16280–16302. doi: 10.3390/ijms140816280
Doxakis, E., Huang, E. J., and Davies, A. M. (2004). Homeodomain-interacting protein kinase-2 regulates apoptosis in developing sensory and sympathetic neurons. Curr. Biol. 14, 1761–1765. doi: 10.1016/j.cub.2004.09.050
Elmen, J., Lindow, M., Schutz, S., Lawrence, M., Petri, A., Obad, S., et al. (2008a). LNA-mediated microRNA silencing in non-human primates. Nature 452, 896–899. doi: 10.1038/nature06783
Elmen, J., Lindow, M., Silahtaroglu, A., Bak, M., Christensen, M., Lind-Thomsen, A., et al. (2008b). Antagonism of microRNA-122 in mice by systemically administered LNA-antimiR leads to upregulation of a large set of predicted target mRNAs in the liver. Nucleic Acids Res. 36, 1153–1162. doi: 10.1093/nar/gkm1113
Fang, Y., Xue, J. L., Shen, Q., Chen, J., and Tian, L. (2012). MicroRNA-7 inhibits tumor growth and metastasis by targeting the phosphoinositide 3-kinase/Akt pathway in hepatocellular carcinoma. Hepatology 55, 1852–1862. doi: 10.1002/hep.25576
Hashimoto, R., Yu, J., Koizumi, H., Ouchi, Y., and Okabe, T. (2012). Ginsenoside Rb1 Prevents MPP(+)-Induced Apoptosis in PC12 Cells by Stimulating Estrogen Receptors with Consequent Activation of ERK1/2, Akt and Inhibition of SAPK/JNK, p38 MAPK. Evid. Based Complement. Alternat. Med. 2012, 693717. doi: 10.1155/2012/693717
Hentges, K. E., Sirry, B., Gingeras, A. C., Sarbassov, D., Sonenberg, N., Sabatini, D., et al. (2001). FRAP/mTOR is required for proliferation and patterning during embryonic development in the mouse. Proc. Natl. Acad. Sci. U.S.A. 98, 13796–13801. doi: 10.1073/pnas.241184198
Hubner, A., Barrett, T., Flavell, R. A., and Davis, R. J. (2008). Multisite phosphorylation regulates Bim stability and apoptotic activity. Mol. Cell 30, 415–425. doi: 10.1016/j.molcel.2008.03.025
Inoki, K., Li, Y., Zhu, T., Wu, J., and Guan, K. L. (2002). TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 4, 648–657. doi: 10.1038/ncb839
Jaworski, J., Spangler, S., Seeburg, D. P., Hoogenraad, C. C., and Sheng, M. (2005). Control of dendritic arborization by the phosphoinositide-3'-kinase-Akt-mammalian target of rapamycin pathway. J. Neurosci. 25, 11300–11312. doi: 10.1523/JNEUROSCI.2270-05.2005
Jia, G., Cheng, G., Gangahar, D. M., and Agrawal, D. K. (2006). Insulin-like growth factor-1 and TNF-alpha regulate autophagy through c-jun N-terminal kinase and Akt pathways in human atherosclerotic vascular smooth cells. Immunol. Cell Biol. 84, 448–454. doi: 10.1111/j.1440-1711.2006.01454.x
Junn, E., Lee, K. W., Jeong, B. S., Chan, T. W., Im, J. Y., and Mouradian, M. M. (2009). Repression of alpha-synuclein expression and toxicity by microRNA-7. Proc. Natl. Acad. Sci. U.S.A. 106, 13052–13057. doi: 10.1073/pnas.0906277106
Junyent, F., Alvira, D., Yeste-Velasco, M., de la Torre, A. V., Beas-Zarate, C., Sureda, F. X., et al. (2010). Prosurvival role of JAK/STAT and Akt signaling pathways in MPP+-induced apoptosis in neurons. Neurochem. Int. 57, 774–782. doi: 10.1016/j.neuint.2010.08.015
Karunakaran, S., Saeed, U., Mishra, M., Valli, R. K., Joshi, S. D., Meka, D. P., et al. (2008). Selective activation of p38 mitogen-activated protein kinase in dopaminergic neurons of substantia nigra leads to nuclear translocation of p53 in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated mice. J. Neurosci. 28, 12500–12509. doi: 10.1523/JNEUROSCI.4511-08.2008
Kefas, B., Godlewski, J., Comeau, L., Li, Y., Abounader, R., Hawkinson, M., et al. (2008). microRNA-7 inhibits the epidermal growth factor receptor and the Akt pathway and is down-regulated in glioblastoma. Cancer Res. 68, 3566–3572. doi: 10.1158/0008-5472.CAN-07-6639
Kim, W. G., Mohney, R. P., Wilson, B., Jeohn, G. H., Liu, B., and Hong, J. S. (2000). Regional difference in susceptibility to lipopolysaccharide-induced neurotoxicity in the rat brain: role of microglia. J. Neurosci. 20, 6309–6316.
Kordower, J. H., and Bjorklund, A. (2013). Trophic factor gene therapy for Parkinson's disease. Mov. Disord. 28, 96–109. doi: 10.1002/mds.25344
Krutzfeldt, J., Rajewsky, N., Braich, R., Rajeev, K. G., Tuschl, T., Manoharan, M., et al. (2005). Silencing of microRNAs in vivo with antagomirs. Nature 438, 685–689. doi: 10.1038/nature04303
Kumar, V., Zhang, M. X., Swank, M. W., Kunz, J., and Wu, G. Y. (2005). Regulation of dendritic morphogenesis by Ras-PI3K-Akt-mTOR and Ras-MAPK signaling pathways. J. Neurosci. 25, 11288–11299. doi: 10.1523/JNEUROSCI.2284-05.2005
Langston, J. W., Ballard, P., Tetrud, J. W., and Irwin, I. (1983). Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 219, 979–980. doi: 10.1126/science.6823561
Langston, J. W., Irwin, I., Langston, E. B., and Forno, L. S. (1984). 1-Methyl-4-phenylpyridinium ion (MPP+): identification of a metabolite of MPTP, a toxin selective to the substantia nigra. Neurosci. Lett. 48, 87–92. doi: 10.1016/0304-3940(84)90293-3
Laplante, M., and Sabatini, D. M. (2013). Regulation of mTORC1 and its impact on gene expression at a glance. J. Cell Sci. 126, 1713–1719. doi: 10.1242/jcs.125773
Lawson, L. J., Perry, V. H., Dri, P., and Gordon, S. (1990). Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 39, 151–170. doi: 10.1016/0306-4522(90)90229-W
Li, Y. H., Werner, H., and Puschel, A. W. (2008). Rheb and mTOR regulate neuronal polarity through Rap1B. J. Biol. Chem. 283, 33784–33792. doi: 10.1074/jbc.M802431200
Liang, Y., Ridzon, D., Wong, L., and Chen, C. (2007). Characterization of microRNA expression profiles in normal human tissues. BMC Genomics 8:166. doi: 10.1186/1471-2164-8-166
Lindow, M., and Kauppinen, S. (2012). Discovering the first microRNA-targeted drug. J. Cell Biol. 199, 407–412. doi: 10.1083/jcb.201208082
Long, J. M., Ray, B., and Lahiri, D. K. (2012). MicroRNA-153 physiologically inhibits expression of amyloid-beta precursor protein in cultured human fetal brain cells and is dysregulated in a subset of Alzheimer disease patients. J. Biol. Chem. 287, 31298–31310. doi: 10.1074/jbc.M112.366336
Malagelada, C., Ryu, E. J., Biswas, S. C., Jackson-Lewis, V., and Greene, L. A. (2006). RTP801 is elevated in Parkinson brain substantia nigral neurons and mediates death in cellular models of Parkinson's disease by a mechanism involving mammalian target of rapamycin inactivation. J. Neurosci. 26, 9996–10005. doi: 10.1523/JNEUROSCI.3292-06.2006
Manning, B. D., Tee, A. R., Logsdon, M. N., Blenis, J., and Cantley, L. C. (2002). Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol. Cell 10, 151–162. doi: 10.1016/S1097-2765(02)00568-3
Mizuno, Y., Sone, N., and Saitoh, T. (1987). Effects of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine and 1-methyl-4-phenylpyridinium ion on activities of the enzymes in the electron transport system in mouse brain. J. Neurochem. 48, 1787–1793. doi: 10.1111/j.1471-4159.1987.tb05737.x
Morfini, G. A., You, Y. M., Pollema, S. L., Kaminska, A., Liu, K., Yoshioka, K., et al. (2009). Pathogenic huntingtin inhibits fast axonal transport by activating JNK3 and phosphorylating kinesin. Nat. Neurosci. 12, 864–871. doi: 10.1038/nn.2346
Mouradian, M. M. (2012). MicroRNAs in Parkinson's disease. Neurobiol. Dis. 46, 279–284. doi: 10.1016/j.nbd.2011.12.046
Mythri, R. B., Harish, G., and Bharath, M. M. (2012). Therapeutic potential of natural products in Parkinson's disease. Recent. Pat. Endocr. Metab. Immune Drug Discov. 6, 181–200. doi: 10.2174/187221412802481793
Nakaso, K., Ito, S., and Nakashima, K. (2008). Caffeine activates the PI3K/Akt pathway and prevents apoptotic cell death in a Parkinson's disease model of SH-SY5Y cells. Neurosci. Lett. 432, 146–150. doi: 10.1016/j.neulet.2007.12.034
Nicklas, W. J., Vyas, I., and Heikkila, R. E. (1985). Inhibition of NADH-linked oxidation in brain mitochondria by 1-methyl-4-phenyl-pyridine, a metabolite of the neurotoxin, 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine. Life Sci. 36, 2503–2508. doi: 10.1016/0024-3205(85)90146-8
Obeso, J. A., Rodriguez-Oroz, M. C., Goetz, C. G., Marin, C., Kordower, J. H., Rodriguez, M., et al. (2010). Missing pieces in the Parkinson's disease puzzle. Nat. Med. 16, 653–661. doi: 10.1038/nm.2165
Park, K. K., Liu, K., Hu, Y., Smith, P. D., Wang, C., Cai, B., et al. (2008). Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science 322, 963–966. doi: 10.1126/science.1161566
Piao, Y., Kim, H. G., Oh, M. S., and Pak, Y. K. (2012). Overexpression of TFAM, NRF-1 and myr-AKT protects the MPP(+)-induced mitochondrial dysfunctions in neuronal cells. Biochim. Biophys. Acta 1820, 577–585. doi: 10.1016/j.bbagen.2011.08.007
Podkowa, M., Zhao, X., Chow, C. W., Coffey, E. T., Davis, R. J., and Attisano, L. (2010). Microtubule stabilization by bone morphogenetic protein receptor-mediated scaffolding of c-Jun N-terminal kinase promotes dendrite formation. Mol. Cell Biol. 30, 2241–2250. doi: 10.1128/MCB.01166-09
Reinhard, J. F. Jr. Daniels, A. J., and Painter, G. R. (1990). Carrier-independent entry of 1-methyl-4-phenylpyridinium (MPP+) into adrenal chromaffin cells as a consequence of charge delocalization. Biochem. Biophys. Res. Commun. 168, 1143–1148. doi: 10.1016/0006-291X(90)91148-L
Reinhardt, P., Schmid, B., Burbulla, L. F., Schondorf, D. C., Wagner, L., Glatza, M., et al. (2013). Genetic correction of a LRRK2 mutation in human iPSCs links parkinsonian neurodegeneration to ERK-dependent changes in gene expression. Cell Stem Cell 12, 354–367. doi: 10.1016/j.stem.2013.01.008
Rodnitzky, R. L. (2012). Upcoming treatments in Parkinson's disease, including gene therapy. Parkinsonism Relat. Disord. 18(Suppl. 1), S37–S40. doi: 10.1016/S1353-8020(11)70014-1
Rodriguez-Blanco, J., Martin, V., Garcia-Santos, G., Herrera, F., Casado-Zapico, S., Antolin, I., et al. (2012). Cooperative action of JNK and AKT/mTOR in 1-methyl-4-phenylpyridinium-induced autophagy of neuronal PC12 cells. J. Neurosci. Res. 90, 1850–1860. doi: 10.1002/jnr.23066
Rosner, M., and Hengstschlager, M. (2008). Cytoplasmic and nuclear distribution of the protein complexes mTORC1 and mTORC2: rapamycin triggers dephosphorylation and delocalization of the mTORC2 components rictor and sin1. Hum. Mol. Genet. 17, 2934–2948. doi: 10.1093/hmg/ddn192
Runden, E., Seglen, P. O., Haug, F. M., Ottersen, O. P., Wieloch, T., Shamloo, M., et al. (1998). Regional selective neuronal degeneration after protein phosphatase inhibition in hippocampal slice cultures: evidence for a MAP kinase-dependent mechanism. J. Neurosci. 18, 7296–7305.
Russ, H., Staust, K., Martel, F., Gliese, M., and Schomig, E. (1996). The extraneuronal transporter for monoamine transmitters exists in cells derived from human central nervous system glia. Eur. J. Neurosci. 8, 1256–1264. doi: 10.1111/j.1460-9568.1996.tb01294.x
Sanchez, N., Gallagher, M., Lao, N., Gallagher, C., Clarke, C., Doolan, P., et al. (2013). MiR-7 triggers cell cycle arrest at the G1/S transition by targeting multiple genes including Skp2 and Psme3. PLoS ONE 8:e65671. doi: 10.1371/journal.pone.0065671
Santos, C. M. (2012). New agents promote neuroprotection in Parkinson's disease models. CNS Neurol. Disord. Drug Targets 11, 410–418. doi: 10.2174/187152712800792820
Sarbassov, D. D., Ali, S. M., Sengupta, S., Sheen, J. H., Hsu, P. P., Bagley, A. F., et al. (2006). Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 22, 159–168. doi: 10.1016/j.molcel.2006.03.029
Sarbassov, D. D., Guertin, D. A., Ali, S. M., and Sabatini, D. M. (2005). Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098–1101. doi: 10.1126/science.1106148
Satoh, T., Nakatsuka, D., Watanabe, Y., Nagata, I., Kikuchi, H., and Namura, S. (2000). Neuroprotection by MAPK/ERK kinase inhibition with U0126 against oxidative stress in a mouse neuronal cell line and rat primary cultured cortical neurons. Neurosci. Lett. 288, 163–166. doi: 10.1016/S0304-3940(00)01229-5
Seidl, S. E., and Potashkin, J. A. (2011). The promise of neuroprotective agents in Parkinson's disease. Front. Neurol. 2:68. doi: 10.3389/fneur.2011.00068
Song, L., Duan, P., Guo, P., Li, D., Li, S., Xu, Y., et al. (2012). Downregulation of miR-223 and miR-153 mediates mechanical stretch-stimulated proliferation of venous smooth muscle cells via activation of the insulin-like growth factor-1 receptor. Arch Biochem. Biophys. 528, 204–211. doi: 10.1016/j.abb.2012.08.015
Stanciu, M., Wang, Y., Kentor, R., Burke, N., Watkins, S., Kress, G., et al. (2000). Persistent activation of ERK contributes to glutamate-induced oxidative toxicity in a neuronal cell line and primary cortical neuron cultures. J. Biol. Chem. 275, 12200–12206. doi: 10.1074/jbc.275.16.12200
Sun, D. S., and Chang, H. H. (2003). Differential regulation of JNK in caspase-3-mediated apoptosis of MPP(+)-treated primary cortical neurons. Cell Biol. Int. 27, 769–777. doi: 10.1016/S1065-6995(03)00165-3
Swiech, L., Perycz, M., Malik, A., and Jaworski, J. (2008). Role of mTOR in physiology and pathology of the nervous system. Biochim. Biophys. Acta 1784, 116–132. doi: 10.1016/j.bbapap.2007.08.015
Takei, N., Kawamura, M., Ishizuka, Y., Kakiya, N., Inamura, N., Namba, H., et al. (2009). Brain-derived neurotrophic factor enhances the basal rate of protein synthesis by increasing active eukaryotic elongation factor 2 levels and promoting translation elongation in cortical neurons. J. Biol. Chem. 284, 26340–26348. doi: 10.1074/jbc.M109.023010
Tararuk, T., Ostman, N., Li, W., Bjorkblom, B., Padzik, A., Zdrojewska, J., et al. (2006). JNK1 phosphorylation of SCG10 determines microtubule dynamics and axodendritic length. J. Cell Biol. 173, 265–277. doi: 10.1083/jcb.200511055
Tavazoie, S. F., Alvarez, V. A., Ridenour, D. A., Kwiatkowski, D. J., and Sabatini, B. L. (2005). Regulation of neuronal morphology and function by the tumor suppressors Tsc1 and Tsc2. Nat. Neurosci. 8, 1727–1734. doi: 10.1038/nn1566
Thomas, G. M., Lin, D. T., Nuriya, M., and Huganir, R. L. (2008a). Rapid and bi-directional regulation of AMPA receptor phosphorylation and trafficking by JNK. EMBO J. 27, 361–372. doi: 10.1038/sj.emboj.7601969
Thomas, T., Timmer, M., Cesnulevicius, K., Hitti, E., Kotlyarov, A., and Gaestel, M. (2008b). MAPKAP kinase 2-deficiency prevents neurons from cell death by reducing neuroinflammation–relevance in a mouse model of Parkinson's disease. J. Neurochem. 105, 2039–2052. doi: 10.1111/j.1471-4159.2008.05310.x
Timmons, S., Coakley, M. F., Moloney, A. M., and O'Neill, C. (2009). Akt signal transduction dysfunction in Parkinson's disease. Neurosci. Lett. 467, 30–35. doi: 10.1016/j.neulet.2009.09.055
Trojanowski, J. Q., Goedert, M., Iwatsubo, T., and Lee, V. M. (1998). Fatal attractions: abnormal protein aggregation and neuron death in Parkinson's disease and Lewy body dementia. Cell Death Differ. 5, 832–837. doi: 10.1038/sj.cdd.4400432
Van der Haar, E., Lee, S. I., Bandhakavi, S., Griffin, T. J., and Kim, D. H. (2007). Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat. Cell Biol. 9, 316–323. doi: 10.1038/ncb1547
Verma, P., Chierzi, S., Codd, A. M., Campbell, D. S., Meyer, R. L., Holt, C. E., et al. (2005). Axonal protein synthesis and degradation are necessary for efficient growth cone regeneration. J. Neurosci. 25, 331–342. doi: 10.1523/JNEUROSCI.3073-04.2005
Wang, G., Pan, J., and Chen, S. D. (2012). Kinases and kinase signaling pathways: potential therapeutic targets in Parkinson's disease. Prog. Neurobiol. 98, 207–221. doi: 10.1016/j.pneurobio.2012.06.003
Wang, L., Yang, H. J., Xia, Y. Y., and Feng, Z. W. (2010). Insulin-like growth factor 1 protects human neuroblastoma cells SH-EP1 against MPP+-induced apoptosis by AKT/GSK-3beta/JNK signaling. Apoptosis 15, 1470–1479. doi: 10.1007/s10495-010-0547-z
Wang, Y., Liu, J., Liu, C., Naji, A., and Stoffers, D. A. (2013). MicroRNA-7 regulates the mTOR pathway and proliferation in adult pancreatic beta-cells. Diabetes 62, 887–895. doi: 10.2337/db12-0451
Wienholds, E., and Plasterk, R. H. (2005). MicroRNA function in animal development. FEBS Lett. 579, 5911–5922. doi: 10.1016/j.febslet.2005.07.070
Willis, S. N., Fletcher, J. I., Kaufmann, T., van Delft, M. F., Chen, L., Czabotar, P. E., et al. (2007). Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science 315, 856–859. doi: 10.1126/science.1133289
Wu, D. C., Teismann, P., Tieu, K., Vila, M., Jackson-Lewis, V., Ischiropoulos, H., et al. (2003). NADPH oxidase mediates oxidative stress in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson's disease. Proc. Natl. Acad. Sci. U.S.A. 100, 6145–6150. doi: 10.1073/pnas.0937239100
Wu, X., Reiter, C. E., Antonetti, D. A., Kimball, S. R., Jefferson, L. S., and Gardner, T. W. (2004). Insulin promotes rat retinal neuronal cell survival in a p70S6K-dependent manner. J. Biol. Chem. 279, 9167–9175. doi: 10.1074/jbc.M312397200
Wu, Z., He, B., He, J., and Mao, X. (2013). Upregulation of miR-153 promotes cell proliferation via downregulation of the PTEN tumor suppressor gene in human prostate cancer. Prostate 73, 596–604. doi: 10.1002/pros.22600
Xu, P., Das, M., Reilly, J., and Davis, R. J. (2011). JNK regulates FoxO-dependent autophagy in neurons. Genes Dev. 25, 310–322. doi: 10.1101/gad.1984311
Yuan, Y., Sun, J., Zhao, M., Hu, J., Wang, X., Du, G., et al. (2010). Overexpression of alpha-synuclein down-regulates BDNF expression. Cell. Mol. Neurobiol. 30, 939–946. doi: 10.1007/s10571-010-9523-y
Zecca, L., Youdim, M. B., Riederer, P., Connor, J. R., and Crichton, R. R. (2004). Iron, brain ageing and neurodegenerative disorders. Nat. Rev. Neurosci. 5, 863–873. doi: 10.1038/nrn1537
Zhang, K., Ma, Z., Wang, J., Xie, A., and Xie, J. (2011). Myricetin attenuated MPP(+)-induced cytotoxicity by anti-oxidation and inhibition of MKK4 and JNK activation in MES23.5 cells. Neuropharmacology 61, 329–335. doi: 10.1016/j.neuropharm.2011.04.021
Zhu, J. H., Guo, F., Shelburne, J., Watkins, S., and Chu, C. T. (2003). Localization of phosphorylated ERK/MAP kinases to mitochondria and autophagosomes in Lewy body diseases. Brain Pathol. 13, 473–481. doi: 10.1111/j.1750-3639.2003.tb00478.x
Zhu, J. H., Gusdon, A. M., Cimen, H., van Houten, B., Koc, E., and Chu, C. T. (2012). Impaired mitochondrial biogenesis contributes to depletion of functional mitochondria in chronic MPP+ toxicity: dual roles for ERK1/2. Cell Death Dis. 3, e312. doi: 10.1038/cddis.2012.46
Zhu, J. H., Horbinski, C., Guo, F., Watkins, S., Uchiyama, Y., and Chu, C. T. (2007). Regulation of autophagy by extracellular signal-regulated protein kinases during 1-methyl-4-phenylpyridinium-induced cell death. Am. J. Pathol. 170, 75–86. doi: 10.2353/ajpath.2007.060524
Zhu, J. H., Kulich, S. M., Oury, T. D., and Chu, C. T. (2002). Cytoplasmic aggregates of phosphorylated extracellular signal-regulated protein kinases in Lewy body diseases. Am. J. Pathol. 161, 2087–2098. doi: 10.1016/S0002-9440(10)64487-2
Zhu, Y., Pak, D., Qin, Y., McCormack, S. G., Kim, M. J., Baumgart, J. P., et al. (2005). Rap2-JNK removes synaptic AMPA receptors during depotentiation. Neuron 46, 905–916. doi: 10.1016/j.neuron.2005.04.037
Keywords: Parkinson's disease, miR-7, miR-153, MPP+, neuron, neuroprotection, rapamycin, mTOR
Citation: Fragkouli A and Doxakis E (2014) miR-7 and miR-153 protect neurons against MPP+-induced cell death via upregulation of mTOR pathway. Front. Cell. Neurosci. 8:182. doi: 10.3389/fncel.2014.00182
Received: 10 September 2013; Accepted: 13 June 2014;
Published online: 03 July 2014.
Edited by:
Tommaso Pizzorusso, University of Florence, ItalyReviewed by:
Riccardo Brambilla, San Raffaele Scientific Institute and University, ItalyVladimir L. Buchman, Cardiff University, UK
Copyright © 2014 Fragkouli and Doxakis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Epaminondas Doxakis, Lab of Molecular and Cellular Neuroscience, Center for Basic Research, Biomedical Research Foundation of the Academy of Athens, Soranou Efesiou 4, Athens 11527, Greece e-mail: edoxakis@bioacademy.gr