Dysregulation of Systemic Immunity in Aging and Dementia
 Jenny Lutshumba
Jenny Lutshumba Barbara S. Nikolajczyk
Barbara S. Nikolajczyk Adam D. Bachstetter
Adam D. Bachstetter- 1Spinal Cord and Brain Injury Research Center, University of Kentucky, Lexington, KY, United States
- 2Department of Neuroscience, University of Kentucky, Lexington, KY, United States
- 3Sanders-Brown Center on Aging, University of Kentucky, Lexington, KY, United States
- 4Department of Pharmacology and Nutritional Science, University of Kentucky, Lexington, KY, United States
- 5Barnstable Brown Diabetes and Obesity Center, University of Kentucky, Lexington, KY, United States
Neuroinflammation and the tissue-resident innate immune cells, the microglia, respond and contribute to neurodegenerative pathology. Although microglia have been the focus of work linking neuroinflammation and associated dementias like Alzheimer’s Disease, the inflammatory milieu of brain is a conglomerate of cross-talk amongst microglia, systemic immune cells and soluble mediators like cytokines. Age-related changes in the inflammatory profile at the levels of both the brain and periphery are largely orchestrated by immune system cells. Strong evidence indicates that both innate and adaptive immune cells, the latter including T cells and B cells, contribute to chronic neuroinflammation and thus dementia. Neurodegenerative hallmarks coupled with more traditional immune system stimuli like infection or injury likely combine to trigger and maintain persistent microglial and thus brain inflammation. This review summarizes age-related changes in immune cell function, with special emphasis on lymphocytes as a source of inflammation, and discusses how such changes may potentiate both systemic and central nervous system inflammation to culminate in dementia. We recap the understudied area of AD-associated changes in systemic lymphocytes in greater detail to provide a unifying perspective of inflammation-fueled dementia, with an eye toward evidence of two-way communication between the brain parenchyma and blood immune cells. We focused our review on human subjects studies, adding key data from animal models as relevant.
Introduction
Dementia is a disease of the mind, but the whole body contributes to brain demise. The corollary is also likely true: the degeneration of the brain likely contributes to the demise of the body. Chronological age is the proverbial thumb on the scale, weighing down the body and the brain until their inevitable end.
As an umbrella term, dementia defines a group of disorders associated with the declining inability to think and remember (cognitive abilities) and negatively affects mood and emotion. Dementia robs a person of themself and impairs their ability to function independently. Alzheimer’s Disease (AD) is the most well-known and well-studied type of dementia, but it is only one of aging’s many neurodegenerative diseases (Kovacs, 2015, 2020; Neltner et al., 2016; Nelson et al., 2019; Karanth et al., 2020; McKee, 2020).
What has become apparent over the last couple of decades is that a pure AD-type of dementia – that is, one associated with only amyloid plaques and neurofibrillary tangles – is rare (Jellinger, 2020). It is true that AD pathological lesions are common and begin to develop even in middle age (Nelson et al., 2011). Most people who die over the age of 70 and are cognitively healthy for their age will have at least some amyloid plaques or neurofibrillary tangles (Price and Morris, 1999). Most individuals with profound cognitive decline will have a high burden of neuropathological changes, including amyloid plaques and neurofibrillary tangles, vascular, synucleinopathy, and TDP-43 pathology (Karanth et al., 2020). We also know that the vascular contribution to cognitive impairment and dementia (VCID) is profound, and here too, we can see the connection between the body and the brain (Ighodaro et al., 2017; Blevins et al., 2020; Zlokovic et al., 2020).
During life, clinical symptoms and biomarkers guide the physician to classifying a person’s dementia as a vascular-type or AD-type dementia, for example. Yet, disease confirmation requires postmortem neuropathological evaluation (Montine et al., 2012; Abner et al., 2017). This distinction is essential for our review. Much of the research we will discuss will be probable AD-type dementia. Few, if any, of the studies will have confirmed that AD neuropathological changes caused dementia. Moreover, the understanding of dementia is ever-evolving.
Neuroinflammation is a prominent feature and a likely contributor to neurodegenerative disease pathogenesis. However, AD and related dementias differ from autoimmune diseases, such as multiple sclerosis, in which T and B lymphocytes invade the central nervous system (CNS) parenchyma. Instead, in AD and related dementias, the brain-resident macrophages, microglia, appear to be the primary component of the immune system acting locally in the CNS tissue (Prinz and Priller, 2017). In healthy brain, parenchyma, lymphocytes, and blood-derived monocytes are absent or rare (Prinz and Priller, 2017). In addition, the CNS barriers establish immune privilege by tightly regulating the movement of cells and fluid into and out of the tissue (Engelhardt et al., 2017). Thus, the interaction of the CNS with the systemic immune system is profoundly different than other tissues.
While the involvement of blood-derived immune cells in CNS tissue is limited in AD and related dementias, it is well-known that a systemic immune response affects the brain. For instance, an immune response to systemic infection can cause delirium without immune cells attacking and killing neurons (Perry et al., 2007). Experimentally, the response to systemic inflammation causes a behavioral response (i.e., lethargy, anhedonia, decreased concentration) termed “sickness behavior” (Hart, 1988; Perry et al., 2007). Multiple direct routes foster communication and functional impacts of a systemic immune response to the CNS. For instance, cytokines and other inflammatory mediators in the blood can enter the brain via the circumventricular organs, or cross the blood–brain barrier (BBB), leading to increased reactive microglia (Perry et al., 2007; Erickson et al., 2020).
In addition to microglia, the brain endothelium is a major cellular target of systemic inflammation. For instance, brain endothelial cells express high levels of the receptor for one of the major proinflammatory cytokines, interleukin-1 (IL-1) (Liu and Quan, 2018; Liu et al., 2019). IL-1 is elevated in AD patients’ brains (Griffin et al., 1989). The brain endothelial cells also alter the permeability and transport of substances across the BBB (Erickson et al., 2020). Systemic inflammation can alter neurovascular coupling (Iadecola, 2017) and increase the adhesion of neutrophils to the brain capillaries, blocking blood flow (Cruz Hernandez et al., 2019). At the extreme, systemic inflammation also causes hypoxic/ischemic changes, including small and larger hemorrhagic infarcts (Thakur et al., 2021). Thus, it is easy to envision how systemic inflammation could synergize and further exacerbate VCID-related pathology in the elderly.
In older individuals, multiple systemic inflammatory events often precede and are hypothesized to contribute to downward spikes in cognitive function (Perry et al., 2007). Thus, while delirium and sickness behavior are transient, there is compelling evidence that systemic immune responses and systemic inflammation have lasting effects on the brain, particularly in older fragile individuals. Evidence for this conjecture comes from aged animals given a systemic immune challenge such as bacterial lipopolysaccharides (LPS) that have prolonged sickness behavior, increased cognitive impairment, and increased reactive microglia (Norden et al., 2015).
Individuals with AD also have higher circulating cytokine amounts compared to healthy controls. For example, across multiple studies, serum/plasma individuals with AD had more IL-1β, IL-6, IL-18, and TNFα than cognitively healthy subjects (Swardfager et al., 2010; Su et al., 2019). Presumably, the circulating cytokines are elevated from some ill-defined baseline and are not generated to counter an active infection or injury, but instead suggest a heightened state of basal inflammation.
Genetics, environment, and systemic diseases such as diabetes are underlying causes of a “primed” state of inflammation. Aging is also a significant driver of inflammation. In addition, nearly every aspect of the immune system is affected by age, collectively termed immunosenescence (Nikolich-Zugich, 2018). Therefore, as diseases of aging, AD, and related dementias must also consider the impact of the aged immune system on the disease.
This review evaluates the evidence for changes in the systemic immune system with age and dementia. We believe changes in the immune system may contribute to dementia, particularly when the immune system responds to disease, infection, or injury. Our central focus is on the understudied systemic cellular arm of the immune system. Aging is the inflammatory background common across dementias; thus, our review starts with an outline of age-related changes in the systemic immune system. We will then describe the sparse studies of systemic immune cell differences in AD that contrast with the extensive understanding of age-related inflammation. As the number of AD-related studies of the systemic immune system is limited, we will describe those studies in greater detail. While animal models are instrumental in testing mechanisms and therapeutics, we have focused our review on studies in people, as these studies can provide the most remarkable insight into the natural history of dementia.
Immunesenescence Changes in People With Age
The progressive decline in immunity contributes to poor responsiveness to vaccines, increased incidences of infections, and decreased immunosurveillance of neoplasms amidst a chronic state of poorly defined inflammation. The decline of the immune system with age is not limited to one element due to interplay between the immune cells (both innate and adaptive), changes to microenvironment (including lymphoid and non-lymphoid tissues), and alterations in circulating inflammatory mediators (cytokines and chemokines, among other soluble factors) (Nikolich-Zugich, 2018). The immune system’s age-related changes are best described as dysregulated and not simplistically skewed pro- or anti-inflammatory as described in many excellent reviews of immunosenescence in general (High et al., 2012; Nikolich-Zugich, 2018; Velardi et al., 2020).
Innate Immune Cell Changes With Age
Innate immunity and macrophage/microglia have traditionally taken center stage in CNS disorders (Bachstetter et al., 2015; Greenhalgh et al., 2020; Madore et al., 2020; Shahidehpour et al., 2021). Therefore, we will begin our discussion of immunosenescence with the innate immune system with a focus on neutrophils, dendritic cells (DCs), and monocytes.
Neutrophils are the first responder and act locally by migrating (chemotaxis) to the site of tissue injury or pathogen. Once the neutrophils leave the circulation by extravasation, they play essential roles in phagocytosis and killing the invading microbes. Neutrophils are short-lived in circulation, with a half-life of 8–12 h that is offset by production of large numbers (1–2 × 1011 neutrophils per day) (Lord et al., 2001). With advanced age, the number of circulating neutrophils in the blood is stable, but they proliferate less in response to G-CSF (Chatta et al., 1993; Born et al., 1995; Wenisch et al., 2000). Neutrophils in older individuals (66–93 years old) are still able to increase in number (neutrophilia) in response to a bacterial infection to reach a normal range (Lord et al., 2001). The neutrophil’s microbicidal function decreases with age, and includes reduced chemotaxis, reduced phagocytosis, decreases in neutrophil extracellular traps, and a slower resolution of inflammation (Shaw et al., 2010; Montgomery and Shaw, 2015).
To the best of our knowledge, it is not known in people if neutrophilia, chemotaxis, and extravasation are maintained in older individuals in response to a brain injury, such as a cerebral microhemorrhages. In response to lung injury in mice, neutrophils migrate to the lung similarly in aged and young adult animals (Nomellini et al., 2008). The neutrophil response resolved in the young adult animals by 24-h after the lung injury, but in aged animals, neutrophil numbers at 24 hrs post-injury were indistinguishable from the 6-h post-injury peak. Aged neutrophil response was resolved by blocking CXCR2 dependent chemokine signaling (MIP2 and KC), which did not otherwise adversely affect wound recovery. These data support the hypothesis that delayed resolution of the neutrophilic response in the brain of aged individuals after an injury may exacerbate the secondary damage caused by the injury.
Monocytes are circulating leukocytes that originate in the bone marrow from a common myeloid progenitor cell. Monocytes account for around 5–10% of the circulating leukocytes in human blood (Guilliams et al., 2018). Monocytes and monocyte-derived macrophages are from a different cellular linage than most tissue-resident macrophages (i.e., microglia), which have an embryonic origin from yolk sac progenitors (Prinz et al., 2017). In humans, monocytes can be classified as classical HLA-DR+CD14+CD16–, intermediate HLA-DR+CD14+CD16+, and non-classical HLA-DR+CD14lowCD16+ (Cros et al., 2010; Hearps et al., 2012; Guilliams et al., 2018; Figure 1). Classical monocytes are about 85% of all monocytes and remain in circulation for around 1 day as they traffic to tissues, such as the intestine, to repopulate the tissue-resident macrophages. Alternatively, classical monocytes can differentiate into non-classical monocytes, which can live for 7 days in humans. However, non-classical monocytes might develop without first becoming a classical monocyte (Guilliams et al., 2018).
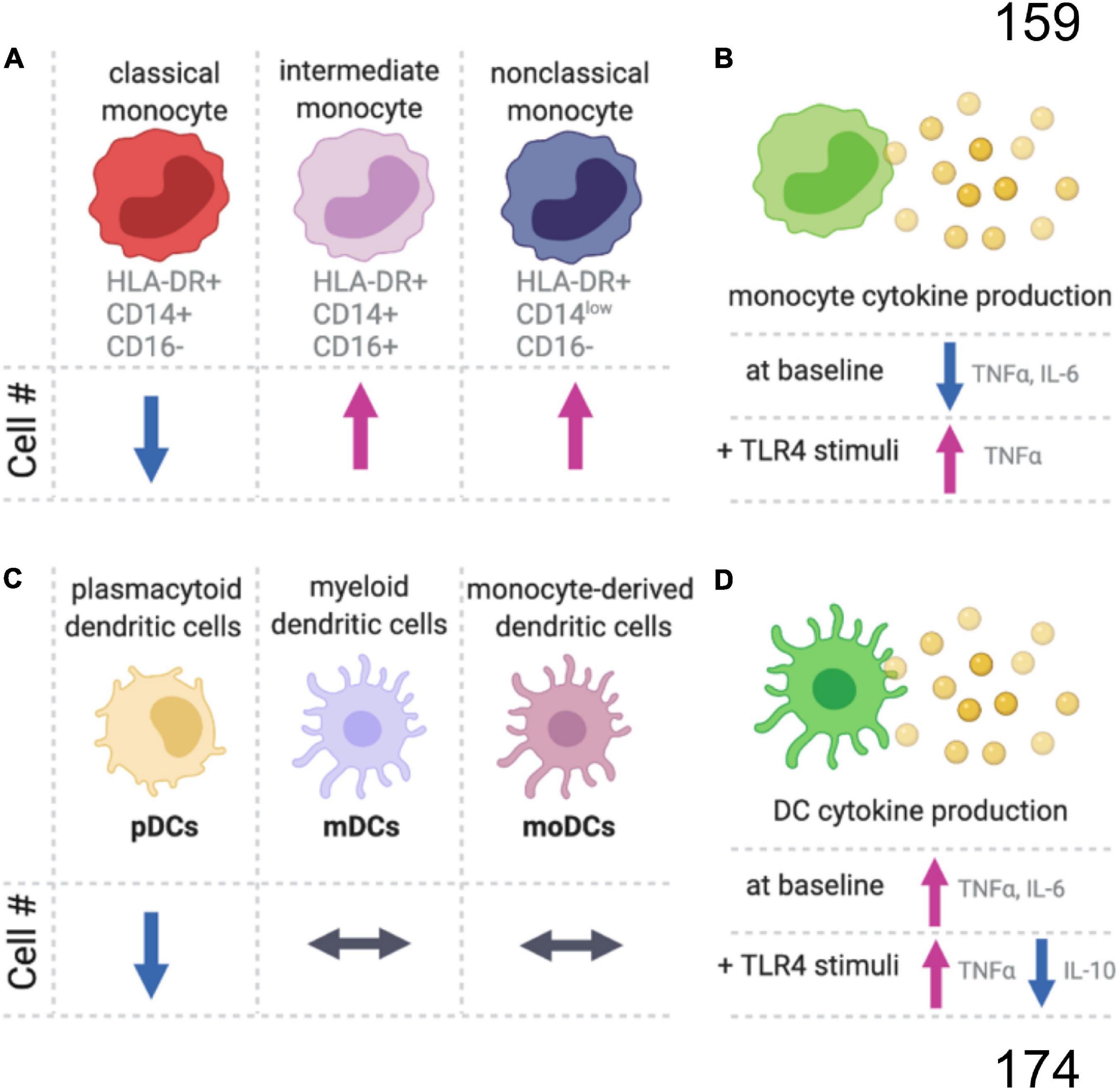
Figure 1. Summary of age-related changes in monocyte and dendritic cells. (A) In the blood of older aged people, there is a decrease in the proportion of classical monocytes, while the intermediate and non-classical monocyte populations are increasing compared to younger adults. (B) Monocytes also have an age-dependent decrease in proinflammatory cytokines at baseline, but with TLR4 stimulation (LPS) the aged monocytes produce more proinflammatory cytokines than monocytes from younger individuals. (C) There are age-related changes in subpopulations for dendritic cells (DC), and (D) the aged DCs produced more proinflammatory and less anti-inflammatory cytokines.
The replacement of tissue macrophages by classical monocytes is not believed to occur in the CNS, at least not in the healthy brain (Waisman et al., 2015; Herz et al., 2017). However, much of what we know about the turnover of tissue macrophages is from rodent studies. There is only limited understanding of microglia replacement in people by monocyte-derived macrophages and the influence of aging, injury, and disease on these processes in people is not well described. The non-classical monocytes have been found to have important functions in patrolling the vasculature, including the arterial, capillary, and venous compartments (Guilliams et al., 2018), and this could be a mechanism for how monocytes might sense changes to the cerebral vasculature as part of VCID.
The number of monocytes increases linearly with age and comparing the number of monocytes in 20- to 30-year-old individuals to the monocyte numbers in 80- to 90-year-old people, there is an approximate tripling of monocyte numbers with age (Della Bella et al., 2007; Alpert et al., 2019). The relative proportion of non-classical and intermediate monocytes also increases with age, while classical monocytes decrease or remain unchanged (Nyugen et al., 2010; Seidler et al., 2010; Hearps et al., 2012; Figure 1). Without stimulation, a decrease in TNFα and IL-6 positive monocytes is seen with age (Nyugen et al., 2010). Upon stimulation of the toll-like receptors (TLRs), intermediate CD11c+ monocytes from aged individuals produce less inflammatory cytokines (IL-6 and TNFα) in response to TLR1/2 ligands, with no difference between young and old monocytes for TLR2/6 heterodimer (LTA), TLR4 (LPS), TLR5 (flagellin), and TLR7/8 (poly(U)) ligands (van Duin et al., 2007). Whereas classical monocytes respond similarly between young and old individuals following TLR4 stimulation, aged intermediated and non-classical monocytes had a greater increase in TNFα than the respective young monocyte populations (Hearps et al., 2012). While the magnitude of the initial response to TLR stimulation may be unaffected by age, aged murine macrophages have an impaired ability to end inflammatory responses (Pattabiraman et al., 2017).
Dendritic cells are the major antigen-presenting cells and play a critical role in bridging innate and adaptive immune responses. DCs can be divided into plasmacytoid DCs (pDCs), myeloid DCs (mDCs), and monocyte-derived DCs (MoDCs) (Figure 1C). Circulating pDC frequencies decrease with age, while the numbers of circulating mDCs and MoDCs are age-independent (Agrawal et al., 2017). However, DC phenotypes change with age. Unstimulated DCs from aged individuals have higher secretion of proinflammatory cytokines that likely contribute to age-related inflammation, or inflammaging (Figure 1D). DCs stimulated with LPS produce higher concentrations of proinflammatory cytokines and lower concentrations of an important anti-inflammatory cytokine, IL-10. For an excellent comprehensive review of DCs changes with age, see Agrawal et al. (2017).
Adaptive Immune Cell Changes With Age
Aging polarizes the immune system in favor of the innate immune response with a profound decline in adaptive immunity (Figure 2). The hematopoietic stem cell (HSC) pool declines with age and is skewed toward the production of myeloid cells (Dykstra et al., 2011). T cells’ progressive decline with age continues with lymphopoiesis and the age-related decline of thymic lymphoid progenitors that in turn reduce T cell generation (Berent-Maoz et al., 2012). The thymus is stable in size with age in humans, but the microenvironment is altered with adipose tissue replacing healthy tissue that supports T cell development (Velardi et al., 2020). As the output of naïve T cells from the thymus slows with age, the body switches to a peripheral proliferation of T cells to maintain the pool of naïve cells in circulation (den Braber et al., 2012).
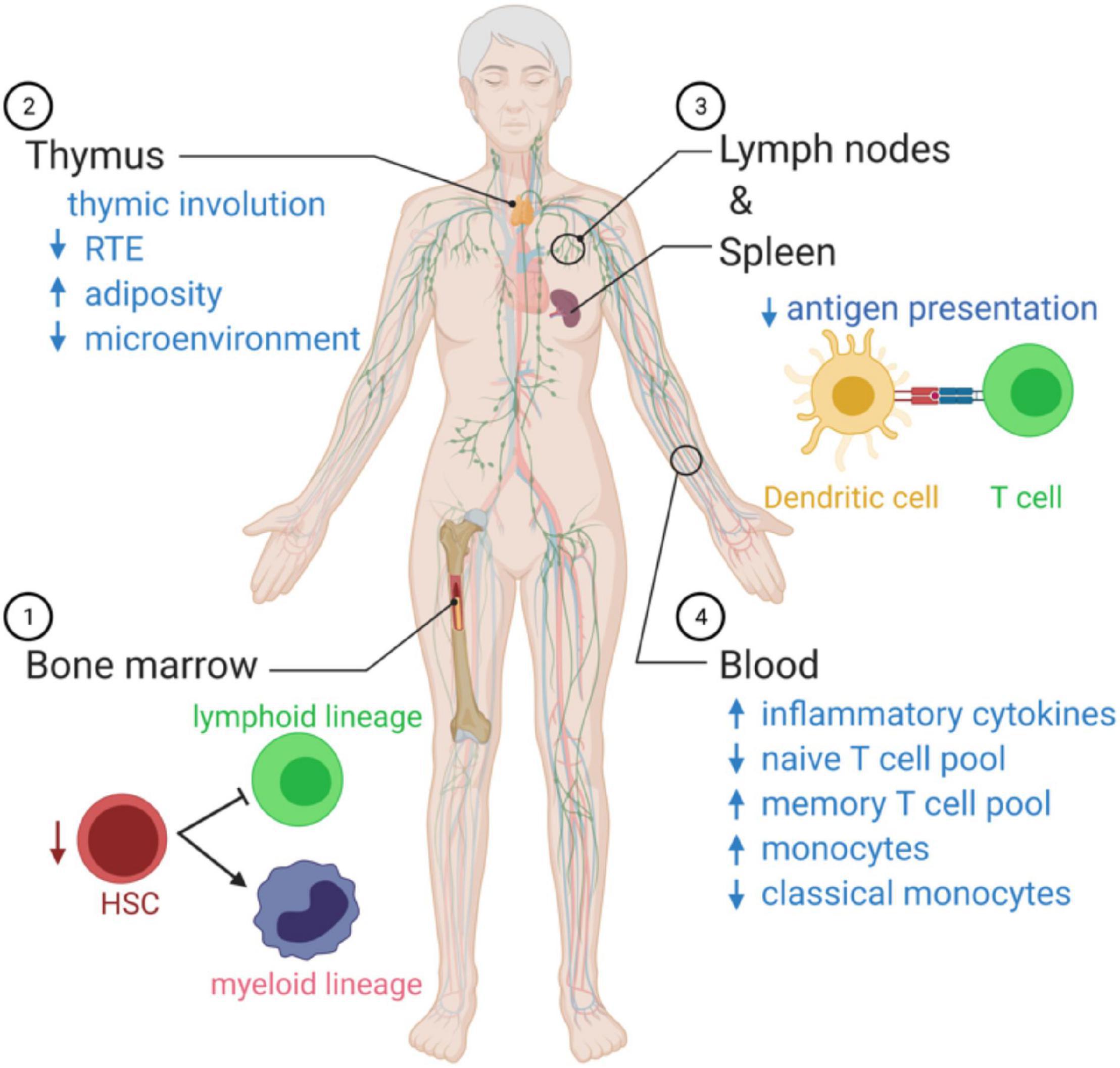
Figure 2. Summary of age-related changes in systemic immune function. (1) As we age, there is an imbalance in the production of innate immune cells and adaptive immune cells that favor cells of the myeloid lineage. (2) The microenvironment decreases with age, and there is a reduction in recent thymic emigrants (RTEs), reducing the naïve T cell poll in the circulation. (3) Antigen presentation by dendritic cells declines with age. (4) In the blood, the immunosenescence leads to a state of chronic inflammation, associated with elevated circulating cytokines, a decrease in naïve T cells able to respond to new pathogens, and monocytes over-produce inflammatory mediators and fail to resolve the inflammatory response.
Episomal DNA fragments are excised during T-cell receptor (TCR) gene rearrangement. These episomal DNA fragments, called TRECs, provide a direct estimate of de novo T cell generation, as they are not regenerated and are thereby diluted in dividing peripheral T cell (Kong et al., 1999; Hazenberg et al., 2000; Ribeiro and Perelson, 2007). By the age of 55 years old, it is estimated that only 5% of naïve T cells are produced in the thymus (Murray et al., 2003), and overall thymic output is decreased by more than 95% in 60-year-old people compared to 25-year-old people as measured by TREC (Naylor et al., 2005). Unlike mice, which produce most naïve T cells through thymic output, humans rely primarily (90% in young adults) on peripheral T cell proliferation (den Braber et al., 2012).
The decline in HSC polarization and thymic function with age is important following immunological insults such as infection and cancer when the immune system can be depleted in older individuals who cannot subsequently recover it (Velardi et al., 2020). Latent persistent infections are also part of the secondary changes leading to immunesenescence (Nikolich-Zugich, 2008).
Overall numbers of T cells in circulation are relatively unaffected by age, despite the decline in recent thymic output. However, there are changes in the distribution of different T cell subsets. The naïve T cells in older individuals have increased turnover, and virtual memory cells’ formation replaces the lost recent thymic emigrants. The virtual memory cells lose some of the naïve T properties, secrete Th1-type cytokines, and have decreased proliferation compared to naïve T cells (Nikolich-Zugich, 2014). Naïve CD8 T cells also decline, while naïve CD4 cells are more stable with age (Wertheimer et al., 2014). While the diversity of CD4 T cells is maintained through much of adulthood, a rapid decline in diversity was reported over the age of 70 years (Naylor et al., 2005). For instance, regulatory T cells (Tregs) and Th17 cells numbers decline with age (Vukmanovic-Stejic et al., 2011; Schmitt et al., 2013; Alpert et al., 2019). Moreover, there is a mismatch of cell number and cell function with age.
Our recent work compared combinatorial cytokine profiles generated by CD4+ T cells from lean, normoglycemic older (average age ∼60) and younger (average age ∼32) subjects and concluded that Th17 cytokines dominate at least part of the inflammatory landscape in older subjects (Figure 3). The age-related Th17 profile is reminiscent of a type 2 diabetes-associated Th17 profile (Ip et al., 2016) and raises the possibility that age-related overexuberance of Th17s foster age-related declines in metabolic control. T cells from older compared to younger subjects had defects in autophagy and mitochondrial bioenergetics that associate with redox imbalance, and thus mirror age-related changes in non-immune cell types. The importance of autophagy defects in age-related inflammation, or inflammaging, were highlighted in cause/effect studies that impaired the autophagy machinery in T cells from young subjects with siRNA. This manipulation disrupted redox balance and activated Th17 profile cytokines in young cells by activating the Th17 master regulator STAT3, which in turn bound IL-17A and F promoters. Surprisingly, mitophagy-targeting siRNA failed to activate the Th17 profile in CD4+ T cells from young subjects, consistent with the idea that non-mitochondrial autophagy plays an independent role in age-related inflammation. Overall, our data support a model in which age-related changes in mitochondrial bioenergetics and non-mitochondrial autophagy converge to promote inflammaging (Bharath et al., 2020). The relationship between the age-associated CD4+ T cell profile and cognitive decline with age remain unexplored.
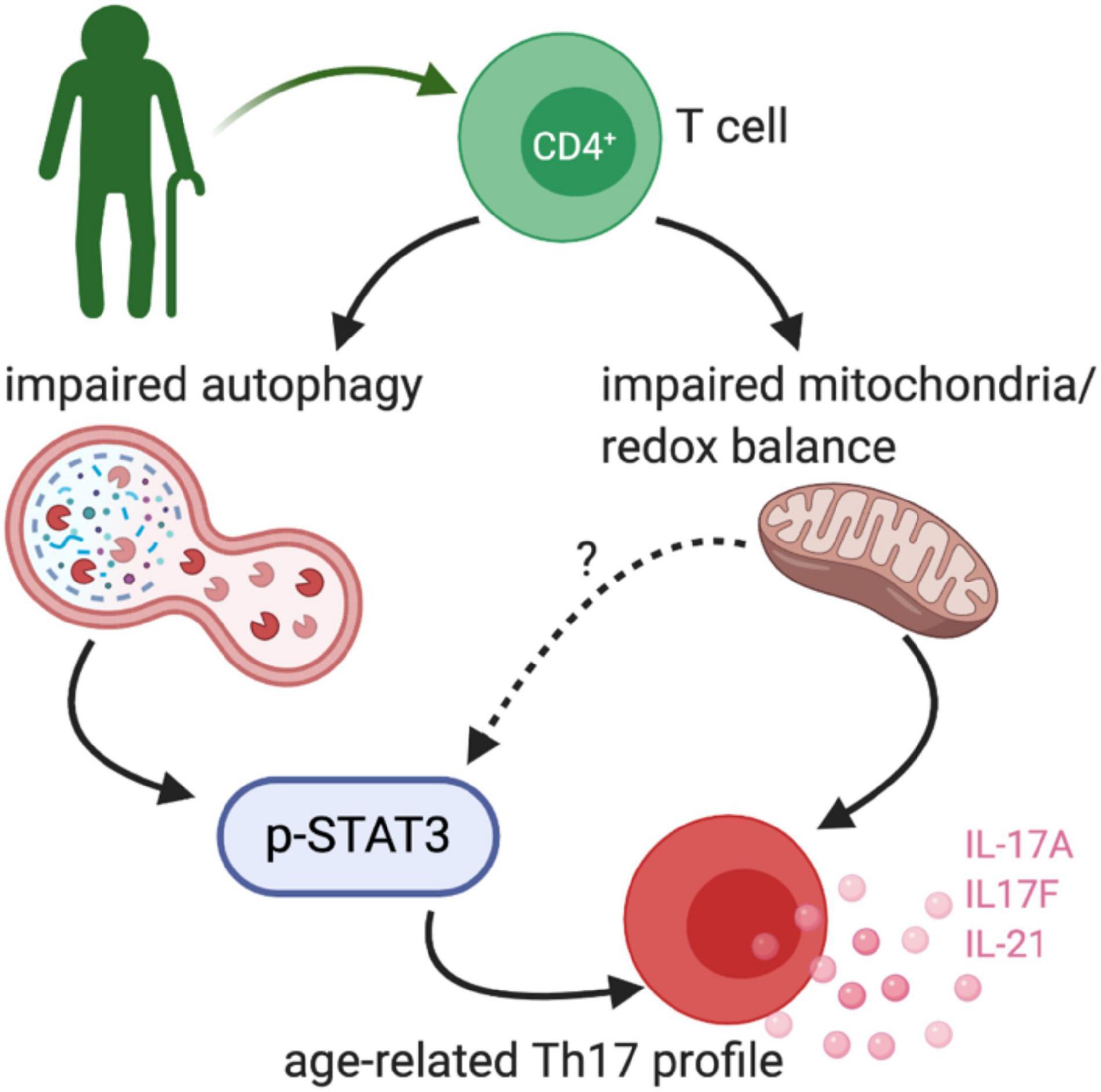
Figure 3. Age-related TH17 changes. Bharath et al. (2020) demonstrated that peripheral blood CD4+ cells from healthy older adults (∼60 years old) had an exaggerated TH17 profile. The TH17 signature was driven by impaired non-mitochondrial autophagy and dysfunctional mitochondria, at least in part via a STAT3 dependent mechanism.
Systemic Immune Changes Seen in People With Dementia
An ever-growing body of evidence shows interaction between the immune and nervous systems (Galea et al., 2007; Prinz and Priller, 2017). Studies dating back to the 1980s provided evidence that both the adaptive and innate immune cells are altered in people with dementia. This section describes changes in systemic immune cells highlighted by this work. The interested reader should also consider previous reviews on the topic (Rezai-Zadeh et al., 2009; Sommer et al., 2017) and reviews of systemic immune changes in animal models of neurodegenerative disease (Nevo et al., 2003; Cao and Zheng, 2018; Mayne et al., 2020).
In the 1980s, studies evaluated both changes in proportions of different immune cell populations (i.e., immunophenotyping) and how immune cells differed in response to a stimulus or mitogen (i.e., immune function). Over the subsequent 40 years, our understanding of the immune system has dramatically expanded. For example, we now have further divided T lymphocytes into smaller, specialized populations of cells using multi-color flow cytometry with parallel functional studies. Also, populations of cells such as “suppressor” T cells, which were dismissed in the late 1980s as artifacts, have now become identified as Tregs, arguably the most intensively studied CD4+ T cell subset in the last decade. We also know that there are many forms of dementia in the community, with a pure AD being relatively rare. So, when going back into the archives of older research, one must be aware of the limitations but also appreciate the strengths of these early studies.
Whole Blood Cell Counts in AD and Related Dementias
One of the primary immunological changes seen with age in humans is the immune system’s polarization in favor of the innate immune response with a profound decline in adaptive immunity. Are further changes seen in the balance between innate and adaptive immunity in people with dementia? van der Willik et al. (2019) addressed this question in a powerful but simple way in the Rotterdam Study – a large prospective population-based cohort study of older adults (Ikram et al., 2017; van der Willik et al., 2019). As part of the Rotterdam study, van der Willik et al. (2019) evaluated blood cell counts in 8313 participants. Longitudinal evaluation of the complete blood counts was done for multiple visits (every 3–5 years) during a 13-year period. The study excluded people with dementia at the start of the study. Participants were screened for dementia at baseline, and each visit using the Mini-Mental State Examination (MMSE) and the Geriatric Mental Schedule score. During the follow-up period, 664 of the cognitively healthy individuals developed dementia, with 82% being probable AD-type dementia and 5% probable vascular dementia (VaD).
To evaluate the balance of innate vs. adaptive immunity, they looked at ratios amongst blood granulocytes, platelets, and lymphocytes, which has recently been proposed to be a validated (albeit simple) measure of changes in immunity (Hu et al., 2014; Templeton et al., 2014a, b; Fest et al., 2018). Three predictive ratios were evaluated: (1) granulocytes-to-lymphocytes; (2) platelets-to-lymphocytes, and (3) a total systemic immune-inflammation index (integrated peripheral lymphocyte, neutrophil, and platelet indicator). van der Willik et al. (2019) found that altered balance between innate (granulocytes) and adaptive immunity (lymphocytes) was associated with an increased risk for developing all-cause dementia over those 13 years (median follow-up 8.6 years) (Figure 4). These results were consistent when correcting only for age and sex or adjusting the model by adding additional variables into the model (education, smoking status, body mass index, diabetes mellitus, history of stroke, and APOE4 carriers). The immune cell balance changes were informative, suggesting that dementia was exacerbating the normal age-related changes in the immune balance – more innate granulocytes and fewer adaptive immune cells. VaD showed greater hazard ratio changes for increased innate activity (granulocyte number, and granulocyte-to-lymphocyte ratio). VaD associated with lower indicators of adaptive immunity (lymphocyte number, and platelet-to-lymphocyte ratio) in comparison to all-cause dementia and AD. These results are in general agreement with four earlier studies that evaluated blood count changes in people with dementia, which found a decrease in lymphocyte number and increase in neutrophil number in the AD vs. the aged-matched control groups (Tollefson et al., 1989; Kalelioglu et al., 2017), and increased neutrophil-to-lymphocyte ratios (Kuyumcu et al., 2012; Rembach et al., 2014; Kalelioglu et al., 2017). In contrast, one study found no difference in lymphocytes between AD and control participants (Dysken et al., 1992). Differences in study design (cross-sectional), smaller sample size, age at the start of the study, and covariate adjustment should all be considered when comparing the results from earlier studies that disagree with van der Willik et al. (2019). Nevertheless, these studies provide compelling evidence for dementia-specific disruption of the immune system atop shifts that characterize healthy aging.
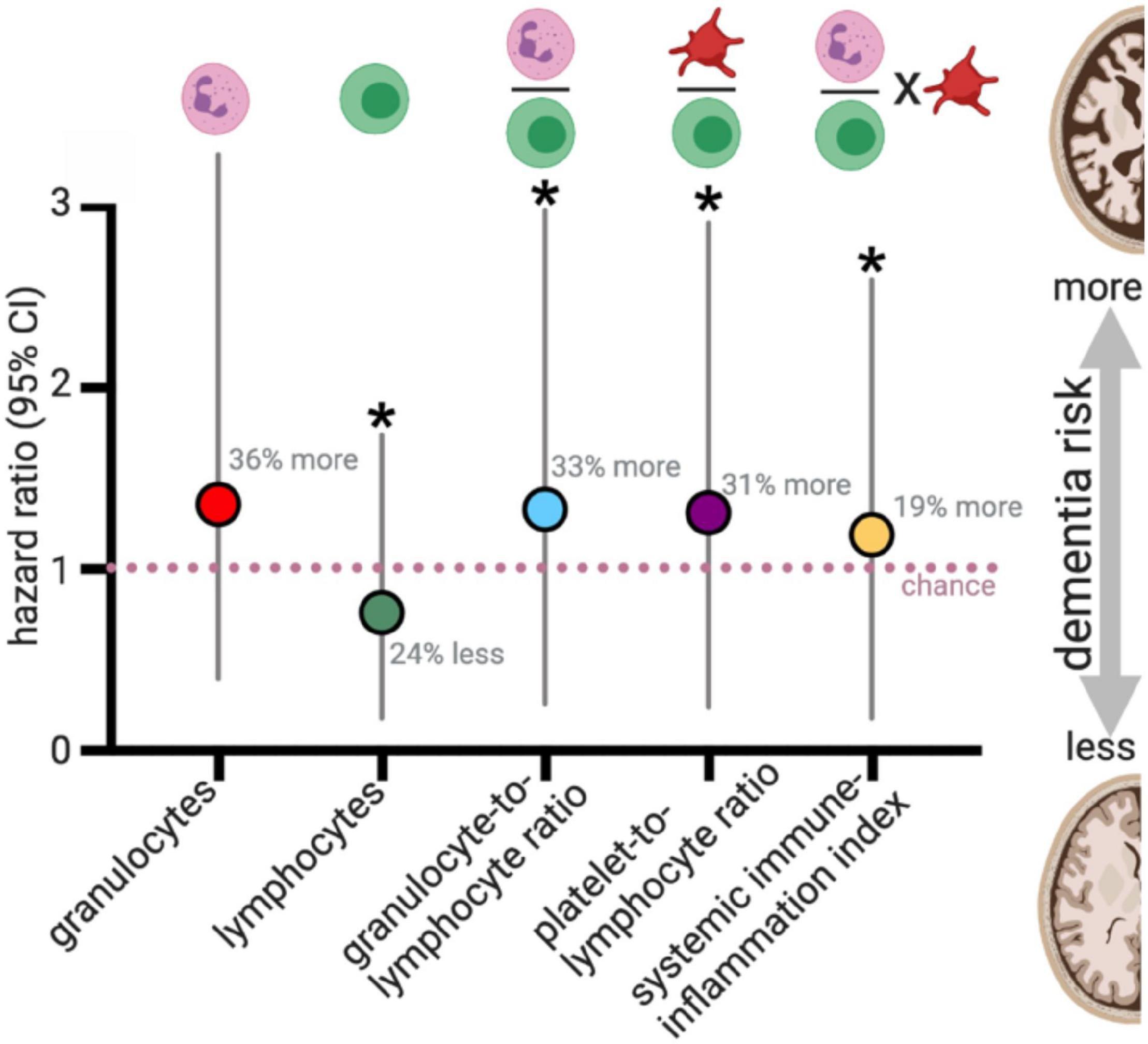
Figure 4. Blood cell counts and risk for all cause dementia. van der Willik et al. (2019) evaluated blood cell counts on 8313 participants who were dementia-free at the start of the study (mean age: 61 years old at the start). During a median follow-up of 8.6 years, 664 developed dementia. For those who did not have a stroke during the study (N = 8008), changes in blood cell counts, and the ratio of blood cell populations were statistically associated with dementia risk. Plotted results are from Table 3 of van der Willik et al. (2019). *p < 0.05.
T Cell Immune Phenotyping in AD and Related Dementias
Leffell et al. (1985) were among the earliest studies to complete immune profiling in people with AD. Their cross-sectional study found no change in total T cells, CD4, CD8, or the ratio of CD4:CD8 cells between the people with probable AD-type dementia and aged-matched controls. In their concluding paragraph they stated, “we believe that there is little, if any, cumulative evidence to support an immunological process in Alzheimer’s disease.” However, this statement needs context, as they, and many studies that followed, explored if AD was caused by an autoimmune attack on neurons similar to multiple sclerosis, or by infectious agents like prions in Creutzfeldt–Jakob disease.
In the past 25 years, changes in the major T cell populations have been evaluated across different stages of dementia progression, spanning from mild cognitive impairment (MCI) to severe AD (Table 1). Throughout the late 1980s and 1990s, a handful of studies evaluated the immune system in people with dementia (Singh et al., 1987; Antonaci et al., 1990; Licastro et al., 1990; Araga et al., 1991; Ikeda et al., 1991; Pirttila et al., 1992; Singh, 1994; Hu et al., 1995; Shalit et al., 1995; Trieb et al., 1996; Lombardi et al., 1999). The majority of studies found no change at any stage of disease progression for total T cells (CD3), or CD4 or CD8 T cells. Some of the studies found no statistical relationship between dementia and immunological functions. For instance, IL-2 was equally effective at inducing proliferation of lymphocytes in AD and control participants (Licastro et al., 1990). While a few studies found no change in CD4, CD8 or CD4:CD8 ratios (Ikeda et al., 1991; Singh, 1994), others found changes in these cell populations (Pirttila et al., 1992). The changes include a decrease in CD8 cells in probable AD dementia and an increase in CD8 cells in probable VaD both compared to age-matched controls (Pirttila et al., 1992; Hu et al., 1995; Shalit et al., 1995). Shalit et al. (1995) found that the severity of the neurodegenerative disease correlated with the immune cell changes including an increase in CD4s and a decrease in CD8s in severe dementia, despite no differences in CD4 or CD8 numbers in mild dementia, both compared to controls. Tregs were found to change by dementia status; however, future studies are needed to replicate these studies using state of the art markers for human Treg cells. A single recent report found that TH17 cells were increased in MCI, but by early AD the increase in TH17 cells was no longer apparent (Oberstein et al., 2018). It will be necessary to replicate these changes in future studies and evaluate these changes in a longitudinal instead of a cross-sectional study.
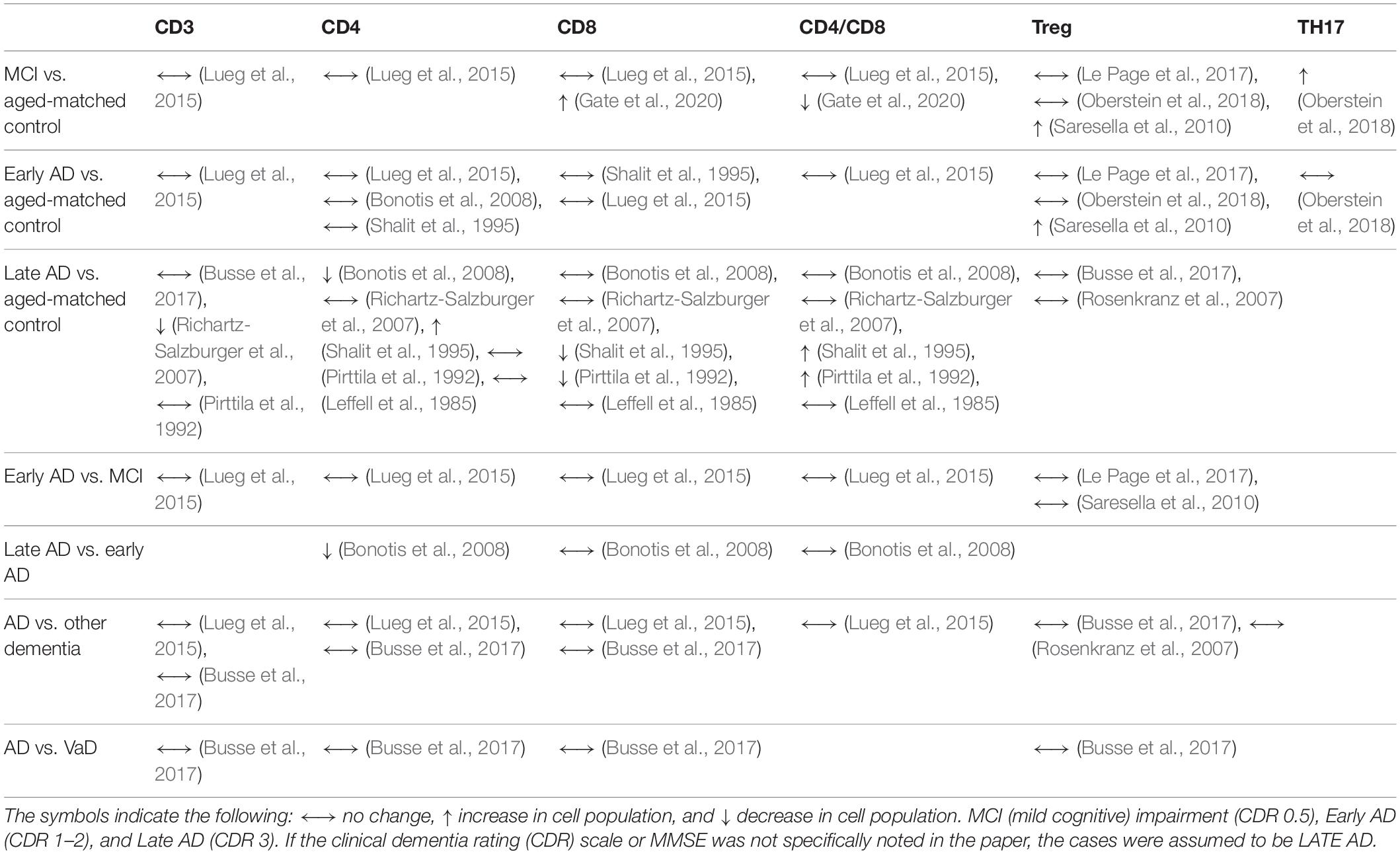
Table 1. Relationship between dementia status and T cell populations.
Two recent studies have evaluated naïve, central memory (TCM), effector memory (TEM), and terminally differentiated (TEMRA) CD4 and CD8 subsets (Busse et al., 2017; Gate et al., 2020; Figure 5). The four subsets of T cells were identified by the expression of lymph node homing molecule receptor, CCR7. Along with the expression pattern of the leukocyte common antigen (CD45) isoforms RA and RO. During the transition from naïve to TEM the expression of CCR7 is lost when the TCM are restimulated by antigen. When the TEM terminally differentiate, they stop expressing CD45RO and re-express CD45RA. It was found that the CD8+ TEMRA were increased in the blood of MCI/AD patients. The percentage of these CD8+ TEMRA cells were strongly correlated with worse cognitive function (Gate et al., 2020). In the AD patient’s cerebrospinal fluid, clonal expansion of the CD8+ TEMRA was found. While the authors identified TCRβ sequence with known specificity for the Herpesviridae Epstein–Barr nuclear antigen 3, they cautioned against suggesting that these finding provided evidence for a link between Epstein–Barr virus and AD, as it has infected up to 95% of American adults, and their caution is well founded.
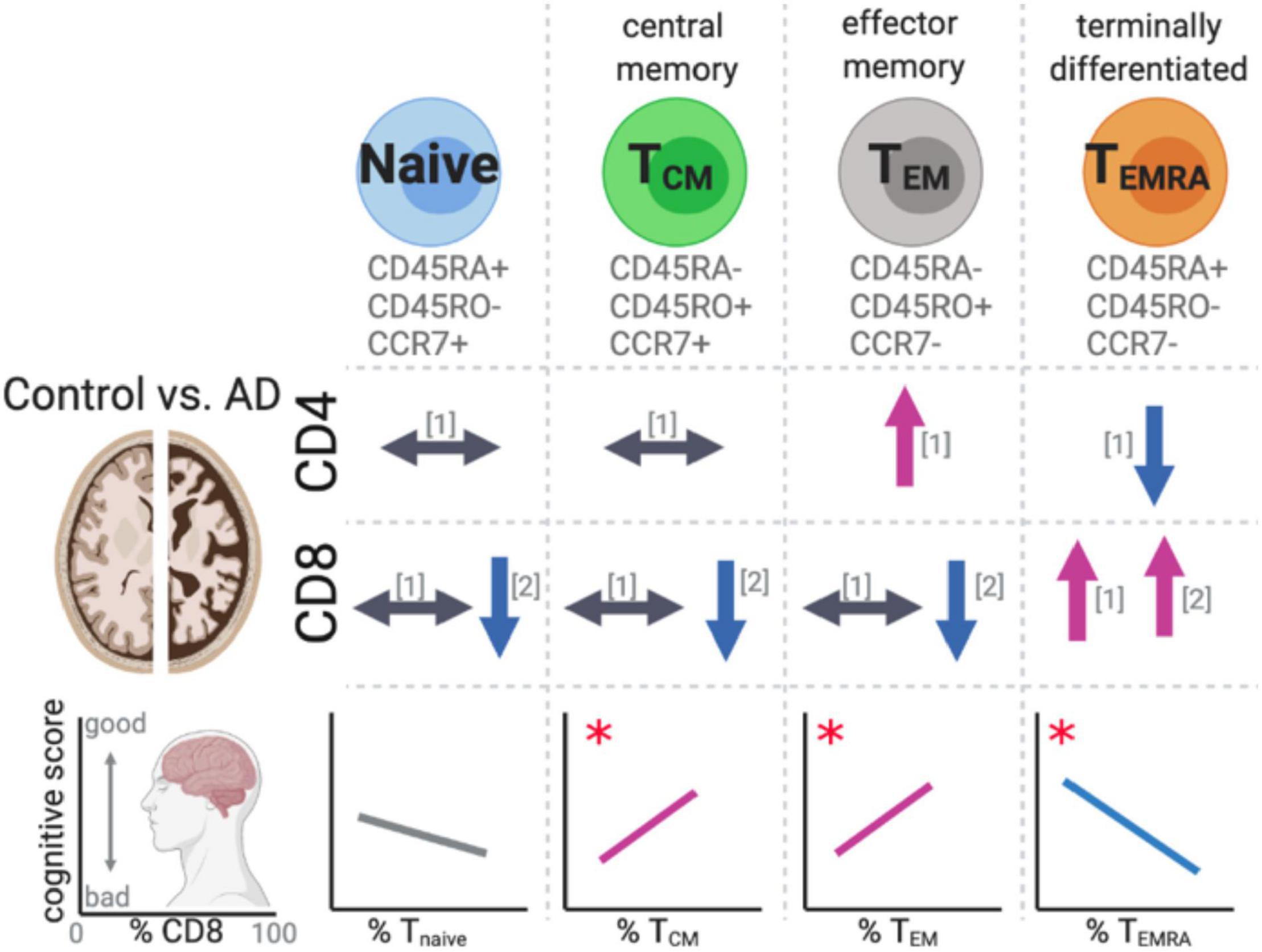
Figure 5. Changes in T cell subsets in AD. Recent studies have identified changes in effector memory and terminally differentiated T cells in AD patients’ peripheral blood compared to aged-matched controls. The changes in the T cells subsets were found to significantly correlate with cognitive scores. References for changes correspond to the following: [1] (Busse et al., 2017) and [2] (Gate et al., 2020). The symbols indicate the following: ⟷ no change, ↑ increase in the cell population, ↓ decrease in the cell population in AD cases compared to aged-matched controls. *p < 0.05.
T Cell Immune Function in AD and Related Dementias
Skias et al. (1985) used immunopanning to enrich for CD4 and CD8 cells from peripheral blood mononuclear cells (PBMCs) to evaluate function of the T cells. In the cross-sectional design they included 16 AD (mean age 75) and 14 age-matched controls (Skias et al., 1985). They found in the CD8 enriched cell fraction that upon stimulation with pokeweed mitogen the CD8 cells from AD participants were less able to suppress IgG production from B cells. Skias et al. (1985) postulated that the changes in immune function in AD could be a form of “accelerated aging” and that the “selective neuronal loss could lead to perturbed neural influences on the immune system.”
Miller et al. (1981) also found that lymphocytes from AD participants compared to aged-matched controls (mean age 75.5) had less response to the mitogen phytohemagglutinin, indicating an overall blunted immune response. They also reported increased immune suppression activity in the AD lymphocytes in response to another non-specific mitogen, concanavalin-A, compared to lymphocytes from age-matched controls. Miller et al. (1981) had a third view on the immune changes in AD. They speculated that AD might be more like a chronic viral infection in the way that it suppresses immunity. Further, they speculated that the “active impairment of a particular immune response has allowed an infectious agent to gain a foot in [dementia patients], resulting in central nervous system damage.” While these findings are 40 years old, and could be more compelling, the ideas proposed are interesting and should be revisited.
Trieb et al. (1996) tested if PBMCs from AD cases would respond to amyloid precursor protein (APP) fragments (Aβ1–28). The study included 20 old healthy controls (mean age 71), 20 young healthy controls (mean age 25), and 10 participants with AD (moderate severity n = 8 and severe n = 2; mean age 73). They found that the AD PMBCs had a decreased response to Aβ1–28 compared to the young or old controls as measured by a proliferation assay, with no effect of age. While it could be speculated that this was some sort of tolerance to Aβ stimulation in AD cases, the effects could also be from a general lack of effector function in PMBC from individuals with AD. Subsequent studies found no difference in the ability of PBMCs to respond to Aβ1–42 between AD vs. control participants (Giubilei et al., 2003; Rocha et al., 2012).
Singh (1994) stimulated PBMCs from probable AD cases or controls with LPS, which activates mainly innate immune cells through TLR4. They found that not only was there a bias toward increased innate immune cell numbers, but those innate immune cells produced 2.3-fold more IL-1 and threefold more IL-6 compared to age-matched control PBMCs (Singh, 1994). Lombardi et al. (1999) expanded upon these findings by measuring a more comprehensive panel of cytokines and including VaD and Parkinson’s disease groups. They found that LPS stimulated whole blood cultures from AD patients showed differences in cytokine production compared to results from aged-match controls and the VaD and Parkinson’s disease groups (Figure 6; Lombardi et al., 1999). The results support the case for an exaggerated innate immune response and also suggest signatures of immune function that are unique to different forms of dementia.
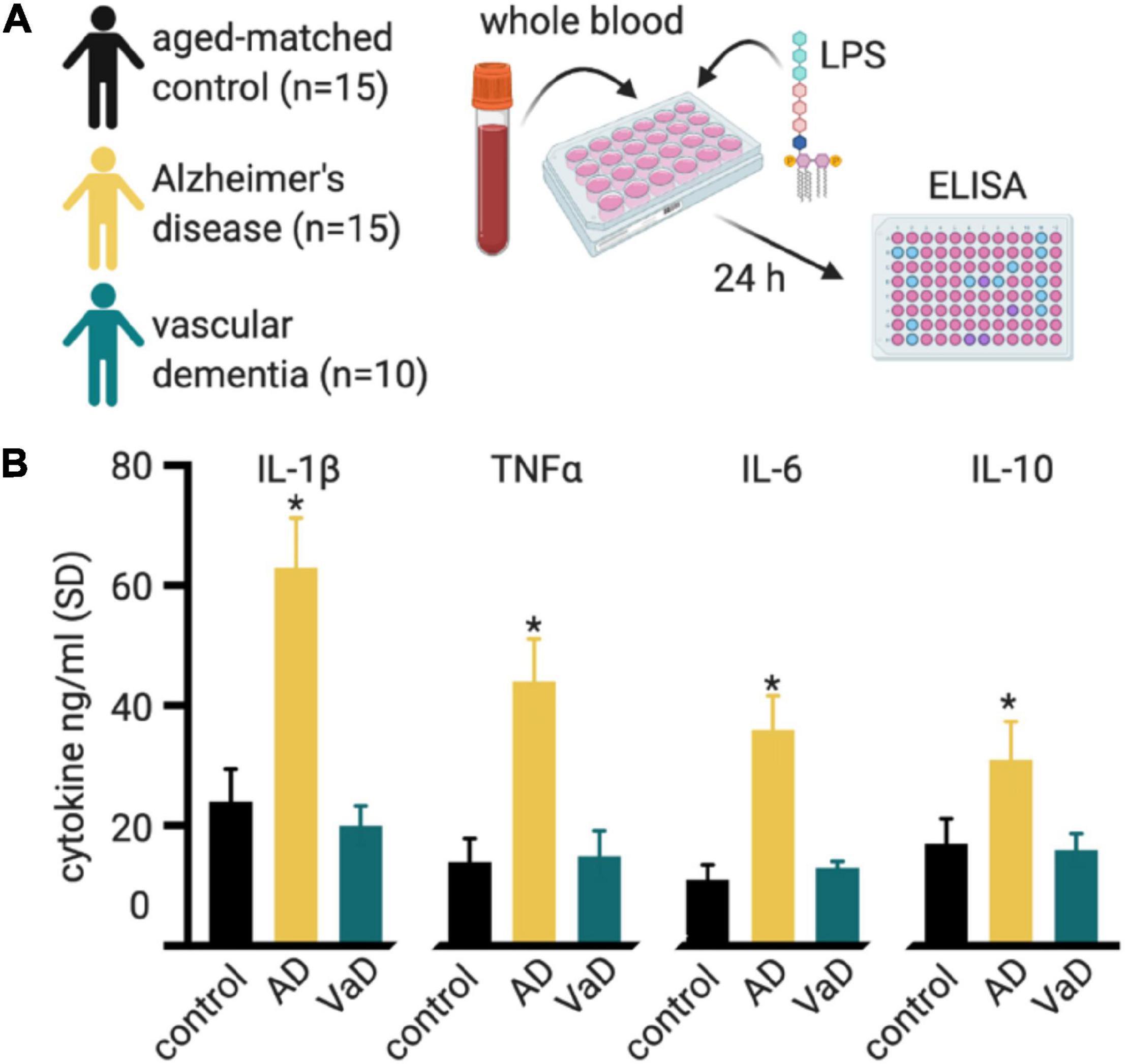
Figure 6. Whole blood cultures show an exaggerated response to LPS. (A) Lombardi et al. (1999) stimulated whole blood cultures from individuals with Alzheimer’s disease (AD), vascular dementia (VaD), and compared these to aged-matched controls. Cell culture supernatant was collected at 24-h intervals for 72 h for ELISA cytokine assays. (B) All cytokines measured showed higher cytokine production in the AD group, compared to the control or VaD groups. The plotted data is from the 24 h post-stimulation time point, from Table 3 of Lombardi et al. (1999). A similar trend was seen for the 48 and 72 h post-stimulation timepoint. *p < 0.05 compared to control.
By the age of 40, individuals with Down’s syndrome will have AD-neuropathological changes in their brains, and the majority will develop dementia. Park et al. (2000) isolated peripheral blood monocytes from individuals (range 26–68 years old, mean 45 years old) with Down syndrome and age-matched controls and stimulated the cells with LPS. They found no difference in TNFα, IL-6, or IL-8. In contrast to Singh (1994) and Lombardi et al. (1999), they found that monocytes from Down’s syndrome individuals produced 30% less IL-1β than the aged match controls.
Stimulating PBMCs from AD patients or aged-matched controls with either PHA or anti-CD3/CD28 it was found that the AD patients had an increased proinflammatory IL-1β response, presumably from the innate immune cells, a decreased in the anti-inflammatory cytokine IL-4, presumably from T cells (Reale et al., 2004; Rocha et al., 2012). Stimulating the PBMCs with PHA-M, a T cell activator, IL-2 was increased in the late AD, but not in the early AD compared to control (Shalit et al., 1995). AD patient CD8 T cells stimulated with PMA and ionomycin produced more IFNγ and TNFα than CD8 T cells from age-matched controls (Gate et al., 2020). In summary, both innate and adaptive immune cells’ products upon activation are dysregulated in people with dementia. More work is needed to understand the inflammatory signature of this dysregulation and the role of immune cell cross-talk, as these areas will be important targets for therapeutic intervention.
B Cell Changes in AD and Related Dementias
Despite the emerging understanding of roles for T cells in AD and related dementias, few studies explored roles for B cells in AD, although B cells coordinate with T cells to define functions of the adaptive immune system. B cells can proliferate in T cell-dependent manner or independent manner with the latter producing immunoglobulins (Ig), also termed antibodies, with low affinity and failure of memory B cell development (Defrance et al., 2011). B cell proliferation in a T cell-dependent manner is facilitated by helper T cells after antigen-presentation to give rise to memory. In addition to producing Ig, B cells act as antigen presenting cells to induce T cell activation through co-stimulation and cytokine production (Chalasani and Rothstein, 2014).
Age-related diseases like chronic lymphocytic leukemia and autoimmune diseases show extremes of B cells changes with age (Candore et al., 1997; Chiorazzi and Ferrarini, 2003). While there is a decrease in total B cells in the elderly, age-related increases in particular B cell populations may be involved in increased inflammation (Colonna-Romano et al., 2009; Bulati et al., 2014). Unfortunately, only a few reports describe B cell status in AD and related dementias. IgG+IgD–CD27– B cells are increased in peripheral blood of patients with moderate to severe AD, compared patients with mild AD or no cognitive impairment (Bulati et al., 2015). B1 cells, which make IgM and are considered more “innate-like” in function, have been implicated in amyloid-beta clearance in people (Agrawal et al., 2018) and mice (Baulch et al., 2020): both B1 and IgM are decreased in an AD-relevant mouse model (Baulch et al., 2020). Using an in vitro model with B cells from AD patients further suggests a role for B cells in amyloid-beta plaque formation (Dezfulian, 2018). Despite the paucity of studies, the available evidence supports the future exploration of B cell function in AD and related dementias.
Conclusion and Future Directions
Aging causes a functional decline and reorganization of the immune system collectively termed immunesenescence, which leads to a chronic state of inflammation that damages many organs, including possibly the brain. Chronic systemic inflammation is greater in people with AD (Holmes et al., 2009). Immunesenescence leaves the body vulnerable to infections and suboptimal vaccine responses through poorly understood mechanisms and leaves the immune system at the risk of being depleted following an infection or injury. In people with AD dementia, and perhaps other dementias, the age-related change in immune system cell distribution and function is exacerbated. The balance of innate vs. adaptive immune systems is further skewed toward innate immune function in people with AD (van der Willik et al., 2019). The decrease in memory T cells and increase in clonal expansion of TEMRA T cells tracks closely with decline in cognitive function in AD (Gate et al., 2020). When the systemic immune cells encounter stimuli they are primed toward an exaggerated proinflammatory response (Singh, 1994; Godbout et al., 2008).
In the neuro-centric view, AD starts in the brain. This could be, as proposed by Leffell et al. (1985), that AD is caused either by a neuron-targeted autoimmune attack or by an infection in the brain that damages the neurons, and that the immune system is altered in response to this infection. Many infectious agents have been proposed over the years (Readhead et al., 2018; Drayman et al., 2019; Abbott, 2020; Allnutt et al., 2020; Gate et al., 2020; Rizzo, 2020). The alternative neuro-centric view is that as neurons are degenerating, there is a loss of neural trophic support, which suppresses the immune system, as postulated by Skias et al. (1985). However, while both of these views have merit, we believe that the current evidence does not support these interpretations.
From the immunological centric standpoint, immunosenescence comes first. As the immune system becomes dysfunctional, we speculate that it is no longer able to protect the brain. The genetic evidence strongly supports the role of the immune system in AD (Kunkle et al., 2019; Andrews et al., 2020; Neuner et al., 2020), as many genetic risk variants associate with innate immunity. Could these genetic risk variants further drive the age-related imbalance in adaptive vs. innate immunity? Or are genetics additive to the effects of aging – that is, more innate cells that are more proinflammatory? Currently, the vast majority of work on innate immune cells in AD is focused on the microglia. Comparatively, much less is known about how AD risk genes affect the systemic innate immune function, and more work is needed in this area.
While it is often assumed that the decline in dementia is steady and progressive, at the individual level, the decline is more zigzag with troughs of decline, which are speculated to correspond to life events (infections, diseases, surgeries, etc.) (Perry et al., 2007). If a dysfunctional immune system leads to more numerous/serious infections, or exaggerated responses to the infection, could these events be driving a neurodegenerative decline? One could speculate that it is not until that immune system is challenged (for example, by a urinary tract infection) that an exaggerated immune response damages the brain. However, more work is needed to test these possibilities.
In conclusion, there is compelling correlational evidence that the systemic immune system is disrupted in people with dementia. There is also strong evidence that genetic polymorphisms in immune function, including HLA and TREM2, modify the risk for developing AD. Still, there are several essential questions. Our review provides compelling evidence that the immune system is altered in AD, but our current knowledge of how the immune system is changed is fragmented. Future studies (Figure 7) are needed to provide a complete picture of how the immune system changes in people with healthy cognitive aging and those who transition into neurodegenerative disease. Such studies are costly, but by partnering and incorporating immunological evaluations as part of ongoing analysis of cognitive state in community cohorts (Schmitt et al., 2012), we can address many unresolved questions in the field.
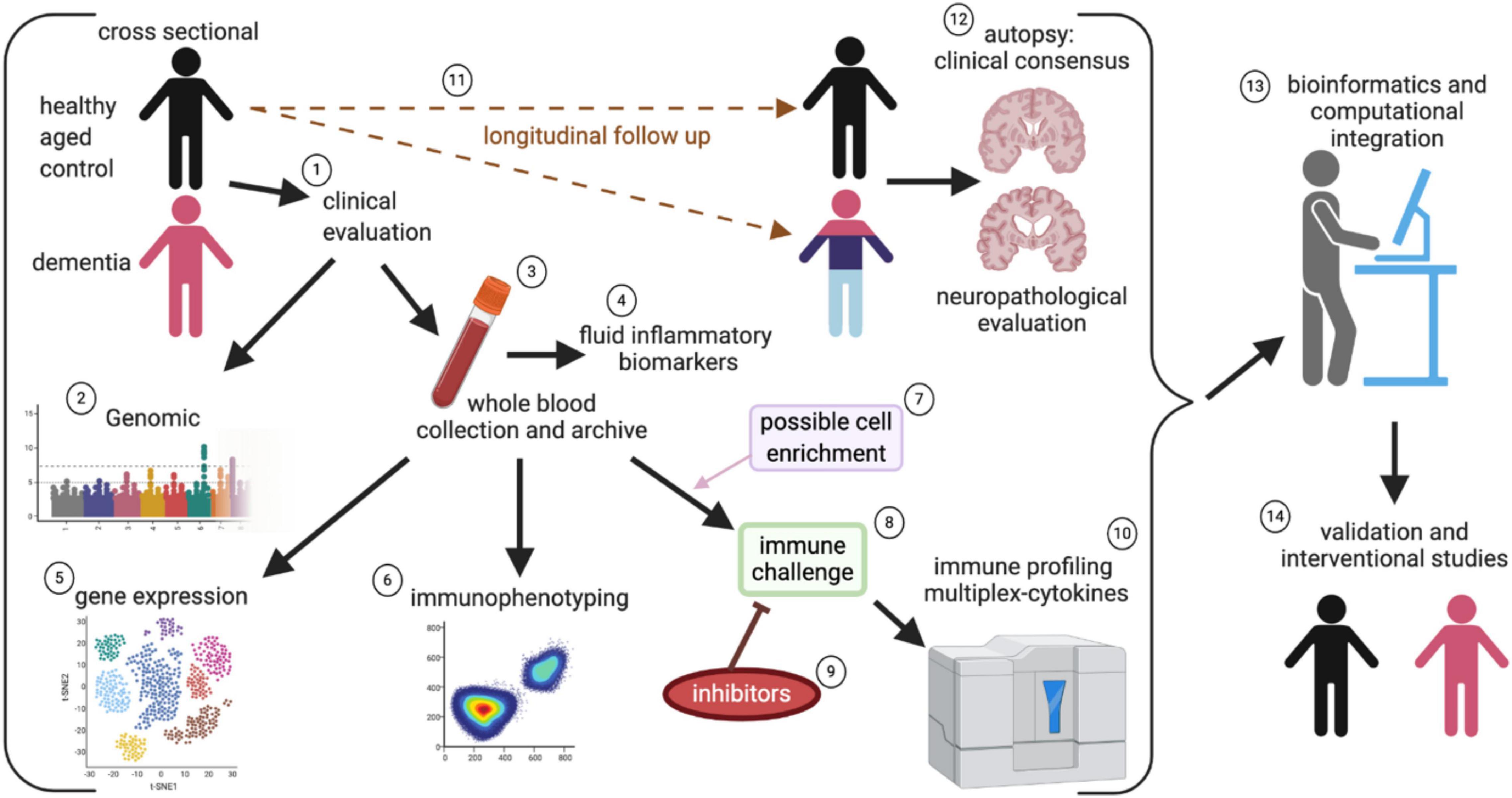
Figure 7. Systems approach to evaluate systemic immune changes in dementia. In a community-based cohort of individuals who are followed longitudinally and have agreed to brain autopsy and donation, the study can begin with a cross-sectional design of participants without cognitive impairment and comparing those with mild cognitive impairment to profound dementia, and incorporating clinical evaluations as part of the study (1). By piggybacking on a larger study that incorporates genomic (2), blood collection (3), and fluid biomarker assessment (including assays for systemic inflammation), (4) it is possible to leverage ongoing cohort studies to better define the role of immune dysfunction in AD. Peripheral blood mononuclear cells (PBMC) are isolated from the fresh whole blood and are archived for future studies (3). The isolated PMBCs can be used for gene expression and gene sequencing to identify changes in immune cell populations and clonal expansion of T cells (5). Immunophenotyping of immune cell subsets can be done to evaluate changes in a population of immune cells (6). Select populations of immune cells can be enriched (7). The PBMC or select cells can be directly stimulated with activators such as LPS or CD3/CD28 (8), to evaluate cellular mechanisms and therapeutic targets, inhibitors can be added along with the mitogens (9). The cell culture supernatant can then be used for cytokines multiplex immune profiling assays (10). By enriching the cross-sectional design for healthy controls, it is possible to evaluate people’s possible conversion into varying neurodegenerative diseases (11), which are ultimately pathologically confirmed at autopsy (12). The wealth of data that is captured will require a very robust statistical and bioinformatical infrastructure to integrate all the data (13). Finally, “hits” will require validation in a subsequent set of study participants, and clinical experimentation can test positive intervention strategies targeted at restoring the immune balance (14).
In addition to understanding the natural history of immune changes associated with AD and related dementias, it will also be important to establish causation, mechanisms of dysfunction, and ultimately therapies. Future studies will require a translational approach using animal models. Example studies from animal models show the influential role of the adaptive immune system as a modifier of brain function. For example, dietary and metabolic manipulations have been shown to lead to an increase in Th17s and Th17-cytokines (IL-17, IL-22) that cause cognitive dysfunction in the absence of amyloid plaques and neurofibrillary tangles (Faraco et al., 2018). Alternatively, boosting adaptive immune system function through blockade of immune checkpoints, such as PD-1, improved outcomes in multiple AD-relevant animal models (Baruch et al., 2015, 2016; Rosenzweig et al., 2019). In contrast, in Rag2, Il2rγ, double-knockout mice that lack T cells, B cells, and natural killer cells, the lack of an adaptive immune system worsens pathology and neuroinflammation in an AD-relevant mouse model (Marsh et al., 2016). These animal studies and others suggest that the adaptive immune system contributes to cognitive decline (Spani et al., 2015; Jevtic et al., 2017; Unger et al., 2020). This interpretation is therapeutically promising as it provides opportunities for manipulation, including approaches such as IL-7 to replenish the depleted immune system and checkpoint inhibitors like PD-1. Much more work is still needed. By gaining a better understanding of the connection between the immune system and the brain there is great promise in obtaining a more holistic perspective of how the demise of mind and body both contribute to dementing diseases.
Author Contributions
AB completed the literature review and wrote the first draft. JL and BN wrote sections, compiled, contributed to the design and conceptualization of the study, and revising the manuscript for intellectual content. All authors read and approved the final manuscript.
Funding
This publication was supported in part by National Institutes of Health under award numbers T32 AG057461, R21AG066865, R21AG059123, R01AG068215, and R01NS103785, and the Department of Defense award number AZ190017. The content is solely the responsibility of the authors and does not represent the official views of the National Institutes of Health or the Department of Defense.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
While, we attempted to provide a comprehensive review of the primary literature, there are likely studies that were involuntarily omitted, or where review articles were used to summarize an extensive body of primary literature. We hope that our colleagues can excuse our omissions of their valuable work. Illustrations were created using BioRender.com.
References
Abbott, A. (2020). Are infections seeding some cases of Alzheimer’s disease? Nature 587, 22–25. doi: 10.1038/d41586-020-03084-9
Abner, E. L., Kryscio, R. J., Schmitt, F. A., Fardo, D. W., Moga, D. C., Ighodaro, E. T., et al. (2017). Outcomes after diagnosis of mild cognitive impairment in a large autopsy series. Ann. Neurol. 81, 549–559. doi: 10.1002/ana.24903
Agrawal, A., Agrawal, S., and Gupta, S. (2017). Role of dendritic cells in inflammation and loss of tolerance in the elderly. Front. Immunol. 8:896. doi: 10.3389/fimmu.2017.00896
Agrawal, S., Abud, E. M., Snigdha, S., and Agrawal, A. (2018). IgM response against amyloid-beta in aging: a potential peripheral protective mechanism. Alzheimers Res. Ther. 10:81.
Allnutt, M. A., Johnson, K., Bennett, D. A., Connor, S. M., Troncoso, J. C., Pletnikova, O., et al. (2020). Human Herpesvirus 6 detection in Alzheimer’s disease cases and controls across multiple cohorts. Neuron 105, 1027–1035.e2.
Alpert, A., Pickman, Y., Leipold, M., Rosenberg-Hasson, Y., Ji, X., Gaujoux, R., et al. (2019). A clinically meaningful metric of immune age derived from high-dimensional longitudinal monitoring. Nat. Med. 25, 487–495. doi: 10.1038/s41591-019-0381-y
Andrews, S. J., Fulton-Howard, B., and Goate, A. (2020). Interpretation of risk loci from genome-wide association studies of Alzheimer’s disease. Lancet Neurol. 19, 326–335. doi: 10.1016/s1474-4422(19)30435-1
Antonaci, S., Garofalo, A. R., Chicco, C., Polignano, A. V., Pugliese, P., Misefari, A., et al. (1990). Senile dementia, Alzheimer type: a distinct entity in the immunosenescence? J. Clin. Lab. Anal. 4, 16–21. doi: 10.1002/jcla.1860040106
Araga, S., Kagimoto, H., Funamoto, K., and Takahashi, K. (1991). Reduced natural killer cell activity in patients with dementia of the Alzheimer type. Acta Neurol. Scand. 84, 259–263. doi: 10.1111/j.1600-0404.1991.tb04948.x
Bachstetter, A. D., Van Eldik, L. J., Schmitt, F. A., Neltner, J. H., Ighodaro, E. T., Webster, S. J., et al. (2015). Disease-related microglia heterogeneity in the hippocampus of Alzheimer’s disease, dementia with Lewy bodies, and hippocampal sclerosis of aging. Acta Neuropathol. Commun. 3:32.
Baruch, K., Deczkowska, A., Rosenzweig, N., Tsitsou-Kampeli, A., Sharif, A. M., Matcovitch-Natan, O., et al. (2016). PD-1 immune checkpoint blockade reduces pathology and improves memory in mouse models of Alzheimer’s disease. Nat. Med. 22, 135–137. doi: 10.1038/nm.4022
Baruch, K., Rosenzweig, N., Kertser, A., Deczkowska, A., Sharif, A. M., Spinrad, A., et al. (2015). Breaking immune tolerance by targeting Foxp3(+) regulatory T cells mitigates Alzheimer’s disease pathology. Nat. Commun. 6:7967.
Baulch, J. E., Acharya, M. M., Agrawal, S., Apodaca, L. A., Monteiro, C., and Agrawal, A. (2020). Immune and inflammatory determinants underlying Alzheimer’s disease pathology. J. Neuroimmune Pharmacol. 15, 852–862. doi: 10.1007/s11481-020-09908-9
Berent-Maoz, B., Montecino-Rodriguez, E., and Dorshkind, K. (2012). Genetic regulation of thymocyte progenitor aging. Semin. Immunol. 24, 303–308. doi: 10.1016/j.smim.2012.04.006
Bharath, L. P., Agrawal, M., Mccambridge, G., Nicholas, D. A., Hasturk, H., Liu, J., et al. (2020). Metformin enhances autophagy and normalizes mitochondrial function to alleviate aging-associated inflammation. Cell Metab. 32, 44–55.e6.
Blevins, B. L., Vinters, H. V., Love, S., Wilcock, D. M., Grinberg, L. T., Schneider, J. A., et al. (2020). Brain arteriolosclerosis. Acta Neuropathol. 141, 1–24.
Bonotis, K., Krikki, E., Holeva, V., Aggouridaki, C., Costa, V., and Baloyannis, S. (2008). Systemic immune aberrations in Alzheimer’s disease patients. J. Neuroimmunol. 193, 183–187. doi: 10.1016/j.jneuroim.2007.10.020
Born, J., Uthgenannt, D., Dodt, C., Nunninghoff, D., Ringvolt, E., Wagner, T., et al. (1995). Cytokine production and lymphocyte subpopulations in aged humans. An assessment during nocturnal sleep. Mech. Ageing Dev. 84, 113–126. doi: 10.1016/0047-6374(95)01638-4
Bulati, M., Buffa, S., Martorana, A., Candore, G., Lio, D., Caruso, C., et al. (2014). Trafficking phenotype and production of granzyme B by double negative B cells (IgG(+)IgD(-)CD27(-)) in the elderly. Exp. Gerontol. 54, 123–129. doi: 10.1016/j.exger.2013.12.011
Bulati, M., Buffa, S., Martorana, A., Gervasi, F., Camarda, C., Azzarello, D. M., et al. (2015). Double negative (IgG+IgD-CD27-) B cells are increased in a cohort of moderate-severe Alzheimer’s disease patients and show a pro-inflammatory trafficking receptor phenotype. J. Alzheimers Dis. 44, 1241–1251. doi: 10.3233/jad-142412
Busse, M., Michler, E., Von Hoff, F., Dobrowolny, H., Hartig, R., Frodl, T., et al. (2017). Alterations in the peripheral immune system in dementia. J. Alzheimers Dis. 58, 1303–1313. doi: 10.3233/jad-161304
Candore, G., Di Lorenzo, G., Mansueto, P., Melluso, M., Frada, G., Li Vecchi, M., et al. (1997). Prevalence of organ-specific and non organ-specific autoantibodies in healthy centenarians. Mech. Ageing Dev. 94, 183–190. doi: 10.1016/s0047-6374(96)01845-3
Cao, W., and Zheng, H. (2018). Peripheral immune system in aging and Alzheimer’s disease. Mol. Neurodegener. 13:51.
Chalasani, G., and Rothstein, D. (2014). Non-antibody mediated roles of B cells in allograft survival. Curr. Transplant. Rep. 1, 155–165. doi: 10.1007/s40472-014-0020-y
Chatta, G. S., Andrews, R. G., Rodger, E., Schrag, M., Hammond, W. P., and Dale, D. C. (1993). Hematopoietic progenitors and aging: alterations in granulocytic precursors and responsiveness to recombinant human G-CSF, GM-CSF, and IL-3. J. Gerontol. 48, M207–M212.
Chiorazzi, N., and Ferrarini, M. (2003). B cell chronic lymphocytic leukemia: lessons learned from studies of the B cell antigen receptor. Annu. Rev. Immunol. 21, 841–894.
Colonna-Romano, G., Bulati, M., Aquino, A., Pellicano, M., Vitello, S., Lio, D., et al. (2009). A double-negative (IgD-CD27-) B cell population is increased in the peripheral blood of elderly people. Mech. Ageing Dev. 130, 681–690. doi: 10.1016/j.mad.2009.08.003
Cros, J., Cagnard, N., Woollard, K., Patey, N., Zhang, S. Y., Senechal, B., et al. (2010). Human CD14dim monocytes patrol and sense nucleic acids and viruses via TLR7 and TLR8 receptors. Immunity 33, 375–386. doi: 10.1016/j.immuni.2010.08.012
Cruz Hernandez, J. C., Bracko, O., Kersbergen, C. J., Muse, V., Haft-Javaherian, M., Berg, M., et al. (2019). Neutrophil adhesion in brain capillaries reduces cortical blood flow and impairs memory function in Alzheimer’s disease mouse models. Nat. Neurosci. 22, 413–420. doi: 10.1038/s41593-018-0329-4
Defrance, T., Taillardet, M., and Genestier, L. (2011). T cell-independent B cell memory. Curr. Opin. Immunol. 23, 330–336. doi: 10.1016/j.coi.2011.03.004
Della Bella, S., Bierti, L., Presicce, P., Arienti, R., Valenti, M., Saresella, M., et al. (2007). Peripheral blood dendritic cells and monocytes are differently regulated in the elderly. Clin. Immunol. 122, 220–228. doi: 10.1016/j.clim.2006.09.012
den Braber, I., Mugwagwa, T., Vrisekoop, N., Westera, L., Mogling, R., De Boer, A. B., et al. (2012). Maintenance of peripheral naive T cells is sustained by thymus output in mice but not humans. Immunity 36, 288–297. doi: 10.1016/j.immuni.2012.02.006
Dezfulian, M. (2018). A new Alzheimer’s disease cell model using B cells to induce beta amyloid plaque formation and increase TNF alpha expression. Int. Immunopharmacol. 59, 106–112. doi: 10.1016/j.intimp.2018.04.012
Drayman, N., Patel, P., Vistain, L., and Tay, S. (2019). HSV-1 single-cell analysis reveals the activation of anti-viral and developmental programs in distinct sub-populations. eLife 8:e46339.
Dykstra, B., Olthof, S., Schreuder, J., Ritsema, M., and De Haan, G. (2011). Clonal analysis reveals multiple functional defects of aged murine hematopoietic stem cells. J. Exp. Med. 208, 2691–2703. doi: 10.1084/jem.20111490
Dysken, M. W., Minichiello, M. D., Hill, J. L., Skare, S., Little, J. T., Molchan, S. E., et al. (1992). Distribution of peripheral lymphocytes in Alzheimer patients and controls. J. Psychiatr. Res. 26, 213–218. doi: 10.1016/0022-3956(92)90024-i
Engelhardt, B., Vajkoczy, P., and Weller, R. O. (2017). The movers and shapers in immune privilege of the CNS. Nat. Immunol. 18, 123–131. doi: 10.1038/ni.3666
Erickson, M. A., Wilson, M. L., and Banks, W. A. (2020). In vitro modeling of blood-brain barrier and interface functions in neuroimmune communication. Fluids Barriers CNS 17:26.
Faraco, G., Brea, D., Garcia-Bonilla, L., Wang, G., Racchumi, G., Chang, H., et al. (2018). Dietary salt promotes neurovascular and cognitive dysfunction through a gut-initiated TH17 response. Nat. Neurosci. 21, 240–249. doi: 10.1038/s41593-017-0059-z
Fest, J., Ruiter, R., Ikram, M. A., Voortman, T., Van Eijck, C. H. J., and Stricker, B. H. (2018). Reference values for white blood-cell-based inflammatory markers in the Rotterdam Study: a population-based prospective cohort study. Sci. Rep. 8:10566.
Galea, I., Bechmann, I., and Perry, V. H. (2007). What is immune privilege (not)? Trends Immunol. 28, 12–18. doi: 10.1016/j.it.2006.11.004
Gate, D., Saligrama, N., Leventhal, O., Yang, A. C., Unger, M. S., Middeldorp, J., et al. (2020). Clonally expanded CD8 T cells patrol the cerebrospinal fluid in Alzheimer’s disease. Nature 577, 399–404. doi: 10.1038/s41586-019-1895-7
Giubilei, F., Antonini, G., Montesperelli, C., Sepe-Monti, M., Cannoni, S., Pichi, A., et al. (2003). T cell response to amyloid-beta and to mitochondrial antigens in Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 16, 35–38.
Godbout, J. P., Moreau, M., Lestage, J., Chen, J., Sparkman, N. L., O’connor, J., et al. (2008). Aging exacerbates depressive-like behavior in mice in response to activation of the peripheral innate immune system. Neuropsychopharmacology 33, 2341–2351. doi: 10.1038/sj.npp.1301649
Greenhalgh, A. D., David, S., and Bennett, F. C. (2020). Immune cell regulation of glia during CNS injury and disease. Nat. Rev. Neurosci. 21, 139–152. doi: 10.1038/s41583-020-0263-9
Griffin, W. S., Stanley, L. C., Ling, C., White, L., Macleod, V., Perrot, L. J., et al. (1989). Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 86, 7611–7615. doi: 10.1073/pnas.86.19.7611
Guilliams, M., Mildner, A., and Yona, S. (2018). Developmental and functional heterogeneity of monocytes. Immunity 49, 595–613. doi: 10.1016/j.immuni.2018.10.005
Hart, B. L. (1988). Biological basis of the behavior of sick animals. Neurosci. Biobehav. Rev. 12, 123–137. doi: 10.1016/s0149-7634(88)80004-6
Hazenberg, M. D., Otto, S. A., Cohen Stuart, J. W., Verschuren, M. C., Borleffs, J. C., Boucher, C. A., et al. (2000). Increased cell division but not thymic dysfunction rapidly affects the T-cell receptor excision circle content of the naive T cell population in HIV-1 infection. Nat. Med. 6, 1036–1042. doi: 10.1038/79549
Hearps, A. C., Martin, G. E., Angelovich, T. A., Cheng, W. J., Maisa, A., Landay, A. L., et al. (2012). Aging is associated with chronic innate immune activation and dysregulation of monocyte phenotype and function. Aging Cell 11, 867–875. doi: 10.1111/j.1474-9726.2012.00851.x
Herz, J., Filiano, A. J., Smith, A., Yogev, N., and Kipnis, J. (2017). Myeloid cells in the central nervous system. Immunity 46, 943–956. doi: 10.1016/j.immuni.2017.06.007
High, K. P., Akbar, A. N., and Nikolich-Zugich, J. (2012). Translational research in immune senescence: assessing the relevance of current models. Semin. Immunol. 24, 373–382. doi: 10.1016/j.smim.2012.04.007
Holmes, C., Cunningham, C., Zotova, E., Woolford, J., Dean, C., Kerr, S., et al. (2009). Systemic inflammation and disease progression in Alzheimer disease. Neurology 73, 768–774. doi: 10.1212/wnl.0b013e3181b6bb95
Hu, B., Yang, X. R., Xu, Y., Sun, Y. F., Sun, C., Guo, W., et al. (2014). Systemic immune-inflammation index predicts prognosis of patients after curative resection for hepatocellular carcinoma. Clin. Cancer Res. 20, 6212–6222. doi: 10.1158/1078-0432.ccr-14-0442
Hu, G. R., Walls, R. S., Creasey, H., Mccusker, E., and Broe, G. A. (1995). Peripheral blood lymphocyte subset distribution and function in patients with Alzheimer’s disease and other dementias. Aust. N.Z. J. Med. 25, 212–217. doi: 10.1111/j.1445-5994.1995.tb01525.x
Iadecola, C. (2017). The neurovascular unit coming of age: a journey through neurovascular coupling in health and disease. Neuron 96, 17–42. doi: 10.1016/j.neuron.2017.07.030
Ighodaro, E. T., Abner, E. L., Fardo, D. W., Lin, A. L., Katsumata, Y., Schmitt, F. A., et al. (2017). Risk factors and global cognitive status related to brain arteriolosclerosis in elderly individuals. J. Cereb. Blood Flow Metab. 37, 201–216. doi: 10.1177/0271678x15621574
Ikeda, T., Yamamoto, K., Takahashi, K., and Yamada, M. (1991). Immune system-associated antigens on the surface of peripheral blood lymphocytes in patients with Alzheimer’s disease. Acta Psychiatr. Scand. 83, 444–448. doi: 10.1111/j.1600-0447.1991.tb05573.x
Ikram, M. A., Brusselle, G. G. O., Murad, S. D., Van Duijn, C. M., Franco, O. H., Goedegebure, A., et al. (2017). The Rotterdam Study: 2018 update on objectives, design and main results. Eur. J. Epidemiol. 32, 807–850. doi: 10.1007/s10654-017-0321-4
Ip, B., Cilfone, N. A., Belkina, A. C., Defuria, J., Jagannathan-Bogdan, M., Zhu, M., et al. (2016). Th17 cytokines differentiate obesity from obesity-associated type 2 diabetes and promote TNFalpha production. Obesity 24, 102–112. doi: 10.1002/oby.21243
Jellinger, K. A. (2020). Pathobiological subtypes of Alzheimer disease. Dement. Geriatr. Cogn. Disord. 49, 321–333. doi: 10.1159/000508625
Jevtic, S., Sengar, A. S., Salter, M. W., and Mclaurin, J. (2017). The role of the immune system in Alzheimer disease: etiology and treatment. Ageing Res. Rev. 40, 84–94. doi: 10.1016/j.arr.2017.08.005
Kalelioglu, T., Yuruyen, M., Gultekin, G., Yavuzer, H., Ozturk, Y., Kurt, M., et al. (2017). Neutrophil and platelet to lymphocyte ratios in people with subjective, mild cognitive impairment and early Alzheimer’s disease. Psychogeriatrics 17, 506–508. doi: 10.1111/psyg.12260
Karanth, S., Nelson, P. T., Katsumata, Y., Kryscio, R. J., Schmitt, F. A., Fardo, D. W., et al. (2020). Prevalence and clinical phenotype of quadruple misfolded proteins in older adults. JAMA Neurol. 77, 1299–1307. doi: 10.1001/jamaneurol.2020.1741
Kong, F. K., Chen, C. L. H., Six, A., Hockett, R. D., and Cooper, M. D. (1999). T cell receptor gene deletion circles identify recent thymic emigrants in the peripheral T cell pool. Proc. Natl. Acad. Sci. U.S.A. 96, 1536–1540. doi: 10.1073/pnas.96.4.1536
Kovacs, G. G. (2015). Neuropathology of tauopathies: principles and practice. Neuropathol. Appl. Neurobiol. 41, 3–23. doi: 10.1111/nan.12208
Kovacs, G. G. (2020). Astroglia and Tau: new perspectives. Front. Aging Neurosci. 12:96. doi: 10.3389/fnagi.2020.00096
Kunkle, B. W., Grenier-Boley, B., Sims, R., Bis, J. C., Damotte, V., Naj, A. C., et al. (2019). Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates A beta, tau, immunity and lipid processing. Nat. Genet. 51, 414–430.
Kuyumcu, M. E., Yesil, Y., Ozturk, Z. A., Kizilarslanoglu, C., Etgul, S., Halil, M., et al. (2012). The evaluation of neutrophil-lymphocyte ratio in Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 34, 69–74.
Le Page, A., Garneau, H., Dupuis, G., Frost, E. H., Larbi, A., Witkowski, J. M., et al. (2017). Differential Phenotypes of Myeloid-Derived suppressor and T regulatory cells and cytokine levels in Amnestic Mild cognitive impairment subjects compared to Mild Alzheimer Diseased Patients. Front. Immunol. 8:783. doi: 10.3389/fimmu.2017.00783
Leffell, M. S., Lumsden, L., and Steiger, W. A. (1985). An analysis of T lymphocyte subpopulations in patients with Alzheimer’s disease. J. Am. Geriatr. Soc. 33, 4–8. doi: 10.1111/j.1532-5415.1985.tb02851.x
Licastro, F., Savorani, G., Sarti, G., Salsi, A., Cavazzuti, F., Zanichelli, L., et al. (1990). Zinc and thymic hormone-dependent immunity in normal ageing and in patients with senile dementia of the Alzheimer type. J. Neuroimmunol. 27, 201–208. doi: 10.1016/0165-5728(90)90070-4
Liu, X., Nemeth, D. P., Mckim, D. B., Zhu, L., Disabato, D. J., Berdysz, O., et al. (2019). Cell-type-specific interleukin 1 receptor 1 signaling in the brain regulates distinct neuroimmune activities. immunity 50, 317–333.e6.
Liu, X., and Quan, N. (2018). Microglia and CNS Interleukin-1: beyond immunological concepts. Front. Neurol. 9:8. doi: 10.3389/fneur.2018.00008
Lombardi, V. R., Garcia, M., Rey, L., and Cacabelos, R. (1999). Characterization of cytokine production, screening of lymphocyte subset patterns and in vitro apoptosis in healthy and Alzheimer’s Disease (AD) individuals. J. Neuroimmunol. 97, 163–171. doi: 10.1016/s0165-5728(99)00046-6
Lord, J. M., Butcher, S., Killampali, V., Lascelles, D., and Salmon, M. (2001). Neutrophil ageing and immunesenescence. Mech. Ageing Dev. 122, 1521–1535. doi: 10.1016/s0047-6374(01)00285-8
Lueg, G., Gross, C. C., Lohmann, H., Johnen, A., Kemmling, A., Deppe, M., et al. (2015). Clinical relevance of specific T-cell activation in the blood and cerebrospinal fluid of patients with mild Alzheimer’s disease. Neurobiol. Aging 36, 81–89. doi: 10.1016/j.neurobiolaging.2014.08.008
Madore, C., Yin, Z., Leibowitz, J., and Butovsky, O. (2020). Microglia, lifestyle stress, and neurodegeneration. Immunity 52, 222–240. doi: 10.1016/j.immuni.2019.12.003
Marsh, S. E., Abud, E. M., Lakatos, A., Karimzadeh, A., Yeung, S. T., Davtyan, H., et al. (2016). The adaptive immune system restrains Alzheimer’s disease pathogenesis by modulating microglial function. Proc. Natl. Acad. Sci. U.S.A. 113, E1316–E1325.
Mayne, K., White, J. A., Mcmurran, C. E., Rivera, F. J., and De La Fuente, A. G. (2020). Aging and neurodegenerative disease: Is the Adaptive immune system a friend or foe? Front. Aging Neurosci. 12:572090. doi: 10.3389/fnagi.2020.572090
McKee, A. C. (2020). The neuropathology of chronic traumatic encephalopathy: the status of the literature. Semin. Neurol. 40, 359–369. doi: 10.1055/s-0040-1713632
Miller, A. E., Neighbour, P. A., Katzman, R., Aronson, M., and Lipkowitz, R. (1981). Immunological studies in senile dementia of the Alzheimer type: evidence for enhanced suppressor cell activity. Ann. Neurol. 10, 506–510. doi: 10.1002/ana.410100603
Montgomery, R. R., and Shaw, A. C. (2015). Paradoxical changes in innate immunity in aging: recent progress and new directions. J. Leukoc. Biol. 98, 937–943. doi: 10.1189/jlb.5mr0315-104r
Montine, T. J., Phelps, C. H., Beach, T. G., Bigio, E. H., Cairns, N. J., Dickson, D. W., et al. (2012). National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol. 123, 1–11.
Murray, J. M., Kaufmann, G. R., Hodgkin, P. D., Lewin, S. R., Kelleher, A. D., Davenport, M. P., et al. (2003). Naive T cells are maintained by thymic output in early ages but by proliferation without phenotypic change after age twenty. Immunol. Cell Biol. 81, 487–495. doi: 10.1046/j.1440-1711.2003.01191.x
Naylor, K., Li, G., Vallejo, A. N., Lee, W. W., Koetz, K., Bryl, E., et al. (2005). The influence of age on T cell generation and TCR diversity. J. Immunol. 174, 7446–7452. doi: 10.4049/jimmunol.174.11.7446
Nelson, P. T., Dickson, D. W., Trojanowski, J. Q., Jack, C. R., Boyle, P. A., Arfanakis, K., et al. (2019). Limbic-predominant age-related TDP-43 encephalopathy (LATE): consensus working group report. Brain 142, 1503–1527.
Nelson, P. T., Head, E., Schmitt, F. A., Davis, P. R., Neltner, J. H., Jicha, G. A., et al. (2011). Alzheimer’s disease is not “brain aging”: neuropathological, genetic, and epidemiological human studies. Acta Neuropathol. 121, 571–587. doi: 10.1007/s00401-011-0826-y
Neltner, J. H., Abner, E. L., Jicha, G. A., Schmitt, F. A., Patel, E., Poon, L. W., et al. (2016). Brain pathologies in extreme old age. Neurobiol. Aging 37, 1–11.
Neuner, S. M., Tcw, J., and Goate, A. M. (2020). Genetic architecture of Alzheimer’s disease. Neurobiol. Dis. 143:104976.
Nevo, U., Kipnis, J., Golding, I., Shaked, I., Neumann, A., Akselrod, S., et al. (2003). Autoimmunity as a special case of immunity: removing threats from within. Trends Mol. Med. 9, 88–93. doi: 10.1016/s1471-4914(03)00024-8
Nikolich-Zugich, J. (2008). Ageing and life-long maintenance of T-cell subsets in the face of latent persistent infections. Nat. Rev. Immunol. 8, 512–522. doi: 10.1038/nri2318
Nikolich-Zugich, J. (2014). Aging of the T cell compartment in mice and humans: from no naive expectations to foggy memories. J. Immunol. 193, 2622–2629. doi: 10.4049/jimmunol.1401174
Nikolich-Zugich, J. (2018). The twilight of immunity: emerging concepts in aging of the immune system. Nat. Immunol. 19, 10–19. doi: 10.1038/s41590-017-0006-x
Nomellini, V., Faunce, D. E., Gomez, C. R., and Kovacs, E. J. (2008). An age-associated increase in pulmonary inflammation after burn injury is abrogated by CXCR2 inhibition. J. Leukoc. Biol. 83, 1493–1501. doi: 10.1189/jlb.1007672
Norden, D. M., Muccigrosso, M. M., and Godbout, J. P. (2015). Microglial priming and enhanced reactivity to secondary insult in aging, and traumatic CNS injury, and neurodegenerative disease. Neuropharmacology 96, 29–41. doi: 10.1016/j.neuropharm.2014.10.028
Nyugen, J., Agrawal, S., Gollapudi, S., and Gupta, S. (2010). Impaired functions of peripheral blood monocyte subpopulations in aged humans. J. Clin. Immunol. 30, 806–813. doi: 10.1007/s10875-010-9448-8
Oberstein, T. J., Taha, L., Spitzer, P., Hellstern, J., Herrmann, M., Komhuber, J., et al. (2018). Imbalance of Circulating T(h)17 and regulatory T cells in Alzheimer’s disease: a case control study. Front. Immunol. 9:1213. doi: 10.3389/fimmu.2018.01213
Park, E., Alberti, J., Mehta, P., Dalton, A., Sersen, E., and Schuller-Levis, G. (2000). Partial impairment of immune functions in peripheral blood leukocytes from aged men with Down’s syndrome. Clin. Immunol. 95, 62–69. doi: 10.1006/clim.2000.4834
Pattabiraman, G., Palasiewicz, K., Galvin, J. P., and Ucker, D. S. (2017). Aging-associated dysregulation of homeostatic immune response termination (and not initiation). Aging Cell 16, 585–593. doi: 10.1111/acel.12589
Perry, V. H., Cunningham, C., and Holmes, C. (2007). Systemic infections and inflammation affect chronic neurodegeneration. Nat. Rev. Immunol. 7, 161–167. doi: 10.1038/nri2015
Pirttila, T., Mattinen, S., and Frey, H. (1992). The decrease of CD8-positive lymphocytes in Alzheimer’s disease. J. Neurol. Sci. 107, 160–165. doi: 10.1016/0022-510x(92)90284-r
Price, J. L., and Morris, J. C. (1999). Tangles and plaques in nondemented aging and “preclinical” Alzheimer’s disease Ann. Neurol. 45, 358–368. doi: 10.1002/1531-8249(199903)45:3<358::aid-ana12>3.0.co;2-x
Prinz, M., Erny, D., and Hagemeyer, N. (2017). Ontogeny and homeostasis of CNS myeloid cells. Nat. Immunol. 18, 385–392. doi: 10.1038/ni.3703
Prinz, M., and Priller, J. (2017). The role of peripheral immune cells in the CNS in steady state and disease. Nat. Neurosci. 20, 136–144. doi: 10.1038/nn.4475
Readhead, B., Haure-Mirande, J. V., Funk, C. C., Richards, M. A., Shannon, P., Haroutunian, V., et al. (2018). Multiscale analysis of independent Alzheimer’s cohorts finds disruption of molecular, genetic, and clinical networks by Human Herpesvirus. Neuron 99, 64–82.e7.
Reale, M., Iarlori, C., Gambi, F., Feliciani, C., Salone, A., Toma, L., et al. (2004). Treatment with an acetylcholinesterase inhibitor in Alzheimer patients modulates the expression and production of the pro-inflammatory and anti-inflammatory cytokines. J. Neuroimmunol. 148, 162–171. doi: 10.1016/j.jneuroim.2003.11.003
Rembach, A., Watt, A. D., Wilson, W. J., Rainey-Smith, S., Ellis, K. A., Rowe, C. C., et al. (2014). An increased neutrophil-lymphocyte ratio in Alzheimer’s disease is a function of age and is weakly correlated with neocortical amyloid accumulation. J. Neuroimmunol. 273, 65–71. doi: 10.1016/j.jneuroim.2014.05.005
Rezai-Zadeh, K., Gate, D., Szekely, C. A., and Town, T. (2009). Can peripheral leukocytes be used as Alzheimer’s disease biomarkers? Expert Rev. Neurother. 9, 1623–1633. doi: 10.1586/ern.09.118
Ribeiro, R. M., and Perelson, A. S. (2007). Determining thymic output quantitatively: using models to interpret experimental T-cell receptor excision circle (TREC) data. Immunol. Rev. 216, 21–34. doi: 10.1111/j.1600-065x.2006.00493.x
Richartz-Salzburger, E., Batra, A., Stransky, E., Laske, C., Kohler, N., Bartels, M., et al. (2007). Altered lymphocyte distribution in Alzheimer’s disease. J. Psychiatr. Res. 41, 174–178. doi: 10.1016/j.jpsychires.2006.01.010
Rizzo, R. (2020). Controversial role of Herpesviruses in Alzheimer’s disease. PLoS Pathog. 16:e1008575. doi: 10.1371/journal.ppat.1008575
Rocha, N. P., Teixeira, A. L., Coelho, F. M., Caramelli, P., Guimaraes, H. C., Barbosa, I. G., et al. (2012). Peripheral blood mono-nuclear cells derived from Alzheimer’s disease patients show elevated baseline levels of secreted cytokines but resist stimulation with beta-amyloid peptide. Mol. Cell. Neurosci. 49, 77–84. doi: 10.1016/j.mcn.2011.09.005
Rosenkranz, D., Weyer, S., Tolosa, E., Gaenslen, A., Berg, D., Leyhe, T., et al. (2007). Higher frequency of regulatory T cells in the elderly and increased suppressive activity in neurodegeneration. J. Neuroimmunol. 188, 117–127. doi: 10.1016/j.jneuroim.2007.05.011
Rosenzweig, N., Dvir-Szternfeld, R., Tsitsou-Kampeli, A., Keren-Shaul, H., Ben-Yehuda, H., Weill-Raynal, P., et al. (2019). PD-1/PD-L1 checkpoint blockade harnesses monocyte-derived macrophages to combat cognitive impairment in a tauopathy mouse model. Nat. Commun. 10:465.
Saresella, M., Calabrese, E., Marventano, I., Piancone, F., Gatti, A., Calvo, M. G., et al. (2010). PD1 negative and PD1 positive CD4+ T regulatory cells in mild cognitive impairment and Alzheimer’s disease. J. Alzheimers Dis. 21, 927–938. doi: 10.3233/jad-2010-091696
Schmitt, F. A., Nelson, P. T., Abner, E., Scheff, S., Jicha, G. A., Smith, C., et al. (2012). University of Kentucky Sanders-Brown healthy brain aging volunteers: donor characteristics, procedures and neuropathology. Curr. Alzheimer Res. 9, 724–733. doi: 10.2174/156720512801322591
Schmitt, V., Rink, L., and Uciechowski, P. (2013). The Th17/Treg balance is disturbed during aging. Exp. Gerontol. 48, 1379–1386. doi: 10.1016/j.exger.2013.09.003
Seidler, S., Zimmermann, H. W., Bartneck, M., Trautwein, C., and Tacke, F. (2010). Age-dependent alterations of monocyte subsets and monocyte-related chemokine pathways in healthy adults. BMC Immunol. 11:30. doi: 10.1186/1471-2172-11-30
Shahidehpour, R. K., Higdon, R. E., Crawford, N. G., Neltner, J. H., Ighodaro, E. T., Patel, E., et al. (2021). Dystrophic microglia are associated with neurodegenerative disease and not healthy aging in the human brain. Neurobiol. Aging 99, 19–27. doi: 10.1016/j.neurobiolaging.2020.12.003
Shalit, F., Sredni, B., Brodie, C., Kott, E., and Huberman, M. (1995). T lymphocyte subpopulations and activation markers correlate with severity of Alzheimer’s disease. Clin. Immunol. Immunopathol. 75, 246–250. doi: 10.1006/clin.1995.1078
Shaw, A. C., Joshi, S., Greenwood, H., Panda, A., and Lord, J. M. (2010). Aging of the innate immune system. Curr. Opin. Immunol. 22, 507–513.
Singh, V. K. (1994). Studies of neuroimmune markers in Alzheimer’s disease. Mol. Neurobiol. 9, 73–81. doi: 10.1007/bf02816106
Singh, V. K., Fudenberg, H. H., and Brown, F. R. (1987). Immunologic dysfunction: simultaneous study of Alzheimer’s and older Down’s patients. Mech. Ageing Dev. 37, 257–264. doi: 10.1016/0047-6374(86)90043-6
Skias, D., Bania, M., Reder, A. T., Luchins, D., and Antel, J. P. (1985). Senile dementia of Alzheimer’s type (SDAT): reduced T8+-cell-mediated suppressor activity. Neurology 35, 1635–1638. doi: 10.1212/wnl.35.11.1635
Sommer, A., Winner, B., and Prots, I. (2017). The Trojan horse - neuroinflammatory impact of T cells in neurodegenerative diseases. Mol. Neurodegener. 12:78.
Spani, C., Suter, T., Derungs, R., Ferretti, M. T., Welt, T., Wirth, F., et al. (2015). Reduced beta-amyloid pathology in an APP transgenic mouse model of Alzheimer’s disease lacking functional B and T cells. Acta Neuropathol. Commun. 3:71.
Su, C., Zhao, K., Xia, H., and Xu, Y. (2019). Peripheral inflammatory biomarkers in Alzheimer’s disease and mild cognitive impairment: a systematic review and meta-analysis. Psychogeriatrics 19, 300–309. doi: 10.1111/psyg.12403
Swardfager, W., Lanctot, K., Rothenburg, L., Wong, A., Cappell, J., and Herrmann, N. (2010). A meta-analysis of cytokines in Alzheimer’s disease. Biol. Psychiatry 68, 930–941.
Templeton, A. J., Ace, O., Mcnamara, M. G., Al-Mubarak, M., Vera-Badillo, F. E., Hermanns, T., et al. (2014a). Prognostic role of platelet to lymphocyte ratio in solid tumors: a systematic review and meta-analysis. Cancer Epidemiol. Biomarkers Prev. 23, 1204–1212. doi: 10.1158/1055-9965.epi-14-0146
Templeton, A. J., Mcnamara, M. G., Seruga, B., Vera-Badillo, F. E., Aneja, P., Ocana, A., et al. (2014b). Prognostic role of neutrophil-to-lymphocyte ratio in solid tumors: a systematic review and meta-analysis. J. Natl. Cancer Inst. 106:dju124.
Thakur, K. T., Miller, E. H., Glendinning, M. D., Al-Dalahmah, O., Banu, M. A., Boehme, A. K., et al. (2021). COVID-19 neuropathology at Columbia University Irving Medical Center/New York Presbyterian Hospital. Brain. doi: 10.1093/brain/awab148 [Epub ahead of print].
Tollefson, G. D., Godes, M., Warren, J. B., Haus, E., Luxenberg, M., and Garvey, M. (1989). Lymphopenia in primary degenerative dementia. J. Psychiatr. Res. 23, 191–199. doi: 10.1016/0022-3956(89)90024-1
Trieb, K., Ransmayr, G., Sgonc, R., Lassmann, H., and Grubeckloebenstein, B. (1996). APP peptides stimulate lymphocyte proliferation in normals, but not in patients with Alzheimer’s disease. Neurobiol. Aging 17, 541–547. doi: 10.1016/0197-4580(96)00068-1
Unger, M. S., Li, E., Scharnagl, L., Poupardin, R., Altendorfer, B., Mrowetz, H., et al. (2020). CD8(+) T-cells infiltrate Alzheimer’s disease brains and regulate neuronal- and synapse-related gene expression in APP-PS1 transgenic mice. Brain Behav. Immun. 89, 67–86. doi: 10.1016/j.bbi.2020.05.070
van der Willik, K. D., Fani, L., Rizopoulos, D., Licher, S., Fest, J., Schagen, S. B., et al. (2019). Balance between innate versus adaptive immune system and the risk of dementia: a population-based cohort study. J. Neuroinflammation 16:68.
van Duin, D., Mohanty, S., Thomas, V., Ginter, S., Montgomery, R. R., Fikrig, E., et al. (2007). Age-associated defect in human TLR-1/2 function. J. Immunol. 178, 970–975. doi: 10.4049/jimmunol.178.2.970
Velardi, E., Tsai, J. J., and Van Den Brink, M. R. M. (2020). T cell regeneration after immunological injury. Nat. Rev. Immunol. 21, 277–291. doi: 10.1038/s41577-020-00457-z
Vukmanovic-Stejic, M., Rustin, M. H., Nikolich-Zugich, J., and Akbar, A. N. (2011). Immune responses in the skin in old age. Curr. Opin. Immunol. 23, 525–531. doi: 10.1016/j.coi.2011.05.008
Waisman, A., Ginhoux, F., Greter, M., and Bruttger, J. (2015). Homeostasis of microglia in the adult brain: review of novel microglia depletion systems. Trends Immunol. 36, 625–636. doi: 10.1016/j.it.2015.08.005
Wenisch, C., Patruta, S., Daxbock, F., Krause, R., and Horl, W. (2000). Effect of age on human neutrophil function. J. Leukoc. Biol. 67, 40–45. doi: 10.1002/jlb.67.1.40
Wertheimer, A. M., Bennett, M. S., Park, B., Uhrlaub, J. L., Martinez, C., Pulko, V., et al. (2014). Aging and cytomegalovirus infection differentially and jointly affect distinct circulating T cell subsets in humans. J. Immunol. 192, 2143–2155. doi: 10.4049/jimmunol.1301721
Zlokovic, B. V., Gottesman, R. F., Bernstein, K. E., Seshadri, S., Mckee, A., Snyder, H., et al. (2020). Vascular contributions to cognitive impairment and dementia (VCID): a report from the 2018 National Heart, Lung, and Blood Institute and National Institute of Neurological Disorders and Stroke Workshop. Alzheimers Dement. 16, 1714–1733. doi: 10.1002/alz.12157
Keywords: monocytes, T cells, Treg, Th17, CD4, CD8, neuroimmunology, neuroinflammation
Citation: Lutshumba J, Nikolajczyk BS and Bachstetter AD (2021) Dysregulation of Systemic Immunity in Aging and Dementia. Front. Cell. Neurosci. 15:652111. doi: 10.3389/fncel.2021.652111
Received: 12 January 2021; Accepted: 28 May 2021;
Published: 22 June 2021.
Edited by:
Axel Montagne, University of Edinburgh, United KingdomReviewed by:
Clive Holmes, University of Southampton, United KingdomJoseph M. Castellano, Icahn School of Medicine at Mount Sinai, United States
Gareth R. Howell, The Jackson Laboratory, United States
Copyright © 2021 Lutshumba, Nikolajczyk and Bachstetter. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Adam D. Bachstetter, adam.bachstetter@uky.edu