
- Center for Neuroscience and Neurological Recovery, Methodist Rehabilitation Center, Jackson, MS, USA
The most common neuromuscular manifestation of West Nile virus (WNV) infection is a poliomyelitis syndrome with asymmetric paralysis variably involving one (monoparesis) to four limbs (quadriparesis), with or without brainstem involvement and respiratory failure. This syndrome of acute flaccid paralysis may occur without overt fever or meningoencephalitis. Although involvement of anterior horn cells in the spinal cord and motor neurons in the brainstem are the major sites of pathology responsible for neuromuscular signs, inflammation also may involve skeletal or cardiac muscle (myositis, myocarditis), motor axons (polyradiculitis), and peripheral nerves [Guillain–Barré syndrome (GBS), brachial plexopathy]. In addition, involvement of spinal sympathetic neurons and ganglia provides an explanation for autonomic instability seen in some patients. Many patients also experience prolonged subjective generalized weakness and disabling fatigue. Despite recent evidence that WNV may persist long-term in the central nervous system or periphery in animals, the evidence in humans is controversial. WNV persistence would be of great concern in immunosuppressed patients or in those with prolonged or recurrent symptoms. Support for the contention that WNV can lead to autoimmune disease arises from reports of patients presenting with various neuromuscular diseases that presumably involve autoimmune mechanisms (GBS, other demyelinating neuropathies, myasthenia gravis, brachial plexopathies, stiff-person syndrome, and delayed or recurrent symptoms). Although there is no specific treatment or vaccine currently approved in humans, and the standard remains supportive care, drugs that can alter the cascade of immunobiochemical events leading to neuronal death may be potentially useful (high-dose corticosteroids, interferon preparations, and intravenous immune globulin containing WNV-specific antibodies). Human experience with these agents seems promising based on anecdotal reports.
Introduction
West Nile virus (WNV), a mosquito-borne RNA flavivirus and human neuropathogen, was first isolated from a febrile woman in the West Nile region of Uganda, Africa in 1937 (Smithburn et al., 1940). During the 1940s and 1950s, transmission of WNV by mosquitoes was demonstrated, the close antigenic relationship with flaviviruses was described, and neutralizing antibody was found in many residents of East-Central Africa (for a historical review see Campbell et al., 2002). The virus also became recognized as a cause of human meningitis or encephalitis in elderly patients during an outbreak in Israel in 1957 (Spigland et al., 1958), although CNS involvement in middle-aged and younger subjects remained unusual and outcome in the younger age-group was generally excellent (Weinberger et al., 2001). During the 1960s to 1970s, birds were identified as a major host, horses were commonly infected, and there were large human epidemics in Africa and the Middle East. Europe also experienced its first WNV outbreak (Murgue et al., 2001). During the 1980s and 1990s, there were major outbreaks in Africa, Middle East, Europe, and Russia, although the Romania epidemic in 1996 marked the geographic transition of WNV epidemics from rural areas to urban industrialized areas (Campbell et al., 2002). In 1999, WNV gained entry into North America in the New York City outbreak (Nash et al., 1999). The viral strain introduced into the United States likely originated from a strain that was circulating in Israel during 1998 (Lanciotti et al., 1999). In the past decade, WNV has spread and is now widely established from Canada to Venezuela (World Health Organization1). In 2011, human cases were reported in Albania, Greece, Israel, Italy, Romania, Russia, and Mexico. Hence, the geographic range of WNV now includes six of seven continents, including Africa, Asia, Europe, Australia (subtype Kunjin), North America, and South America.
Before 1996, WNV was known to cause high fever, chills, malaise, headache, backache, arthralgia, myalgias, retro-orbital pain, and a maculopapular rash, but neurological symptoms were uncommon. However, since the New York City outbreak, severe neurological illness, including encephalitis and meningitis, has been reported much more frequently, together with neuromuscular manifestations. The diagnosis of WNV infection should be considered in any patient with an unexplained acute febrile or neurological illness during the summer months, particularly if recently exposed to mosquitoes. In such cases, serum should be tested for class M immunoglobulin (IgM) antibody to WNV, which indicates a recent infection. If there are signs of CNS involvement, cerebrospinal fluid (CSF) should be analyzed and also tested for WNV IgM antibody. CSF findings typically show increased leukocytes (usually >200 cells/mm3), increased protein, and normal glucose. Almost half of WNV meningitis patients may have at least 50% neutrophils in their initial CSF specimen (Tyler et al., 2006), followed by a shift to lymphocytosis. Imaging studies in WNV infection are frequently normal, although they may be useful in excluding other etiologies of acute myelomeningoencephalitis. When abnormal, findings are generally non-specific and without mass effect. T2-weighted magnetic resonance signal abnormalities have been reported in brainstem, deep gray structures (basal ganglia or thalami), and cerebellum (Petropoulou et al., 2005). However, other imaging series have found no definite predilection for any specific area of the brain parenchyma (Ali et al., 2005). In patients with WNV-associated limb paralysis, abnormal signal intensity may be more pronounced in the spinal cord ventral horns with enhancement around the conus medullaris and cauda equina (Petropoulou et al., 2005). However, follow-up MRIs may show complete resolution of signal abnormalities.
Neuromuscular manifestations are now recognized as a prominent feature in patients with WNV neuroinvasive disease (encephalitis, meningitis). In the 1999 New York City outbreak, more than 50% of patients with confirmed WNV encephalitis had severe muscle weakness as a cardinal sign (Nash et al., 1999). Weakness was an apparent risk factor predicting death in patients with WNV encephalitis (Nash et al., 1999; Petersen and Marfin, 2002). In the 2002 and 2003 WNV epidemics in the United States, neuromuscular manifestations were a well-recognized feature associated with increased morbidity and mortality (Jeha et al., 2003). In Colorado, the state with the most reported cases of WNV infection (2,943) and fatalities (63) during the 2003 epidemic, as many as 50% of patients with encephalitis had evidence of acute flaccid paralysis (Tyler, 2004).
West Nile Virus Poliomyelitis
In the original New York City outbreak, several case series attributed neuromuscular complications, particularly acute flaccid paralysis, to peripheral neuronal processes, namely Guillain–Barré syndrome (GBS), motor axonopathy, or severe axonal polyneuropathy (Nash et al., 1999; Asnis et al., 2000; Sampson et al., 2000). In the 2002 epidemic, more cases of WNV-associated acute flaccid paralysis were also seen across the Southern United States. These patients had asymmetric acute flaccid paralysis, absent deep tendon reflexes in affected limbs, preserved sensation, bowel, or bladder dysfunction, and respiratory distress. Electrodiagnostic studies in these subjects revealed markedly decreased or absent motor responses in the paretic limbs, preserved sensory responses, and widespread asymmetric muscle denervation, without evidence of demyelination or myopathy. The clinical and electrodiagnostic findings were classic for poliomyelitis and inconsistent with GBS or other peripheral nerve disorders. These cases were reported in the Centers for Disease Control Morbidity and Mortality Weekly Report on September 20, 2002 (Centers for Disease Control and Prevention, 2002), with clinical, laboratory, and electrophysiologic guidelines to help physicians discern poliomyelitis from GBS (Table 1). Subsequently, clinical, laboratory, and neurophysiologic findings suggested that WNV-associated acute flaccid paralysis was a poliomyelitis syndrome with involvement of anterior horn cells of the spinal cord (Glass et al., 2002; Leis et al., 2002; Li et al., 2003; Al-Shekhlee and Katirji, 2004). Soon thereafter pathologic confirmation of WNV poliomyelitis (Figures 1A–C) was provided by several groups (Doron et al., 2003; Jeha et al., 2003; Kelly et al., 2003; Leis et al., 2003a; Fratkin et al., 2004; Guarner et al., 2004). Our postmortem examinations of four patients with WNV infection from the 2002 epidemic, who developed muscle weakness and acute respiratory distress, showed that poliomyelitis was the major CNS finding in each case (Leis et al., 2003a; Fratkin et al., 2004). This is in agreement with the neuropathology of experimental or naturally occurring WNV infection in monkeys (Manuelidis, 1956), horses (Cantile et al., 2000, 2001), and birds (Steele et al., 2000). In these vertebrates, WNV shows a pronounced tropism for gray matter of the spinal cord, causing poliomyelitis. In contrast, peripheral nerves are not commonly involved.
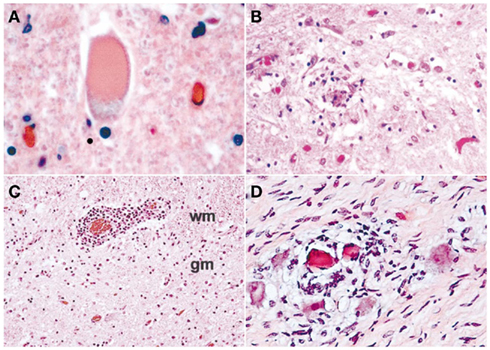
Figure 1. Spectrum of pathological findings in West Nile virus poliomyelitis: (A) Chromatolytic neuron with eccentric nucleus and distended cell body. Chromatolysis is usually triggered by damage to the cell body or axon. Neuronal recovery through regeneration can occur after chromatolysis, but most often it is a precursor of cell death (apoptosis). The event of chromatolysis is also characterized by a prominent migration of the nucleus toward the periphery of the cell and increase in the size of the cell body (hematoxylin–eosin, original magnification ×400); (B) Activated microglial cells surround and ingest a dead neuron (neuronophagia), which is typical of viral infections (hematoxylin–eosin, original magnification ×400); (C) Blood vessel at the interface between ventral horn gray matter and adjacent white matter surrounded by a dense cuff of chronic inflammatory cells (perivascular inflammation); wm, white matter; gm, gray matter (hematoxylin–eosin, original magnification ×100); (D) Cervical sympathetic ganglia. Microglial nodules are clustered around eosinophilic husks of dying ganglion cells. Microglial cells are consuming the pyknotic sympathetic neurons (neuronophagia; hematoxylin–eosin, original magnification ×400).

Table 1. Characteristics in West Nile virus-associated poliomyelitis compared with typical Guillain–Barré syndrome.
Most investigators actively involved in WNV clinical research now accept poliomyelitis to be the most common cause of WNV-associated acute flaccid paralysis in humans. Moreover, the CDC now classifies WNV infection into WNV fever or neuroinvasive disease, with further subdivision of the latter group into encephalitis, meningitis, and poliomyelitis. Patients with the poliomyelitis presentation commonly have associated signs of meningitis, encephalitis, or respiratory distress from involvement of spinal motor neurons supplying the phrenic nerves to the diaphragm, but acute flaccid paralysis also may occur in the absence of fever or meningoencephalitis (Leis et al., 2003b; Li et al., 2003; Sejvar et al., 2003). Recovery from neurological sequelae of WNV neuroinvasive disease may be slow and incomplete (Davis et al., 2006), with a poorer prognosis for recovery of physical function in patients with acute flaccid paralysis (Johnstone et al., 2011). Our initial four patients in 2002 with acute flaccid paralysis and profound loss of anterior horn cells in paretic limbs based on electrodiagnostic studies (CDC 2002; Leis et al., 2002) remain weak in affected limbs almost a decade after the acute WNV illness. However, each of these patients had needle electromyographic (EMG) evidence of profound denervation in muscles of the most affected limbs with few or no voluntarily recruited motor unit potentials (MUPs) and absent or markedly reduced motor responses with normal sensory responses on nerve conduction studies. Table 2 shows motor and sensory amplitudes in one of these patients, a 50-year-old man with WNV poliomyelitis limited to the right upper limb. Follow-up nerve conduction studies at 3-, 6-, and 12-month continued to show markedly reduced motor responses in proximal muscles with normal sensory responses. Follow-up needle EMG examinations also showed persistent profound denervation in shoulder girdle muscles with only a single voluntarily recruited MUP in deltoid and biceps (Figure 2). This patient never regained the ability to abduct or forward elevate the arm past the horizontal position. In contrast, limbs with lesser degrees of anterior horn cell loss, based on the presence of recordable motor responses and preserved innervation on needle EMG, had better recovery of function. Indeed, our observations after performing electrodiagnostic evaluations on many WNV patients with varying degrees of acute flaccid paralysis suggest that prognosis for recovery of function is greatly dependent on the degree of motor neuron loss; limbs with absent motor responses and no voluntary EMG activity have a poorer long-term prognosis while those with relatively preserved motor responses and some voluntary activity have a more favorable outcome.
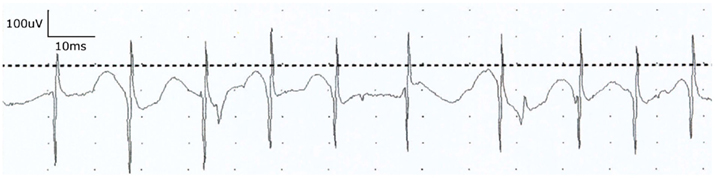
Figure 2. Electromyographic (EMG) examination in a 50-year-old man with WNV poliomyelitis limited to the right upper limb. Needle examination of the biceps muscle at 1 year follow-up showed persistent profound denervation with only a single voluntarily recruited rapidly firing motor unit potential. Similar denervation was noted in other shoulder girdle muscles. Clinical follow-up 5 years later revealed persistent severe weakness in proximal upper limb muscles.
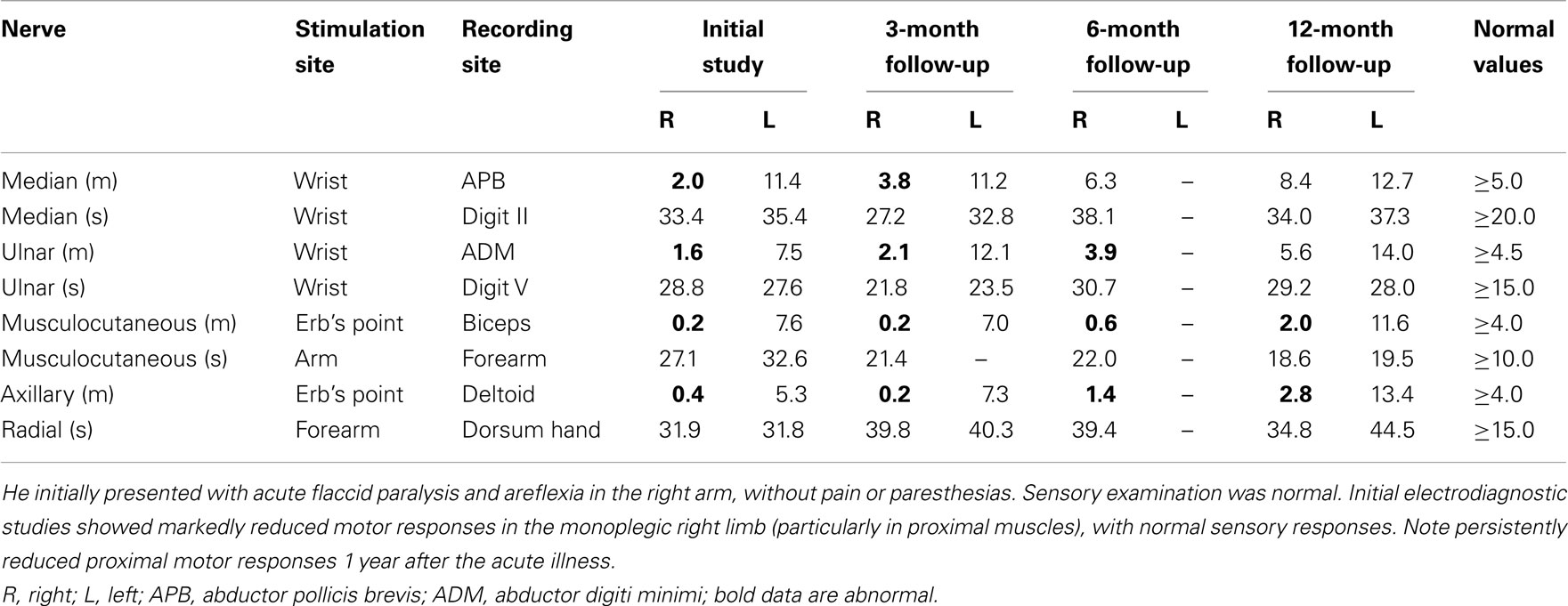
Table 2. Baseline-to-peak amplitudes of motor (m, in mV) and sensory (s, in μV) responses in a 50 year-old man with West Nile poliomyelitis of right upper limb.
Other Spinal Cord Neuropathology in West Nile Virus Infection
Although the anterior horns are the major site of spinal cord pathology (Kelly et al., 2003; Leis et al., 2003a; Fratkin et al., 2004), autopsy series show that pathologic changes may extend beyond the spinal cord gray matter with focal inflammatory changes involving the adjacent white matter (Fratkin et al., 2004). This may explain the infrequent occurrence of WNV-associated transverse myelitis with clinical involvement of spinal sensory and motor pathways (Leis, personal observation). In addition, neuronophagia, neuronal disappearance, and pathologic alterations have been described in dorsal root ganglia and sympathetic ganglia (Fratkin et al., 2004). Involvement of dorsal root ganglia may explain some of the sensory deficits and reduced sensory nerve action potentials on electrodiagnostic testing that occasionally are reported in patients with WNV infection (Doron et al., 2003; Jeha et al., 2003). However, sensory loss attributed to WNV has not been a prominent clinical finding. Another often overlooked finding is the disappearance of neurons in the sympathetic ganglia (Figure 1D), which offers a plausible explanation for the autonomic instability observed in some patients, including labile vital signs, hypotension, and potentially lethal cardiac arrhythmias (Pepperell et al., 2003; Fratkin et al., 2004; Bode et al., 2006). In rodents, WNV can lead to autonomic dysfunction by infecting neurons controlling cardiac and gastrointestinal function (Wang et al., 2011). These data lead to hypothesis that autonomic instability may play a role in the morbidity and mortality of human WNV infection.
West Nile Virus Spinal Nerve Root Involvement
Inflammatory changes in spinal cord gray matter also may extend into the spinal nerve roots to cause a myeloradiculitis (Jeha et al., 2003; Bouffard et al., 2004). Involvement of ventral spinal roots may contribute to the asymmetric acute flaccid paralysis seen in many patients with WNV infection. Magnetic resonance imaging findings showing apparent enhancement of ventral nerve roots support the concept that anterior radiculopathy should be considered, in addition to anterior horn cell pathology, when assessing patients with WNV-associated acute flaccid paralysis (Park et al., 2003). However, acute isolated radiculopathy as the sole neuromuscular manifestation of WNV infection is not common, although confusion may arise when acute flaccid paralysis attributable to poliomyelitis is limited to one limb (monoparesis; Leis et al., 2003b). In one series that included 10 patients with WNV poliomyelitis, 4 patients had monoparesis (Emig and Apple, 2004). We have also encountered several patients with WNV and asymmetric weakness in the arms or legs who initially were thought to have cervical or lumbosacral radiculopathies (Leis et al., 2002; Leis and Stokic, 2005).
West Nile Virus Peripheral Nerve Involvement
West Nile virus can involve peripheral nerves although this manifestation is much less frequent than originally assumed during the 1999 outbreak, when weakness and acute flaccid paralysis were attributed to GBS or axonal polyneuropathy (Nash et al., 1999; Asnis et al., 2000; Sampson et al., 2000). In 1 series of 64 patients with WNV infection, 3 patients had mixed axonal degenerating and demyelinating processes, and 1 had a pure demyelinating neuropathy (Pepperell et al., 2003). In addition, there are case reports of patients with true GBS (Ahmed et al., 2000; Xu et al., 2003; Sejvar et al., 2004), unilateral brachial plexopathy (Almhanna et al., 2003; Sejvar et al., 2004), and bilateral diaphragmatic paralysis attributed to loss of motor neurons or motor axons supplying the phrenic nerves (Betensley et al., 2004). Lymphocytic infiltration of nerves and occasional degenerating axons also has been described (Li et al., 2003; Smith et al., 2004), prompting a suggestion that WNV may reach the CNS via peripheral nerves (Smith et al., 2004). Indeed, recent evidence suggests that axonal transport mediates WNV entry into the CNS and induces acute flaccid paralysis (Samuel et al., 2007). However, most autopsy series do not suggest that WNV exhibits a predilection for peripheral nerves. Similarly, WNV infection in monkeys (Manuelidis, 1956), horses (Cantile et al., 2000, 2001), and birds (Steele et al., 2000) does not commonly involve peripheral nerves. In addition, it should be recognized that prolonged critical illness can be associated with an axonal sensorimotor polyneuropathy, termed critical illness polyneuropathy (Hund, 2001), which can confound the interpretation of WNV-associated polyneuropathy.
Neuromuscular Junction and Skeletal Muscle in West Nile Virus Infection
West Nile virus has not been reported to cause a defect in neuromuscular transmission, but we have encountered three patients with WNV poliomyelitis who developed classic myasthenia gravis with positive acetylcholine receptor antibodies and a marked decremental response on repetitive nerve stimulation studies (initial case reported in Leis and Stokic, 2005). We also are aware of the association of WNV and myasthenia gravis in two other patients (personal communication). Myopathy also is an uncommon manifestation of WNV infection, although there are several reports of rhabdomyolysis with creatine kinase levels as high as 45,000 U/L (normal <220 U/L; Doron et al., 2003; Jeha et al., 2003; Gupta et al., 2008). However, in one of these reports, postmortem examination confirmed poliomyelitis with striking loss of motor neurons in the anterior horn and brainstem, and only mild inflammation without necrosis in skeletal muscle (Doron et al., 2003). In another series, muscle biopsies on four patients with WNV with acute asymmetric paralysis showed scattered necrotic muscle fibers invaded by macrophages (two patients), normal muscle fibers with inflammatory cells surrounding small blood vessels (one patient), and a normal muscle biopsy (one patient; Li et al., 2003). In one of the patients with scattered necrotic muscle fibers, immunohistochemistry with polyclonal antibodies against flaviviruses did not detect WNV on biopsied muscle. These investigators acknowledged that the scattered necrotic muscle fibers were an unlikely explanation for the severe paralysis observed (Li et al., 2003). In the most comprehensive series of rhabdomyolysis in patients with WNV neuroinvasive disease, 9 of 244 hospitalized patients had rhabdomyolysis (median age 70 years, CK levels ranged from 1,153 to 42,113 IU; Montgomery et al., 2005). However, six of nine patients had history of recent falls prior to admission. The authors concluded that although the temporal relationship of rhabdomyolysis and WNV illness suggested a common etiology, these patients presented with complex clinical conditions that may have led to development of rhabdomyolysis from other causes (Montgomery et al., 2005). In addition, it is now commonly recognized that critical illness can be associated with a diffuse myopathy, termed critical illness myopathy, which can cause generalized weakness, respiratory failure, and inability to wean from the respirator (Lacomis et al., 2000). Accordingly, the role played by direct WNV invasion of muscles and the clinical significance of WNV-associated myositis remains to be elucidated.
West Nile virus has also been reported to cause myocarditis (Braun et al., 2006; Kushawaha et al., 2009) and cardiomyopathy (Khouzam, 2009), which can predispose to fatal arrhythmia. However, cardiac arrhythmias have also been reported to occur across the spectrum of WNV disease, including in cases of WNV fever not associated with myocarditis (Bode et al., 2006). Since statistics on the incidence of WNV myocarditis as the cause of cardiac arrhythmias are lacking, it is possible that autonomic instability caused by direct WNV infection of neurons controlling cardiac function (Fratkin et al., 2004; Wang et al., 2011) may also have precipitated cardiac arrhythmias.
West Nile Virus and Autoimmune Disease
An unresolved issue that may have important neuromuscular implications is whether WNV infection can induce autoimmune disease (Leis and Stokic, 2005). Support for this contention arises from the numerous reports of WNV patients with various neuromuscular diseases that have a presumed autoimmune mechanism, including GBS (Ahmed et al., 2000; Xu et al., 2003; Sejvar et al., 2004, 2005), other demyelinating neuropathies (Pepperell et al., 2003; Sumner and Jones, 2008), myasthenia gravis (Leis and Stokic, 2005), brachial plexopathies (Almhanna et al., 2003; Sejvar et al., 2004), and stiff-person syndrome (Hassin-Baer et al., 2004). In the latter case, stiff-person syndrome with antibodies to glutamic acid decarboxylase developed several weeks after WNV fever. CNS vasculitis with acute stroke or loss of vision due to WNV infection has also been reported (Kaiser et al., 2003; Kulstad and Wichter, 2003; Lowe et al., 2005). There is also experimental evidence that WNV causes a significant upregulation of class I and II major histocompatibility complex (MHC) molecules in Lewis rat Schwann cells (Argall et al., 1991). Moreover, irradiated medium from WNV-infected Schwann cell cultures upregulated class I molecule expression on dissociated Schwann cell cultures. These findings were considered to have implications for virally triggered autoimmune disease of nervous tissue. Infection of human embryonic myoblasts by WNV also causes a significant upregulation of class I and II MHC expression (Bao et al., 1992). MHC class I was increased after exposure to virus-inactivated supernatant from infected cells, indicating the presence of additional factors contributing to the MHC class I increase. The investigators concluded that these findings may be important in establishing a link between viral infection of human cells and induction of inflammatory autoimmune myositis (Bao et al., 1992). Other studies have shown that WNV can regulate the cell surface expression of numerous immune recognition molecules, resulting in increased recognition by WNV-specific and allo-specific cytotoxic T cells (Kesson et al., 2002). However, long-term follow-up of patients with WNV infection should clarify whether there is an increased incidence of autoimmune diseases.
Persistence of West Nile Virus in the Human Body
Acute WNV infection is usually cleared by an effective immune response after several days of viremia. Recent evidence, however, suggests that WNV may persist long-term in animals (Appler et al., 2010) and humans (Murray et al., 2010). Immunocompetent mice inoculated with WNV showed infectious virus or WNV RNA in the CNS and periphery for up to 6 months post-inoculation (Appler et al., 2010). Viral persistence occurred in the face of a robust antibody response and in the presence of inflammation in the brain. Furthermore, persistence in the CNS and pathological evidence of encephalitis were observed even in mice with subclinical infections. In such cases, the CNS immune response appears to be ineffective in clearing the virus (Stewart et al., 2011). In humans, WNV RNA was recently demonstrated in 5 (20%) of 25 urine samples collected from convalescent WNV patients diagnosed with WNV neuroinvasive disease between 2002 and 2007 (Murray et al., 2010). However, attempts to grow the virus from urine of these five patients were not successful. These findings suggest that in some individuals, WNV RNA may persist for several years after infection. These results may also have implications for some WNV-infected humans, particularly immunosuppressed patients or those with long-term or recurrent sequelae. Conversely, a subsequent study failed to demonstrate WNV RNA in urine samples collected from an established cohort of 40 persons over 6 years after initial infection with WNV. Urine from all participants tested negative for WNV RNA by reverse-transcription polymerase chain reaction and transcription-mediated amplification (Gibney et al., 2011). These conflicting findings suggest that prospective studies are needed to determine if and for how long WNV may persist in urine or other tissues following acute infection.
Spectrum of Neuromuscular Symptoms and Signs in West Nile Virus Infection
In our initial series of 54 WNV patients from 2002 to 2006, who had extensive electrodiagnostic evaluation and four autopsies, neuromuscular manifestations included: acute flaccid paralysis with electrophysiologic or pathologic features of poliomyelitis (n = 23), clinical findings of autonomic instability (cardiac dysrhythmias, marked fluctuations in blood pressure, gastrointestinal complications including gastroparesis) or pathologic alterations in sympathetic ganglia (n = 8), brainstem dysfunction (n = 4) including three cases of seventh nerve palsies (two delayed several weeks after acute illness and one with acute illness), myopathy (n = 3, in two attributed to critical illness myopathy and one case of rhabdomyolysis with acute renal failure), new onset myasthenia gravis (n = 3, one case diagnosed in 2011), diffuse axonal polyneuropathy (n = 2, one attributed to critical illness polyneuropathy and one to acute polyneuropathy associated with WNV), transverse myelitis with involvement of sensory and motor pathways (n = 1), gait apraxia (n = 1), and optic nerve involvement (n = 1). In the latter case, full-field pattern reversal visual evoked potential studies showed a monocular abnormality that suggested a conduction defect in the visual pathways anterior to the optic chiasm. In nine patients, the chief complaint was severe or disabling fatigue without objective muscle weakness on clinical examination or abnormalities on neurophysiologic studies. The specific physiologic mechanism for WNV fatigue remains unknown. Aside from the psycho-social factors, residual dysfunction in anterior horn cells and involvement of brainstem reticular activating system may play a role. In the post-polio syndrome, poliovirus-induced lesions in the reticular activating system are thought to contribute to the subjective fatigue (Bruno et al., 1994). In patients with WNV infection, prolonged fatigue is common after the acute illness (Campbell et al., 2002), affecting nearly two-thirds of patients. At 1 year follow-up, fatigue was the most common persistent symptom in patients hospitalized from the 1999 New York outbreak, affecting 67% of patients whereas muscle weakness was found in 44% of patients (New York Department of Health, 2001; Petersen and Marfin, 2002). Thus, physicians should be aware that fatigue and subjective weakness may be the major complaint in patients with WNV infection, particularly in those with WNV fever.
In addition, we observed two patients that developed severe, but reversible, muscle weakness that recovered completely within weeks. Both patients were hospitalized for their weakness. Weakness involved both lower limbs in one patient (paraparesis) and one upper limb in the other (monoparesis). Their neurophysiologic studies were unremarkable after recovery of function, in agreement with the clinical examination. The reversible muscle weakness likely reflects transient anterior horn cell dysfunction during the phase of acute WN illness, with full recovery of function occurring within several weeks. Although rapid reversal of paralysis has rarely been reported with WNV infection, this phenomenon is not new, and was first observed in patients with poliomyelitis caused by the poliovirus (Paul, 1971). Jacob von Heine (1799–1878) recognized the transitory nature of some attacks of paralysis early in the nineteenth century, and attributed the rapid improvement to fluid exudate and edema in the spinal cord that was reabsorbed (Paul, 1971). Therefore, in acute WNV infection, as in acute poliovirus infection, reversible paralysis may reflect transient anterior horn cell dysfunction. Two other patients developed exaggerated weakness in the distribution of preexisting lower motor neuron dysfunction after WNV infection. One of these patients developed persistent weakness limited to the same leg that had been symptomatic 14 years earlier from spinal stenosis caused by a synovial cyst. Another patient had a four-level lumbar laminectomy in 2001 for spinal stenosis that caused weakness, pain, and altered sensation in the right more than left leg. After surgery, symptoms improved although tingling persisted in the right leg. After acute WNV illness 11 months later, the patient reported disabling fatigue and exaggerated weakness in the same distribution as the preexisting weakness. In these patients, the observation that WNV infection caused exaggerated weakness in the previously weak limbs suggests that preexisting dysfunction may predispose anterior horn cells to additional injury during acute WNV infection. It is speculated that anterior horn cells that have survived an initial insult, or incorporated too many muscle fibers from denervated motor units beyond the metabolic capability, may be especially prone to injury (Leis et al., 2003b).
West Nile Virus Immunity and Pathogenesis
Although postmortem examinations have confirmed WNV poliomyelitis and encephalitis, including patchy infection of neurons in the cerebral cortex, hippocampus, basal ganglia, cerebellum, and brain stem (Doron et al., 2003; Kelly et al., 2003; Leis et al., 2003a; Fratkin et al., 2004; Guarner et al., 2004), the precise mechanisms that underlie destruction of neurons remain to be fully elucidated. The increased risk of severe WNV infection in immunosuppressed and elderly patients suggests that an intact immune system is essential for control of WNV. WNV-specific antibodies are responsible for reducing viremia and preventing development of severe disease, while different T-lymphocyte populations play an important role in clearing infection from tissues and preventing viral persistence (Lim et al., 2011). However, the pathologic effect of the immune system in WNV neuroinvasive disease cannot be overlooked. Studies with a neuroadapted strain of Sindbis virus, a related RNA neurotropic virus capable of inducing death of spinal motor neurons in rodents resulting in acute flaccid hind-limb paralysis, revealed that many degenerating neurons were not directly infected with the virus (Jackson et al., 1988; Havert et al., 2000; Darman et al., 2004). Rather, bystander neuronal death occurred as a result of a cascade of immunobiochemical events in the spinal cord that impaired glutamate transport and allowed excess glutamate to accumulate extracellularly around motor neurons. This excess synaptic glutamate is bound by calcium permeable glutamate receptors on post-synaptic motor neurons, causing the excitotoxic destruction of that neuron, whether it harbored the virus or not (Darman et al., 2004). WNV neuroinvasive disease is also characterized by increased production of pro-inflammatory cytokines derived from infected cells (Kumar et al., 2010) and upregulation of other pro-inflammatory genes (van Marle et al., 2007). These cytokines and other pro-inflammatory factors are either neurotoxic or attract leukocytes into the affected area, which further contribute to WNV-induced neurotoxicity. Hence, WNV-induced inflammation is now recognized as a major contributor of neuropathogenesis. The concept of a pathologic effect of the immune system in WNV neuroinvasive disease provides a framework for the development of anti-inflammatory drugs as much needed interventions to limit the cascade of immunobiochemical events leading to neurotoxicity (Kumar et al., 2010).
Treatment for Neuroinvasive West Nile Infection
At present, no specific therapy has been approved for human use in WNV infection. However, the merging knowledge about the pathogenesis of WNV infection has direct therapeutic implications. Treatment strategies that control the previously described cascade of events leading to neuronal death may prove beneficial. Promising therapies include the use of interferon and interferon inducers, which have been shown to reduce mortality in mice infected by subcutaneous injection of WNV (Morrey et al., 2004). A preliminary report described three WNV patients with neurologic syndromes who showed improvement after receiving interferon alpha, thereby increasing optimism toward this treatment (Sayao et al., 2004). Interferon gamma producing gamma-delta T cells also have been shown to prevent mortality from murine WNV infection (Wang et al., 2003). Other potential therapies include ribavirin, nucleic acids, RNA interference, antisense oligomers, peptides, imino sugars, and mycophenolic acid (for a review, see Diamond, 2009). These agents act through distinct mechanisms and are moving through various stages of pre-clinical development.
The role of corticosteroids in WNV neuroinvasive disease is controversial, with concern that immunosuppressive effects may worsen outcome. However, high-dose steroids have been used to successfully treat a patient with WNV-associated acute flaccid paralysis (Pyrgos and Younus, 2004). In addition, findings in 14 patients with acute infection suggest that intravenous dexamethasone may be the reason for shortening the acute phase of WNV meningoencephalitis and hastening patient recovery (Narayanaswami et al., 2004). We also have administered high-dose intravenous steroids to two patients with acute flaccid paralysis and brainstem involvement, including progressive seventh nerve palsies, who showed clear improvement in brainstem symptoms and facial paralysis within 24 h of treatment. However, both cases were characterized by an atypical temporal pattern of progressive symptoms or new deficits occurring several weeks after the onset of the acute illness. In most cases, WNV is thought to be cleared by an effective immune response after only several days of viremia. Accordingly, this relatively delayed progression of symptoms is more likely to reflect secondary injury from the downstream cascade of excitotoxic events and a secondary wave of inflammation. In cases where the temporal course suggests that indirect immune-mediated mechanisms may be contributing to neuronal injury, a trial of high-dose corticosteroids seems justified.
There also has been great interest in passive immunization with intravenous immune globulin (IVIG) for the treatment of patients with acute WNV infection. Immune serum frequently was used in the pre-antibiotic era to treat infectious diseases, and animal data indicate an important role for humoral immunity in controlling WNV infection (Gea-Banacloche et al., 2004). Given the endemic nature of WNV in the Middle East, IVIG obtained from Israeli blood donors that contains WNV-specific antibodies provided clear-cut protection to animals treated before or shortly after infectious challenge with WNV (Ben-Nathan et al., 2003). In contrast, IVIG obtained from blood donors from the United States without WNV-specific antibodies had no similar protective effect (Ben-Nathan et al., 2003). Several case reports also show efficacy of IVIG containing WNV-specific antibodies in aborting human WNV infection (Hamdan et al., 2002; Agrawal and Peterson, 2003), although other case studies found no clear benefit (Haley et al., 2003). A double-blind placebo-controlled multicenter trial was initiated in 2003 to compare the efficacy of an Israeli IVIG preparation that had a high-titer anti-WNV antibody (Omr-IgG-am; Omrix, Tel Aviv) with that of U.S. IVIG (Polygam; Baxter Healthcare Corp., Deerfield, IL, USA), which lacked detectable anti-WNV antibody. However, the study did not enroll enough cases to permit data analysis in the 2003 epidemic season. Nonetheless, neutralizing antibody therapeutics show promise in inhibiting WNV infection and preventing acute flaccid paralysis in vivo (Diamond, 2009), which justifies phase I and II studies using humanized or human monoclonal antibodies.
Differential Diagnosis of Neuroinvasive West Nile Infection
Our observations and literature review suggest that patients with WNV infection who have muscle weakness or other neuromuscular signs and symptoms often are given erroneous diagnoses and may receive inappropriate, potentially injurious treatments. Among patients referred to our rehabilitation center with WNV neuroinvasive disease, initial diagnoses that ultimately proved to be erroneous included evolving stroke, GBS, myopathy, food poisoning, endocarditis, sepsis, heat stroke, malingering, gastroenteritis, drug reaction, spinal cord compression, diabetic amyotrophy, and myocardial infarction. Diagnostic studies and therapies directed at these erroneous diagnoses are typically ineffective and can produce significant morbidity. Hence, physicians should consider a diagnosis of WNV infection in any patient who presents with a febrile illness that progresses over several days associated with neurological signs or symptoms, especially during the summer months (i.e., “summer flu” plus neurological symptoms). Health care providers also need to be aware that the spectrum of neuromuscular manifestations may range from a poliomyelitis syndrome in the absence of overt fever or meningoencephalitis to subjective weakness and disabling fatigue. This awareness will help to avoid less tenable diagnoses and inappropriate treatment.
The differential diagnoses of WNV infection include other arbovirus encephalitides (e.g., St. Louis encephalitis virus, Japanese encephalitis virus, Eastern, Western, and Venezuelan equine encephalitis viruses, tick borne encephalitis virus), other viral meningoencephalitides (La Crosse virus, Murray Valley virus, coxsackievirus, echovirus, enterovirus), bacterial meningitis or encephalitis (including Lyme disease and Leptospirosis), tick paralysis, and non-infectious conditions that affect brain or spinal cord (e.g., stroke, brain or spinal cord tumors, spinal cord compression). Guillian–Barre syndrome and other immune-mediated neuropathies are also diagnostic considerations, since in some cases WNV poliomyelitis may mimic these disorders (see Table 1). Poliovirus poliomyelitis should also be considered in the differential diagnosis if the patient resides in or travels to a polio-endemic region. However, in 2011 only four countries (India, Afghanistan, Nigeria, and Pakistan) remain polio-endemic (see text footnote 1). In addition, poliovirus infection mainly affects infants or young children.
Concluding Remarks
Although anterior horns are the major site of spinal cord pathology, inflammatory changes may also involve muscle fibers, peripheral nerves, spinal roots, and spinal sympathetic neurons and ganglia, contributing to the wide spectrum of neuromuscular manifestations of WNV infection. Unresolved issues with important implications include whether WNV may persist in the CNS or periphery, and whether WNV infection may lead to autoimmune disease. Although there is no specific treatment or vaccine approved for human WNV infection, several drugs that can alter the cascade of immunobiochemical events leading to neuronal death may be potentially useful.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnote
References
Agrawal, A. G., and Peterson, L. R. (2003). Human immunoglobulin as a treatment for West Nile virus infection. J. Infect. Dis. 188, 1–4.
Ahmed, S., Libman, R., Wesson, K., Ahmed, F., and Einberg, K. (2000). Guillain-Barre syndrome: an unusual presentation of West Nile virus infection. Neurology 55, 144–146.
Ali, M., Safriel, Y., Sohi, J., Llave, A., and Weathers, S. (2005). West Nile virus infection: MR imaging findings in the nervous system. AJNR Am. J. Neuroradiol. 26, 289–297.
Almhanna, K., Palanichamy, N., Sharma, M., Hobbs, R., and Sil, A. (2003). Unilateral brachial plexopathy associated with West Nile virus meningoencephalitis. Clin. Infect. Dis. 36, 1629–1630.
Al-Shekhlee, A., and Katirji, B. (2004). Electrodiagnostic features of acute paralytic poliomyelitis associated with West Nile virus infection. Muscle Nerve 29, 376–380.
Appler, K. K., Brown, A. N., Stewart, B. S., Behr, M. J., Demarest, V. L., Wong, S. J., and Bernard, K. A. (2010). Persistence of West Nile virus in the central nervous system and periphery of mice. PLoS ONE 5, e10649. doi:10.1371/journal.pone.0010649
Argall, K. G., Armati, P. J., King, N. J., and Douglas, M. W. (1991). The effects of West Nile virus on major histocompatibility complex class I and II molecule expression by Lewis rat Schwann cells in vitro. J. Neuroimmol. 35, 273–284.
Asnis, D. S., Conetta, R., Teixeira, A. A., Waldman, G., and Sampson, B. A. (2000). The West Nile virus outbreak of 1999 in New York City: the flushing hospital experience. Clin. Infect. Dis. 30, 413–418.
Bao, S., King, N. J., and Dos Remedios, C. G. (1992). Flavivirus induces MHC antigen on human myoblasts: a model of autoimmune myositis? Muscle Nerve 15, 1271–1277.
Ben-Nathan, D., Lustig, S., Tam, G., Robinzon, S., Segal, S., and Rager-Zisman, B. (2003). Prophylactic and therapeutic efficacy of human intravenous immunoglobulin in treating West Nile virus infection in mice. J. Infect. Dis. 188, 5–12.
Betensley, A. D., Jaffery, S. H., Collins, H., Sripathi, N., and Alabi, F. (2004). Bilateral diaphragmatic paralysis and related respiratory complications in a patient with West Nile virus infection. Thorax 59, 268–269.
Bode, A. V., Sejvar, J. J., Pape, W. J., Campbell, G. L., and Marfin, A. A. (2006). West Nile virus disease: a descriptive study of 228 patients hospitalized in a 4-county region of Colorado in 2003. Clin. Infect. Dis. 42, 1234–1240.
Bouffard, J. P., Riudavets, M. A., Holman, R., and Rushing, E. J. (2004). Neuropathology of the brain and spinal cord in human West Nile virus infection. Clin. Neuropathol. 23, 59–61.
Braun, L. E., Tsuchida, T., and Spiegel, H. (2006). Meningoencephalitis in a child complicated by myocarditis, quadriparesis and respiratory failure. Pediatr. Infect. Dis. J. 25, 853, 855–856.
Bruno, R. L., Cohen, J. M., Galski, T., and Frick, N. M. (1994). The neuroanatomy of post-polio fatigue. Arch. Phys. Med. Rehabil. 75, 498–504.
Campbell, G. L., Marfin, A. A., Lanciotti, R. S., and Gubler, D. J. (2002). West Nile virus. Lancet. Infect. Dis. 2, 519–529.
Cantile, C., Di Guardo, G., Eleni, C., and Arispici, M. (2000). Clinical and neuropathological features of West Nile virus equine encephalomyelitis in Italy. Equine Vet. J. 32, 31–35.
Cantile, C., Del Piero, F., Di Guardo, G., and Arispici, M. (2001). Pathologic and immunohistochemical findings in naturally occuring West Nile virus infection in horses. Vet. Pathol. 38, 414–421.
Centers for Disease Control and Prevention. (2002). Acute flaccid paralysis syndrome associated with West Nile virus infection- Mississippi and Louisiana, July-August 2002. MMWR 51, 825–828.
Darman, J., Backovic, S., Dike, S., Maragakis, N. J., Krishnan, C., Rothstein, J. D., Irani, D. N., and Kerr, D. A. (2004). Viral-induced spinal motor neuron death is non-cell-autonomous and involves glutamate excitotoxicity. J. Neurosci. 34, 7566–7575.
Davis, L. E., DeBiasi, R., Goade, D. E., Haaland, K. Y., Harrington, J. A., Harnar, J. B., Pergam, S. A., King, M. K., DeMasters, B. K., and Tyler, K. L. (2006). West Nile virus neuroinvasive disease. Ann. Neurol. 60, 286–300.
Diamond, M. S. (2009). Progress on the development of therapeutics against West Nile virus. Antiviral Res. 83, 214–227.
Doron, S. I., Dashe, J. F., Adelman, L. S., Brown, W. F., Werner, B. G., and Hadley, S. (2003). Histopathologically proven poliomyelitis with quadriplegia and loss of brainstem function due to West Nile infection. Clin. Infect. Dis. 37, e74–e77.
Emig, M., and Apple, D. J. (2004). Severe West Nile virus disease in healthy adults. Clin. Infect. Dis. 38, 289–292.
Fratkin, J. D., Leis, A. A., Stokic, D. S., Slavinski, S. A., and Geiss, R. W. (2004). Spinal cord neuropathology in human West Nile virus infection. Arch. Pathol. Lab. Med. 128, 533–537.
Gea-Banacloche, J., Johnson, R. T., Bagic, A., Butman, J. A., Murray, P. R., and Agrawal, A. G. (2004). West Nile virus: pathogenesis and therapeutic options. Ann. Intern. Med. 140, 545–553.
Gibney, K. B., Lanciotti, R. S., Sejvar, J. J., Nugent, C. T., Linnen, J. M., Delorey, M. J., Lehman, J. A., Boswell, E. N., Staples, J. E., and Fischer, M. (2011). West nile virus RNA not detected in urine of 40 people tested 6 years after acute West Nile virus disease. J. Infect. Dis. 203, 344–347.
Glass, J. D., Samuels, O., and Rich, M. M. (2002). Polio myelitis due to West Nile virus. N. Engl. J. Med. 347, 1280–1281.
Guarner, J., Shieh, W. J., Hunter, S., Paddock, C. D., Morken, T., Campbell, G. L., Marfin, A. A., and Zaki, S. R. (2004). Clinicopathologic study and laboratory diagnosis of cases with West Nile virus encephalomyelitis. Hum. Pathol. 35, 983–990.
Gupta, M., Ghaffari, M., and Freire, A. X. (2008). Rhabdomyolysis in a patient with West Nile encephalitis and flaccid paralysis. Tenn. Med. 101, 45–47.
Haley, M., Retter, A. S., Fowler, D., Gea-Banacloche, J., and O’Grady, N. P. (2003). The role for intravenous immunoglobulin in the treatment of West Nile virus encephalitis. Clin. Infect. Dis. 37, e88–e90.
Hamdan, A., Green, P., Mendelson, E., Kramer, M. R., Pitlik, S., and Weinberger, M. (2002). Possible benefit of intravenous immunoglobulin therapy in a lung transplant recipient with West Nile virus encephalitis. Transpl. Infect. Dis. 4, 160–162.
Hassin-Baer, S., Kirson, E. D., Shulman, L., Buchman, A. S., Bin, H., Hindiyeh, M., Markevich, L., and Mendelson, E. (2004). Stiff-person syndrome following West Nile fever. Arch. Neurol. 61, 938–941.
Havert, M. B., Schofield, B., Griffin, D. E., and Irani, D. N. (2000). Activation of divergent neuronal cell death pathways in different target cell populations during neuroadapted Sindbis virus infection of mice. J. Virol. 4, 5352–5356.
Jackson, A. C., Moench, T. R., Trapp, B. D., and Griffin, D. E. (1988). Basis of neurovirulence in Sindbis virus encephalomyelitis of mice. Lab. Invest. 58, 503–509.
Jeha, L. E., Sila, C. A., Lederman, R. J., Prayson, R. A., Isada, C. M., and Gordon, S. M. (2003). West Nile virus infection. A new acute paralytic illness. Neurology 61, 55–59.
Johnstone, J., Hanna, S. E., Nicolle, L. E., Drebot, M. A., Neupane, B., Mahony, J. B., and Loeb, M. B. (2011). Prognosis of West Nile virus associated acute flaccid paralysis: a case series. J. Med. Case Reports 5, 395.
Kaiser, P. K., Lee, M. S., and Martin, D. A. (2003). Occlusive vasculitis in a patient with concomitant West Nile virus infection. Am. J. Ophthalmol. 136, 928–930.
Kelly, T. W., Prayson, R. A., and Isada, C. M. (2003). Spinal cord pathology in West Nile virus infection. N. Engl. J. Med. 348, 564–566.
Kesson, A. M., Cheng, Y., and King, N. J. (2002). Regulation of immune recognition molecules by flavivirus, West Nile. Viral Immunol. 15, 273–283.
Khouzam, R. N. (2009). Significant cardiomyopathy secondary to West Nile virus infection. South. Med. J. 102, 527–528.
Kulstad, E. B., and Wichter, M. D. (2003). West Nile encephalitis presenting as a stroke. Ann. Emerg. Med. 41, 283.
Kumar, M., Verma, S., and Nerurkar, V. R. (2010). Pro-inflammatory cytokines derived from West Nile virus (WNV)-infected SK-N-SH cells mediate neuroinflammatory markers and neuronal death. J. Neuroinflammation 31, 7–73.
Kushawaha, A., Jadonath, S., and Mobarakai, N. (2009). West Nile virus myocarditis causing a fatal arrhythmia: a case report. Cases J. 2, 7147.
Lacomis, D., Zochodne, D. W., and Bird, S. J. (2000). Critical illness myopathy. Muscle Nerve 23, 1785–1788.
Lanciotti, R. S., Roehrig, J. T., Deubel, V., Smith, J., Parker, M., Steele, K., Crise, B., Volpe, K. E., Crabtree, M. B., Scherret, J. H., Hall, R. A., MacKenzie, J. S., Cropp, C. B., Panigrahy, B., Ostlund, E., Schmitt, B., Malkinson, M., Banet, C., Weissman, J., Komar, N., Savage, H. M., Stone, W., McNamara, T., and Gubler, D. J. (1999). Origin of the West Nile virus responsible for an outbreak of encephalitis in the northeastern United States. Science 286, 2333–2337.
Leis, A. A., Fratkin, J., Stokic, D. S., Harrington, T., Webb, R. M., and Slavinski, S. A. (2003a). West Nile poliomyelitis. Lancet. Infect. Dis. 3, 9–10.
Leis, A. A., Stokic, D. S., Webb, R. M., Slavinski, S. A., and Fratkin, J. (2003b). Clinical spectrum of muscle weakness in human West Nile virus infection. Muscle Nerve 28, 302–308.
Leis, A. A., and Stokic, D. S. (2005). Neuromuscular manifestations of human West Nile virus infection. Curr. Treat. Options Neurol. 7, 15–22.
Leis, A. A., Stokic, D. S., Polk, J. L., Dostrow, V., and Winkelmann, M. (2002). Poliomyelitis-like syndrome from West Nile virus infection. N. Engl. J. Med. 347, 1279–1280.
Li, J., Loeb, J. A., Shy, M. E., Shah, A. K., Tselis, A. C., Kupski, W. J., and Lewis, R. A. (2003). Asymmetric flaccid paralysis: a neuro muscular presentation of West Nile virus infection. Ann. Neurol. 53, 703–710.
Lim, S. M., Koraka, P., Osterhaus, A. D. M. E., and Martina, B. E. E. (2011). West Nile virus: immunity and pathogenesis. Viruses 3, 811–828.
Lowe, L. H., Morello, F. P., Jackson, M. A., and Lasky, A. (2005). Application of transcranial Doppler sonography in children with acute neurologic events due to primary cerebral and West Nile vasculitis. AJNR Am. J. Neuroradiol. 26, 1698–1701.
Manuelidis, E. E. (1956). Neuropathology of experimental West Nile virus infection in monkeys. J. Neuropathol. Exp. Neurol. 15, 448–460.
Montgomery, S. P., Chow, C. C., Smith, S. W., Marfin, A. A., O’Leary, D. R., and Campbell, G. L. (2005). Rhabdomyolysis in patients with west nile encephalitis and meningitis. Vector Borne Zoonotic Dis. 5, 252–257.
Morrey, J. D., Day, C. W., Julander, J. G., Blatt, L. M., Smee, D. F., and Sidwell, R. W. (2004). Effect of interferon-alpha and interferon-inducers on West Nile virus is mouse and hamster animal models. Antivir. Chem. Chemother. 15, 101–109.
Murgue, B., Murri, S., Triki, H., Deubel, V., and Zeller, H. G. (2001). West Nile in the Mediterranean basin: 1950-2000. Ann. N. Y. Acad. Sci. 951, 117–126.
Murray, K., Walker, C., Herrington, E., Lewis, J. A., McCormick, J., Beasley, D. W., Tesh, R. B., and Fisher-Hoch, S. (2010). Persistent infection with West Nile virus years after initial infection. J. Infect. Dis. 201, 2–4.
Narayanaswami, P., Edwards, L., Hyde, C., Page, C., and Hastings, N. E. (2004). West Nile meningitis/encephalitis: experience with cortico steroid therapy. Neurology 62, A404.
Nash, D., Mostashari, F., Fine, A., Miller, J., O’Leary, D., Murray, K., Huang, A., Rosenberg, A., Greenberg, A., Sherman, M., Wong, S., Layton, M., and West Nile Outbreak Response Working Group. (1999). The outbreak of West Nile virus infection in the New York City area in 1999. N. Engl. J. Med. 344, 1807–1814.
New York Department of Health. (2001). West Nile virus surveillance, and control: an update for healthcare providers in New York City. City Health Inf. 20, 1–7.
Park, M., Hui, J. S., and Bartt, R. E. (2003). Acute anterior radiculitis associated with West Nile virus infection. J. Neurol. Neurosurg. Psychiatr. 74, 823–825.
Pepperell, C., Rau, N., Krajden, S., Kern, R., Humar, A., Mederski, B., Simor, A., Low, D. E., McGeer, A., Mazzulli, T., Burton, J., Jaigobin, C., Fearon, M., Artsob, H., Drebot, M. A., Halliday, W., and Brunton, J. (2003). West Nile virus infection in 2002: morbidity and mortality among patients admitted to hospital in southcentral Ontario. CMAJ 168, 1399–1405.
Petersen, L. R., and Marfin, A. A. (2002). West Nile virus: a primer for the clinician. Ann. Intern. Med. 137, 173–179.
Petropoulou, K. A., Gordon, S. M., Prayson, R. A., and Ruggierri, P. M. (2005). West Nile virus meningoencephalitis: MR imaging findings. AJNR Am. J. Neuroradiol. 8, 1986–1995.
Pyrgos, V., and Younus, F. (2004). High-dose steroids in the management of acute flaccid paralysis due to West Nile virus infection. Scand. J. Infect. Dis. 36, 509–512.
Sampson, B. A., Ambrosi, C., Charlot, A., Reiber, K., Veress, J. F., and Armbrustmacher, V. (2000). The pathology of human West Nile virus infection. Hum. Pathol. 31, 527–531.
Samuel, M. A., Wang, H., Siddharthan, V., Morrey, J. D., and Diamond, M. S. (2007). Axonal transport mediates West Nile virus entry into the central nervous system and induces acute flaccid paralysis. Proc. Natl. Acad. Sci. U.S.A. 104, 17140–17145.
Sayao, A., Patry, D., Suchowersky, O., Al-Khathaami, A., and Klassen, B. (2004). A report of seven cases of West Nile virus neurological syndrome in Calgary, Alberta: clinical presentations and treatment with interferon alpha-2B. Neurology 62, A404.
Sejvar, J., Bode, A., Ewing, D., Mazowiecki, M., Pavot, P., Marfin, A., and Petersen, L. (2004). The spectrum of West Nile virus-associated weakness. Neurology 62, A446.
Sejvar, J. J., Bode, A. V., Marfin, A. A., Campbell, G. L., Ewing, D., Mazowiecki, M., Pavot, P. V., Schmitt, J., Pape, J., Biggerstaff, B. J., and Petersen, L. R. (2005). West Nile virus-associated flaccid paralysis. Emerging Infect. Dis. 11, 1021–1027.
Sejvar, J. J., Leis, A. A., Stokic, D. S., Van Gerpen, J. A., Marfin, A. A., Webb, R., Haddad, M. B., Tierney, B. C., Slavinski, S. A., Polk, J. L., Dostrow, V., Winkelmann, M., and Petersen, L. R. (2003). Acute flaccid paralysis and West Nile virus infection. Emerging Infect. Dis. 9, 788–793.
Smith, R. D., Konoplev, S., DeCourten-Myers, G., and Brown, T. (2004). West Nile virus encephalitis with myositis and orchitis. Hum. Pathol. 35, 254–258.
Smithburn, K. C., Hughes, T. P., Burke, A. W., and Paul, J. H. (1940). A neurotropic virus isolated from the blood of a native of Uganda. Am. J. Trop. Med. 20, 471–492.
Spigland, I., Jasinska-Klinberg, W., Hofshi, E., and Goldblum, N. (1958). Clinical and laboratory observations in an outbreak of West Nile fever in Israel. Harefuah 54, 275–281.
Steele, K. E., Linn, M. J., Schoepp, R. J., Komar, N., Geisbert, T. W., Manduca, R. M., Calle, P. P., Raphael, B. L., Clippinger, T. L., Larsen, T., Smith, J., Lanciotti, R. S., Panella, N. A., and McNamara, T. S. (2000). Pathology of fatal West Nile virus infection in native and exotic birds during the 1999 outbreak in New York City, New York. Vet. Pathol. 37, 208–224.
Stewart, B. S., Demarest, V. L., Wong, S. J., Green, S., and Bernard, KA. (2011). Persistence of virus-specific immune responses in the central nervous system of mice after West Nile virus infection. BMC Immunol. 12, 6. doi:10.1186/1471-2172-12-6
Sumner, N., and Jones, L. (2008). Multifocal neuropathy associated with West Nile virus infection. Neurology 71, 1123.
Tyler, K. L., Pape, J., Goody, R. J., Corkill, M., and Kleinschmidt-DeMasters, B. K. (2006). CSF findings in 250 patients with serologically confirmed West Nile virus meningitis and encephalitis. Neurology 66, 361–365.
van Marle, G., Antony, J., Ostermann, H., Dunham, C., Hunt, T., Halliday, W., Maingat, F., Urbanowski, M. D., Hobman, T., Peeling, J., and Power, C. (2007). West Nile virus-induced neuroinflammation: glial infection and capsid protein-mediated neurovirulence. J. Virol. 81, 10933–10949.
Wang, H., Siddharthan, V., Hall, J. O., and Morrey, J. D. (2011). Autonomic nervous dysfunction in hamsters infected with West Nile virus. PLoS ONE 6, e19575. doi:10.1371/journal.pone.0019575
Wang, T., Scully, E., Yin, Z., Kim, J. H., Wang, S., Yan, J., Mamula, M., Anderson, J. F., Craft, J., and Fikrig, E. (2003). IFN-gamma-producing gamma delta T cells help control murine West Nile virus infection. J. Immunol. 171, 2524–2531.
Weinberger, M., Pitlik, S. D., Gandacu, D., Lang, R., Nassar, F., Ben David, D., Rubinstein, E., Izthaki, A., Mishal, J., Kitzes, R., Siegman-Igra, Y., Giladi, M., Pick, N., Mendelson, E., Bin, H., and Shohat, T. (2001). West Nile fever outbreak, Israel, 2000: epidemiologic aspects. Emerging Infect. Dis. 7, 686–691.
Keywords: West Nile virus, infection, poliomyelitis, fever
Citation: Leis AA and Stokic DS (2012) Neuromuscular manifestations of west nile virus infection. Front. Neur. 3:37. doi: 10.3389/fneur.2012.00037
Received: 13 October 2011; Accepted: 26 February 2012;
Published online: 21 March 2012.
Edited by:
Marianne De Visser, Academic Medical Centre, NetherlandsReviewed by:
Marianne De Visser, Academic Medical Centre, NetherlandsS. H. Subramony, University of Florida College of Medicine, USA
Copyright: © 2012 Leis and Stokic. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: A. Arturo Leis, Methodist Rehabilitation Center and University of Mississippi Medical Center, Center for Neuroscience and Neurological Recovery, 1350 East WoodrowWilson, Suite 2, Jackson, MS 39216, USA. e-mail: aleis@mmrcrehab.org