 Adrianne S. Chesser
Adrianne S. Chesser- Neuroscience Graduate Program, Department of Anesthesiology, University of Rochester, Rochester, NY, USA
One of the defining pathological features of Alzheimer disease (AD) is the intraneuronal accumulation of tau. The tau that forms these accumulations is altered both posttranslationally and conformationally, and there is now significant evidence that soluble forms of these modified tau species are the toxic entities rather than the insoluble neurofibrillary tangles. However there is still noteworthy debate concerning which specific pathological forms of tau are the contributors to neuronal dysfunction and death in AD. Given that increases in aberrant forms of tau play a role in the neurodegeneration process in AD, there is growing interest in understanding the degradative pathways that remove tau from the cell, and the selectivity of these different pathways for various forms of tau. Indeed, one can speculate that deficits in a pathway that selectively removes certain pathological forms of tau could play a pivotal role in AD. In this review we will discuss the different proteolytic and degradative machineries that may be involved in removing tau from the cell. How deficits in these different degradative pathways may contribute to abnormal accumulation of tau in AD will also be considered. In addition, the issue of the selective targeting of specific tau species to a given degradative pathway for clearance from the cell will be addressed.
Introduction
Insoluble, fibrillar intraneuronal accumulations of pathological forms of the tau protein called neurofibrillary tangles (NFTs) are important and defining hallmarks of the Alzheimer disease (AD) brain. Indeed, the progression of AD can be neuropathologically staged based on the location and extent of tau pathology (1). The predominant post-translational modification of tau in the NFTs is phosphorylation; however numerous modifications have been noted including truncation, acetylation, nitration, and several others (2–4). Historically the NFTs were considered to be the toxic entities, however over the past decade a new conceptual framework has developed in which pathologically modified monomeric and/or soluble oligomeric forms of tau are considered to be the harmful species (5, 6). Nevertheless, determining exactly which forms of tau compromise neuronal function is still an area of significant investigation. Even though the modifications of tau that are the primary contributors to toxicity have not been conclusively determined, it is clear that tau plays an essential role in the pathogenesis of AD. Given that in animal models of AD reducing tau levels attenuates neuronal dysfunction (7, 8), and in humans the extent of tau pathology correlates with cognitive decline (9), there is a growing interest in defining the degradative pathways that remove tau from the cell. Also of importance is understanding the role of non-degradative cleavage in influencing the eventual clearance of tau. Numerous proteases have been shown to proteolyze tau including aminopeptidases (10–12), thrombin (13–15), human high temperature requirement serine protease A1 (HTRA1) (16), calpain (17–20), and caspases (21–24). Overall, however, most of these enzymes do not appear to be principally responsible for tau clearance. Instead, they are able to generate modified tau species which may then contribute to developing tau pathology, enhanced tau clearance, or both. The bulk of clearance of both physiological and pathological forms of tau is instead mediated by the proteasomal and autophagic degradative systems (25). The contribution of each of these pathways in the turnover of tau, and which forms of tau – including various proteolytic forms – are degraded by each pathway, is an area of significant interest. Our understanding of this issue to date will be reviewed below, and the role of tau proteolysis on subsequent degradation will be discussed. Delineating how these pathways may be compromised in AD and how this contributes to tau pathology is of great importance and could have significance for informing new therapeutic approaches.
Tau Proteolysis
Tau is a cytosolic, dynamically regulated protein. In differentiated PC12 cells, a pulse-chase experiment showed that ∼90% of the tau was degraded in 18 h (26). Normal, monomeric tau is likely a proteasomal substrate. However, there is evidence that tau is also a substrate for a wide range of proteases as indicated above. This is significant as tau proteolysis could be beneficial in disease by helping to enhance removal of abnormal tau from the cell. Alternately, it could be detrimental by generating toxic fragments. Below we will discuss the different proteases that have been shown to act on tau, at least in vitro, and the possible involvement of these proteolytic events in AD.
Aminopeptidases
Aminopeptidases are a group of enzymes that cleave from the N-terminal end of a protein. The family includes alanyl, arginyl, and glutamyl peptidases. Puromycin sensitive aminopeptidase (PSA) is an alanyl peptidase that is responsible for ∼90% of the aminopeptidase activity in the brain (10). PSA was identified as a potential player in tau pathology through a microarray analysis of gene expression in disease-vulnerable vs. disease-resistant brain regions in JNPL3 mice that overexpress a mutant form of tau (P301L) found in the disease frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17). These mice develop neurodegeneration in the cortex while the cerebellum is relatively spared [although in the original description of these animals pathology was found in the deep cerebellar nuclei (27)]. Interestingly, PSA was found to be elevated in the cerebellum of these TAUP301L mice (10). The levels of PSA are also higher in human cerebellum compared to cortex in both controls and FTD cases. A slight elevation in PSA was also observed in FTD cortices compared to controls. In addition, a non-functional PSA mutant exacerbated tau pathology in a Drosophila model of tauopathy, while overexpressing PSA ameliorated the tau phenotype and diminished tau levels (10). Overexpressing PSA had a similar effect in the TAUP301L mice, reducing the pathologic phenotype (delaying paralysis, increasing motor neuron density in the spinal cord, decreasing gliosis) and decreasing tau levels (12). PSA was able to cleave recombinant tau in vitro, as well as tau from control human brain (11). However, the data presented in this study suggest that PSA is cleaving tau from both the C- and N-terminal ends, which is not expected from an aminopeptidase. Additionally, other studies failed to demonstrate tau cleavage by PSA (28, 29). One explanation for these discrepancies may be the limitations of in vitro assays and experimental techniques. For example, the FTDP-17 mutant tau used in many studies, while relevant for human tauopathy, is not found in AD. Additionally, this form of tau may be processed differently than tau without this mutation. For example, it has been shown that the isomerase Pin1, which has been implicated in AD (30), had opposite effects on P301L and wild-type tau degradation (31). An alternative explanation for the effects of PSA may be that PSA is indirectly regulating tau degradation. PSA has been shown to be involved in the induction of autophagy and specifically the formation of autophagosomes, in a model of overexpressed mutant huntingtin (32). Thus, the in vivo effects of PSA on promoting tau clearance may relate to its ability to modulate the key clearance pathway for abnormal and aggregated proteins (to be described in more detail below).
Thrombin
Thrombin is a serine protease that is a well characterized component of the coagulation cascade. It is typically produced and secreted by endothelial cells, including those in the brain in response to hemodynamic injury. Thrombin may be inappropriately expressed in AD brain. A recent study showed that thrombin is elevated in microvessels isolated from AD brain compared to microvessels from control brain (33). Additionally, thrombin was present in the CSF of AD patients but not in that of controls (33). This is important, as thrombin can act as a neurotoxin by activating intracellular signaling cascades causing neurite retraction and stimulating apoptosis (34–36). Thrombin may also be influencing tau pathology, as treatment of immortalized hippocampal neuronal cells (HT22 cells) with thrombin resulted in the formation of thioflavin-S positive tau aggregates within 24 h, followed by an increase in cell death at 72 h (37). It is unclear how this exogenously applied thrombin may be altering tau within the cells. There are also data to suggest that thrombin may act intracellularly to mediate tau pathology. Thrombin is expressed within neurons and astrocytes in both normal and AD brain (38). In AD brain the staining pattern for thrombin and prothrombin was characteristic of the pattern of NFTs, although these structures were not colabeled with antibodies for tau (38). Evidence supporting a role for thrombin in tau proteolysis came initially from an in vitro study showing that thrombin degraded recombinant full-length tau from the N-terminus yielding a 25-kDa fragment, while preserving the microtubule binding repeat domain (13). A later study, however, showed that in N2a neuroblastoma cells expressing a construct of only the tau repeat domain, thrombin cleavage could still occur, indicating additional cleavage sites (15). Similar results were observed in an in vitro assay (15).
The products of thrombin proteolysis are potentially pathogenic. Thrombin cleavage of the repeat domain construct yielded fragments that rapidly aggregated, which closely correlated with toxicity in cell culture (15). These fragments can also induce the aggregation of full-length tau (39). A final point of interest relates to potential upstream modifications of tau. Endogenous tau is phosphorylated, and in AD, tau phosphorylation becomes dysregulated. This may interfere with subsequent processes including cleavage and degradation. For example, tau that is in the cis-conformation at T231 appears resistant to degradation, as cis-tau is found in dystrophic neurites while trans-tau is not. Additionally cis-tau partitions to the insoluble fraction (30). Phosphorylation at T231 prevents the isomerase Pin1 from converting cis-tau to trans-tau (30). Interestingly, phosphorylation of tau also appears to disrupt some thrombin cleavage sites, changing the pattern of cleavage without impeding the thrombin-mediated proteolysis (14, 28). It has yet to be determined whether there is a difference in toxicity potential between fragments generated from phosphorylated vs. unphosphorylated tau. Nonetheless, thrombin is a potential candidate for contributing to tau proteolysis and pathology.
Human High Temperature Requirement Serine Protease A1
Another serine protease recently implicated in tau processing is HTRA1. This is a ubiquitously expressed, ATP-independent intracellular protease. Expression is detectable in many tissues, including the nervous system, although expression is low (40). Nonetheless, this enzyme was initially implicated in AD because it may play a role in amyloid processing (41). Tubulin was later identified as a substrate for HTRA1, suggesting HTRA1 may be involved in mediating microtubule function (42, 43). A more recent study showed that HTRA1 can cleave recombinant tau in vitro into multiple fragments of varying sizes, and furthermore can degrade insoluble and fibrillarized tau (16). This ability to degrade aggregates is particularly intriguing, especially in light of the fact that HTRA1 has potential chaperone activity due to its C-terminal PDZ domains and has a preference for misfolded substrates (44). While more work needs to be done on the role this enzyme plays in tau proteolysis, these findings further indicate the complexity and likely involvement of multiple players in this process.
Calpains
Calpains are calcium-activated cytosolic cysteine proteases. Two isoforms differentiated and named by their sensitivities to calcium (i.e., μ-calpain and m-calpain, also called calpain-1 and calpain-2) are abundant in the central nervous system, and respond to micromolar and millimolar concentrations of calcium, respectively (45). Calpain has been implicated in a number of neurodegenerative diseases [for a review, see (46)]. The active form of calpain-2 is found in 50–75% of NFTs in tauopathies including AD, but not in protein aggregates found in other diseases (47). This is consistent with another study that found equivalent calpain levels between control and AD cases, but the activity level of the enzyme isolated from AD brain tissue was increased (48). Excitotoxicity leading to elevated intracellular calcium is a common feature of neurodegenerative diseases, and is implicated in AD (49, 50). This process may lead to enhanced activation of calpains (51). This in turn could influence a number of pathologic processes, including tau proteolysis. Indeed, tau has a number of putative calpain cleavage sites, and incubation of recombinant tau with calpain generates specific fragments, including one that is ∼35 kDa and one that is ∼17 kDa (19, 20). Increasing intracellular calcium levels in PC12 cells leads to calpain-induced cleavage of tau (18). This may reflect a potential effect of excitotoxicity in AD. Inducing apoptosis in cerebellar granule cells yields calpain-mediated tau fragments, including a dominant ∼17 kDa fragment (17). Also, treating primary hippocampal neurons with pre-aggregated amyloid β (Aβ) led to the generation of tau fragments of ∼35, ∼24, and ∼17 kDa, which was blocked by addition of a calpain inhibitor (52, 53). Tau fragments of the same size were also found in AD brain tissue (19).
The pathological role of this calpain-cleaved tau is unclear. While some studies demonstrate toxicity resulting from calpain proteolysis of tau, other studies do not support this conclusion. On the one hand, expressing a 17-kDa fragment of tau based on calpain cleavage site mapping in hippocampal neurons led to neurite retraction and the appearance of varicosities after 48 h (52). Additionally, suppressing calpain activity in a fly model of tauopathy prevented neurodegeneration, as did expressing a calpain-resistant form of tau (54). In contrast, another study used mass spectroscopy and sequencing to identify the “17 kDa” tau cleavage product and found it did not correspond to the recombinant fragment utilized in the above studies (19). Expression of a recombinant form of the mass spectroscopy-identified fragment in hippocampal neurons was not toxic (19). Further studies are needed to clarify the contribution of calpain-mediated proteolysis of tau to AD pathology.
Caspases
There is significant evidence that tau is a caspase substrate and that caspase-mediated tau cleavage may play a role in AD pathology. Early in vitro studies demonstrated that tau is cleaved in the C-terminus by several caspases including caspase-3 and caspase-6 (21–23). Caspase-6 was also shown to cleave the N-terminus of tau in vitro (24). Caspase-3, which is a key effector in the apoptotic cascade, cleaves tau predominantly at the C-terminal D421 site generating a fragment often referred to as tauC3 (22, 23). There may be reciprocity with the apoptosis pathway as activating caspase-3 by inducing apoptosis in cortical neuronal culture led to tau cleavage (22), and selectively expressing tauC3 led to apoptosis in NT2 and COS cells (21). This might represent a feed-forward loop of neurotoxicity. Furthermore, expressing a cleavage resistant form of tau (D421E) protects cells from apoptotic cell death (22). Another potential mechanism of inducing caspase-3 cleavage of tau is the presence of Aβ peptides. TauC3 is formed in primary cortical neurons after treatment with Aβ (23).
Caspase cleavage of tau may play a role in stimulating the tau aggregation seen in AD. Indeed, in vitro polymerization assays demonstrate that caspase-cleaved tau has a greater propensity to aggregate compared to full-length tau (23, 55). Intriguingly, caspase activation was shown to immediately and consistently precede the formation of tangles (56). This group used in vivo multiphoton imaging in Tg4510 TAUP301L mice to simultaneously image activated caspases and Thioflavin-S positive tau tangles. There was a strong correlation between active caspases and the presence of tangles within viable neurons. In the few cells found that were caspase-positive and tangle-negative, 88% had tangles within 24 h (56). This seems to further support a role for caspase cleavage in the evolution of tau pathology.
In order for caspase to cleave tau in the AD brain, it needs to be present in its active form. The active forms of both caspase-3 and caspase-6 are elevated in AD-specific brain regions (temporal and frontal lobes) compared to unaffected regions (cerebellum) and control brains (57, 58). Furthermore, active caspase co-localizes to NFTs (58), and caspase-cleaved tau is found in AD-affected brain regions, particularly in neurons displaying tangle pathology (59, 60). This includes tau cleaved by caspase-6 in the C-terminus (58–60) as well as in the N-terminus (24). TauC3 is present in AD brain – in neurons and co-localized with NFTs – and inversely correlates with cognitive function (55, 60, 61).
The activation of caspases and the subsequent cleavage of tau is likely to occur independent of apoptotic cell death (56). The processes that may result in the activation of caspases in an apoptosis independent manner have not been clearly delineated; however several possibilities have been suggested. First, inflammation, which is a common feature of AD, may contribute to tau pathology by activating caspases. Treating cells with the prostaglandin cyclopentenone byproduct PGJ2 increased caspase activity and increased cleaved tau (62). Thrombin signaling can also activate caspases (36). Proteasomal impairment appears to be upstream of caspase activation, as inhibiting the proteasome with epoxomicin (EPX) led to activation of caspase-3 in primary neurons (63) and in a neuroblastoma cell line expressing wild-type tau (64). In both studies caspase activation correlated with the appearance and increase over time of caspase-cleaved tau species, which appeared to subsequently form aggregates in the neurons (63). While the mechanism is unclear, a possibility is that accumulating proteins might be a factor in initiating caspase activation.
Proteolysis vs. Degradation
As discussed above, a number of enzymes have been shown to act on tau, under potentially pathological, as well as physiological conditions. Many of these enzymes cleave tau at discrete sites, generating specific fragments. Some of these fragments, such as those generated by thrombin, calpain, and caspase, are potentially toxic to the cell if they accumulate due to inefficient clearance mechanisms. Figure 1 illustrates the potential contribution of these different proteases to the processing of tau. These proteolytically generated tau fragments can show an increased propensity for self-association, prior to the formation of overt aggregates. Thus, in the context of enhanced proteolysis (for example by caspases) there may be increased low-order oligomers formed by cleaved tau species. These oligomers may be unable to be cleared as effectively by the cell and contribute to neuronal dysfunction. Therefore coordination between proteolytic processing of tau and clearance by degradative pathways is essential for maintaining the appropriate levels of tau in a functional state. Below we will discuss the main degradative pathways of the cell-the proteasome and autophagy-which likely clear full-length tau as well as proteolytically generated tau fragments.
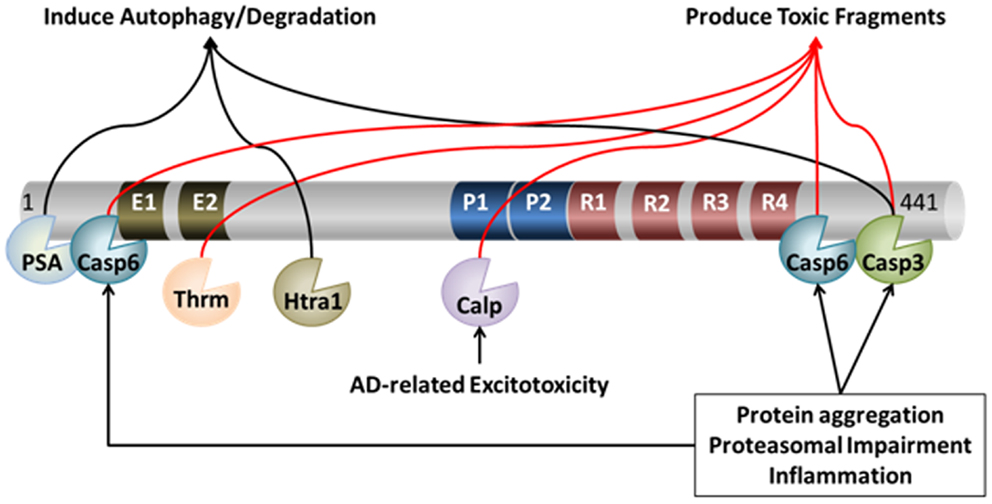
Figure 1. Proteolytic processing of tau. Under pathological and physiological conditions, tau undergoes cleavage at many distinct proteolytic sites by a myriad of proteases. The action of these proteases can lead to both protection and/or exacerbation of pathology. For example, cleavage of tau by caspase (Casp) 3, caspase-6, calpain (Calp), and thrombin (Thrm) leads to the production of toxic fragments of tau that exacerbate pathology. On the other hand, cleavage of tau by PSA, Htra1, and – in some circumstances – caspase-3, may facilitate its degradation, which may protect neurons from AD-related neuronal death.
The Proteasome
The proteasome is a multimeric barrel-shaped structure that is a key complex for clearing soluble cytosolic proteins. The 26S proteasome has a regulatory cap (19S, or alternatively the 11S regulatory particle) on either end of its catalytic core (20S), which contains the proteolytic activities and degrades substrates tagged with poly-ubiquitin chains as the targeting sequence. The regulatory cap in conjunction with chaperone proteins unfolds the protein substrate and removes the ubiquitin tag in an ATP-dependent process prior to feeding the protein into the catalytic core, where it is systematically degraded by the enzymatic properties of the proteasome. The 20S proteasome, which is the catalytic core without its regulatory caps, is also able to degrade natively unfolded substrates directly through an ATP- and ubiquitin-independent process. As shown in Figure 2, tau is an ideal proteasomal substrate for either form of the proteasome because it is a relatively small, unfolded, short-lived cytosolic protein (64–67).
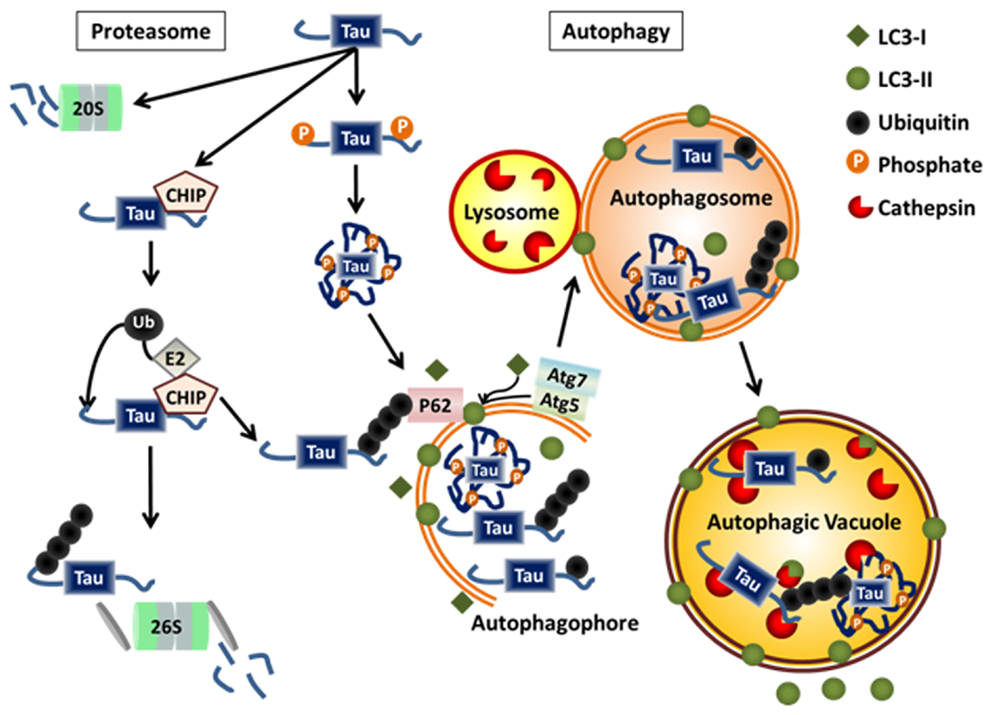
Figure 2. Physiological degradation of tau. Tau is degraded by both the proteasome and autophagy systems. Targeting of tau to either system may be determined by the extent and nature of post-translational modifications, the folding state, the level of aggregation, and its interaction with chaperone proteins or ubiquitin ligases. Monomeric tau is natively unfolded making it a likely target for the 20S proteasome. Monomeric tau also interacts with the E3 ligase, CHIP, which can lead to its ubiquitylation and degradation via the 26S proteasome or autophagy. Certain cleavage products and phosphorylated forms of tau, as well as, monoubiquitylated tau and tau aggregates are selectively degraded by autophagy.
The accumulation of proteins in AD patients’ brains generated interest in the role of proteasomal function. There is evidence suggesting that proteasomal activity, but not protein level, is decreased in AD-sensitive brain regions specifically compared to unaffected regions (68, 69). Additionally, tau appears to be physically associated with the proteasome in brain tissue from AD cases. When tau was immunoprecipitated it pulled down both the 26S and 20S proteasomes, while immunoprecipitating for the 20S catalytic core pulled down tau (69). This suggests tau is being targeted to the proteasome, but may also indicate impaired ability to complete degradation; hence it is remaining associated with the proteasome. Further, there was an inverse correlation between proteasomal activity and high molecular weight forms of tau (69). This may suggest that abnormal proteins themselves may interfere with proteasomal degradative processes. Indeed, in vitro aggregated paired helical filament tau could inhibit proteasome activity (69).
Evidence that Tau is Degraded by the Proteasome
A number of studies have used various in vitro techniques to analyze proteasomal degradation of tau. These include cell culture and cell free studies. Not surprisingly, if recombinant tau is incubated with isolated 20S proteasomal complexes, degradation occurs (65). In this system proteolysis is bidirectional. Also, if tau is first ubiquitylated in an in vitro reaction and then incubated with isolated 26S proteasomes supplemented with MgCl2 and ATP, degradation proceeds (66). These data indicate tau can be a substrate for both forms of the proteasome. Similar data has been obtained from studies using various cell culture systems as well as animal tissue and primary cultures with a variety of proteasomal inhibitors. When HEK cells are co-transfected with tau and ubiquitin, tau accumulates in the insoluble fraction. Its accumulation in the insoluble fraction is enhanced by proteasomal inhibition (using ALLN or MG-132) suggesting that tau is degraded by the proteasome (66). In SH-SY5Y neuroblastoma cells, treatment with lactacystin, a selective inhibitor of the 20S catalytic core, maintained levels of transfected wild-type full-length tau (4R0N) after cycloheximide treatment halted protein synthesis (65). Similarly, overexpressing the FTDP-17 mutant P301L tau in SH-SY5Y cells and then treating with lactacystin led to significantly increased tau levels (70). Lactacystin also caused accumulation of endogenous tau in the HT22 murine neuronal cell line (71). In immortalized mouse cortical neuronal cells inducibly expressing full-length wild-type tau, EPX slowed the degradation of full-length tau (72). In M1C neuroblastoma cells that inducibly express wild-type full-length tau (4R0N), EPX, and MG-132 induced accumulation of full-length tau but there was a concomitant loss of C-terminus immunoreactivity (64). This was attributed to caspase cleavage, as activated caspase-3 was detected, and a caspase inhibitor preserved C-terminal immunoreactivity (64). Additionally, incubation of rat brain extract (containing endogenous tau and proteasomal enzymes) with the proteasome activators Mg2+ and ATP resulted in lower total tau levels with an increase in smaller forms, compared to extract not supplemented with Mg2+ and ATP (73). The loss of tau was blocked by lactacystin giving further evidence that the proteasome was degrading tau (73). The story is more complex, however, as proteasomal inhibition under physiological conditions does not consistently lead to tau accumulation. For example, treatment of primary neurons with an Hsp90 inhibitor to interrupt the proper chaperoning of tau leads to decreased levels of tau. Adding MG-132 to block the proteasome prevented the Hsp90 inhibitor-induced reduction in total tau. MG-132 alone had no effect on tau levels (67). This might suggest that under normal circumstances, if proteasomal impairment occurs, tau levels are maintained by autophagic degradation. But when the system is pushed to promote proteasomal degradation over autophagy – such as by inhibiting Hsp90 – then the homeostatic maintenance of tau levels is disrupted and tau degradation does not occur when the proteasome is inhibited.
Autophagy
Autophagy is the process of “self-eating.” Under starvation conditions, bulk autophagy can be induced to catabolize cellular substrates to generate energy. However it is now evident that autophagy is an ongoing clearance mechanism for larger, longer-lived proteins and aggregates, as well as organelles such as mitochondria and peroxisomes (74) and pathogenic bacteria (75–77). There are three forms of autophagy: microautophagy, macroautophagy, and chaperone-mediated autophagy. The most common and well understood is macroautophagy, hereafter referred to simply as autophagy. For a more complete review of autophagy, see (78). Briefly, a double membrane autophagophore is initiated and subsequently expanded to engulf a region of cytoplasm containing the substrate/substrates to be degraded, such as tau (see Figure 2). Once fully formed into an enclosed vesicle called an autophagosome, it is trafficked to a lysosome where it undergoes fusion to become an autophagic vacuole (AV). The lysosomal enzymes degrade the inner membrane of the autophagosome as well as the delivered contents. The enzymes responsible for degrading protein substrates of autophagy are the cathepsins. Once the contents are fully degraded the lysosome is regenerated via acidification through vacuolar ATPases. There are 15 core autophagy related genes (Atgs) that are involved in the process of autophagy. Many of these have E1, E2, or E3 ligase activity to catalyze the reactions necessary for the initiation and expansion of the autophagosomal membrane. Critical early steps in the formation of the autophagopore require a complex of Atg proteins that conjugate phosphatidylethanolamine onto Atg8 family members (including LC3), a process that is critical for allowing expansion of the autophagosomal membrane. Conjugated LC3, called LC3-II, is the canonical marker of autophagosomes. Atg7 is a critical E1 ligase for several of the reactions necessary for autophagy (74).
Evidence that Autophagy is Impaired in AD
There is significant support for the possibility of defective autophagy in AD. Electron microscopic analysis of brain tissue from confirmed AD cases revealed that AVs accumulated in dystrophic neurites and correlated with the presence of filamentous tau (79). However, this correlation was not quantified (79). Similar results were observed in mouse models of AD. For example, in a presenilin 1 (PS1)/Amyloid Precursor Protein (APP) double transgenic mouse, AVs were prevalent in dystrophic neurites at as early as 4.5 months without a similar accumulation of other structures such as lysosomes (80, 81). In these transgenic mice LC3-positive bodies were particularly apparent in neurites surrounding amyloid plaques, and immunoblotting of hippocampi from 6 month old transgenic PS1/APP mice revealed increased levels of LC3-II compared to wild-type mice (81). It is well established that mutations in PS1 result in familial AD, and until recently it was thought that this was only due to alterations in APP processing. However PS1 has a number of non-secretase functions, including acting as the chaperone for the vacuolar-ATPase used to acidify the lysosomal lumen (82, 83). Mutations in PS1 were shown to impair the acidification of lysosomes, which is necessary for activating the proteolytic enzymes in this compartment. Improper acidification and impaired proteolysis of substrates would compromise the autophagy system and result in the accumulation of AVs as described above. However, another mouse model, the TgCRND8 mouse, which expresses mutant APP only, also has increased staining for LC3-II, as well as an increase in cathepsin D-positive lysosomes (84). This demonstrates that in the absence of mutant PS1, AD-associated impairment in autophagy occurs and thus is due to other factors. Treatment of ex vivo hippocampal slice cultures with lysosomal disruptors causes the formation of enlarged, dystrophic neurites filled with AVs and lysosomes, similar to what is seen in mouse AD models and human AD tissue (85, 86). It has also been suggested that specific cathepsins may become extralysosomal in certain diseases, including AD (87, 88). Together these observations implicate a possible failure of autophagy as part of AD pathogenesis.
Evidence Tau Can be Degraded by Autophagy
As indicated above, a functioning lysosomal compartment is critical for the completion of autophagy. Given the possibility of a defect at this level of autophagy, numerous studies have directly assessed the effects of impairing lysosomal function on tau turnover, including specifically targeting the cathepsins. In an early study the direct cleavage of tau by cathepsin D was investigated in an in vitro assay using tau partially purified from rat brain in combination with cathepsin D from human liver. Incubation of tau with cathepsin D at pH 4.0 resulted in a decrease in full-length tau and a concomitant increase in cleaved fragments of varying sizes (89). Similarly, adding exogenous cathepsin D to homogenates of rat cortex at a neutral pH also generated tau fragments. Intriguingly, if a cysteine protease inhibitor was added to the assay, tau cleavage stopped at the 29-kDa fragment, suggesting that cathepsin D (an aspartyl protease) could cleave tau to a 29-kDa fragment after which other proteases may act to further degrade the protein. This also suggests if cathepsin D was able to escape from the lysosome, for example in the context of an AD-related stressor, it could still function in the neutral environment of the cytosol. However, the activity of cathepsin D at the more neutral pH may be more impeded than appears, as a previous study found cathepsin D’s proteolytic activity was significantly reduced above pH 6.0 (90). Treating hippocampal slices with chloroquine (CQ), which raises the pH of lysosomes to impair enzymatic function, was associated with increased levels of full-length tau (89, 91). This was in conjunction with an accumulation of intracellular PHF1 immunopositive tau (91). In M1C neuroblastoma cells that inducibly express full-length wild-type tau (4R0N), treatment with CQ also significantly slowed down tau degradation, and caused its accumulation (92). Treatment of hippocampal slices with the cathepsin modulator ZPAD (which stimulates cathepsin D very strongly) appears to increase the proteolysis of full-length tau resulting in the production of smaller fragments, including a phosphorylated 29 kDa fragment (86, 89). This partial degradation of tau was inhibited by inclusion of a selective cathepsin D inhibitor (86). Cathepsin D seems particularly important for degrading tau, as its expression was neuroprotective in a Drosophila tauopathy model. Levels of cathepsin D are elevated in flies expressing mutant human tau. If cathepsin D is genetically ablated, these tau flies exhibit enhanced neurotoxicity and a shorter lifespan (93).
Modulating autophagy through other approaches also indicates that tau can be degraded through this pathway. Overexpressing only the repeat domain of tau containing an FTDP-17 mutation in neuroblastoma cells leads to tau aggregation as well as the appearance of smaller proteolytic fragments. Using the autophagy inhibitor 3-methyladenine (3-MA) to block the formation of autophagosomes led to an increase in both soluble and insoluble tau (94). Directly activating autophagy through a variety of mechanisms leads consistently to enhanced tau clearance – either pathological forms or total tau. In a hippocampal slice preparation methylene blue was used to induce autophagy, which resulted in a decrease in phosphorylated tau and insoluble tau, specifically (95). In a cell line expressing the repeat domain of tau containing the FTDP-17ΔK280 mutant, treatment with the disaccharide trehalose, an mTor-independent autophagy activator, significantly reduced aggregated tau as measured by Thioflavin-S staining, as well as total tau levels both soluble and insoluble as detected by western blotting (96). Stimulating autophagy either through serum withdrawal or rapamycin treatment in SH-SY5Y cells overexpressing P301L tau that had been induced to aggregate led to substantial reduction in aggregates that was prevented by 3-MA (70). In a mouse model expressing the FTDP-17 mutant P301S, promoting autophagy with trehalose treatment beginning at weaning significantly reduced insoluble tau, as well as tau phosphorylated at T212/S214 (AT100) (97). However, no other phosphorylation sites were assessed. This effect was correlated with improved neuronal survival in cortical layers I–III (97). Stimulating autophagy via genetic manipulation of the mTor pathway decreased total and phosphorylated tau in the same mouse model (98). Conversely, inhibiting autophagy (also via mTor) lead to increased total and AT8-positive phosphorylated tau (98). Mice in which the critical autophagy gene Atg7 is knocked out in forebrain neurons develop age-dependent neurodegeneration with accumulation of phosphorylated tau within intracellular inclusions (99). These inclusions specifically contained tau phosphorylated at AT8, AT100, and TG3 epitopes, but not PHF1. Significantly, if tau was also knocked out in these autophagy-deficient mice, neurodegeneration was reduced (99).
Interestingly, other evidence for the role of autophagy in clearing tau was the result of attempting to elucidate the role of the proteasome in tau degradation. Treating rat primary neurons with the proteasomal inhibitor MG-132 actually led to a reduction in total tau. This effect was likely due to a compensatory upregulation of autophagy, as evidenced by increased LC3-II protein and an increased number of autophagosomes in treated cells (96). This will be discussed in more detail below, as it has important ramifications for the intersection of these two degradative pathways.
Interplay between Autophagy and the Proteasome
There is compelling evidence for significant and extensive interplay between the autophagy and proteasomal systems. This has intriguing implications for disease processes and specifically tau degradation in AD. First, while each system preferentially degrades specific substrates, there are many substrates that can be degraded by both systems, tau being a prime example (25). For instance, a particular substrate may be degraded by the proteasome under normal conditions, but if that system is impaired and/or there is an excess of that substrate it may be degraded in a compensatory manner by autophagy. Another possibility is that particular forms of a substrate may be shuttled to one pathway or another. In the case of tau, as a monomer it is natively unfolded and hence a likely proteasomal substrate, as discussed above. However, any of the numerous modifications tau undergoes during AD pathogenesis may render it less able to do so, for example, by inducing conformational changes to a more ordered structure as suggested by several conformation-specific antibodies that label tau in AD brain (Alz-50, MC-1, etc.). Additionally, oligomerized or aggregated tau may not be a preferred proteasomal target. It has been suggested that this change in state is part of the signal for tau to be degraded by autophagy. This is supported by evidence that full-length tau, which has a lower propensity for aggregating, is cleared by the proteasome while caspase-cleaved tau, which is more aggregate prone, goes through autophagy (72). Also, aggregated tau can be cleared by inducing autophagy (70, 96).
Ubiquitin is implicated in targeting substrates to both pathways. Historically, poly-ubiquitin chains generated through lysine 48 (K48) linkages were viewed as the prototypical proteasomal targeting sequence, while K63 chains appeared more specific for autophagy. However, the experimental evidence indicates a more complex picture. For example, if HEK cells are transfected with tau and ubiquitin, tau is readily ubiquitylated and degraded by the proteasome (66). However, if a ubiquitin construct containing a site mutation at K63 is transfected in, no ubiquitylation of tau occurs. Other mutated forms of ubiquitin, including ubiquitin unable to form linkages at K48, still resulted in tau ubiquitylation. This indicates that in this experimental overexpression paradigm, K63 linkages are the primary ubiquitin linkage for tau, and they target tau to the proteasome rather than to autophagy (66). However, another study, also using HEK cells and ubiquitin K48 and K63 mutants, demonstrated that in the presence of the E3 ligase CHIP, tau could be ubiquitylated by both K48 and K63 linkages (100). The likelihood that in vivo tau can be ubiquitylated in multiple ways is supported by studies showing tau isolated from NFTs in human brain has several forms of ubiquitin linkages as well as mono-ubiquitylation (101, 102). These data suggest that the physical structure of the ubiquitin chain is unlikely to be a sufficient signal for selectively targeting tau to either the proteasome or autophagy. An alternate mechanism for specifically targeting substrates is the involvement of chaperone proteins. The chaperones involved in proteasomal targeting are not well characterized, although it is known that ubiquitin-tagged substrates are trafficked to the organelle. Currently identified chaperones include p62 and Hsp70 (66, 100). Slightly more is known about autophagy adaptors, and there is significant overlap, as both p62 and Hsp70 are adaptors for this pathway as well (103, 104). This further complicates the understanding of how a substrate is selectively targeted to one path or the other. For example, a ubiquitylated substrate can be bound by p62 and either delivered to the proteasome (66) or engulfed by an autophagosome via p62 binding to LC3 (105). These findings suggest the involvement of a currently unidentified chaperones and/or targeting signals, or undetermined additional factors.
Other characteristics of the substrate are likely to also play a role in successfully targeting the protein either to the proteasome or to autophagy. In the case of tau, two modifications seem to be critical for this process: phosphorylation and truncation. For example, in the study where rat brain extract was incubated with Mg2+ and ATP, there was an overall decrease in tau due to proteasomal activity; however tau phosphorylated at the PHF1 and Tau-1 epitopes seemed to be preferentially degraded as they were non-detectable within 3 h (73). The preferential degradation of specific phospho-forms of tau by a particular pathway has been reported in other studies as well. In CHO cells overexpressing P301L mutant tau, treatment with the Hsp90 inhibitor geldanamycin led to a more pronounced proteasome-mediated reduction in tau phosphorylated at proline-directed S/T sites compared to total tau (67). However, the levels of tau phosphorylated at KXGS sites within the repeat domain were not reduced by geldanamycin treatment. In agreement with those findings, inhibiting autophagy in primary rat cortical neurons with 3-MA resulted in the selective accumulation of tau phosphorylated at the KXGS motif S262 (recognized by the 12E8 antibody) (106). Additionally, in a hippocampal slice preparation, induction of autophagy by treatment with methylene blue led to a decrease in phosphorylated tau and insoluble tau without an effect on total tau (95). Activating autophagy with trehalose in rat cortical neurons demonstrated certain phospho-epitopes (AT8, PHF1, and 12E8) were reduced more significantly than total tau – up to 80% compared to the 20% reduction in total tau (96). Finally, caspase-3 cleaved tau has a shorter half-life than full-length tau and is preferentially degraded by autophagy (72). Additional modified forms of tau have yet to be fully examined for their preferred route of degradation.
As specific substrates are targeted to one degradative pathway or the other, the function of each system can also directly impact the functioning of the other. It is well documented that blocking the proteasome with small molecule inhibitors causes an increase in autophagic flux (107). This can be seen both as increased autophagosome formation and maturation as well as enhanced degradation of autophagy substrates (96). However the converse is not true; autophagy impairment does not elevate proteasomal function and, in fact, rather strikingly inhibits it. There are several possible mechanisms for this inhibition. The accumulation of large aggregated substrates might impair the proteasome, as seen for PHF tau (69). Also, reduced recycling of p62 by impairing autophagy (causing its accumulation) will impair proteasomal processing, potentially by p62 competing with other chaperones for proteasomal targets and impeding their delivery (107). The degradation of tau is thus a complex process mediated by multiple factors. While much is known about how tau can be cleared, additional studies are needed to clarify what actually happens in both the normal brain and in the context of AD. This information will yield critical insights into potential therapeutics.
Role of Oligomers in Affecting Tau Degradation Decisions
Given the data indicating tau can be processed by both autophagy and the proteasome, and furthermore that the signaling mechanisms directing substrates to either path are shared, it is unclear how decisions regarding which way tau is degraded are made. One possibility could be tau’s physical state of oligomerization. Soluble, monomeric tau is an ideal proteasomal substrate. Indeed, it has been clearly demonstrated that tau can be degraded by the proteasome (65–67, 73). It thus can be suggested that under physiologic circumstances much of tau is degraded in this manner, with select modified forms being cleared by autophagy. However, within the context of the AD milieu, additional tau modifications and degradative impairments may cause the balance to shift away from proteasomal degradation toward autophagy. For example, as discussed above, certain modified forms of tau, such as caspase-cleaved tau, have a stronger tendency to aggregate. As tau begins to assemble into oligomers, it may become increasingly undesirable as a proteasomal substrate. These low-order, soluble oligomers may be preferentially degraded by autophagy. However, as previously discussed, autophagy is likely impaired in AD. The tendency for certain phospho-epitopes to show preferential clearance by certain pathways may also relate to their propensity for aggregation. As tau oligomers increase in size, density, and modifications during the development of filaments and tangles, not only will they be unable to undergo proteasomal degradation, they may directly impair proteasomal function (69). This proteasomal impairment could have multiple effects. For example, autophagy may initially be activated as a compensatory response. Caspase-3 and possibly other proteases may also be activated as well. However, this may result in an accumulation of potentially toxic cleaved forms of tau. Additionally, given the significant evidence that autophagy is impaired in AD, possibly at the level of the lysosomes, proteasomally mediated activation may serve to further obstruct the autophagy system (see Figure 3).
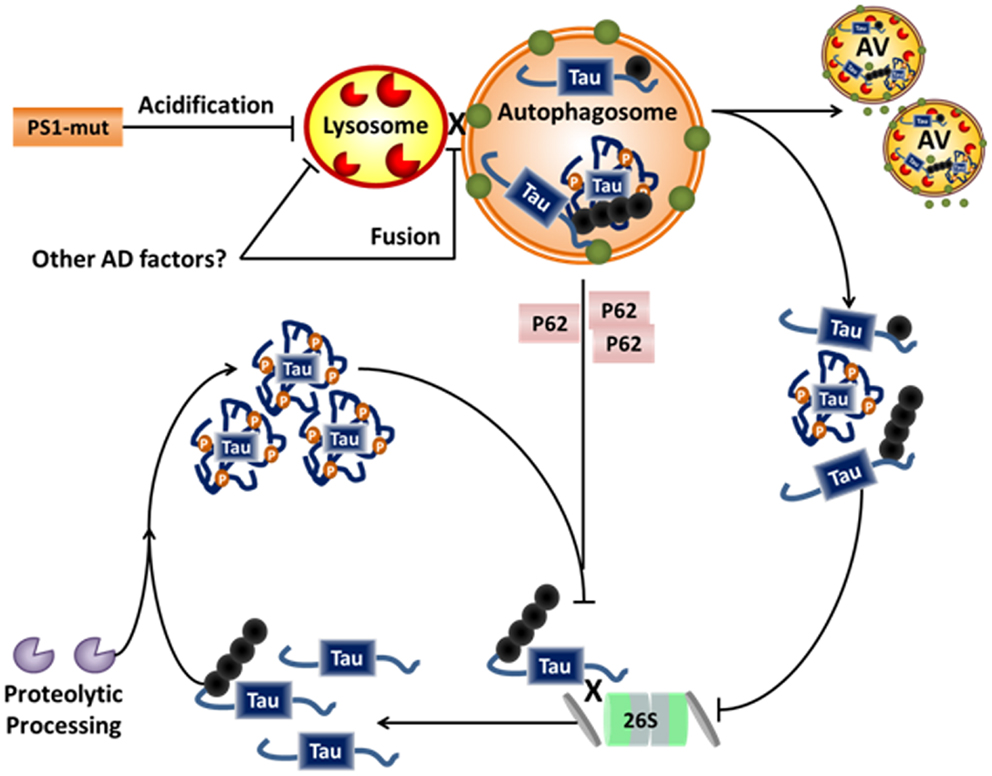
Figure 3. Alzheimer disease-related disruption of tau degradation. Impairment of protein degradation is a known component of both familial and sporadic AD. Familial AD-related mutations of PS1 are linked to impairment of lysosomal acidification and/or fusion to the autophagosome, while sporadic factors leading to similar impairments have not yet been elucidated. Impairment of the lysosome/autophagosome leads to accumulation of dysfunctional autophagic vacuoles (AVs), cytosolic p62, and aggregates of tau. Dysfunction of autophagy further inhibits the proteasome, possibly via accumulation of p62, which can be a chaperone for both systems. P62 is usually recycled via autophagy and accumulates in the cytosol when autophagy is impaired. Hypothetically, this allows p62 to compete with other proteasome chaperones thus inhibiting proteasomal degradation. Further, accumulation of aggregated proteins has also been shown to inhibit the proteasome. Taken together, this could lead to accumulation of soluble tau, and thus more proteolytic processing and further aggregate. Hence, a vicious cycle of degradative pathway impairment and tau accumulation/aggregation may contribute to the neurodegenerative processes in AD.
Conclusion
It is clear that tau plays a significant role in AD pathology, although the mechanisms involved have not been clearly delineated. Tau is a normal neuronal protein that modulates microtubule-based functions, and becomes increasingly hyperphosphorylated, truncated, and otherwise modified in AD. These modifications not only impair tau’s normal function, but also appear to promote its oligomerization. These oligomers eventually accumulate to form the NFTs which are pathognomonic for AD. While the NFTs may be harmful to the cell in some ways, it is now believed that the principal toxicity results from pre-aggregated, soluble tau oligomers. Thus, understanding how these tau species can be cleared may allow for the development of effective therapeutic approaches. It is clear from the data that certain species of tau are preferentially degraded by the proteasome and others by autophagy. There is evidence that both of these degradative systems are likely impaired at some level in AD. Additionally, there is a complex interplay between the proteolytic and degradative pathways that suggests a cycle of pathology may develop in AD whereby alterations in tau processing, including by cytosolic proteases, pushes more tau toward the autophagy system. Decreased autophagic function would result in accumulation of these autophagy-cleared tau species. The combination of impaired autophagy and accumulating substrates has the potential to lead to proteasomal inhibition, in addition to other factors (such as Aβ) that may impair the proteasome in AD (108). This then further promotes cytosolic accumulation of tau leading to its aggregation. Additionally, modifications including caspase-3 cleavage and hyperphosphorylation promote aggregation even of full-length tau, reducing the pool of functional tau.
Understanding how tau is cleared may enable us to identify potential mechanisms for enhancing clearance of pathological forms of tau. Ameliorating the deficit in autophagy is a likely target for this process, and initial results of stimulating autophagy show promise for clearing tau. Indeed, several studies aimed at stimulating autophagy have demonstrated efficacy in reducing phosphorylated and aggregated tau in both in vitro and in vivo models (95–98). These studies are an important initial step toward elucidating the exact role of tau degradation in modulating neurodegeneration in AD. Further studies that better gauge the contribution of each degradative pathway will be necessary. Due to the complexity of the cellular environment, in vitro studies that can tightly control for variables including tau modifications and proteolytic pathway function will likely be instrumental. Ultimately, a more complete understanding of the differential contribution of various proteolytic and degradative pathways will provide critical opportunities for therapeutically addressing the tau pathology associated with neurodegeneration in AD.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Work from the authors’ laboratory was supported by NIH grants NS076789 (Gail V. W. Johnson) and ES020081 (Adrianne S. Chesser). The authors would like to thank Dr. Soner Gundemir for his critical reading of the manuscript.
References
1. Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol (1991) 82:239–59. doi:10.1007/BF00308809
2. Arnold CS, Johnson GV, Cole RN, Dong DL, Lee M, Hart GW. The microtubule-associated protein tau is extensively modified with O-linked N-acetylglucosamine. J Biol Chem (1996) 271:28741–4. doi:10.1074/jbc.271.46.28741
3. Reynolds MR, Berry RW, Binder LI. Nitration in neurodegeneration: deciphering the “Hows” “nYs.” Biochemistry (2007) 46:7325–36. doi:10.1021/bi700430y
4. Cohen TJ, Guo JL, Hurtado DE, Kwong LK, Mills IP, Trojanowski JQ, et al. The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat Commun (2011) 2:252. doi:10.1038/ncomms1255
5. Lasagna-Reeves CA, Castillo-Carranza DL, Sengupta U, Sarmiento J, Troncoso J, Jackson GR, et al. Identification of oligomers at early stages of tau aggregation in Alzheimer’s disease. FASEB J (2012) 26:1946–59. doi:10.1096/fj.11-199851
6. Ward SM, Himmelstein DS, Lancia JK, Binder LI. Tau oligomers and tau toxicity in neurodegenerative disease. Biochem Soc Trans (2012) 40:667–71. doi:10.1042/BST20120134
7. Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, Van Eersel J, et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell (2010) 142:387–97. doi:10.1016/j.cell.2010.06.036
8. Roberson ED, Halabisky B, Yoo JW, Yao J, Chin J, Yan F, et al. Amyloid-beta/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer’s disease. J Neurosci (2011) 31:700–11. doi:10.1523/JNEUROSCI.4152-10.2011
9. Guillozet AL, Weintraub S, Mash DC, Mesulam MM. Neurofibrillary tangles, amyloid, and memory in aging and mild cognitive impairment. Arch Neurol (2003) 60:729–36. doi:10.1001/archneur.60.5.729
10. Karsten SL, Sang TK, Gehman LT, Chatterjee S, Liu J, Lawless GM, et al. A genomic screen for modifiers of tauopathy identifies puromycin-sensitive aminopeptidase as an inhibitor of tau-induced neurodegeneration. Neuron (2006) 51:549–60. doi:10.1016/j.neuron.2006.07.019
11. Sengupta S, Horowitz PM, Karsten SL, Jackson GR, Geschwind DH, Fu Y, et al. Degradation of tau protein by puromycin-sensitive aminopeptidase in vitro. Biochemistry (2006) 45:15111–9. doi:10.1021/bi061830d
12. Kudo LC, Parfenova L, Ren G, Vi N, Hui M, Ma Z, et al. Puromycin-sensitive aminopeptidase (PSA/NPEPPS) impedes development of neuropathology in hPSA/TAU(P301L) double-transgenic mice. Hum Mol Genet (2011) 20:1820–33. doi:10.1093/hmg/ddr065
13. Olesen OF. Proteolytic degradation of microtubule associated protein tau by thrombin. Biochem Biophys Res Commun (1994) 201:716–21. doi:10.1006/bbrc.1994.1759
14. Wang X, An S, Wu JM. Specific processing of native and phosphorylated tau protein by proteases. Biochem Biophys Res Commun (1996) 219:591–7. doi:10.1006/bbrc.1996.0278
15. Khlistunova I, Biernat J, Wang Y, Pickhardt M, Von Bergen M, Gazova Z, et al. Inducible expression of tau repeat domain in cell models of tauopathy: aggregation is toxic to cells but can be reversed by inhibitor drugs. J Biol Chem (2006) 281:1205–14. doi:10.1074/jbc.M507753200
16. Tennstaedt A, Popsel S, Truebestein L, Hauske P, Brockmann A, Schmidt N, et al. Human high temperature requirement serine protease A1 (HTRA1) degrades tau protein aggregates. J Biol Chem (2012) 287:20931–41. doi:10.1074/jbc.M111.316232
17. Canu N, Dus L, Barbato C, Ciotti MT, Brancolini C, Rinaldi AM, et al. Tau cleavage and dephosphorylation in cerebellar granule neurons undergoing apoptosis. J Neurosci (1998) 18:7061–74.
18. Xie HQ, Johnson GV. Calcineurin inhibition prevents calpain-mediated proteolysis of tau in differentiated PC12 cells. J Neurosci Res (1998) 53:153–64. doi:10.1002/(SICI)1097-4547(19980715)53:2<153::AID-JNR4>3.3.CO;2-5
19. Garg S, Timm T, Mandelkow EM, Mandelkow E, Wang Y. Cleavage of tau by calpain in Alzheimer’s disease: the quest for the toxic 17 kD fragment. Neurobiol Aging (2011) 32:1–14. doi:10.1016/j.neurobiolaging.2010.09.008
20. Liu MC, Kobeissy F, Zheng W, Zhang Z, Hayes RL, Wang KK. Dual vulnerability of tau to calpains and caspase-3 proteolysis under neurotoxic and neurodegenerative conditions. ASN Neuro (2011) 3:e00051. doi:10.1042/AN20100012
21. Fasulo L, Ugolini G, Visintin M, Bradbury A, Brancolini C, Verzillo V, et al. The neuronal microtubule-associated protein tau is a substrate for caspase-3 and an effector of apoptosis. J Neurochem (2000) 75:624–33. doi:10.1046/j.1471-4159.2000.0750624.x
22. Chung CW, Song YH, Kim IK, Yoon WJ, Ryu BR, Jo DG, et al. Proapoptotic effects of tau cleavage product generated by caspase-3. Neurobiol Dis (2001) 8:162–72. doi:10.1006/nbdi.2000.0335
23. Gamblin TC, Chen F, Zambrano A, Abraha A, Lagalwar S, Guillozet AL, et al. Caspase cleavage of tau: linking amyloid and neurofibrillary tangles in Alzheimer’s disease. Proc Natl Acad Sci U S A (2003) 100:10032–7. doi:10.1073/pnas.1630428100
24. Horowitz PM, Patterson KR, Guillozet-Bongaarts AL, Reynolds MR, Carroll CA, Weintraub ST, et al. Early N-terminal changes and caspase-6 cleavage of tau in Alzheimer’s disease. J Neurosci (2004) 24:7895–902. doi:10.1523/JNEUROSCI.1988-04.2004
25. Lee MJ, Lee JH, Rubinsztein DC. Tau degradation: the ubiquitin-proteasome system versus the autophagy-lysosome system. Prog Neurobiol (2013) 105:49–59. doi:10.1016/j.pneurobio.2013.03.001
26. Drubin D, Kobayashi S, Kellogg D, Kirschner M. Regulation of microtubule protein levels during cellular morphogenesis in nerve growth factor-treated PC12 cells. J Cell Biol (1988) 106:1583–91. doi:10.1083/jcb.106.5.1583
27. Lewis J, McGowan E, Rockwood J, Melrose H, Nacharaju P, Van Slegtenhorst M, et al. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet (2000) 25:402–5. doi:10.1038/78078
28. Arai T, Guo JP, McGeer PL. Proteolysis of non-phosphorylated and phosphorylated tau by thrombin. J Biol Chem (2005) 280:5145–53. doi:10.1074/jbc.M409234200
29. Chow KM, Guan H, Hersh LB. Aminopeptidases do not directly degrade tau protein. Mol Neurodegener (2010) 5:48. doi:10.1186/1750-1326-5-48
30. Nakamura K, Greenwood A, Binder L, Bigio EH, Denial S, Nicholson L, et al. Proline isomer-specific antibodies reveal the early pathogenic tau conformation in Alzheimer’s disease. Cell (2012) 149:232–44. doi:10.1016/j.cell.2012.02.016
31. Lim J, Balastik M, Lee TH, Nakamura K, Liou YC, Sun A, et al. Pin1 has opposite effects on wild-type and P301L tau stability and tauopathy. J Clin Invest (2008) 118:1877–89. doi:10.1172/JCI34308
32. Menzies FM, Hourez R, Imarisio S, Raspe M, Sadiq O, Chandraratna D, et al. Puromycin-sensitive aminopeptidase protects against aggregation-prone proteins via autophagy. Hum Mol Genet (2010) 19:4573–86. doi:10.1093/hmg/ddq385
33. Yin X, Wright J, Wall T, Grammas P. Brain endothelial cells synthesize neurotoxic thrombin in Alzheimer’s disease. Am J Pathol (2010) 176:1600–6. doi:10.2353/ajpath.2010.090406
34. Gill JS, Pitts K, Rusnak FM, Owen WG, Windebank AJ. Thrombin induced inhibition of neurite outgrowth from dorsal root ganglion neurons. Brain Res (1998) 797:321–7. doi:10.1016/S0006-8993(98)00344-8
35. Smirnova IV, Zhang SX, Citron BA, Arnold PM, Festoff BW. Thrombin is an extracellular signal that activates intracellular death protease pathways inducing apoptosis in model motor neurons. J Neurobiol (1998) 36:64–80. doi:10.1002/(SICI)1097-4695(199807)36:1<64::AID-NEU6>3.0.CO;2-8
36. Turgeon VL, Lloyd ED, Wang S, Festoff BW, Houenou LJ. Thrombin perturbs neurite outgrowth and induces apoptotic cell death in enriched chick spinal motoneuron cultures through caspase activation. J Neurosci (1998) 18:6882–91.
37. Suo Z, Wu M, Citron BA, Palazzo RE, Festoff BW. Rapid tau aggregation and delayed hippocampal neuronal death induced by persistent thrombin signaling. J Biol Chem (2003) 278:37681–9. doi:10.1074/jbc.M301406200
38. Arai T, Miklossy J, Klegeris A, Guo JP, McGeer PL. Thrombin and prothrombin are expressed by neurons and glial cells and accumulate in neurofibrillary tangles in Alzheimer disease brain. J Neuropathol Exp Neurol (2006) 65:19–25. doi:10.1097/01.jnen.0000196133.74087.cb
39. Wang YP, Biernat J, Pickhardt M, Mandelkow E, Mandelkow EM. Stepwise proteolysis liberates tau fragments that nucleate the Alzheimer-like aggregation of full-length tau in a neuronal cell model. Proc Natl Acad Sci U S A (2007) 104:10252–7. doi:10.1073/pnas.0703676104
40. De Luca A, De Falco M, Severino A, Campioni M, Santini D, Baldi F, et al. Distribution of the serine protease HtrA1 in normal human tissues. J Histochem Cytochem (2003) 51:1279–84. doi:10.1177/002215540305101004
41. Grau S, Baldi A, Bussani R, Tian X, Stefanescu R, Przybylski M, et al. Implications of the serine protease HtrA1 in amyloid precursor protein processing. Proc Natl Acad Sci U S A (2005) 102:6021–6. doi:10.1073/pnas.0501823102
42. Chien J, He X, Shridhar V. Identification of tubulins as substrates of serine protease HtrA1 by mixture-based oriented peptide library screening. J Cell Biochem (2009) 107:253–63. doi:10.1002/jcb.22121
43. Chien J, Ota T, Aletti G, Shridhar R, Boccellino M, Quagliuolo L, et al. Serine protease HtrA1 associates with microtubules and inhibits cell migration. Mol Cell Biol (2009) 29:4177–87. doi:10.1128/MCB.00035-09
44. Clausen T, Kaiser M, Huber R, Ehrmann M. HTRA proteases: regulated proteolysis in protein quality control. Nat Rev Mol Cell Biol (2011) 12:152–62. doi:10.1038/nrm3065
45. Liu J, Liu MC, Wang KK. Calpain in the CNS: from synaptic function to neurotoxicity. Sci Signal (2008) 1:re1. doi:10.1126/stke.114re1
46. Vosler PS, Brennan CS, Chen J. Calpain-mediated signaling mechanisms in neuronal injury and neurodegeneration. Mol Neurobiol (2008) 38:78–100. doi:10.1007/s12035-008-8036-x
47. Adamec E, Mohan P, Vonsattel JP, Nixon RA. Calpain activation in neurodegenerative diseases: confocal immunofluorescence study with antibodies specifically recognizing the active form of calpain 2. Acta Neuropathol (2002) 104:92–104. doi:10.1007/s00401-002-0528-6
48. Ferreira A, Bigio EH. Calpain-mediated tau cleavage: a mechanism leading to neurodegeneration shared by multiple tauopathies. Mol Med (2011) 17:676–85. doi:10.2119/molmed.2010.00220
49. Olney JW, Wozniak DF, Farber NB. Excitotoxic neurodegeneration in Alzheimer disease. New hypothesis and new therapeutic strategies. Arch Neurol (1997) 54:1234–40. doi:10.1001/archneur.1997.00550220042012
50. Wang Q, Yu S, Simonyi A, Sun GY, Sun AY. Kainic acid-mediated excitotoxicity as a model for neurodegeneration. Mol Neurobiol (2005) 31:3–16. doi:10.1385/MN:31:1-3:003
51. D’Orsi B, Bonner H, Tuffy LP, Dussmann H, Woods I, Courtney MJ, et al. Calpains are downstream effectors of bax-dependent excitotoxic apoptosis. J Neurosci (2012) 32:1847–58. doi:10.1523/JNEUROSCI.2345-11.2012
52. Park SY, Ferreira A. The generation of a 17 kDa neurotoxic fragment: an alternative mechanism by which tau mediates beta-amyloid-induced neurodegeneration. J Neurosci (2005) 25:5365–75. doi:10.1523/JNEUROSCI.1125-05.2005
53. Reifert J, Hartung-Cranston D, Feinstein SC. Amyloid beta-mediated cell death of cultured hippocampal neurons reveals extensive tau fragmentation without increased full-length tau phosphorylation. J Biol Chem (2011) 286:20797–811. doi:10.1074/jbc.M111.234674
54. Reinecke JB, Devos SL, McGrath JP, Shepard AM, Goncharoff DK, Tait DN, et al. Implicating calpain in tau-mediated toxicity in vivo. PLoS ONE (2011) 6:e23865. doi:10.1371/journal.pone.0023865
55. Cotman CW, Poon WW, Rissman RA, Blurton-Jones M. The role of caspase cleavage of tau in Alzheimer disease neuropathology. J Neuropathol Exp Neurol (2005) 64:104–12.
56. de Calignon A, Fox LM, Pitstick R, Carlson GA, Bacskai BJ, Spires-Jones TL, et al. Caspase activation precedes and leads to tangles. Nature (2010) 464:1201–4. doi:10.1038/nature08890
57. Su JH, Zhao M, Anderson AJ, Srinivasan A, Cotman CW. Activated caspase-3 expression in Alzheimer’s and aged control brain: correlation with Alzheimer pathology. Brain Res (2001) 898:350–7. doi:10.1016/S0006-8993(01)02018-2
58. Guo H, Albrecht S, Bourdeau M, Petzke T, Bergeron C, Leblanc AC. Active caspase-6 and caspase-6-cleaved tau in neuropil threads, neuritic plaques, and neurofibrillary tangles of Alzheimer’s disease. Am J Pathol (2004) 165:523–31. doi:10.1016/S0002-9440(10)63317-2
59. Guillozet-Bongaarts AL, Garcia-Sierra F, Reynolds MR, Horowitz PM, Fu Y, Wang T, et al. Tau truncation during neurofibrillary tangle evolution in Alzheimer’s disease. Neurobiol Aging (2005) 26:1015–22. doi:10.1016/j.neurobiolaging.2004.09.019
60. Basurto-Islas G, Luna-Munoz J, Guillozet-Bongaarts AL, Binder LI, Mena R, Garcia-Sierra F. Accumulation of aspartic acid421- and glutamic acid391-cleaved tau in neurofibrillary tangles correlates with progression in Alzheimer disease. J Neuropathol Exp Neurol (2008) 67:470–83. doi:10.1097/NEN.0b013e31817275c7
61. Rissman RA, Poon WW, Blurton-Jones M, Oddo S, Torp R, Vitek MP, et al. Caspase-cleavage of tau is an early event in Alzheimer disease tangle pathology. J Clin Invest (2004) 114:121–30. doi:10.1172/JCI20640
62. Arnaud LT, Myeku N, Figueiredo-Pereira ME. Proteasome-caspase-cathepsin sequence leading to tau pathology induced by prostaglandin J2 in neuronal cells. J Neurochem (2009) 110:328–42. doi:10.1111/j.1471-4159.2009.06142.x
63. Metcalfe MJ, Huang Q, Figueiredo-Pereira ME. Coordination between proteasome impairment and caspase activation leading to TAU pathology: neuroprotection by cAMP. Cell Death Dis (2012) 3:e326. doi:10.1038/cddis.2012.70
64. Hamano T, Gendron TF, Ko LW, Yen SH. Concentration-dependent effects of proteasomal inhibition on tau processing in a cellular model of tauopathy. Int J Clin Exp Pathol (2009) 2:561–73.
65. David DC, Layfield R, Serpell L, Narain Y, Goedert M, Spillantini MG. Proteasomal degradation of tau protein. J Neurochem (2002) 83:176–85. doi:10.1046/j.1471-4159.2002.01137.x
66. Babu JR, Geetha T, Wooten MW. Sequestosome 1/p62 shuttles polyubiquitinated tau for proteasomal degradation. J Neurochem (2005) 94:192–203. doi:10.1111/j.1471-4159.2005.03181.x
67. Dickey CA, Dunmore J, Lu B, Wang JW, Lee WC, Kamal A, et al. HSP induction mediates selective clearance of tau phosphorylated at proline-directed Ser/Thr sites but not KXGS (MARK) sites. FASEB J (2006) 20:753–5.
68. Keller JN, Hanni KB, Markesbery WR. Impaired proteasome function in Alzheimer’s disease. J Neurochem (2000) 75:436–9. doi:10.1046/j.1471-4159.2000.0750436.x
69. Keck S, Nitsch R, Grune T, Ullrich O. Proteasome inhibition by paired helical filament-tau in brains of patients with Alzheimer’s disease. J Neurochem (2003) 85:115–22. doi:10.1046/j.1471-4159.2003.01642.x
70. Wong ES, Tan JM, Soong WE, Hussein K, Nukina N, Dawson VL, et al. Autophagy-mediated clearance of aggresomes is not a universal phenomenon. Hum Mol Genet (2008) 17:2570–82. doi:10.1093/hmg/ddn157
71. Grune T, Botzen D, Engels M, Voss P, Kaiser B, Jung T, et al. Tau protein degradation is catalyzed by the ATP/ubiquitin-independent 20S proteasome under normal cell conditions. Arch Biochem Biophys (2010) 500:181–8. doi:10.1016/j.abb.2010.05.008
72. Dolan PJ, Johnson GV. A caspase cleaved form of tau is preferentially degraded through the autophagy pathway. J Biol Chem (2010) 285:21978–87. doi:10.1074/jbc.M110.110940
73. Zhang JY, Liu SJ, Li HL, Wang JZ. Microtubule-associated protein tau is a substrate of ATP/Mg(2+)-dependent proteasome protease system. J Neural Transm (2005) 112:547–55. doi:10.1007/s00702-004-0196-x
74. Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy (2011) 7:279–96. doi:10.4161/auto.7.3.14487
75. Ivanov S, Roy CR. NDP52: the missing link between ubiquitinated bacteria and autophagy. Nat Immunol (2009) 10:1137–9. doi:10.1038/ni1109-1137
76. von Muhlinen N, Akutsu M, Ravenhill BJ, Foeglein A, Bloor S, Rutherford TJ, et al. LC3C, bound selectively by a noncanonical LIR motif in NDP52, is required for antibacterial autophagy. Mol Cell (2012) 48:329–42. doi:10.1016/j.molcel.2012.08.024
77. Watson RO, Manzanillo PS, Cox JS. Extracellular M. tuberculosis DNA targets bacteria for autophagy by activating the host DNA-sensing pathway. Cell (2012) 150:803–15. doi:10.1016/j.cell.2012.06.040
78. Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev (2010) 90:1383–435. doi:10.1152/physrev.00030.2009
79. Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A, et al. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol (2005) 64:113–22.
80. Yang DS, Kumar A, Stavrides P, Peterson J, Peterhoff CM, Pawlik M, et al. Neuronal apoptosis and autophagy cross talk in aging PS/APP mice, a model of Alzheimer’s disease. Am J Pathol (2008) 173:665–81. doi:10.2353/ajpath.2008.071176
81. Sanchez-Varo R, Trujillo-Estrada L, Sanchez-Mejias E, Torres M, Baglietto-Vargas D, Moreno-Gonzalez I, et al. Abnormal accumulation of autophagic vesicles correlates with axonal and synaptic pathology in young Alzheimer’s mice hippocampus. Acta Neuropathol (2012) 123:53–70. doi:10.1007/s00401-011-0896-x
82. Nixon RA, Yang DS. Autophagy failure in Alzheimer’s disease – locating the primary defect. Neurobiol Dis (2011) 43:38–45. doi:10.1016/j.nbd.2011.01.021
83. Nixon RA, Yang DS. Autophagy and neuronal cell death in neurological disorders. Cold Spring Harb Perspect Biol (2012) 4:a008839. doi:10.1101/cshperspect.a008839
84. Yang DS, Stavrides P, Mohan PS, Kaushik S, Kumar A, Ohno M, et al. Reversal of autophagy dysfunction in the TgCRND8 mouse model of Alzheimer’s disease ameliorates amyloid pathologies and memory deficits. Brain (2011) 134:258–77. doi:10.1093/brain/awq341
85. Bi X, Zhou J, Lynch G. Lysosomal protease inhibitors induce meganeurites and tangle-like structures in entorhinohippocampal regions vulnerable to Alzheimer’s disease. Exp Neurol (1999) 158:312–27. doi:10.1006/exnr.1999.7087
86. Bi X, Haque TS, Zhou J, Skillman AG, Lin B, Lee CE, et al. Novel cathepsin D inhibitors block the formation of hyperphosphorylated tau fragments in hippocampus. J Neurochem (2000) 74:1469–77. doi:10.1046/j.1471-4159.2000.0741469.x
87. Yang AJ, Chandswangbhuvana D, Margol L, Glabe CG. Loss of endosomal/lysosomal membrane impermeability is an early event in amyloid Abeta1-42 pathogenesis. J Neurosci Res (1998) 52:691–8. doi:10.1002/(SICI)1097-4547(19980615)52:6<691::AID-JNR8>3.0.CO;2-3
88. Ditaranto K, Tekirian TL, Yang AJ. Lysosomal membrane damage in soluble Abeta-mediated cell death in Alzheimer’s disease. Neurobiol Dis (2001) 8:19–31. doi:10.1006/nbdi.2000.0364
89. Bednarski E, Lynch G. Cytosolic proteolysis of tau by cathepsin D in hippocampus following suppression of cathepsins B and L. J Neurochem (1996) 67:1846–55. doi:10.1046/j.1471-4159.1996.67051846.x
90. Johnson GV, Litersky JM, Whitaker JN. Proteolysis of microtubule-associated protein 2 and tubulin by cathepsin D. J Neurochem (1991) 57:1577–83. doi:10.1111/j.1471-4159.1991.tb06354.x
91. Bendiske J, Bahr BA. Lysosomal activation is a compensatory response against protein accumulation and associated synaptopathogenesis – an approach for slowing Alzheimer disease? J Neuropathol Exp Neurol (2003) 62:451–63.
92. Hamano T, Gendron TF, Causevic E, Yen SH, Lin WL, Isidoro C, et al. Autophagic-lysosomal perturbation enhances tau aggregation in transfectants with induced wild-type tau expression. Eur J Neurosci (2008) 27:1119–30. doi:10.1111/j.1460-9568.2008.06084.x
93. Khurana V, Elson-Schwab I, Fulga TA, Sharp KA, Loewen CA, Mulkearns E, et al. Lysosomal dysfunction promotes cleavage and neurotoxicity of tau in vivo. PLoS Genet (2010) 6:e1001026. doi:10.1371/journal.pgen.1001026
94. Wang Y, Martinez-Vicente M, Kruger U, Kaushik S, Wong E, Mandelkow EM, et al. Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum Mol Genet (2009) 18:4153–70. doi:10.1093/hmg/ddp367
95. Congdon EE, Wu JW, Myeku N, Figueroa YH, Herman M, Marinec PS, et al. Methylthioninium chloride (methylene blue) induces autophagy and attenuates tauopathy in vitro and in vivo. Autophagy (2012) 8:609–22. doi:10.4161/auto.19048
96. Kruger U, Wang Y, Kumar S, Mandelkow EM. Autophagic degradation of tau in primary neurons and its enhancement by trehalose. Neurobiol Aging (2012) 33:2291–305. doi:10.1016/j.neurobiolaging.2011.11.009
97. Schaeffer V, Lavenir I, Ozcelik S, Tolnay M, Winkler DT, Goedert M. Stimulation of autophagy reduces neurodegeneration in a mouse model of human tauopathy. Brain (2012) 135:2169–77. doi:10.1093/brain/aws143
98. Caccamo A, Magri A, Medina DX, Wisely EV, Lopez-Aranda MF, Silva AJ, et al. mTor regulates tau phosphorylation and degradation: implications for Alzheimer’s disease and other tauopathies. Aging Cell (2013) 12:370–80. doi:10.1111/acel.12057
99. Inoue K, Rispoli J, Kaphzan H, Klann E, Chen EI, Kim J, et al. Macroautophagy deficiency mediates age-dependent neurodegeneration through a phospho-tau pathway. Mol Neurodegener (2012) 7:48. doi:10.1186/1750-1326-7-48
100. Petrucelli L, Dickson D, Kehoe K, Taylor J, Snyder H, Grover A, et al. CHIP and Hsp70 regulate tau ubiquitination, degradation and aggregation. Hum Mol Genet (2004) 13:703–14. doi:10.1093/hmg/ddh083
101. Morishima-Kawashima M, Hasegawa M, Takio K, Suzuki M, Titani K, Ihara Y. Ubiquitin is conjugated with amino-terminally processed tau in paired helical filaments. Neuron (1993) 10:1151–60. doi:10.1016/0896-6273(93)90063-W
102. Cripps D, Thomas SN, Jeng Y, Yang F, Davies P, Yang AJ. Alzheimer disease-specific conformation of hyperphosphorylated paired helical filament-tau is polyubiquitinated through Lys-48, Lys-11, and Lys-6 ubiquitin conjugation. J Biol Chem (2006) 281:10825–38. doi:10.1074/jbc.M512786200
104. Knaevelsrud H, Simonsen A. Fighting disease by selective autophagy of aggregate-prone proteins. FEBS Lett (2010) 584:2635–45. doi:10.1016/j.febslet.2010.04.041
105. Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem (2007) 282:24131–45. doi:10.1074/jbc.M702824200
106. Dolan PJ, Jin YN, Hwang W, Johnson GV. Decreases in valosin-containing protein result in increased levels of tau phosphorylated at Ser262/356. FEBS Lett (2011) 585:3424–9. doi:10.1016/j.febslet.2011.09.032
107. Korolchuk VI, Menzies FM, Rubinsztein DC. Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett (2010) 584:1393–8. doi:10.1016/j.febslet.2009.12.047
Keywords: tau, proteasome, autophagy, proteolysis, degradation
Citation: Chesser AS, Pritchard SM and Johnson GVW (2013) Tau clearance mechanisms and their possible role in the pathogenesis of Alzheimer disease. Front. Neurol. 4:122. doi: 10.3389/fneur.2013.00122
Received: 13 May 2013; Paper pending published: 29 May 2013;
Accepted: 15 August 2013; Published online: 03 September 2013.
Edited by:
Jesus Avila, Universidad Autónoma de Madrid, SpainReviewed by:
Cheng-Xin Gong, The City University of New York, USAIllana Gozes, Tel Aviv University, Israel
Copyright: © 2013 Chesser, Pritchard and Johnson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gail V. W. Johnson, Neuroscience Graduate Program, Department of Anesthesiology, University of Rochester, 601 Elmwood Avenue, Box 604, Rochester, NY 14642, USA e-mail: gail_johnsonvoll@urmc.rochester.edu