 Devin W. McBride
Devin W. McBride Spiros L. Blackburn1
Spiros L. Blackburn1- 1The Vivian L. Smith Department of Neurosurgery, McGovern Medical School, The University of Texas Health Science Center at Houston, Houston, TX, United States
- 2Department of Physiology and Pharmacology, Loma Linda School of Medicine, Loma Linda University, Loma Linda, CA, United States
- 3Department of Neurosurgery, Loma Linda School of Medicine, Loma Linda University, Loma Linda, CA, United States
Delayed cerebral ischemia (DCI) is a major determinant of patient outcome following aneurysmal subarachnoid hemorrhage. Although the exact mechanisms leading to DCI are not fully known, inflammation, cerebral vasospasm, and microthrombi may all function together to mediate the onset of DCI. Indeed, inflammation is tightly linked with activation of coagulation and microthrombi formation. Thromboinflammation is the intersection at which inflammation and thrombosis regulate one another in a feedforward manner, potentiating the formation of thrombi and pro-inflammatory signaling. In this review, we will explore the role(s) of inflammation and microthrombi in subarachnoid hemorrhage (SAH) pathophysiology and DCI, and discuss the potential of targeting thromboinflammation to prevent DCI after SAH.
Introduction
Aneurysmal subarachnoid hemorrhage (aSAH) affects 9 per 100,000 individuals per year in the United States (1) and has high rates of morbidity and mortality (2). Since the mean affected age is 45–55 years old (3), the socioeconomic burden is great, and despite advances in aneurysmal securement technique and management of aSAH in the last 20 years, many of those who survive the initial insult develop significant neurological and cognitive disabilities (1, 4–6).
A major contributor to poor outcomes after aSAH is delayed cerebral ischemia (DCI) (1, 5, 6). DCI affects 20–30% of survivors and occurs between 4 and 10 days after subarachnoid hemorrhage (SAH), leading to cognitive decline (7–9), ultimately causing severe disability and worse quality of life or death (10–12). DCI is the clinical syndrome used to describe delayed development of neurological impairment (9). Historically, DCI was thought to be caused by cerebral vasospasm. However, multiple studies indicate that DCI is multifactorial, with vasospasm being just a contributing factor (13–17) along with cortical spreading depression, disrupted/altered cerebral autoregulation, microthrombosis, and inflammation (6, 18). Due to the multi-faceted nature of DCI involving both inflammation and microthrombosis, in this review, we will provide evidence that thromboinflammation is an unexplored therapeutic target for preventing DCI in aSAH patients.
SAH Pathophysiology
Following the initial bleed and subsequent treatment of the ruptured aneurysm, the accumulated blood within the subarachnoid space triggers several response mechanisms, as well as pathological events. In response to extravascular blood, endogenous mechanisms of repair are initiated, including removal of the red blood cells (RBCs) (19). To this end, specific cell adhesion molecules are rapidly expressed on the luminal endothelial cell surface (19), which allows circulating macrophages and neutrophils to enter into the subarachnoid space. Extravasated blood is phagocytosed by infiltrating macrophages (20), and as a result, inflammatory cytokines are released by the infiltrated leukocytes and activated resident microglia, stimulating the immune cascade. Indeed, inflammation has been shown to be a major player in early brain injury (21, 22), and may be a factor leading to DCI (23, 24), contributing to long-term deficits (25–27).
A second pathological factor triggered after SAH is hypercoagulability (28). Although the coagulation cascade is immediately activated following aneurysm rupture to stop bleeding, the hypercoagulable state does not end when the bleeding ceases; a sub-set of aSAH patients will continue to have hypercoagulability for several days post-SAH which has been shown to correlate with the development of DCI (28) and result in poorer outcome (28, 29).
Mechanism of Thromboinflammation after SAH
While there are distinct mechanisms by which both inflammation and thrombosis can lead to deleterious events after SAH, both of these pathological events have cross talk by which they potentiate each other (30). The overlap between inflammation and thrombosis, called thromboinflammation, has been reported to play roles in a number of brain diseases/damage, including ischemia (31–34). Since ischemia also plays a role in patient outcome as a downstream deleterious event from SAH, thromboinflammation may be an overlooked pathophysiology after SAH.
Following SAH, activated macrophages release pro-inflammatory cytokines/chemokines for the recruitment of circulating macrophages to aid in RBC clearance (19). Phagocytosis of RBCs and subsequent death of the infiltrated leukocytes (occurring 2–4 days post-ictus) in the cerebrospinal fluid leads to release of a plethora of toxic molecules (35) including endothelins, oxygen free radicals, and hemoglobin/heme (and byproducts), thereby potentiating pro-inflammatory pathways and endothelial cell-mediated thrombosis. The latter event induces a positive feedforward loop, potentiating thromboinflammation by continuing to recruit systemic immune cells for debris clean-up, causing cell adhesion molecules to continuedly activate platelets, leading to microthrombi formation (36) (Figure 1).
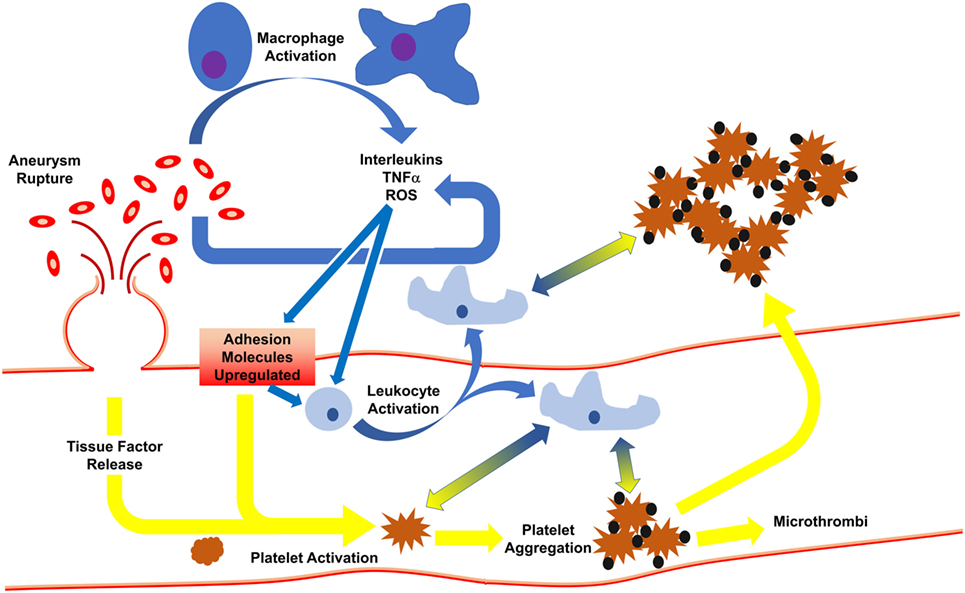
Figure 1. Initiation of inflammation and thrombosis after subarachnoid hemorrhage. Following rupture of an aneurysm, red blood cells spill into the subarachnoid space, activating macrophages which release cytokines. In response, endothelial cells express adhesion molecules to activate circulating leukocytes and platelets. This results in leukocyte infiltration, phagocytosis of red blood cells, and release of more cytokines. Activated platelets develop into thrombi which may travel downstream to occlude distal microvessels or may cross into the brain parenchyma. Platelet activation and aggregation signal for leukocyte activation, and vice versa, such that even when the aneurysm is treated, thromboinflammation continues via feedforward mechanisms.
Production/release of reactive oxygen species and nitric oxide adds to endothelial damage (37), stimulating endothelial expression of adhesion molecules (e.g., ICAM-1, P-selectin, VCAM, collagen, fibrinogen, etc.) (37, 38), and release of tissue factor and vWF, thereby activating platelets. This process is controlled by ADAMTS13 (a disintegrin-like metalloprotease with thrombospondin type 1 repeats-13), which acts to control clot formation by cleaving hyperactive ultralarge vWF to prevent platelet adherence (39). Interestingly, aSAH patients who go on to develop DCI are more hypercoagulable than aSAH patients who will not develop DCI (40, 41). Auxiliary to greater/sustained microthrombi formation, continued activation of coagulation by endothelial cells and platelets further promote inflammation via activation of circulating leukocytes through protease activated receptor-1 and toll-like receptor 4 signaling (36).
Finally, emerging evidence further displays the delicate balance that inflammation plays in modulating coagulation (36). In the non-diseased state, systemic leukocytes aid in preventing coagulation by expressing several anticoagulant factors (42–44). Yet, when pro-inflammatory signaling is initiated, leukocytes change phenotypes, thereby aiding in coagulation through the release of procoagulant factors (45, 46) and reduced expression and degradation of anti-coagulation factors (47–49). While leukocytes play a critical role in inducing and propagating inflammation, leukocytes also promote a hypercoagulable state after SAH (36) and may play a crucial role in DCI via thromboinflammation.
Clinical Evidence
To date, the exact role thromboinflammation plays in patients suffering from aSAH is not known. Yet, the following clinical studies suggest that thromboinflammation may be a key contributor to DCI after aSAH, and thus should be considered as a therapeutic target.
First, microthrombi have been reported as an associative cause of DCI and worse neurological outcome (50, 51). In human autopsy studies, microthrombi, reported to be throughout the brain, are associated with regions of infarct (50, 51), and also develop with a timing similar to DCI (6). Following aSAH, platelet activation is over-stimulated, resulting in microthrombi within small arterioles (51), peaking within 2 days and then again from 1 to 2 weeks post-ictus (50). Platelet-derived thromboxane B2 is higher and platelet count is lower in DCI patients compared to non-DCI patients (52, 53), suggesting platelet aggregation as a potential cause of microthrombi. In addition, aSAH patients are hypercoagulable with changes in platelet-activating factor (54), vWF (40, 54), and tissue plasminogen activator (55, 56) correlating with the incidence of DCI (54, 55) and poor patient outcome (28). Specific polymorphisms of plasminogen activator inhibitor (PAI) have higher PAI activity (57) leading to a higher incidence of DCI (41). Finally, in addition to an over active coagulation cascade, fibrinolysis after aSAH is impaired (40, 56). ADAMTS13, proposed as a critical link between inflammation and thrombosis (39), has been observed to have low activity after aSAH, and is associated with an increased risk of ischemic stroke (58) and may potentiate microthrombi formation. The inherent roles of leukocytes and platelets in inflammation and thrombosis, as well as the involvement of ADAMTS13, argue for thromboinflammation being considered as a therapeutic target after SAH.
Not only is the coagulation cascade not functioning properly, so too is inflammation continually hyperactive. Several pro-inflammatory cytokines have been linked with worse clinical outcome after aSAH (25–27, 59, 60). Increases in tumor necrosis factor α, interleukin-1α, interleukin-1β, interleukin-6, and interleukin-8 have all been found in the cerebrospinal fluid and blood (59, 61–68), with several factors also being associated with poor outcome and DCI (59, 61). Additionally, elevated adhesion molecules in the blood have also been observed after aSAH in patients, remaining elevated for 6–8 days, and are associated with DCI (69–71). Blood P-selectin is elevated in patients with DCI, suggesting that inflammation and platelet activation adhesion are associated with DCI (72, 73). Indeed, adhesion molecules are well documented to be at the crux of thromboinflammation (39, 74–76). These findings, which suggest thromboinflammation is active, are also supported by increased leukocyte infiltration into the brain following SAH (77–80) and also correlate with ischemia after SAH (81). Thus, the so-called “leukocyte–endothelial cell interaction” (78) may play a role in SAH pathophysiology (i.e., thromboinflammation).
A recent study on 106 aSAH patients showed that platelet activation and inflammation (via C-reactive protein measurement) occurred simultaneously and were associated with worse early brain injury and 3-month outcome (82). Similar to others (40, 54), Frontera et al. found that platelet activation and C-reactive protein levels were associated with DCI, but even more interesting, the authors reported that higher platelet activation and C-reactive protein levels at 72 h post-SAH correlate with DCI (82). The findings by Frontera et al. further support the potential role of thromboinflammation in DCI after aSAH, suggesting thromboinflammation may be an unexplored therapeutic target for improving aSAH outcome.
Evidence from Experimental Models
Thromboinflammation has also not been specifically studied in experimental models of SAH, nor has it been examined for correlation with microthrombi, ischemia, or DCI in animals. While DCI is unmeasurable in animals, several pathological measures are used as correlates for DCI (e.g., vasospasm, microthrombi formation, ischemia/infarct/cell death).
Several groups have reported the existence of microthrombi within the vasculature both locally, as well as distally from the SAH insult in experimental SAH models (83–86). In rodent models of SAH using autologous blood infusion, microthrombi and occluded blood vessels have been reported to occur throughout the brain at 2 days post-SAH (84) and can be observed for up to 7 days post-ictus (86). While microthrombi have been observed in puncture models of SAH (87, 88), it is the blood infusion models which offer the most insight into the potential role of thromboinflammation in clot formation after experimental SAH. Indeed, thrombi may be thrown downstream from the vessel rupture in endovascular perforation models as the mechanism by which microthrombi occur in the vasculature; however, no such vessel rupture is present in the autologous blood infusion models. In the blood injection SAH models, the likely cause of microthrombi formation and deposition within the cerebral vasculature distal from the SAH insult is inflammation.
Furthermore, following experimental SAH, endothelial damage resulting from leukocyte infiltration may lead to the downward spiral that is thromboinflammation. Indeed, post-SAH inflammation is devastating (19) and includes cytokine/chemokine upregulation (89, 90), increased expression of adhesion molecules (91, 92), and leukocyte infiltration (93–95). In fact, either depleting neutrophils or reducing neutrophil activity decreases microvascular injury following SAH in rats (93).
Similar to clinical findings, P-selectin has been observed to increase after experimental SAH, leading to activation of platelets and thrombi formation (84). Interestingly, platelet aggregates have been observed to extravasate into the brain parenchyma in the rodent SAH autologous blood injection model which likely propagates pro-inflammatory signaling (22), further showing that there is cross talk between inflammation and thrombosis after SAH.
Evidence from experimental models of SAH, similar to clinical findings, suggest that significant overlap between inflammation and thrombosis occurs post-SAH and may be involved in secondary injury and neurological deterioration. Future studies are warranted to investigate the exact role(s) thromboinflammation plays in SAH and its potential as a therapeutic target.
Thromboinflammation as a Therapeutic Target to Prevent DCI
The detrimental roles thromboinflammation play in brain pathologies has led to thromboinflammation to be suggested as a therapeutic target (32, 96), but not for SAH. Based on the review presented above, thromboinflammation should also be considered a therapeutic target for patients of SAH (54). Potential targets of thromboinflammation may include expression of adhesion molecules, release of factors activating leukocytes and platelets simultaneously (e.g., selectins), disrupting platelet-leukocyte formations, and targeting ADAMTS13. To date, no clinical trials have studied these factors as specific therapeutic targets. Below we review some of the clinical trials which have targeted either coagulation or inflammation, and show that targeting these physiological events separately does not seem to hold the answer for SAH treatment.
Previous Trials
Targeting Coagulation and Clot Clearance
Several early clinical trials investigated therapeutics directed at targeting coagulation following SAH, many of which used antiplatelet therapies. However, meta-analysis of antiplatelet therapy indicates that antiplatelet therapy has a trend for improving outcome in aSAH patients, with tendencies to decrease secondary ischemia (except for acetylsalicylic acid which shows no benefit) (97). Some of the antiplatelet therapies thus far investigated are acetylsalicylic acid, ticlopidine (adenosine diphosphate receptor inhibitor), dipyridamole (phosphodiesterase inhibitor), OKY-046/Calaclot (thromboxane synthetase inhibitor), a thromboxane A2 antagonist, and E5880 (inhibitor of platelet-activating factor receptor) (97). Of these agents, Calaclot and E5880 seemed to improve functional outcome, but additional studies need to be undertaken to determine the therapeutic benefit associated with antiplatelet therapies.
Studies have also examined the benefit of targeting clot clearance. In this regard, recombinant tissue plasminogen activator (administered intraventricularly) has been investigated in several clinical trials which have shown that although recombinant tissue plasminogen activator can improve clot clearance, it fails to attenuate cerebral vasospasm or DCI (98), reduce mortality (99), or improve functional outcome (100). These findings indicate that clot clearance alone may not be enough for aSAH.
Targeting Inflammation
Trials targeting inflammation have had varied success. Erythropoietin-β, despite having mechanisms of action against vasospasm and inflammation (101), had no effect on cerebral vasospasm, but did reduce DCI and improve functional outcome (102). This was a small study and is at high risk for bias (103), so additional trials are needed. Another anti-inflammatory agent, methylprednisolone, was found to improve functional outcome at 1 year despite having no effect on cerebral vasospasm or hypodensity on CT scans (104). It should be noted that methylprednisolone is an anti-inflammatory and thus may have no effect on vasospasm, although the lack of change in CT hypodensity is puzzling. Regardless, these trials suggest that inflammation indeed plays a major role in SAH outcome. Since there is considerable feedforward/back mechanisms between thrombosis and inflammation, anti-inflammatory agents may indirectly reduce thromboinflammation. While previous trials have shown promise, additional studies are needed to determine any benefit to patient outcome for anti-inflammatory agents as well as the relationship to thromboinflammation.
Current Trials
The lack of promising data for therapeutics targeting either cerebral vasospasm (100, 103) or clot clearance suggest that SAH prognosis may lie within inflammation, platelet activation, or at the intersection of the two (i.e., thromboinflammation). To date, the only FDA approved treatment after aSAH is nimodipine. While the initial mechanism of action was purported to be via anti-vasospasm (105, 106), its effects seem to be more than just preventing vasospasm. Indeed, nimodipine can improve patient outcome irrespective of vasospasm attenuation (107). Although the exact mechanism of nimodipine is unknown, it may reduce microthrombosis, be neuroprotective, and inhibit cortical spreading ischemia (14, 108). Furthermore, nimodipine has been implicated as being anti-inflammatory (109), and it is important to note that calcium channel blockers may reduce leukocyte infiltration (110), and therefore reduce thromboinflammation. Mechanistic studies for the role(s) of nimodipine are needed to clarify the mechanisms by which nimodipine improves patient outcome after SAH. These studies may also shed light onto the therapeutic targets for thromboinflammation.
Fasudil is currently used in Asia instead of nimodipine, because the latter is not commercially available. Fasudil is a rho-kinase inhibitor which is reported to reduce hemodynamic dysfunction (i.e., vasoconstriction, endothelial injury) and inflammation through downstream signaling pathways via inhibition of rho-kinase (111). Initially, fasudil was used to target cerebral vasospasm, but fasudil was found to also reduce cerebral infarction, and improve functional outcome after aSAH (112). Thus, fasudil also seems to provide therapeutic benefit for aSAH patients, but still requires validation in large randomized controlled clinical trials (113). Being a rho-kinase inhibitor, fasudil may prevent a number of downstream signaling pathways involved in SAH pathophysiology [smooth muscle cell contraction and endothelial dysfunction (95, 114), inflammation, and leukocyte activation (115)], arguing that drugs with pleiotropic effects may be an answer. Similar to nimodipine, the exact mechanisms by which fasudil improves outcome following SAH remain a mystery, and translational studies for SAH will benefit for additional studies on the mechanism(s) of fasudil.
The recent study by Wessell et al. found that infusion of low-dose heparin administered with nimodipine is associated with increased odds of patient discharge (116), and, in a preliminary trial, heparin reduced cerebral vasospasm and infarct (117). These studies support the notion that combining therapies which reduce distinct injury mechanisms can provide increased effectiveness. Furthermore, these studies indicate that there may be therapeutic potential in anti-coagulation and anti-inflammation (103). Currently, heparin is currently being explored in the ASTROH trial (NCT02501434) which will examine the efficacy of heparin for reducing vasospasm and delayed neurological deficits in SAH patients.
Specific information regarding the role of thromboinflammation and its potential as a therapeutic target may be elucidated through the SoSTART trial (NCT03153150) and the etanercept (TNFα receptor antagonist) trial (NCT01865630). The SoSTART trial is set to investigate seven anti-coagulants for therapeutic benefit in patients of cerebral hemorrhage, including aSAH. These proposed drugs are known to be reduce coagulability and prevent inflammation (118, 119), which may reduce the overall thromboinflammation.
Challenges in Targeting Thromboinflammation
Several challenges exist when considering thromboinflammation as a therapeutic target for preventing DCI. First and foremost, special care needs to be taken when identifying potential therapies for thromboinflammation since the ideal candidate will only target newly formed microthrombi and not have any significant effect on the clot formed at the site of aneurysm rupture. Once the aneurysm has been secured with clip or coil, the aneurysm is of less concern, but potential complications such as post-operative bleeding or bleeding along the ventriculostomy remain a risk.
In most pathologies, including SAH, both pro- and anti-inflammatory pathways are activated to promote debris/toxin clearance and repair of injured tissue, respectively. Since pro-inflammatory cytokines and downstream signaling is linked with thromboinflammation (34), it is critical to pursue agents which mitigate pro-inflammatory cytokines. So far research has largely focused on this, however, anti-inflammatory signaling aids in repair and healing, thus this arm of inflammation should remain active (or upregulated) after reducing thromboinflammation.
Finally, although experimental studies are required to understand pathophysiology, uncover signaling mechanisms, and investigate novel drugs and potential therapeutic targets, animals (including non-human primates) have differences when it comes to coagulation (120, 121), as well as inflammation (122, 123). Therefore, it follows that animals may have distinct differences when it comes to thromboinflammation and its treatment. Future studies should examine these differences to aid in therapeutic development against thromboinflammation.
Conclusion
Thromboinflammation, the cross talk between the thrombotic and inflammatory pathways, likely plays a critical role in the development of DCI after SAH, thereby having significant impact on overall patient outcome. The current understanding of thromboinflammation and its role in SAH pathophysiology is only beginning to emerge, but evidence from experimental and clinical studies suggest that preventing thromboinflammation has the potential for benefiting patients of aSAH.
Author Contributions
All authors contributed to the conception and drafting of this article.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by a NIH grant (JZ, NS081740) and by a seed grant provided by The Vivian L. Smith Department of Neurosurgery at The University of Texas Health Science Center at Houston (DM).
References
1. Connolly ES Jr, Rabinstein AA, Carhuapoma JR, Derdeyn CP, Dion J, Higashida RT, et al. Guidelines for the management of aneurysmal subarachnoid hemorrhage: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke (2012) 43(6):1711–37. doi:10.1161/STR.0b013e3182587839
2. Grunwald IQ, Kuhn AL, Schmitt AJ, Balami JS. Aneurysmal SAH: current management and complications associated with treatment and disease. J Invasive Cardiol (2014) 26(1):30–7.
3. de Rooij NK, Linn FH, van der Plas JA, Algra A, Rinkel GJ. Incidence of subarachnoid haemorrhage: a systematic review with emphasis on region, age, gender and time trends. J Neurol Neurosurg Psychiatry (2007) 78(12):1365–72. doi:10.1136/jnnp.2007.117655
4. Cossu G, Messerer M, Oddo M, Daniel RT. To look beyond vasospasm in aneurysmal subarachnoid hemorrhage. Biomed Res Int (2014) 2014:628597. doi:10.1155/2014/628597
5. Foreman B. The pathophysiology of delayed cerebral ischemia. J Clin Neurophysiol (2016) 33(3):174–82. doi:10.1097/WNP.0000000000000273
6. Macdonald RL. Delayed neurological deterioration after subarachnoid haemorrhage. Nat Rev Neurol (2014) 10(1):44–58. doi:10.1038/nrneurol.2013.246
7. Brilstra EH, Rinkel GJ, Algra A, van Gijn J. Rebleeding, secondary ischemia, and timing of operation in patients with subarachnoid hemorrhage. Neurology (2000) 55(11):1656–60. doi:10.1212/WNL.55.11.1656
8. Schweizer TA, Al-Khindi T, Macdonald RL. Mini-mental state examination versus montreal cognitive assessment: rapid assessment tools for cognitive and functional outcome after aneurysmal subarachnoid hemorrhage. J Neurol Sci (2012) 316(1–2):137–40. doi:10.1016/j.jns.2012.01.003
9. Vergouwen MD, Vermeulen M, van Gijn J, Rinkel GJ, Wijdicks EF, Muizelaar JP, et al. Definition of delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage as an outcome event in clinical trials and observational studies: proposal of a multidisciplinary research group. Stroke (2010) 41(10):2391–5. doi:10.1161/STROKEAHA.110.589275
10. Frontera JA, Fernandez A, Schmidt JM, Claassen J, Wartenberg KE, Badjatia N, et al. Defining vasospasm after subarachnoid hemorrhage: what is the most clinically relevant definition? Stroke (2009) 40(6):1963–8. doi:10.1161/STROKEAHA.108.544700
11. Schmidt JM, Wartenberg KE, Fernandez A, Claassen J, Rincon F, Ostapkovich ND, et al. Frequency and clinical impact of asymptomatic cerebral infarction due to vasospasm after subarachnoid hemorrhage. J Neurosurg (2008) 109(6):1052–9. doi:10.3171/JNS.2008.109.12.1052
12. Springer MV, Schmidt JM, Wartenberg KE, Frontera JA, Badjatia N, Mayer SA. Predictors of global cognitive impairment 1 year after subarachnoid hemorrhage. Neurosurgery (2009) 65(6):1043–50; discussion 50–1. doi:10.1227/01.NEU.0000359317.15269.20
13. Dankbaar JW, Rijsdijk M, van der Schaaf IC, Velthuis BK, Wermer MJH, Rinkel GJE. Relationship between vasospasm, cerebral perfusion, and delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage. Neuroradiology (2009) 51(12):813–9. doi:10.1007/s00234-009-0575-y
14. Etminan N, Vergouwen MDI, Ilodigwe D, Macdonald RL. Effect of pharmaceutical treatment on vasospasm, delayed cerebral ischemia, and clinical outcome in patients with aneurysmal subarachnoid hemorrhage: a systematic review and meta-analysis. J Cereb Blood Flow Metab (2011) 31(6):1443–51. doi:10.1038/jcbfm.2011.7
15. Kramer A, Fletcher J. Do endothelin-receptor antagonists prevent delayed neurological deficits and poor outcomes after aneurysmal subarachnoid hemorrhage? A meta-analysis. Stroke (2009) 40(10):3403–6. doi:10.1161/STROKEAHA.109.560243
16. Macdonald RL, Higashida RT, Keller E, Mayer SA, Molyneux A, Raabe A, et al. Randomised trial of clazosentan, an endothelin receptor antagonist, in patients with aneurysmal subarachnoid hemorrhage undergoing surgical clipping (CONSCIOUS-2). Acta Neurochir Suppl (2013) 115:27–31. doi:10.1007/978-3-7091-1192-5_7
17. Rabinstein AA, Friedman JA, Nichols DA, Pichelmann MA, McClelland RL, Manno EM, et al. Predictors of outcome after endovascular treatment of cerebral vasospasm. Am J Neuroradiol (2004) 25(10):1778–82.
18. Chamling B, Gross S, Stoffel-Wagner B, Schubert GA, Clusmann H, Coburn M, et al. Early diagnosis of delayed cerebral ischemia: possible relevance for inflammatory biomarkers in routine clinical practice? World Neurosurg (2017) 104:152–7. doi:10.1016/j.wneu.2017.05.021
19. Lucke-Wold BP, Logsdon AF, Manoranjan B, Turner RC, McConnell E, Vates GE, et al. Aneurysmal subarachnoid hemorrhage and neuroinflammation: a comprehensive review. Int J Mol Sci (2016) 17(4):497. doi:10.3390/ijms17040497
20. Ascenzi P, Bocedi A, Visca P, Altruda F, Tolosano E, Beringhelli T, et al. Hemoglobin and heme scavenging. IUBMB Life (2005) 57(11):749–59. doi:10.1080/15216540500380871
21. Fujii M, Yan J, Rolland WB, Soejima Y, Caner B, Zhang JH. Early brain injury, an evolving frontier in subarachnoid hemorrhage research. Trans Stroke Res (2013) 4(4):432–46. doi:10.1007/s12975-013-0257-2
22. Sehba FA, Hou J, Pluta RM, Zhang JH. The importance of early brain injury after subarachnoid hemorrhage. Prog Neurobiol (2012) 97(1):14–37. doi:10.1016/j.pneurobio.2012.02.003
23. McMahon CJ, Hopkins S, Vail A, King AT, Smith D, Illingworth KJ, et al. Inflammation as a predictor for delayed cerebral ischemia after aneurysmal subarachnoid haemorrhage. J Neurointerv Surg (2013) 5(6):512–7. doi:10.1136/neurintsurg-2012-010386
24. Provencio JJ. Inflammation in subarachnoid hemorrhage and delayed deterioration associated with vasospasm: a review. Acta Neurochir Suppl (2013) 115:233–8. doi:10.1007/978-3-7091-1192-5_42
25. Chou SH, Feske SK, Atherton J, Konigsberg RG, De Jager PL, Du R, et al. Early elevation of serum tumor necrosis factor-alpha is associated with poor outcome in subarachnoid hemorrhage. J Investig Med (2012) 60(7):1054–8. doi:10.2310/JIM.0b013e3182686932
26. Gruber A, Rossler K, Graninger W, Donner A, Illievich MU, Czech T. Ventricular cerebrospinal fluid and serum concentrations of sTNFR-I, IL-1ra, and IL-6 after aneurysmal subarachnoid hemorrhage. J Neurosurg Anesthesiol (2000) 12(4):297–306. doi:10.1097/00008506-200010000-00001
27. Hendryk S, Jarzab B, Josko J. Increase of the IL-1 beta and IL-6 levels in CSF in patients with vasospasm following aneurysmal SAH. Neuro Endocrinol Lett (2004) 25(1–2):141–7.
28. Ramchand P, Nyirjesy S, Frangos S, Doerfler S, Nawalinski K, Quattrone F, et al. Thromboelastography parameter predicts outcome after subarachnoid hemorrhage: an exploratory analysis. World Neurosurg (2016) 96:215–21. doi:10.1016/j.wneu.2016.04.002
29. Vergouwen MD, Vermeulen M, Coert BA, Stroes ES, Roos YB. Microthrombosis after aneurysmal subarachnoid hemorrhage: an additional explanation for delayed cerebral ischemia. J Cereb Blood Flow Metab (2008) 28(11):1761–70. doi:10.1038/jcbfm.2008.74
30. Salvarani C, Brown RD Jr, Hunder GG. Adult primary central nervous system vasculitis. Lancet (2012) 380(9843):767–77. doi:10.1016/S0140-6736(12)60069-5
31. Albert-Weissenberger C, Mencl S, Schuhmann MK, Salur I, Gob E, Langhauser F, et al. C1-Inhibitor protects from focal brain trauma in a cortical cryolesion mice model by reducing thrombo-inflammation. Front Cell Neurosci (2014) 8:269. doi:10.3389/fncel.2014.00269
32. De Meyer SF, Denorme F, Langhauser F, Geuss E, Fluri F, Kleinschnitz C. Thromboinflammation in stroke brain damage. Stroke (2016) 47(4):1165–72. doi:10.1161/STROKEAHA.115.011238
33. Gragnano F, Sperlongano S, Golia E, Natale F, Bianchi R, Crisci M, et al. The role of von Willebrand factor in vascular inflammation: from pathogenesis to targeted therapy. Mediators Inflamm (2017) 2017:5620314. doi:10.1155/2017/5620314
34. Kehrel BE, Fender AC. Resolving thromboinflammation in the brain after ischemic stroke? Circulation (2016) 133(22):2128–31. doi:10.1161/CIRCULATIONAHA.116.022858
35. Hartz S, Menart B, Tschoepe D. Leukocyte apoptosis in whole blood involves platelet-dependent coaggregation. Cytometry A (2003) 52(2):117–21. doi:10.1002/cyto.a.10026
36. Swystun LL, Liaw PC. The role of leukocytes in thrombosis. Blood (2016) 128(6):753–62. doi:10.1182/blood-2016-05-718114
37. da Fonseca ACC, Matias D, Garcia C, Amaral R, Geraldo LH, Freitas C, et al. The impact of microglial activation on blood-brain barrier in brain diseases. Front Cell Neurosci (2014) 8:362. doi:10.3389/fncel.2014.00362
38. Munakata A, Naraoka M, Katagai T, Shimamura N, Ohkuma H. Role of cyclooxygenase-2 in relation to nitric oxide and endothelin-1 on pathogenesis of cerebral vasospasm after subarachnoid hemorrhage in rabbit. Trans Stroke Res (2016) 7(3):220–7. doi:10.1007/s12975-016-0466-6
39. Chauhan AK, Kisucka J, Brill A, Walsh MT, Scheiflinger F, Wagner DD. ADAMTS13: a new link between thrombosis and inflammation. J Exp Med (2008) 205(9):2065–74. doi:10.1084/jem.20080130
40. Vergouwen MD, Bakhtiari K, van Geloven N, Vermeulen M, Roos YB, Meijers JC. Reduced ADAMTS13 activity in delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage. J Cereb Blood Flow Metab (2009) 29(10):1734–41. doi:10.1038/jcbfm.2009.88
41. Hendrix P, Foreman PM, Harrigan MR, Fisher WSR, Vyas NA, Lipsky RH, et al. Association of plasminogen activator inhibitor 1 (SERPINE1 gene) polymorphisms and aneurysmal subarachnoid hemorrhage. World Neurosurg (2017) 105:672–77. doi:10.3171/2017.2.JNS162933
42. McGee MP, Foster S, Wang X. Simultaneous expression of tissue factor and tissue factor pathway inhibitor by human monocytes. A potential mechanism for localized control of blood coagulation. J Exp Med (1994) 179(6):1847–54. doi:10.1084/jem.179.6.1847
43. McCachren SS, Diggs J, Weinberg JB, Dittman WA. Thrombomodulin expression by human blood monocytes and by human synovial tissue lining macrophages. Blood (1991) 78:3128–32.
44. Galligan L, Livingstone W, Volkov Y, Hokamp K, Murphy C, Lawler M, et al. Characterization of protein C receptor expression in monocytes. Br J Haematol (2001) 115(2):408–14. doi:10.1046/j.1365-2141.2001.03187.x
45. Shantsila E, Lip GY. The role of monocytes in thrombotic disorders. Insights from tissue factor, monocyte-platelet aggregates and novel mechanisms. Thromb Haemost (2009) 102(5):916–24. doi:10.1160/TH09-01-0023
46. Pham CT. Neutrophil serine proteases: specific regulators of inflammation. Nat Rev Immunol (2006) 6(7):541–50. doi:10.1038/nri1841
47. Basavaraj MG, Gruber FX, Sovershaev M, Appelbom HI, Osterud B, Petersen LC, et al. The role of TFPI in regulation of TF-induced thrombogenicity on the surface of human monocytes. Thromb Res (2010) 126(5):418–25. doi:10.1016/j.thromres.2010.07.014
48. Belaaouaj AA, Li A, Wun TC, Welgus HG, Shapiro SD. Matrix metalloproteinases cleave tissue factor pathway inhibitor. Effects on coagulation. J Biol Chem (2000) 275(35):27123–8. doi:10.1074/jbc.M004218200
49. Massberg S, Grahl L, von Bruehl ML, Manukyan D, Pfeiler S, Goosmann C, et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat Med (2010) 16(8):887–96. doi:10.1038/nm.2184
50. Stein SC, Browne KD, Chen XH, Smith DH, Graham DI. Thromboembolism and delayed cerebral ischemia after subarachnoid hemorrhage: an autopsy study. Neurosurgery (2006) 59(4):781–7; discussion 7–8. doi:10.1227/01.NEU.0000227519.27569.45
51. Suzuki S, Kimura M, Souma M, Ohkima H, Shimizu T, Iwabuchi T. Cerebral microthrombosis in symptomatic cerebral vasospasm – a quantitative histological study in autopsy cases. Neurol Med Chir (Tokyo) (1990) 30(5):309–16. doi:10.2176/nmc.30.309
52. Hirashima Y, Hamada H, Kurimoto M, Origasa H, Endo S. Decrease in platelet count as an independent risk factor for symptomatic vasospasm following aneurysmal subarachnoid hemorrhage. J Neurosurg (2005) 102(5):882–7. doi:10.3171/jns.2005.102.5.0882
53. Vinge E, Brandt L, Ljunggren B, Andersson KE. Thromboxane B2 levels in serum during continuous administration of nimodipine to patients with aneurysmal subarachnoid hemorrhage. Stroke (1988) 19(5):644–7. doi:10.1161/01.STR.19.5.644
54. Boluijt J, Meijers JC, Rinkel GJ, Vergouwen MD. Hemostasis and fibrinolysis in delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage: a systematic review. J Cereb Blood Flow Metab (2015) 35(5):724–33. doi:10.1038/jcbfm.2015.13
55. Ji Y, Meng QH, Wang ZG. Changes in the coagulation and fibrinolytic system of patients with subarachnoid hemorrhage. Neurol Med Chir (Tokyo) (2014) 54(6):457–64. doi:10.2176/nmc.oa2013-0006
56. Peltonen S, Juvela S, Kaste M, Lassila R. Hemostasis and fibrinolysis activation after subarachnoid hemorrhage. J Neurosurg (1997) 87(2):207–14. doi:10.3171/jns.1997.87.2.0207
57. Al-Hamodi ZH, Saif-Ali R, Ismail IS, Ahmed KA, Muniandy S. Plasminogen activator inhibitor-1 4G/5G polymorphism is associated with metabolic syndrome parameters in Malaysian subjects. J Clin Biochem Nutr (2012) 50(3):184–9. doi:10.3164/jcbn.11-48
58. Sonneveld MA, de Maat MP, Portegies ML, Kavousi M, Hofman A, Turecek PL, et al. Low ADAMTS13 activity is associated with an increased risk of ischemic stroke. Blood (2015) 126(25):2739–46. doi:10.1182/blood-2015-05-643338
59. Mathiesen T, Edner G, Ulfarsson E, Andersson B. Cerebrospinal fluid interleukin-1 receptor antagonist and tumor necrosis factor-alpha following subarachnoid hemorrhage. J Neurosurg (1997) 87(2):215–20. doi:10.3171/jns.1997.87.2.0215
60. Graetz D, Nagel A, Schlenk F, Sakowitz O, Vajkoczy P, Sarrafzadeh A. High ICP as trigger of proinflammatory IL-6 cytokine activation in aneurysmal subarachnoid hemorrhage. Neurol Res (2010) 32(7):728–35. doi:10.1179/016164109X12464612122650
61. Fassbender K, Hodapp B, Rossol S, Bertsch T, Schmeck J, Schutt S, et al. Inflammatory cytokines in subarachnoid haemorrhage: association with abnormal blood flow velocities in basal cerebral arteries. J Neurol Neurosurg Psychiatry (2001) 70(4):534–7. doi:10.1136/jnnp.70.4.534
62. Gaetani P, Tartara F, Pignatti P, Tancioni F, Rodriguez y Baena R, De Benedetti F. Cisternal CSF levels of cytokines after subarachnoid hemorrhage. Neurol Res (1998) 20(4):337–42. doi:10.1080/01616412.1998.11740528
63. Hirashima Y, Nakamura S, Endo S, Kuwayama N, Naruse Y, Takaku A. Elevation of platelet activating factor, inflammatory cytokines, and coagulation factors in the internal jugular vein of patients with subarachnoid hemorrhage. Neurochem Res (1997) 22(10):1249–55. doi:10.1023/A:1021985030331
64. Kikuchi T, Okuda Y, Kaito N, Abe T. Cytokine production in cerebrospinal fluid after subarachnoid haemorrhage. Neurol Res (1995) 17(2):106–8. doi:10.1080/01616412.1995.11740296
65. Mathiesen T, Andersson B, Loftenius A, von Holst H. Increased interleukin-6 levels in cerebrospinal fluid following subarachnoid hemorrhage. J Neurosurg (1993) 78(4):562–7. doi:10.3171/jns.1993.78.4.0562
66. Osuka K, Suzuki Y, Tanazawa T, Hattori K, Yamamoto N, Takayasu M, et al. Interleukin-6 and development of vasospasm after subarachnoid haemorrhage. Acta Neurochir (Wien) (1998) 140(9):943–51. doi:10.1007/s007010050197
67. Osuka K, Watanabe Y, Aoyama M, Nakura T, Matsuo N, Takayasu M. Expression of suppressor of cytokine signaling 3 in cerebrospinal fluid after subarachnoid hemorrhage. J Neuroinflammation (2014) 11:142. doi:10.1186/s12974-014-0142-2
68. Takizawa T, Tada T, Kitazawa K, Tanaka Y, Hongo K, Kameko M, et al. Inflammatory cytokine cascade released by leukocytes in cerebrospinal fluid after subarachnoid hemorrhage. Neurol Res (2001) 23(7):724–30. doi:10.1179/016164101101199243
69. Kubo Y, Ogasawara K, Kakino S, Kashimura H, Tomitsuka N, Sugawara A, et al. Serum inflammatory adhesion molecules and high-sensitivity C-reactive protein correlates with delayed ischemic neurologic deficits after subarachnoid hemorrhage. Surg Neurol (2008) 69(6):592–6; discussion 6. doi:10.1016/j.surneu.2008.02.014
70. Mack WJ, Mocco J, Hoh DJ, Huang J, Choudhri TF, Kreiter KT, et al. Outcome prediction with serum intercellular adhesion molecule-1 levels after aneurysmal subarachnoid hemorrhage. J Neurosurg (2002) 96(1):71–5. doi:10.3171/jns.2002.96.1.0071
71. Mocco J, Mack WJ, Kim GH, Lozier AP, Laufer I, Kreiter KT, et al. Rise in serum soluble intercellular adhesion molecule-1 levels with vasospasm following aneurysmal subarachnoid hemorrhage. J Neurosurg (2002) 97(3):537–41. doi:10.3171/jns.2002.97.3.0537
72. Frijns CJ, Kasius KM, Algra A, Fijnheer R, Rinkel GJ. Endothelial cell activation markers and delayed cerebral ischaemia in patients with subarachnoid haemorrhage. J Neurol Neurosurg Psychiatry (2006) 77(7):863–7. doi:10.1136/jnnp.2005.064956
73. Nissen JJ, Mantle D, Gregson B, Mendelow AD. Serum concentration of adhesion molecules in patients with delayed ischaemic neurological deficit after aneurysmal subarachnoid haemorrhage: the immunoglobulin and selectin superfamilies. J Neurol Neurosurg Psychiatry (2001) 71(3):329–33. doi:10.1136/jnnp.71.3.329
74. Denis CV, Wagner DD. Platelet adhesion receptors and their ligands in mouse models of thrombosis. Arterioscler Thromb Vasc Biol (2007) 27(4):728–39. doi:10.1161/01.ATV.0000259359.52265.62
75. Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol (2007) 7(9):678–89. doi:10.1038/nri2156
76. Wagner DD. New links between inflammation and thrombosis. Arterioscler Thromb Vasc Biol (2005) 25(7):1321–4. doi:10.1161/01.ATV.0000166521.90532.44
77. Crompton MR. The pathogenesis of cerebral infarction following the rupture of cerebral berry aneurysms. Brain (1964) 87:491–510. doi:10.1093/brain/87.3.491
78. Gallia GL, Tamargo RJ. Leukocyte-endothelial cell interactions in chronic vasospasm after subarachnoid hemorrhage. Neurol Res (2006) 28(7):750–8. doi:10.1179/016164106X152025
79. Hughes JT, Schianchi PM. Cerebral artery spasm. A histological study at necropsy of the blood vessels in cases of subarachnoid hemorrhage. J Neurosurg (1978) 48(4):515–25. doi:10.3171/jns.1978.48.4.0515
80. Pluta RM, Zauner A, Morgan JK, Muraszko KM, Oldfield EH. Is vasospasm related to proliferative arteriopathy? J Neurosurg (1992) 77(5):740–8. doi:10.3171/jns.1992.77.5.0740
81. Spallone A, Acqui M, Pastore FS, Guidetti B. Relationship between leukocytosis and ischemic complications following aneurysmal subarachnoid hemorrhage. Surg Neurol (1987) 27(3):253–8. doi:10.1016/0090-3019(87)90038-3
82. Frontera JA, Provencio JJ, Sehba FA, McIntyre TM, Nowacki AS, Gordon E, et al. The role of platelet activation and inflammation in early brain injury following subarachnoid hemorrhage. Neurocrit Care (2017) 26(1):48–57. doi:10.1007/s12028-016-0292-4
83. Andereggen L, Neuschmelting V, von Gunten M, Widmer HR, Fandino J, Marbacher S. The role of microclot formation in an acute subarachnoid hemorrhage model in the rabbit. Biomed Res Int (2014) 2014:161702. doi:10.1155/2014/161702
84. Sabri M, Ai J, Lakovic K, D’Abbondanza J, Ilodigwe D, Macdonald RL. Mechanisms of microthrombi formation after experimental subarachnoid hemorrhage. Neuroscience (2012) 224:26–37. doi:10.1016/j.neuroscience.2012.08.002
85. Tso MK, Macdonald RL. Acute microvascular changes after subarachnoid hemorrhage and transient global cerebral ischemia. Stroke Res Treat (2013) 2013:425281. doi:10.1155/2013/425281
86. Wang Z, Chen G, Zhu WW, Bian JY, Shen XO, Zhou D. Influence of simvastatin on microthrombosis in the brain after subarachnoid hemorrhage in rats: a preliminary study. Ann Clin Lab Sci (2010) 40(1):32–42.
87. Herz DA, Baez S, Shulman K. Pial microcirculation in subarachnoid hemorrhage. Stroke (1975) 6(4):417–24. doi:10.1161/01.STR.6.4.417
88. Sehba FA, Mostafa G, Friedrich V Jr, Bederson JB. Acute microvascular platelet aggregation after subarachnoid hemorrhage. J Neurosurg (2005) 102(6):1094–100. doi:10.3171/jns.2005.102.6.1094
89. Maddahi A, Ansar S, Chen Q, Edvinsson L. Blockade of the MEK/ERK pathway with a raf inhibitor prevents activation of pro-inflammatory mediators in cerebral arteries and reduction in cerebral blood flow after subarachnoid hemorrhage in a rat model. J Cereb Blood Flow Metab (2011) 31(1):144–54. doi:10.1038/jcbfm.2010.62
90. Maddahi A, Povlsen G, Edvinsson L. Regulation of enhanced cerebrovascular expression of proinflammatory mediators in experimental subarachnoid hemorrhage via the mitogen-activated protein kinase kinase/extracellular signal-regulated kinase pathway. J Neuroinflammation (2012) 9:274. doi:10.1186/1742-2094-9-274
91. Handa Y, Kubota T, Kaneko M, Tsuchida A, Kobayashi H, Kawano H, et al. Expression of intercellular adhesion molecule 1 (ICAM-1) on the cerebral artery following subarachnoid haemorrhage in rats. Acta Neurochir (1995) 132(1):92–7. doi:10.1007/BF01404854
92. Lin CL, Dumont AS, Calisaneller T, Kwan AL, Hwong SL, Lee KS. Monoclonal antibody against E selectin attenuates subarachnoid hemorrhage-induced cerebral vasospasm. Surg Neurol (2005) 64(3):201–5; discussion 5–6. doi:10.1016/j.surneu.2005.04.038
93. Friedrich V, Flores R, Muller A, Bi W, Peerschke EIB, Sehba FA. Reduction of neutrophil activity decreases early microvascular injury after subarachnoid haemorrhage. J Neuroinflammation (2011) 8:103. doi:10.1186/1742-2094-8-103
94. Hayman EG, Patel AP, James RF, Simard JM. Heparin and heparin-derivatives in post-subarachnoid hemorrhage brain injury: a multimodal therapy for a multimodal disease. Molecules (2017) 22(5):724. doi:10.3390/molecules22050724
95. Satoh S-I, Yamamoto Y, Toshima Y, Ikegaki I, Asano T, Suzuki Y, et al. Fasudil, a protein kinase inhibitor, prevents the development of endothelial injury and neutrophil infiltration in a two-haemorrhage canine subarachnoid model. J Clin Neurosci (1999) 6(5):394–9. doi:10.1016/S0967-5868(99)90034-6
96. Stokes KY, Granger DN. Platelets: a critical link between inflammation and microvascular dysfunction. J Physiol (2012) 590(5):1023–34. doi:10.1113/jphysiol.2011.225417
97. Dorhout Mees SM, van den Bergh WM, Algra A, Rinkel GJ. Antiplatelet therapy for aneurysmal subarachnoid haemorrhage. Cochrane Database Syst Rev (2007) 4:CD006184. doi:10.1002/14651858.CD006184.pub2
98. Kramer AH, Roberts DJ, Holodinsky J, Todd S, Hill MD, Zygun DA, et al. Intraventricular tissue plasminogen activator in subarachnoid hemorrhage patients: a prospective, randomized, placebo-controlled pilot trial. Neurocrit Care (2014) 21(2):275–84. doi:10.1007/s12028-014-9965-z
99. Litrico S, Almairac F, Gaberel T, Ramakrishna R, Fontaine D, Sedat J, et al. Intraventricular fibrinolysis for severe aneurysmal intraventricular hemorrhage: a randomized controlled trial and meta-analysis. Neurosurg Rev (2013) 36(4):523–30; discussion 30–1. doi:10.1007/s10143-013-0469-7
100. Etminan N, Beseoglu K, Eicker SO, Turowski B, Steiger H-J, Hanggi D. Prospective, randomized, open-label phase ii trial on concomitant intraventricular fibrinolysis and low-frequency rotation after severe subarachnoid hemorrhage. Stroke (2013) 44:2162–8. doi:10.1161/STROKEAHA.113.001790
101. Grasso G, Tomasello G, Noto M, Alafaci C, Cappello F. Erythropoietin for the treatment of subarachnoid hemorrhage: a feasible ingredient for a successful medical recipe. Mol Med (2015) 21(1):979–87. doi:10.2119/molmed.2015.00177
102. Tseng MY, Hutchinson PJ, Richards HK, Czosnyka M, Pickard JD, Erber WN, et al. Acute systemic erythropoietin therapy to reduce delayed ischemic deficits following aneurysmal subarachnoid hemorrhage: a phase II randomized, double-blind, placebo-controlled trial. Clinical article. J Neurosurg (2009) 111(1):171–80. doi:10.3171/2009.3.JNS081332
103. Veldeman M, Höllig A, Clusmann H, Stevanovic A, Rossaint R, Coburn M. Delayed cerebral ischaemia prevention and treatment after aneurysmal subarachnoid haemorrhage: a systematic review. Br J Anaesth (2016) 117(1):17–40. doi:10.1093/bja/aew095
104. Gomis P, Graftieaux JP, Sercombe R, Hettler D, Scherpereel B, Rousseaux P. Randomized, double-blind, placebo-controlled, pilot trial of high-dose methylprednisolone in aneurysmal subarachnoid hemorrhage. J Neurosurg (2010) 112(3):681–8. doi:10.3171/2009.4.JNS081377
105. Mijailovic M, Lukic S, Laudanovic D, Folic M, Folic N, Jankovic S. Effects of nimodipine on cerebral vasospasm in patients with aneurysmal subarachnoid hemorrhage treated by endovascular coiling. Adv Clin Exp Med (2013) 22(1):101–9.
106. Castanares-Zapatero D, Hantson P. Pharmacological treatment of delayed cerebral ischemia and vasospasm in subarachnoid hemorrhage. Ann Intensive Care (2011) 1(1):12. doi:10.1186/2110-5820-1-12
107. Raya AK, Diringer MN. Treatment of subarachnoid hemorrhage. Crit Care Clin (2014) 30(4):719–33. doi:10.1016/j.ccc.2014.06.004
108. Etminan N, Macdonald RL. Management of aneurysmal subarachnoid hemorrhage. Handb Clin Neurol (2017) 140:195–228. doi:10.1016/B978-0-444-63600-3.00012-X
109. Sanz JM, Chiozzi P, Colaianna M, Zotti M, Ferrari D, Trabace L, et al. Nimodipine inhibits IL-1beta release stimulated by amyloid beta from microglia. Br J Pharmacol (2012) 167(8):1702–11. doi:10.1111/j.1476-5381.2012.02112.x
110. Martinez LL, Aparecida De Oliveira M, Fortes ZB. Influence of verapamil and diclofenac on leukocyte migration in rats. Hypertension (1999) 34(4 Pt 2):997–1001. doi:10.1161/01.HYP.34.4.997
111. Zhao J, Zhou D, Guo J, Ren Z, Zhou L, Wang S, et al. Efficacy and safety of fasudil in patients with subarachnoid hemorrhage: final results of a randomized trial of fasudil versus nimodipine. Neurol Med Chir (Tokyo) (2011) 51(10):679–83. doi:10.2176/nmc.51.679
112. Liu GJ, Wang ZJ, Wang YF, Xu LL, Wang XL, Liu Y, et al. Systematic assessment and meta-analysis of the efficacy and safety of fasudil in the treatment of cerebral vasospasm in patients with subarachnoid hemorrhage. Eur J Clin Pharmacol (2012) 68(2):131–9. doi:10.1007/s00228-011-1100-x
113. Liu Y-F, Qiu H-Q, Su J, Jiang W-J. Drug treatment of cerebral vasospasm after subarachnoid hemorrhage following aneurysms. Chinese Neurosurg J (2016) 2:4. doi:10.1186/s41016-016-0023-x
114. Liu AJ, Ling F, Wang D, Wang Q, Lu XD, Liu YL. Fasudil inhibits platelet-derived growth factor-induced human pulmonary artery smooth muscle cell proliferation by up-regulation of p27kip(1) via the ERK signal pathway. Chinese Med J (2011) 124(19):3098–104.
115. Slotta JE, Braun OO, Menger MD, Thorlacius H. Fasudil, a Rho-kinase inhibitor, inhibits leukocyte adhesion in inflamed large blood vessels in vivo. Inflamm Res (2006) 55(9):364–7. doi:10.1007/s00011-006-6013-2
116. Wessell A, Kole MJ, Badjatia N, Parikh G, Albrecht JS, Schreibman DL, et al. High compliance with scheduled nimodipine is associated with better outcome in aneurysmal subarachnoid hemorrhage patients cotreated with heparin infusion. Front Neurol (2017) 8:268. doi:10.3389/fneur.2017.00268
117. Simard JM, Aldrich EF, Schreibman D, James RF, Polifka A, Beaty N. Low-dose intravenous heparin infusion in patients with aneurysmal subarachnoid hemorrhage: a preliminary assessment. J Neurosurg (2013) 119(6):1611–9. doi:10.3171/2013.8.JNS1337
118. Stapler S, Farris D, Luke C, Diaz J, Henke P, Wakefield T, et al. Efficacy of apixaban on thrombus resolution and anti-inflammation in a murine model of acute deep venous thrombosis. J Vascular Surg Venous Lymphat Disord (2016) 4(1):139–40. doi:10.1016/j.jvsv.2015.10.019
119. Zemer-Wassercug N, Haim M, Leshem-Lev D, Orvin KL, Vaduganathan M, Gutstein A, et al. The effect of dabigatran and rivaroxaban on platelet reactivity and inflammatory markers. J Thromb Thrombolysis (2015) 40(3):340–6. doi:10.1007/s11239-015-1245-z
120. Neville B, Fareed J, Florian-Kujawski M, Cera L, Duff R, Valero A, et al. Coagulation profiling of human, non-human, primate, pig, dog, rabbit, and rat plasma: pharmacologic implications. FASEB J (2006) 20:A655–6.
121. Siller-Matula JM, Plasenzotti R, Spiel A, Quehenberger P, Jilma B. Interspecies differences in coagulation profile. Thromb Haemost (2008) 100(3):397–404. doi:10.1160/TH08-02-0103
122. Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol (2005) 5(12):953–64. doi:10.1038/nri1733
Keywords: thromboinflammation, delayed cerebral ischemia, subarachnoid hemorrhage, thrombosis, inflammation, cerebral vasospasm
Citation: McBride DW, Blackburn SL, Peeyush KT, Matsumura K and Zhang JH (2017) The Role of Thromboinflammation in Delayed Cerebral Ischemia after Subarachnoid Hemorrhage. Front. Neurol. 8:555. doi: 10.3389/fneur.2017.00555
Received: 16 August 2017; Accepted: 02 October 2017;
Published: 23 October 2017
Edited by:
J. Marc Simard, University of Maryland, Baltimore, United StatesReviewed by:
Rick Gill, Hospital of the University of Pennsylvania, United StatesBaback Arshi, University of Illinois at Chicago, United States
Jose Javier Provencio, Cleveland Clinic, United States
Copyright: © 2017 McBride, Blackburn, Peeyush, Matsumura and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Devin W. McBride, devin.w.mcbride@uth.tmc.edu