 Xu Yan
Xu Yan Yue Hu
Yue Hu Biyao Wang
Biyao Wang Sijian Wang
Sijian Wang Xinwen Zhang
Xinwen Zhang- 1The VIP Department, School and Hospital of Stomatology, China Medical University, Liaoning Provincial Key Laboratory of Oral Diseases, Shenyang, China
- 2Center of Implant Dentistry, School and Hospital of Stomatology, China Medical University, Liaoning Provincial Key Laboratory of Oral Diseases, Shenyang, China
Alzheimer’s disease (AD) is an incurable neurodegenerative disease. Numerous studies have demonstrated a critical role for dysregulated glucose metabolism in its pathogenesis. In this review, we summarize metabolic alterations in aging brain and AD-related metabolic deficits associated with glucose metabolism dysregulation, glycolysis dysfunction, tricarboxylic acid (TCA) cycle, oxidative phosphorylation (OXPHOS) deficits, and pentose phosphate pathway impairment. Additionally, we discuss recent treatment strategies targeting metabolic defects in AD, including their limitations, in an effort to encourage the development of novel therapeutic strategies.
Introduction
Alzheimer’s disease (AD) is an age-related neurodegenerative disease characterized by a progressive loss of neuronal structure and function. It is the most common type of dementia worldwide. The condition involves a progressive deterioration in memory, cognition, and mobility (Alzheimer’s Association, 2012). Its pathophysiology is extremely complex and heterogeneous, entailing accumulation of senile plaques caused by abnormal amyloid β (Aβ) metabolism, and neurofibrillary tangles caused by tau hyperphosphorylation (i.e., the formation of p-tau). Furthermore, the cerebrovascular system is seriously damaged, including the disturbance of the blood-brain barrier (BBB) and cerebral amyloid angiopathy (Viswanathan and Greenberg, 2011). Increased levels of reactive oxygen species (ROS) induce the transcription of pro-inflammatory genes and the release of cytokines (e.g., interleukin-1β [IL-1β], IL-6, and tumor necrosis factor-alpha [TNF-α]) and chemokines that cause neuroinflammation. In addition, reactive microglia and astrocytes and other pathological events also contribute to the dysfunction and deprivation of synapses and, ultimately, neuronal death (Martins et al., 2018). Aging is a major risk factor for AD. Some similarities and differences occur in glucose metabolism-related proteins have been observed in the brain during normal aging and AD (Klosinski et al., 2015). The apolipoprotein E (ApoE) gene is responsible for synaptic repair and neuronal structure maintenance and is a major risk factor for the sporadic form of AD. Of the three major isoforms (i.e., ApoE2, ApoE3, and ApoE4), people with the ApoE4 allele are at higher risk of developing AD than the others (Munoz and Feldman, 2000).
The brain consumes the greatest amount of energy of all the organs in the body. Because neurons require large amounts of energy to maintain their normal activities, a metabolic decline in the aging brain contributes to cognitive impairment (Boveris and Navarro, 2008). There is an age-related decrease in glucose utilization in most human brains (Petit-Taboué et al., 1998). In addition, reduced O2 uptake has been observed in the aging rodent brain (Navarro and Boveris, 2008). The pathological metabolic alterations in aging (e.g., cerebral glucose hypometabolism) are early and consistent events in the progression of AD. Glucose, the main transportation form of carbohydrate in our blood, is also the crucial and primary energy substrate for the brain under physiological conditions (Bouzier-Sore et al., 2006). However, alternative substrates, such as glycogen, ketone bodies, and amino acids, are also critical. Energy hypometabolism, particularly a decline in glucose metabolism, is one of the earliest and most common anomalies observed in patients with AD (Small et al., 2006). Indeed, insulin and insulin growth factor-1 (IGF-1) signaling help to maintain and control metabolism and cognition in the central nervous system (CNS) (de la Monte and Wands, 2005), and insulin resistance is one of the main risk factors for AD (Diehl et al., 2017). The main intracellular energy metabolism pathways occurring in our brains are complicated and include anaerobic glycolysis and the pentose phosphate pathway (PPP) in the cytoplasm, as well as oxidative phosphorylation (OXPHOS) in mitochondria and the tricarboxylic acid (TCA) cycle (also known as the citric acid cycle and Krebs cycle) (Dienel, 2019).
Metabolic processes are regulated by a series of key enzymes. Indeed, a growing body of evidence suggests the presence of organic impairment of mitochondria (Macdonald et al., 2018) and damage to related metabolic enzymes (Bubber et al., 2005). In addition, oxygen and glucose metabolic rates are drastically changed in many neurodegenerative diseases, including AD due to marked alterations in the glycolytic pathway and TCA cycle (Hoyer, 1982; Arias et al., 2002; van Gijsel-Bonnello et al., 2017). To add insult to injury, metabolism dysregulation is related to inflammatory responses, particularly in microglia. Baik et al. (2019) reported that Aβ could directly activate microglia to produce inflammatory factors by shifting their metabolism from OXPHOS to aerobic glycolysis. Thus, understanding the relationship between dysfunctional metabolism and AD could provide new insights into the pathogenesis of AD to support the development of new therapies. In this review, we discuss normal metabolic processes, aging, AD-related dysregulation, and relevant treatment strategies.
Glucose Metabolism Dysregulation
Normal Glucose Metabolism
As the major energy source for the brain, glucose is metabolized to ATP, an unstable high-energy compound. Glucose metabolism in the brain involves several stages. First, a signal is received by the brain to trigger glucose uptake (e.g., insulin signaling). Next, the physiological process of glucose uptake occurs. This process is dependent on glucose transporters (GLUTs) spread throughout the brain that allow glucose to cross the BBB and reach the neurocytes (e.g., astrocytes) (Molofsky et al., 2012). There are several different GLUT subtypes in the brain. GLUT1 (55 kDa) transfers glucose from the blood into the extracellular space of the brain through the BBB endothelium, while GLUT1 (45 kDa) and GLUT3 take up glucose into astrocytes and neurons, respectively (Cunnane et al., 2011). The insulin-sensitive GLUT4 is found in discrete subsets of neurons (Choeiri et al., 2002). GLUT8 is located in the intracellular compartment of hippocampal and cerebellar neurons regulated by hormone, while its exact location and function are still undefined (Gomez et al., 2010). After uptake, glucose is metabolized through the glycolysis pathway to pyruvate, generating ATP. Finally, pyruvate is converted to acetyl coenzyme A (acetyl-CoA) via the TCA cycle, which eventually forms an electro-gradient to drive the rotation of the V-ATPase machinery, and energy is transferred from the electro-potential to the bound ATP (Adeva-Andany et al., 2016). The hydrogen generated from this oxidation is transformed into water and ATP through complexes I, II, III, and IV of the electron transport chain (ETC). This OXPHOS reaction occurs on the inner mitochondrial membrane (IMM) (Hauptmann et al., 2009). Concurrently, the carbon dioxide produced by decarboxylation is transported through the blood to the respiratory system and expelled.
Blood glucose accesses the brain via the GLUTs with the help of insulin (Arnold et al., 2018). Decreased expression of insulin-sensitive GLUTs is strongly associated with a decline in glucose uptake (Taguchi et al., 2007). Insulin and the insulin receptor (IR) are vital factors in regulating glucose utilization and energy homeostasis between the CNS and peripheral circulatory system. Insulin signaling is reciprocally linked to the mammalian target of rapamycin (mTOR) pathway via the phosphoinositide-3-kinase (PI3K)/Akt axis (O’Neill, 2013). As an intracellular energy sensor, mTOR is activated by growth factors, including amino acids and high cellular energy status. Thus, the mTOR signaling pathway plays a major role in regulating cell growth and lipid and glucose metabolism (Di Domenico et al., 2018). Additionally, both inhibited insulin signaling and altered protein homeostasis in early AD can lead to aberrant mTOR activation (Dennis et al., 2001). The coupling of PI3K/Akt to IGF-1 and IR can be eliminated through serine phosphorylation of insulin receptor substrate-1 (IRS-1), mediated by mTOR, and IRS-1 inactivation and degradation, which is a prominent trigger of brain insulin resistance (BIR) (Tanti and Jager, 2009). Insulin also downregulates mTOR due to continuous activation of IRS-1 by mTOR (Yoon, 2017). The insulin signaling cascade is also regulated by the unique Ser/Thr/Tyr kinase biliverdin reductase-A (BVR-A). Oxidative stress-induced impairment of BVR-A kinase activity is an early event. Moreover, glucose starvation (hypoglycemia) reduces the intracellular ATP/AMP ratio, activating AMP-activated protein kinase (AMPK). As the main sensor of intracellular fuel status activated by energy stress (Gowans and Hardie, 2014), AMPK participates in the induction of several genes responsible for the growth, maintenance, and repair of neuronal cells and synapses. In addition, AMPK can regulate the plasticity of the hippocampal synapse, the cornerstone of learning and memory (Akter et al., 2011). To sum up, GLUTs are regulated by multiple pathways, and a balance among these pathways is crucial to maintaining the stability of energy metabolism.
Altered Glucose Metabolism in Aging and AD
The brain is mainly composed of terminally differentiated neurons. During aging, neurons with relatively low regenerative ability are unable to adapt to alterations in the basal metabolic rate, and the energy-driven state is decreased or degenerated, which may contribute to various neurodegenerative diseases (Swerdlow, 2007; Isaev et al., 2019). 18F-fluorodeoxyglucose (FDG) and Pittsburgh Compound B (PIB) positron emission tomography (PET) (PIB-PET and FDG-PET, respectively) are suitable for detecting brain glucose uptake. Micro-FDG-PET scans show that cerebral glucose uptake in normal-aging humans (Pardo et al., 2007) and aged rats (Jiang et al., 2013) is declined compared to the youth. Moreover, the role of reduced cerebral glucose uptake in age-related cognitive impairment has been verified in clinical studies (Gejl et al., 2017). The expression of neuronal GLUTs, such as GLUT3 and GLUT4, significantly decreases with aging, while GLUT1 (55 KDa) expression in vascular endothelial cells decreases only slightly (Jais et al., 2016). BIR, resulting in the partial inactivation of insulin signaling and the impairment of PI3K/Akt and several downstream pathways, has been linked to aging (Barzilai and Ferrucci, 2012). Ultimately, these changes contribute to a variety of characteristics of normal aging in the body.
Compared to normal aging, a strong reduction in glucose consumption can be observed in AD (Kato et al., 2016). Alterations in glucose metabolism can affect the maintenance of neurotransmission and neuronal function and impact the ability to learn and memorize. Specific damage to cerebral glucose metabolism has been detected in the posterior cingulum cortex and temporoparietal cortices using FDG-PET (Bohnen et al., 2012). In contrast, glucose metabolism is relatively constant in areas of primary sensorimotor and visual cortices, basal ganglia, and the cerebellum in AD patients (Womack et al., 2011). Decreased glucose utilization may occur even before the clinical symptoms of AD based on a study involving mild cognitive impairment (MCI) subjects (Drzezga et al., 2003). Glucose uptake in the precuneus, an area of early Aβ deposition, is significantly decreased in individuals with the disease 10 years before the appearance of symptoms (Bateman, 2012). Tau mutants regulate mitochondrial trafficking by altering the fragmentation of mitochondria in neuronal cells (Kausar et al., 2018). Two different sites in human islet amyloid polypeptide sequence are sensitive to BACE1-mediated APP cleavage (Rulifson et al., 2016). Aβ may be relevant to the interaction of IR and the GLUTs (Rulifson et al., 2016). The disturbance of neurogenic glucose metabolism caused by impaired insulin signaling results in AD characteristics that parallel the pathophysiology of non-nervous tissues in type 2 diabetes mellitus. The density of the neuronal GLUT3 transporter is associated with local cerebral glucose utilization (Duelli and Kuschinsky, 2001). A reduction in GLUT1 and GLUT3 in AD patient brains (Simpson et al., 1994; An et al., 2018) is correlated with a decline in brain glucose consumption and cognition impairment (Landau et al., 2010). The decline may result from the abnormal hyperphosphorylation of tau and decreased hypoxia-inducible factor-1 α (HIF-1α) levels, which contribute to the transcriptional activation of GLUT (Liu et al., 2008). GLUT4 is reduced in male 3xTG-AD mice (Sancheti et al., 2013); however, cerebral glucose uptake does not coincide with GLUT4 expression in female mice. In addition, astrocyte activation may contribute to the GLUT2 upregulation observed in postmortem brain tissue from AD patients (Liu et al., 2008).
The insulin and IGF-1 signaling (IIS) pathway has considerable effects on metabolism regulation and cognitive function (de la Monte and Wands, 2005). IIS binds to tyrosine kinase receptors and IGF-1 receptor (IGF-1R), which are widely distributed in the hippocampus and cerebral cortex in AD (Freude et al., 2009). The insulin signal is inhibited in the AD brain, which is closely connected to inefficiency in glucose metabolism (Neth and Craft, 2017). The impairment of insulin signaling also contributes to abnormalities in mitochondrial structure and function (Cheng et al., 2010). Moreover, significant gene expression alterations observed in the AD brain are related to the generation and transmission of insulin signals (Hokama et al., 2014). For example, the post-synaptic β-aminobutyric acid (GABA) - a receptor accumulates on the cell surface rapidly when the insulin pathway is activated; however, the genes encoding GABA receptors are markedly reduced in AD brain tissue (Luscher et al., 2011). Insulin competitively inhibits the insulin-degrading enzyme (IDE) to degrade Aβ and elevates extracellular Aβ levels by promoting its secretion (Gasparini et al., 2001). IDE is involved in insulin function/resistance and metabolism-related processes and plays an important role in the degradation of Aβ monomers (Farris et al., 2004). However, its expression is reduced in AD patients (Craft et al., 2003). Energy-transducing pathways always occur in the mitochondria, and the initial activation of IIS requires mitochondria to produce low-level H2O2, which reflects the energy state of mitochondria and is involved in the regulation of redox (Storozhevykh et al., 2007). The insulin receptor (IR) and insulin receptor substrate (IRS) renders them susceptible to oxidation (and activation) by H2O2 (Yin et al., 2014). Excess H2O2 can give rise to BIR (Swerdlow, 2011), which is a risk factor for diabetes mellitus and AD (Luchsinger et al., 2001; Bedse et al., 2015). Thus, the regulation of redox-sensitive signals in the mitochondria should also be considered (Yin et al., 2014). BIR compromises the intracellular translocation of low-density lipoprotein receptor-related protein 1 (LRP1, a receptor for ApoE) to the plasma membrane of hepatocytes, potentially hindering the hepatic elimination of circulating Aβ (Tamaki et al., 2007). In a 3xTg-AD mouse model, oxidative stress first causes consistent activation of IRS-1 and then activates negative feedback mechanisms (e.g., mTOR) to disable IRS-1 hyperactivity and cause BIR (Barone et al., 2016). Overall, BIR is regulated by aberrant insulin signaling and may contribute to disordered glucose metabolism, oxidative stress, BBB dysfunction, and energy supply insufficiency, which are pathological features of diabetes mellitus that can further affect Aβ generation and clearance (Leuner et al., 2012; Salameh et al., 2016). Thus, insulin resistance that induces glucose hypometabolism might be the main cause of energy deficiency in AD brains, which resemble, but is distinct from, the manifestation of diabetes. Thus, it is hypothesized that AD is a neuroendocrine disorder that may be identified as “Type 3 diabetes,” reflecting a new mechanism of neurodegeneration (Steen et al., 2005).
As mentioned above, insulin mediates the phosphorylation of downstream mTOR by activating PI3K/Akt signal pathways in our brains. As one of the cellular degradation pathways of misfolded and unfolded proteins in neurodegeneration (Garcia-Arencibia et al., 2010), autophagy is regulated by mTOR, which affects ATP levels and biosynthetic pathways (Ulland et al., 2017). Because the maturation of autolysosomes and their retrograde transport are impeded in AD, a massive accumulation of defective autophagosomes can lead to autophagy intermediates (autophagic and lysosomal vesicles) within large swellings along dystrophic and degenerating neurites (Nixon, 2007). Defective autophagy leads to the overproduction, aggregation, and diminished clearance of Aβ and p-tau, oxidative damage, and mitochondrial dysfunction, which, in turn, contribute to the impairment of the metabolic pathways controlled by insulin and mTOR (Perluigi et al., 2015). Moreover, mTOR signaling hyperactivity inhibits the induction of the autophagy clearance system (Majumder et al., 2011; Orr and Oddo, 2013) and increases the accumulation of Aβ in AD (Cai et al., 2012; Perluigi et al., 2014). Thus, defective autophagy causes various effects in AD.
Triggering receptor expressed on myeloid cells 2 (TREM2) is one of the immune receptors expressed in the plasma membrane of microglia in the brain that can recognize phospholipids, lipoproteins, and apoptotic cells (Wolfe et al., 2018). Studies have shown that TREM2 defects can lead to impaired mTOR activation and the enhancement of AMPK activation in microglia in AD patients and an AD mouse model. This effect can result in hyperactive autophagy and microglial energy impairment, which can be compensated by energy repletion (Ulland et al., 2017). Thus, defective autophagy caused by abnormal mTOR regulation plays an important role in the development of AD. Thus, TREM2 represents a risk factor for AD (Ulland et al., 2017); however, the mechanism is unknown.
Moreover, decreased metabolic levels are more likely to be detected in the brains of ApoE4 carriers (Mosconi et al., 2004). ApoE4 is a crucial risk factor for late-onset AD (Zhao et al., 2018). There are key structural differences between the three isoforms of ApoE. ApoE2 has two cysteine residues (Cys, residues 112 and 158), while ApoE3 has a positively charged arginine residue at position 158, and ApoE4 has positively charged arginine residues at both positions 112 and 158 (Muñoz et al., 2019). The differences in the protein structures of the three ApoE isoforms affect their interactions with other proteins and peptides. Accumulating evidence has shown that ApoE, particularly ApoE4, binds to residues 12–28 of Aβ, and this binding promotes Aβ accumulation and strengthens Aβ pathology, especially oxidative stress (Ma et al., 1996; Potter and Wisniewski, 2012; Huang and Mahley, 2014; Liao et al., 2017). Oxidative stress-related modification analysis of plasma and cerebrospinal fluid (CSF) proteins demonstrated that ApoE oxidation could affect the antioxidant activity of thiol, which allows the formation of lipoprotein particles caused by excessive oxidative damage (Di Domenico et al., 2016). Under high levels of oxidative stress, lipid peroxidation produces the highly reactive and neurotoxic molecule, 4-hydroxynonenal (HNE), which covalently binds to Cys residues. Covalently modified Cys residues significantly alter the structure and function of modified proteins. HNE binds to Cys residues in ApoE2 and ApoE3, protecting other proteins from HNE damage. However, ApoE4 lacks Cys residues. Therefore, it cannot scavenge HNE, permitting this neurotoxic molecule to modify neuronal proteins and induce cell death (Sultana et al., 2013; Di Domenico et al., 2017). ApoE2 appears to have neuroprotective effects in the AD patient brain (Keeney et al., 2015) and exhibits the most robust metabolic profile for glucose uptake and glycolysis (Wu et al., 2018). In contrast, ApoE4 causes the most detrimental effects on aging and AD brains. There is reduced cerebral glucose metabolism in cognitively normal individuals carrying ApoE4 (Reiman et al., 2004, 2005). The brains of ApoE4 mice have lower GLUT3 mRNA levels compared to ApoE3 mice, which contributes to inadequate energy supplies in the ApoE4 brain (Wu et al., 2018). Understanding the metabolic process and its effects on the early or progressive alterations in AD may provide new treatment strategies for this irreversible neurodegenerative disease.
Treatment
Several strategies are available for the development of therapeutics to prevent or slow down the progression of AD. Given that BIR is the main risk factor for AD, Reger et al. (2006) showed that a single dose of intranasal insulin can significantly improve the memory of patients with AD or mild cognitive impairment. Based on PET findings, intranasal insulin increases 18F-fluorodeoxyglucose uptake in the precuneus, frontal, cuneus, and parietotemporal regions of the brain (Craft et al., 2012). A Phase 2/3 multi-site clinical trial with intranasal insulin conducted between 2014 and 2018 showed no significant adverse reactions, and treatment improved cognitive function (Craft et al., 2020). Therefore, the specific regimen for intranasal insulin injection and its feasibility require further study. Moreover, one of the common drugs used in diabetes (peroxisome proliferator-activated receptors [PPAR]-γ agonists) increases metabolic efficiency by enhancing insulin sensitivity to reduce Aβ levels (Nicolakakis et al., 2008). Because the mTOR signaling pathway participates in multiple processes that regulate neuronal functions, rapamycin can be used to improve learning and memory and reduce Aβ and tau pathology (Caccamo et al., 2010). In addition, oral administration of rapamycin in a 3xTg-AD mouse model relieves memory symptoms (Spilman et al., 2010). Moreover, rapamycin analogs (Rapalogs), which have been approved by the FDA when used concurrently with metformin, are recommended to pharmacologically address the impaired glucose metabolism (Kezic et al., 2018).
Evidence suggests that short- and long-acting intranasal insulin therapy can improve memory in AD patients in an ApoE4-dependent manner (Reger et al., 2008; Claxton et al., 2015). In response to reduced glucose metabolism, mammalian cells promote the synthesis and utilization of ketone bodies, circulating energy sources for tissues in times of fasting or prolonged exercise (Akram, 2013). Targeted replacement of ApoE in mice with the human ApoE genes demonstrated that brains from ApoE2 and ApoE4 mice have stronger absorption and metabolism of ketone bodies than brains of ApoE3 mice (Wu et al., 2018). Moreover, ApoE–/– mice (an AD model) exhibit circadian rhythm disturbances and increased tau deposition. These phenomena are related to energy shortage and degeneration of the suprachiasmatic nucleus, which can be alleviated by supplementing with ketone bodies in the absence of glucose (Zhou et al., 2016). Therefore, ApoE and ketone bodies may represent new therapeutic targets for improving brain energy metabolism in patients with AD. The specific mechanisms associated with ApoE and ketone bodies and their related treatment pathways need further investigation.
Earlier, we defined the important proteins needed for glucose to flow from the blood into the brain and subsequently regulate metabolic changes under normal conditions. We then summarized a series of changes in these proteins and other risk factors (e.g., ApoE4) in aging and AD. The insulin signaling pathway is closely linked to metabolic change, and the manifestation of decreased brain energy metabolism caused by insulin resistance is similar to diabetes. Thus, the “Type 3 diabetes” hypothesis is well supported. Furthermore, PET is a method used clinically to detect glucose intake and a reference for AD diagnosis. Given these metabolic changes, we described several potential clinical treatments; however, an effective treatment still requires further study.
Glycolysis Dysfunction
Normal Mechanism and Biological Function
Glycolysis, as the first step of glycometabolism and one of the main energy sources, plays an essential role in CNS metabolism. GLUT1 and GLUT3, mainly expressed on astrocytes and neurons, mediate the entry of glucose into these cells. Glucose can then be converted into glucose-6-phosphate (G6P) and then into fructose-6-phosphate (F6P) by hexokinase (Goncalves et al., 2018). F6P, which continues to participate in the subsequent steps of glycolysis, is converted into pyruvate. On the other hand, G6P not only participates in both glycogen synthesis and the PPP, but also non-competitively inhibits hexokinase (DiNuzzo et al., 2015). Phosphofructokinase (PFK), which takes part in the second step of glycolysis, catalyzes F6P into fructose 1,6-bisphosphate (F1,6BP). Fructose 2,6-bisphosphate (F2,6BP), which upregulates PFK-1, is generated via a reaction catalyzed by fructose 2,6-bisphosphatase isoform 3 (PFKFB) (Herrero-Mendez et al., 2009). Astrocytes are reported to have higher glycolysis activity due to higher PFK1 activity. In contrast, neurons exhibit lower glycolytic capacity due to lower levels of PFK1 and 6-phosphofructo-2-kinase and PFKFB (Bolanos et al., 2010). Thus, astrocytes have a significant effect on glycolysis in the CNS. In addition, the astrocytic levels of the abovementioned enzymes are elevated when rat hippocampal astrocytes are co-cultured with neurons, which suggests that neurons can affect astrocyte glycolysis (Mamczur et al., 2015).
Microglia maintain normal brain function by providing trophic support, respond to changes in CNS metabolism, and carry out classic immune cell functions that promote phagocyte clearance (Sarlus and Heneka, 2017; Ghosh et al., 2018). An in vivo study reported that the immune response of microglia is based on glycolysis metabolism (Baik et al., 2019). As first reported in 2002, the inflammasome, a multi-protein complex assembled by intracytoplasmic pattern recognition receptors, plays an important role in the natural immune system (Martinon et al., 2002). NLRP3 is the most studied component of the inflammasome. While the function of the NLRP3 inflammasome in astrocytes is controversial, it is known to be expressed and activated in microglia (Gustin et al., 2015; Freeman and Guo, 2017; Slowik et al., 2018). Evidence shows that pentabromophenol (PBP) and tetrabromobisphenol A (TBBPA) improves the metabolic rate of glycolysis in mouse microglia and promotes the activation of NLRP3 inflammasome (Bowen et al., 2020). Hexokinase is one of the key glycolytic enzymes. Wolf et al. (2016) reported that N-acetylglucosamine activates the inflammasome, particularly the NLRP3 inflammasome, through hexokinase inhibition by promoting its disassociation from the outer mitochondrial membrane (OMM).
Altered Glycolysis in Aging and AD
Studies in mouse models of different ages found that the levels of glycolysis products (e.g., G6P and F1,6BP) decreased in adult mice compared to young mice (Hoyer, 1985). However, recent studies demonstrated that glycolysis increased with a decline in the resting cerebral blood flow of the aged brain (Lourenço et al., 2017). It also increased in naturally aging astrocytes (Cao et al., 2019). Furthermore, glycolysis dysfunction can lead to age-related neurodegeneration (Hipkiss, 2019). Therefore, glycolysis has a complex relationship with growth and aging in the brain.
Considerable research has examined the associations between AD and the enzymes intimately linked to glycolysis (i.e., hexokinase, glyceraldehyde 3-phosphate dehydrogenase [GAPDH], and pyruvate kinase [PK]) (Vallee et al., 2018b; Wu et al., 2018; Butterfield and Halliwell, 2019). Cisternas et al. (2019) reported that Wnt signaling promotes glucose metabolism by increasing the expression of hexokinase and PFK, which causes neuroprotective effects. However, decreased hexokinase and PFK expression and dysregulated Wnt signaling are observed in AD. The accumulation of G6P can reduce hexokinase activity by competitively inhibiting ATP binding to the active site of the enzyme (Liu et al., 1999). In addition, an unbiased metabolomics approach demonstrated G6P accumulation in humans and mice with AD, which restrains glycolysis (Demarest et al., 2020). Hexokinase binds to the OMM via the voltage-dependent anion channel (VDAC), which controls the mitochondrial permeability transition pore (MPTP) (Harris et al., 2014). When hexokinase associates with VDAC, the MPTP tends to be closed. In the postmortem brain tissue of AD mice and patients, hexokinase levels were decreased while VDAC1 levels were elevated (Cuadrado-Tejedor et al., 2011). Furthermore, the interaction of VDAC1 with hexokinase I can generate outer membrane potential in brain mitochondria. Outer membrane potential metabolic-dependent homeostasis can prevent the mitochondrial permeability transition, which leads to Ca2+ activation and neuronal cell death. In addition, this may be involved in resistance to neuronal death and neurodegenerative disorders such as AD (Lemeshko, 2018). Glycogen synthase kinase 3 (GSK3), a serine/threonine protein kinase, disassociates VDAC1 from hexokinase (Reddy, 2013). The activation of GSK3 promotes apoptosis in neuroblastoma cells by reducing the level of cyclin D1 (Kunnimalaiyaan et al., 2018), which inhibits the extrinsic apoptotic signaling pathway mediated by death receptor (Beurel and Jope, 2006). Tau phosphorylation, one of the characteristics of AD, is regulated by GSK3 (Hernandez et al., 2013). Notably, GSK3 inhibition attenuates the symptoms of mild cognitive impairment (Nunes et al., 2013) and restrains oxidative stress in AD (Vallee et al., 2017). Activation of the NLRP3 inflammasome resulting from mitochondrial DNA synthesis in macrophages results in damage to macrophage mitochondria (Zhong et al., 2018). The NLRP3 inflammasome-driven proinflammatory cascade in microglia is augmented by impaired mitochondrial function (Sarkar et al., 2017). Furthermore, NLRP3–/– or caspase-1–/– mice with AD-related mutations largely avoid spatial memory loss and other sequelae associated with AD, suggesting a key role for the NLRP3 inflammasome in the pathogenesis of AD (Heneka et al., 2013). Overall, the hexokinase dysfunction observed in AD brain samples is mediated by the accumulation of G6P that dissociates hexokinase from mitochondria. This process activates the NLRP3 inflammasome, indicating that G6P accumulation may contribute to neuroinflammation in AD. Therefore, the effect of hexokinase activity on glycolysis in the CNS and mitochondrial function is a starting point to study the metabolic mechanism of AD.
GAPDH participates in the sixth step of glycolysis, catalyzing glyceraldehyde 3-phosphate to 1,3-bisphosphoglycerate and increases the NADH:NAD+ ratio (Diaz-Garcia et al., 2017). 2-Deoxyglucose (2DG), which inhibits glycolytic processes (Kim et al., 2017), prevents neurodegeneration by eliminating microglia (microglia can damage neurons in an inflammatory situation). In addition, it reduces the impact of Aβ on neuron cells (Vilalta and Brown, 2014). Moreover, an increased NADH concentration reverses the abovementioned effects of 2DG (Shen et al., 2017). Hou et al. (Hou et al., 2018) found that a decreased NADH:NAD+ ratio is a possible way to reduce AD-associated pathology. Recent research also reported many other functions of GAPDH beyond glycolysis, including DNA repair (Kosova et al., 2017), control of the activity of kinase and phosphotransferase (Tisdale, 2002), and an integral association with membrane ion pumps participating in Ca2+ release (Patterson et al., 2005). Hyperphosphorylated tau protein leads to microtubule depolymerization, resulting in neuron dysfunction (Higuchi et al., 2005; Iqbal et al., 2005). Chen et al. (2000) showed that tau bound to denatured GAPDH but not the native molecule and that the aggregation of the non-native GAPDH in solution was ameliorated by tau. Nakajima et al. (2017) further reported that GAPDH aggregation induced by nitric oxide led to MPTP opening and cell death. An increase in GAPDH disulfide bonding in AD patients and transgenic AD mice compared with controls suggests a potential relationship between GAPDH disulfide bonding and protein aggregation (Cumming and Schubert, 2005). In addition, GAPDH promotes Aβ amyloidogenesis in vitro (Itakura et al., 2015). A recent study found that the concentration of S-glutathionylated-GAPDH in the blood of patients with AD was significantly higher than in the control group, indicating that this indicator is related to the degree of neuronal apoptosis during the progression of AD (Tsai et al., 2020). Thus, GAPDH and its substrates are associated with neuronal cell death in AD. Moreover, Aβ causes microglial inflammation and induces a shift in the metabolic pathway from OXPHOS to glycolysis in the 5XFAD mouse model. During this process, Aβ induces Akt phosphorylation to activate the mTOR-HIF-1α pathway. HIF-1α then increases GAPDH expression. Inhibition of this pathway decreases the levels of pro-inflammatory cytokines, including IL-1β and TNF-α. These data demonstrate the relationship between glycolysis and inflammation in microglia induced by Aβ (Baik et al., 2019).
PK, a rate-limiting enzyme in glycolysis, has four isomers: M1, M2, L, and R. PKM2, which controls the levels of glycolytic intermediates as well as ATP, is linked to neurodegenerative diseases (Vallee et al., 2018a). As described by the Warburg effect, upregulation of the Wnt/β-catenin pathway can promote glycolysis, which is connected to PKM2. In AD, the Wnt/β-catenin pathway is downregulated (partially via inactivation of PKM2) and this results in oxidative stress and cell death (Vallee et al., 2018b). An increase in PK2 expression has been reported in AD transgenic mouse models (Martire et al., 2016). PKM2 promotes cell proliferation by binding to receptors of activated growth factors and then induces dimer formation through phosphorylation (Christofk et al., 2008). In addition, a study that induced yeast pyruvate kinase Cdc19 to form amyloid-like aggregates in vitro found similar characteristics between Cdc19 and PKM2, its mammalian counterpart (Cereghetti et al., 2018). Thus, the association between PK and AD might be worthy of investigation.
Treatment
The latest AD patient symptom management guidelines consider pimavanserin and citalopram to be the two most promising medications for AD (Kales et al., 2019). Pimavanserin selectively and inversely excites 5-hydroxytryptamine 2A (5-HT2A) receptor (Hunter et al., 2015). Citalopram prevents the reuptake of 5-hydroxytryptamine (5-HT) by inhibiting 5-HT transporter, blocking reuptake on the presynaptic membrane (El-Armouche et al., 2003). Thus, both pimavanserin and citalopram increase the concentration of 5-HT in the synaptic cleft. Tiritilli (2000) found that oxygen deficit and glucose deprivation inhibit the contractive response of umbilical arteries to 5-HT, which implies that aerobic glycolysis can increase sensitivity to 5-HT. Research into human pancreatic ductal adenocarcinoma also indicates that the levels of enzymes participating in glycolysis increase after 5-HT stimulation (Jiang et al., 2017). Furthermore, conscious mice injected with 5-HT exhibit an increased brain glucose concentration (Leonard, 1975). In addition, 5-HT facilitates glycolysis through PKM2 upregulation in breast cancer cells (Sola-Penna et al., 2019). Therefore, an increased concentration of 5-HT stimulates glycolysis, although the specific glycolysis-related mechanism in the CNS remains unclear.
Besides the international guidelines for AD treatment, other recent guidelines, such as Chinese guidelines for the treatment of AD, propose cholinesterase inhibitors and excitatory amino acid receptor antagonist as well as certain traditional Chinese medicines for the treatment of AD (Group Chinese Dementia and Cognitive Impairment Writing Group, Association Special Committee on Cognitive Disorders of the Chinese Medical Doctors Association, 2018). The relationship between cholinesterase and glycolysis in CNS has been studied in the last century (Peiss et al., 1949). A study in 2013 indicated that carbachol (a cholinergic agonist) could increase fluxes in both glycolysis and OXPHOS in SH-SY5Y neuroblastoma cells (Lu et al., 2013). Glucose metabolism dysregulation can influence acetyl coenzyme A and indirectly retard the synthesis of acetylcholine (Slotkin et al., 1990). In addition, the activity of choline acetyltransferase (formed from choline and acetyl coenzyme A) is below normal in AD patients’ brains (Bowen et al., 1976). In AD treatment, cholinesterase inhibitors, such as donepezil, may help patients to control the symptoms of the disease (Matsunaga et al., 2018; Ruthirakuhan et al., 2018).
Excitatory neurotransmission, particularly that of glutamate, as well as the receptors involved, play essential roles in synaptic plasticity (Riedel et al., 2003) via N-methyl-D-aspartic acid (NMDA) receptors (NMDARs) (Collingridge et al., 2009). NMDAR stimulation facilitates glucose uptake and glycolysis in oligodendroglial cells (Saab et al., 2016). However, intense signal stimulation by glutamate would produce excitotoxicity, leading to CNS damage (Rothman and Olney, 1986) via excessive Ca2+ entry (Choi, 1987). A study of mouse astrocytes provided evidence that glutamate promotes glycolysis and damages mitochondrial aerobic capacity (Yan et al., 2017). Aβ has two forms, a soluble oligomeric form and an insoluble aggregate form. The former is primarily responsible for neurodegeneration and diminished synaptic function in AD (Barghorn et al., 2005), and it can interact with NMDARs, leading to mitochondrial Ca2+ overload and cell apoptosis (Alberdi et al., 2010). Memantine, as one of the uncompetitive NMDAR antagonists, has a strong voltage dependency (Alam et al., 2017). A study in a male Wistar rat model showed that memantine and lithium could reverse the decreased in IL-4 induced by oligomeric Aβ1–42, whereas lithium alone had no effect (Budni et al., 2017). Meta-analyses also indicate that memantine, particularly in conjunction with cholinesterase inhibitors, inhibits or slows the progress of AD symptoms (Tan et al., 2014; Kishi et al., 2017; Matsunaga et al., 2018). Therefore, NMDAR antagonism by memantine might prevent excessive glycolysis mediated by excitatory neurotransmission and the resultant excitotoxicity in AD.
In short, aging and AD are closely related to abnormal glycolysis in the nervous system. Dysfunction of the key enzymes involved in glycolysis can affect the generation of Aβ and tau, the activation of the NLRP3 inflammasome, and even mitochondrial function. A variety of drugs for the treatment of AD, backed by the existing guidelines, can regulate glycolysis in the nervous system. Moreover, controlling the activity of enzymes and products that regulate glycolysis (e.g., hexokinase, GAPDH, and G6P) could significantly improve the nervous system and may represent a new direction for AD treatment.
TCA Cycle and Oxphos Deficits
Normal Mechanism and Biological Function
The TCA cycle is the central hub for energy metabolism, macromolecule synthesis, and redox balance. In normal aerobic organisms, most cellular glucose is converted into pyruvate through glycolysis. Pyruvate is subsequently oxidized by PDH to acetyl-CoA, which is fed into the TCA cycle. One acetyl-CoA molecule can generate six molecules of NADH and two molecules of FADH through this pathway. The coenzyme NADH contains many electrons, which are transferred to the ETC on the IMM. Finally, ATP and H2O are generated. This process is called OXPHOS and also generates ROS as a byproduct to maintain cellular homeostasis. However, excessive ROS production may contribute to oxidative stress, with mitochondria as the first target (Rego and Oliveira, 2003). The body utilizes antioxidants (e.g., superoxide dismutase [SOD] and glutathione [GSH] to offset the adverse effects of ROS, Indo et al., 2015). However, the brain is an organ with high oxygen consumption and low antioxidant defenses. Thus, it is vulnerable to oxidative stress (Cobley et al., 2018). In addition, α-ketoglutarate (α-KG) is a TCA cycle intermediate that is transformed into glutamate and GABA by glutamate decarboxylase (GAD) and glutamate dehydrogenase. Reducing equivalents (i.e., NADH) generated from OXPHOS synthesize ATP in conjunction with glutamate and GABA, which helps maintain synaptic plasticity (Bak et al., 2006).
In addition to antioxidants, the TCA cycle products themselves affect the mitochondrial redox balance. Nicotinamide nucleotide transhydrogenase (NNT), NADP+-dependent isocitrate dehydrogenase (ICDH) 2, and malic enzyme can generate NADPH using the electrons from NADH (Metherell et al., 2016; Navarro et al., 2017; Meimaridou et al., 2018). All these proteins can affect redox activity in mitochondria (Yin et al., 2012). The redox state of mitochondria can regulate energy metabolism via the oxidization of several metabolic enzymes, including aconitase, α-ketoglutarate dehydrogenase (α-KGDH), malate dehydrogenase (Reed et al., 2008), and succinyl-CoA-3-oxoacid CoA transferase (SCOT) (Garcia et al., 2010), and complexes I (Cortes-Rojo et al., 2020), II (Jones et al., 2019), and V (Kramer et al., 2018). It can also regulate S-glutathione glycosylation, a modification that reflects the redox state of mitochondria by reversibly forming mixed disulfide bonds between protein cysteine sulfhydryl and GSH (Schafer and Buettner, 2001; Hill and Bhatnagar, 2012). Therefore, NADPH plays a crucial role in a series of processes mediating glucose metabolism as part of the redox-energy metabolism axis.
Altered TCA Cycle and OXPHOS in Aging and AD
Proteomics analysis found that the dysregulation of TCA enzyme levels during aging includes the upregulation of malate dehydrogenase 1 (MDH1), fumarate dehydrogenase 1 (FH1), and subunits of NADH dehydrogenase, succinate dehydrogenase, and pyruvate dehydrogenase (PDH). Impaired TCA cycle metabolism is also associated with the downregulation of ICDH 1/2 and a subunit of succinyl-CoA ligase in the brains of aged mouse brains (Guo et al., 2020). Furthermore, a reduction in the metabolites from the TCA cycle has been measured in both aging and AD mouse models, while increases in the levels of the ceramides and sphingosine-1 phosphate (i.e., inflammatory metabolites) occurred in the aging group. The study also found that the levels of NADH and acetyl-CoA were positively correlated with age and the degree of AD, while glutamine and GABA concentrations were negatively correlated (Dong and Brewer, 2019). A decline in glutamine and GABA levels is associated with impaired neurotransmitter circulation (Dedeoglu et al., 2004; Tiwari and Patel, 2012). Moreover, acetyl-CoA and NADH+ have allosteric inhibitory effects on PDH, which subsequently affects the TCA cycle (Jha et al., 2016). Many studies have demonstrated that in the aging brain, as mutations in mitochondrial DNA (mtDNA) increase, the expression of respiratory chain complexes I, III, and IV are suppressed (Navarro and Boveris, 2007). The oxidants (i.e., H2O2) are produced (Petrosillo et al., 2008) while NADPH is insufficient (Pinto and Moraes, 2015; DeBalsi et al., 2017; Golpich et al., 2017). In addition, the high H2O2 levels alter the redox environment in cells and can be partially released into the cytoplasm through VDAC (Zou et al., 2018). Redox homeostasis is not only one of the characteristics of aging but also of neurodegenerative diseases, such as AD (Cenini et al., 2019). A clinical study on the quantitative detection of GSH in the occipital cortex showed that GSH levels in the elderly are significantly lower than those in the young (Emir et al., 2011). This age-dependent decline seems to be associated with cognitive impairment (Falls et al., 2018). Recent studies have shown that GSH depletion may cause metabolic stress in neurons by generating more ROS, which may eventually contribute to cognitive impairment (Gonzalez-Fraguela et al., 2018). As an important factor in regulating the energy-redox axis, NNT may also be a potential regulator in aging. Indeed, NNT overexpression can restore the mitochondrial NAD+ levels, enhance the reprogramming efficiency of senescent cells, and prolong the life-span of mesenchymal stem cells by delaying senescence (Son et al., 2016). Also, glucose metabolism is decreased and the life span is shortened in NNT–/– mice (Kim et al., 2010). Therefore, there is a relationship between the TCA cycle and aging-related changes in the nervous system.
The majority of the ATP required by the brain is generated from the TCA cycle. Metabolomics analysis revealed that the utilization rate of glucose is altered in astrocytes extracted from 5xTg-AD mouse (van Gijsel-Bonnello et al., 2017). Several proteins and TCA cycle enzymes involved in glucose metabolism are also altered in AD brain tissue (Blass et al., 2000). Pyruvate is taken up into mitochondria via the mitochondrial pyruvate carrier, as the final metabolite of glycolysis (McCommis and Finck, 2015). Reduced mitochondrial pyruvate carrier activity might contribute to the inactivation of PDH caused by p-tau and alterative ketone body metabolism (Ding et al., 2013). Furthermore, pyruvate is the substrate for PDH and reduced in rodent models of AD (Sheu et al., 1985). Acetyl-CoA is made by PDH complex-catalyzed oxidative decarboxylation and flows into the TCA cycle. Acetyl-CoA levels in the synaptosomes of Tg2576 AD mice model are reduced (Bielarczyk et al., 2015). Moreover, acetyl-CoA is a substrate for the acetylation of the lysine group in AD pathological marker proteins, including β-site amyloid precursor protein (APP) cleaving enzyme 1 (BACE1) and APP (Jonas et al., 2010). Aβ causes a reduction in acetyl-CoA levels in neurons and glial cells (Bielarczyk et al., 2006). Both acetyl-CoA and succinyl-CoA are essential components for the formation of precursors of acetyl-choline, a neurotransmitter closely related to cognitive function (Kann and Kovacs, 2007). In addition, an examination of brains from deceased AD patients showed that the activities of the PDH complex, ICDH, and the α-KGDH complex are reduced (Sheu et al., 1994), while the activities of SDH and MDH are increased (Bubber et al., 2005). Citrate synthase (CS) activity appears to be negatively regulated by ApoE4 (Wilkins et al., 2017) and also decreased in AD patients (Fisar et al., 2016). As a downstream product of α-KG, succinyl-CoA may be reduced due to the downregulation of the pathway enzymes influencing the TCA cycle. However, there may be upregulation of succinate and aspartate (Zhou et al., 2018). Other enzymes and metabolites related to the metabolic process (e.g., citric acid, cis-aconitic acid, and fumaric acid) are decreased (Zheng et al., 2018). Thus, the changes in energy acquisition caused by these factors have various effects on many aspects of the body.
Under aerobic conditions, the TCA cycle is mainly active in mitochondria. Hence, the integrality of mitochondrial morphology and function are particularly essential for this process. Impaired mitochondria are observed in AD (Golpich et al., 2017) and lead to glucose hypometabolism, OXPHOS damage, excessive ROS accumulation, elevated oxidative stress (Wang et al., 2005), and disruption of the main pathway of glucose metabolism. Mitochondria are not only the main source of ROS but also the target of its attack. 8-oxoguanine (8-oxoG) accumulates in mtDNA (Bradley-Whitman et al., 2014) and the cytoplasm of hippocampal neurons (Song et al., 2011), which is an obvious sign of oxidative stress in AD patients. A vicious cycle occurs because oxidative stress can further aggravate mitochondrial dysfunction. The activity of the ETC complex is significantly reduced in AD (Holper et al., 2019), suggesting impaired OXPHOS. This phenomenon has been confirmed in mitochondria isolated from 3-month-old AD mice (Yao et al., 2009) and brain tissue from AD patients (Kim et al., 2000). The impairment of mitochondrial respiratory function in AD patients is also negatively correlated with Aβ levels and may be caused by the effect of Aβ on mitochondrial OXPHOS (Dragicevic et al., 2010). Similarly, another study demonstrated that Aβ could inhibit mitochondrial complexes I and IV (Bobba et al., 2013).
Mitochondria-driven glucose metabolism abnormalities have been recorded by magnetic resonance spectroscopy (MRS) or nuclear magnetic resonance (NMR) in several studies with AD rodent models. Decreased phosphomonoester levels and increased levels of phosphocreatine and adenosine diphosphate reflect changes in the oxidative metabolic rate, suggesting that oxidative stress occurs in the AD brain (Pettegrew et al., 1997). A reduction in glutamate, GSH, and GABA (Dedeoglu et al., 2004; Tiwari and Patel, 2012) suggests a damaged glutamatergic and GABAergic glucose oxidation and neurotransmitter cycle, which is also present in these mouse models. Thus, glutamine synthase impairment and decreased glutamate flux through the GABA pathway may be caused by mitochondrial dysfunction (Doert et al., 2015). The depletion of mitochondrial GSH leads to increased H2O2 levels and decreased mitochondrial membrane potential in neurons and astrocytes (Muyderman et al., 2007). Aβ can induce the internalization of the glutamate A2 (GluA2) subunit, which is highly Ca2+ impermeable and contributes to the production of proinflammatory cytokines by microglia that accelerates neurotoxicity in AD patients (Beppu et al., 2013; Noda, 2016). Therefore, altered levels of various enzymes and metabolites of the TCA cycle have an impact on mitochondrial function, resulting in abnormal redox homeostasis, reduced ATP production, and increased ROS production that injures surrounding tissues.
Treatment
The cognitive impairment observed in AD can be improved by upregulating acetyl-CoA levels to attain normal mitochondrial function. This view is supported by experiments using CMS121 and J147 as candidate drugs against AD in mice (Currais et al., 2019) and long-term oral administration of acetyl-L-carnitine to AD patients, which provides additional acetyl-CoA (Epis et al., 2008). Specific therapeutic drugs for other metabolites require further research.
Nutraceuticals appear to be a feasible approach for protecting mitochondria. Micronutrients are key co-factors that sustain mitochondrial metabolic hemostasis, such as the generation of ATP, construction of the electron transport complex, and oxygen detoxification (Atamna, 2004). Thus, micronutrient deficiencies may cause drops in essential enzymatic activities and subsequently increase ROS production, decrease cellular energy metabolism, and aggravate mitochondrial abnormalities, promoting Aβ toxicity and AD progression (Young et al., 2007). Coenzyme Q10 (CoQ10), an antioxidant forming part of the ETC, can enhance mitochondrial function and promote ATP synthesis (Kumar and Singh, 2015). CoQ10 also inhibits nerve cell death resulting from oxidative stress and neurotoxins and has neuroprotective effects in double transgenic AD mouse models (Muthukumaran et al., 2018). Moreover, CoQ10 stabilizes mitochondria and reduces ROS production in fibroblasts from AD patients (Naderi et al., 2006; Ma et al., 2014; Muthukumaran et al., 2018). Thus, mitochondria-specific nutraceuticals (e.g., vitamins and CoQ10) can be beneficial for AD patients (Beal, 2004).
In summary, the TCA cycle and OXPHOS in the mitochondria can produce the greatest amount of energy for the body. The enzymes and metabolites involved in these reactions undergo various changes during aging and AD development. The imbalance in mitochondrial redox and resulting by-products (e.g., ROS) generated by these changes may cause inflammation in the surrounding tissues. Thus, to a certain extent, metabolic dysfunction could affect the occurrence and development of neurodegenerative disorders. However, most current treatments target symptom attenuation, and there is still no feasible research on specific treatments involving these altered intermediate products.
Pentose Phosphate Pathway Impairment
Normal Mechanism and Biological Function
The PPP is a significant component of intracellular oxidative catabolism, which is vitally important for oxidative stress resistance and the production of essential material for biological synthesis (Palmer, 1999; Russell et al., 1999). Although the pathway does not generate ATP, it can produce NADPH to maintain the reduced form of GSH (Cenini et al., 2019). When functioning as one of the most effective antioxidants, GSH is oxidized by ROS and is converted into the oxidized glutathione (GSSG), which subsequently enters a loop along with GSH peroxidase and GSH reductase.
The PPP is regulated by enzymes and the NADPH/NADP+ ratio. Alterations in either of the two factors will critically affect the pathway. A crucial enzyme system supports the phosphopentose pathway, among which G6P dehydrogenase (G6PD) and transketolase are particularly important. G6PD and transketolase are both rate-limiting enzymes, responsible for the redox equilibrium and the non-oxidative branch in the PPP, respectively (Gibson and Blass, 2007). In addition, transketolase plays a pivotal role in the material communication between glycolysis and the PPP (Kauffman, 1972). Moreover, an elevated NADPH/NADP+ ratio significantly inhibits G6PD, helping to disrupt G6P flux into the pathway. In conclusion, the PPP maintains redox hemostasis to prevent the initiation and development of oxidative stress in the brain.
Altered PPP in Aging and AD
During aging, damaged mitochondria that produce less ATP and more ROS accumulate, leading to oxidative stress. G6PD plays a vital role in protecting neurons against endogenous ROS-mediated neurodegeneration in aging mice (Jeng et al., 2013). In the cerebral cortex of aged mice, there is a decline in the levels and activities of G6PD and other GSH-regenerating enzymes (Dukhande et al., 2009). Moreover, G6PD appears to be neuroprotective against endogenous ROS in the aged human brain (Sbodio et al., 2019). However, alterations in G6PD levels in the human brain have not been identified. Furthermore, decreased NADPase levels and increased NAD kinases (NADK) levels may alleviate oxidative stress during human aging by promoting the synthesis of NADPH and inhibiting the production of NADH (Clement et al., 2019).
Multiple risk factors, such as Aβ peptide, tau aggregation, and ApoE, play crucial roles in the PPP impairment contributing to AD. Oxidative stress and chronic inflammation are two critical factors demonstrated to trigger an elevation in the level of Aβ (Verdile et al., 2015) and subsequently the aggregation of toxic oligomers, particularly the Aβ42 forms. Later, aggregation of the Aβ peptides disturbs the redox balance, finally establishing a toxic cycle in AD patients (Hardy and Higgins, 1992; Musiek and Holtzman, 2015). Tau phosphorylation, aggregation, and accumulation are closely linked to APP malfunction, which will inhibit cellular metabolism, including the PPP (Takahashi et al., 2015; Kametani and Hasegawa, 2018). Mutant ApoE not only is involved in Aβ clearance and aggregation, but also reduces the rate of glucose metabolism before AD symptom onset (Reiman et al., 2001, 2005; Johnson et al., 2017), along with the low flux of glucose into the PPP.
NADPH, a reducing cofactor prominently recycled in the PPP, participates in the conversion of the oxidized form of GSH into the reduced form (Dringen, 2000). Hence, the PPP establishes a crucial relationship between glucose metabolism and the redox equilibrium. Notably, Aβ not only triggers ROS generation but also decreases GSH levels in astrocytes (Abramov et al., 2004; Canevari et al., 2004). These effects lead to disequilibrium and aggravate oxidative stress. Interestingly, various groups have found that there is increased glucose flux through the PPP to counteract Aβ toxicity (Hakim et al., 1976; Soucek et al., 2003; Sun et al., 2006; Allaman et al., 2010; de Bari et al., 2019). Research has identified a decreased level of ribose-5-phosphate and an elevated level of lactic acid, which indicates upregulation of the PPP (Oresic et al., 2011). Moreover, the dramatic degradation of phosphofructokinase B3 results in a higher level of glucose metabolism via the PPP than via glycolysis in neuronal cells (Herrero-Mendez et al., 2009).
Accumulating evidence indicates that there is an alteration of the enzymes in the phosphate pentose shunt, which could significantly impact the antioxidant system in AD. G6PD is involved in generating NADPH for the reduction of the oxidized GSH, which helps to maintain the redox hemostasis. Several studies reported elevated levels of sulfhydryls and upregulation of G6PD in AD (Martins et al., 1986; Russell et al., 1999; Scheltens et al., 2016), which may be neuroprotective. However, the mechanism underlying this protective effect remains elusive (Tang, 2019). Notably, another report found that G6PD activity was decreased in the hippocampi of human AD brains (Bigl et al., 1999). In addition to the enzymatic activity, antioxidants are generated in astrocytes to resist oxidative stress (Ben-Yoseph et al., 1994; Rahman et al., 2000). In response to the aggregation of Aβ, ROS levels increase and there is a decrease in GSH in astrocytes in both AD and mild cognitive impairment (Sultana et al., 2008; Mandal et al., 2012).
Furthermore, the activities of transketolase and their common coenzymes thiamine diphosphate, GSH peroxidase, glutathione-S-transferase, and δ-aminolevulinate dehydratase (δ-ALA-D) have all been demonstrated to be decreased in AD (Sheu et al., 1988; Mastrogiacomo et al., 1996; Ansari and Scheff, 2010; Yu et al., 2018). Multiple metabolites altered by ApoE are identified within the PPP, including gluconolactone, gluconate, and G6P (Johnson et al., 2017). These findings may provide a novel strategy to increase GSH production by regulating the activity of these enzymes in the PPP.
Treatment
AD therapies concentrate on oxidative stress, mainly targeting the PPP. For example, Krautwald and Munch (2010) emphasized that advanced glycation end products and their methylglyoxal precursors are both biomarkers and pathogenic factors in AD, with direct neurotoxicity related to oxidative stress and apoptosis. Meanwhile, they proposed that the formation of advanced glycation end products occurred through a lower methylglyoxal concentration, which could be achieved by permitting a higher flux through the PPP. Cai et al. (2020) hypothesized that hollow manganese Prussian white nanocapsules (HMPWCs) participated in the resistance to the harmful effects of tau by relieving neuronal inflammation, eliminating ROS, and inhibiting tau hyperphosphorylation. Targeting of astrocytic NRF2, a regulator of GSH synthesis, could be a potent therapeutic strategy in AD (Oksanen et al., 2019).
Because NADPH is essential for reducing GSSG to GSH, PPP is a pivotal part of the oxidative stress observed in AD pathology. Nevertheless, the upregulation of glucose flux into the PPP increases the synthesis of NADPH. However, it is controversial whether the activity of G6PD is activated or suppressed in AD. Hence, it is uncertain whether NADPH production is increased or decreased, and further investigation is needed. Despite the uncertain mechanism, there are still strategies to relieve PPP dysfunction and oxidative stress in AD.
Conclusion and Future Perspectives
In this review, we summarized the metabolic deficits, including glucose metabolism dysregulation, glycolysis dysfunction, and PPP impairment, in AD (Figure 1). These deficits cause significant effects on AD pathogenesis. One direct consequence of these deficits is the inhibition of ATP generation, which leads to insufficient energy to support the normal neuronal functions and, ultimately, neurodegeneration. In addition, the metabolic deficits indirectly trigger neuronal death via mitochondrial dysfunction, oxidative stress (increased ROS, decreased NADPH), and inflammation.
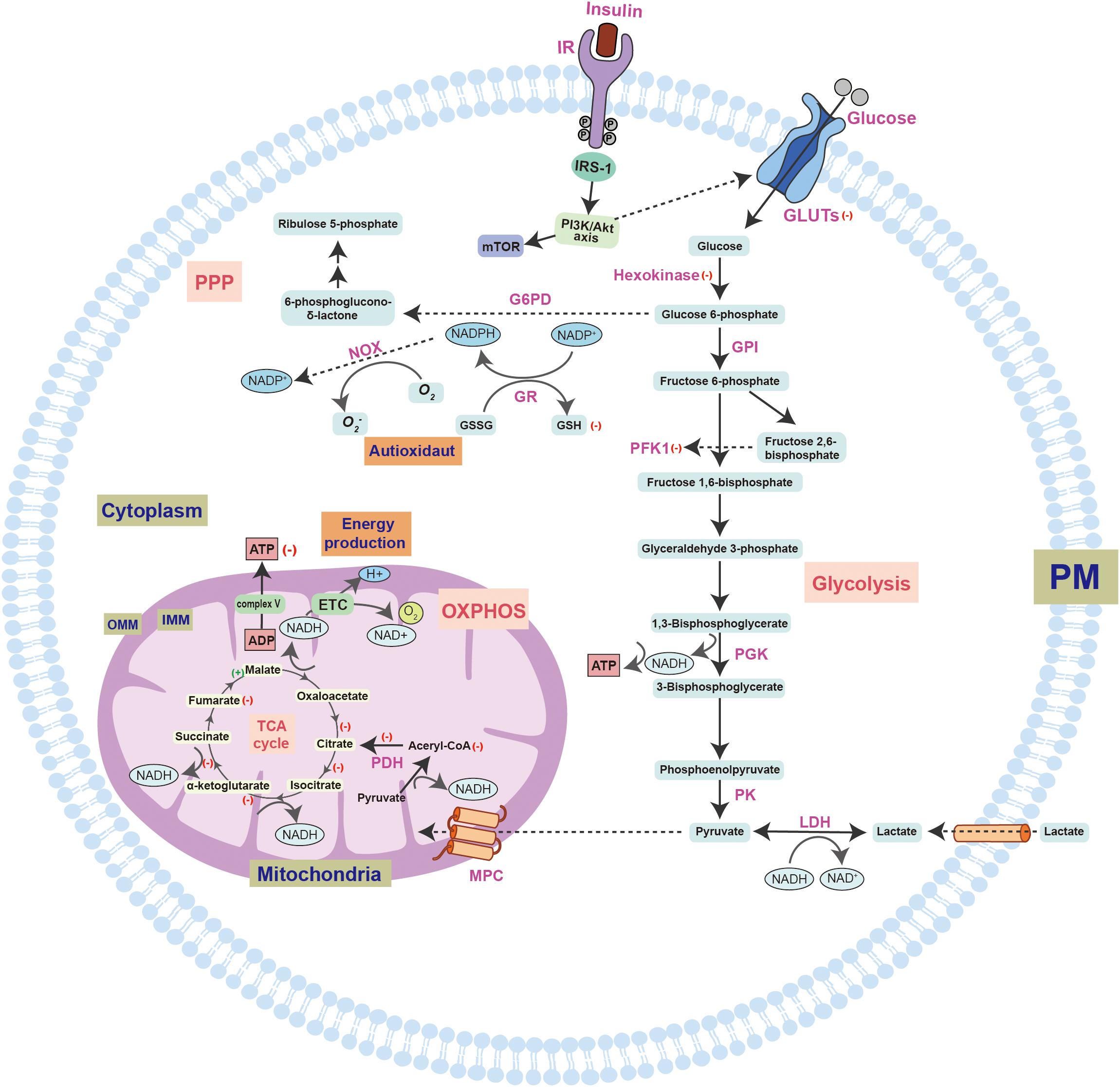
Figure 1. A schematic overview of insulin signaling and glucose utilization pathways in the brain. Glucose enters the cell through the synergistic action of a variety of GLUTs and is further catalyzed by different enzymes. However, the most important factor regulating glucose transport is insulin and its receptors. Insulin promotes the activation of IR through autophosphorylation of specific tyrosine residues, which can bind to IRS-1/2 and activate it, thereby regulating the cascade of energy metabolism signals, such as the PI3K-Akt-mTOR signal pathway. In the brains of early AD patients with MCI, alterations in the gene expression profiles will lead to increased insulin consumption. The symptoms of cerebral diabetes, such as decreased glucose metabolism and energy exhaustion, have been found in MCI samples. The decrease in glucose metabolism may aggravate the impairment of cognitive function. Furthermore, the brain in the early stage of AD under diabetic conditions can aggravate the symptoms of peripheral diabetes. The biochemical diagram of the changes in normal intracellular glucose catabolism and ATP synthesis includes the glycolysis pathway that initially occurs in the cytoplasm, the TCA cycle and OXPHOS pathway that occurs in the mitochondria, and the PPP, which provides raw materials for biosynthesis and regulates redox. Changes in these biological processes in the AD brain are shown by the following symbols: (+), increased expression; (–), decreased expression. The structural and functional disorders of mitochondria eventually lead to decreased ATP production, increased ROS production, and the occurrence of oxidative stress. Moreover, further mitochondrial damage will also lead to a vicious cycle that aggravates neurodegeneration. GLUT, glucose transporter; G6PD, glucose 6-phosphate dehydrogenase; F6BP, Fructose 6-bisphosphate; PFK1, phosphofructokinase 1; PGK, phosphoglycerate kinase; LDH, lactate dehydrogenase; PK, pyruvate kinase; MPC, mitochondrial pyruvate carrier; ETC, electron transport chain; PDH, pyruvate dehydrogenase; OXPHOS, oxidative phosphorylation; TCA, tricarboxylic acid; IMM, inner mitochondrial membrane; OMM, outer mitochondrial membrane; NADP+, nicotinamide adenine dinucleotide phosphate; GR, glutathione reductase; NOX, NADPH-oxidase; GSH, glutathione; GSSG, glutathione; O2-, superoxide anion. (+): indicates an increase in expression, (–): indicates a decrease in expression.
Compared to previous review articles (Atamna and Frey II, 2007; Yin et al., 2016; Butterfield and Halliwell, 2019), we not only described changes in the regulation of signal pathways but also discussed in detail, the changes of implicated enzymes and metabolites during energy metabolism in AD. Moreover, we highlighted the intimate relationship between AD pathogenesis and glucose metabolism dysregulation, implying that a focus on the preclinical and clinical manifestations of glucose metabolic dysregulation in AD might be a promising strategy to diagnose and prevent or slow the progression of this disease.
Peripheral diabetes appears to aggravate these metabolic changes rather than cause them. Moreover, peripheral diabetes-related abnormalities do not directly influence the expression of diabetes-related genes in the brain (Hokama et al., 2014), suggesting that AD pathology may play a key role in altering gene expression, which is correlative with diabetes in AD patients. The GLUT isoforms have variable effects on glucose utilization. Although altered cerebral glucose uptake is currently considered a predictive method for diagnosing AD, there is still a lack of adequate research to support its widespread use. Because of the constantly changing pathological processes in AD (Varma et al., 2018), the metabolite signal changes in the blood cannot reflect the changes in the brain in time, and there is still no accurate and easy-to-use index for preclinical and clinical diagnosis and treatment. Thus, it is difficult to assess whether peripheral signals related to the state of the disease are also reflected in the brain. The combination of multi-omics analysis and multi-type data will increase our understanding of the profound mechanisms of AD and identify potential biomarkers for diagnosis, prognosis and monitoring treatment of this disease. Therefore, further research is needed to develop more accurate and convenient diagnostic techniques for clinical use.
Alzheimer’s disease treatment currently concentrates on symptom attenuation, targeting the specific enzymes or intermediate products. Different types of medications are used to recover the normal function of the proteins or ameliorate the intermediate outcomes, including some exogenous antioxidant interventions (vitamin E, polyphenol, and deuterated lipids). Although symptom treatment is the most widespread method for managing AD, it is not the best strategy to slow or reverse the neuronal degeneration because it only targets the manifestations of the disease and not its essence. Targeting these pathways may lead to the development of effective treatments (Table 1). We suggest that future AD treatments focus on early metabolic changes, clinical predictions, diagnosis, prevention, and the combined treatment of multiple pathogens at the pre-clinical and clinical stages using personalized drugs to prevent or delay the progression of AD. Further efforts are still needed to understand the metabolic basis of the etiology and pathogenesis of AD.

Table 1. Defective metabolic pathways and treatments in AD.
Author Contributions
XY and XZ contributed to the conceptualization and methodology. XY, YH, BW, SW, and XZ wrote the first draft of the manuscript. YH created the figure. BW prepared the table. All the authors approved the final manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (no. 81700977 to XY; no. 81500858 to XZ).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abramov, A. Y., Canevari, L., and Duchen, M. R. (2004). Beta-amyloid peptides induce mitochondrial dysfunction and oxidative stress in astrocytes and death of neurons through activation of NADPH oxidase. J. Neurosci. 24, 565–575. doi: 10.1523/jneurosci.4042-03.2004
Adeva-Andany, M. M., Perez-Felpete, N., Fernandez-Fernandez, C., Donapetry-Garcia, C., and Pazos-Garcia, C. (2016). Liver glucose metabolism in humans. Biosci. Rep. 36:e00416.
Akram, M. (2013). A focused review of the role of ketone bodies in health and disease. J. Med. Food 16, 965–967. doi: 10.1089/jmf.2012.2592
Akter, K., Lanza, E. A., Martin, S. A., Myronyuk, N., Rua, M., and Raffa, R. B. (2011). Diabetes mellitus and Alzheimer’s disease: shared pathology and treatment? Br. J. Clin. Pharmacol. 71, 365–376. doi: 10.1111/j.1365-2125.2010.03830.x
Alam, S., Lingenfelter, K. S., Bender, A. M., and Lindsley, C. W. (2017). Classics in chemical neuroscience: memantine. ACS Chem. Neurosci. 8, 1823–1829. doi: 10.1021/acschemneuro.7b00270
Alberdi, E., Sanchez-Gomez, M. V., Cavaliere, F., Perez-Samartin, A., Zugaza, J. L., Trullas, R., et al. (2010). Amyloid beta oligomers induce Ca2+ dysregulation and neuronal death through activation of ionotropic glutamate receptors. Cell Calc. 47, 264–272. doi: 10.1016/j.ceca.2009.12.010
Allaman, I., Gavillet, M., Belanger, M., Laroche, T., Viertl, D., Lashuel, H. A., et al. (2010). Amyloid-beta aggregates cause alterations of astrocytic metabolic phenotype: impact on neuronal viability. J. Neurosci. 30, 3326–3338. doi: 10.1523/jneurosci.5098-09.2010
Alzheimer’s Association (2012). 2012 Alzheimer’s disease facts and figures. Alzheimers Dement. 8, 131–168. doi: 10.1016/j.jalz.2012.02.001
An, Y., Varma, V. R., Varma, S., Casanova, R., Dammer, E., Pletnikova, O., et al. (2018). Evidence for brain glucose dysregulation in Alzheimer’s disease. Alzheimers Dement. 14, 318–329.
Ansari, M. A., and Scheff, S. W. (2010). Oxidative stress in the progression of Alzheimer disease in the frontal cortex. J. Neuropathol. Exper. Neurol. 69, 155–167. doi: 10.1097/nen.0b013e3181cb5af4
Arias, C., Montiel, T., Quiroz-Baez, R., and Massieu, L. (2002). beta-Amyloid neurotoxicity is exacerbated during glycolysis inhibition and mitochondrial impairment in the rat hippocampus in vivo and in isolated nerve terminals: implications for Alzheimer’s disease. Exp. Neurol. 176, 163–174. doi: 10.1006/exnr.2002.7912
Arnold, S. E., Arvanitakis, Z., Macauley-Rambach, S. L., Koenig, A. M., Wang, H. Y., Ahima, R. S., et al. (2018). Brain insulin resistance in type 2 diabetes and Alzheimer disease: concepts and conundrums. Nat. Rev. Neurol. 14, 168–181. doi: 10.1038/nrneurol.2017.185
Atamna, H. (2004). Heme, iron, and the mitochondrial decay of ageing. Age. Res. Rev. 3, 303–318. doi: 10.1016/j.arr.2004.02.002
Atamna, H., and Frey, W. H. II (2007). Mechanisms of mitochondrial dysfunction and energy deficiency in Alzheimer’s disease. Mitochondrion 7, 297–310. doi: 10.1016/j.mito.2007.06.001
Baik, S. H., Kang, S., Lee, W., Choi, H., Chung, S., Kim, J. I., et al. (2019). A breakdown in metabolic reprogramming causes microglia dysfunction in Alzheimer’s disease. Cell Metab. 30, 493–507.e6.
Bak, L. K., Schousboe, A., and Waagepetersen, H. S. (2006). The glutamate/GABA-glutamine cycle: aspects of transport, neurotransmitter homeostasis and ammonia transfer. J. Neurochem. 98, 641–653. doi: 10.1111/j.1471-4159.2006.03913.x
Barghorn, S., Nimmrich, V., Striebinger, A., Krantz, C., Keller, P., Janson, B., et al. (2005). Globular amyloid beta-peptide oligomer - a homogenous and stable neuropathological protein in Alzheimer’s disease. J. Neurochem. 95, 834–847. doi: 10.1111/j.1471-4159.2005.03407.x
Barone, E., Di Domenico, F., Cassano, T., Arena, A., Tramutola, A., Lavecchia, M. A., et al. (2016). Impairment of biliverdin reductase-A promotes brain insulin resistance in Alzheimer disease: a new paradigm. Free Radic. Biol. Med. 91, 127–142. doi: 10.1016/j.freeradbiomed.2015.12.012
Barzilai, N., and Ferrucci, L. (2012). Insulin resistance and aging: a cause or a protective response? J. Gerontol. Ser. A Biol. Sci. Med. Sci. 67, 1329–1331. doi: 10.1093/gerona/gls145
Bateman, T. M. (2012). Advantages and disadvantages of PET and SPECT in a busy clinical practice. J. Nucl. Cardiol. 19(Suppl. 1), S3–S11.
Beal, M. F. (2004). Mitochondrial dysfunction and oxidative damage in Alzheimer’s and Parkinson’s diseases and coenzyme Q10 as a potential treatment. J. Bioenerg. Biomembr. 36, 381–386. doi: 10.1023/b:jobb.0000041772.74810.92
Bedse, G., Di Domenico, F., Serviddio, G., and Cassano, T. (2015). Aberrant insulin signaling in Alzheimer’s disease: current knowledge. Front. Neurosci. 9:204. doi: 10.3389/fnagi.2017.00204
Ben-Yoseph, O., Boxer, P. A., and Ross, B. D. (1994). Oxidative stress in the central nervous system: monitoring the metabolic response using the pentose phosphate pathway. Dev. Neurosci. 16, 328–336. doi: 10.1159/000112127
Beppu, K., Kosai, Y., Kido, M. A., Akimoto, N., Mori, Y., Kojima, Y., et al. (2013). Expression, subunit composition, and function of AMPA-type glutamate receptors are changed in activated microglia; possible contribution of GluA2 (GluR-B)-deficiency under pathological conditions. Glia 61, 881–891. doi: 10.1002/glia.22481
Beurel, E., and Jope, R. S. (2006). The paradoxical pro- and anti-apoptotic actions of GSK3 in the intrinsic and extrinsic apoptosis signaling pathways. Prog. Neurobiol. 79, 173–189. doi: 10.1016/j.pneurobio.2006.07.006
Bielarczyk, H., Gul, S., Ronowska, A., Bizon-Zygmanska, D., Pawelczyk, T., and Szutowicz, A. (2006). RS-alpha-lipoic acid protects cholinergic cells against sodium nitroprusside and amyloid-beta neurotoxicity through restoration of acetyl-CoA level. J. Neurochem. 98, 1242–1251. doi: 10.1111/j.1471-4159.2006.03966.x
Bielarczyk, H., Jankowska-Kulawy, A., Hofling, C., Ronowska, A., Gul-Hinc, S., Rossner, S., et al. (2015). AbetaPP-transgenic 2576 mice mimic cell type-specific aspects of Acetyl-CoA-linked metabolic deficits in Alzheimer’s disease. J. Alzheimers Dis. 48, 1083–1094. doi: 10.3233/jad-150327
Bigl, M., Brückner, M. K., Arendt, T., Bigl, V., and Eschrich, K. (1999). Activities of key glycolytic enzymes in the brains of patients with Alzheimer’s disease. J. Neural Trans. 106, 499–511. doi: 10.1007/s007020050174
Blass, J. P., Sheu, R. K., and Gibson, G. E. (2000). Inherent abnormalities in energy metabolism in Alzheimer disease. Interaction with cerebrovascular compromise. Ann. N. Y. Acad. Sci. 903, 204–221. doi: 10.1111/j.1749-6632.2000.tb06370.x
Bobba, A., Amadoro, G., Valenti, D., Corsetti, V., Lassandro, R., and Atlante, A. (2013). Mitochondrial respiratory chain complexes I and IV are impaired by beta-amyloid via direct interaction and through complex I-dependent ROS production, respectively. Mitochondrion 13, 298–311. doi: 10.1016/j.mito.2013.03.008
Bohnen, N. I., Djang, D. S., Herholz, K., Anzai, Y., and Minoshima, S. (2012). Effectiveness and safety of 18F-FDG PET in the evaluation of dementia: a review of the recent literature. J. Nucl. Med. 53, 59–71. doi: 10.2967/jnumed.111.096578
Bolanos, J. P., Almeida, A., and Moncada, S. (2010). Glycolysis: a bioenergetic or a survival pathway? Trends Biochem. Sci. 35, 145–149. doi: 10.1016/j.tibs.2009.10.006
Bouzier-Sore, A. K., Voisin, P., Bouchaud, V., Bezancon, E., Franconi, J. M., and Pellerin, L. (2006). Competition between glucose and lactate as oxidative energy substrates in both neurons and astrocytes: a comparative NMR study. Eur. J. Neurosci. 24, 1687–1694. doi: 10.1111/j.1460-9568.2006.05056.x
Boveris, A., and Navarro, A. (2008). Brain mitochondrial dysfunction in aging. IUBMB Life 60, 308–314. doi: 10.1002/iub.46
Bowen, C., Childers, G., Perry, C., Martin, N., McPherson, C. A., Lauten, T., et al. (2020). Mitochondrial-related effects of pentabromophenol, tetrabromobisphenol A, and triphenyl phosphate on murine BV-2 microglia cells. Chemosphere 255:126919. doi: 10.1016/j.chemosphere.2020.126919
Bowen, D. M., Smith, C. B., White, P., and Davison, A. N. (1976). Neurotransmitter-related enzymes and indices of hypoxia in senile dementia and other abiotrophies. Brain 99, 459–496. doi: 10.1093/brain/99.3.459
Bradley-Whitman, M. A., Timmons, M. D., Beckett, T. L., Murphy, M. P., Lynn, B. C., and Lovell, M. A. (2014). Nucleic acid oxidation: an early feature of Alzheimer’s disease. J. Neurochem. 128, 294–304.
Bubber, P., Haroutunian, V., Fisch, G., Blass, J. P., and Gibson, G. E. (2005). Mitochondrial abnormalities in Alzheimer brain: mechanistic implications. Ann. Neurol. 57, 695–703. doi: 10.1002/ana.20474
Budni, J., Feijo, D. P., Batista-Silva, H., Garcez, M. L., Mina, F., Belletini-Santos, T., et al. (2017). Lithium and memantine improve spatial memory impairment and neuroinflammation induced by beta-amyloid 1-42 oligomers in rats. Neurobiol. Learn. Mem. 141, 84–92. doi: 10.1016/j.nlm.2017.03.017
Butterfield, D. A., and Halliwell, B. (2019). Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 20, 148–160. doi: 10.1038/s41583-019-0132-6
Caccamo, A., Majumder, S., Richardson, A., Strong, R., and Oddo, S. (2010). Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and Tau: effects on cognitive impairments. J. Biol. Chem. 285, 13107–13120. doi: 10.1074/jbc.m110.100420
Cai, X., Zhang, K., Xie, X., Zhu, X., Feng, J., Jin, Z., et al. (2020). Self-assembly hollow manganese Prussian white nanocapsules attenuate Tau-related neuropathology and cognitive decline. Biomaterials 231:119678. doi: 10.1016/j.biomaterials.2019.119678
Cai, Z., Zhao, B., Li, K., Zhang, L., Li, C., Quazi, S. H., et al. (2012). Mammalian target of rapamycin: a valid therapeutic target through the autophagy pathway for Alzheimer’s disease? J. Neurosci. Res. 90, 1105–1118. doi: 10.1002/jnr.23011
Canevari, L., Abramov, A. Y., and Duchen, M. R. (2004). Toxicity of amyloid beta peptide: tales of calcium, mitochondria, and oxidative stress. Neurochem. Res. 29, 637–650. doi: 10.1023/b:nere.0000014834.06405.af
Cao, P., Zhang, J., Huang, Y., Fang, Y., Lyu, J., and Shen, Y. (2019). The age-related changes and differences in energy metabolism and glutamate-glutamine recycling in the d-gal-induced and naturally occurring senescent astrocytes in vitro. Exper. Gerontol. 118, 9–18. doi: 10.1016/j.exger.2018.12.018
Cenini, G., Lloret, A., and Cascella, R. (2019). Oxidative stress in neurodegenerative diseases: from a mitochondrial point of view. Oxid. Med. Cell Longev. 2019:2105607.
Cereghetti, G., Saad, S., Dechant, R., and Peter, M. (2018). Reversible, functional amyloids: towards an understanding of their regulation in yeast and humans. Cell Cycle 17, 1545–1558. doi: 10.1080/15384101.2018.1480220
Chen, Y. H., He, R. Q., Liu, Y., Liu, Y., and Xue, Z. G. (2000). Effect of human neuronal tau on denaturation and reactivation of rabbit muscle D-glyceraldehyde-3-phosphate dehydrogenase. Biochem. J. 351(Pt 1), 233–240. doi: 10.1042/0264-6021:3510233
Cheng, Z., Tseng, Y., and White, M. F. (2010). Insulin signaling meets mitochondria in metabolism. Trends Endocrinol. Metab. 21, 589–598. doi: 10.1016/j.tem.2010.06.005
Choeiri, C., Staines, W., and Messier, C. (2002). Immunohistochemical localization and quantification of glucose transporters in the mouse brain. Neuroscience 111, 19–34. doi: 10.1016/s0306-4522(01)00619-4
Choi, D. W. (1987). Ionic dependence of glutamate neurotoxicity. J. Neurosci. 7, 369–379. doi: 10.1523/jneurosci.07-02-00369.1987
Christofk, H. R., Vander Heiden, M. G., Wu, N., Asara, J. M., and Cantley, L. C. (2008). Pyruvate kinase M2 is a phosphotyrosine-binding protein. Nature 452, 181–186. doi: 10.1038/nature06667
Cisternas, P., Zolezzi, J. M., Martinez, M., Torres, V. I., Wong, G. W., and Inestrosa, N. C. (2019). Wnt-induced activation of glucose metabolism mediates the in vivo neuroprotective roles of Wnt signaling in Alzheimer disease. J. Neurochem. 149, 54–72. doi: 10.1111/jnc.14608
Claxton, A., Baker, L. D., Hanson, A., Trittschuh, E. H., Cholerton, B., Morgan, A., et al. (2015). Long-acting intranasal insulin detemir improves cognition for adults with mild cognitive impairment or early-stage Alzheimer’s disease dementia. J. Alzheimer Dis. 44, 897–906. doi: 10.3233/jad-141791
Clement, J., Wong, M., Poljak, A., Sachdev, P., and Braidy, N. (2019). The plasma NAD(+) metabolome is dysregulated in “Normal” aging. Rejuven. Res. 22, 121–130. doi: 10.1089/rej.2018.2077
Cobley, J. N., Fiorello, M. L., and Bailey, D. M. (2018). 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 15, 490–503. doi: 10.1016/j.redox.2018.01.008
Collingridge, G. L., Olsen, R. W., Peters, J., and Spedding, M. (2009). A nomenclature for ligand-gated ion channels. Neuropharmacology 56, 2–5. doi: 10.1016/j.neuropharm.2008.06.063
Cortes-Rojo, C., Vargas-Vargas, M. A., Olmos-Orizaba, B. E., Rodriguez-Orozco, A. R., and Calderon-Cortes, E. (2020). Interplay between NADH oxidation by complex I, glutathione redox state and sirtuin-3, and its role in the development of insulin resistance. Biochim. Biophys. Acta Mol. Basis Dis. 1866:165801. doi: 10.1016/j.bbadis.2020.165801
Craft, S., Asthana, S., Cook, D. G., Baker, L. D., Cherrier, M., Purganan, K., et al. (2003). Insulin dose-response effects on memory and plasma amyloid precursor protein in Alzheimer’s disease: interactions with apolipoprotein E genotype. Psychoneuroendocrinology 28, 809–822. doi: 10.1016/s0306-4530(02)00087-2
Craft, S., Baker, L. D., Montine, T. J., Minoshima, S., Watson, G. S., Claxton, A., et al. (2012). Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: a pilot clinical trial. Arch. Neurol. 69, 29–38. doi: 10.1001/archneurol.2011.233
Craft, S., Raman, R., Chow, T. W., Rafii, M. S., Sun, C. K., Rissman, R. A., et al. (2020). Safety, efficacy, and feasibility of intranasal insulin for the treatment of mild cognitive impairment and alzheimer disease dementia: a randomized clinical trial. JAMA Neurol. 77, 1–11. doi: 10.1097/00045391-900000000-98514
Cuadrado-Tejedor, M., Vilarino, M., Cabodevilla, F., Del Rio, J., Frechilla, D., and Perez-Mediavilla, A. (2011). Enhanced expression of the voltage-dependent anion channel 1 (VDAC1) in Alzheimer’s disease transgenic mice: an insight into the pathogenic effects of amyloid-beta. J. Alzheimer Dis. 23, 195–206. doi: 10.3233/jad-2010-100966
Cumming, R. C., and Schubert, D. (2005). Amyloid-beta induces disulfide bonding and aggregation of GAPDH in Alzheimer’s disease. FASEB J. 19, 2060–2062. doi: 10.1096/fj.05-4195fje
Cunnane, S., Nugent, S., Roy, M., Courchesne-Loyer, A., Croteau, E., Tremblay, S., et al. (2011). Brain fuel metabolism, aging, and Alzheimer’s disease. Nutrition 27, 3–20.
Currais, A., Huang, L., Goldberg, J., Petrascheck, M., Ates, G., Pinto-Duarte, A., et al. (2019). Elevating acetyl-CoA levels reduces aspects of brain aging. eLife 8:e47866.
de Bari, L., Atlante, A., Armeni, T., and Kalapos, M. P. (2019). Synthesis and metabolism of methylglyoxal, S-D-lactoylglutathione and D-lactate in cancer and Alzheimer’s disease. Exploring the crossroad of eternal youth and premature aging. Age. Res. Rev. 53:100915. doi: 10.1016/j.arr.2019.100915
de la Monte, S. M., and Wands, J. R. (2005). Review of insulin and insulin-like growth factor expression, signaling, and malfunction in the central nervous system: relevance to Alzheimer’s disease. J. Alzheimers Dis. 7, 45–61. doi: 10.3233/jad-2005-7106
DeBalsi, K. L., Hoff, K. E., and Copeland, W. C. (2017). Role of the mitochondrial DNA replication machinery in mitochondrial DNA mutagenesis, aging and age-related diseases. Age. Res. Rev. 33, 89–104. doi: 10.1016/j.arr.2016.04.006
Dedeoglu, A., Choi, J. K., Cormier, K., Kowall, N. W., and Jenkins, B. G. (2004). Magnetic resonance spectroscopic analysis of Alzheimer’s disease mouse brain that express mutant human APP shows altered neurochemical profile. Brain Res. 1012, 60–65. doi: 10.1016/j.brainres.2004.02.079
Demarest, T. G., Varma, V. R., Estrada, D., Babbar, M., Basu, S., Mahajan, U. V., et al. (2020). Biological sex and DNA repair deficiency drive Alzheimer’s disease via systemic metabolic remodeling and brain mitochondrial dysfunction. Acta Neuropathol. 140, 25–47. doi: 10.1007/s00401-020-02152-8
Dennis, P. B., Jaeschke, A., Saitoh, M., Fowler, B., Kozma, S. C., and Thomas, G. (2001). Mammalian TOR: a homeostatic ATP sensor. Science 294, 1102–1105. doi: 10.1126/science.1063518
Di Domenico, F., Pupo, G., Giraldo, E., Badia, M. C., Monllor, P., Lloret, A., et al. (2016). Oxidative signature of cerebrospinal fluid from mild cognitive impairment and Alzheimer disease patients. Free Radic. Biol. Med. 91, 1–9. doi: 10.1016/j.freeradbiomed.2015.12.004
Di Domenico, F., Tramutola, A., and Butterfield, D. A. (2017). Role of 4-hydroxy-2-nonenal (HNE) in the pathogenesis of alzheimer disease and other selected age-related neurodegenerative disorders. Free Radic. Biol. Med. 111, 253–261. doi: 10.1016/j.freeradbiomed.2016.10.490
Di Domenico, F., Tramutola, A., Foppoli, C., Head, E., Perluigi, M., and Butterfield, D. A. (2018). mTOR in down syndrome: role in ass and tau neuropathology and transition to Alzheimer disease-like dementia. Free Radic. Biol. Med. 114, 94–101. doi: 10.1016/j.freeradbiomed.2017.08.009
Diaz-Garcia, C. M., Mongeon, R., Lahmann, C., Koveal, D., Zucker, H., and Yellen, G. (2017). Neuronal stimulation triggers neuronal Glycolysis and not lactate uptake. Cell Metab. 26, 361–374.e4.
Diehl, T., Mullins, R., and Kapogiannis, D. (2017). Insulin resistance in Alzheimer’s disease. Transl. Res. 183, 26–40.
Dienel, G. A. (2019). Brain glucose metabolism: integration of energetics with function. Physiol. Rev. 99, 949–1045. doi: 10.1152/physrev.00062.2017
Ding, F., Yao, J., Rettberg, J. R., Chen, S., and Brinton, R. D. (2013). Early decline in glucose transport and metabolism precedes shift to ketogenic system in female aging and Alzheimer’s mouse brain: implication for bioenergetic intervention. PLoS One 8:e79977. doi: 10.1371/journal.pone.079977
DiNuzzo, M., Giove, F., Maraviglia, B., and Mangia, S. (2015). Monoaminergic control of cellular glucose utilization by glycogenolysis in neocortex and hippocampus. Neurochem. Res. 40, 2493–2504. doi: 10.1007/s11064-015-1656-4
Doert, A., Pilatus, U., Zanella, F., Muller, W. E., and Eckert, G. P. (2015). (1)H- and (1)(3)C-NMR spectroscopy of Thy-1-APPSL mice brain extracts indicates metabolic changes in Alzheimer’s disease. J. Neural Transm. 122, 541–550. doi: 10.1007/s00702-015-1387-3
Dong, Y., and Brewer, G. J. (2019). Global metabolic shifts in age and Alzheimer’s disease mouse brains pivot at NAD+/NADH redox sites. J. Alzheimer Dis. 71, 119–140. doi: 10.3233/jad-190408
Dragicevic, N., Mamcarz, M., Zhu, Y., Buzzeo, R., Tan, J., Arendash, G. W., et al. (2010). Mitochondrial amyloid-beta levels are associated with the extent of mitochondrial dysfunction in different brain regions and the degree of cognitive impairment in Alzheimer’s transgenic mice. J. Alzheimers Dis. 20(Suppl. 2), S535–S550.
Dringen, R. (2000). Metabolism and functions of glutathione in brain. Prog. Neurobiol. 62, 649–671. doi: 10.1016/s0301-0082(99)00060-x
Drzezga, A., Lautenschlager, N., Siebner, H., Riemenschneider, M., Willoch, F., Minoshima, S., et al. (2003). Cerebral metabolic changes accompanying conversion of mild cognitive impairment into Alzheimer’s disease: a PET follow-up study. Eur. J. Nucl. Med. Mol. Imag. 30, 1104–1113. doi: 10.1007/s00259-003-1194-1
Duelli, R., and Kuschinsky, W. (2001). Brain glucose transporters: relationship to local energy demand. News Physiol. Sci. 16, 71–76. doi: 10.1152/physiologyonline.2001.16.2.71
Dukhande, V. V., Isaac, A. O., Chatterji, T., and Lai, J. C. (2009). Reduced glutathione regenerating enzymes undergo developmental decline and sexual dimorphism in the rat cerebral cortex. Brain Res. 1286, 19–24. doi: 10.1016/j.brainres.2009.05.029
El-Armouche, A., Zolk, O., and Eschenhagen, T. (2003). Citalopram. Deutsche Med. Wochenschrift 128, 2253–2256.
Emir, U. E., Raatz, S., McPherson, S., Hodges, J. S., Torkelson, C., Tawfik, P., et al. (2011). Noninvasive quantification of ascorbate and glutathione concentration in the elderly human brain. NMR Biomed. 24, 888–894. doi: 10.1002/nbm.1646
Epis, R., Marcello, E., Gardoni, F., Longhi, A., Calvani, M., Iannuccelli, M., et al. (2008). Modulatory effect of acetyl-L-carnitine on amyloid precursor protein metabolism in hippocampal neurons. Eur. J. Pharmacol. 597, 51–56. doi: 10.1016/j.ejphar.2008.09.001
Falls, N., Singh, D., Anwar, F., Verma, A., and Kumar, V. (2018). Amelioration of neurodegeneration and cognitive impairment by Lemon oil in experimental model of Stressed mice. Biomed. Pharmacother. 106, 575–583. doi: 10.1016/j.biopha.2018.06.160
Farris, W., Mansourian, S., Leissring, M. A., Eckman, E. A., Bertram, L., Eckman, C. B., et al. (2004). Partial loss-of-function mutations in insulin-degrading enzyme that induce diabetes also impair degradation of amyloid beta-protein. Am. J. Pathol. 164, 1425–1434. doi: 10.1016/s0002-9440(10)63229-4
Fisar, Z., Hroudova, J., Hansikova, H., Spacilova, J., Lelkova, P., Wenchich, L., et al. (2016). Mitochondrial respiration in the platelets of patients with Alzheimer’s disease. Curr. Alzheimer Res. 13, 930–941. doi: 10.2174/1567205013666160314150856
Freeman, L., and Guo, H. (2017). NLR members NLRC4 and NLRP3 mediate sterile inflammasome activation in microglia and astrocytes. J. Exp. Med. 214, 1351–1370. doi: 10.1084/jem.20150237
Freude, S., Schilbach, K., and Schubert, M. (2009). The role of IGF-1 receptor and insulin receptor signaling for the pathogenesis of Alzheimer’s disease: from model organisms to human disease. Curr. Alzheimer Res. 6, 213–223. doi: 10.2174/156720509788486527
Garcia, J., Han, D., Sancheti, H., Yap, L. P., Kaplowitz, N., and Cadenas, E. (2010). Regulation of mitochondrial glutathione redox status and protein glutathionylation by respiratory substrates. J. Biol. Chem. 285, 39646–39654. doi: 10.1074/jbc.m110.164160
Garcia-Arencibia, M., Hochfeld, W. E., Toh, P. P., and Rubinsztein, D. C. (2010). Autophagy, a guardian against neurodegeneration. Semin. Cell Dev. Biol. 21, 691–698. doi: 10.1016/j.semcdb.2010.02.008
Gasparini, L., Gouras, G. K., Wang, R., Gross, R. S., Beal, M. F., Greengard, P., et al. (2001). Stimulation of beta-amyloid precursor protein trafficking by insulin reduces intraneuronal beta-amyloid and requires mitogen-activated protein kinase signaling. J. Neurosci. 21, 2561–2570. doi: 10.1523/jneurosci.21-08-02561.2001
Gejl, M., Brock, B., Egefjord, L., Vang, K., Rungby, J., and Gjedde, A. (2017). Blood-brain glucose transfer in Alzheimer’s disease: effect of GLP-1 analog treatment. Sci. Rep. 7:17490.
Ghosh, S., Castillo, E., Frias, E. S., and Swanson, R. A. (2018). Bioenergetic regulation of microglia. Cell 66, 1200–1212. doi: 10.1002/glia.23271
Gibson, G. E., and Blass, J. P. (2007). Thiamine-dependent processes and treatment strategies in neurodegeneration. Antioxid. Redox Signal. 9, 1605–1620. doi: 10.1089/ars.2007.1766
Golpich, M., Amini, E., Mohamed, Z., Azman Ali, R., Mohamed Ibrahim, N., and Ahmadiani, A. (2017). Mitochondrial dysfunction and biogenesis in neurodegenerative diseases: pathogenesis and treatment. CNS Neurosci. Ther. 23, 5–22. doi: 10.1111/cns.12655
Gomez, O., Ballester-Lurbe, B., Poch, E., Mesonero, J. E., and Terrado, J. (2010). Developmental regulation of glucose transporters GLUT3, GLUT4 and GLUT8 in the mouse cerebellar cortex. J. Anat. 217, 616–623. doi: 10.1111/j.1469-7580.2010.01291.x
Goncalves, C. A., Rodrigues, L., Bobermin, L. D., Zanotto, C., Vizuete, A., Quincozes-Santos, A., et al. (2018). Glycolysis-derived compounds from astrocytes that modulate synaptic communication. Front. Neurosci. 12:1035. doi: 10.3389/fnagi.2017.1035
Gonzalez-Fraguela, M. E., Blanco, L., Fernandez, C. I., Lorigados, L., Serrano, T., and Fernandez, J. L. (2018). Glutathione depletion: Starting point of brain metabolic stress, neuroinflammation and cognitive impairment in rats. Brain Res. Bull. 137, 120–131. doi: 10.1016/j.brainresbull.2017.11.015
Gowans, G. J., and Hardie, D. G. (2014). AMPK: a cellular energy sensor primarily regulated by AMP. Biochem. Soc. Trans. 42, 71–75. doi: 10.1042/bst20130244
Group Cognitive Impairment Writing Group, Association Special Committee on Cognitive Disorders of the Chinese Medical Doctors Association (2018). 2018 guidelines for the diagnosis and treatment of dementia and cognitive impairment in China (2): guidelines for the diagnosis and treatment of Alzheimer’s disease. Natl. Med. J. China 98, 971–977.
Guo, X., Park, J. E., Gallart-Palau, X., and Sze, S. K. (2020). Oxidative damage to the TCA cycle enzyme MDH1 dysregulates bioenergetic enzymatic activity in the aged murine brain. J. Proteom. Res. 19, 1706–1717. doi: 10.1021/acs.jproteome.9b00861
Gustin, A., Kirchmeyer, M., Koncina, E., Felten, P., Losciuto, S., Heurtaux, T., et al. (2015). NLRP3 inflammasome is expressed and functional in mouse brain microglia but not in Astrocytes. PLoS One 10:e0130624. doi: 10.1371/journal.pone.0130624
Hakim, A. M., Moss, G., and Gollomp, S. M. (1976). The effect of hypoxia on the pentose phosphate pathway in brain. J. Neurochem. 26, 683–688. doi: 10.1111/j.1471-4159.1976.tb04437.x
Hardy, J., and Higgins, G. (1992). Alzheimer’s disease: the amyloid cascade hypothesis. Science 256, 184–185.
Harris, R. A., Tindale, L., and Cumming, R. C. (2014). Age-dependent metabolic dysregulation in cancer and Alzheimer’s disease. Biogerontology 15, 559–577. doi: 10.1007/s10522-014-9534-z
Hauptmann, S., Scherping, I., Drose, S., Brandt, U., Schulz, K. L., Jendrach, M., et al. (2009). Mitochondrial dysfunction: an early event in Alzheimer pathology accumulates with age in AD transgenic mice. Neurobiol. Aging 30, 1574–1586. doi: 10.1016/j.neurobiolaging.2007.12.005
Heneka, M. T., Kummer, M. P., Stutz, A., Delekate, A., Schwartz, S., Vieira-Saecker, A., et al. (2013). NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 493, 674–678. doi: 10.1038/nature11729
Hernandez, F., Lucas, J. J., and Avila, J. (2013). GSK3 and tau: two convergence points in Alzheimer’s disease. J. Alzheimer Dis. 33(Suppl. 1), S141–S144.
Herrero-Mendez, A., Almeida, A., Fernandez, E., Maestre, C., Moncada, S., and Bolanos, J. P. (2009). The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C-Cdh1. Nat. Cell Biol. 11, 747–752. doi: 10.1038/ncb1881
Higuchi, M., Zhang, B., Forman, M. S., Yoshiyama, Y., Trojanowski, J. Q., and Lee, V. M. (2005). Axonal degeneration induced by targeted expression of mutant human tau in oligodendrocytes of transgenic mice that model glial tauopathies. J. Neurosci. 25, 9434–9443. doi: 10.1523/jneurosci.2691-05.2005
Hill, B. G., and Bhatnagar, A. (2012). Protein S-glutathiolation: redox-sensitive regulation of protein function. J. Mol. Cell Cardiol. 52, 559–567. doi: 10.1016/j.yjmcc.2011.07.009
Hipkiss, A. R. (2019). Aging, Alzheimer’s disease and dysfunctional glycolysis; similar effects of too much and too little. Aging Dis. 10, 1328–1331. doi: 10.14336/ad.2019.0611
Hokama, M., Oka, S., Leon, J., Ninomiya, T., Honda, H., Sasaki, K., et al. (2014). Altered expression of diabetes-related genes in Alzheimer’s disease brains: the Hisayama study. Cereb. Cortex 24, 2476–2488. doi: 10.1093/cercor/bht101
Holper, L., Ben-Shachar, D., and Mann, J. J. (2019). Multivariate meta-analyses of mitochondrial complex I and IV in major depressive disorder, bipolar disorder, schizophrenia, Alzheimer disease, and Parkinson disease. Neuropsychopharmacology 44, 837–849. doi: 10.1038/s41386-018-0090-0
Hou, Y., Lautrup, S., Cordonnier, S., Wang, Y., Croteau, D. L., Zavala, E., et al. (2018). NAD(+) supplementation normalizes key Alzheimer’s features and DNA damage responses in a new AD mouse model with introduced DNA repair deficiency. Proc. Natl. Acad. Sci. U.S.A. 115, E1876–E1885.
Hoyer, S. (1982). The young-adult and normally aged brain. Its blood flow and oxidative metabolism. A review–part I. Arch. Gerontol. Geriatr. 1, 101–116. doi: 10.1016/0167-4943(82)90010-3
Hoyer, S. (1985). The effect of age on glucose and energy metabolism in brain cortex of rats. Archiv. Gerontol. Geriatr. 4, 193–203. doi: 10.1016/0167-4943(85)90001-9
Huang, Y., and Mahley, R. W. (2014). Apolipoprotein E: structure and function in lipid metabolism, neurobiology, and Alzheimer’s diseases. Neurobiol. Dis. 72(Pt A), 3–12. doi: 10.1016/j.nbd.2014.08.025
Hunter, N. S., Anderson, K. C., and Cox, A. (2015). Pimavanserin. Drugs Today 51, 645–652. doi: 10.1358/dot.2015.51.11.2404001
Indo, H. P., Yen, H. C., Nakanishi, I., Matsumoto, K., Tamura, M., Nagano, Y., et al. (2015). A mitochondrial superoxide theory for oxidative stress diseases and aging. J. Clin. Biochem. Nutr. 56, 1–7.
Iqbal, K., Alonso Adel, C., Chen, S., Chohan, M. O., El-Akkad, E., Gong, C. X., et al. (2005). Tau pathology in Alzheimer disease and other tauopathies. Biochim. Biophys. Acta 1739, 198–210.
Isaev, N. K., Stelmashook, E. V., and Genrikhs, E. E. (2019). Neurogenesis and brain aging. Rev. Neurosci. 30, 573–580.
Itakura, M., Nakajima, H., Kubo, T., Semi, Y., Kume, S., Higashida, S., et al. (2015). Glyceraldehyde-3-phosphate dehydrogenase aggregates accelerate Amyloid-beta Amyloidogenesis in Alzheimer disease. J. Biol. Chem. 290, 26072–26087. doi: 10.1074/jbc.m115.669291
Jais, A., Solas, M., Backes, H., Chaurasia, B., Kleinridders, A., Theurich, S., et al. (2016). Myeloid-cell-derived VEGF maintains brain glucose uptake and limits cognitive impairment in obesity. Cell 165, 882–895. doi: 10.1016/j.cell.2016.03.033
Jeng, W., Loniewska, M. M., and Wells, P. G. (2013). Brain glucose-6-phosphate dehydrogenase protects against endogenous oxidative DNA damage and neurodegeneration in aged mice. ACS Chem. Neurosci. 4, 1123–1132. doi: 10.1021/cn400079y
Jha, M. K., Lee, I. K., and Suk, K. (2016). Metabolic reprogramming by the pyruvate dehydrogenase kinase-lactic acid axis: linking metabolism and diverse neuropathophysiologies. Neurosci. Biobehav. Rev. 68, 1–19. doi: 10.1016/j.neubiorev.2016.05.006
Jiang, S. H., Li, J., Dong, F. Y., Yang, J. Y., Liu, D. J., Yang, X. M., et al. (2017). Increased serotonin signaling contributes to the warburg effect in pancreatic tumor cells under metabolic stress and promotes growth of pancreatic tumors in mice. Gastroenterology 153, 277–291.e19.
Jiang, T., Yin, F., Yao, J., Brinton, R. D., and Cadenas, E. (2013). Lipoic acid restores age-associated impairment of brain energy metabolism through the modulation of Akt/JNK signaling and PGC1alpha transcriptional pathway. Aging Cell 12, 1021–1031. doi: 10.1111/acel.12127
Johnson, L. A., Torres, E. R. S., Impey, S., Stevens, J. F., and Raber, J. (2017). Apolipoprotein E4 and insulin resistance interact to impair cognition and alter the Epigenome and Metabolome. Sci. Rep. 7:43701.
Jonas, M. C., Pehar, M., and Puglielli, L. (2010). AT-1 is the ER membrane acetyl-CoA transporter and is essential for cell viability. J. Cell Sci. 123(Pt 19), 3378–3388. doi: 10.1242/jcs.068841
Jones, C. L., Stevens, B. M., D’Alessandro, A., Culp-Hill, R., Reisz, J. A., Pei, S., et al. (2019). Cysteine depletion targets leukemia stem cells through inhibition of electron transport complex II. Blood 134, 389–394. doi: 10.1182/blood.2019898114
Kales, H. C., Lyketsos, C. G., Miller, E. M., and Ballard, C. (2019). Management of behavioral and psychological symptoms in people with Alzheimer’s disease: an international Delphi consensus. Intern. Psychogeriatr. 31, 83–90. doi: 10.1017/s1041610218000534
Kametani, F., and Hasegawa, M. (2018). Reconsideration of Amyloid hypothesis and Tau hypothesis in Alzheimer’s disease. Front. Neurosci. 12:25. doi: 10.3389/fnagi.2017.00025
Kann, O., and Kovacs, R. (2007). Mitochondria and neuronal activity. Am. J. Physiol. Cell Physiol. 292, C641–C657.
Kato, T., Inui, Y., Nakamura, A., and Ito, K. (2016). Brain fluorodeoxyglucose (FDG) PET in dementia. Age. Res. Rev. 30, 73–84. doi: 10.1016/j.arr.2016.02.003
Kauffman, F. C. (1972). The quantitative histochemistry of enzymes of the pentose phosphate pathway in the central nervous system of the Rat1. J. Neurochem. 19, 1–9. doi: 10.1111/j.1471-4159.1972.tb01247.x
Kausar, S., Wang, F., and Cui, H. (2018). The role of mitochondria in reactive oxygen species generation and its implications for neurodegenerative diseases. Cells 17:274. doi: 10.3390/cells7120274
Keeney, J. T., Ibrahimi, S., and Zhao, L. (2015). Human ApoE isoforms differentially modulate glucose and Amyloid metabolic pathways in female brain: evidence of the mechanism of neuroprotection by ApoE2 and implications for Alzheimer’s disease prevention and early intervention. J. Alzheimers Dis. 48, 411–424. doi: 10.3233/jad-150348
Kezic, A., Popovic, L., and Lalic, K. (2018). mTOR inhibitor therapy and metabolic consequences: where do we stand? Oxid. Med. Cell Longev. 2018: 2640342.
Kim, A., Chen, C. H., Ursell, P., and Huang, T. T. (2010). Genetic modifier of mitochondrial superoxide dismutase-deficient mice delays heart failure and prolongs survival. Mamm. Genome 21, 534–542. doi: 10.1007/s00335-010-9299-x
Kim, E. H., Lee, J. H., Oh, Y., Koh, I., Shim, J. K., Park, J., et al. (2017). Inhibition of glioblastoma tumorspheres by combined treatment with 2-deoxyglucose and metformin. Neurol. Oncol. 19, 197–207.
Kim, S. H., Vlkolinsky, R., Cairns, N., and Lubec, G. (2000). Decreased levels of complex III core protein 1 and complex V beta chain in brains from patients with Alzheimer’s disease and down syndrome. Cell Mol. Life Sci. 57, 1810–1816. doi: 10.1007/pl00000661
Kishi, T., Matsunaga, S., Oya, K., Nomura, I., Ikuta, T., and Iwata, N. (2017). Memantine for Alzheimer’s disease: an updated systematic review and meta-analysis. J. Alzheimer Dis. 60, 401–425. doi: 10.3233/jad-170424
Klosinski, L. P., Yao, J., Yin, F., Fonteh, A. N., Harrington, M. G., Christensen, T. A., et al. (2015). White matter lipids as a ketogenic fuel supply in aging female brain: implications for Alzheimer’s disease. eBio Med. 2, 1888–1904. doi: 10.1016/j.ebiom.2015.11.002
Kosova, A. A., Khodyreva, S. N., and Lavrik, O. I. (2017). Role of glyceraldehyde-3-Phosphate dehydrogenase (GAPDH) in DNA repair. Biochem. Biokhimiia 82, 643–654. doi: 10.1134/s0006297917060013
Kramer, P. A., Duan, J., Gaffrey, M. J., Shukla, A. K., Wang, L., Bammler, T. K., et al. (2018). Fatiguing contractions increase protein S-glutathionylation occupancy in mouse skeletal muscle. Redox Biol. 17, 367–376. doi: 10.1016/j.redox.2018.05.011
Krautwald, M., and Munch, G. (2010). Advanced glycation end products as biomarkers and gerontotoxins - A basis to explore methylglyoxal-lowering agents for Alzheimer’s disease? Exp. Gerontol. 45, 744–751. doi: 10.1016/j.exger.2010.03.001
Kumar, A., and Singh, A. (2015). A review on mitochondrial restorative mechanism of antioxidants in Alzheimer’s disease and other neurological conditions. Front. Pharmacol. 6:206. doi: 10.3389/fnagi.2017.0206
Kunnimalaiyaan, S., Schwartz, V. K., Jackson, I. A., Clark Gamblin, T., and Kunnimalaiyaan, M. (2018). Antiproliferative and apoptotic effect of LY2090314, a GSK-3 inhibitor, in neuroblastoma in vitro. BMC Cancer 18:560. doi: 10.1186/s12885-018-4474-7
Landau, S. M., Harvey, D., Madison, C. M., Reiman, E. M., Foster, N. L., Aisen, P. S., et al. (2010). Comparing predictors of conversion and decline in mild cognitive impairment. Neurology 75, 230–238.
Lemeshko, V. V. (2018). VDAC electronics: 5. Mechanism and computational model of hexokinase-dependent generation of the outer membrane potential in brain mitochondria. Biochim. Biophys. Acta Biomembr. 1860, 2599–2607. doi: 10.1016/j.bbamem.2018.10.004
Leonard, B. E. (1975). The effect of 5-hydroxytryptamine and histamine on glycolysis in the mouse brain. Zeitschrift Naturforschung Sect. C Biosci. 30, 113–116. doi: 10.1515/znc-1975-1-222
Leuner, K., Schutt, T., Kurz, C., Eckert, S. H., Schiller, C., Occhipinti, A., et al. (2012). Mitochondrion-derived reactive oxygen species lead to enhanced amyloid beta formation. Antioxid. Redox Signal. 16, 1421–1433. doi: 10.1089/ars.2011.4173
Liao, F., Yoon, H., and Kim, J. (2017). Apolipoprotein E metabolism and functions in brain and its role in Alzheimer’s disease. Curr. Opin. Lipidol. 28, 60–67.
Liu, X., Kim, C. S., Kurbanov, F. T., Honzatko, R. B., and Fromm, H. J. (1999). Dual mechanisms for glucose 6-phosphate inhibition of human brain hexokinase. J. Biol. Chem. 274, 31155–31159. doi: 10.1074/jbc.274.44.31155
Liu, Y., Liu, F., Iqbal, K., Grundke-Iqbal, I., and Gong, C. X. (2008). Decreased glucose transporters correlate to abnormal hyperphosphorylation of Tau in Alzheimer disease. FEBS Lett. 582, 359–364. doi: 10.1016/j.febslet.2007.12.035
Lourenço, C. F., Ledo, A., Barbosa, R. M., and Laranjinha, J. (2017). Neurovascular-neuroenergetic coupling axis in the brain: master regulation by nitric oxide and consequences in aging and neurodegeneration. Free Rad. Biol. Med. 108, 668–682. doi: 10.1016/j.freeradbiomed.2017.04.026
Lu, J., Lezi, E., Roy, N., Hutfles, L., Selfridge, E., Funk, E., et al. (2013). Effect of cholinergic signaling on neuronal cell bioenergetics. J. Alzheimer Dis. 33, 1135–1146. doi: 10.3233/jad-2012-121822
Luchsinger, J. A., Tang, M. X., Stern, Y., Shea, S., and Mayeux, R. (2001). Diabetes mellitus and risk of Alzheimer’s disease and dementia with stroke in a multiethnic cohort. Am. J. Epidemiol. 154, 635–641. doi: 10.1093/aje/154.7.635
Luscher, B., Fuchs, T., and Kilpatrick, C. L. (2011). GABAA receptor trafficking-mediated plasticity of inhibitory synapses. Neuron 70, 385–409. doi: 10.1016/j.neuron.2011.03.024
Ma, D., Stokes, K., Mahngar, K., Domazet-Damjanov, D., Sikorska, M., and Pandey, S. (2014). Inhibition of stress induced premature senescence in presenilin-1 mutated cells with water soluble Coenzyme Q10. Mitochondrion 17, 106–115. doi: 10.1016/j.mito.2014.07.004
Ma, J., Brewer, H. B. Jr., and Potter, H. (1996). Alzheimer A beta neurotoxicity: promotion by antichymotrypsin, ApoE4; inhibition by A beta-related peptides. Neurobiol. Aging 17, 773–780. doi: 10.1016/0197-4580(96)00112-1
Macdonald, R., Barnes, K., Hastings, C., and Mortiboys, H. (2018). Mitochondrial abnormalities in Parkinson’s disease and Alzheimer’s disease: can mitochondria be targeted therapeutically? Biochem. Soc. Trans. 46, 891–909. doi: 10.1042/bst20170501
Majumder, S., Richardson, A., Strong, R., and Oddo, S. (2011). Inducing autophagy by rapamycin before, but not after, the formation of plaques and tangles ameliorates cognitive deficits. PLoS One 6:e25416. doi: 10.1371/journal.pone.025416
Mamczur, P., Borsuk, B., Paszko, J., Sas, Z., Mozrzymas, J., Wisniewski, J. R., et al. (2015). Astrocyte-neuron crosstalk regulates the expression and subcellular localization of carbohydrate metabolism enzymes. Glia 63, 328–340. doi: 10.1002/glia.22753
Mandal, P. K., Tripathi, M., and Sugunan, S. (2012). Brain oxidative stress: detection and mapping of anti-oxidant marker ‘Glutathione’ in different brain regions of healthy male/female, MCI and Alzheimer patients using non-invasive magnetic resonance spectroscopy. Biochem. Biophys. Res. Commun. 417, 43–48. doi: 10.1016/j.bbrc.2011.11.047
Martinon, F., Burns, K., and Tschopp, J. (2002). The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 10, 417–426.
Martins, R. N., Harper, C. G., Stokes, G. B., and Masters, C. L. (1986). Increased cerebral glucose-6-phosphate dehydrogenase activity in Alzheimer’s disease may reflect oxidative stress. J. Neurochem. 46, 1042–1045. doi: 10.1111/j.1471-4159.1986.tb00615.x
Martins, R. N., Villemagne, V., Sohrabi, H. R., Chatterjee, P., Shah, T. M., Verdile, G., et al. (2018). Alzheimer’s disease: a journey from amyloid peptides and oxidative stress, to biomarker technologies and disease prevention strategies-gains from AIBL and DIAN cohort studies. J. Alzheimers Dis. 62, 965–992. doi: 10.3233/jad-171145
Martire, S., Fuso, A., Mosca, L., Forte, E., Correani, V., Fontana, M., et al. (2016). Bioenergetic impairment in animal and cellular models of Alzheimer’s disease: PARP-1 inhibition rescues metabolic dysfunctions. J. Alzheimer Dis. 54, 307–324. doi: 10.3233/jad-151040
Mastrogiacomo, F., Bettendorff, L., Grisar, T., and Kish, S. J. (1996). Brain thiamine, its phosphate esters, and its metabolizing enzymes in Alzheimer’s disease. Ann. Neurol. 39, 585–591. doi: 10.1002/ana.410390507
Matsunaga, S., Kishi, T., Nomura, I., Sakuma, K., Okuya, M., Ikuta, T., et al. (2018). The efficacy and safety of memantine for the treatment of Alzheimer’s disease. Expert Opin. Drug Saf. 17, 1053–1061.
McCommis, K. S., and Finck, B. N. (2015). Mitochondrial pyruvate transport: a historical perspective and future research directions. Biochem. J. 466, 443–454. doi: 10.1042/bj20141171
Meimaridou, E., Goldsworthy, M., Chortis, V., Fragouli, E., Foster, P. A., Arlt, W., et al. (2018). NNT is a key regulator of adrenal redox homeostasis and steroidogenesis in male mice. J. Endocrinol. 236, 13–28. doi: 10.1530/joe-16-0638
Metherell, L. A., Guerra-Assuncao, J. A., Sternberg, M. J., and David, A. (2016). Three-dimensional model of human nicotinamide nucleotide Transhydrogenase (NNT) and sequence-structure analysis of its disease-causing variations. Hum. Mutat. 37, 1074–1084. doi: 10.1002/humu.23046
Molofsky, A. V., Krencik, R., Ullian, E. M., Tsai, H. H., Deneen, B., Richardson, W. D., et al. (2012). Astrocytes and disease: a neurodevelopmental perspective. Genes Dev. 26, 891–907. doi: 10.1101/gad.188326.112
Mosconi, L., Nacmias, B., Sorbi, S., De Cristofaro, M. T., Fayazz, M., Tedde, A., et al. (2004). Brain metabolic decreases related to the dose of the ApoE e4 allele in Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 75, 370–376. doi: 10.1136/jnnp.2003.014993
Munoz, D. G., and Feldman, H. (2000). Causes of Alzheimer’s disease. CMAJ Can. Med. Assoc. J. 162, 65–72.
Muñoz, S. S., Garner, B., and Ooi, L. (2019). Understanding the role of ApoE fragments in Alzheimer’s disease. Neurochem. Res. 44, 1297–1305. doi: 10.1007/s11064-018-2629-1
Musiek, E. S., and Holtzman, D. M. (2015). Three dimensions of the amyloid hypothesis: time, space and ‘wingmen’. Nat. Neurosci. 18, 800–806. doi: 10.1038/nn.4018
Muthukumaran, K., Kanwar, A., Vegh, C., Marginean, A., Elliott, A., Guilbeault, N., et al. (2018). Ubisol-Q10 (a Nanomicellar water-soluble formulation of CoQ10) treatment inhibits Alzheimer-type behavioral and pathological symptoms in a double transgenic mouse (TgAPEswe, PSEN1dE9) model of Alzheimer’s disease. J. Alzheimer Dis. 61, 221–236. doi: 10.3233/jad-170275
Muyderman, H., Wadey, A. L., Nilsson, M., and Sims, N. R. (2007). Mitochondrial glutathione protects against cell death induced by oxidative and nitrative stress in astrocytes. J. Neurochem. 102, 1369–1382. doi: 10.1111/j.1471-4159.2007.04641.x
Naderi, J., Lopez, C., and Pandey, S. (2006). Chronically increased oxidative stress in fibroblasts from Alzheimer’s disease patients causes early senescence and renders resistance to apoptosis by oxidative stress. Mech. Age. Dev. 127, 25–35. doi: 10.1016/j.mad.2005.08.006
Nakajima, H., Itakura, M., Kubo, T., Kaneshige, A., Harada, N., Izawa, T., et al. (2017). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) aggregation causes mitochondrial dysfunction during oxidative stress-induced Cell death. J. Biol. Chem. 292, 4727–4742. doi: 10.1074/jbc.m116.759084
Navarro, A., and Boveris, A. (2007). Brain mitochondrial dysfunction in aging: conditions that improve survival, neurological performance and mitochondrial function. Front. Biosci. 12, 1154–1163. doi: 10.2741/2133
Navarro, A., and Boveris, A. (2008). Mitochondrial nitric oxide synthase, mitochondrial brain dysfunction in aging, and mitochondria-targeted antioxidants. Adv. Drug Deliv. Rev. 60, 1534–1544. doi: 10.1016/j.addr.2008.05.002
Navarro, C. D. C., Figueira, T. R., Francisco, A., Dal’Bo, G. A., Ronchi, J. A., Rovani, J. C., et al. (2017). Redox imbalance due to the loss of mitochondrial NAD(P)-transhydrogenase markedly aggravates high fat diet-induced fatty liver disease in mice. Free Radic. Biol. Med. 113, 190–202. doi: 10.1016/j.freeradbiomed.2017.09.026
Neth, B. J., and Craft, S. (2017). Insulin resistance and Alzheimer’s disease: bioenergetic linkages. Front. Aging Neurosci. 9:345. doi: 10.3389/fnagi.2017.00345
Nicolakakis, N., Aboulkassim, T., Ongali, B., Lecrux, C., Fernandes, P., Rosa-Neto, P., et al. (2008). Complete rescue of cerebrovascular function in aged Alzheimer’s disease transgenic mice by antioxidants and pioglitazone, a peroxisome proliferator-activated receptor gamma agonist. J. Neurosci. 28, 9287–9296. doi: 10.1523/jneurosci.3348-08.2008
Nixon, R. A. (2007). Autophagy, amyloidogenesis and Alzheimer disease. J. Cell Sci. 120(Pt 23), 4081–4091. doi: 10.1242/jcs.019265
Noda, M. (2016). Dysfunction of glutamate receptors in microglia may cause neurodegeneration. Curr. Alzheimer Res. 13, 381–386. doi: 10.2174/1567205013666151116125810
Nunes, M. A., Viel, T. A., and Buck, H. S. (2013). Microdose lithium treatment stabilized cognitive impairment in patients with Alzheimer’s disease. Curr. Alzheimer Res. 10, 104–107. doi: 10.2174/156720513804871354
O’Neill, C. (2013). PI3-kinase/Akt/mTOR signaling: impaired on/off switches in aging, cognitive decline and Alzheimer’s disease. Exp. Gerontol. 48, 647–653. doi: 10.1016/j.exger.2013.02.025
Oksanen, M., Hyötyläinen, I., Trontti, K., Rolova, T., Wojciechowski, S., Koskuvi, M., et al. (2019). NF-E2-related factor 2 activation boosts antioxidant defenses and ameliorates inflammatory and amyloid properties in human Presenilin-1 mutated Alzheimer’s disease astrocytes. Glia 68, 589–599. doi: 10.1002/glia.23741
Oresic, M., Hyotylainen, T., Herukka, S. K., Sysi-Aho, M., Mattila, I., Seppanan-Laakso, T., et al. (2011). Metabolome in progression to Alzheimer’s disease. Transl. Psychiatry 1:e57.
Orr, M. E., and Oddo, S. (2013). Autophagic/lysosomal dysfunction in Alzheimer’s disease. Alzheimers Res. Ther. 5:53. doi: 10.1186/alzrt217
Palmer, A. M. (1999). The activity of the pentose phosphate pathway is increased in response to oxidative stress in Alzheimer’s disease. J. Neural Trans. 106, 317–328. doi: 10.1007/s007020050161
Pardo, J. V., Lee, J. T., Sheikh, S. A., Surerus-Johnson, C., Shah, H., Munch, K. R., et al. (2007). Where the brain grows old: decline in anterior cingulate and medial prefrontal function with normal aging. Neuroimage 35, 1231–1237. doi: 10.1016/j.neuroimage.2006.12.044
Patterson, R. L., van Rossum, D. B., Kaplin, A. I., Barrow, R. K., and Snyder, S. H. (2005). Inositol 1,4,5-trisphosphate receptor/GAPDH complex augments Ca2+ release via locally derived NADH. Proc. Natl. Acad. Sci. U.S.A. 102, 1357–1359. doi: 10.1073/pnas.0409657102
Peiss, C. N., Hall, V. E., and Field, J. (1949). The influence of magnesium on respiration glycolysis and cholinesterase activity in rat brain. J. Physiol. 108, 365–373. doi: 10.1113/jphysiol.1949.sp004341
Perluigi, M., Di Domenico, F., and Butterfield, D. A. (2015). mTOR signaling in aging and neurodegeneration: at the crossroad between metabolism dysfunction and impairment of autophagy. Neurobiol. Dis. 84, 39–49. doi: 10.1016/j.nbd.2015.03.014
Perluigi, M., Pupo, G., Tramutola, A., Cini, C., Coccia, R., Barone, E., et al. (2014). Neuropathological role of PI3K/Akt/mTOR axis in Down syndrome brain. Biochim. Biophys. Acta 1842, 1144–1153. doi: 10.1016/j.bbadis.2014.04.007
Petit-Taboué, M. C., Landeau, B., Desson, J. F., Desgranges, B., and Baron, J. C. (1998). Effects of healthy aging on the regional cerebral metabolic rate of glucose assessed with statistical parametric mapping. Neuroimage 7, 176–184. doi: 10.1006/nimg.1997.0318
Petrosillo, G., Matera, M., Casanova, G., Ruggiero, F. M., and Paradies, G. (2008). Mitochondrial dysfunction in rat brain with aging involvement of complex I, reactive oxygen species and cardiolipin. Neurochem. Int. 53, 126–131. doi: 10.1016/j.neuint.2008.07.001
Pettegrew, J. W., Klunk, W. E., Panchalingam, K., McClure, R. J., and Stanley, J. A. (1997). Magnetic resonance spectroscopic changes in Alzheimer’s disease. Ann. N. Y. Acad. Sci. 826, 282–306.
Pinto, M., and Moraes, C. T. (2015). Mechanisms linking mtDNA damage and aging. Free Radic. Biol. Med. 85, 250–258. doi: 10.1016/j.freeradbiomed.2015.05.005
Potter, H., and Wisniewski, T. (2012). Apolipoprotein e: essential catalyst of the Alzheimer amyloid cascade. Intern. J. Alzheimer Dis. 2012:489428.
Rahman, B., Kussmaul, L., Hamprecht, B., and Dringen, R. (2000). Glycogen is mobilized during the disposal of peroxides by cultured astroglial cells from rat brain. Neurosci. Lett. 290, 169–172. doi: 10.1016/s0304-3940(00)01369-0
Reddy, P. H. (2013). Amyloid beta-induced glycogen synthase kinase 3beta phosphorylated VDAC1 in Alzheimer’s disease: implications for synaptic dysfunction and neuronal damage. Biochim. Biophys. Acta 1832, 1913–1921. doi: 10.1016/j.bbadis.2013.06.012
Reed, T., Perluigi, M., Sultana, R., Pierce, W. M., Klein, J. B., Turner, D. M., et al. (2008). Redox proteomic identification of 4-hydroxy-2-nonenal-modified brain proteins in amnestic mild cognitive impairment: insight into the role of lipid peroxidation in the progression and pathogenesis of Alzheimer’s disease. Neurobiol. Dis. 30, 107–120. doi: 10.1016/j.nbd.2007.12.007
Reger, M. A., Watson, G. S., Frey, W. H. II, Baker, L. D., Cholerton, B., Keeling, M. L., et al. (2006). Effects of intranasal insulin on cognition in memory-impaired older adults: modulation by APOE genotype. Neurobiol. Aging 27, 451–458. doi: 10.1016/j.neurobiolaging.2005.03.016
Reger, M. A., Watson, G. S., Green, P. S., Baker, L. D., Cholerton, B., Fishel, M. A., et al. (2008). Intranasal insulin administration dose-dependently modulates verbal memory and plasma amyloid-beta in memory-impaired older adults. J. Alzheimer Dis. 13, 323–331. doi: 10.3233/jad-2008-13309
Rego, A. C., and Oliveira, C. R. (2003). Mitochondrial dysfunction and reactive oxygen species in excitotoxicity and apoptosis: implications for the pathogenesis of neurodegenerative diseases. Neurochem. Res. 28, 1563–1574.
Reiman, E. M., Caselli, R. J., Chen, K., Alexander, G. E., Bandy, D., and Frost, J. (2001). Declining brain activity in cognitively normal apolipoprotein E ϵ4 heterozygotes: a foundation for using positron emission tomography to efficiently test treatments to prevent Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 98, 3334–3339. doi: 10.1073/pnas.061509598
Reiman, E. M., Chen, K., Alexander, G. E., Caselli, R. J., Bandy, D., Osborne, D., et al. (2004). Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer’s dementia. Proc. Natl. Acad. Sci. U.S.A. 101, 284–289. doi: 10.1073/pnas.2635903100
Reiman, E. M., Chen, K., Alexander, G. E., Caselli, R. J., Bandy, D., Osborne, D., et al. (2005). Correlations between apolipoprotein E ε4 gene dose and brain-imaging measurements of regional hypometabolism. Proc. Natl. Acad. Sci. U.S.A. 102, 8299–8302. doi: 10.1073/pnas.0500579102
Riedel, G., Platt, B., and Micheau, J. (2003). Glutamate receptor function in learning and memory. Behav. Brain Res. 140, 1–47. doi: 10.1016/s0166-4328(02)00272-3
Rothman, S. M., and Olney, J. W. (1986). Glutamate and the pathophysiology of hypoxic–ischemic brain damage. Ann. Neurol. 19, 105–111. doi: 10.1002/ana.410190202
Rulifson, I. C., Cao, P., Miao, L., Kopecky, D., Huang, L., White, R. D., et al. (2016). Identification of human islet amyloid polypeptide as a BACE2 substrate. PLoS One 11:e0147254. doi: 10.1371/journal.pone.0147254
Russell, R. L., Siedlak, S. L., Raina, A. K., Bautista, J. M., Smith, M. A., and Perry, G. (1999). Increased neuronal Glucose-6-phosphate Dehydrogenase and Sulfhydryl levels indicate reductive compensation to oxidative stress in Alzheimer disease. Archiv. Biochem. Biophys. 370, 236–239. doi: 10.1006/abbi.1999.1404
Ruthirakuhan, M. T., Herrmann, N., Abraham, E. H., Chan, S., and Lanctot, K. L. (2018). Pharmacological interventions for apathy in Alzheimer’s disease. Cochrane Database Syst. Rev. 5:Cd012197.
Saab, A. S., Tzvetavona, I. D., Trevisiol, A., Baltan, S., Dibaj, P., Kusch, K., et al. (2016). Oligodendroglial NMDA receptors regulate glucose import and axonal energy metabolism. Neuron 91, 119–132. doi: 10.1016/j.neuron.2016.05.016
Salameh, T. S., Shah, G. N., Price, T. O., Hayden, M. R., and Banks, W. A. (2016). Blood-brain barrier disruption and neurovascular unit dysfunction in diabetic mice: protection with the mitochondrial carbonic Anhydrase inhibitor Topiramate. J. Pharmacol. Exp. Ther. 359, 452–459. doi: 10.1124/jpet.116.237057
Sancheti, H., Akopian, G., Yin, F., Brinton, R. D., Walsh, J. P., and Cadenas, E. (2013). Age-dependent modulation of synaptic plasticity and insulin mimetic effect of lipoic acid on a mouse model of Alzheimer’s disease. PLoS One 8:e69830. doi: 10.1371/journal.pone.069830
Sarkar, S., Malovic, E., and Harishchandra, D. S. (2017). Mitochondrial impairment in microglia amplifies NLRP3 inflammasome proinflammatory signaling in cell culture and animal models of Parkinson’s disease. NPJ Parkinsons Dis. 3:30.
Sarlus, H., and Heneka, M. T. (2017). Microglia in Alzheimer’s disease. J. Clin. Invest. 127, 3240–3249.
Sbodio, J. I., Snyder, S. H., and Paul, B. D. (2019). Redox mechanisms in neurodegeneration: from disease outcomes to therapeutic opportunities. Antioxid. Redox Signal. 30, 1450–1499. doi: 10.1089/ars.2017.7321
Schafer, F. Q., and Buettner, G. R. (2001). Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic. Biol. Med. 30, 1191–1212. doi: 10.1016/s0891-5849(01)00480-4
Scheltens, P., Blennow, K., Breteler, M. M., de Strooper, B., Frisoni, G. B., Salloway, S., et al. (2016). Alzheimer’s disease. Lancet 388, 505–517.
Shen, Y., Kapfhamer, D., Minnella, A. M., Kim, J. E., Won, S. J., Chen, Y., et al. (2017). Bioenergetic state regulates innate inflammatory responses through the transcriptional co-repressor CtBP. Nat. Commun. 8:624.
Sheu, K. F., Cooper, A. J., Koike, K., Koike, M., Lindsay, J. G., and Blass, J. P. (1994). Abnormality of the alpha-ketoglutarate dehydrogenase complex in fibroblasts from familial Alzheimer’s disease. Ann. Neurol. 35, 312–318. doi: 10.1002/ana.410350311
Sheu, K. F., Kim, Y. T., Blass, J. P., and Weksler, M. E. (1985). An immunochemical study of the pyruvate dehydrogenase deficit in Alzheimer’s disease brain. Ann. Neurol. 17, 444–449. doi: 10.1002/ana.410170505
Sheu, K.-F. R., Clarke, D. D., Kim, Y.-T., Blass, J. P., Harding, B. J., and DeCicco, J. (1988). Studies of transketolase abnormality in Alzheimer’s disease. Archiv. Neurol. 45, 841–845. doi: 10.1001/archneur.1988.00520320027010
Simpson, I. A., Vannucci, S. J., and Maher, F. (1994). Glucose transporters in mammalian brain. Biochem. Soc. Trans. 22, 671–675. doi: 10.1042/bst0220671
Slotkin, T. A., Seidler, F. J., Crain, B. J., Bell, J. M., Bissette, G., and Nemeroff, C. B. (1990). Regulatory changes in presynaptic cholinergic function assessed in rapid autopsy material from patients with Alzheimer disease: implications for etiology and therapy. Proc. Natl. Acad. Sci. U.S.A. 87, 2452–2455. doi: 10.1073/pnas.87.7.2452
Slowik, A., Lammerding, L., Zendedel, A., Habib, P., and Beyer, C. (2018). Impact of steroid hormones E2 and P on the NLRP3/ASC/Casp1 axis in primary mouse astroglia and BV-2 cells after in vitro hypoxia. J. Steroid Biochem. Mol. Biol. 183, 18–26. doi: 10.1016/j.jsbmb.2018.05.003
Small, G. W., Kepe, V., and Barrio, J. R. (2006). Seeing is believing: neuroimaging adds to our understanding of cerebral pathology. Curr. Opin. Psychiatry 19, 564–569. doi: 10.1097/01.yco.0000245747.53008.e2
Sola-Penna, M., Paixao, L. P., Branco, J. R., Ochioni, A. C., Albanese, J. M., Mundim, D. M., et al. (2019). Serotonin activates glycolysis and mitochondria biogenesis in human breast cancer cells through activation of the Jak1/STAT3/ERK1/2 and adenylate cyclase/PKA, respectively. Br. J. Cancer 122, 194–208. doi: 10.1038/s41416-019-0640-1
Son, M. J., Kwon, Y., Son, T., and Cho, Y. S. (2016). Restoration of mitochondrial NAD(+) levels delays stem cell senescence and facilitates reprogramming of aged somatic cells. Stem Cells 34, 2840–2851. doi: 10.1002/stem.2460
Song, X. N., Zhang, L. Q., Liu, D. G., Lin, J., Zheng, J. D., Dai, D. P., et al. (2011). Oxidative damage to RNA and expression patterns of MTH1 in the hippocampi of senescence-accelerated SAMP8 mice and Alzheimer’s disease patients. Neurochem. Res. 36, 1558–1565. doi: 10.1007/s11064-011-0484-4
Soucek, T., Cumming, R., Dargusch, R., Maher, P., and Schubert, D. (2003). The regulation of glucose metabolism by HIF-1 mediates a neuroprotective response to Amyloid beta peptide. Neuron 39, 43–56. doi: 10.1016/s0896-6273(03)00367-2
Spilman, P., Podlutskaya, N., Hart, M. J., Debnath, J., Gorostiza, O., Bredesen, D., et al. (2010). Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer’s disease. PLoS One 5:e9979. doi: 10.1371/journal.pone.009979
Steen, E., Terry, B. M., Rivera, E. J., Cannon, J. L., Neely, T. R., Tavares, R., et al. (2005). Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease–is this type 3 diabetes? J. Alzheimers Dis. 7, 63–80. doi: 10.3233/jad-2005-7107
Storozhevykh, T. P., Senilova, Y. E., Persiyantseva, N. A., Pinelis, V. G., and Pomytkin, I. A. (2007). Mitochondrial respiratory chain is involved in insulin-stimulated hydrogen peroxide production and plays an integral role in insulin receptor autophosphorylation in neurons. BMC Neurosci. 8:84. doi: 10.1186/1471-2202-8-84
Sultana, R., Perluigi, M., and Butterfield, D. A. (2013). Lipid peroxidation triggers neurodegeneration: a redox proteomics view into the Alzheimer disease brain. Free Radic. Biol. Med. 62, 157–169. doi: 10.1016/j.freeradbiomed.2012.09.027
Sultana, R., Piroddi, M., Galli, F., and Butterfield, D. A. (2008). Protein levels and activity of some antioxidant enzymes in hippocampus of subjects with Amnestic mild cognitive impairment. Neurochem. Res. 33, 2540–2546. doi: 10.1007/s11064-008-9593-0
Sun, X., He, G., Qing, H., Zhou, W., Dobie, F., Cai, F., et al. (2006). Hypoxia facilitates Alzheimer’s disease pathogenesis by up-regulating BACE1 gene expression. Proc. Natl. Acad. Sci. U.S.A. 103, 18727–18732. doi: 10.1073/pnas.0606298103
Swerdlow, R. H. (2007). Treating neurodegeneration by modifying mitochondria: potential solutions to a “complex” problem. Antioxid. Redox Signal. 9, 1591–1603. doi: 10.1089/ars.2007.1676
Swerdlow, R. H. (2011). Brain aging, Alzheimer’s disease, and mitochondria. Biochim. Biophys. Acta 1812, 1630–1639.
Taguchi, A., Wartschow, L. M., and White, M. F. (2007). Brain IRS2 signaling coordinates life span and nutrient homeostasis. Science 317, 369–372. doi: 10.1126/science.1142179
Takahashi, M., Miyata, H., Kametani, F., Nonaka, T., Akiyama, H., Hisanaga, S.-I., et al. (2015). Extracellular association of APP and tau fibrils induces intracellular aggregate formation of tau. Acta Neuropathol. 129, 895–907. doi: 10.1007/s00401-015-1415-2
Tamaki, C., Ohtsuki, S., and Terasaki, T. (2007). Insulin facilitates the hepatic clearance of plasma amyloid beta-peptide (LABEL:1_40) by intracellular translocation of low-density lipoprotein receptor-related protein 1 (LRP-1) to the plasma membrane in hepatocytes. Mol. Pharmacol. 72, 850–855. doi: 10.1124/mol.107.036913
Tan, C. C., Yu, J. T., Wang, H. F., Tan, M. S., Meng, X. F., Wang, C., et al. (2014). Efficacy and safety of donepezil, galantamine, rivastigmine, and memantine for the treatment of Alzheimer’s disease: a systematic review and meta-analysis. J. Alzheimer Dis. 41, 615–631. doi: 10.3233/jad-132690
Tang, B. L. (2019). Neuroprotection by glucose-6-phosphate dehydrogenase and the pentose phosphate pathway. J. Cell. Biochem. 120, 14285–14295. doi: 10.1002/jcb.29004
Tanti, J. F., and Jager, J. (2009). Cellular mechanisms of insulin resistance: role of stress-regulated serine kinases and insulin receptor substrates (IRS) serine phosphorylation. Curr. Opin. Pharmacol. 9, 753–762. doi: 10.1016/j.coph.2009.07.004
Tiritilli, A. (2000). 5-hydroxytryptamine induces vasoconstriction of the human umbilical artery: effects of hypoxia and nicorandil. Gynecol. Obstetr. Invest. 50, 77–83. doi: 10.1159/000010286
Tisdale, E. J. (2002). Glyceraldehyde-3-phosphate dehydrogenase is phosphorylated by protein kinase Ciota/lambda and plays a role in microtubule dynamics in the early secretory pathway. J. Biol. Chem. 277, 3334–3341. doi: 10.1074/jbc.m109744200
Tiwari, V., and Patel, A. B. (2012). Impaired glutamatergic and GABAergic function at early age in AbetaPPswe-PS1dE9 mice: implications for Alzheimer’s disease. J. Alzheimers Dis. 28, 765–769. doi: 10.3233/jad-2011-111502
Tsai, C. W., Tsai, C. F., Lin, K. H., Chen, W. J., and Lin, M. S. (2020). An investigation of the correlation between the S-glutathionylated GAPDH levels in blood and Alzheimer’s disease progression. PLoS One 15:e0233289. doi: 10.1371/journal.pone.0233289
Ulland, T. K., Song, W. M., Huang, S. C., Ulrich, J. D., Sergushichev, A., Beatty, W. L., et al. (2017). TREM2 maintains microglial metabolic fitness in Alzheimer’s disease. Cell 170, 649–663.e13.
Vallee, A., Lecarpentier, Y., Guillevin, R., and Vallee, J. N. (2018a). Reprogramming energetic metabolism in Alzheimer’s disease. Life Sci. 193, 141–152. doi: 10.1016/j.lfs.2017.10.033
Vallee, A., Lecarpentier, Y., Guillevin, R., and Vallee, J. N. (2018b). Thermodynamics in neurodegenerative diseases: interplay between canonical WNT/Beta-catenin pathway-PPAR gamma, energy metabolism and circadian rhythms. Neuromol. Med. 20, 174–204. doi: 10.1007/s12017-018-8486-x
Vallee, A., Lecarpentier, Y., Guillevin, R., and Vallee, J. N. (2017). Effects of cannabidiol interactions with Wnt/beta-catenin pathway and PPARgamma on oxidative stress and neuroinflammation in Alzheimer’s disease. Acta Biochim. Biophys. Sin. 49, 853–866. doi: 10.1093/abbs/gmx073
van Gijsel-Bonnello, M., Baranger, K., Benech, P., Rivera, S., Khrestchatisky, M., de Reggi, M., et al. (2017). Metabolic changes and inflammation in cultured astrocytes from the 5xFAD mouse model of Alzheimer’s disease: alleviation by pantethine. PLoS One 12:e0175369. doi: 10.1371/journal.pone.0175369
Varma, V. R., Oommen, A. M., Varma, S., Casanova, R., An, Y., Andrews, R. M., et al. (2018). Brain and blood metabolite signatures of pathology and progression in Alzheimer disease: a targeted metabolomics study. PLoS Med. 15:e1002482. doi: 10.1371/journal.pone.1002482
Verdile, G., Keane, K. N., Cruzat, V. F., Medic, S., Sabale, M., Rowles, J., et al. (2015). Inflammation and oxidative stress: the molecular connectivity between insulin resistance, obesity, and Alzheimer’s disease. Med. Inflamm. 2015:105828.
Vilalta, A., and Brown, G. C. (2014). Deoxyglucose prevents neurodegeneration in culture by eliminating microglia. J. Neuroinflamm. 11:58. doi: 10.1186/1742-2094-11-58
Viswanathan, A., and Greenberg, S. M. (2011). Cerebral amyloid angiopathy in the elderly. Ann. Neurol. 70, 871–880.
Wang, J., Xiong, S., Xie, C., Markesbery, W. R., and Lovell, M. A. (2005). Increased oxidative damage in nuclear and mitochondrial DNA in Alzheimer’s disease. J. Neurochem. 93, 953–962. doi: 10.1111/j.1471-4159.2005.03053.x
Wilkins, H. M., Koppel, S. J., Bothwell, R., Mahnken, J., Burns, J. M., and Swerdlow, R. H. (2017). Platelet cytochrome oxidase and citrate synthase activities in APOE epsilon4 carrier and non-carrier Alzheimer’s disease patients. Redox Biol. 12, 828–832. doi: 10.1016/j.redox.2017.04.010
Wolf, A. J., Reyes, C. N., Liang, W., Becker, C., Shimada, K., Wheeler, M. L., et al. (2016). Hexokinase is an innate immune receptor for the detection of bacterial peptidoglycan. Cell 166, 624–636. doi: 10.1016/j.cell.2016.05.076
Wolfe, C. M., Fitz, N. F., Nam, K. N., Lefterov, I., and Koldamova, R. (2018). The role of APOE and TREM2 in Alzheimer’s disease-current understanding and perspectives. Intern. J. Mol. Sci. 20:81. doi: 10.3390/ijms20010081
Womack, K. B., Diaz-Arrastia, R., Aizenstein, H. J., Arnold, S. E., Barbas, N. R., Boeve, B. F., et al. (2011). Temporoparietal hypometabolism in frontotemporal lobar degeneration and associated imaging diagnostic errors. Arch. Neurol. 68, 329–337.
Wu, L., Zhang, X., and Zhao, L. (2018). Human ApoE isoforms differentially modulate brain glucose and Ketone body metabolism: implications for Alzheimer’s disease risk reduction and early intervention. J. Neurosci. 38, 6665–6681. doi: 10.1523/jneurosci.2262-17.2018
Yan, X., Shi, Z. F., Xu, L. X., Li, J. X., Wu, M., Wang, X. X., et al. (2017). Glutamate impairs mitochondria aerobic respiration capacity and enhances Glycolysis in cultured rat Astrocytes. Biomed. Environ. Sci. 30, 44–51.
Yao, J., Irwin, R. W., Zhao, L., Nilsen, J., Hamilton, R. T., and Brinton, R. D. (2009). Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 106, 14670–14675. doi: 10.1073/pnas.0903563106
Yin, F., Boveris, A., and Cadenas, E. (2014). Mitochondrial energy metabolism and redox signaling in brain aging and neurodegeneration. Antioxid. Redox Signal. 20, 353–371. doi: 10.1089/ars.2012.4774
Yin, F., Sancheti, H., and Cadenas, E. (2012). Silencing of nicotinamide nucleotide transhydrogenase impairs cellular redox homeostasis and energy metabolism in PC12 cells. Biochim. Biophys. Acta 1817, 401–409. doi: 10.1016/j.bbabio.2011.12.004
Yin, F., Sancheti, H., Patil, I., and Cadenas, E. (2016). Energy metabolism and inflammation in brain aging and Alzheimer’s disease. Free Radic. Biol. Med. 100, 108–122.
Yoon, M. S. (2017). The role of mammalian target of Rapamycin (mTOR) in insulin signaling. Nutrients 9:1176. doi: 10.3390/nu9111176
Young, A. J., Johnson, S., Steffens, D. C., and Doraiswamy, P. M. (2007). Coenzyme Q10: a review of its promise as a neuroprotectant. CNS Spectr. 12, 62–68. doi: 10.1017/s1092852900020538
Yu, Q., Liu, H., Sang, S., Chen, L., Zhao, Y., Wang, Y., et al. (2018). Thiamine deficiency contributes to synapse and neural circuit defects. Biol. Res. 51:35.
Zhao, N., Liu, C. C., Qiao, W., and Bu, G. (2018). Apolipoprotein E, receptors, and modulation of Alzheimer’s disease. Biol. Psychiatry 83, 347–357.
Zheng, Y., Zhang, X., Chen, J., Zhou, Q., and Gao, H. (2018). Metabonomics studies of urine from APP/PS1 mice with early-stage Alzheimer’s disease. Zhejiang Da Xue Xue Bao Yi Xue Ban 47, 636–642.
Zhong, Z., Liang, S., Sanchez-Lopez, E., He, F., Shalapour, S., Lin, X. J., et al. (2018). New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 560, 198–203. doi: 10.1038/s41586-018-0372-z
Zhou, L., Gao, Q., Nie, M., Gu, J. L., Hao, W., Wang, L., et al. (2016). Degeneration and energy shortage in the suprachiasmatic nucleus underlies the circadian rhythm disturbance in ApoE(-/-) mice: implications for Alzheimer’s disease. Sci. Rep. 6:36335.
Zhou, Q., Zheng, H., Chen, J., Li, C., Du, Y., Xia, H., et al. (2018). Metabolic fate of glucose in the brain of APP/PS1 transgenic mice at 10 months of age: a (13)C NMR metabolomic study. Metab. Brain Dis. 33, 1661–1668. doi: 10.1007/s11011-018-0274-7
Keywords: Alzheimer’s disease, glucose metabolism dysregulation, glycolysis dysfunction, TCA cycle and OXPHOS deficits, pentose phosphate pathway impairment
Citation: Yan X, Hu Y, Wang B, Wang S and Zhang X (2020) Metabolic Dysregulation Contributes to the Progression of Alzheimer’s Disease. Front. Neurosci. 14:530219. doi: 10.3389/fnins.2020.530219
Received: 28 January 2020; Accepted: 25 September 2020;
Published: 05 November 2020.
Edited by:
Kristine Freude, University of Copenhagen, DenmarkReviewed by:
Natalia P. Rocha, University of Texas Health Science Center at Houston, United StatesGhulam Md Ashraf, King Abdulaziz University, Saudi Arabia
Tim Huang, Sanford Burnham Prebys Medical Discovery Institute, United States
LuLin Jiang, Sanford Burnham Prebys Medical Discovery Institute, United States
Charlene Supnet, University of Texas Southwestern Medical Center, United States
Copyright © 2020 Yan, Hu, Wang, Wang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xinwen Zhang, zhangxinwen@cmu.edu.cn
†These authors have contributed equally to this work