Activin Signaling in the Pathogenesis and Therapy of Neuropsychiatric Diseases
 Andrea S. Link
Andrea S. Link  Christian Alzheimer
Christian Alzheimer- Institute of Physiology and Pathophysiology, Friedrich-Alexander-Universität Erlangen-Nürnberg, Erlangen, Germany
Activins are members of the transforming growth factor β (TGFβ) family and serve as multifunctional regulatory proteins in many tissues and organs. In the brain, activin A, which is formed by two disulfide-linked βA subunits, is recognized as the predominant player in activin signaling. Over the last years, considerable progress has been made in elucidating novel and unexpected functions of activin in the normal and diseased brain and in deciphering the underlying molecular mechanisms. Initially identified as a neurotrophic and protective factor during development and in several forms of acute injury, the scope of effects of activin A in the adult central nervous system (CNS) has been considerably broadened by now. Here, we will highlight recent findings that bear significance for a better understanding of the pathogenesis of various neuropsychiatric diseases and might hold promise for novel therapeutic strategies. While the basal level of activin A in the adult brain is low, significant short-term up-regulation occurs in response to increased neuronal activity. In fact, brief exposure to an enriched environment (EE) is already sufficient to considerably strengthen activin signaling. Enhancement of this pathway tunes the performance of glutamatergic and GABAergic synapses in a fashion that impacts on cognitive functions and affective behavior, counteracts death-inducing signals through extrasynaptic NMDA receptors (NMDARs), and stimulates adult neurogenesis in the hippocampus. We will discuss how impaired activin signaling is involved in anxiety disorders, depression, drug dependence, and neurodegenerative diseases such as Alzheimer’s and Parkinson’s, and how reinforcement of activin signaling might be exploited for therapeutic interventions.
Introduction
Activins are members of the transforming growth factor β (TGFβ) family of growth and differentiation factors. They serve as multifunctional regulatory proteins in many tissues and organ systems, with particular emphasis on proliferation, differentiation, apoptosis, inflammation, immunoregulation and repair (Werner and Alzheimer, 2006; Hedger et al., 2011). Structurally, activins are homo- or heterodimeric proteins containing two disulfide-linked βA (encoded by the Inhba gene) and/or βB (encoded by the Inhbb gene) subunits. After proteolytic processing of their precursor proteins, activins are secreted as mature bioactive proteins. Activin A (βA/βA) is the most abundant and best characterized member of the activin family and plays the predominant role in activin signaling in the central nervous system (CNS). In the developing brain, activin A exerts distinct neurotrophic effects and is involved in proper cortical layering and corticostriatal wiring (Andreasson and Worley, 1995). Importantly, a recent study provided evidence that activin A supports neuronal differentiation of cortical neuronal progenitor cells (Rodríguez-Martinez et al., 2012). In the adult brain, activin A was originally identified as a neuroprotective factor in various forms of acute brain injury, including stroke (Wu et al., 1999; Tretter et al., 2000; Mukerji et al., 2009). In addition to its release from neurons, activin A can be also of glial origin, as demonstrated in a CNS demyelination model, where M2 microglia-derived activin A promotes oligodendrocyte differentiation and remyelination (Miron et al., 2013). The observation that the expression of activin A is strongly up-regulated in response to brain lesion and that both endogenous and recombinant activin A are capable of affording neuroprotection, led originally to the concept that, in the adult brain, activin A is a neuroprotective factor that is called to arms in emergency situations, when an initial damaging event, such as stroke, threatens to cause a massive neuronal loss.
Over the last decade, this somewhat narrow view on the role of activin has been substantially revised and extended. Our current understanding of the functions of activin A in the normal and diseased brain encompasses an astonishingly broad spectrum. On the physiological side of this range, activin regulates the daily operations of central synapses in a behaviorally relevant fashion. Major findings were that: (i) activin enhances cognitive performance by augmenting synaptic plasticity of excitatory (glutamatergic) synapses; and that (ii) by tuning inhibitory (GABAergic) synapses, activin sets the level of anxiety-like behavior. These findings are testimony to the impact of activin on mental faculties as well as on affective behavior. Since the effects of activin on central synapses of the healthy adult brain and their behavioral consequences have been reviewed elsewhere (Krieglstein et al., 2011), we will focus here on the role of activin signaling in neuropsychiatric disorders.
Activin Signaling
The activin signaling pathway is schematically depicted in Figure 1. Activins signal through heteromeric complexes of type II (ActRIIA, ActRIIB) and type I receptors (predominantly ActRIB, but also ActRIA and ActRIC). Type II receptors bind activin and recruit type I receptors, which then phosphorylate the intracellular signaling proteins SMAD2/3. These assemble with SMAD4, translocate to the nucleus, and bind to specific target genes to modulate their expression (Chen et al., 2006; Xia and Schneyer, 2009). It is important to stress that the strength and duration of activin signaling are tightly controlled at several nodes along the signaling pathway (Choi and Han, 2011). In the schematic drawing of Figure 1, negative regulators of activin signaling are colored in red. Well-known inhibitors are the secreted proteins follistatin and inhibin, which preclude activin receptor binding through direct physical interaction and receptor competition, respectively. Betaglycan promotes the interaction between inhibin and ActRII, thereby counteracting activin binding and signaling (Hedger et al., 2011). In the cell membrane, the pseudoreceptor BAMBI (BMP and activin membrane-bound inhibitor) inhibits transmission in a ligand-independent manner. PMEPA1 (prostate transmembrane protein, androgen induced 1) directly binds to SMAD2/3 and thus prevents their phosphorylation by activated type I receptors. Notably, PMEPA1 was recently identified as an activin target gene in the brain (Link et al., 2015), suggesting that it might serve as a feed-back inhibitor of activin signaling. The intracellular inhibitor SMAD7 competitively blocks SMAD2/3 binding sites at type I receptors. In the nucleus, the transcriptional co-factors SKI, SKIL and TGIF2 act as transcriptional repressors. In addition to this canonical signaling pathway, activins can also activate other pathways, including mitogen-activated protein kinase (MAPK) signaling (Moustakas and Heldin, 2005).
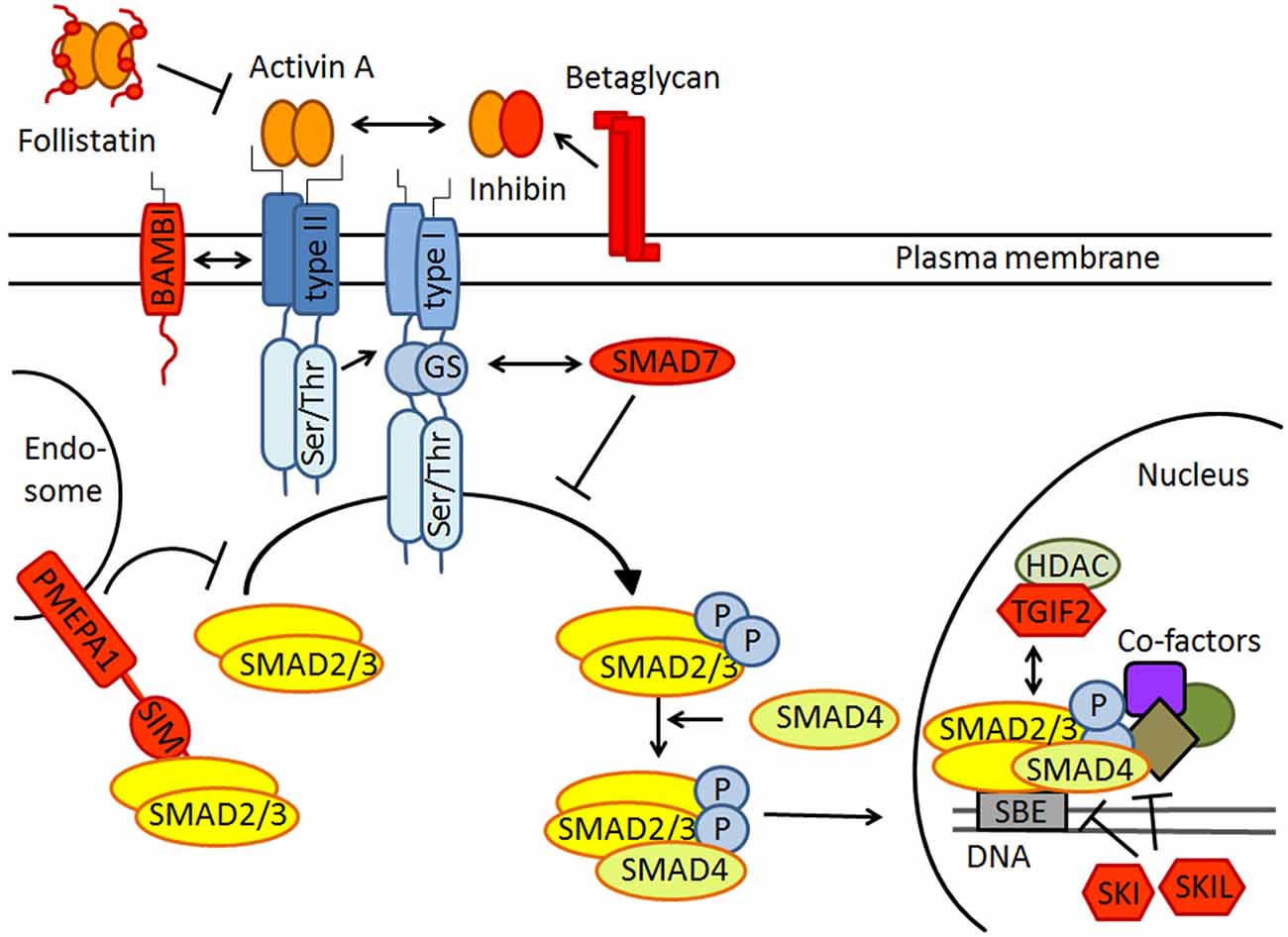
Figure 1. Schematic drawing of canonical activin receptor signaling via SMAD2/3 proteins. Note that SMAD2/3 signaling is strictly regulated by extracellular inhibitors like follistatin and inhibin, transmembrane antagonists such as BAMBI and PMEPA1, the cytoplasmatic inhibitory SMAD7 and several transcriptional repressors including SKI, SKIL and TGIF2 in the nucleus. All inhibitors are colored in red. For further explanation see text.
Activin Signaling in Conditioned Fear, Anxiety, and Drug Dependence
Although anxiety and fear both describe forms of affective behavior, which occur in anticipation of threatening stimuli, be they real or envisioned, it is conceptually helpful to make a distinction between these terms. We will refer to fear responses as conditioned behaviors elicited by a particular cue or environmental context. By contrast, anxiety manifests itself as a form of behavioral inhibition in conditions of conflict, when, for example, a novel, but potentially threatening object or environment requires a choice between approach vs. avoidance. Conditioned fear has been linked to long-term synaptic plasticity of glutamatergic synapses in the amygdala (Pape and Pare, 2010), with the contextual information provided by the hippocampus. Anxiety-like behavior appears to be primarily controlled by the (ventral) hippocampus (Bannerman et al., 2004). As will be detailed in the next section, activin signaling impinges on both, conditioned fear and anxiety-like behavior.
Conditioned Fear
Ageta et al. (2010) generated forebrain-specific activin- or follistatin-transgenic mice in which the transgene expression was regulated by the Tet-OFF system, allowing temporally controlled over-expression of activin or its inhibitor follistatin. If already over-expressed during training in a contextual fear-conditioning test, follistatin reduced freezing behavior tested 24 h later. When follistatin or activin were over-expressed after training, reconsolidation of fear memory was weakened or strengthened, respectively (Ageta et al., 2010; Ageta and Tsuchida, 2011). The finding that inhibition of activin signaling during memory retrieval is capable of suppressing previously consolidated fear memories is of particular translational interest, because it advances activin signaling as a putative therapeutic target to extinguish traumatic memories and fear-laden stimuli in human anxiety disorders such as posttraumatic stress disorder (PTSD) and phobias.
How does activin signaling interfere with acquisition, retrieval and extinction of conditioned fear? In view of the dependence of contextual fear conditioning on synaptic plasticity in amygdalar and hippocampal circuits, the known effects of activin signaling on hippocampal long-term potentiation (LTP) offer a plausible clue. Andreasson and Worley (1995) were the first to demonstrate up-regulation of activin βA mRNA in the dentate gyrus (DG) in vivo in response to LTP-inducing electrical stimulation of the perforant path. Underscoring the functionality of this up-regulation, later studies found that forebrain-specific disruption of activin receptor signaling and over-expression of follistatin impaired LTP in area CA1 and DG (Muller et al., 2006; Ageta et al., 2010). Mechanistically, activin augments LTP by targeting several of its essential mechanisms, including NMDA receptor (NMDAR) currents and spine density and morphology (Muller et al., 2006; Shoji-Kasai et al., 2007; Kurisaki et al., 2008; Hasegawa et al., 2014).
Anxiety, Alcohol and GABAA Receptors
Transgenic mice expressing a dominant-negative ActRIB in forebrain neurons display a low-anxiety phenotype in typical conflict paradigms in which the innate drive to explore a novel environment is opposed by the tendency to stay in safe surroundings (Zheng et al., 2009). The decrease in anxiety-like behavior in these mice has been linked to concomitant alterations in GABAergic inhibition, based on a number of conspicuous parallels: (i) Tonic inhibition by extrasynaptic GABAA receptors (GABAARs), which is thought to exert anxiolytic effects (Brickley and Mody, 2012; Whissell et al., 2015), is enhanced in the mutant mice; (ii) GABAARs of the mutant mice show substantially less sensitivity to diazepam, concomitant with a loss of the anxiolytic drug effect at the behavioral level; and (iii) GABABR function is augmented in the transgenic mice, consistent with reports that positive GABABR modulators reduce anxiety-like behavior (Zheng et al., 2009).
Given that disruption of activin receptor signaling reduces allosteric modulation of GABAARs by diazepam, it came as a surprise that ethanol potentiation of GABAAR-mediated inhibitory synaptic transmission was enhanced in the same transgenic mice (Zheng et al., 2015). As a consequence, low concentrations of ethanol (≤30 mM) are now capable of potentiating the inhibitory effects of synaptic GABAARs. Loss of neuronal activin receptor signaling renders not only GABAARs more sensitive to ethanol, but also produces significantly stronger sedation at the behavioral level (Zheng et al., 2015). These data suggest that activin regulates behavioral effects of ethanol by controlling ethanol potentiation of GABAAR function, arguably the pre-eminent target of ethanol in the brain. At the molecular level, activin appears to adjust ethanol sensitivity of GABAARs through a non-canonical, i.e., SMAD2/3-independent signaling pathway involving PKCε (Zheng et al., 2015). Notably, this PKC isoform has been previously shown to regulate the responsiveness of GABAARs to ethanol (Hodge et al., 1999).
Cocaine Craving
While activin is involved in the sedating, but not in the reinforcing effects of ethanol (Zheng et al., 2015), a recent study implicated activin signaling in maladaptive processes leading to drug craving and relapse after cocaine withdrawal (Gancarz et al., 2015). After 7 days, but not after 1 day of withdrawal following cocaine self-administration in rats, phosphorylation of SMAD3 was specifically increased in the shell of the nucleus accumbens (NAc). Intra-accumbal microinjection of activin A increased drug self-administration, whereas viral over-expression of a dominant-negative mutant of SMAD3 (dnSMAD3) in the NAc had the opposite effect. Moreover, dnSMAD3 abrogated the increase in the density of dendritic spines on medium spiny neurons in the NAc, which typically accompanies drug-induced reinstatement of cocaine self-administration (Gancarz et al., 2015). Such structural plasticity following withdrawal is considered essential for the endurance of cocaine-seeking behavior, and activin signaling appears to be a driving force behind this rewiring process. As pointed out earlier, changes in spine morphology have been also proposed as a mechanism through which activin promotes hippocampal LTP and memory formation. This leaves us with the intriguing idea that, depending on whether it is recruited endogenously (e.g., by learning processes in the hippocampus) or hijacked exogenously (e.g., by drugs of abuse in the NAc), activin signaling can operate both ways, increasing cognitive performance as well as instituting drug-seeking behavior and relapse.
Activin Signaling and Depression
Activin Signaling as a Target in Antidepressant Treatment
Given the co-morbidity of mood and anxiety disorders, it should not come unexpected that activin signaling has also been assigned a role in depression. Specifically, several proteins of the activin signaling pathway were found to be up-regulated in rodent brain after chronic administration of clinically used antidepressants. Chronic and subchronic paroxetine produced an increased expression of Inhba mRNA and of ActRIA mRNA in circumscribed regions of mouse hippocampus (Ganea et al., 2012). While not influencing Inhba expression, chronic fluoxetine and desipramine did increase SMAD2 phosphorylation in rat frontal cortex (Dow et al., 2005). Furthermore, genetic polymorphism in TGFBR3 which codes for betaglycan, a functional inhibitor of activin binding and signaling (v.s.), has been associated with the clinical response to antidepressant drugs (Ganea et al., 2012). Electroconvulsive therapy (ECT), the second mainstay of antidepressant treatment, engenders a strong and rapid (within 2 h) increase of Inhba mRNA in hippocampus, amygdala and several neocortical areas, followed by enhanced SMAD2 phosphorylation, indicating subsequent activation of the canonical activin signaling pathway in this rodent model of ECT (Dow et al., 2005; Link et al., 2015). Compared to other members of the TGFβ family, which also signal via SMAD2/3, only Inhba expression was significantly increased, strongly suggesting that ECT gives rise exclusively to activin A protein (Link et al., 2015). Taken by themselves, these findings do not necessarily indicate a role of activin as a mediator of the therapeutic effects of antidepressant drugs or ECT. It was therefore reassuring that stereotactic infusion of recombinant activin A into rat and mouse hippocampus produced antidepressant-like effects in the forced swim test, a behavioral model of depression (Dow et al., 2005; Ganea et al., 2012). Notably, the antidepressant-like effects were only observed when activin A was injected into the DG, but not when injected into the CA1 region or into the amygdala (Dow et al., 2005; Ganea et al., 2012).
Activin, Depression, Adult Neurogenesis and Epigenetic Modification
The DG clearly emerges as the essential brain region linking activin signaling to its antidepressant effects. In view of the (not undisputed) neurogenic hypothesis of depression, which posits that adult-generated neurons of the DG are required for balanced mood and antidepressant efficacy (Eisch and Petrik, 2012), an obvious question is whether the therapeutic effects of activin can be causally related to increased adult neurogenesis. Whereas it remains controversial whether the basal level of endogenous activin in the intact adult brain is sufficient to influence baseline neurogenesis, administration of activin A to the normal hippocampus did enhance neural stem/precursor cell proliferation (Ageta et al., 2008; Abdipranoto-Cowley et al., 2009). These findings would be consistent with the concept that the dramatic up-regulation of activin by antidepressant treatment contributes to disease remission by promoting hippocampal neurogenesis.
With the identification of the histone demethylase KDM6B as the product of an activin target gene in the brain, epigenetic regulation might emerge as a new effect of activin signaling that could contribute to its antidepressant effects (Link et al., 2015). According to this hypothesis, enhanced activin signaling following ECT or antidepressant pharmacotherapy would lead to chromatin remodeling and increased transcriptional activity as schematically depicted in Figure 2.
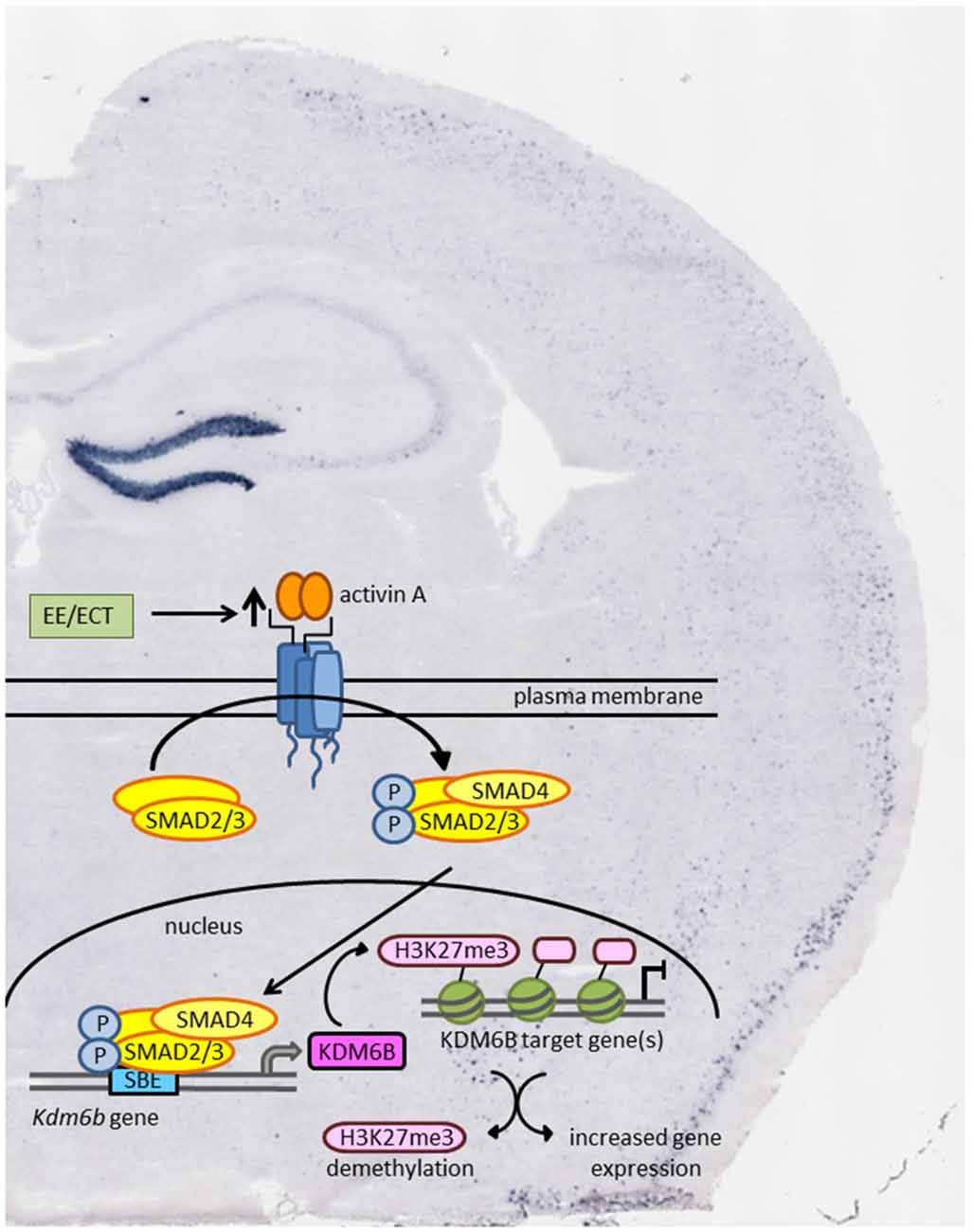
Figure 2. Schematic model of how environmental enrichment (EE) and electroconvulsive therapy (ECT) may produce epigenetic modification through enhanced activin signaling and transcription of the activin target gene Kdm6b. KDM6B acts as a histone demethylase thereby promoting gene expression of transcriptionally repressed genes. Background: in situ hybridization of coronal mouse brain section showing pronounced increase in Inhba mRNA level in dentate gyrus (DG) 2 h after ECT-like electrical stimulation.
Finally, activin A might also serve as an endogenous antidepressant in its own right, independent of its up-regulation by antidepressant treatment. Strong support for this notion comes from experiments with mice placed into an enriched environment (EE). Compared to mice housed in standard cages, EE exposure produced a marked up-regulation of Inhba mRNA in the DG and the CA3 region of the hippocampus, accompanied by increased SMAD2/3 phosphorylation (Link et al., 2015). These findings suggest that the mood-elevating and cognition-enhancing effects attributed to EE might be mediated, at least in part, by enhanced activin signaling.
Activin and Neurodegenerative Diseases
In an excitotoxic rodent model of Huntington’s disease, intrastriatal administration of recombinant activin A for 1 week rescued striatal interneurons and projection neurons from death (Hughes et al., 1999). Administration of recombinant activin A also protected dopaminergic midbrain neurons against MPP+-induced cell death in vitro (Krieglstein et al., 1995) and in the 6-OHDA mouse model of Parkinson’s disease (Stayte et al., 2015). While these studies suggest that activin signaling might be targeted to fight off gradual neuronal demise in chronic neurodegenerative disease, the underlying mechanism had been elusive until recently. A widely held concept posits that a proper balance between signals transmitted by synaptic vs. extrasynaptic NMDARs is an essential determinant of neuronal integrity (Hardingham and Bading, 2010). Activation of synaptic NMDARs triggers synaptic plasticity and fosters genetic programs for stress resilience and survival. By contrast, activation of extrasynaptic NMDARs can incur lethal consequences for neurons, as it initiates pro-apoptotic pathways. Lau et al. (2015) have now revealed an intriguing interplay between brain-derived neurotrophic factor (BDNF) and activin signaling that is instrumental to shift the delicate balance between extrasynaptic and synaptic NMDARs in favor of the latter, thereby strengthening neuronal health and long-term survival. In brief, this survival-promoting scheme comprises the following sequence of events. Activity-dependent increase in Bdnf transcription or administration of exogenous BDNF promotes activation of synaptic NMDARs, thereby inducing nuclear calcium signaling, which in turn activates transcription of Inhba. Secreted activin A then reduces potentially neurotoxic Ca2+ influx through extrasynaptic NMDARs, thereby counteracting pro-death pathways (Lau et al., 2015). Based on such a scenario, it seems plausible to assume that any perturbation of the neuroprotective interaction between BDNF and activin A should render neurons more vulnerable to excitotoxic damage and might thus represent an important pathogenetic factor in protracted neurodegenerative diseases such as Huntington’s, Parkinson’s or Alzheimer’s.
Outlook
The characterization of Inhba as a BDNF-induced gene and the identification of the sequential and interdependent activation of BNDF and activin signaling as a health-promoting mechanism offer an intriguing new perspective on possibly shared functions of these two factors that might well go beyond their involvement in combating neuronal loss in neurodegenerative diseases. A quick review of the multiple roles of BDNF in the adult brain reveals a broad spectrum of effects that show a striking overlap with those of activin. Like activin, BDNF has been implicated in activity-dependent regulation of synaptic plasticity (Minichiello, 2009; Edelmann et al., 2014). In fact, the effects of BDNF bear a strong resemblance with those of activin in that both factors alter key morphological and electrophysiological features of glutamatergic synapses, thereby providing a double underpinning for neuronal processes intimately involved in learning and memory. Moreover, several lines of evidence show that BDNF, like activin, crucially modulates the strength of GABAergic inhibition (Gottmann et al., 2009). In addition to their involvement in anxiety disorders (Liu et al., 2004; Martinowich et al., 2007) and drug addiction (Li and Wolf, 2015), activin and BDNF appear to be endowed with antidepressant efficacy (Martinowich et al., 2007). Both signaling pathways are stimulated by EE and, much more so, by ECT, as well as by antidepressant drug treatment (Nibuya et al., 1995; Link et al., 2015). With BDNF and activin being up-regulated strongest in the DG, both factors were found to stimulate adult neurogenesis, which is thought to support remission of depression (Martinowich et al., 2007). Thus, several lines of evidence indicate that BDNF and activin synergistically co-regulate neuronal responses, suggesting essential cross-talk of their signaling pathways.
In contrast to BDNF, which has long since taken center stage as a master regulator of various neuronal properties and functions during development and in adulthood, the multiple roles of activin are only gradually being uncovered. It seems that, in many aspects, activin and BDNF are two players with very similar overall targets, which join forces in neuroprotection, synaptic plasticity and neuropsychiatric diseases. The commonalities and differences in the strategies these two factors pursue to achieve these goals and the extent to which the underlying mechanisms are complementary and interdependent should be exciting topics for future research.
Author Contributions
The manuscript was written by CA, with contributions of ASL and FZ. Schematic drawings were designed by ASL.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Dr. Sabine Werner, Institute of Molecular Health Sciences, ETH Zurich, for helpful discussions and comments on the manuscript. Research in our laboratory was supported by the Deutsche Forschungsgemeinschaft (DFG AL294/9, AL294/10 to CA and INST 90/675-1 FUGG to CA), the Johannes und Frieda Marohn-Stiftung (to FZ and CA), the Neurotrition Project of the FAU Emerging Field Initiative (to CA), the Dr. Ernst und Anita Bauer Stiftung (to ASL), and the Jürgen Manchot Stiftung (to ASL).
References
Abdipranoto-Cowley, A., Park, J. S., Croucher, D., Daniel, J., Henshall, S., Galbraith, S., et al. (2009). Activin A is essential for neurogenesis following neurodegeneration. Stem Cells 27, 1330–1346. doi: 10.1002/stem.80
Ageta, H., Ikegami, S., Miura, M., Masuda, M., Migishima, R., Hino, T., et al. (2010). Activin plays a key role in the maintenance of long-term memory and late-LTP. Learn. Mem. 17, 176–185. doi: 10.1101/lm.16659010
Ageta, H., Murayama, A., Migishima, R., Kida, S., Tsuchida, K., Yokoyama, M., et al. (2008). Activin in the brain modulates anxiety-related behavior and adult neurogenesis. PLoS One 3:e1869. doi: 10.1371/journal.pone.0001869
Ageta, H., and Tsuchida, K. (2011). Multifunctional roles of activins in the brain. Vitam. Horm. 85, 185–206. doi: 10.1016/B978-0-12-385961-7.00009-3
Andreasson, K., and Worley, P. F. (1995). Induction of β-A activin expression by synaptic activity and during neocortical development. Neuroscience 69, 781–796. doi: 10.1016/0306-4522(95)00245-e
Bannerman, D. M., Rawlins, J. N., McHugh, S. B., Deacon, R. M., Yee, B. K., Bast, T., et al. (2004). Regional dissociations within the hippocampus—memory and anxiety. Neurosci. Biobehav. Rev. 28, 273–283. doi: 10.1016/j.neubiorev.2004.03.004
Brickley, S. G., and Mody, I. (2012). Extrasynaptic GABAA receptors: their function in the CNS and implications for disease. Neuron 73, 23–34. doi: 10.1016/j.neuron.2011.12.012
Chen, Y. G., Wang, Q., Lin, S. L., Chang, C. D., Chuang, J., and Ying, S. Y. (2006). Activin signaling and its role in regulation of cell proliferation, apoptosis and carcinogenesis. Exp. Biol. Med. (Maywood.) 231, 534–544.
Choi, S. C., and Han, J. K. (2011). Negative regulation of activin signal transduction. Vitam. Horm. 85, 79–104. doi: 10.1016/B978-0-12-385961-7.00005-6
Dow, A. L., Russell, D. S., and Duman, R. S. (2005). Regulation of activin mRNA and Smad2 phosphorylation by antidepressant treatment in the rat brain: effects in behavioral models. J. Neurosci. 25, 4908–4916. doi: 10.1523/JNEUROSCI.5155-04.2005
Edelmann, E., Lessmann, V., and Brigadski, T. (2014). Pre- and postsynaptic twists in BDNF secretion and action in synaptic plasticity. Neuropharmacology 76, 610–627. doi: 10.1016/j.neuropharm.2013.05.043
Eisch, A. J., and Petrik, D. (2012). Depression and hippocampal neurogenesis: a road to remission? Science 338, 72–75. doi: 10.1126/science.1222941
Gancarz, A. M., Wang, Z. J., Schroeder, G. L., Damez-Werno, D., Braunscheidel, K. M., Mueller, L. E., et al. (2015). Activin receptor signaling regulates cocaine-primed behavioral and morphological plasticity. Nat. Neurosci. 18, 959–961. doi: 10.1038/nn.4036
Ganea, K., Menke, A., Schmidt, M. V., Lucae, S., Rammes, G., Liebl, C., et al. (2012). Convergent animal and human evidence suggests the activin/inhibin pathway to be involved in antidepressant response. Transl. Psychiatry 2:e177. doi: 10.1038/tp.2012.104
Gottmann, K., Mittmann, T., and Lessmann, V. (2009). BDNF signaling in the formation, maturation and plasticity of glutamatergic and GABAergic synapses. Exp. Brain Res. 199, 203–234. doi: 10.1007/s00221-009-1994-z
Hardingham, G. E., and Bading, H. (2010). Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat. Rev. Neurosci. 11, 682–696. doi: 10.1038/nrn2911
Hasegawa, Y., Mukai, H., Asashima, M., Hojo, Y., Ikeda, M., Komatsuzaki, Y., et al. (2014). Acute modulation of synaptic plasticity of pyramidal neurons by activin in adult hippocampus. Front. Neural Circuits 8:56. doi: 10.3389/fncir.2014.00056
Hedger, M. P., Winnall, W. R., Phillips, D. J., and de Kretser, D. M. (2011). The regulation and functions of activin and follistatin in inflammation and immunity. Vitam. Horm. 85, 255–297. doi: 10.1016/B978-0-12-385961-7.00013-5
Hodge, C. W., Mehmert, K. K., Kelley, S. P., McMahon, T., Haywood, A., Olive, M. F., et al. (1999). Supersensitivity to allosteric GABAA receptor modulators and alcohol in mice lacking PKCepsilon. Nat. Neurosci. 2, 997–1002. doi: 10.1038/14795
Hughes, P. E., Alexi, T., Williams, C. E., Clark, R. G., and Gluckman, P. D. (1999). Administration of recombinant human Activin-A has powerful neurotrophic effects on select striatal phenotypes in the quinolinic acid lesion model of Huntington’s disease. Neuroscience 92, 197–209. doi: 10.1016/s0306-4522(98)00724-6
Krieglstein, K., Suter-Crazzolara, C., Fischer, W. H., and Unsicker, K. (1995). TGF-beta superfamily members promote survival of midbrain dopaminergic neurons and protect them against MPP+ toxicity. EMBO J. 14, 736–742.
Krieglstein, K., Zheng, F., Unsicker, K., and Alzheimer, C. (2011). More than being protective: functional roles for TGF-β/activin signaling pathways at central synapses. Trends Neurosci. 34, 421–429. doi: 10.1016/j.tins.2011.06.002
Kurisaki, A., Inoue, I., Kurisaki, K., Yamakawa, N., Tsuchida, K., and Sugino, H. (2008). Activin induces long-lasting N-methyl-D-aspartate receptor activation via scaffolding PDZ protein activin receptor interacting protein 1. Neuroscience 151, 1225–1235. doi: 10.1016/j.neuroscience.2007.12.012
Lau, D., Bengtson, C. P., Buchthal, B., and Bading, H. (2015). BDNF reduces toxic extrasynaptic NMDA receptor signaling via synaptic NMDA receptors and nuclear-calcium-induced transcription of inhba/activin A. Cell Rep. 12, 1353–1366. doi: 10.1016/j.celrep.2015.07.038
Li, X., and Wolf, M. E. (2015). Multiple faces of BDNF in cocaine addiction. Behav. Brain Res. 279, 240–254. doi: 10.1016/j.bbr.2014.11.018
Link, A. S., Kurinna, S., Havlicek, S., Lehnert, S., Reichel, M., Kornhuber, J., et al. (2015). Kdm6b and pmepa1 as targets of bioelectrically and behaviorally induced activin A signaling. Mol. Neurobiol. doi: 10.1007/s12035-015-9363-3 [Epub ahead of print].
Liu, I. Y., Lyons, W. E., Mamounas, L. A., and Thompson, R. F. (2004). Brain-derived neurotrophic factor plays a critical role in contextual fear conditioning. J. Neurosci. 24, 7958–7963. doi: 10.1523/JNEUROSCI.1948-04.2004
Martinowich, K., Manji, H., and Lu, B. (2007). New insights into BDNF function in depression and anxiety. Nat. Neurosci. 10, 1089–1093. doi: 10.1038/nn1971
Minichiello, L. (2009). TrkB signalling pathways in LTP and learning. Nat. Rev. Neurosci. 10, 850–860. doi: 10.1038/nrn2738
Miron, V. E., Boyd, A., Zhao, J. W., Yuen, T. J., Ruckh, J. M., Shadrach, J. L., et al. (2013). M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat. Neurosci. 16, 1211–1218. doi: 10.1038/nn.3469
Moustakas, A., and Heldin, C. H. (2005). Non-Smad TGF-β signals. J. Cell Sci. 118, 3573–3584. doi: 10.1242/jcs.02554
Mukerji, S. S., Rainey, R. N., Rhodes, J. L., and Hall, A. K. (2009). Delayed activin A administration attenuates tissue death after transient focal cerebral ischemia and is associated with decreased stress-responsive kinase activation. J. Neurochem. 111, 1138–1148. doi: 10.1111/j.1471-4159.2009.06406.x
Muller, M. R., Zheng, F., Werner, S., and Alzheimer, C. (2006). Transgenic mice expressing dominant-negative activin receptor IB in forebrain neurons reveal novel functions of activin at glutamatergic synapses. J. Biol. Chem. 281, 29076–29084. doi: 10.1074/jbc.m604959200
Nibuya, M., Morinobu, S., and Duman, R. S. (1995). Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J. Neurosci. 15, 7539–7547.
Pape, H. C., and Pare, D. (2010). Plastic synaptic networks of the amygdala for the acquisition, expression and extinction of conditioned fear. Physiol Rev. 90, 419–463. doi: 10.1152/physrev.00037.2009
Rodríguez-Martinez, G., Molina-Hernández, A., and Velasco, I. (2012). Activin A promotes neuronal differentiation of cerebrocortical neural progenitor cells. PLoS One 7:e43797. doi: 10.1371/journal.pone.0043797
Shoji-Kasai, Y., Ageta, H., Hasegawa, Y., Tsuchida, K., Sugino, H., and Inokuchi, K. (2007). Activin increases the number of synaptic contacts and the length of dendritic spine necks by modulating spinal actin dynamics. J. Cell Sci. 120, 3830–3837. doi: 10.1242/jcs.012450
Stayte, S., Rentsch, P., Li, K. M., and Vissel, B. (2015). Activin A protects midbrain neurons in the 6-hydroxydopamine mouse model of Parkinson’s disease. PLoS One 10:e0124325. doi: 10.1371/journal.pone.0124325
Tretter, Y. P., Hertel, M., Munz, B., ten Bruggencate, G., Werner, S., and Alzheimer, C. (2000). Induction of activin A is essential for the neuroprotective action of basic fibroblast growth factor in vivo. Nat. Med. 6, 812–815. doi: 10.1038/77548
Werner, S., and Alzheimer, C. (2006). Roles of activin in tissue repair, fibrosis and inflammatory disease. Cytokine Growth Factor Rev. 17, 157–171. doi: 10.1016/j.cytogfr.2006.01.001
Whissell, P. D., Lecker, I., Wang, D.-S., Yu, J., and Orser, B. A. (2015). Altered expression of δGABAA receptors in health and disease. Neuropharmacology 88, 24–35. doi: 10.1016/j.neuropharm.2014.08.003
Wu, D. D., Lai, M., Hughes, P. E., Sirimanne, E., Gluckman, P. D., and Williams, C. E. (1999). Expression of the activin axis and neuronal rescue effects of recombinant activin A following hypoxic-ischemic brain injury in the infant rat. Brain Res. 835, 369–378. doi: 10.1016/s0006-8993(99)01638-8
Xia, Y., and Schneyer, A. L. (2009). The biology of activin: recent advances in structure, regulation and function. J. Endocrinol. 202, 1–12. doi: 10.1677/JOE-08-0549
Zheng, F., Adelsberger, H., Muller, M. R., Fritschy, J. M., Werner, S., and Alzheimer, C. (2009). Activin tunes GABAergic neurotransmission and modulates anxiety-like behavior. Mol. Psychiatry 14, 332–346. doi: 10.1038/sj.mp.4002131
Zheng, F., Puppel, A., Huber, S. E., Link, A. S., Eulenburg, V., van Brederode, J. F., et al. (2015). Activin controls ethanol potentiation of inhibitory synaptic transmission through GABAA Receptors and concomitant behavioral sedation. Neuropsychopharmacology doi: 10.1038/npp.2015.372 [Epub ahead of print].
Keywords: activin, depression, anxiety disorders, drug craving, neurodegenerative disease
Citation: Link AS, Zheng F and Alzheimer C (2016) Activin Signaling in the Pathogenesis and Therapy of Neuropsychiatric Diseases. Front. Mol. Neurosci. 9:32. doi: 10.3389/fnmol.2016.00032
Received: 25 February 2016; Accepted: 25 April 2016;
Published: 10 May 2016.
Edited by:
Andreas Vlachos, Heinrich-Heine-University, GermanyReviewed by:
Thomas Mittmann, Johannes Gutenberg University Mainz, GermanyHartmut Lüddens, University Medical Centre Mainz, Germany
Copyright © 2016 Link, Zheng and Alzheimer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christian Alzheimer, christian.alzheimer@fau.de