 Stephan Lindsey
Stephan Lindsey Sigrid A. Langhans
Sigrid A. Langhans- Nemours Center for Childhood Cancer Research, Alfred I. duPont Hospital for Children, Wilmington, DE, USA
Epithelial–mesenchymal transition (EMT) and cell transformation have been well-documented in multiple cancer cell models and are believed to be one of the earliest events in tumor progression. Genetic and epigenetic modifications shift cells toward either end of the EMT spectrum, and can be influenced by the microenvironment surrounding a tumor. EMT and mesenchymal–epithelial transition are critical to normal function and development and an intricate network of transcription factors and transcriptional regulators tightly regulates these processes. As evidenced in normal and transformed cell lines, many signaling pathways trigger EMT during development and differentiation. The signaling pathways include those triggered by different members of the transforming growth factor superfamily, epidermal growth factor, fibroblast growth factor, hepatocyte growth factor, hypoxia-inducible factor, Wnt, Notch, and many others. Functional redundancies allow cells to undergo EMT even if these key transcriptional regulators are lacking, but these same redundancies also make these pathways particularly susceptible to gain-of-function mutations or constitutive signal activation; the “forced” transition toward either a mesenchymal or epithelial phenotype.
Introduction
Historically, signaling pathways were studied in isolation and treated as linear entities that never interacted; however, studies in the emerging field of systems biology have provided a growing appreciation of the importance of pathway crosstalk and emphasized the complexity of signaling webs during development and tumor progression. This is especially true for the process known as epithelial–mesenchymal transition (EMT). EMT was first described in the 1980s because of its pivotal role during embryonic development and was later implicated in the physiological response to injury (1). EMT is critically involved in normal embryogenesis and development and epithelial cells have developed an intricate network of signaling pathways that include redundancies that safeguard and ensure proper functioning even in the event of a genetic lesion. However, these functional redundancies also leave the signaling network particularly susceptible to gain-of-function mutations and inappropriate signal amplification, eventually leading to tumor progression. Thus, EMT is not only a key biological process during embryonic morphogenesis but also a defining characteristic. EMT is also one of the earliest steps of solid tumor progression, associated with tumor growth, invasion, and metastasis, and contributes to the conversion of tumors from low- to high-grade malignancy (2, 3).
During EMT, epithelial cells undergo a developmental switch that results in decreased adhesion and loss of cell polarity, increased proliferation, and increased motility and invasiveness (4). Invasion is a key step to progression toward a malignant phenotype, and occurs when tumor cells translocate from the relatively constrained initial neoplastic mass into neighboring host tissues. To accomplish this, cancer cells must somehow detach from the primary tumor and migrate through surrounding tissues, opening up the opportunity to penetrate the basal-membrane surrounding a blood or lymphatic vessel, travel throughout the body via the circulatory system, and colonize distant sites where metastatic foci can be formed. Growing evidence suggests that in order for benign cancer to progress toward malignant disease tumor cells undergo EMT (5). The EMT process is associated with a number of morphological and biochemical changes where polarized and basal-membrane anchored epithelial cells acquire a mesenchymal, fibroblastoid phenotype. Morphologically, during the onset of EMT cells transform from a cuboidal epithelial-like cell to a spindle-shaped mesenchymal-like cell. These changes are associated with the down-regulation of epithelial cell surface markers and cytoskeleton components [e.g., E-cadherin, zonula occludens (ZO)-1, claudins, occludins, cytokeratins] and the up-regulation of mesenchymal markers (e.g., vimentin and α-smooth muscle actin) and extracellular matrix components (e.g., collagens and fibronectin) (6). The essential features of EMT as it relates to tumor progression are disruption of intercellular contacts and enhanced migration, the capability of matrix remodeling and tumor tissue remodeling, invasion into and migration through the extracellular matrix without the assistance of cell–cell contacts, and apoptotic resistance. Although the molecular basis of EMT have not been completely elucidated, in vitro and in vivo model systems have identified five main interconnected transduction pathways that lead to EMT and EMT-like phenotypes, many of which connect EMT to the extracellular matrix and the microenvironment surrounding tumors: tyrosine kinase receptors including the receptors for platelet-derived growth factor (PDGF), epidermal growth factor (EGF), insulin-like growth factor (IGF), hepatocyte growth factor (HGF), and fibroblast growth factor (FGF); nuclear factor kappa-light-chain-enhancer of activated B-cells (NF-κB); integrins; transforming growth factor (TGF)-β; Wnt; and many others (7). Many of these pathways share common downstream signaling effectors, highlighting the complexity of the signaling networks involved in EMT (8). In this review, we summarize some of the most prominent EMT-inducing networks and the associated molecular events leading to the transition of differentiated, polarized epithelial cells to a fibroblastic, mesenchymal cell.
EMT-Related Signaling Networks That Regulate E-Cadherin
Most signaling pathways involved in the initiation of EMT result in the down-regulation of E-cadherin, an epithelial cell adhesion molecule that serves as a “master programmer” of EMT [recently reviewed in Ref. (9)]. A critical mediator of EMT, E-cadherin has often been described as the gatekeeper of EMT (10, 11) and in most cell types, the loss of functional E-cadherin results in loss of cell adhesion, leading to rapid cell growth and metastasis (9). In addition to its role in cell adhesion, E-cadherin is involved in transmitting signals within cells that control cell maturation, differentiation, motility, and growth. E-cadherin also acts as a tumor suppressor protein, preventing cells from growing and dividing too rapidly or in an uncontrolled way; E-cadherin down-regulation has been implicated in cell migration and invasion in murine models of mammary, prostate, and pancreatic cancer (12). Providing further correlative support for a role of E-cadherin during tumor formation, E-cadherin is inactivated in many diffuse-type cancers such as lobular breast carcinoma and gastric carcinoma, in which cells in a tumor mass lose epithelial characteristics and exhibit a highly invasive EMT-derived histological pattern. E-cadherin down-regulation occurs in solid, non-diffuse-type cancers at the tumor-stroma boundary where single EMT-derived tumor cells invade otherwise healthy tissue. In the case of single cell infiltration, E-cadherin loss and subsequent resulting EMT could be a transient, reversible process, possibly regulated by the tumor microenvironment; neoplastic cells that have undergone EMT during invasion seem to regain E-cadherin expression and their epithelial, cohesive characteristics at the secondary foci (13).
Molecular events during EMT result in transcriptional regulation of the transcription factors Snail (Snail1), Slug (Snail2), zinc-finger E-box-binding homeobox (ZEB)1/2, and Twist1/2, leading to a molecular fingerprint that serves as a phenotypic marker during EMT (14). In particular, Snail, Slug, Twist, SIP1/ZEB, and E47 negatively regulate E-cadherin expression (14, 15) and display overlapping functional redundancy, in part through their common recognition of E-box sequences (Figure 1). Snail and Slug initiate EMT during development, fibrosis, and the initial invasion of cancer by repressing epithelial genes like E-cadherin by binding to E-box DNA sequences through their carboxy-terminal zinc-finger domains (16). While ZEB1/2 also binds to E-box sequences, ZEB-mediated transcriptional repression often involves the recruitment of a C-terminal-binding protein (CTBP) co-repressor (16). Twist1/2 belongs to the basic helix–loop–helix (bHLH) family of transcription factors and represses E-cadherin expression independently of Snail, probably through interactions with co-repressors (16). E47 also binds to the E-cadherin E-box, but appears to independently promote angiogenesis during tumor growth (17). Involved in most physiological EMT situations, overexpression of Snail, Slug, ZEB1/2, or Twist1/2 in epithelial cell lines typically induces EMT (18–20). These transcription factors also regulate genes other than E-cadherin. Twist and Snail have emerged as promising candidates of EMT “master genes” because they regulate genes involved in motility, proliferation, differentiation, and survival, including matrix metalloproteinases, N-cadherin, and E-cadherin in in vitro and in vivo experiments (19, 21). The signaling pathways involved in EMT should not be viewed in isolation, for evidence of interactions and crosstalk between multiple pathways exists. For example, Snail and Slug both repress E-cadherin levels and are co-expressed in various carcinomas, including breast and ovarian cancer (22). Nevertheless, by employing multiple signaling cascades, Snail and Slug could have both overlapping and simultaneously distinct roles during tumor progression, similar to what has been described for Dnmt3a/b and Vav1/2 during hematopoiesis (22–25). β-Catenin not only interacts with E-cadherin to maintain cell–cell adhesion but is also shuttled to the nucleus where Wnt serves as the transcription effector of the Wnt signaling pathway to promote proliferation and cell survival (26). This is particularly relevant during EMT because Wnt gene mutations and aberrant activation of β-catenin are considered critical events in tumor cell maintenance and growth (27). Glycogen synthase kinase (GSK)-3β-mediated stabilization of Snail is not only part of the Wnt signaling cascade but is also required in colorectal cancer cells for EMT induced by the pro-inflammatory cytokine tumor necrosis factor (TNF)-α (28). Slug was identified as a downstream Wnt signaling pathway effector in a basal-like carcinoma model that also linked the Wnt pathway to tumor proliferation and self-renewal (29). These finding suggest that Slug and Wnt play important roles in maintaining the stemness of human mammary tumor cells.
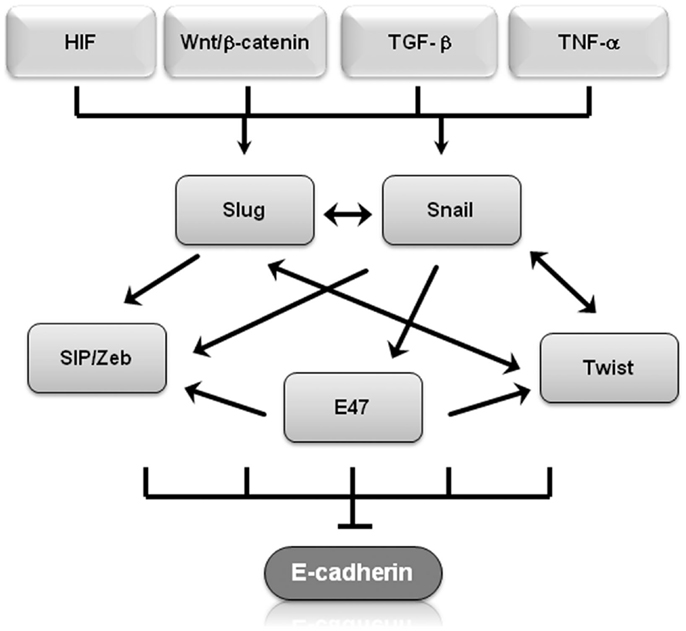
Figure 1. Transcriptional regulators of EMT regulate E-cadherin. Shown is a graphical representation of how several of transcriptional regulators of EMT regulate E-cadherin expression. Arrows represent up-regulation, T-bars represent inhibition.
EMT and Circulating Cancer Cells
It is believed that some cells slough off the outer edges of a tumor as it proliferates and are swept away by the bloodstream or lymphatic system (30). These rare so-called circulating tumor cells (CTCs) from a primary tumor have the capacity to be shed into the vasculature, where they circulate throughout the bloodstream and eventually find a suitable location to colonize and form distant metastases in new tissues (30). One of the key features of the CTC theory of metastasis is that these cells are able to become quiescent until microenvironmental conditions favor growth. Interestingly, two EMT-inducers, Snail and Twist, are upregulated in CTCs and allow cancer cell populations to revert to a stem-cell-like quiescent state (31). Further supporting a link between EMT and CTCs, more than 80% of CTCs collected based on the expression of an epithelial marker (EpCAM) also expressed a mixture of epithelial and mesenchymal markers, suggesting that these CTC were currently transitioning along the EMT axis (32). Indeed, these cells expressed many molecular markers only seen in cells undergoing EMT, including epithelial proteins such as cytokeratin and E-cadherin; mesenchymal proteins including vimentin, N-cadherin, and O-cadherin; and the stem cell marker CD133 (32). Additionally, CTC-like cells increased after immortalized human mammary epithelial cells were transfected with Ras to initiate EMT (33, 34). Further implicating EMT in CTC production, CTCs obtained from metastatic breast cancer patients express a much higher rate of Twist and vimentin, two markers of EMT, than patients with early breast cancer (35). Many of the same microenvironmental cues that regulate EMT also seem to be upregulated in CTCs, especially hypoxia and inflammatory mediators such as NFκB and TGFβ (36). Inflammatory signaling mediators emanating from the microenvironment also play a critical role in the maintenance of CTCs. The receptor for stromal-derived-factor-1 (SDF-1), C–X–C chemokine receptor 4 (CXCR4) enhances the survival of CTCs as they circulate throughout the body (37). Microenvironmental signals also serve as cues to tell CTCs to leave the circulation and to colonize distant organs. In breast cancer, bone marrow cells secrete parathyroid hormone-related protein (PTHRP), TNF-α, interleukin 6 (IL-6), and/or IL-11 stimulate the release of the receptor activator of NF-κB ligand (RANKL) from osteoblasts and suppress the release of the RANKL antagonist osteoprotegerin, allowing for breast tumor CTCs to colonize the bone marrow (37). TNF-related apoptosis-inducing ligand (TRAIL) has also been recently shown to contribute to EMT by miR-221-induced suppression of phosphatase and tensin homolog (PTEN) (38). Similarly, interactions between endothelial selectins and selectin ligands expressed on CTCs, up-regulation of adhesion molecules, and interactions between adherent neutrophils within inflamed sinusoids and CTCs contribute metastatic colonization of the liver (39). Together, these examples paint a complex picture of signaling crosstalk that intersect at the CTC and play a critical role in tumor progression.
MicroRNA Regulation of EMT
MicroRNAs (miRNAs) are small non-coding RNA molecules that play key roles in the regulation of transcriptional and post-transcriptional gene expression (40). In addition to their important roles in healthy individuals, miRNAs are important players during EMT and are differentially expressed in a broad range of cancers (41). Because a single miRNA can target several messenger RNAs, dysregulation of miRNAs can influence multiple signaling pathways leading to tumor formation and metastasis. For example, miR-138 controls EMT by targeting at least three genes: vimentin, ZEB2, and enhancer of zeste homolog EZH2 (42). Similarly, the miR-200 micro RNA family targets at least two transcriptional repressors of E-cadherin, ZEB1 and ZEB2; altering miR-200 in transformed cell lines induced changes consistent with either inducing EMT or the reverse process, mesenchymal–epithelial transition (MET) (43). Reduced expression of miR-30a promotes TGF-β-induced EMT by targeting SNAI1 (44). In addition to the control of transcription factors, miRNA also affects multiple aspects of the EMT process, including increased motility and invasiveness, cell adhesion, disassembly of epithelial cell junctions, and destabilization of tight junctions (45).
Genetic Lesions and Epigenetic Modifications in EMT
Many signaling pathways associated with EMT result in increased cellular proliferation and create feedback loops, resulting in a perpetual proliferative state during the initial stages of EMT. In normal, healthy cells, genomic integrity during cell division is ensured by DNA repair and cell cycle checkpoints that respond to DNA damage by inhibiting critical cell cycle events (46). However, the increased proliferation rate in cells undergoing EMT provides tumor cells an opportunity to proceed through mitosis without high-fidelity proofreading and/or repair, consequently, resulting in the potential of increased mutation rates (47). However, increased proliferation during EMT is not sufficient for tumor development on its own and additional genetic lesions are required to move past the initial cellular dysplasia toward a malignant tumor. Consistent with this model, the carcinogenic potential of estradiol, for example, is thought to be mediated by a combination of proliferation and increased mutation rate [reviewed in Ref. (48, 49)].
Epigenetic deregulation of gene expression is involved in the initiation and progression of multiple cancers and an important initiator of EMT. Similar to its role in differentiation, development, and malignant transformation, epigenetic reprograming during EMT is largely mediated by chromatin remodeling (50). DNA methylation patterns are preserved during EMT and sustained EMT activation leads to epigenetic alterations, inducing heritable changes that maintain the mesenchymal phenotype even after EMT-initiating signals are removed. Epigenetic modifications, especially histone and DNA methylation, are critical to gene regulation and establish patterns of gene repression during development and EMT (51). Snail represses E-cadherin expression by forming a co-repressor complex with histone deacetylase HDAC1 and HDAC2, resulting in E-cadherin silencing and in vivo pancreatic cancer progression (52). Overexpression of the histone methyltransferase MMSET (multiple myeloma SET domain) in prostate cancer influences histone 3 lysine 36 dimethylation (H3K36me2) and lysine 27 tri-methylation (H3K27me3). MMSET overexpression in immortalized prostatic epithelial cells leads to increased migration, increased invasion, morphological changes, and altered gene expression consistent with transition from an epithelial cell-like state to a mesenchymal cell-like state (53). Mediated by the ability of MMSET-mediated activation of TWIST1, a gene implicated in tumor-associated EMT and invasion (19), these data suggest that deregulated MMSET results in aberrant epigenetic gene regulation, leading to tumor progression and metastasis. Genome-wide histone maps focusing on H3 lysine 4 and lysine 27 tri-methylation (H3K4me3 and H3K27me3) identified differentially expressed genes in embryonic stem cells (54–57), hematopoietic stem cells/progenitor cells (58), T cells (59), and in prostate cancer cells (60). Although DNA methylation has been implicated in the transition from EMT to MET, reversible histone modifications are the predominant factors in reactivation of E-cadherin expression during the transition from EMT to MET (61).
Genome-scale mapping revealed that most chromatin changes are heterochromatin K9-modifications, suggesting that EMT is characterized by the epigenetic reprograming of specific, large chromatin domains across the genome (50). Similarly, clustered chromatin profiles using combinatorial patterns of posttranslational histone modifications and covalent changes to genomic DNA discovered a distinct chromatin signature among genes in well-established EMT pathways including the epidermal growth factor receptor (EGFR), suggesting that chromatin remodeling of EGFR plays an important role in EMT (62). Acetylation, regulated mainly through HDACs also affects EGFR expression and downstream signaling. HDAC6 up-regulation slows EGFR endocytic trafficking from early endosomes to late endosomes in renal epithelial cells and HDAC6 inhibition results in decreased phosphorylation of extracellular signal-regulated kinase (ERK) 1/2, a downstream target of EGFR (63). Future experiments should determine if these findings are common to EMT and determine if similar epigenetic reprograming occurs in other physiological contexts. Aside from this role in epigenetic reprograming, crosstalk between the more traditionally known EGFR signaling cascade and components of other signaling pathways frequently leads to abnormal activation of pro-proliferative and anti-apoptotic pathways. The most common signaling cascades activated by EGFR are the phosphatidylinositol-3-kinase (PI3K)/Akt, Ras/Raf/Mek/extracellular signal-regulated kinase, and the Jak/Stat pathways (64) that both contribute to the development of malignancies by impacting cell cycle progression, inhibition of apoptosis, angiogenesis, tumor cell motility, and metastases (65). Crosstalk between EGFR and other signaling pathways impact cancer treatment as well as the initiation of EMT. For example, one well-known mechanism of resistance to the selective EGFR inhibitor gefitinib/erlotinib is HGF receptor tyrosine kinase gene amplification. HGF receptor tyrosine kinase gene amplification bypasses normal EGFR signaling to instead activate AKT through HER3-mediated activation of PI3K in the presence of EGFR tyrosine kinase inhibitors (66).
Crosstalk between TGFβ and Other Signaling Pathways Mediating EMT
Signaling pathways are not independent from each other, but rather interact to form complex signaling networks; the TGFβ signaling pathway is no exception. Most likely, due to its involvement during many cellular processes including proliferation, differentiation, apoptosis, and cellular homeostasis, the TGFβ pathway interacts with many other signaling pathways during EMT (Figure 2). One mechanism by which TGFβ initiates EMT is by removing β-catenin from adherens junctions in a process that involves TGFβ-dependent PTEN dissociation from β-catenin and Akt activation (67). Depending on the context, Notch can either synergize with TGFβ/bone morphogenetic protein (BMP) signals to induce target genes or inhibit TGFβ/BMP signaling (68–71). In the presence of other growth factors, TGFβ/BMP signaling generally stimulates migration and blocks endothelial cell proliferation, but Notch signaling inhibits the migratory affect of BMP (72). Stimulation of endothelial cells with BMP alone promoted cell migration, but in the presence of Notch signaling, cell migration was inhibited (72). Interestingly, the dominance of Notch signaling over BMP signaling was cell–cell contact-dependent, suggesting that endothelial cells not in contact with surrounding cells are stimulated by TGFβ to migrate until new cell–cell contacts are established, at which point Notch induces gene expression changes and arrests further migration (72). Similarly, Notch signaling is necessary for growth arrest by TGFβ in epithelium; over 30% of TGFβ-induced epithelial genes require Notch signaling for full expression (68). TGFβ leads to increased jagged-1 expression and siRNA-mediated knockdown of jagged-1 leads to reduced TGFβ-induced p21 expression, rescuing TGFβ-inhibited proliferation (68). Therefore, TGFβ induces both c-myc, which stimulates cell cycle progression, and jagged-1, which blocks cell cycle progression through stimulation of Notch and induction of p21. Jagged-1 induction is rapid and transient, so a balance between TGFβ/Notch-induced p21 and TGFβ/Smad-induced c-myc may act as a switch to regulate cell proliferation (72).
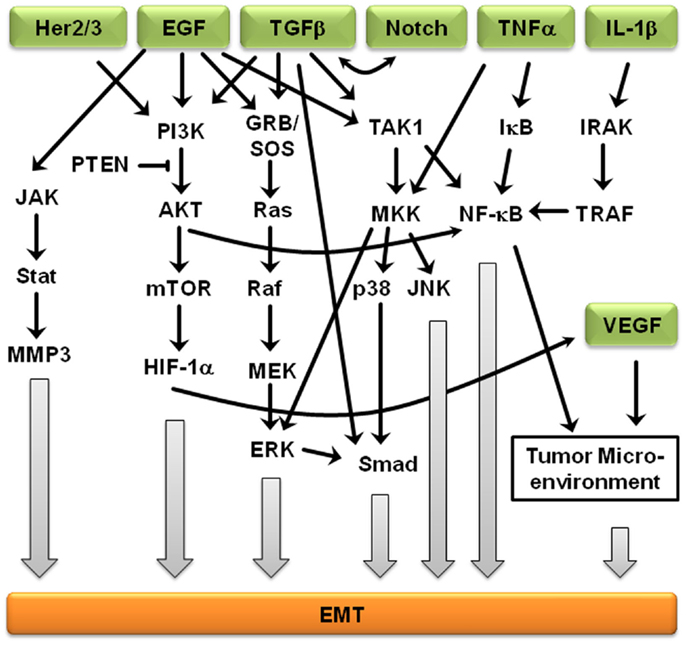
Figure 2. Crosstalk between EMT-inducing signaling pathways. Representation of some of the points of intersection between various EMT-inducing signaling pathways. For clarity, branches that did not result in crosstalk are not shown. Arrows represent up-regulation.
Further supporting the importance of crosstalk during EMT, Erk, c-Jun N-terminal kinase (JNK), and p38 indirectly regulate TGFβ signaling, but TGFβ treatment leads to activation of Erk1/2 and mitogen-activated protein kinase (MAPK) signaling (73). Smad-dependent signaling and MAPK-mediated Erk1/2 activation is believed to result in cross-talk between the TGFβ and EGF signaling pathways (74). The MAPK/Erk signaling pathway also mediates Smad2/3 phosphorylation and nuclear exclusion, which is believed to be important for the attenuation of TGFβ-induced nuclear translocation of MAPK (74). MAPK signaling also results in Smad1/5 phosphorylation, leading to an inability to translocate specific Smads into the nucleus (74). Erk-mediated Smad1 phosphorylation creates a docking site for the Smad1/5-specific E3 ubiquitin ligase, Smurf1. Smurf1 binding results in Smad ubiquitination and eventual degradation and blocks Smad interactions with the nuclear pore complex. MAPKs also regulate the protein stability of Smad4 and the inhibitory Smad7 (74), suggesting that Smad3 is indispensable to the mediation of the pro-apoptotic effects of TGFβ, Smad3, but not the closely related Smad2, is the primary target of PI3K/Akt-mediated inhibition (74). In addition, TGFβ regulates Akt activity and phosphatase and tensin homolog (PTEN) function during EMT initiation. In addition to activating the MAPK and PI3K/Akt pathways, ErbB signaling interacts with TGFβ/Smad during development and breast cancer progression (75). The PI3K/Akt pathway is also subjected to TGFβ regulation. Akt activity increases in response to TGFβ treatment, which seems to be required for a variety of TGFβ-induced activities, such as cell migration of HER2-expressing breast cancer cells, EMT of normal mammary epithelial cells, cell survival of mouse hippocampal neurons and mesenchymal cells, as well as growth stimulation of certain fibroblasts (74, 75). EGFR and IL-6R signaling cross-talk through JAK2/STAT3 to mediate EMT in ovarian carcinomas; activated STAT3 in high-grade ovarian carcinomas may occur directly through activation of EGFR/IL-6R or indirectly through induction of IL-6R signaling (76). Another ligand of EGFR, TNF-α, also induces EMT through NF-κB-mediated transcriptional up-regulation of Twist1 (77). In breast cancer-related EMT, HER2/Ras antagonizes TGFβ-induced apoptosis and cell cycle arrest while simultaneously enhancing the pro-migratory and pro-invasive functions of TGFβ (78). TGFβ transcriptionally downregulates PTEN in Smad4 null pancreatic cancer cells and relies on the function of the Ras/MAPK pathway (73–75). EMT-related crosstalk is also clinically relevant; pharmacological blockade of IGF-1R fully prevented TGFβ’s ability to activate an EMT protein signature (79).
Involvement of the Microenvironment during EMT
The tumor microenvironment plays a crucial role in tumor progression and metastasis, and as tumors develop, the integrity of the surrounding basement membrane plays a critical role in invasion and metastasis. The tumor microenvironment is composed of inflammatory and immune cells, physical interactions with neighboring cells, oxygen and nutritional gradients, stromal extracellular matrix, and soluble factors. Cells neighboring the developing tumor secrete growth factors and inhibitory molecules that regulate tumor proliferation and apoptosis, while tumor cells simultaneously secrete factors to neighboring cells that regulate adhesion. The temporal–spatial changes within the microenvironment surrounding tumors and the interaction between tumor cells and their microenvironment are crucial to tumor initiation and development, and are especially critical to cancer cell quiescence, tumor progression, invasion, tumor metastasis, and drug resistance (Figure 3) (80).
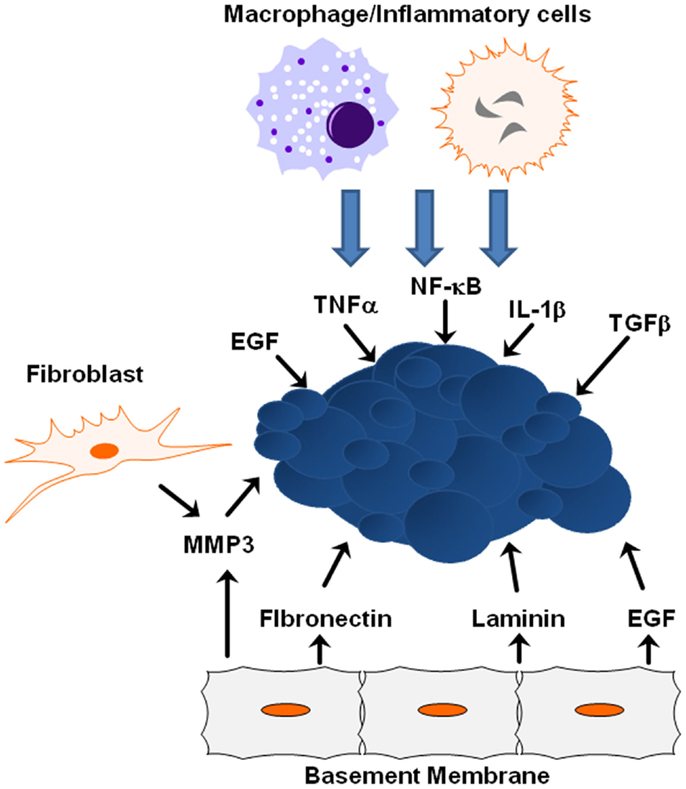
Figure 3. ECM/growth factor involvement in EMT. Shown are several molecular mechanisms by which the cells within the tumor microenvironment can influence tumor progression and EMT initiation.
Many signals received from the tumor microenvironment can initiate EMT including TGFβ, hypoxia-inducible factor (HIF)-1α, EGF, WNTs, and Notch (21). Various signals trigger expression of these transcription factors including heterotypic interactions with neighboring cancer cells and interactions with adjacent tumor-associated stromal cells. Epithelial–mesenchymal interactions within the tumor microenvironment integrate several important signaling molecules that are critical for tumor growth and metastasis, including integrins, cytokines, and growth factors (81). Crosstalk between the TGFβ and HER2/Ras/MAPK signaling pathways often leads to secretion of additional growth factors and cytokines, including TGFβ itself, which in turn promotes EMT and cell invasion, whereas JNK kinases negatively regulate the autocrine expression of TGFβ1 (73, 75). ErbB receptors and their ligands are also involved in cross-talk between cancer cells and the tumor microenvironment. EGFR is activated in tumor-associated endothelial cells, but not in endothelial cells within uninvolved organ regions, suggesting that EGFR activation and expression is partially determined by the tumor microenvironment (82). As time goes on, the importance of the microenvironment to pathogenesis is becoming clearer, from the role the ECM and matrix rigidity plays in determining polarity, to the extracellular metabolism of growth factors and matrix molecules during cancer progression and metastasis.
Extracellular matrix proteins and physical properties within the microenvironment can lead to tumor progression by activating EMT-inducing pathways within tumors. One key extracellular matrix protein is matrix metalloproteinase-3 (MMP3), a matrix-degrading enzyme secreted by stromal fibroblasts known to induce in vitro and in vivo EMT in mammary epithelial cells (83). When tumor cells are exposed to MMP3, transcription of a splice variant of Rac1 called Rac1b increases stimulating the production of reactive oxygen species and expression of Snail1 (84). EGFR activation in human carcinoma cell lines increases MMP-9 activity and is associated with increased in vitro cell invasion (85). Synthetic low-molecular weight or endogenous MMP inhibitors or an anti-catalytic MMP-9 antibody blocked increased invasive activity after EGF-mediated induction, indicating EGFR activation results in enhanced MMP-9 expression and may facilitate the removal of extracellular matrix barriers to tumor invasion. Additional proteins within the basement membrane influence EMT induction from ectopic exposure of MMP3. For example, the laminin suppresses EMT in MMP3-treated cells, while fibronectin promotes EMT due to interactions with specific integrin receptors (7). During this process, α6-integrin sequesters Rac1b from the basement membrane and is required for inhibition of EMT by laminin; α5-integrin maintains Rac1b at the membrane and is required for the promotion of EMT by fibronectin (7). Additionally, matrix rigidity may also play an important role during EMT. The microenvironmental stiffness surrounding cells impacts differentiation and response to external molecular cues, while epithelial cells treated with MMP3 undergo EMT when cultured on plastic or glass, cells cultured on soft matrices do not undergo EMT in response to treatment with MMP3 (7).
Involvement of Inflammatory Signaling in EMT
The microenvironment surrounding a tumor is often dominated by inflammatory cytokines that promote tumor initiation by leading to increased angiogenesis, tumor growth, and tumor progression (86). Tumor-associated macrophages secrete EGF to neighboring cancer cells, which in turn stimulate macrophages to facilitate intravasation and metastatic dissemination of the cancer cells (87, 88). Together, these findings substantiate a role of EGF-mediated signaling not only in EMT and proliferative signaling itself but also in the cross-talk between tumor cells and the microenvironment. The tumor microenvironment is largely orchestrated by inflammatory cells, which facilitate extracellular matrix breakdown, angiogenesis, and tissue remodeling, thus, promoting tumor cell motility (89). Inflammatory cells play a major role in secreting activating factors that lead to NF-κB activation; NF-κB is a key regulator of the inflammatory response shown to regulate Slug and Snail (90). TGF-β activity is deregulated during malignant cancer progression, and plays an important role in EMT (91). Similarly, both TNFα and interleukin-1β (IL-1β) are expressed at low levels in normal breast epithelial cells, but are upregulated in the majority of breast cancer patients, with pronounced expression of both cytokines in over 80% of patients who experience breast tumor relapse (92). In vivo murine breast models suggest increased expression and activity of TNFα results in many cancer-promoting functions and that inhibition of TNFα expression leads to reduced breast cancer malignancy (93). Chronic TNFα expression in the tumor microenvironment is correlated with a more aggressive tumor phenotype (93). IL-1β upregulates a variety of processes that contribute to higher angiogenesis, tumor growth, and tumor progression and is considered a strong and causative pro-inflammatory factor whose expression is associated with advanced cancer (94). TNFα impacts cell morphology and may cooperate with TGFβ to lead to EMT in non-transformed breast epithelial cells (95). Sustained co-expression of TNFα and IL-1β acts through the complex regulatory processes of the EMT activators Zeb1, Snail, and Twist to result in morphologic changes including cell spreading, protrusion formation, decreased E-cadherin expression, and increased expression of vimentin, all consistent with EMT (96).
Hypoxia and EMT
When microenvironmental cues are favorable for growth, rapid cell growth with a tumor results in local hypoxia and nutrient deficits, regardless of the oxygen tension surrounding the tumor (97). Therefore, sustained tumor growth requires increased local vasculature to provide oxygen and metabolites to feed the growing tumor (98) and the nutritionally impoverished and hypoxic environment within tumors results in local changes in hypoxia-related gene expression, contributing to tumor heterogeneity (99). Tumor cells adjust to hypoxia and lack of nutrients not only by activating specific pathways associated with angiogenesis but also associated with hypermetabolism, glycolysis, and resistance to acidosis-induced toxicity (100). Hypoxia genes, especially HIF-1α, are frequently upregulated within many solid tumors and promote tumor progression (101, 102). HIF-1α induces EMT and self-renewal of cancer stem cells, and facilitates metastasis; knockdown of HIF-1α inhibits or even reverses the EMT-like phenotype (103, 104). Hypoxia-induced EMT is mediated by HIF-1α via up-regulation of transcription effectors such as Snail, Twist, and ZEB1/2 and results in the suppression of E-cadherin expression (105–107). Several additional signaling pathways that are critical for embryonic development including Notch, Wnt, and TGFβ are also involved in hypoxia-induced EMT. Demonstrating a complex integration of hypoxic signals into EMT, Notch signaling directly upregulates Snail, and potentiates HIF-1α recruitment to the lysyl oxidase promoter, resulting in stabilization of Snail, increased cell motility, and increased invasiveness (108). Similarly, Wnt/β-catenin signaling enhances hypoxia-induced EMT by increasing the EMT-associated activity of HIF-1α and preventing cell death (109). Hypoxia also inhibits Wnt signaling by interfering with β-catenin acetylation (110), blocking secretion of Wnt (111), and activating Siah-1 in a p53-dependent manner (112).
Depending on cell type, Wnt/β-catenin signaling also enhances hypoxia-induced EMT by increasing the EMT-associated activity of HIF-1α and preventing tumor cell death (109). HIF-1α also competes with T-cell factor-4 (TCF-4) to bind β-catenin and form a HIF-1α/β-catenin complex, accompanied by increased HIF-1α transcriptional activity in colorectal tumors (113). TGFβ signaling also integrates hypoxia-related cues, for TGFβ/Smad3 inhibit vascular smooth muscle cell apoptosis through an autocrine signaling mechanism involving VEGF (114). Adding further complexity to the impact of TGFβ signaling, TGFβ not only activates the Notch signaling pathway but Notch signaling also activates TGFβ in rat mesangial cells under high-glucose conditions (68, 115). It is important to note that this example also highlights how signals from the microenvironment can influence signaling outcomes.
Conclusion
Epithelial–mesenchymal transition is a key physiological process during normal development and regulated by an intricate network of signaling pathways that allows for the integration of signaling cues during embryonic morphogenesis. While these signaling networks allow for the precise control required for a major switch from a differentiated epithelial cell into mesenchymal cell, it also opens up the possibility of deregulation on multiple levels during pathological processes such as cancer and fibrosis. Owing to the complex interactions between these signaling pathways, activating mutations in signaling molecules can be amplified. Many of these potentially deregulated pathways converge on a few master regulatory molecules or parallel pathways can induce changes on various levels. Thus, it is plausible that EMT contributes to cancer progression in various ways, including tumor growth, invasion, and metastasis. Moreover, depending on the nature of the genetic lesions, EMT can become a very individualized process, adding to the complexity of cancer, while also opening up the possibility of personalized medicine. Thus, our improved understanding of EMT signaling networks and their association with therapeutic resistance is imperative for future development of novel anti-tumor drug and treatment strategies, especially in high-grade tumors and tumors that have developed therapeutic resistance.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Support was provided by the National Institute of General Medical Sciences of the National Institutes of Health Awards Number NIGMS-P20GM103464 (Stephan Lindsey and Sigrid A. Langhans), the American Cancer Society Grant Number RSG-09-021-01-CNE (Sigrid A. Langhans), the DO Believe Foundation (Sigrid A. Langhans), and the Nemours Foundation.
Abbreviations
bHLH, basic helix–loop–helix; BMP, bone morphogenetic protein; CTBP, C-terminal binding protein; CTC, circulating tumor cell; CXCR4, C–X–C, chemokine receptor 4; EGFR, epidermal growth factor receptor; EMT, epithelial–mesenchymal transition; ERK, extracellular signal-regulated kinase; FGF, fibroblast growth factor; GRB2, growth factor receptor-bound protein 2; GSK, glycogen synthase kinase; HDAC, histone deacetylase; HGF, hepatocyte growth factor; HIF, hypoxia-inducible factor; IGF, insulin-like growth factor; IκB, nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha; IL-6 interleukin 6; IRAK, interleukin-1 receptor-associated kinase; JNK, c-Jun N-terminal kinase; MAPK, mitogen-activated protein kinase; MET, mesenchymal–epithelial transition; MMP3, matrix metalloproteinase-3; MMSET, multiple myeloma SET domain; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B-cells; PDGF, platelet-derived growth factor; PI3K, phosphatidylinositol-3-kinase; PTEN, phosphatase and tensin homolog; PTHRP, parathyroid hormone-related protein; RANKL, receptor activator of NF-κB ligand; SDF-1, stromal-derived-factor-1; SOS, son of sevenless; TGF, transforming growth factor; TNF, tumor necrosis factor; TRAIL, tumor necrosis factor-related apoptosis-inducing ligand; TRAF, TNF receptor-associated factor; ZEB, zinc-finger E-box-binding homeobox; ZO, zonula occludens.
References
1. Mjaatvedt CH, Markwald RR. Induction of an epithelial-mesenchymal transition by an in vivo adheron-like complex. Dev Biol (1989) 136(1):118–28. doi:10.1016/0012-1606(89)90135-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Thompson EW, Torri J, Sabol M, Sommers CL, Byers S, Valverius EM, et al. Oncogene-induced basement membrane invasiveness in human mammary epithelial cells. Clin Exp Metastasis (1994) 12(3):181–94. doi:10.1007/BF01753886
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Thiery JP. Epithelial-mesenchymal transitions in development and pathologies. Curr Opin Cell Biol (2003) 15(6):740–6. doi:10.1016/j.ceb.2003.10.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Acloque H, Adams MS, Fishwick K, Bronner-Fraser M, Nieto MA. Epithelial-mesenchymal transitions: the importance of changing cell state in development and disease. J Clin Invest (2009) 119(6):1438–49. doi:10.1172/JCI38019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Puisieux A, Brabletz T, Caramel J. Oncogenic roles of EMT-inducing transcription factors. Nat Cell Biol (2014) 16(6):488–94. doi:10.1038/ncb2976
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Lee JM, Dedhar S, Kalluri R, Thompson EW. The epithelial-mesenchymal transition: new insights in signaling, development, and disease. J Cell Biol (2006) 172(7):973–81. doi:10.1083/jcb.200601018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Chen QK, Lee K, Radisky DC, Nelson CM. Extracellular matrix proteins regulate epithelial-mesenchymal transition in mammary epithelial cells. Differentiation (2013) 86(3):126–32. doi:10.1016/j.diff.2013.03.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Savagner P. Leaving the neighborhood: molecular mechanisms involved during epithelial-mesenchymal transition. Bioessays (2001) 23(10):912–23. doi:10.1002/bies.1132
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Nagathihalli NS, Merchant NB. Src-mediated regulation of E-cadherin and EMT in pancreatic cancer. Front Biosci (Landmark Ed) (2012) 17:2059–69. doi:10.2741/4037
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Batlle E, Sancho E, Franci C, Dominguez D, Monfar M, Baulida J, et al. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol (2000) 2(2):84–9. doi:10.1038/35000034
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Cano A, Perez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, et al. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol (2000) 2(2):76–83. doi:10.1038/35010506
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Derksen PW, Liu X, Saridin F, van der Gulden H, Zevenhoven J, Evers B, et al. Somatic inactivation of E-cadherin and p53 in mice leads to metastatic lobular mammary carcinoma through induction of anoikis resistance and angiogenesis. Cancer Cell (2006) 10(5):437–49. doi:10.1016/j.ccr.2006.09.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Guarino M, Rubino B, Ballabio G. The role of epithelial-mesenchymal transition in cancer pathology. Pathology (2007) 39(3):305–18. doi:10.1080/00313020701329914
14. Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer (2007) 7(6):415–28. doi:10.1038/nrc2131
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Bolos V, Peinado H, Perez-Moreno MA, Fraga MF, Esteller M, Cano A. The transcription factor Slug represses E-cadherin expression and induces epithelial to mesenchymal transitions: a comparison with Snail and E47 repressors. J Cell Sci (2003) 116(Pt 3):499–511. doi:10.1242/jcs.00224
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol (2014) 15(3):178–96. doi:10.1038/nrm3758
17. Perez-Moreno MA, Locascio A, Rodrigo I, Dhondt G, Portillo F, Nieto MA, et al. A new role for E12/E47 in the repression of E-cadherin expression and epithelial-mesenchymal transitions. J Biol Chem (2001) 276(29):27424–31. doi:10.1074/jbc.M100827200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Savagner P, Yamada KM, Thiery JP. The zinc-finger protein slug causes desmosome dissociation, an initial and necessary step for growth factor-induced epithelial-mesenchymal transition. J Cell Biol (1997) 137(6):1403–19. doi:10.1083/jcb.137.6.1403
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell (2004) 117(7):927–39. doi:10.1016/j.cell.2004.06.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Nieto MA. The snail superfamily of zinc-finger transcription factors. Nat Rev Mol Cell Biol (2002) 3(3):155–66. doi:10.1038/nrm757
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Barrallo-Gimeno A, Nieto MA. The Snail genes as inducers of cell movement and survival: implications in development and cancer. Development (2005) 132(14):3151–61. doi:10.1242/dev.01907
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Elloul S, Elstrand MB, Nesland JM, Trope CG, Kvalheim G, Goldberg I, et al. Snail, Slug, and Smad-interacting protein 1 as novel parameters of disease aggressiveness in metastatic ovarian and breast carcinoma. Cancer (2005) 103(8):1631–43. doi:10.1002/cncr.20946
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Come C, Magnino F, Bibeau F, De Santa Barbara P, Becker KF, Theillet C, et al. Snail and slug play distinct roles during breast carcinoma progression. Clin Cancer Res (2006) 12(18):5395–402. doi:10.1158/1078-0432.CCR-06-0478
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Challen GA, Sun D, Mayle A, Jeong M, Luo M, Rodriguez B, et al. Dnmt3a and dnmt3b have overlapping and distinct functions in hematopoietic stem cells. Cell Stem Cell (2014) 15(3):350–64. doi:10.1016/j.stem.2014.06.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Lindsey S, Jiang J, Woulfe D, Papoutsakis ET. Platelets from mice lacking the aryl hydrocarbon receptor exhibit defective collagen-dependent signaling. J Thromb Haemost (2014) 12(3):383–94. doi:10.1111/jth.12490
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell (2009) 17(1):9–26. doi:10.1016/j.devcel.2009.06.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature (2005) 434(7035):843–50. doi:10.1038/nature03319
28. Wang H, Wang HS, Zhou BH, Li CL, Zhang F, Wang XF, et al. Epithelial-mesenchymal transition (EMT) induced by TNF-alpha requires AKT/GSK-3beta-mediated stabilization of snail in colorectal cancer. PLoS One (2013) 8(2):e56664. doi:10.1371/journal.pone.0056664
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. DiMeo TA, Anderson K, Phadke P, Fan C, Perou CM, Naber S, et al. A novel lung metastasis signature links Wnt signaling with cancer cell self-renewal and epithelial-mesenchymal transition in basal-like breast cancer. Cancer Res (2009) 69(13):5364–73. doi:10.1158/0008-5472.CAN-08-4135
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Friedlander TW, Premasekharan G, Paris PL. Looking back, to the future of circulating tumor cells. Pharmacol Ther (2014) 142(3):271–80. doi:10.1016/j.pharmthera.2013.12.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Li YM, Xu SC, Li J, Han KQ, Pi HF, Zheng L, et al. Epithelial-mesenchymal transition markers expressed in circulating tumor cells in hepatocellular carcinoma patients with different stages of disease. Cell Death Dis (2013) 4:e831. doi:10.1038/cddis.2013.347
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Armstrong AJ, Marengo MS, Oltean S, Kemeny G, Bitting RL, Turnbull JD, et al. Circulating tumor cells from patients with advanced prostate and breast cancer display both epithelial and mesenchymal markers. Mol Cancer Res (2011) 9(8):997–1007. doi:10.1158/1541-7786.MCR-10-0490
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell (2008) 133(4):704–15. doi:10.1016/j.cell.2008.03.027
34. Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene (2010) 29(34):4741–51. doi:10.1038/onc.2010.215
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Kallergi G, Papadaki MA, Politaki E, Mavroudis D, Georgoulias V, Agelaki S. Epithelial to mesenchymal transition markers expressed in circulating tumour cells of early and metastatic breast cancer patients. Breast Cancer Res (2011) 13(3):R59. doi:10.1186/bcr2896
36. Fazilaty H, Gardaneh M, Bahrami T, Salmaninejad A, Behnam B. Crosstalk between breast cancer stem cells and metastatic niche: emerging molecular metastasis pathway? Tumour Biol (2013) 34(4):2019–30. doi:10.1007/s13277-013-0831-y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Nguyen DX, Bos PD, Massague J. Metastasis: from dissemination to organ-specific colonization. Nat Rev Cancer (2009) 9(4):274–84. doi:10.1038/nrc2622
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Wang H, Xu C, Kong X, Li X, Wang Y, Ding X, et al. Trail resistance induces epithelial-mesenchymal transition and enhances invasiveness by suppressing PTEN via miR-221 in breast cancer. PLoS One (2014) 9(6):e99067. doi:10.1371/journal.pone.0099067
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. McDonald B, Spicer J, Giannais B, Fallavollita L, Brodt P, Ferri LE. Systemic inflammation increases cancer cell adhesion to hepatic sinusoids by neutrophil mediated mechanisms. Int J Cancer (2009) 125(6):1298–305. doi:10.1002/ijc.24409
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell (2004) 116(2):281–97. doi:10.1016/S0092-8674(04)00045-5
41. Gregory PA, Bracken CP, Bert AG, Goodall GJ. MicroRNAs as regulators of epithelial-mesenchymal transition. Cell Cycle (2008) 7(20):3112–8. doi:10.4161/cc.7.20.6851
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Liu X, Wang C, Chen Z, Jin Y, Wang Y, Kolokythas A, et al. MicroRNA-138 suppresses epithelial-mesenchymal transition in squamous cell carcinoma cell lines. Biochem J (2011) 440(1):23–31. doi:10.1042/BJ20111006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Park SM, Gaur AB, Lengyel E, Peter ME. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev (2008) 22(7):894–907. doi:10.1101/gad.1640608
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Zhang J, Zhang H, Liu J, Tu X, Zang Y, Zhu J, et al. miR-30 inhibits TGF-beta1-induced epithelial-to-mesenchymal transition in hepatocyte by targeting Snail1. Biochem Biophys Res Commun (2012) 417(3):1100–5. doi:10.1016/j.bbrc.2011.12.121
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Hao J, Zhang Y, Deng M, Ye R, Zhao S, Wang Y, et al. MicroRNA control of epithelial-mesenchymal transition in cancer stem cells. Int J Cancer (2014) 135(5):1019–27. doi:10.1002/ijc.28761
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Weinert T. DNA damage and checkpoint pathways: molecular anatomy and interactions with repair. Cell (1998) 94(5):555–8. doi:10.1016/S0092-8674(00)81597-4
47. Feigelson HS, Ross RK, Yu MC, Coetzee GA, Reichardt JK, Henderson BE. Genetic susceptibility to cancer from exogenous and endogenous exposures. J Cell Biochem Suppl (1996) 25:15–22. doi:10.1002/(SICI)1097-4644(1996)25+<15::AID-JCB2>3.0.CO;2-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Roy D, Liehr JG. Estrogen, DNA damage and mutations. Mutat Res (1999) 424(1–2):107–15. doi:10.1016/S0027-5107(99)00012-3
49. Liehr JG. Is estradiol a genotoxic mutagenic carcinogen? Endocr Rev (2000) 21(1):40–54. doi:10.1210/edrv.21.1.0386
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. McDonald OG, Wu H, Timp W, Doi A, Feinberg AP. Genome-scale epigenetic reprogramming during epithelial-to-mesenchymal transition. Nat Struct Mol Biol (2011) 18(8):867–74. doi:10.1038/nsmb.2084
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Cedar H, Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet (2009) 10(5):295–304. doi:10.1038/nrg2540
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. von Burstin J, Eser S, Paul MC, Seidler B, Brandl M, Messer M, et al. E-cadherin regulates metastasis of pancreatic cancer in vivo and is suppressed by a SNAIL/HDAC1/HDAC2 repressor complex. Gastroenterology (2009) 137(1):361–71. doi:10.1053/j.gastro.2009.04.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Ezponda T, Popovic R, Shah MY, Martinez-Garcia E, Zheng Y, Min DJ, et al. The histone methyltransferase MMSET/WHSC1 activates TWIST1 to promote an epithelial-mesenchymal transition and invasive properties of prostate cancer. Oncogene (2013) 32(23):2882–90. doi:10.1038/onc.2012.297
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell (2006) 125(2):315–26. doi:10.1016/j.cell.2006.02.041
55. Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature (2007) 448(7153):553–60. doi:10.1038/nature06008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Pan G, Tian S, Nie J, Yang C, Ruotti V, Wei H, et al. Whole-genome analysis of histone H3 lysine 4 and lysine 27 methylation in human embryonic stem cells. Cell Stem Cell (2007) 1(3):299–312. doi:10.1016/j.stem.2007.08.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Zhao XD, Han X, Chew JL, Liu J, Chiu KP, Choo A, et al. Whole-genome mapping of histone H3 Lys4 and 27 trimethylations reveals distinct genomic compartments in human embryonic stem cells. Cell Stem Cell (2007) 1(3):286–98. doi:10.1016/j.stem.2007.08.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Cui K, Zang C, Roh TY, Schones DE, Childs RW, Peng W, et al. Chromatin signatures in multipotent human hematopoietic stem cells indicate the fate of bivalent genes during differentiation. Cell Stem Cell (2009) 4(1):80–93. doi:10.1016/j.stem.2008.11.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Wei G, Wei L, Zhu J, Zang C, Hu-Li J, Yao Z, et al. Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity (2009) 30(1):155–67. doi:10.1016/j.immuni.2008.12.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Ke XS, Qu Y, Rostad K, Li WC, Lin B, Halvorsen OJ, et al. Genome-wide profiling of histone h3 lysine 4 and lysine 27 trimethylation reveals an epigenetic signature in prostate carcinogenesis. PLoS One (2009) 4(3):e4687. doi:10.1371/journal.pone.0004687
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Ke XS, Qu Y, Cheng Y, Li WC, Rotter V, Oyan AM, et al. Global profiling of histone and DNA methylation reveals epigenetic-based regulation of gene expression during epithelial to mesenchymal transition in prostate cells. BMC Genomics (2010) 11:669. doi:10.1186/1471-2164-11-669
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Cieslik M, Hoang SA, Baranova N, Chodaparambil S, Kumar M, Allison DF, et al. Epigenetic coordination of signaling pathways during the epithelial-mesenchymal transition. Epigenetics Chromatin (2013) 6(1):28. doi:10.1186/1756-8935-6-28
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Liu W, Fan LX, Zhou X, Sweeney WE Jr, Avner ED, Li X. HDAC6 regulates epidermal growth factor receptor (EGFR) endocytic trafficking and degradation in renal epithelial cells. PLoS One (2012) 7(11):e49418. doi:10.1371/journal.pone.0049418
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Holbro T, Hynes NE. ErbB receptors: directing key signaling networks throughout life. Annu Rev Pharmacol Toxicol (2004) 44:195–217. doi:10.1146/annurev.pharmtox.44.101802.121440
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Al Moustafa AE, Achkhar A, Yasmeen A. EGF-receptor signaling and epithelial-mesenchymal transition in human carcinomas. Front Biosci (Schol Ed) (2012) 4:671–84. doi:10.2741/S292
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science (2007) 316(5827):1039–43. doi:10.1126/science.1141478
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Yu M, Trobridge P, Wang Y, Kanngurn S, Morris SM, Knoblaugh S, et al. Inactivation of TGF-beta signaling and loss of PTEN cooperate to induce colon cancer in vivo. Oncogene (2014) 33(12):1538–47. doi:10.1038/onc.2013.102
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Niimi H, Pardali K, Vanlandewijck M, Heldin CH, Moustakas A. Notch signaling is necessary for epithelial growth arrest by TGF-beta. J Cell Biol (2007) 176(5):695–707. doi:10.1083/jcb.200612129
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Kluppel M, Wrana JL. Turning it up a Notch: cross-talk between TGF beta and Notch signaling. Bioessays (2005) 27(2):115–8. doi:10.1002/bies.20187
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Masuda S, Kumano K, Shimizu K, Imai Y, Kurokawa M, Ogawa S, et al. Notch1 oncoprotein antagonizes TGF-beta/Smad-mediated cell growth suppression via sequestration of coactivator p300. Cancer Sci (2005) 96(5):274–82. doi:10.1111/j.1349-7006.2005.00048.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Zavadil J, Cermak L, Soto-Nieves N, Bottinger EP. Integration of TGF-beta/Smad and Jagged1/Notch signalling in epithelial-to-mesenchymal transition. EMBO J (2004) 23(5):1155–65. doi:10.1038/sj.emboj.7600069
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Holderfield MT, Hughes CC. Crosstalk between vascular endothelial growth factor, notch, and transforming growth factor-beta in vascular morphogenesis. Circ Res (2008) 102(6):637–52. doi:10.1161/CIRCRESAHA.107.167171
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Bhaskaran N, Souchelnytskyi S. Systemic analysis of TGFbeta proteomics revealed involvement of Plag1/CNK1/RASSF1A/Src network in TGFbeta1-dependent activation of Erk1/2 and cell proliferation. Proteomics (2008) 8(21):4507–20. doi:10.1002/pmic.200700960
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Guo X, Wang XF. Signaling cross-talk between TGF-beta/BMP and other pathways. Cell Res (2009) 19(1):71–88. doi:10.1038/cr.2008.302
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Kretzschmar M, Doody J, Timokhina I, Massague J. A mechanism of repression of TGFbeta/Smad signaling by oncogenic Ras. Genes Dev (1999) 13(7):804–16. doi:10.1101/gad.13.7.804
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Colomiere M, Ward AC, Riley C, Trenerry MK, Cameron-Smith D, Findlay J, et al. Cross talk of signals between EGFR and IL-6R through JAK2/STAT3 mediate epithelial-mesenchymal transition in ovarian carcinomas. Br J Cancer (2009) 100(1):134–44. doi:10.1038/sj.bjc.6604794
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Li CW, Xia W, Huo L, Lim SO, Wu Y, Hsu JL, et al. Epithelial-mesenchymal transition induced by TNF-alpha requires NF-kappaB-mediated transcriptional upregulation of Twist1. Cancer Res (2012) 72(5):1290–300. doi:10.1158/0008-5472.CAN-11-3123
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Ueda Y, Wang S, Dumont N, Yi JY, Koh Y, Arteaga CL. Overexpression of HER2 (erbB2) in human breast epithelial cells unmasks transforming growth factor beta-induced cell motility. J Biol Chem (2004) 279(23):24505–13. doi:10.1074/jbc.M400081200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Vazquez-Martin A, Cufi S, Oliveras-Ferraros C, Torres-Garcia VZ, Corominas-Faja B, Cuyas E, et al. IGF-1R/epithelial-to-mesenchymal transition (EMT) crosstalk suppresses the erlotinib-sensitizing effect of EGFR exon 19 deletion mutations. Sci Rep (2013) 3:2560. doi:10.1038/srep02560
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144(5):646–74. doi:10.1016/j.cell.2011.02.013
81. Tlsty TD, Coussens LM. Tumor stroma and regulation of cancer development. Annu Rev Pathol (2006) 1:119–50. doi:10.1146/annurev.pathol.1.110304.100224
82. Kim SJ, Uehara H, Karashima T, Shepherd DL, Killion JJ, Fidler IJ. Blockade of epidermal growth factor receptor signaling in tumor cells and tumor-associated endothelial cells for therapy of androgen-independent human prostate cancer growing in the bone of nude mice. Clin Cancer Res (2003) 9(3):1200–10.
83. Sternlicht MD, Lochter A, Sympson CJ, Huey B, Rougier JP, Gray JW, et al. The stromal proteinase MMP3/stromelysin-1 promotes mammary carcinogenesis. Cell (1999) 98(2):137–46. doi:10.1016/S0092-8674(00)81009-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Radisky DC, Levy DD, Littlepage LE, Liu H, Nelson CM, Fata JE, et al. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature (2005) 436(7047):123–7. doi:10.1038/nature03688
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Zuo JH, Zhu W, Li MY, Li XH, Yi H, Zeng GQ, et al. Activation of EGFR promotes squamous carcinoma SCC10A cell migration and invasion via inducing EMT-like phenotype change and MMP-9-mediated degradation of E-cadherin. J Cell Biochem (2011) 112(9):2508–17. doi:10.1002/jcb.23175
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Wu Y, Zhou BP. Inflammation: a driving force speeds cancer metastasis. Cell Cycle (2009) 8(20):3267–73. doi:10.4161/cc.8.20.9699
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Allavena P, Sica A, Solinas G, Porta C, Mantovani A. The inflammatory micro-environment in tumor progression: the role of tumor-associated macrophages. Crit Rev Oncol Hematol (2008) 66(1):1–9. doi:10.1016/j.critrevonc.2007.07.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Zheng Y, Yang W, Aldape K, He J, Lu Z. Epidermal growth factor (EGF)-enhanced vascular cell adhesion molecule-1 (VCAM-1) expression promotes macrophage and glioblastoma cell interaction and tumor cell invasion. J Biol Chem (2013) 288(44):31488–95. doi:10.1074/jbc.M113.499020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell (2006) 124(2):263–6. doi:10.1016/j.cell.2006.01.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Zhang C, Carl TF, Trudeau ED, Simmet T, Klymkowsky MW. An NF-kappaB and slug regulatory loop active in early vertebrate mesoderm. PLoS One (2006) 1:e106. doi:10.1371/journal.pone.0000106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Kurtzman SH, Anderson KH, Wang Y, Miller LJ, Renna M, Stankus M, et al. Cytokines in human breast cancer: IL-1alpha and IL-1beta expression. Oncol Rep (1999) 6(1):65–70.
93. Ben-Baruch A. The tumor-promoting flow of cells into, within and out of the tumor site: regulation by the inflammatory axis of TNFalpha and chemokines. Cancer Microenviron (2012) 5(2):151–64. doi:10.1007/s12307-011-0094-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Pantschenko AG, Pushkar I, Anderson KH, Wang Y, Miller LJ, Kurtzman SH, et al. The interleukin-1 family of cytokines and receptors in human breast cancer: implications for tumor progression. Int J Oncol (2003) 23(2):269–84. doi:10.3892/ijo.23.2.269
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Asiedu MK, Ingle JN, Behrens MD, Radisky DC, Knutson KL. TGFbeta/TNF(alpha)-mediated epithelial-mesenchymal transition generates breast cancer stem cells with a claudin-low phenotype. Cancer Res (2011) 71(13):4707–19. doi:10.1158/0008-5472.CAN-10-4554
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Leibovich-Rivkin T, Liubomirski Y, Bernstein B, Meshel T, Ben-Baruch A. Inflammatory factors of the tumor microenvironment induce plasticity in nontransformed breast epithelial cells: EMT, invasion, and collapse of normally organized breast textures. Neoplasia (2013) 15(12):1330–46.
97. Wilson WR, Hay MP. Targeting hypoxia in cancer therapy. Nat Rev Cancer (2011) 11(6):393–410. doi:10.1038/nrc3064
99. Harris AL. Hypoxia – a key regulatory factor in tumour growth. Nat Rev Cancer (2002) 2(1):38–47. doi:10.1038/nrc704
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Zhu QC, Gao RY, Wu W, Qin HL. Epithelial-mesenchymal transition and its role in the pathogenesis of colorectal cancer. Asian Pac J Cancer Prev (2013) 14(5):2689–98. doi:10.7314/APJCP.2013.14.5.2689
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. Vaupel P. The role of hypoxia-induced factors in tumor progression. Oncologist (2004) 9(Suppl 5):10–7. doi:10.1634/theoncologist.9-90005-10
102. Axelson H, Fredlund E, Ovenberger M, Landberg G, Pahlman S. Hypoxia-induced dedifferentiation of tumor cells – a mechanism behind heterogeneity and aggressiveness of solid tumors. Semin Cell Dev Biol (2005) 16(4–5):554–63. doi:10.1016/j.semcdb.2005.03.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
103. Moen I, Oyan AM, Kalland KH, Tronstad KJ, Akslen LA, Chekenya M, et al. Hyperoxic treatment induces mesenchymal-to-epithelial transition in a rat adenocarcinoma model. PLoS One (2009) 4(7):e6381. doi:10.1371/journal.pone.0006381
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
104. Zeisberg M, Neilson EG. Biomarkers for epithelial-mesenchymal transitions. J Clin Invest (2009) 119(6):1429–37. doi:10.1172/JCI36183
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
105. Lu X, Kang Y. Hypoxia and hypoxia-inducible factors: master regulators of metastasis. Clin Cancer Res (2010) 16(24):5928–35. doi:10.1158/1078-0432.CCR-10-1360
106. Martin A, Cano A. Tumorigenesis: Twist1 links EMT to self-renewal. Nat Cell Biol (2010) 12(10):924–5. doi:10.1038/ncb1010-924
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
107. Yang MH, Wu MZ, Chiou SH, Chen PM, Chang SY, Liu CJ, et al. Direct regulation of TWIST by HIF-1alpha promotes metastasis. Nat Cell Biol (2008) 10(3):295–305. doi:10.1038/ncb1691
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. Sahlgren C, Gustafsson MV, Jin S, Poellinger L, Lendahl U. Notch signaling mediates hypoxia-induced tumor cell migration and invasion. Proc Natl Acad Sci U S A (2008) 105(17):6392–7. doi:10.1073/pnas.0802047105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
109. Zhang Q, Bai X, Chen W, Ma T, Hu Q, Liang C, et al. Wnt/beta-catenin signaling enhances hypoxia-induced epithelial-mesenchymal transition in hepatocellular carcinoma via crosstalk with hif-1alpha signaling. Carcinogenesis (2013) 34(5):962–73. doi:10.1093/carcin/bgt027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
110. Lim JH, Chun YS, Park JW. Hypoxia-inducible factor-1alpha obstructs a Wnt signaling pathway by inhibiting the hARD1-mediated activation of beta-catenin. Cancer Res (2008) 68(13):5177–84. doi:10.1158/0008-5472.CAN-07-6234
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
111. Verras M, Papandreou I, Lim AL, Denko NC. Tumor hypoxia blocks Wnt processing and secretion through the induction of endoplasmic reticulum stress. Mol Cell Biol (2008) 28(23):7212–24. doi:10.1128/MCB.00947-08
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
112. Wang D, Wang Y, Kong T, Fan F, Jiang Y. Hypoxia-induced beta-catenin downregulation involves p53-dependent activation of Siah-1. Cancer Sci (2011) 102(7):1322–8. doi:10.1111/j.1349-7006.2011.01950.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
113. Kaidi A, Williams AC, Paraskeva C. Interaction between beta-catenin and HIF-1 promotes cellular adaptation to hypoxia. Nat Cell Biol (2007) 9(2):210–7. doi:10.1038/ncb1534
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
114. Shi X, Guo LW, Seedial SM, Si Y, Wang B, Takayama T, et al. TGF-beta/Smad3 inhibit vascular smooth muscle cell apoptosis through an autocrine signaling mechanism involving VEGF-A. Cell Death Dis (2014) 5:e1317. doi:10.1038/cddis.2014.282
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
115. Liu L, Gao C, Chen G, Li X, Li J, Wan Q, et al. Notch signaling molecules activate TGF-beta in rat mesangial cells under high glucose conditions. J Diabetes Res (2013) 2013:979702. doi:10.1155/2013/979702
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: epithelial–mesenchymal transition, microenvironment, invasion, motility, transforming growth factor-beta
Citation: Lindsey S and Langhans SA (2014) Crosstalk of oncogenic signaling pathways during epithelial–mesenchymal transition. Front. Oncol. 4:358. doi: 10.3389/fonc.2014.00358
Received: 02 October 2014; Paper pending published: 31 October 2014;
Accepted: 27 November 2014; Published online: 11 December 2014.
Edited by:
Andreas Behren, Ludwig Institute for Cancer Research, AustraliaReviewed by:
Frederique Gaits-Iacovoni, Institut National de la Santé et de la Recherche Médicale, FranceHolger Kalthoff, Institute for Experimental Cancer Research, Germany
Aparna Jayachandran, Ludwig Institute for Cancer Research, Australia
Copyright: © 2014 Lindsey and Langhans. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sigrid A. Langhans, Nemours Center for Childhood Cancer Research, Alfred I. duPont Hospital for Children, Rockland Center I, 1701 Rockland Road, Wilmington, DE 19803, USA e-mail: langhans@nemoursresearch.org