 Huifeng Niu
Huifeng Niu Mark Manfredi
Mark Manfredi Jeffrey A. Ecsedy
Jeffrey A. Ecsedy- 1Department of Translational Medicine, Takeda Pharmaceuticals International Co, Cambridge, MA, USA
- 2Department of Oncology Biology, Takeda Pharmaceuticals International Co, Cambridge, MA, USA
Alisertib (MLN8237) is a selective small molecule inhibitor of Aurora A kinase that is being developed in multiple cancer indications as a single agent and in combination with other therapies. A significant amount of research has elucidated a role for Aurora A in orchestrating numerous activities of cells transiting through mitosis and has begun to shed light on potential non-mitotic roles for Aurora A as well. These biological insights laid the foundation for multiple clinical trials evaluating the antitumor activity of alisertib in both solid cancers and heme-lymphatic malignancies. Several key facets of Aurora A biology as well as empirical data collected in experimental systems and early clinical trials have directed the development of alisertib toward certain cancer types, including neuroblastoma, small cell lung cancer, neuroendocrine prostate cancer, atypical teratoid/rhabdoid tumors, and breast cancer among others. In addition, these scientific insights provided the rationale for combining alisertib with other therapies, including microtubule perturbing agents, such as taxanes, EGFR inhibitors, hormonal therapies, platinums, and HDAC inhibitors among others. Here, we link the key aspects of the current clinical development of alisertib to the originating scientific rationale and provide an overview of the alisertib clinical experience to date.
Alisertib: A Highly Selective Aurora A Kinase Inhibitor
Early interest in targeting Aurora A for cancer treatment stemmed in part from the fact that the gene, localized to chromosome 20q13.2, is commonly amplified and overexpressed in a diversity of cancer types (1–7). Aurora A amplification and overexpression is correlated to a worsened prognosis for patients. For example, a meta-analysis study of 5523 cancer patients from thirty-nine studies demonstrated that patients with higher Aurora A expression levels had a significantly worsened survival outcome irrespective of disease type or stage (8). Aurora A overexpression is also thought to drive oncogenesis by causing genomic instability; this proposal is supported by evidence demonstrating that Aurora A overexpression transforms normal cells into cancer cells in experimental studies (7, 9–13). As such, Aurora A has been considered an attractive target for treating cancer and multiple Aurora kinase inhibitors have been developed and tested in cancer patients, including alisertib (MLN8237).
Alisertib is a benzazepine containing small molecule inhibitor of Aurora A (14). In enzymatic, cell and in vivo assays, alisertib has proven to selectively inhibit Aurora A (14). For example, alisertib demonstrated selectivity for Aurora A relative to other kinases in an in vitro screen of 205 kinases, and was >200-fold more potent against Aurora A than the structurally related kinase Aurora B in cellular assays. The selectivity for Aurora A was substantiated by mechanism of action studies in cultured cancer cells and tumors grown as xenografts in immunocompromised mice. Alisertib concentrations that lead to cell cycle arrest and death are preceded by phenotypic changes consistent with Aurora A inhibition; including increased incidence of tetraploid (4N) cells as well as mitotic cells with abnormal mitotic spindles and misaligned chromosomes (Figure 1). Furthermore, alisertib did not affect the viability of cancer cell lines expressing a drug-resistant Aurora A mutation, suggesting that its antitumor activity occurs predominantly through Aurora A inhibition (15).
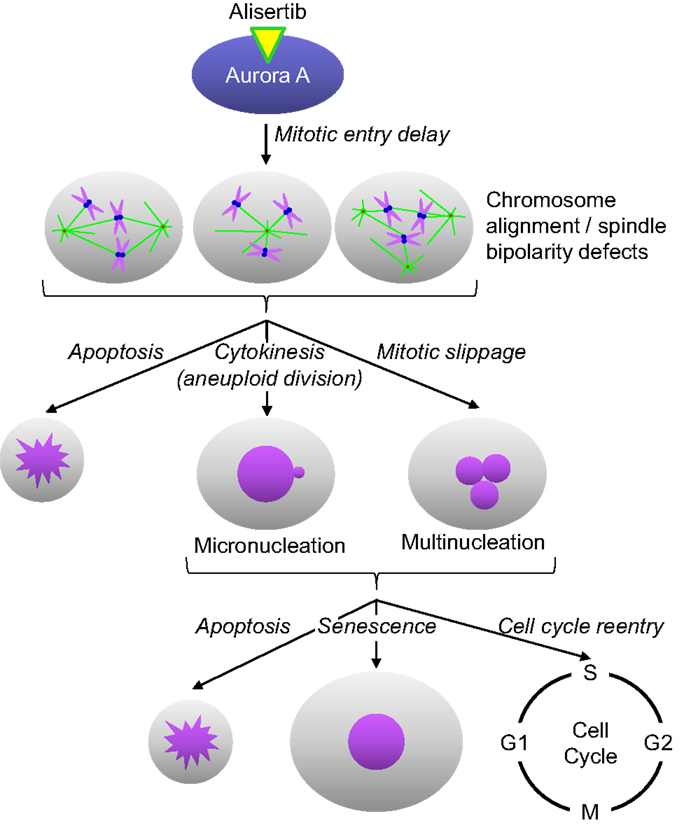
Figure 1. Alisertib mechanism of action. Alisertib selectively binds to and inhibits Aurora A kinase in cells. Inhibition of Aurora A results in delayed mitotic entry and progression through mitosis leading to an accumulation of cells with a tetraploid DNA content. Mitotic cells treated with alisertib display a variety of defects, including monopolar, bipolar, and multipolar spindles, all with misaligned chromosomes. These cells can die directly out of mitosis via apoptosis, undergo aneuploid cytokinesis or exit mitosis without undergoing cytokenesis through a process known as mitotic slippage. The resulting interphase cells typically display gross nuclear defects including micronucleation and multinucleation. These cells will then undergo apoptosis, senescence or reenter the cell cycle; the specific fate is likely dictated by the extent of DNA damage/aneuploidy that occurred in any cells as a result of the abnormal mitotic division as well as other underlying genetic factors.
Alisertib has demonstrated antitumor activity across a broad array of solid cancers and heme-lymphatic experimental tumor models when grown in vitro and in vivo (14, 16–24). In addition, single-agent alisertib has been evaluated in multiple clinical trials and has shown clinical activity across a diversity of cancer types, including solid and hematological cancers in adult and pediatric populations. Though alisertib displays differential antitumor activity across experimental tumor models and in cancer patients, the biological underpinnings for alisertib sensitivity remain unclear. Multiple hypotheses have been developed based on Aurora A biology and data collected in experimental models that predict which cancers will most likely respond to alisertib as a single agent or in combination with other therapeutic agents. In this review, the data supporting some of these concepts is shared.
Early Clinical Studies for Dose/Schedule Selection and Proof of Mechanism
Alisertib has been formulated for oral administration in patients and is available as an enteric-coated tablet and as a liquid solution for pediatric cancers. In two phase 1 studies of alisertib in adults with advanced solid malignancies (25, 26), and in one phase 1 study of alisertib in adults with hematological cancers (27), the single agent maximum tolerated dose was determined to be 50 mg dosed orally twice daily for 7 days followed by 14 days of non-treatment. This dose was selected for further single-agent alisertib evaluation in additional clinical trials of adult cancer patients. Alisertib was also evaluated once daily for 21 days followed by 14 days of non-treatment; 50 mg was the maximum tolerated dose on this schedule (25, 26). The most common dose limiting toxicities (DLTs) observed with alisertib were fatigue, nausea, neutropenia, and stomatitis. These toxicities reflect the pharmacologic activity of alisertib as a cell cycle inhibitor in highly proliferative tissues. Other common alisertib-associated toxicities included alopecia, anorexia, leukopenia, anemia, thrombocytopenia, asthenia, vomiting, diarrhea, and somnolence. The occurrence of somnolence was likely due to the benzodiazepine-like structure of alisertib.
Alisertib has also been evaluated in pediatric cancer patients. This was in part based on the observation that alisertib was active against a range of pediatric tumors grown in vitro and in vivo, in particular, neuroblastoma and acute lymphocytic leukemia (28, 29). In a phase 1 study of children with solid tumors, the maximum tolerated dose of alisertib in children with solid tumors was 80 mg/m2 administered orally once daily for 7 days followed by 14 days of non-treatment (30). The exposures achieved with this dose is approximately 1.5-fold greater than the adult maximum tolerated dose of 50 mg twice daily. Mucositis/stomatitis, mood alteration/depression, neutropenia, and elevated alkaline phosphatase were the DLTs in these patients; neutropenia being the most frequently occurring dose-limiting toxicity. In addition to depression, other mood alterations included impaired memory, agitation, euphoria, and somnolence, predominantly grade 1 and 2. Hand–foot–skin reactions were also observed in these patients.
The selectivity of alisertib for Aurora A relative to Aurora B observed in non-clinical experimental models also translated into the cancer patients. Pharmacodynamic studies performed on tumor biopsies obtained from patients prior to and after alisertib dosing demonstrated an exposure-related decreases in tumor mitotic cells with aligned chromosomes and bipolar spindles in the post-dose samples; phenotypes consistent with Aurora A inhibition (25, 31). Moreover, skin and tumor biopsies taken prior to and after alisertib dosing had increased in mitotic cells in the post-treatment biopsies with serine 10 phosphorylated Histone H3. As serine 10 phosphorylation of histone H3 is catalyzed by Aurora B in cells, these data demonstrate that alisertib does not significantly inhibit Aurora B at the single agent maximum tolerated dose (25, 26, 31). Confirmation of alisertib’s functional selectivity for Aurora A in cancer patients allows for its rational development for treating multiple types of cancers as single agent or in combination with other therapeutic agents.
Population-based pharmacokinetic–pharmacodynamic modeling demonstrate that alisertib steady-state exposures achieved with 50 mg twice daily for 7 days is associated with pharmacodynamic activity in tumors and a low probability for DLTs (31). Moreover, patients with intolerable treatment related toxicities at 50 mg twice daily can be dose reduced to 40 or 30 mg on the same schedule and still maintain tumor pharmacodynamic effects. Overall, multiple tolerated and pharmacodynamically active dose/schedules have been identified in adult and pediatric patients allowing for sufficient flexibility in alisertib dosing that can be taken advantage of for single-agent evaluation and for combining with multiple other therapeutic agents.
Alisertib Single-Agent Rationale
Neuroblastoma
Interest for developing alisertib in neuroblastoma initially derived from an evaluation of alisertib antitumor activity in a large set of pediatric cancer models executed by the Pediatric Preclinical Testing Program which maintains the mission for identifying novel therapies for treating childhood cancers. Alisertib potently inhibited the growth of neuroblastoma cells in vitro and resulted in maintained complete responses in three of seven neuroblastoma xenograft models grown in immunocompromised mice; responses which surpassed the activity of other agents tested in these models (29). Subsequent to these findings it was proposed that Aurora A is essential for the growth and survival of MYCN-amplified neuroblastoma cells. Aurora A binds to and stabilizes N-MYC by protecting it from FBXW7 E3 ubiquitin ligase-mediated proteasomal degradation in a manner that is independent from Aurora A catalytic activity (Figure 2) (32). Furthermore, alisertib and the structurally related molecule MLN8054 bind to Aurora A’s catalytic domain in manner that causes an allosteric shift in the protein thereby disrupting its’ interaction with N-Myc (33, 34). Interestingly, the allosteric shift at the Aurora A/N-Myc interaction site caused by alisertib does not occur with all catalytic inhibitors of Aurora A kinase. Several studies have also demonstrated antitumor activity of Aurora A inhibition in MYCN-amplified neuroblastoma models. For example, treatment of TH-MYCN neuroblastoma mice with MLN8054 resulted in decreased N-Myc protein expression, diminished expression of N-Myc target genes, tumor regressions and increased survival (33). Other Aurora A inhibitors also decreased N-Myc expression resulting in inhibited tumor growth of other MYCN-amplified tumors (34, 35).
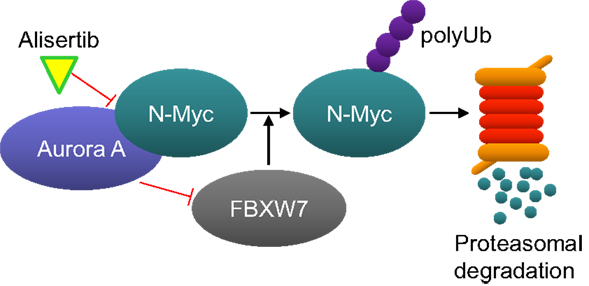
Figure 2. Aurora A protects N-Myc from proteasome-mediated degradation. Aurora A binds to N-Myc, thereby preventing it from being ubiquitinated by the E3-ligase FBXW7. Alisertib binds to Aurora A and changes its confirmation in a manner that disrupts its interaction with N-Myc. N-Myc is than ubiquitinated by FBXW7 and subsequently degraded by the proteasome.
As a result of these findings, the Children’s Oncology Group led a phase 1 study of single-agent alisertib in children with relapsed/recurrent solid tumors including neuroblastoma to determine the maximum tolerated dose, safety profile and pharmacokinetics of alisertib. In this study, 4 out of 11 evaluable neuroblastoma patients treated with alisertib had stable disease (≥6 cycles) (30). As described above, the DLTs in these patients was mucositis, neutropenia, and mood alteration. A phase 2 study of alisertib in young patients with recurrent or refractory solid tumors or leukemias including neuroblastoma has also recently been completed (NCT01154816). Currently, there is an ongoing study being led by the New Approaches to Neuroblastoma Therapy (NANT) consortium in recurrent or resistant neuroblastoma patients combining alisertib with the FDA-approved drugs for neuroblastoma treatment, irinotecan and temozolomide (NCT01601535). In this study, there is a plan to compare MYCN status to patient outcome.
Small Cell Lung Cancer
Similar to neuroblastoma, SCLC has an etiological link to Myc-family of oncogenes including MYC (c-Myc), MYCN (N-Myc) and MYCL1 (L-Myc). Amplification and overexpression of these genes is thought to constitute 18–31% of SCLCs (36–38). Multiple preclinical studies have suggested that SCLCs with Myc activation or amplification are notably sensitive to Aurora kinase inhibitors. For example, SCLC cell lines with MYC, MYCN, and MYCL1 activation or amplification were the most sensitive in a viability screen of 87 cell lines using the dual Aurora A and Aurora B kinase inhibitor PF-03814735 (39). In a separate screen of 34 SCLC cell lines, four structurally diverse Aurora kinase inhibitors VX680, alisertib, PHA680632, and ZM447439 were most effective against the MYC-amplified cell lines (37). Studies with the dual Aurora A and Aurora B kinase inhibitor VX680 demonstrated that it selectively killed human retinal pigment epithelial cells that overexpress c-Myc (40).
In a phase 2 study of single-agent alisertib in five types of advanced refractory or relapsed solid cancers, encouraging activity was seen in SCLC (41). Objective partial responses were observed in 10 of the 48 (21%) SCLC enrolled in this study; these responses occurred in both chemotherapy-sensitive and chemotherapy-resistant disease, the latter which has a worse prognosis. The most common grade 3–4 adverse events in the SCLC patients from this phase 2 study were neutropenia, anemia, leucopenia, and thrombocytopenia, which are consistent with those noted in earlier trials of alisertib. Currently, a phase 2 study of alisertib in combination with paclitaxel compared to placebo in combination with paclitaxel in patients with second line relapsed or refractory SCLC is ongoing (NCT02038647).
Neuroendocrine Prostate Cancer
Neuroendocrine prostate cancer is thought to evolve from late stage prostate adenocarcinoma concurrent to become resistant to hormonal therapy (42, 43). As part of that transition, neuroendocrine prostate cancers become more genomically unstable than prostate adenocarcinoma and include co-amplification of MYCN and Aurora A (44, 45). Given this observation, the relative sensitivity of several prostate adenocarcinoma and neuroendocrine cancer models to the pan-Aurora inhibitor danusertib was tested (44). In a viability screen of four cell lines grown in cell culture, the one neuroendocrine prostate cancer model was significantly more sensitive to danusertib than the three adenocarcinoma cell lines. Danusertib also displayed greater antitumor activity in LNCaP cells transfected with MYCN than vector–control LNCaP cells and was more effective in inhibiting the growth in vivo of a neuroendocrine prostate cancer model relative to an adenocarcinoma model. As a result of these observations, a phase 2 in NEPC is ongoing with single-agent alisertib (NCT01799278).
Atypical Teratoid/Rhabdoid Tumors
Aurora A is a promising target for therapy in ATRT and alisertib has demonstrated to be a potent radiosensitizer in ATRT experimental models (46). ATRT is a rare and highly malignant central nervous system (CNS) tumor usually diagnosed in childhood. ATRT represents around 3% of CNS pediatric cancers and has a high mortality rate with a very poor prognosis. Mutation or deletion of the tumor suppressor gene INI1/hSNF5 occurs in the majority of ATRTs. hSNF5/INI1 is a component of the chromatin remodeling SWI/SNF complex which regulates many proteins involved in chromatin structure. Aurora A is a direct downstream target of hSNF5/INI1. hSNF5/INI1 acts to repress Aurora A expression; as such, loss of INI1/hSNF5 in rhabdoid tumors leads to aberrant overexpression of Aurora A which is required for tumor survival in non-clinical cancer models (47). These preclinical findings supported the use of alisertib for ATRT patients. Wetmore et al. reported an encouraging result for clinical use of alisertib as single agent in recurrent ATRT in four children (48). Patients with recurrent or progressive ATRT received oral administration of alisertib 80 mg/m2 once daily for 7 days of a 21-day treatment cycle. Disease burden was evaluated by brain and spine MRI and by evaluation of spinal fluid cytology (lumbar puncture) after two cycles of alisertib and every 2–3 cycles thereafter for as long as the patients remained free from tumor progression. All four patients had disease stabilization and/or regression after three cycles of alisertib therapy. Two patients on therapy showed stable disease regression for 1 and 2 years. Consistent with other pediatric studies, alisertib in these patients had moderate but manageable toxicities, including neutropenia, leukopenia, thrombocytopenia, anemia, somnolence, and alopecia. Alisertib appears a promising therapeutic agent in this pediatric population. A phase 2 study is ongoing to further evaluate alisertib in the treatment of children with ATRT.
Breast Cancer
Single-agent alisertib efficacy was evaluated in a phase 2 study that comprised five advanced solid tumor indications including breast cancer (41). Among response-evaluable breast cancer patients, objective response (all partial responses) was observed in 9 [18%, 95% confidence interval (CI) = 9–32%] of 49 women with breast cancer. The most common grade 3–4 adverse events in the breast cancer patients from this study included neutropenia, fatigue, leucopenia, and stomatitis. The antitumor activity of alisertib was particularly encouraging in the hormone receptor-positive and HER2-negative subgroups. Median progression-free survival in this subgroup was 7.9 months (95% CI 4.2–12.2). This clinical finding is supported by previously reported preclinical results. D’Assoro et al. demonstrated that Aurora A drives the transition of estrogen receptor α-positive (ERα+) breast cancer cells from an epithelial to a highly invasive mesenchymal phenotype (49). The transition from an epithelial-like to a mesenchymal-like phenotype was characterized by reduced expression of ERα, HER-2/Neu overexpression and loss of CD24 surface receptor (CD24–/low) and overexpression of Aurora A (Figure 3). Aurora A overexpression induces epithelial–mesenchymal transition (EMT) and a cancer stem cell-like phenotype. Inhibition of Aurora A by alisertib in vitro reverses EMT and suppresses the self-renewal ability of CD24–/low breast cancer. Moreover, molecular targeting of Aurora A by shRNA in vivo restores a CD24+ epithelial phenotype and inhibits the development of distant metastases. Other studies demonstrated that increased Aurora A activity may result in anti-hormonal therapy resistance in breast cancer (50). Aurora A induces endocrine resistance through down-regulation of ERα expression in initially ERα+ breast cancer cells (51). In breast cancer patients, high Aurora A expression is associated with poor survival particularly in node-negative ER-positive breast cancer patients (50). Taken together, alisertib could be a novel promising therapeutic agent to selectively eliminate highly invasive cancer cells and improve the disease-free and overall survival of ER-positive breast cancer patients resistant to conventional endocrine therapy.
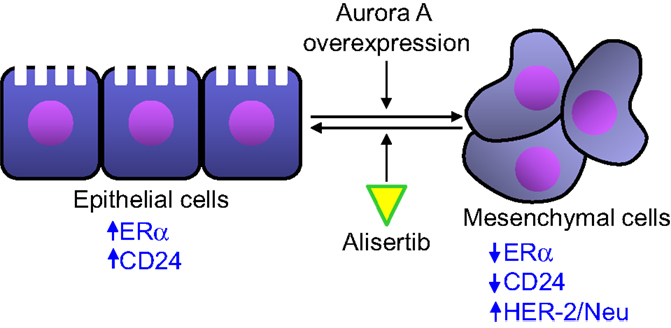
Figure 3. Aurora A activity affects epithelial to mesenchymal transition in breast cancer cells. Aurora A overexpression leads to the transition of breast cancer cell from an epithelial phenotype to a mesenchymal phenotype, leading to decreased ERα and CD24 expression and HER-2/Neu overexpression. Alisertib counteracts the effects of Aurora A overexpression leading to an epithelial to mesenchymal transition reversion.
Alisertib Combination Development Rationale
Taxanes
A considerable amount of data has accumulated in preclinical studies suggesting the benefit of combining Aurora kinase inhibitors with antimicrotubule perturbing agents. This class of anticancer therapies which comprises the taxanes, vinka alkaloids, and the epothilones is among the most commonly used for treating both solid and hematological cancers. Multiple preclinical studies have demonstrated the beneficial combination of inhibiting Aurora kinase with this class of agents (52–60). For example, alisertib combined with the taxanes paclitaxel and docetaxel in triple-negative breast cancer tumors grown as xenografts in immunocompromised mice led to additive or synergistic antitumor activity with prolonged tumor growth delay and in some cases durable complete responses after discontinuing treatment (53). Though the underlying biological underpinnings explaining the beneficial combination between antimicrotubule agents remains uncertain, it has been shown that Aurora A inhibition using MLN8054 or RNA interference in the presence of paclitaxel caused cells to rapidly exit mitosis without completing cytokinesis, presumably due to a disruption of the spindle assembly checkpoint (61).
Alisertib administered as a single agent was evaluated in patients with platinum-resistant or -refractory epithelial ovarian, fallopian tube, or primary peritoneal carcinoma (62). Though active in these diseases as a single agent (overall response rate of 10%, durable for 6.9–11.1 months), the activity was not considered sufficient for further development in ovarian cancer as a single agent. Therefore, alisertib was tested in combination with paclitaxel in relapsed and refractory ovarian cancer (NCT01091428). During the phase 1b portion of this study weekly paclitaxel (QWx3) at 80 mg/m2 and 60 mg/m2 was administered with alisertib dosed twice daily on a 3 days on, 4 days off schedule for three consecutive weeks over 28-day cycles (63). Exposure efficacy modeling was used for selecting the phase 2 dose for this study (53). In addition, alisertib and paclitaxel are being tested in metastatic or locally recurrent breast cancer (NCT02187991) and SCLC (NCT02038647). Numerous other studies have been completed or are ongoing testing alisertib in combination with other microtubule perturbing agents, including Abraxane (nab-paclitaxel) in patients with advanced solid cancers (NCT01677559), docetaxel in patients with advanced solid tumors (NCT01094288), and vincristine and rituximab in patients with relapsed or refractory B-Cell lymphomas (NCT01397825).
EGFR Inhibitors
Epidermal growth factor receptor (EGFR)-targeting antibodies or small molecular EGFR inhibitors are widely used to treat patients with gastrointestinal (GI), breast, head and neck, and lung cancers. However, the clinical efficacy of these agents is limited by intrinsic and acquired resistance factors. Astsaturov and colleagues employed a synthetic lethal screening method and identified Aurora A as a promising hit necessary for cells to survive in the presence of an EGFR inhibitor (64). In addition, they observed synergistic activity of combined inhibition of the EGFR and Aurora A pathways in cancer cells. Combination of erlotinib and alisertib showed synergistic antitumor activity in vitro and in vivo in lung cancer models (65). Furthermore, Aurora A and EGFR protein expression were assessed by immunohistochemistry in patients with squamous cell cancer of the head and neck (SCCHN) (n = 180). Co-expression of elevated levels of Aurora A and EGFR was a poor prognostic factor in SCCHN (66). Recently, Crystal and colleagues established patient-derived resistant NSCLC models to identify effective drug combinations (67). Aurora kinase inhibitors were active in combination with EGFR inhibition in a number of EGFR-mutant cell lines. These data together suggest a potential benefit of such combination therapy in patients. Currently, there is an ongoing phase 1 study evaluating the safety and tolerability of combining alisertib with erlotinib in patients with non-SCLC (NCT01471964).
Hormonal Therapy in Breast Cancer
A number of evidences suggest alisertib may be a rationale combination partner for hormonal therapy. First, promising alisertib single-agent activity was observed in ER-positive and HER2-negative patients as described above (41); second, Aurora A plays a role in the development of endocrine resistance through activation of SMAD5 nuclear signaling and down-regulation of ERα expression in initially ERα+ breast cancer cells (51); and third, aromatase inhibitors (AIs) are used for treatment of ER-positive breast cancer though resistance to AI is a major obstacle to optimal patient outcome. Aurora A is upregulated in AI-resistant cell lines and knockdown studies of Aurora A have shown that it is essential for AI-resistant cell growth. In AI-resistant cell lines, alisertib blocked cell cycle progression at the G2/M phase, interfered with chromosome alignment and spindle pole formation, and preferentially inhibited AI-resistant cell growth compared with parental control cells (68). Furthermore, combination of Aurora inhibitors (alisertib, JNJ-7706621, or danusertib) with fulvestrant is superior to treatment with either of the compounds alone, particularly in AI-resistant cell lines (68). Importantly, this combination may have minimal overlapping toxicities in breast cancer patients. A phase 1/2 trial of alisertib in combination with fulvestrant in patients with hormone receptor-positive metastatic or locally advanced breast cancer is ongoing (NCT02219789).
Platinums
Platinum-based drugs continue to be the mainstay of therapy for many cancers, such as ovarian and lung cancers; however, chemoresistance (intrinsic or acquired) is a major limitation for platinums as it is for other therapies. Increasing evidence suggests a role of Aurora A in platinum resistance. Elevated expression of Aurora A is associated with poor prognosis in epithelial ovarian cancer patients (69) and high Aurora A expression is correlated with cisplatin-based chemotherapeutic resistance and predicts poor patient overall survival (OS) and progression-free survival in NSCLC (70). Moreover, forced expression of Aurora A increased the resistance of the lung cancer cells to cisplatin and knocked down of Aurora A expression in the cisplatin resistant cells by siRNA resulted in a significantly enhanced sensitivity to cisplatin (70). In addition, combination of alisertib and cisplatin resulted in enhanced antitumor activity in vivo in multiple preclinical models (21). In a recent phase 2 clinical trial, alisertib exhibits encouraging single-agent activity in SCLCs, particularly in refractory or chemotherapy-resistant/relapsed patients as described above. Three of twelve patients with refractory or chemotherapy-resistant disease had objective responses to alisertib (41). In earlier studies, alisertib also showed modest single-agent antitumor activity in patients with platinum-resistant ovarian cancers (62). Combination of alisertib with platinums may be a viable strategy for the treatment of patients with platinum-resistant recurrent SCLC and ovarian cancers.
HDAC Inhibitors
Alisertib has shown promising single-agent antitumor efficacy in a phase 2 trial for the treatment of various hematological malignances (71). The overall response rate was 27% (10% CRs) including 100% (1/1) in Burkitt lymphoma (BL), 29% (6/21) in diffuse large B cell lymphoma (DLBCL), and 50% (4/8) in peripheral T-cell lymphoma (PTCL). Recent data from a phase 2 study of alisertib in PTCL led by the South West Oncology Group (SWOG) showed two complete responses and seven partial responses and a response rate (ORR) of 24%. Among the most common subtypes (PTCL NOS, AITL, and ALCL), the ORR was 33% (72). Similar to previously described data with alisertib, myelosuppression was a common adverse effect and constituted the predominant toxicity requiring dose reduction. Mucositis, anorexia, and diarrhea occurred in less than one-quarter of patients and were largely grade 1 or 2 in severity. Grade 1 or 2 fatigue was also common, being observed in nearly half of patients. Nonetheless, two responding patients in this trial received alisertib for 1 year. On the basis of these results, a global phase 3, randomized registration-enabling trial (NCT01482962) was initiated comparing alisertib with investigator’s choice (gemcitabine, pralatrexate, or romidepsin) in patients with relapsed/refractory PTCL. This study was discontinued as a pre-specified interim analysis indicated that the study was unlikely to meet the primary endpoint of superior progression-free survival (PFS) over the standard of care in this treatment setting, although single-agent activity of alisertib was confirmed. In this phase 3 study, alisertib showed a similar ORR compared to the control arm.
The histone deacetylase (HDAC) inhibitors (vorinostat and romidepsin) were approved in the United States for the treatment of cutaneous T-cell lymphoma and romidepsin for the treatment of PTCL. Preclinical data support combining Aurora A inhibitors with HDAC inhibitors. For example, several studies demonstrated that HDAC inhibitors reduce Aurora A expression leading to arrest in the G2/M portion of the cell cycle, abnormal mitotic spindles and followed by apoptosis (73–75). The pan-Aurora kinase inhibitor MK-0457 in combination with the vorinostat enhanced lymphoma cell death through repression of c-Myc and c-Myc responsive micro RNAs (76). Alisertib also demonstrated synergistic antitumor activity when combined with romidepsin in experimental models of T-cell lymphoma (77). Alisertib selectively synergizes with romidepsin by inducing cytokinesis failure in T-cell lymphoma. Cytokinesis failure was confirmed after a corresponding post-treatment increase in CENP-A protein levels. CENP-A is a chromatin-associated protein and plays a role in the final stages of cytokinesis. Overall, these collective data provide a rationale for evaluating alisertib in combination with romidepsin in patients with multiple lymphoma subtypes. A phase 1 trial of alisertib plus romidepsin for relapsed/refractory aggressive B- and T-cell lymphoma is ongoing (NCT01897012).
Summary
To date, many clinical studies have been conducted to evaluate antitumor efficacy of alisertib in patients with diverse solid tumors or hematologic malignancies. Treatment related adverse events (in ≥10% of patients) of single-agent alisertib are summarized in Table 1 (25, 26, 41). Although alisertib has shown single-agent clinical activity in multiple tumor settings, identification of appropriate combination partners and sensitive patient populations is required to ensure that an acceptable risk/benefit profile can be achieved. Aurora A has been implicated in the development of resistance to multiple chemotherapies and targeted agents and preclinical data suggest that alisertib can be combined with multiple therapies to yield additive or synergistic antitumor activity. Furthermore, combinations with targeted therapies might yield more favorable clinical risk/benefit profile than combinations with chemotherapeutic partners due to decreased risk for overlapping toxicities. Lastly, identification of potential predictive biomarkers for alisertib will significantly increase the likelihood of expanding the clinical risk/benefit profile. As such, many correlative studies are ongoing to identify predictive biomarkers which could lead to a precision medicine strategy for alisertib.
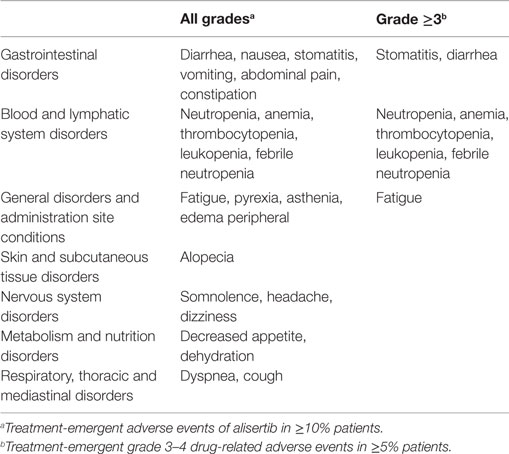
Table 1. Most common treatment-emergent adverse events of alisertib dosed at 50 mg orally twice daily for 7 days followed by 14 days of non-treatment.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Bischoff JR, Anderson L, Zhu Y, Mossie K, Ng L, Souza B, et al. A homologue of Drosophila Aurora kinase is oncogenic and amplified in human colorectal cancers. EMBO J (1998) 17:3052–65. doi:10.1093/emboj/17.11.3052
2. Camacho E, Bea S, Salaverria I, Lopez-Guillermo A, Puig X, Benavente Y, et al. Analysis of Aurora-A and hMPS1 mitotic kinases in mantle cell lymphoma. Int J Cancer (2006) 118:357–63. doi:10.1002/ijc.21370
3. Ikezoe T, Yang J, Nishioka C, Tasaka T, Taniguchi A, Kuwayama Y, et al. A novel treatment strategy targeting Aurora kinases in acute myelogenous leukemia. Mol Cancer Ther (2007) 6:1851–7. doi:10.1158/1535-7163.MCT-07-0067
4. Miyoshi Y, Iwao K, Egawa C, Noguchi S. Association of centrosomal kinase STK15/BTAK mRNA expression with chromosomal instability in human breast cancers. Int J Cancer (2001) 92:370–3. doi:10.1002/ijc.1200
5. Sen S, Zhou H, Zhang RD, Yoon DS, Vakar-Lopez F, Ito S, et al. Amplification/overexpression of a mitotic kinase gene in human bladder cancer. J Natl Cancer Inst (2002) 94:1320–9. doi:10.1093/jnci/94.17.1320
6. Tanaka T, Kimura M, Matsunaga K, Fukada D, Mori H, Okano Y. Centrosomal kinase AIK1 is overexpressed in invasive ductal carcinoma of the breast. Cancer Res (1999) 59:2041–4.
7. Zhou H, Kuang J, Zhong L, Kuo WL, Gray JW, Sahin A, et al. Tumour amplified kinase STK15/BTAK induces centrosome amplification, aneuploidy and transformation. Nat Genet (1998) 20:189–93. doi:10.1038/2496
8. Zhang J, Li B, Yang Q, Zhang P, Wang H. Prognostic value of Aurora kinase A (AURKA) expression among solid tumor patients: a systematic review and meta-analysis. Jpn J Clin Oncol (2015) 45:629–36. doi:10.1093/jjco/hyv058
9. Goepfert TM, Adigun YE, Zhong L, Gay J, Medina D, Brinkley WR. Centrosome amplification and overexpression of Aurora A are early events in rat mammary carcinogenesis. Cancer Res (2002) 62:4115–22.
10. Li CC, Chu HY, Yang CW, Chou CK, Tsai TF. Aurora-A overexpression in mouse liver causes p53-dependent premitotic arrest during liver regeneration. Mol Cancer Res (2009) 7:678–88. doi:10.1158/1541-7786.MCR-08-0483
11. Wang X, Zhou YX, Qiao W, Tominaga Y, Ouchi M, Ouchi T, et al. Overexpression of Aurora kinase A in mouse mammary epithelium induces genetic instability preceding mammary tumor formation. Oncogene (2006) 25:7148–58. doi:10.1038/sj.onc.1209707
12. Wang XX, Liu R, Jin SQ, Fan FY, Zhan QM. Overexpression of Aurora-A kinase promotes tumor cell proliferation and inhibits apoptosis in esophageal squamous cell carcinoma cell line. Cell Res (2006) 16:356–66. doi:10.1038/sj.cr.7310046
13. Zhang D, Shimizu T, Araki N, Hirota T, Yoshie M, Ogawa K, et al. Aurora A overexpression induces cellular senescence in mammary gland hyperplastic tumors developed in p53-deficient mice. Oncogene (2008) 27:4305–14. doi:10.1038/onc.2008.76
14. Manfredi MG, Ecsedy JA, Chakravarty A, Silverman L, Zhang M, Hoar KM, et al. Characterization of Alisertib (MLN8237), an investigational small-molecule inhibitor of Aurora A kinase using novel in vivo pharmacodynamic assays. Clin Cancer Res (2011) 17:7614–24. doi:10.1158/1078-0432.CCR-11-1536
15. Sloane DA, Trikic MZ, Chu ML, Lamers MB, Mason CS, Mueller I, et al. Drug-resistant Aurora A mutants for cellular target validation of the small molecule kinase inhibitors MLN8054 and MLN8237. ACS Chem Biol (2010) 5:563–76. doi:10.1021/cb100053q
16. Do TV, Xiao F, Bickel LE, Klein-Szanto AJ, Pathak HB, Hua X, et al. Aurora kinase A mediates epithelial ovarian cancer cell migration and adhesion. Oncogene (2014) 33:539–49. doi:10.1038/onc.2012.632
17. Gorgun G, Calabrese E, Hideshima T, Ecsedy J, Perrone G, Mani M, et al. A novel Aurora-A kinase inhibitor MLN8237 induces cytotoxicity and cell-cycle arrest in multiple myeloma. Blood (2010) 115:5202–13. doi:10.1182/blood-2009-12-259523
18. Kelly KR, Ecsedy J, Medina E, Mahalingam D, Padmanabhan S, Nawrocki ST, et al. The novel Aurora A kinase inhibitor MLN8237 is active in resistant chronic myeloid leukaemia and significantly increases the efficacy of nilotinib. J Cell Mol Med (2011) 15:2057–70. doi:10.1111/j.1582-4934.2010.01218.x
19. Muscal JA, Scorsone KA, Zhang L, Ecsedy JA, Berg SL. Additive effects of vorinostat and MLN8237 in pediatric leukemia, medulloblastoma, and neuroblastoma cell lines. Invest New Drugs (2013) 31:39–45. doi:10.1007/s10637-012-9831-9
20. Neel NF, Stratford JK, Shinde V, Ecsedy JA, Martin TD, Der CJ, et al. Response to MLN8237 in pancreatic cancer is not dependent on RalA phosphorylation. Mol Cancer Ther (2014) 13:122–33. doi:10.1158/1535-7163.MCT-12-1232
21. Sehdev V, Peng D, Soutto M, Washington MK, Revetta F, Ecsedy J, et al. The Aurora kinase A inhibitor MLN8237 enhances cisplatin-induced cell death in esophageal adenocarcinoma cells. Mol Cancer Ther (2012) 11:763–74. doi:10.1158/1535-7163.MCT-11-0623
22. Van Brocklyn JR, Wojton J, Meisen WH, Kellough DA, Ecsedy JA, Kaur B, et al. Aurora-A inhibition offers a novel therapy effective against intracranial glioblastoma. Cancer Res (2014) 74:5364–70. doi:10.1158/0008-5472.CAN-14-0386
23. Vilgelm AE, Pawlikowski JS, Liu Y, Hawkins OE, Davis TA, Smith J, et al. Mdm2 and Aurora kinase a inhibitors synergize to block melanoma growth by driving apoptosis and immune clearance of tumor cells. Cancer Res (2015) 75:181–93. doi:10.1158/0008-5472.CAN-14-2405
24. Zhou N, Singh K, Mir MC, Parker Y, Lindner D, Dreicer R, et al. The investigational Aurora kinase A inhibitor MLN8237 induces defects in cell viability and cell-cycle progression in malignant bladder cancer cells in vitro and in vivo. Clin Cancer Res (2013) 19:1717–28. doi:10.1158/1078-0432.CCR-12-2383
25. Cervantes A, Elez E, Roda D, Ecsedy J, Macarulla T, Venkatakrishnan K, et al. Phase I pharmacokinetic/pharmacodynamic study of MLN8237, an investigational, oral, selective Aurora A kinase inhibitor, in patients with advanced solid tumors. Clin Cancer Res (2012) 18:4764–74. doi:10.1158/1078-0432.CCR-12-0571
26. Dees EC, Cohen RB, von Mehren M, Stinchcombe TE, Liu H, Venkatakrishnan K, et al. Phase I study of Aurora A kinase inhibitor MLN8237 in advanced solid tumors: safety, pharmacokinetics, pharmacodynamics, and bioavailability of two oral formulations. Clin Cancer Res (2012) 18:4775–84. doi:10.1158/1078-0432.CCR-12-0589
27. Kelly KR, Shea TC, Goy A, Berdeja JG, Reeder CB, McDonagh KT, et al. Phase I study of MLN8237 – investigational Aurora A kinase inhibitor – in relapsed/refractory multiple myeloma, non-Hodgkin lymphoma and chronic lymphocytic leukemia. Invest New Drugs (2014) 32:489–99. doi:10.1007/s10637-013-0050-9
28. Carol H, Boehm I, Reynolds CP, Kang MH, Maris JM, Morton CL, et al. Efficacy and pharmacokinetic/pharmacodynamic evaluation of the Aurora kinase A inhibitor MLN8237 against preclinical models of pediatric cancer. Cancer Chemother Pharmacol (2011) 68:1291–304. doi:10.1007/s00280-011-1618-8
29. Maris JM, Morton CL, Gorlick R, Kolb EA, Lock R, Carol H, et al. Initial testing of the Aurora kinase A inhibitor MLN8237 by the pediatric preclinical testing program (PPTP). Pediatr Blood Cancer (2010) 55:26–34. doi:10.1002/pbc.22430
30. Mosse YP, Lipsitz E, Fox E, Teachey DT, Maris JM, Weigel B, et al. Pediatric phase I trial and pharmacokinetic study of MLN8237, an investigational oral selective small-molecule inhibitor of Aurora kinase A: a Children’s Oncology Group Phase I Consortium study. Clin Cancer Res (2012) 18:6058–64. doi:10.1158/1078-0432.CCR-11-3251
31. Venkatakrishnan K, Zhou X, Ecsedy J, Mould DR, Liu H, Danaee H, et al. Dose selection for the investigational anticancer agent alisertib (MLN8237): pharmacokinetics, pharmacodynamics, and exposure-safety relationships. J Clin Pharmacol (2015) 55(3):336–47. doi:10.1002/jcph.410
32. Otto T, Horn S, Brockmann M, Eilers U, Schuttrumpf L, Popov N, et al. Stabilization of N-Myc is a critical function of Aurora A in human neuroblastoma. Cancer Cell (2009) 15:67–78. doi:10.1016/j.ccr.2008.12.005
33. Brockmann M, Poon E, Berry T, Carstensen A, Deubzer HE, Rycak L, et al. Small molecule inhibitors of Aurora-A induce proteasomal degradation of N-myc in childhood neuroblastoma. Cancer Cell (2013) 24:75–89. doi:10.1016/j.ccr.2013.05.005
34. Gustafson WC, Meyerowitz JG, Nekritz EA, Chen J, Benes C, Charron E, et al. Drugging MYCN through an allosteric transition in Aurora kinase A. Cancer Cell (2014) 26:414–27. doi:10.1016/j.ccr.2014.07.015
35. Faisal A, Vaughan L, Bavetsias V, Sun C, Atrash B, Avery S, et al. The Aurora kinase inhibitor CCT137690 downregulates MYCN and sensitizes MYCN-amplified neuroblastoma in vivo. Mol Cancer Ther (2011) 10:2115–23. doi:10.1158/1535-7163.MCT-11-0333
36. Peifer M, Fernandez-Cuesta L, Sos ML, George J, Seidel D, Kasper LH, et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat Genet (2012) 44:1104–10. doi:10.1038/ng.2396
37. Sos ML, Dietlein F, Peifer M, Schottle J, Balke-Want H, Muller C, et al. A framework for identification of actionable cancer genome dependencies in small cell lung cancer. Proc Natl Acad Sci U S A (2012) 109:17034–9. doi:10.1073/pnas.1207310109
38. Wistuba II, Gazdar AF, Minna JD. Molecular genetics of small cell lung carcinoma. Semin Oncol (2001) 28:3–13. doi:10.1053/sonc.2001.25738
39. Hook KE, Garza SJ, Lira ME, Ching KA, Lee NV, Cao J, et al. An integrated genomic approach to identify predictive biomarkers of response to the Aurora kinase inhibitor PF-03814735. Mol Cancer Ther (2012) 11:710–9. doi:10.1158/1535-7163.MCT-11-0184
40. Yang D, Liu H, Goga A, Kim S, Yuneva M, Bishop JM. Therapeutic potential of a synthetic lethal interaction between the MYC proto-oncogene and inhibition of Aurora-B kinase. Proc Natl Acad Sci U S A (2010) 107:13836–41. doi:10.1073/pnas.1008366107
41. Melichar B, Adenis A, Lockhart AC, Bennouna J, Dees EC, Kayaleh O, et al. Safety and activity of alisertib, an investigational Aurora kinase A inhibitor, in patients with breast cancer, small-cell lung cancer, non-small-cell lung cancer, head and neck squamous-cell carcinoma, and gastro-oesophageal adenocarcinoma: a five-arm phase 2 study. Lancet Oncol (2015) 16:395–405. doi:10.1016/S1470-2045(15)70051-3
42. Grigore AD, Ben-Jacob E, Farach-Carson MC. Prostate cancer and neuroendocrine differentiation: more neuronal, less endocrine? Front Oncol (2015) 5:37. doi:10.3389/fonc.2015.00037
43. Hirano D, Okada Y, Minei S, Takimoto Y, Nemoto N. Neuroendocrine differentiation in hormone refractory prostate cancer following androgen deprivation therapy. Eur Urol (2004) 45:586–92. doi:10.1016/j.eururo.2003.11.032
44. Beltran H, Rickman DS, Park K, Chae SS, Sboner A, MacDonald TY, et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov (2011) 1:487–95. doi:10.1158/2159-8290.CD-11-0130
45. Mosquera JM, Beltran H, Park K, MacDonald TY, Robinson BD, Tagawa ST, et al. Concurrent AURKA and MYCN gene amplifications are harbingers of lethal treatment-related neuroendocrine prostate cancer. Neoplasia (2013) 15:1–10. doi:10.1593/neo.121550
46. Venkataraman S, Alimova I, Tello T, Harris PS, Knipstein JA, Donson AM, et al. Targeting Aurora kinase A enhances radiation sensitivity of atypical teratoid rhabdoid tumor cells. J Neurooncol (2012) 107:517–26. doi:10.1007/s11060-011-0795-y
47. Lee S, Cimica V, Ramachandra N, Zagzag D, Kalpana GV. Aurora A is a repressed effector target of the chromatin remodeling protein INI1/hSNF5 required for rhabdoid tumor cell survival. Cancer Res (2011) 71:3225–35. doi:10.1158/0008-5472.CAN-10-2167
48. Wetmore C, Boyett J, Li S, Lin T, Bendel A, Gajjar A, et al. Alisertib is active as single agent in recurrent atypical teratoid rhabdoid tumors in 4 children. Neuro Oncol (2015) 17:882–8. doi:10.1093/neuonc/nov017
49. D’Assoro AB, Liu T, Quatraro C, Amato A, Opyrchal M, Leontovich A, et al. The mitotic kinase Aurora – a promotes distant metastases by inducing epithelial-to-mesenchymal transition in ERalpha(+) breast cancer cells. Oncogene (2014) 33:599–610. doi:10.1038/onc.2012.628
50. Siggelkow W, Boehm D, Gebhard S, Battista M, Sicking I, Lebrecht A, et al. Expression of Aurora kinase A is associated with metastasis-free survival in node-negative breast cancer patients. BMC Cancer (2012) 12:562. doi:10.1186/1471-2407-12-562
51. Opyrchal M, Salisbury JL, Zhang S, McCubrey J, Hawse J, Goetz MP, et al. Aurora-A mitotic kinase induces endocrine resistance through down-regulation of ERalpha expression in initially ERalpha+ breast cancer cells. PLoS One (2014) 9:e96995. doi:10.1371/journal.pone.0096995
52. Hata T, Furukawa T, Sunamura M, Egawa S, Motoi F, Ohmura N, et al. RNA interference targeting Aurora kinase a suppresses tumor growth and enhances the taxane chemosensitivity in human pancreatic cancer cells. Cancer Res (2005) 65:2899–905. doi:10.1158/0008-5472.CAN-04-3981
53. Huck JJ, Zhang M, Mettetal J, Chakravarty A, Venkatakrishnan K, Zhou X, et al. Translational exposure-efficacy modeling to optimize the dose and schedule of taxanes combined with the investigational Aurora A kinase inhibitor MLN8237 (alisertib). Mol Cancer Ther (2014) 13:2170–83. doi:10.1158/1535-7163.MCT-14-0027
54. Lentini L, Amato A, Schillaci T, Insalaco L, Di Leonardo A. Aurora-A transcriptional silencing and vincristine treatment show a synergistic effect in human tumor cells. Oncol Res (2008) 17:115–25.
55. Mahadevan D, Stejskal A, Cooke LS, Manziello A, Morales C, Persky DO, et al. Aurora A inhibitor (MLN8237) plus vincristine plus rituximab is synthetic lethal and a potential curative therapy in aggressive B-cell non-Hodgkin lymphoma. Clin Cancer Res (2012) 18:2210–9. doi:10.1158/1078-0432.CCR-11-2413
56. Qi W, Cooke LS, Liu X, Rimsza L, Roe DJ, Manziolli A, et al. Aurora inhibitor MLN8237 in combination with docetaxel enhances apoptosis and anti-tumor activity in mantle cell lymphoma. Biochem Pharmacol (2011) 81:881–90. doi:10.1016/j.bcp.2011.01.017
57. Sehdev V, Katsha A, Ecsedy J, Zaika A, Belkhiri A, El-Rifai W. The combination of alisertib, an investigational Aurora kinase A inhibitor, and docetaxel promotes cell death and reduces tumor growth in preclinical cell models of upper gastrointestinal adenocarcinomas. Cancer (2013) 119:904–14. doi:10.1002/cncr.27801
58. Shimomura T, Hasako S, Nakatsuru Y, Mita T, Ichikawa K, Kodera T, et al. MK-5108, a highly selective Aurora-A kinase inhibitor, shows antitumor activity alone and in combination with docetaxel. Mol Cancer Ther (2010) 9:157–66. doi:10.1158/1535-7163.MCT-09-0609
59. Tanaka E, Hashimoto Y, Ito T, Kondo K, Higashiyama M, Tsunoda S, et al. The suppression of Aurora-A/STK15/BTAK expression enhances chemosensitivity to docetaxel in human esophageal squamous cell carcinoma. Clin Cancer Res (2007) 13:1331–40. doi:10.1158/1078-0432.CCR-06-1192
60. Yang J, Ikezoe T, Nishioka C, Tasaka T, Taniguchi A, Kuwayama Y, et al. AZD1152, a novel and selective Aurora B kinase inhibitor, induces growth arrest, apoptosis, and sensitization for tubulin depolymerizing agent or topoisomerase II inhibitor in human acute leukemia cells in vitro and in vivo. Blood (2007) 110:2034–40. doi:10.1182/blood-2007-02-073700
61. Wysong DR, Chakravarty A, Hoar K, Ecsedy JA. The inhibition of Aurora A abrogates the mitotic delay induced by microtubule perturbing agents. Cell Cycle (2009) 8:876–88. doi:10.4161/cc.8.6.7897
62. Matulonis UA, Sharma S, Ghamande S, Gordon MS, Del Prete SA, Ray-Coquard I, et al. Phase II study of MLN8237 (alisertib), an investigational Aurora A kinase inhibitor, in patients with platinum-resistant or -refractory epithelial ovarian, fallopian tube, or primary peritoneal carcinoma. Gynecol Oncol (2012) 127:63–9. doi:10.1016/j.ygyno.2012.06.040
63. Falchook G, Goff B, Kurzrock R, Martin LP, Gray HJ, Coleman RL, et al. Oral MLN8237 with weekly paclitaxel in patients with recurrent epithelial ovarian, fallopian tube, primary peritoneal (OC) or breast cancer (BRC): phase 1 results. Int J Gynecol Cancer (2013) 23:131–2.
64. Astsaturov I, Ratushny V, Sukhanova A, Einarson MB, Bagnyukova T, Zhou Y, et al. Synthetic lethal screen of an EGFR-centered network to improve targeted therapies. Sci Signal (2010) 3:ra67. doi:10.1126/scisignal.2001083
65. Zhang J, Pan X, Astsaturov A, Egleston B, Borghaei H. Relationship of Aurora-A kinase and EGFR inhibition in non-small cell lung cancer (NSCLC) cell lines. J Clin Oncol (2011) 29:e13038.
66. Hoellein A, Pickhard A, von Keitz F, Schoeffmann S, Piontek G, Rudelius M, et al. Aurora kinase inhibition overcomes cetuximab resistance in squamous cell cancer of the head and neck. Oncotarget (2011) 2:599–609.
67. Crystal AS, Shaw AT, Sequist LV, Friboulet L, Niederst MJ, Lockerman EL, et al. Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science (2014) 346:1480–6. doi:10.1126/science.1254721
68. Hole S, Pedersen AM, Lykkesfeldt AE, Yde CW. Aurora kinase A and B as new treatment targets in aromatase inhibitor-resistant breast cancer cells. Breast Cancer Res Treat (2015) 149:715–26. doi:10.1007/s10549-015-3284-8
69. Landen CN Jr, Lin YG, Immaneni A, Deavers MT, Merritt WM, Spannuth WA, et al. Overexpression of the centrosomal protein Aurora-A kinase is associated with poor prognosis in epithelial ovarian cancer patients. Clin Cancer Res (2007) 13:4098–104. doi:10.1158/1078-0432.CCR-07-0431
70. Xu J, Yue CF, Zhou WH, Qian YM, Zhang Y, Wang SW, et al. Aurora-A contributes to cisplatin resistance and lymphatic metastasis in non-small cell lung cancer and predicts poor prognosis. J Transl Med (2014) 12:200. doi:10.1186/1479-5876-12-200
71. Friedberg JW, Mahadevan D, Cebula E, Persky D, Lossos I, Agarwal AB, et al. Phase II study of alisertib, a selective Aurora A kinase inhibitor, in relapsed and refractory aggressive B- and T-cell non-Hodgkin lymphomas. J Clin Oncol (2014) 32:44–50. doi:10.1200/JCO.2012.46.8793
72. Barr PM, Li H, Spier CM, Mahadevan D, Friedberg JW, LeBlanc ML, et al. U.S. Intergroup phase II trial (SWOG 1108) of alisertib, and investigational Aurora A kinase (AAK) inhibitor, in patients with peripheral T-cell lymphoma (PTCL; NCT01466881). J Clin Oncol (2014) 32:8523. doi:10.1200/JCO.2014.60.6327
73. Cha TL, Chuang MJ, Wu ST, Sun GH, Chang SY, Yu DS, et al. Dual degradation of Aurora A and B kinases by the histone deacetylase inhibitor LBH589 induces G2-M arrest and apoptosis of renal cancer cells. Clin Cancer Res (2009) 15:840–50. doi:10.1158/1078-0432.CCR-08-1918
74. Park JH, Jong HS, Kim SG, Jung Y, Lee KW, Lee JH, et al. Inhibitors of histone deacetylases induce tumor-selective cytotoxicity through modulating Aurora-A kinase. J Mol Med (2008) 86:117–28. doi:10.1007/s00109-007-0260-8
75. Zhang XH, Rao M, Loprieato JA, Hong JA, Zhao M, Chen GZ, et al. Aurora A, Aurora B and survivin are novel targets of transcriptional regulation by histone deacetylase inhibitors in non-small cell lung cancer. Cancer Biol Ther (2008) 7:1388–97. doi:10.4161/cbt.7.9.6415
76. Kretzner L, Scuto A, Dino PM, Kowolik CM, Wu J, Ventura P, et al. Combining histone deacetylase inhibitor vorinostat with Aurora kinase inhibitors enhances lymphoma cell killing with repression of c-Myc, hTERT, and microRNA levels. Cancer Res (2011) 71:3912–20. doi:10.1158/0008-5472.CAN-10-2259
Keywords: Aurora, combination therapy, biomarkers, alisertib, mitosis
Citation: Niu H, Manfredi M and Ecsedy JA (2015) Scientific rationale supporting the clinical development strategy for the investigational Aurora A kinase inhibitor alisertib in cancer. Front. Oncol. 5:189. doi: 10.3389/fonc.2015.00189
Received: 28 May 2015; Accepted: 07 August 2015;
Published: 24 August 2015
Edited by:
Ignacio Perez De Castro, Spanish National Cancer Research Centre, SpainReviewed by:
Valerio Donato, New York University Medical Center, USAMaria Jose Lallena, Eli Lilly and Company, USA
Copyright: © 2015 Niu, Manfredi and Ecsedy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jeffrey A. Ecsedy, Takeda Pharmaceuticals International Co, 35 Landsdowne Street, Cambridge, MA 02139, USA, jeffrey.ecsedy@takeda.com