 Jun Zhang
Jun Zhang L. Jeffrey Medeiros
L. Jeffrey Medeiros Ken H. Young
Ken H. Young- 1Department of Hematopathology, The University of Texas MD Anderson Cancer Center, Houston, TX, United States
- 2Graduate School of Biomedical Sciences, University of Texas Health Science Center, Houston, TX, United States
Remarkable progress has been made in the field of cancer immunotherapy in the past few years. Immunotherapy has become a standard treatment option for patients with various cancers, including melanoma, lymphoma, and carcinomas of the lungs, kidneys, bladder, and head and neck. Promising immunotherapy approaches, such as chimeric antigen receptor (CAR) T cell therapy and therapeutic blockade of immune checkpoints, in particular cytotoxic T lymphocyte-associated protein 4 (CTLA-4) and programmed cell death protein 1 pathway (PD-1/PD-L1), have boosted the development of new therapeutic regimens for patients with cancer. Immunotherapeutic strategies for diffuse large B-cell lymphoma (DLBCL) include monoclonal anti-CD20 antibody (rituximab), monoclonal anti-PD-1 antibodies (nivolumab and pembrolizumab), monoclonal anti-PD-L1 antibodies (avelumab, durvalumab, and atezolizumab) and chimeric antigen receptor (CAR) T cell therapy. In this review, we outline the latest highlights and progress in using immunotherapy to treat patients with DLBCL, with a focus on the therapeutic blockade of PD-1/PD-L1 and CAR T cell therapy in DLBCL. We also discuss current clinical trials of PD-1/PD-L1 and CAR T cell therapy and review the challenges and opportunities of using immunotherapy for the treatment of DLBCL.
Introduction
Diffuse large B-cell lymphoma (DLBCL) is the most common type of non-Hodgkin lymphoma. Approximately 60% of DLBCL patients are cured using standard chemotherapy that includes monoclonal anti-CD20 antibody (rituximab), cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP). However, 30–40% of DLBCL patients will develop relapse or have refractory disease that cannot be cured with the standard R-CHOP therapy, indicating the need for more effective therapies for this patient subset. For patients with high-risk DLBCL who often fail R-CHOP therapy, especially patients with high-grade B-cell lymphoma with MYC and BCL2 or BCL6 translocation, dose-adjusted rituximab, etoposide, prednisone, vincristine, cyclophosphamide, and doxorubicin (DA.R-EPOCH) regimen is a commonly used high intensity regimen.
The development of rituximab was an early step in the application of immunotherapy for the treatment of lymphoma, as it was the first monoclonal antibody (mAb) approved by the US Food and Drug Administration (FDA) for the treatment of patients with advanced stage or relapsed low-grade non-Hodgkin lymphoma, in 1997 (1). See comment in PubMed Commons below Rituximab is a chimeric (mouse and human) monoclonal antibody directed against the B-cell antigen CD20. Rituximab acts via a number of mechanisms including direct antibody dependent cellular cytotoxicity, apoptosis induction, and complement mediated cell death (2). Other monoclonal antibodies that target B-cell antigens, such as CD19 and CD22, also have been developed. CD19 is a specific B cell marker widely expressed during all phases of B cell development until terminal differentiation into plasma cells, with a potential efficacy on a large panel of B cell malignancies. Although initial attempts to target CD19 were unsuccessful, accumulated studies demonstrated targeting CD19 has a therapeutic potential for patients with B cell malignancies (3, 4).
More recently, a number of innovative immunotherapy approaches have shown promising results in patients with relapsed or refractory DLBCL, leading to numerous ongoing clinical trials. CTLA-4 is a negative regulator of T-cell activation, which inhibits anti-tumor immune responses. Blockade of CTLA-4 using the monoclonal antibody ipilimumab improves anti-tumor activity. Ipilimumab was the first immune checkpoint inhibitor approved by the US FDA for the treatment of patients with malignant melanoma. However, the role of the CTLA-4 pathway in DLBCL remains to be elucidated. A phase I clinical trial of ipilimumab in 18 patients with relapsed/refractory B-cell NHL included 3 patients with DLBCL (NCT00089076). Two of these patients had clinical responses and 1 achieved a complete response that lasted more than 31 months. In this study, investigators reported that ipilimumab was well tolerated at the doses used, and that ipilimumab has anti-tumor activity resulting in durable responses in a minority of DLBCL patients (5).
Two highly promising strategies designed to harness the immune system to treat patients with DLBCL are therapeutic blockade of the PD-1/PD-L1 pathway and chimeric antigen receptor (CAR) T cell therapy. These approaches are triggering a paradigm shift in cancer immunotherapy.
PD-1/PD-L1 Signaling Pathway
PD-1/PD-L1 pathway blockade with nivolumab, pembrolizumab, atezolizumab, avelumab, and durvalumab has demonstrated activity in multiple solid tumor malignancies (6–17). Monoclonal anti-PD-1 antibody (nivolumab) was granted designation as a breakthrough therapy for the treatment of patients with relapsed or refractory classical Hodgkin lymphoma on May 17, 2016. The FDA recently granted accelerated approval to another monoclonal anti-PD-1 antibody (pembrolizumab) for the treatment of adult and pediatric patients with refractory primary mediastinal large B-cell lymphoma, or who have relapsed after two or more prior lines of therapy (June 13, 2018). More clinical trials of PD-1 and PD-L1 monoclonal antibodies are currently ongoing (Figure 1). Despite the potential activity of PD-1–blocking antibodies in DLBCL, a subset of patients experiences progressive disease after an initial, often short response (18, 19). Additional research is therefore needed to better understand the reasons for host resistance and to prevent immune-related adverse events.
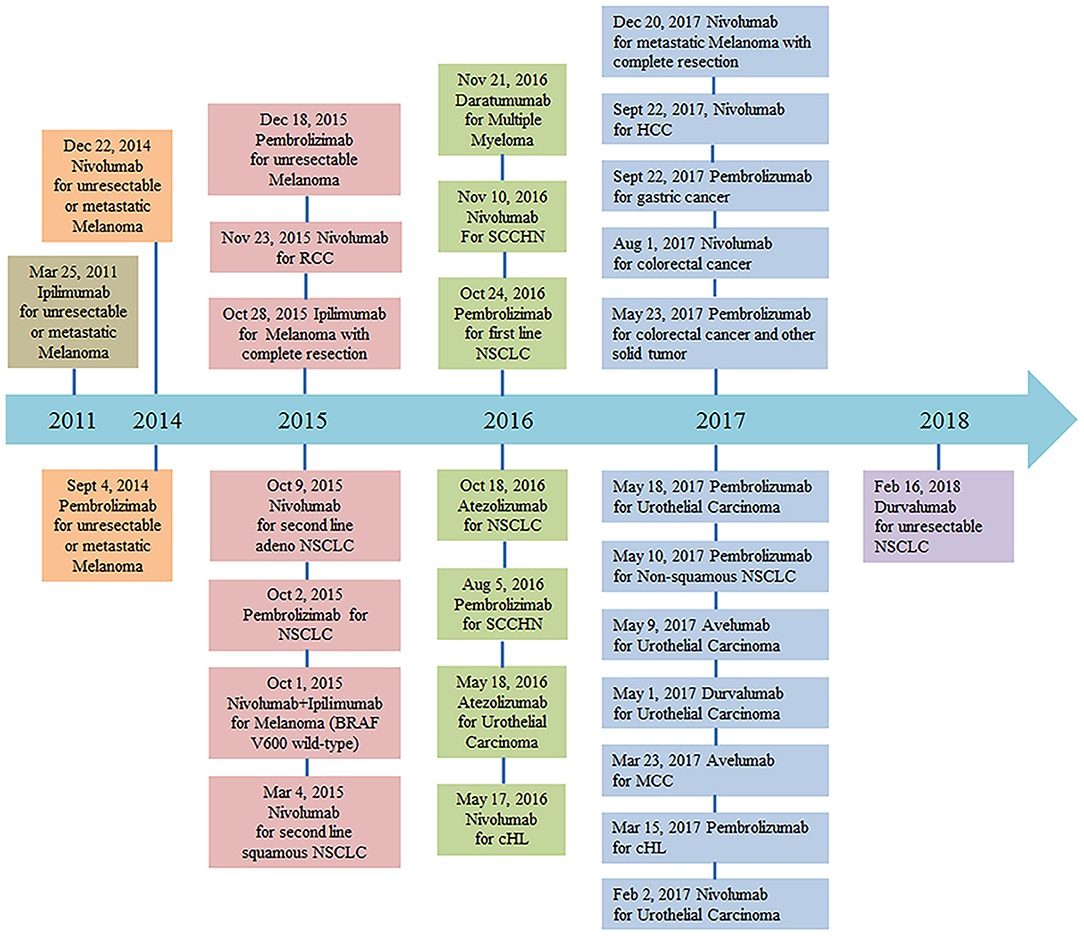
Figure 1. FDA approval timeline of immune checkpoint inhibitors for the treatment of malignancies (https://www.fda.gov/drugs, retrieved Mar 7, 2018). Abbreviations: NSCLC, non–small cell lung cancer; RCC, renal cell carcinoma; cHL, classical Hodgkin Lymphoma; SCCHN, squamous cell carcinoma of the head and neck; MCC, merkel cell carcinoma; HCC, hepatocellular carcinoma.
Mechanisms of PD-1/PD-L1 Signal Pathway Blockade
The immune system protects the body against illness and infection by bacteria, viruses, fungi, or parasites. Simultaneously, the immune system has the capacity to recognize tumors, inhibit tumor development, and eliminate malignant cells. Cancer cells, however, can evolve and therefore escape from immune surveillance and attack. The mechanisms of cancer immune escape mainly include: reducing the expression of tumor antigens; increasing co-inhibitor expression (e.g., PD-L1, CTLA-4) (20) (Figure 2); secreting suppressive cytokines (e.g., TGF-β and IL-10); and lastly orchestrating an immunosuppressive microenvironment (21, 22).
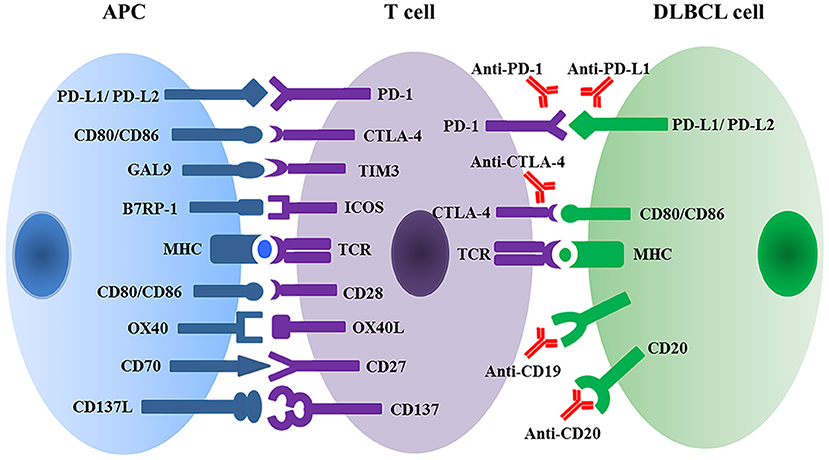
Figure 2. Multiple immune checkpoint and ligand-receptor interactions between T cell and APC or DLBCL malignant cells regulate T cell activation and anti-tumor activity. APC, antigen presenting cell; DLBCL, diffuse large B cell lymphoma; PD1, programmed cell death protein 1; PD-L, programmed cell death ligand; GAL9, galectin 9; TIM3, T cell membrane protein 3; B7RP1, B7-related protein 1; ICOS, inducible T cell co-stimulator; MHC, major histocompatibility complex; TCR, T cell receptor.
PD-1 (CD279), a member of the CD28 and CTLA-4 immunoglobulin superfamily, interacts with two B7 family ligands: PD-L1 (CD274 and also known as B7-H1) and PD-L2 (CD273 and also known as B7-DC). PD-1 is expressed on the surface of activated T cells, B cells, natural killer cells, and macrophages as well as by a large proportion of tumor infiltrating lymphocytes (TILs) (15). PD-1 exerts an important immune checkpoint function in the regulation of T-cell mediated immune responses. PD-1 delivers inhibitory signals that regulate T-cell activation, exhaustion, and tolerance through binding to its ligands PD-L1 and PD-L2. PD-L1 and PD-L2 have distinct patterns of expression (23). PD-L1 is expressed primarily by antigen-presenting cells (APC), as well as by a variety of non-hematopoietic cells and tumor cells. PD-L1 expression is induced by pro-inflammatory cytokines, including type I and type II interferons, tumor necrosis factor α (TNF-α) and vascular endothelial growth factor (VEGF) (24, 25). PD-L2 is expressed primarily by dendritic cells and macrophages, and is induced by IL-4 and granulocyte-macrophage colony-stimulating factor (GM-CSF) (26).
In addition to PD-1, PD-L1 also interacts with CD80 expressed on T cells and inhibits T cell responses, whereas PD-L2 also binds to a novel partner repulsive guidance molecule b (RGMb), and plays an important role in pulmonary tolerance (27). Further investigation is needed to explore how these novel pathways are involved in anti-tumor immune responses.
Negative regulation of the PD-1 pathway may be accomplished via multiple mechanisms. The engagement of PD-1 with PD-L1/PD-L2 may suppress T cell activation by competing directly with CD28 for CD80/CD86 binding, resulting in impaired T cell activation and decreased IL-2 production (28). PD-1 binding to PD-L1/PD-L2 results in tyrosine phosphorylation of the PD-1 cytoplasmic regions ITIM and ITSM, which bind the phosphatases SHP-1 and SHP-2, leading to decreased T cell activation and cytokine production (29). PD-1 signaling also inhibits CD28-mediated activation of phosphatidylinositol 3-kinase (PI3K), leading to decreased activation of Akt and reduced expression of transcription factors associated with cell effector functions including GATA3, T-bet, and Eomes (30). Signaling through PD-1 decreases tyrosine phosphorylation of the TCR ζ chain and ZAP-70 (31). PD-1 signaling inhibits the expression of transcription factors associated with effector cell functions, including GATA-3, T-bet, and Eomes (32).
Clinical Immunotherapy of PD-1/PD-L1 Inhibitors in DLBCL
As already mentioned, 30–40% of DLBCL patients fail standard therapy and have relapsed or refractory disease (33). PD-1 and PD-L1 expression are not usually a striking feature of patients with cancer (34–36), although several studies have reported over-expression of PD-L1 in specific lymphoma subsets (37, 38). Immune blockade of the PD-1/PD-L1 interaction by monoclonal antibodies can restore the antitumor activity of cytotoxic T cells. Immunotherapy using PD-1/PD-L1 inhibitors has become a clinically validated treatment and has produced durable objective responses and improved overall survival (OS) in patients with solid and hematologic neoplasms. Several monoclonal antibodies targeting the PD-1/PD-L1 pathway are currently in early clinical development including two anti-PD-1 antibodies (nivolumab and pembrolizumab) (Table 1), and three anti-PD-L1 antibodies (avelumab, durvalumab, and atezolizumab) (Table 2).

Table 1. Ongoing PD-1 inhibitors trials in DLBCL.
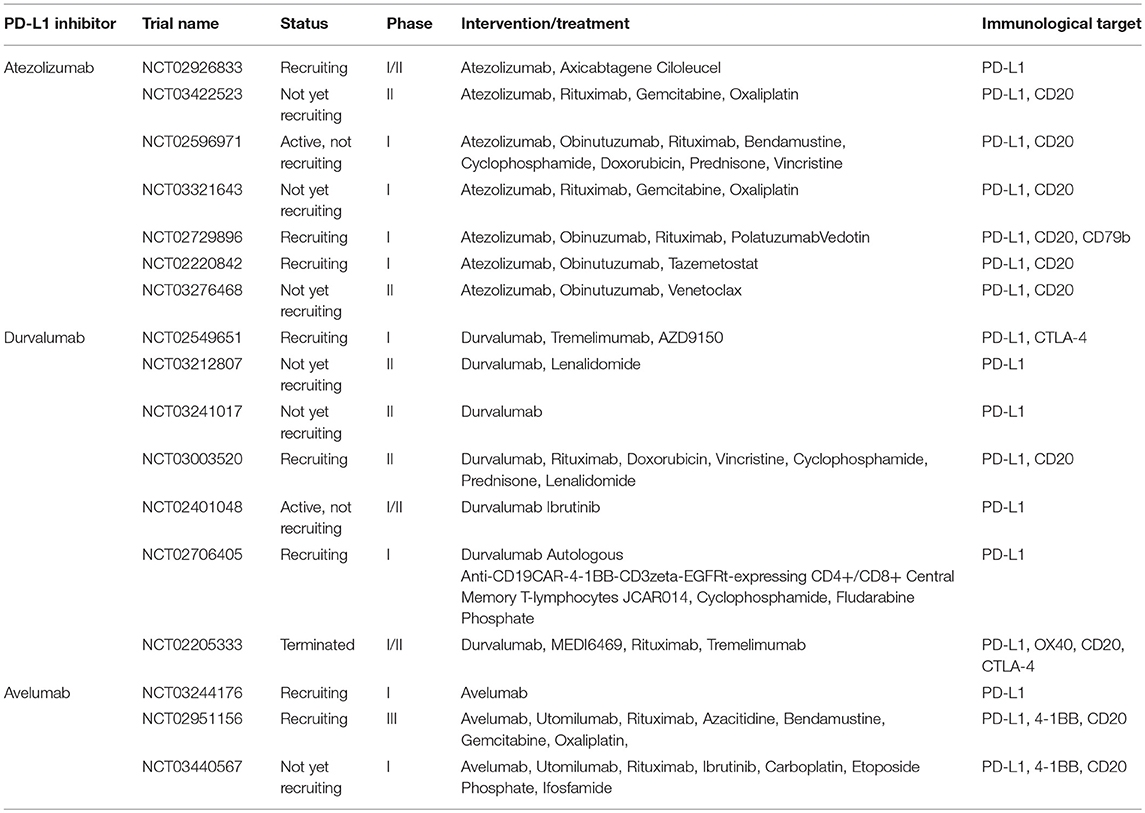
Table 2. Ongoing PD-L1 inhibitors trials in DLBCL.
Nivolumab is a human IgG4 anti-PD-1 monoclonal antibody. Multiple phase I/II studies of nivolumab are evaluating (or planning to evaluate) its efficacy in combination with agents such as ipilimumab (NCT03305445), rituximab and chemotherapy (NCT03259529), varlilumab (anti-CD27) (NCT03038672), the IDO1 inhibitor epacadostat (NCT02327078), and lenalidomide (NCT03015896) in participants with DLBCL.
Although early results in phase I studies were promising, only one phase II study has been reported for the use of PD-1/PD-L1 inhibitors in DLBCL patients, “A single-arm, open-label, phase 2 study of nivolumab (BMS-936558) in subjects with relapsed or refractory DLBCL after failure of autologous stem cell transplant (ASCT) or after failure of at least two prior multi-agent chemotherapy regimens in subjects who are not candidates for ASCT”. In this study, 161 participants were enrolled and 121 participants entered the treatment period. Participants were enrolled, but not treated due to adverse events (n = 2), withdrawal of consent (n = 2), death (n = 2), or they no longer met study criteria (n = 34). Finally, 102 participants completed the treatment period. Nivolumab 3 mg/kg administered as an IV infusion on treatment day 1 of each 14 day cycle until disease progression or discontinuation due to toxicity, withdrawal of study consent, or the study ends. Nivolumab therapy resulted in an overall response rate (ORR) of 10.3% in the ASCT-failed group (complete response [CR], 3.4%; partial response [PR], 6.9%) and 2.9% in the ASCT ineligible group (CR, 0%; PR, 2.9%). The median duration of response was 11.4 months in the ASCT-failed group and 8.3 months in the ASCT-ineligible group (NCT02038933).
A phase I trial of nivolumab monotherapy recruited patients with heavily pretreated relapsed or refractory lymphoid malignancies including 11 patients with DLBCL. Four (36%) patients responded (2 CR and 2 PR). The median follow-up duration for patients with DLBCL was 22.7 weeks; 1 of 4 patients with DLBCL has had an ongoing response, and 2 patients continue to be followed (18).
Pembrolizumab is another humanized IgG4 anti-PD-1 monoclonal antibody. Various phase I/II studies of the PD-1 antibody pembrolizumab are still ongoing, either as a single agent or in combination with antibodies, small molecular inhibitors, immunotherapeutic vaccine, dendritic cell therapy, and CAR T cell treatment in participants with DLBCL.
Atezolizumab is a human IgG1 monoclonal antibody that targets PD-L1. Seven phase I/II studies of atezolizumab are ongoing to evaluate its efficacy in combination with other agents such as CAR T cells, antibodies, small molecular inhibitors, and chemotherapy in participants with DLBCL.
Durvalumab is a human IgG1 monoclonal antibody that targets PD-L1. One phase II clinical trial of durvalumab as a single agent is ongoing to assess the progression-free survival (PFS) two years after ASCT in high-risk DLBCL patients. Five phase I/II studies are underway to evaluate the efficacy of durvalumab in combination with antibodies, small molecular inhibitors, and chemotherapy as well as CAR T cell therapy in participants with DLBCL. Another phase II clinical trial of durvalumab in combination with monoclonal antibodies directed against CD20, OX40 and CTLA4 designed to determine the optimal dose of MEDI6469 (anti-OX40) that is safe and tolerable in participants with DLBCL was terminated early at the sponsor's discretion due technical problems (NCT02205333).
Another human IgG1 anti-PD-L1 monoclonal antibody is avelumab. An early phase I study of avelumab as a single agent is ongoing to evaluate the feasibility of adding induction and maintenance avelumab to standard R-CHOP therapy in patients with stage II, III, and IV DLBCL (NCT03244176). An ongoing phase III study is evaluating the efficacy of avelumab in combination with a variety of agents for relapsed or refractory DLBCL patients; these agents include utomilumab (anti-4-1BB/CD137), rituximab, azacitidine, bendamustine, gemcitabine, and oxaliplatin, (NCT02951156). A recent phase I trial is studying the side effects and optimal dosing of avelumab, utomilumab, rituximab, ibrutinib, and combination chemotherapy for treating patients with DLBCL or relapsed/refractory mantle cell lymphoma, but is not yet recruiting (NCT03440567).
Pidilizumab (MDV9300, Medivation, Inc) was originally considered a monoclonal antibody binding to PD-1. This agent yielded encouraging results in phase II clinical trials for DLBCL. However, recent evidence suggests that PD-1 is not the target of pidilizumab. The FDA has lifted its partial clinical hold on the investigational new drug (IND) application for pidilizumab (MDV9300) in hematological malignancies and has confirmed that the phase II clinical trial in patients with relapsed or refractory DLBCL, as well as other studies that cross reference the IND, may now proceed. The partial clinical hold was not related to any safety concerns. The investigator brochure, protocols, and informed consent documents related to the phase II trial have satisfactorily been revised to reflect that the manufacturer's understands that PD-1 is not the target of pidilizumab. No patients had yet been enrolled in the trial which commenced in late 2015. Patients who were receiving pidilizumab through investigator-sponsored trials have continued to receive treatment and the investigators have been informed to update their protocols and informed consent documents to state that pidilizumab is not an anti-PD-1 antibody, but an anti-Delta-like ligand 1 antibody (39, 40).
Immune checkpoint blockade has promising potential in DLBCL therapy. A subgroup of patients with advanced cancers may respond to single-agent immune checkpoint blockade, however, most patients do not respond to monotherapy (41). In order to enhance the antitumor efficacy, a combination of multiple therapeutic approaches is urgently needed. Many clinical trials are ongoing to evaluate the synergistic efficacy of immune checkpoint inhibitors in combination with other agents, which mainly includes co-inhibitory blockade (anti-CTLA-4), co-stimulatory agonists (anti-OX40, anti-4-1BB), rituximab (anti-CD20) and conventional chemotherapy. Both PD-1 and CTLA-4 are expressed on T cells, but they play different regulatory functions via different signaling pathways in suppressing T cell activation and proliferation. The combined therapy of anti-PD-1 and anti-CTLA-4 has demonstrated synergistic efficacy and improve antitumor activities. In contrast, both OX40 and 4-1BB are members of the tumor necrosis factor (TNF) family of co-stimulatory receptors, expressed on the surface of CD4+ and CD8+ T cells. Agonist antibodies anti-OX40 and anti-4-1BB promote T cell activation, growth, and survival and enhance antitumor functions. Conventional chemotherapy in combination with immune checkpoint blockade has shown synergistic efficacy by releasing multiple tumor neoantigens or modifying the tumor microenvironment (Tables 1, 2).
Challenges and Opportunities for Blocking the PD-1/PD-L1 Pathway
Targeting the PD-1/PD-L1 pathway in patients with DLBCL is a promising treatment strategy. However, there are adverse events associated with PD-1/PD-L1 inhibitors that reflect the actions of the PD-1 pathway in the regulation of immune responses. PD-1 pathway blockade can cause immune-related adverse events that may affect almost all tissues. Toxicities related to immune checkpoint inhibitors typically include dermatologic manifestations, diarrhea, colitis, hepatotoxicity, endocrinopathies, and pneumonitis (42–44). Based on the experience of immune-checkpoint inhibitors in patients with solid tumors, the occurrence of grade 3–4 immune-related adverse events is approximately 20% with ipilimumab, compared with 5–10% with nivolumab or pembrolizumab (45). Generally, PD-1 pathway blockade is associated with fewer and less severe toxicities compared with CTLA-4 blockade. Toxicities can be managed with immune-modulating agents including corticosteroids and infliximab. Early studies suggest that combination therapy with CTLA-4 and PD-1 inhibitors may increase efficacy, but at the cost of increased toxicity (46). However, the combination of anti-CTLA-4 and anti-PD-1 antibodies demonstrated a similar safety and efficacy profile compared to a previous report for anti-PD-1 monotherapy in Hodgkin lymphoma, non-Hodgkin lymphoma (NHL), and multiple myeloma (19).
In patients with NHL, severe immune-related adverse events have been rare to date. A phase I trial of ipilimumab in patients with relapsed/refractory B-cell lymphoma is designed to evaluate safety, immunologic activity, and potential clinical efficacy. Diarrhea has been reported frequently among patients receiving ipilimumab, in 56%, with 28% of these patients developing grade 3–4 adverse events (5). Among patients with relapsed NHL receiving nivolumab within a phase Ib trial, 4% developed grade 3–5 pneumonitis (18). Another adverse event is fatigue, reported to occur in 13–56% of patients, mostly grade 1–2 (5). In clinical practice, adverse events associated with nivolumabhave been well tolerated and this agent has exhibited antitumor activity in extensively pretreated patients with relapsed or refractory B- and T-cell lymphomas (5, 18).
Biomarker data might be useful in guiding dose and regimen selection in early clinical development. However, a correlation between the expression of PD-L1 by DLBCL cells and response to PD-1 inhibitors has not been confirmed and remains controversial (39). Evaluation of PD-L1 expression by tumor-cells as a predictive marker has been inconclusive. This observation might be due to complex dynamics of expression depending on the tumor microenvironment and the lack of standardized immunohistochemical assessment of PD-L1 expression (47).
CAR T Cell Therapy
CAR T-cells are autologous, polyclonal T lymphocytes genetically engineered to express a tumor-targeting receptor, directing the T cells to bind to a specific tumor-associated antigen. CAR T cells are composed of an extracellular single chain variable fragment (scFv) and intracellular signaling domains that allow T cells to effect functions independent of major histocompatibility complex (MHC) antigens. Depending on differences in the intracellular signaling domains and cytokine secretion, CAR T cells have been classified as first-, second-, third- and fourth-generation. First-generation CAR T cells consisted of an extracellular scFv and a single intracellular signaling domain CD3ζ. The limited activity of this generation was probably attributable to their inability to adequately activate T cells, especially in cases where tumor cells did not express T cell co-stimulatory molecules (48). Subsequently, second (and third and fourth)-generation CAR T cells included co-stimulatory domains, such as CD28 or CD137 (4-1BB), to improve expansion and persistence of T cells (49, 50). Kochenderfer first reported the anti-tumor efficacy of an anti-CD19 CAR T cell containing the CD28 costimulatory domain in aggressive lymphoma (51). In order to enhance the activation of CAR T cells, third-generation CAR T cells were designed by combining two signaling domains among CD28, CD27, 4-1BB, ICOS, and OX40 (52–56). Including two co-stimulatory domains into CAR T cells can improve the tumor cell-killing efficacy. However, because of the activation of multiple intracellular signaling caused by the co-stimulatory domains of third-generation CAR T cells, abundant cytokines might be released which may result in a life-threatening cytokine storm (57). In order to enhance their tumor cell-killing efficacy and impact local suppressive cells, fourth-generation CAR T cells were engineered with an inducible expression component, such as cytokine IL-12, and also are known as T cells redirected for universal cytokine-mediated killing (TRUCKs). TRUCKs not only increase the activation of CAR T cells, they also induce cytokines and attract innate immune cells to eliminate antigen-negative cancer cells (58). In addition, for safety considerations, an inducible caspase 9 self-withdrawal genetic design allows for rapid elimination of infused CAR T cells once the anti-tumor mission is accomplished (59, 60).
Clinical Trials of CAR T Cells as Therapy in DLBCL
CAR T cell therapies have been most efficacious in patients with B-cell acute lymphoblastic leukemia; less data are available for patients with DLBCL. According to the American Cancer Society, ~72,000 children and adults in the US will be diagnosed with non-Hodgkin lymphoma in 2017; 60% of these cases are aggressive neoplasms with the most common type being DLBCL. The typical survival duration of patients with DLBCL who have disease progression after chemotherapy or ASCT is 9 months. The cumulative promising data indicate that immunotherapy using CAR T cells offers hope for achieving long-term survival in patients with relapse/refractory DLBCL or follicular lymphoma (FL).
Investigators from Kite Pharma developed a clinical trial of CD19-CAR T cells (NCT02348216) that was approved on October 2017, becoming the first CAR T therapy approved by the FDA for the treatment of adults with relapsed or refractory DLBCL after two or more lines of systemic therapy. CD19-targeting CAR T cell therapy showed that 42% of patients with refractory DLBCL remained in remission at 15 months following treatment with axi-cel (marketed as Yescarta). Axi-cel CAR T cell therapy is the second gene therapy approved by the FDA and the first for adult patients with DLBCL after failing at least two other kinds of treatment; the types of large B-cell lymphoma in this study include DLBCL not otherwise specified (NOS), primary mediastinal large B-cell lymphoma, DLBCL arising from follicular lymphoma, and cases that fit into the new World Health organization category of high grade B-cell lymphoma (e.g., DLBCL with double hit genetics). This study, named ZUMA-1, also reported measurable responses in 82% of patients and complete responses in 54%. Over half (56%) of patients were alive at 15 months following therapy, with some remaining cancer-free for 2 years post-treatment. Among the 111 patients who were enrolled, axi-cel was successfully manufactured for 110 and administered to 101. The median age of these patients was 58 years (range, 23–76 years). Most (85%) patients in the study group had stage III or IV disease; 77% had disease that was resistant to second line or subsequent therapies, 21% had disease relapse after transplantation, 69% had received at least three previous therapies, and 26% had a history of primary refractory disease. Among the 101 patients who received axi-cel, the ORR was 82%, with a 54% CR. With a median follow-up of 15.4 months, 42% of patients continued to have a response, with 40% in CR. The overall rate of survival at 18 months was 52%. The most common adverse events of grade 3 or higher during treatment were neutropenia (78% of the patients), anemia (43%), and thrombocytopenia (38%). Grade 3 or higher cytokine release syndrome (CRS) and neurologic events occurred in 13 and 28% of patients, respectively. Three patients died during treatment. In this multicenter study, patients with refractory DLBCL who received CAR T-cell therapy with axi-cel had high levels of durable response, with a safety profile that included myelosuppression, CRS, and neurologic events (61).
Kochenderfer and colleagues at the National Cancer Institute were the first to report a partial response (PR) lasting 32 weeks after infusing autologous T cells directed against CD19 in a patient with FL (62). This group later published seven patients with DLBCL: four patients achieved a CR, two achieved a PR, and one had stable disease (SD) (57). Recently, Kochenderfer et al. reported results for 22 patients with advanced-stage lymphoma in a clinical trial of CAR-19 T cells preceded by low-dose chemotherapy, including 19 patients with DLBCL, two patients with FL, and one patient with mantle cell lymphoma. Patients received a single dose of CAR-19 T cells 2 days after a low-dose chemotherapy conditioning regimen of cyclophosphamide plus fludarabine. This study showed that CAR-19 T cells are an effective therapy for lymphoma patients and with lower doses of chemotherapy than they previously used; the ORR was 73%, with 55% achieving CR and 18% achieving PR. Eleven of 12 patients remain in CR and grade 3 or 4 neurologic toxicities in about half of the patients resolved completely (51).
Investigators from the University of Pennsylvania Medical Center have collaborated with Novartis to develop a second-generation CD19-CAR T cell named CTL019. This CAR consists of a murine anti-CD19 scFv, a CD8 hinge, a trans-membrane domain, 4-1BB (co-stimulatory molecule), and CD3ζ. This group has conducted a phase IIa clinical trial of CTL019 cells in patients with relapsed or refractory CD19+ non-Hodgkin lymphomas (NCT02030834); 29 patients (19 DLBCL; 8 FL; 2 MCL) enrolled and 20 patients received CTL019 per protocol dose (12 DLBCL; 7 FL; 1 MCL). Pre-infusion chemotherapy regimens were EPOCH (n = 2); cyclophosphamide (n = 9); radiation + cyclophosphamide (n = 2); bendamustine (n = 6); cyclophosphamide-fludarabine (n = 1). Cytokine release syndrome occurred in 15 patients (13 grade 2; 2 grade 3). Neurologic toxicity occurred in 3 patients: transient delirium (1 grade 2, 1 grade 3) and 1 possibly related, grade 5 encephalopathy. For 18 patients evaluable for response at 3 months (12 DLBCL; 6 FL), the ORR was 67% (DLBCL 50%; FL 100%). At a median follow up 6 months, progression-free survival for evaluable patients was 59% (DLBCL 37%; FL 100%). This report shows that CTL019 cells induce durable responses in patients with relapsed/refractory DLBCL and FL with acceptable toxicity (63).
Recently, interim results from a global, pivotal multi-center phase II JULIET trial (NCT02445248) of CTL019 (tisagenlecleucel) showed durable complete responses in adults with relapsed/refractory DLBCL. The ORR at 3 months was 45% (23 of 51 patients evaluated), with 37% achieving CR and 8% achieving PR. The patients with CR remained stable from 3 months through data cutoff among the study cohort (64).
Investigators from the Fred Hutchinson Cancer Research Center, Memorial Sloan Kettering Cancer Center, and Seattle Children's Research Institute have collaborated with Juno Therapeutics to conduct several clinical trials of CD19-CAR T cell products: JCAR014, JCAR015, JCAR017, JCAR021, and others. Among them, updated results from the ongoing TRANSCEND study of JCAR017, which contains the 4-1BB costimulatory domain, in patients with relapsed or refractory aggressive non-Hodgkin lymphoma were presented during 2017 American Society of Hematology meeting. The core group (n = 49) included patients with DLBCL (NOS and transformed from follicular lymphoma) who were ECOG performance status 0–1. These patients represented a highly refractory population based on factors associated with a poor prognosis, including older patient age, double, or triple hit genetics (MYC and BCL2 and/or BCL6 rearrangement), and the DLBCL being refractory to chemotherapy. Dose level 1 (DL1 = 50 million cells) showed a 3 month ORR of 52% (11/21 patients) and a 3 month CR rate of 33% (7/21). Dose level 2 (DL2 = 100 million cells), the dose in the pivotal cohort of the TRANSCEND study, showed a 3 month overall response rate (ORR) of 80% (12/15) and a 3 month complete response (CR) rate of 73% (11/15) in the core group. These data support a dose response relationship. Across both doses in the core group, the best overall response was 84% (41/49 patients) and the best overall CR rate was 61% (30/49). There was no increase in CRS or neurotoxicity (NT) rates associated with the higher dose or between the full and core groups. Across doses in the full group, 1 of 69 (1%) patients experienced severe CRS and 10 (14%) patients experienced severe NT. Twenty-one of 69 (30%) patients had any grade CRS and 14 (20%) patients had any grade NT. 64% (44/69) of patients had no evidence of CRS or NT. The most common treatment-emergent adverse events other than CRS and NT that occurred at ≥25% in the full group included neutropenia (41%), fatigue (30%), thrombocytopenia (30%), and anemia (26%) (65–67).
Challenges and Opportunities for CAR T Cell Therapy
CAR T cells have shown promising efficacy in patients with DLBCL, including those with relapsed or refractory DLBCL. However, this therapy can be associated with unexpected toxicities that can be life-threatening, including CRS, NT, and “on-target off-tumor” recognition. The challenges are to reduce toxicity, prolong disease-free survival, and to determine which factors can predict relapse of DLBCL after successful CAR T cell therapy.
Cytokine release syndrome is a systemic inflammatory response to the activation and proliferation of CAR T cells. The clinical features of CRS include high fever, fatigue, nausea, malaise, hypotension, cardiac dysfunction, renal impairment, hepatic failure, capillary leak, and disseminated intravascular coagulation (68). CRS is associated with a dramatic elevation of inflammatory cytokines in the serum including C reactive protein (CRP), interferon-γ, ferritin, granulocyte macrophage colony-stimulating factor, IL-10, and IL-6 following CAR T-cell infusion (69–72). CRS occurs most frequently within the first 2 weeks after CAR T cell infusion. Clinical management schemes of CRS include administration of steroids and the IL-6 receptor blocking antibody, tocilizumab (68, 73). However, steroids blunt the anti-tumor function of CAR T cells and the long-term impact of tocilizumab on CAR T cell function remains unclear. It remains a challenge to control CRS without inhibiting the anti-tumor efficacy of CAR T cell therapy.
Neurologic adverse events have been observed in many patients receiving CD19-CAR T cell therapy. Reversible symptoms of NT, including confusion, delirium, expressive aphasia, encephalopathy, and seizures, have been reported in several studies (51, 69, 74–77). In some patients, CD19-CAR T cells have been found in cerebrospinal fluid (74, 76). Whether neurological toxicities are solely restricted to CD19-specific CAR T cells or are associated generally with CAR T cell therapy remains unclear and the potential causes of NT remain to be elucidated. The postulated pathophysiological mechanisms include cytokine diffusion and/or translocation of activated CAR T cells across the blood brain barrier.
On-target off-tumor recognition side effects caused by depletion of healthy CD19-positive B-cells by CAR T cells are also an issue. B cell aplasia is a common adverse event in CAR T cells trials targeting B cell malignancies (75, 77, 78). Off-tumor recognition side effects in CAR T cell treated patients also can occur as a result of cross-reactivity of the engineered antigen binding domain with a non-related surface protein.
Selective depletion of CAR T cells can be approached by the use of “self-withdrawal CARs” in which is inserted an inducible caspase 9 (ICasp9) (79, 80). Current T-cell engineering approaches redirect patient T cells to tumors by transducing them with antigen-specific T-cell receptors (TCRs) or CARs that target a single antigen. However, healthy tissues that express the targeted antigen may undergo CAR T cell-mediated damage. A novel strategy that combines antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells (81). In trials using CD19-targeting T-cells, CD19-negative clones have expanded and caused progressive disease (82). The approach of increasing the specificity of CARs is to combine more CAR T cells to recognize multiple targets. This treatment strategy may help broaden the applicability and avoid some of the side effects of targeted T-cell therapies. In addition, a novel agent that blocks IL-35 may support CAR T cell therapy by reducing the inhibitory effect of regulatory T cells that may be of value in the future (83). Furthermore, several small molecule inhibitors, such as ibrutinib (Bruton tyrosine kinase inhibitor) (84), ABT-199 (Bcl-2 inhibitor) (85), and JQ-1 (bromodomain inhibitor) (86), has shown impressive potential for treating DLBCL patients. CAR T immunotherapy in combination with a small molecule inhibitor is likely to provide greater benefit for the treatment of patients with DLBCL.
Conclusions
Cancer immunotherapy that harnesses the host immune system in novel ways to kill tumor cells is emerging. Immunotherapy offers promising opportunities with the potential to induce sustained remissions, and is expected to become a “game changer” for the treatment of patients with cancer. Novel immunotherapy regimens, PD-1/PD-L1, and CTLA-4 checkpoint inhibitors, and CAR T cells have shown promising potential in the treatment of patients with DLBCL.
Early clinical trials using PD-1/PD-L1 checkpoint inhibitors including two anti-PD-1 antibodies (nivolumab and pembrolizumab), and three anti-PD-L1 antibodies (avelumab, durvalumab, and atezolizumab), have shown great promise. CAR T cell therapy also has shown remarkable activity in patients with refractory DLBCL. Yescarta, a CAR T cell immunotherapy, has been approved by the FDA for use in adults with large B-cell lymphoma after at least two other kinds of treatment have failed. Numerous ongoing clinical trials will undoubtedly offer the hope of achieving long-term survival in patients with relapsed or refractory disease.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by National Cancer Institute/National Institutes of Health grants R01CA138688, R01CA187415, and 1RC1CA146299 to KHY. KHY is also supported by The University of Texas MD Anderson Cancer Center Institutional Research and Development Fund, Gundersen Lutheran Medical Foundation Award, Hagemeister Lymphoma Foundation Award, and the University Cancer Foundation via the Sister Institution Network Fund at The University of Texas MD Anderson Cancer Center. KHY receives research support from Roche Molecular System, Gilead Sciences, Seattle Genetics, Dai Sanyo, Adaptive Biotechnology, Incyte Pharmaceutical, and HTG Molecular Diagnostics. This work was also partially funded by National Cancer Institute and National Institutes of Health grants P50CA136411 and P50CA142509, and by the MD Anderson Cancer Center Support Grant CA016672.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Leget GA, Czuczman MS. Use of rituximab, the new FDA-approved antibody. Curr Opin Oncol. (1998) 10:548–51. doi: 10.1097/00001622-199811000-00012
2. Maloney DG, Grillo-Lopez AJ, White CA, Bodkin D, Schilder RJ, Neidhart JA, et al. IDEC-C2B8 (Rituximab) anti-CD20 monoclonal antibody therapy in patients with relapsed low-grade non-Hodgkin's lymphoma. Blood (1997) 90:2188–95.
3. Breton CS, Nahimana A, Aubry D, Macoin J, Moretti P, Bertschinger M, et al. A novel anti-CD19 monoclonal antibody (GBR 401) with high killing activity against B cell malignancies. J Hematol Oncol. (2014) 7:33. doi: 10.1186/1756-8722-7-33
4. Scheuermann RH, Racila E. CD19 antigen in leukemia and lymphoma diagnosis and immunotherapy. Leuk Lymphoma (1995) 18:385–97. doi: 10.3109/10428199509059636
5. Ansell SM, Hurvitz SA, Koenig PA, LaPlant BR, Kabat BF, Fernando D, et al. Phase I study of ipilimumab, an anti-CTLA-4 monoclonal antibody, in patients with relapsed and refractory B-cell non-Hodgkin lymphoma. Clin Cancer Res. (2009) 15:6446–53. doi: 10.1158/1078-0432.CCR-09-1339
6. Rizvi NA, Mazieres J, Planchard D, Stinchcombe TE, Dy GK, Antonia SJ, et al. Activity and safety of nivolumab, an anti-PD-1 immune checkpoint inhibitor, for patients with advanced, refractory squamous non-small-cell lung cancer (CheckMate 063): a phase 2, single-arm trial. Lancet Oncol. (2015) 16:257–65. doi: 10.1016/S1470-2045(15)70054-9
7. Larkin J, Hodi FS, Wolchok JD. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. (2015) 373:1270–1. doi: 10.1056/NEJMoa1504030
8. Hodi FS, Chesney J, Pavlick AC, Robert C, Grossmann KF, McDermott DF, et al. Combined nivolumab and ipilimumab versus ipilimumab alone in patients with advanced melanoma: 2-year overall survival outcomes in a multicentre, randomised, controlled, phase 2 trial. Lancet Oncol. (2016) 17:1558–68. doi: 10.1016/S1470-2045(16)30366-7
9. Gettinger S, Rizvi NA, Chow LQ, Borghaei H, Brahmer J, Ready N, et al. Nivolumab monotherapy for first-line treatment of advanced non-small-cell lung cancer. J Clin Oncol. (2016) 34:2980–7. doi: 10.1200/JCO.2016.66.9929
10. Burki TK. Pembrolizumab for classical Hodgkin's lymphoma. Lancet Oncol. (2016) 17:e324. doi: 10.1016/S1470-2045(16)30301-1
11. Herbst RS, Baas P, Kim DW, Felip E, Perez-Gracia JL, Han JY, et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet (2016) 387:1540–50. doi: 10.1016/S0140-6736(15)01281-7
12. Goldberg SB, Gettinger SN, Mahajan A, Chiang AC, Herbst RS, Sznol M, et al. Pembrolizumab for patients with melanoma or non-small-cell lung cancer and untreated brain metastases: early analysis of a non-randomised, open-label, phase 2 trial. Lancet Oncol. (2016) 17:976–83. doi: 10.1016/S1470-2045(16)30053-5
13. Rosenberg JE, Hoffman-Censits J, Powles T, van der Heijden MS, Balar AV, Necchi A, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet (2016) 387:1909–20. doi: 10.1016/S0140-6736(16)00561-4
14. Balar AV, Galsky MD, Rosenberg JE, Powles T, Petrylak DP, Bellmunt J, et al. Atezolizumab as first-line treatment in cisplatin-ineligible patients with locally advanced and metastatic urothelial carcinoma: a single-arm, multicentre, phase 2 trial. Lancet (2017) 389:67–76. doi: 10.1016/S0140-6736(16)32455-2
15. Kaufman HL, Russell J, Hamid O, Bhatia S, Terheyden P, D'Angelo SP, et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. Lancet Oncol. (2016) 17:1374–85. doi: 10.1016/S1470-2045(16)30364-3
16. Massard C, Gordon MS, Sharma S, Rafii S, Wainberg ZA, Luke J, et al. Safety and efficacy of durvalumab (MEDI4736), an anti-programmed cell death ligand-1 immune checkpoint inhibitor, in patients with advanced urothelial bladder cancer. J Clin Oncol. (2016) 34:3119–25. doi: 10.1200/JCO.2016.67.9761
17. Antonia S, Goldberg SB, Balmanoukian A, Chaft JE, Sanborn RE, Gupta A, et al. Safety and antitumour activity of durvalumab plus tremelimumab in non-small cell lung cancer: a multicentre, phase 1b study. Lancet Oncol. (2016) 17:299–308. doi: 10.1016/S1470-2045(15)00544-6
18. Lesokhin AM, Ansell SM, Armand P, Scott EC, Halwani A, Gutierrez M, et al. Nivolumab in patients with relapsed or refractory hematologic malignancy: preliminary results of a phase Ib study. J Clin Oncol. (2016) 34:2698–704. doi: 10.1200/JCO.2015.65.9789
19. Ansell S, Gutierrez ME, Shipp MA, Gladstone D, Moskowitz A, Borello I, et al. A Phase 1 study of nivolumab in combination with ipilimumab for relapsed or refractory hematologic malignancies (CheckMate 039). In: ASH 58th Annual Meeting and Exposition. San Diego, CA. Oral Abstract # 183 (2016).
20. Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci USA. (2002) 99:12293–7. doi: 10.1073/pnas.192461099
21. Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol. (2007) 25:267–96. doi: 10.1146/annurev.immunol.25.022106.141609
22. Trotta AM, Pacelli R, Scala S. Predictive immune biomarkers: an unattainable chimera? Cell Mol Immunol. (2018). doi: 10.1038/cmi.2017.162
23. Liang SC, Latchman YE, Buhlmann JE, Tomczak MF, Horwitz BH, Freeman GJ, et al. Regulation of PD-1, PD-L1, and PD-L2 expression during normal and autoimmune responses. Eur J Immunol. (2003) 33:2706–16. doi: 10.1002/eji.200324228
24. Ou JN, Wiedeman AE, Stevens AM. TNF-alpha and TGF-beta counter-regulate PD-L1 expression on monocytes in systemic lupus erythematosus. Sci Rep. (2012) 2:295. doi: 10.1038/srep00295
25. He J, Hu Y, Hu M, Li B. Development of PD-1/PD-L1 pathway in tumor immune microenvironment and treatment for non-small cell lung cancer. Sci Rep. (2015) 5:13110. doi: 10.1038/srep13110
26. Loke P, Allison JP. PD-L1 and PD-L2 are differentially regulated by Th1 and Th2 cells. Proc Natl Acad Sci USA. (2003) 100:5336–41. doi: 10.1073/pnas.0931259100
27. Xiao Y, Yu S, Zhu B, Bedoret D, Bu X, Francisco LM, et al. RGMb is a novel binding partner for PD-L2 and its engagement with PD-L2 promotes respiratory tolerance. J Exp Med. (2014) 211:943–59. doi: 10.1084/jem.20130790
28. Rudd CE, Taylor A, Schneider H. CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol Rev. (2009) 229:12–26. doi: 10.1111/j.1600-065X.2009.00770.x
29. Yokosuka T, Takamatsu M, Kobayashi-Imanishi W, Hashimoto-Tane A, Azuma M, Saito T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J Exp Med. (2012) 209:1201–17. doi: 10.1084/jem.20112741
30. Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol. (2005) 25:9543–53. doi: 10.1128/MCB.25.21.9543-9553.2005
31. Sheppard KA, Fitz LJ, Lee JM, Benander C, George JA, Wooters J, et al. PD-1 inhibits T-cell receptor induced phosphorylation of the ZAP70/CD3zeta signalosome and downstream signaling to PKCtheta. FEBS Lett. (2004) 574:37–41. doi: 10.1016/j.febslet.2004.07.083
32. Kao C, Oestreich KJ, Paley MA, Crawford A, Angelosanto JM, Ali MA, et al. Transcription factor T-bet represses expression of the inhibitory receptor PD-1 and sustains virus-specific CD8+ T cell responses during chronic infection. Nat Immunol. (2011) 12:663–71. doi: 10.1038/ni.2046
33. Crump M, Neelapu SS, Farooq U, Van Den Neste E, Kuruvilla J, Westin J, et al. Outcomes in refractory diffuse large B-cell lymphoma: results from the international SCHOLAR-1 study. Blood (2017) 130:1800–8. doi: 10.1182/blood-2017-03-769620
34. Kwon D, Kim S, Kim PJ, Go H, Nam SJ, Paik JH, et al. Clinicopathological analysis of programmed cell death 1 and programmed cell death ligand 1 expression in the tumour microenvironments of diffuse large B cell lymphomas. Histopathology (2016) 68:1079–89. doi: 10.1111/his.12882
35. Menter T, Bodmer-Haecki A, Dirnhofer S, Tzankov A. Evaluation of the diagnostic and prognostic value of PDL1 expression in Hodgkin and B-cell lymphomas. Hum Pathol. (2016) 54:17–24. doi: 10.1016/j.humpath.2016.03.005
36. Rossille D, Azzaoui I, Feldman AL, Maurer MJ, Laboure G, Parrens M, et al. Soluble programmed death-ligand 1 as a prognostic biomarker for overall survival in patients with diffuse large B-cell lymphoma: a replication study and combined analysis of 508 patients. Leukemia (2017) 31:988–91. doi: 10.1038/leu.2016.385
37. Kiyasu J, Miyoshi H, Hirata A, Arakawa F, Ichikawa A, Niino D, et al. Expression of programmed cell death ligand 1 is associated with poor overall survival in patients with diffuse large B-cell lymphoma. Blood (2015) 126:2193–201. doi: 10.1182/blood-2015-02-629600
38. Kwiecinska A, Tsesmetzis N, Ghaderi M, Kis L, Saft L, Rassidakis GZ. CD274 (PD-L1)/PDCD1 (PD-1) expression in de novo and transformed diffuse large B-cell lymphoma. Br J Haematol. (2018) 180:744–8. doi: 10.1111/bjh.14432
39. Xu-Monette ZY, Zhou J, Young KH. PD-1 expression and clinical PD-1 blockade in B-cell lymphomas. Blood (2018) 131:68–83. doi: 10.1182/blood-2017-07-740993
40. Xu D, Hu J, Xu S, De Bruyne E, Menu E, Van Camp B, et al. Dll1/Notch activation accelerates multiple myeloma disease development by promoting CD138+ MM-cell proliferation. Leukemia (2012) 26:1402–5. doi: 10.1038/leu.2011.332
41. Mahoney KM, Rennert PD, Freeman GJ. Combination cancer immunotherapy and new immunomodulatory targets. Nat Rev Drug Discov. (2015) 14:561–84. doi: 10.1038/nrd4591
42. Weber JS. Practical management of immune-related adverse events from immune checkpoint protein antibodies for the oncologist. Am Soc Clin Oncol Educ Book (2012) 2012:174–7. doi: 10.14694/EdBook_AM.2012.32.174
43. Brahmer JR, Lacchetti C, Schneider BJ, Atkins MB, Brassil KJ, Caterino JM, et al. Management of immune-related adverse events in patients treated with immune checkpoint inhibitor therapy: american society of clinical oncology clinical practice guideline. J Clin Oncol. (2018) 2018:JCO2017776385. doi: 10.1200/JCO.2017.77.6385
44. Villadolid J, Amin A. Immune checkpoint inhibitors in clinical practice: update on management of immune-related toxicities. Transl Lung Cancer Res. (2015) 4:560–75. doi: 10.3978/j.issn.2218-6751.2015.06.06
45. Weber JS, Yang JC, Atkins MB, Disis ML. Toxicities of immunotherapy for the practitioner. J Clin Oncol. (2015) 33:2092–9. doi: 10.1200/JCO.2014.60.0379
46. Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. (2013) 369:122–33. doi: 10.1056/NEJMoa1302369
47. Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature (2014) 515:563–67. doi: 10.1038/nature14011
48. Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci USA. (1993) 90:720–4. doi: 10.1073/pnas.90.2.720
49. Kowolik CM, Topp MS, Gonzalez S, Pfeiffer T, Olivares S, Gonzalez N, et al. CD28 costimulation provided through a CD19-specific chimeric antigen receptor enhances in vivo persistence and antitumor efficacy of adoptively transferred T cells. Cancer Res. (2006) 66:10995–1004. doi: 10.1158/0008-5472.CAN-06-0160
50. Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther. (2009) 17:1453–64. doi: 10.1038/mt.2009.83
51. Kochenderfer JN, Dudley ME, Kassim SH, Somerville RP, Carpenter RO, Stetler-Stevenson M, et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol. (2015) 33:540–9. doi: 10.1200/JCO.2014.56.2025
52. Carpenito C, Milone MC, Hassan R, Simonet JC, Lakhal M, Suhoski MM, et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci USA. (2009) 106:3360–5. doi: 10.1073/pnas.0813101106
53. Zhong XS, Matsushita M, Plotkin J, Riviere I, Sadelain M. Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication. Mol Ther. (2010) 18:413–20. doi: 10.1038/mt.2009.210
54. Song DG, Ye Q, Poussin M, Harms GM, Figini M, Powell DJ Jr. CD27 costimulation augments the survival and antitumor activity of redirected human T cells in vivo. Blood (2012) 119:696–706. doi: 10.1182/blood-2011-03-344275
55. Guedan S, Chen X, Madar A, Carpenito C, McGettigan SE, Frigault MJ, et al. ICOS-based chimeric antigen receptors program bipolar TH17/TH1 cells. Blood (2014) 124:1070–80. doi: 10.1182/blood-2013-10-535245
56. Pule MA, Straathof KC, Dotti G, Heslop HE, Rooney CM, Brenner MK. A chimeric T cell antigen receptor that augments cytokine release and supports clonal expansion of primary human T cells. Mol Ther. (2005) 12:933–41. doi: 10.1016/j.ymthe.2005.04.016
57. Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood (2012) 119:2709–20. doi: 10.1182/blood-2011-10-384388
58. Pegram HJ, Lee JC, Hayman EG, Imperato GH, Tedder TF, Sadelain M, et al. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood (2012) 119:4133–41. doi: 10.1182/blood-2011-12-400044
59. Hoyos V, Savoldo B, Quintarelli C, Mahendravada A, Zhang M, Vera J, et al. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia (2010) 24:1160–70. doi: 10.1038/leu.2010.75
60. Diaconu I, Ballard B, Zhang M, Chen Y, West J, Dotti G, et al. Inducible caspase-9 selectively modulates the toxicities of CD19-specific chimeric antigen receptor-modified T cells. Mol Ther. (2017) 25:580–92. doi: 10.1016/j.ymthe.2017.01.011
61. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. (2017) 377:2531–44. doi: 10.1056/NEJMoa1707447
62. Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler-Stevenson M, Feldman SA, et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood (2010) 116:4099–102. doi: 10.1182/blood-2010-04-281931
63. Schuster SJ, Svoboda J, Nasta S, Porter DL, Mato A, Shah GD, et al. Phase IIa trial of chimeric antigen receptor modified T cells directed against CD19 (CTL019) in patients with relapsed or refractory CD19+ lymphomas. J Clin Oncol. (2015) 33:8516. doi: 10.1200/jco.2015.33.15_suppl.8516
64. Schuster SJ, Svoboda J, Chong EA, Nasta SD, Mato AR, Anak O, et al. Chimeric antigen receptor T cells in refractory B-cell lymphomas. N Engl J Med. (2017) 377:2545–54. doi: 10.1056/NEJMoa1708566
65. Siddiqi T, Abramson JS, Li D, Brown W, Devries T, Dave K, et al. Patient characteristics and pre-infusion biomarkers of inflammation correlate with clinical outcomes after treatment with the defined composition, CD19-targeted CAR T cell product, JCAR017. In: ASH 59th Annual Meeting and Exposition. Atlanta, GA. (2017). p. 193.
66. Swanson C, Do T, Merrigan S, Lonning S, Merriam K, Prentiss J, et al. Predicting clinical response and safety of JCAR017 in B-NHL patients: potential importance of tumor microenvironment biomarkers and CAR T-cell tumor infiltration. In: ASH 59th Annual Meeting and Exposition. Atlanta, GA. (2017). p. 194.
67. Abramson JS, Palomba ML, Gordon LI, Lunning MA, Arnason JE, Wang M, et al. High durable CR rates in relapsed/refractory (R/R) aggressive B-NHL treated with the CD19-directed CAR T cell product JCAR017 (TRANSCEND NHL 001): defined composition allows for dose-finding and definition of pivotal cohort. In: ASH 59th Annual Meeting and Exposition. Atlanta, GA. (2017). p. 581.
68. Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood (2014) 124:188–95. doi: 10.1182/blood-2014-05-552729
69. Porter DL, Hwang WT, Frey NV, Lacey SF, Shaw PA, Loren AW, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med. (2015) 7:303ra139. doi: 10.1126/scitranslmed.aac5415
70. Hombach AA, Heiders J, Foppe M, Chmielewski M, Abken H. OX40 costimulation by a chimeric antigen receptor abrogates CD28 and IL-2 induced IL-10 secretion by redirected CD4(+) T cells. Oncoimmunology (2012) 1:458–66. doi: 10.4161/onci.19855
71. Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. (2011) 365:725–33. doi: 10.1056/NEJMoa1103849
72. Klinger M, Brandl C, Zugmaier G, Hijazi Y, Bargou RC, Topp MS, et al. Immunopharmacologic response of patients with B-lineage acute lymphoblastic leukemia to continuous infusion of T cell-engaging CD19/CD3-bispecific BiTE antibody blinatumomab. Blood (2012) 119:6226–33. doi: 10.1182/blood-2012-01-400515
73. Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. (2014) 6:224ra225. doi: 10.1126/scitranslmed.3008226
74. Brentjens RJ, Riviere I, Park JH, Davila ML, Wang X, Stefanski J, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood (2011) 118:4817–28. doi: 10.1182/blood-2011-04-348540
75. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. (2014) 371:1507–17. doi: 10.1056/NEJMoa1407222
76. Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet (2015) 385:517–28. doi: 10.1016/S0140-6736(14)61403-3
77. Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest. (2016) 126:2123–38. doi: 10.1172/JCI85309
78. Kochenderfer JN, Dudley ME, Carpenter RO, Kassim SH, Rose JJ, Telford WG, et al. Donor-derived CD19-targeted T cells cause regression of malignancy persisting after allogeneic hematopoietic stem cell transplantation. Blood (2013) 122:4129–39. doi: 10.1182/blood-2013-08-519413
79. Straathof KC, Pule MA, Yotnda P, Dotti G, Vanin EF, Brenner MK, et al. An inducible caspase 9 safety switch for T-cell therapy. Blood (2005) 105:4247–54. doi: 10.1182/blood-2004-11-4564
80. Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med. (2011) 365:1673–83. doi: 10.1056/NEJMoa1106152
81. Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol. (2013) 31:71–5. doi: 10.1038/nbt.2459
82. Gill S, June CH. Going viral: chimeric antigen receptor T-cell therapy for hematological malignancies. Immunol Rev. (2015) 263:68–89. doi: 10.1111/imr.12243
83. Collison LW, Chaturvedi V, Henderson AL, Giacomin PR, Guy C, Bankoti J, et al. IL-35-mediated induction of a potent regulatory T cell population. Nat Immunol. (2010) 11:1093–101. doi: 10.1038/ni.1952
84. Wilson WH, Young RM, Schmitz R, Yang Y, Pittaluga S, Wright G, et al. Targeting B cell receptor signaling with ibrutinib in diffuse large B cell lymphoma. Nat Med. (2015) 21:922–6. doi: 10.1038/nm.3884
85. Gerecitano JF, Roberts AW, Seymour JF, Wierda WG, Kahl BS, Pagel JM, et al. A Phase 1 study of venetoclax (ABT-199 / GDC-0199) monotherapy in patients with relapsed/refractory non-hodgkin lymphoma. In: ASH 57th Annual Meeting and Exposition. Orlando, FL. (2015). p. 254.
86. Emadali A, Rousseaux S, Bruder-Costa J, Rome C, Duley S, Hamaidia S, et al. Identification of a novel BET bromodomain inhibitor-sensitive, gene regulatory circuit that controls Rituximab response and tumour growth in aggressive lymphoid cancers. EMBO Mol Med. (2013) 5:1180–95. doi: 10.1002/emmm.201202034
Keywords: DLBCL, NHL, immunotheray, PD-1, PD-L1, CTLA-4, Chimeric antigen receptor (CAR) T cells therapy, immune checkpoint
Citation: Zhang J, Medeiros LJ and Young KH (2018) Cancer Immunotherapy in Diffuse Large B-Cell Lymphoma. Front. Oncol. 8:351. doi: 10.3389/fonc.2018.00351
Received: 21 March 2018; Accepted: 09 August 2018;
Published: 10 September 2018.
Edited by:
Catherine Sautes-Fridman, INSERM U1138 Centre de Recherche des Cordeliers, FranceReviewed by:
Daniel Olive, Aix Marseille Université, FranceJohn Timmerman, UCLA David Geffen School of Medicine, United States
Copyright © 2018 Zhang, Medeiros and Young. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ken H. Young, khyoung@mdanderson.org