Update on Genetic Conditions Affecting the Skin and the Kidneys
 Antonia Reimer
Antonia Reimer Yinghong He1
Yinghong He1  Cristina Has
Cristina Has- 1Department of Dermatology, Faculty of Medicine, Medical Center, University of Freiburg, Freiburg, Germany
- 2Berta-Ottenstein-Programme, Faculty of Medicine, University of Freiburg, Freiburg, Germany
Genetic conditions affecting the skin and kidney are clinically and genetically heterogeneous, and target molecular components present in both organs. The molecular pathology involves defects of cell–matrix adhesion, metabolic or signaling pathways, as well as tumor suppressor genes. This article gives a clinically oriented overview of this group of disorders, highlighting entities which have been recently described, as well as the progress made in understanding well-known entities. The genetic bases as well as molecular cell biological mechanisms are described, with therapeutic applications.
Introduction
Anomalies of both skin and kidney occur in a vast number of genetic conditions. There are two major reasons for this concomitant occurrence of clinical manifestations. First, skin and kidney share a common embryological background represented by mesoderm for dermal connective tissue and kidneys. Second, various molecules involved in adhesion (e.g., integrin α3, CD151), cholesterol biosynthesis (e.g., NSDHL), or signaling pathways [e.g., Wnt, hedgehog (Hh), Ras/MAPK] are highly relevant for the development, structure, and function of both organs. In some syndromes, cutaneous and renal involvements are among the striking, pathognomonic features. Many of these disorders are recognizable at birth or early childhood.
Renal anomalies include congenital abnormalities of the kidney and urinary tract (CAKUT) (e.g., renal hypoplasia or aplasia, horseshoe deformations, anomalies of the urine collection system), malfunctioning of glomerular filtration, and the predisposition for tumors. The spectrum of skin anomalies is wide including pigmentation anomalies, skin dryness and ichthyosis, vascular anomalies (e.g., nevi flammei and hemangiomas), benign and malign skin tumors, abnormal hair, and nail dystrophy.
In this overview, genetic conditions affecting the skin and the kidneys are divided into three main groups:
1. Monogenic disorders with skin and renal involvement
2. Disorders due to postzygotic mosaicism
3. Chromosomal aberrations.
Monogenic Disorders with Skin and Renal Involvement
In certain monogenic disorders, such as epidermolysis bullosa (EB), RASopathies or disorders with tumor predisposition, cutaneous, as well as primary or secondary renal involvement may occur. In this section, we update the clinical and molecular features of the most relevant disorders of this vast group. In a large number of other genodermatoses and genetic syndromes, renal involvement may occur, but is not a defining feature. The clinical and molecular characteristics of these rare disorders are updated in Tables 1 and 2.
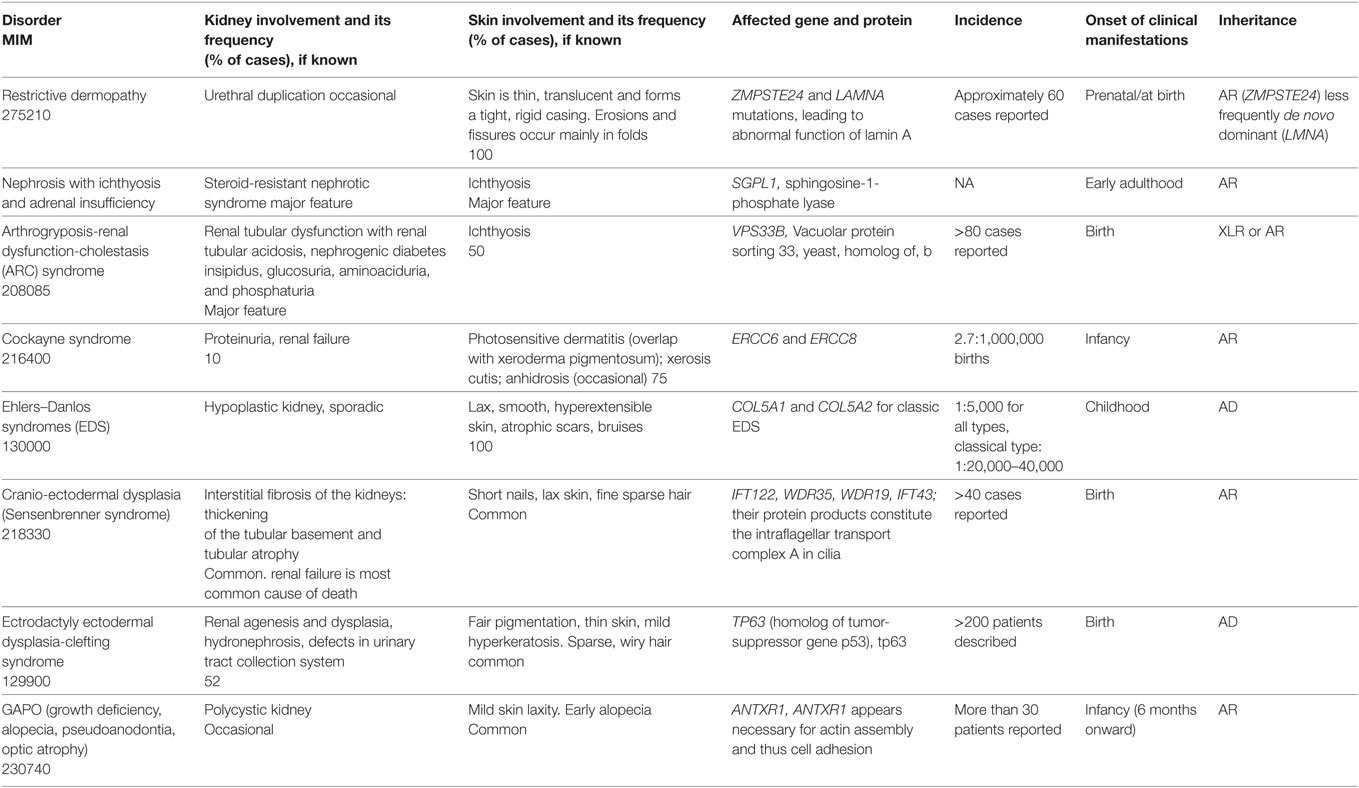
Table 1. Genodermatoses with reno-urinary involvement.
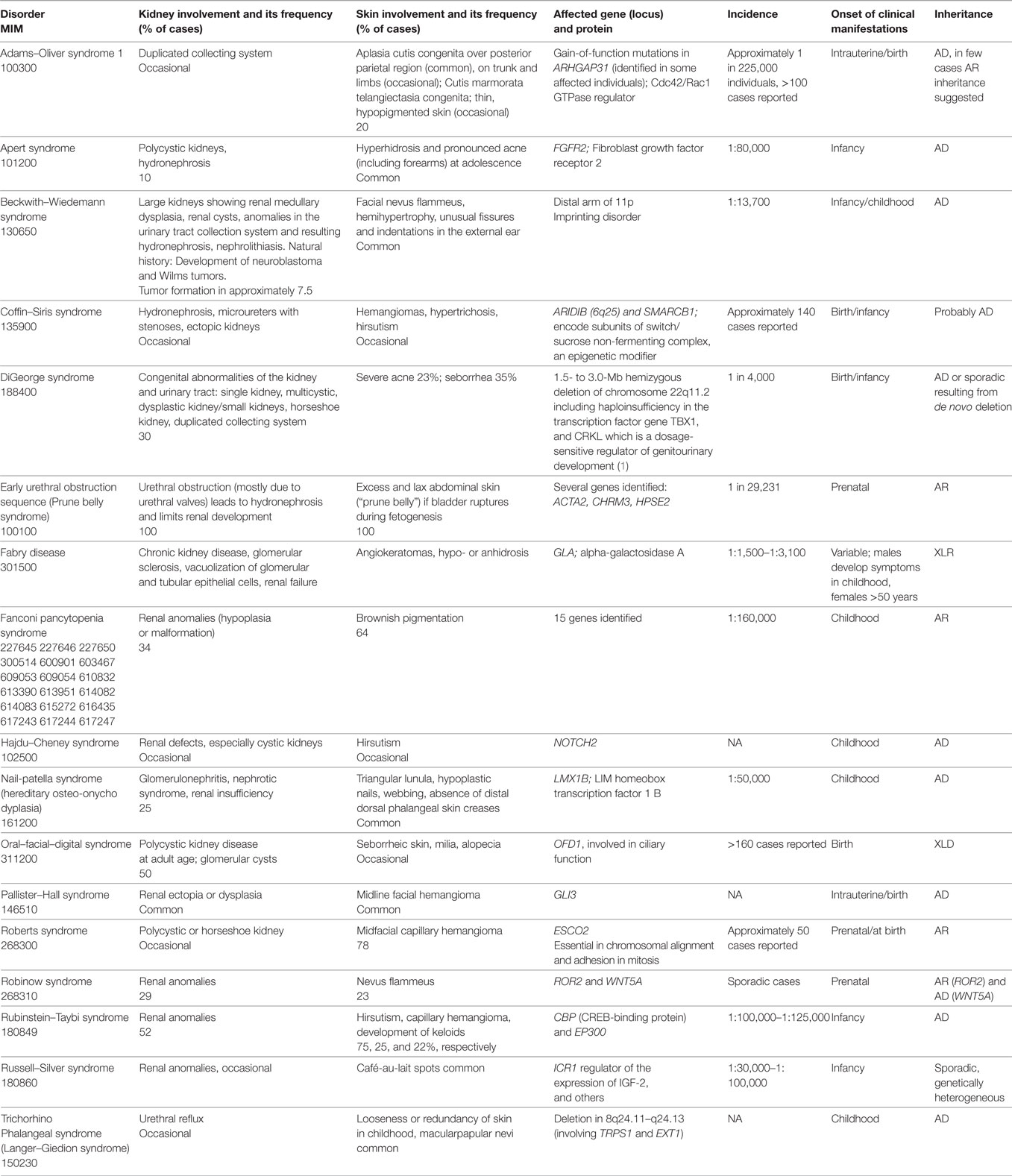
Table 2. Syndromes with cutaneous and reno-urinary involvement.
Epidermolysis Bullosa
Epidermolysis bullosa encompasses disorders defined by cutaneous and mucosal fragility. Classification into four major EB types, EB simplex, junctional EB, dystrophic EB, and the Kindler syndrome, is based on the ultrastructural level of skin cleavage (2, 3). Renal and urinary tract anomalies may occur in all EB types, in particular in junctional and dystrophic EB. In patients with severe dystrophic EB due to absence of collagen VII various renal pathologies may occur and lead to chronic renal failure. Hydronephrosis, poststreptococcal glomerulonephritis, IgA mesangial disease, or renal amyloidosis has been reported in dystrophic EB case series (4, 5). The mechanisms may include repetitive vesiculation within the lining epithelia of the urinary tract, and chronic systemic inflammation (6). Only EB types for which reno-urinary involvement is a primary feature will be described here.
Interstitial Lung Disease, Nephrotic Syndrome, and EB (ILNEB; MIM 614748)
Clinical Features
ILNEB is a rare autosomal recessive multiorgan disorder affecting the skin, kidneys and lungs. So far, 11 cases have been identified [reviewed in Ref. (5), and own unreported data], but the disease may be under recognized.
The clinical manifestations of ILNEB encompass the triad of early onset interstitial lung disease with respiratory distress, variable renal anomalies, and skin fragility. Since integrin α3 is widely expressed, clinical manifestations may occur in other organs, but are not characterized yet, because of the small number of cases and the early lethality. Skin involvement may include blistering, erosions or nail dystrophies, or may remain clinically unrecognized. The following renal anomalies were reported: congenital nephrotic syndrome, focal–segmental glomerulosclerosis, bilateral renal cysts, and a spectrum of CAKUT, including renal hypodysplasia, unilateral kidney hypoplasia, and ectopic conjoint kidney (7–12). Recently, two siblings of 13 and 9 years with viable ILNEB phenotypes presenting with growth retardation, severe pulmonary fibrosis, skin atrophy and erythema, scarce eyelashes/eyebrows, and nail anomalies (pachyonychia) but without renal features were described (13).
Genetics and Molecular Pathology
This disease is caused by mutations in the gene for integrin α3 (ITGA3) (7). Thus far, 10 ITGA3 mutations have been reported: 2 frameshift, 2 splicing, and 6 missense mutations (5). Loss-of-function mutations were associated with lethality before the age of 2 years. The consequences of missense mutations cannot be easily predicted. Some of them were shown to disturb the posttranslational modifications of integrin α3, which proved to be critical for the heterodimerization with integrin β1 and localization to the cell membrane (8, 9, 14).
Integrin α3 is the main integrin linking podocyte foot processes to the glomerular basement membrane [reviewed in Ref. (15, 16)]. In keratinocytes, it is located at cell–matrix adhesions, promoting epidermal adhesion primarily by maintaining the integrity of the basement membrane (17). The integrin α3 subunit is a widely expressed type I transmembrane protein consisting of a large extracellular region, a single transmembrane domain, and a short cytoplasmic tail (18). It forms obligate heterodimers with β1 integrin serving as a receptor for laminins, the major components of epithelial basement membranes (19). Integrin α3 is reduced or lost in several acquired conditions with glomerular disease, in which it is associated with reduction in podocyte adhesion to the glomerular basement membrane. For example, in podocytes of early-stage diabetic nephropathy integrin α3 expression was upregulated (20), while expression was suppressed with progression of the disease (21). In patients with primary focal segmental glomerulosclerosis, podocyte depletion was accompanied by reduced podocyte expression of α3β1 integrins (22). Moreover, integrin α3 is involved in podocyte foot process effacement during nephrotic syndrome (23).
Nephropathy with Pretibial EB and Deafness (MIM 609057)
Clinical Features
Two siblings with congenital nephrotic syndrome and pretibial EB were first described in 1988 (24). The disease-causing mutation in the gene for the tetraspanin CD151 was identified in 2004 (25), and very recently an additional case was reported (26). The first two cases had proteinuria in the nephrotic range and end-stage renal failure requiring hemodialysis or peritoneal dialysis from the age of 14 or 16 years on, respectively (24). The third case was a 33-year-old male with nephropathy manifesting with proteinuria below the nephrotic range, multiple episodes of pyelonephritis, and urinary incontinence, manifesting as a combination of overflow incontinence and intermittent urge incontinence (26). Additional manifestations included pretibial or extensive skin blistering, poikiloderma, nail dystrophy, hair loss, dystrophic teeth, involvement of the ocular, oral, gastrointestinal, and urogenital mucosal membranes (25, 26).
Genetics and Molecular Pathology
A homozygous single-nucleotide duplication in the CD151 gene leading to frameshift and a premature stop codon was identified in the first two cases (25). Flow cytometry analysis demonstrated absence of reactivity for CD151, suggesting that the predicted truncated polypeptide was not functional. In the third case, a homozygous CD151 splice site mutation, affecting a canonical donor splice site junction was found (26). Immunofluorescence staining and western blot analysis confirmed that the splice site mutation led to absence of CD151 in the cells of the patient (26).
CD151 (syn. Raph blood group, TSPAN24) is a member of the tetraspanin family of cell surface proteins and acts as a stabilizer of integrins (27). CD151 forms complexes with integrin α3β1 in cell culture and in vivo (28, 29). These complexes are assembled early during the integrin biosynthesis and precede the interaction of CD151 with other tetraspanins (30). CD151 also regulates glycosylation of α3β1 (31). CD151 is widely expressed in epithelia, endothelia, muscle cells, renal glomerular podocytes, Schwann and dendritic cells, in platelets and megakaryocytes. CD151 is involved in the formation and/or maintenance of the glomerular basement membrane (32).
Junctional EB with Pyloric Atresia (MIM 226730)
Clinical Features
Junctional EB with pyloric atresia manifests with aplasia cutis congenita (Figure 1), generalized skin blistering, and pyloric atresia. Several acquired complications of the reno-urinary system are reported, including pyelonephritis, hydronephrosis, urinary retention, development of bladder hypertrophy, and urethral meatal stenosis (4, 33, 34).
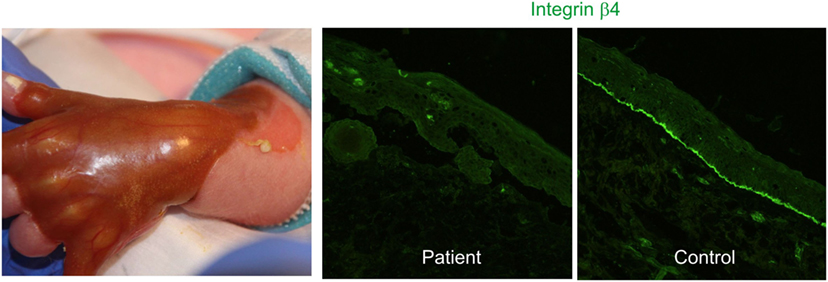
Figure 1. Congenital absence of the skin, also known as aplasia cutis congenita, in a newborn with hydronephrosis and pyloric atresia due to integrin α6β4 deficiency (right panel) [clinical pictures, courtesy of Dr. P. Häusermann (Department of Dermatology Basel)].
Genetics and Molecular Pathology
The disease is caused by mutations in the genes coding for the α6 or β4 integrin subunits, most mutations residing in ITGB4. Absence of α6β4 integrin is associated with a high rate of lethality in the first months of life, while missense and splicing mutations lead to moderate disease severity and reno-urinary manifestations.
Integrin α6β4 is a heterodimer composed of two type I transmembrane subunits localized in hemidesmosomes’, which anchor keratin intermediate filaments to the cell membrane and extracellular matrix. The intracellular region of α6β4 consists of the short tail of α6 and a large β4 cytoplasmic domain, which interacts with plectin and collagen XVII in keratinocytes. The ligands of α6 integrin are CD151, collagen XVII and laminin 332. Integrin α6β4 has a major adhesive function and promotes polarization of the cells (35). α6β4 is expressed in the epithelial cells within the medulla of the kidney. In a mouse model, α6β4 was not required for morphogenesis of the urinary tract, but for maintaining the integrity of the kidney collecting system. Collecting duct anomalies appeared as the animals aged. α6-null collecting duct cells were not able to withstand mechanical stress and detached from the basement membrane (36, 37).
Junctional EB Generalized Severe (Formerly: Herlitz EB; MIM 226700)
Clinical Features
Junctional EB generalized severe is caused by complete lack of laminin 332, the major laminin expressed in the cutaneous basement membrane. Laminin 332 is a heterotrimeric glycoprotein consisting of three polypeptide chains: laminin α3, β3, and γ2, encoded by LAMA3, LAMB3, and LAMC2, respectively. The clinical picture is dominated by mucocutaneous blistering from birth onward. Extensive generalized blistering leads to loss of fluids and protein and failure to thrive. The most common complications are anemia, dyspnea, infections, and sepsis. Affected children show multiorgan involvement and commonly die before the age of 2 years (38). In an infant with LAMB3 mutations, nephrotic syndrome with albuminuria due to failure of the glomerular filtration barrier, and high urinary N-acetylglucosaminidase levels, also indicating renal tubular involvement were reported (39).
Genetics and Molecular Pathology
Laminin 332 is the major laminin expressed by keratinocytes, but is also present in multiple epithelial basement membranes, including those of kidney. Like all laminins, it is a glycoprotein composed of three chains (α3, β3, and γ2) bound through disulfide bonds (5). In junctional EB generalized severe, mutations are found in one of the three genes encoding the laminin 332 chains. In the majority of cases, mutations are located in LAMB3 and lead to premature termination codons, mRNA decay, and absence of laminin 332.
RASopathies
RASopathies represent an expanding common group of neurodevelopmental disorders caused by germline mutations in genes encoding components of the Ras/MAPK pathway (40). Collectively, they affect >1 in 1,000 individuals (41). The Ras/MAPK pathway is a conserved omnipresent intracellular signaling pathway that is critical in regulating cell cycle, differentiation, growth, apoptosis, and senescence (40). The group of RASopathies includes neurofibromatosis type 1 (NF1), Noonan syndrome (NS), NS with multiple lentigines, Legius syndrome, Costello syndrome, cardio-facio-cutaneous syndrome, capillary malformation-arteriovenous malformation, and autosomal dominant intellectual disability type 5. Because of the common molecular mechanisms, phenotypic features of these syndromes are overlapping.
NF1 (von Recklinghausen Disease, MIM 162200)
Clinical Features
With an incidence of 1:2,500–3,000 (42), NF1 is one of the most common disorders of this group. NF1 follows autosomal dominant inheritance, about half of all cases occur due to spontaneous mutations. Diagnosis of NF1 is established following a set of clinical diagnostic criteria established in 1988 [Table 3, diagnosis of NF1 is probable when more than two criteria are present (43)]. Most cases are diagnosed in childhood, but when the complete set of criteria is not yet evident, follow-up and re-evaluation are necessary. Cutaneous features include café-au-lait macules, cutaneous and internal neurofibromas, or plexiform neurofibromas and axillary freckling. Renal involvement occurs sporadically, manifestations include hypertension due to renal artery stenosis, renal neurofibromas, and renal metastases of malignant schwannomas. The cooccurrence of NF1 and Wilms’ tumor has been reported in some cohorts (44, 45).

Table 3. Diagnostic criteria for neurofibromatosis 1 (NF1) (43).
Individuals with NF1 have a high risk of developing malignancies, especially malignant peripheral nerve sheath tumors (46). Life expectancy has been found to be approximately 8 years lower than in the normal population (47).
The cutaneous features progress with age. Neurofibromas as the main cutaneous finding in NF1 can be itchy, lead to disfigurement, and cause psychological strain. They can be treated with excisions or laser ablation (Er:YAG or CO2 laser) (48, 49), both with risk for hypertrophic scarring and recurrence (42).
In uncomplicated cases, clinical evaluation in childhood should be performed annually and include auxologic measurements, cardiovascular assessment, skin examination, and developmental progress (42). In childhood, visual assessment should be performed every 6–12 months for early detection of optical pathway glioma until the age of 7 years (42).
Genetics and Molecular Pathology
Monoallelic loss-of-function variants in NF1 coding for neurofibromin 1 are disease-causing in NF1. Neurofibromin is a multifunctional tumor suppressive protein which functions as a GTPase-activating protein. Neurofibromin inhibits cell proliferation and growth by blocking RAS-mediated signal transduction and modulates cell motility and adhesion (50).
The mutational spectrum is highly heterogeneous including nonsense and missense mutations, splice site mutations (about 30% of cases), small insertion–deletions, whole-gene deletions (4–5%), and structural rearrangements (51). Penetrance is complete after childhood, but NF1 is characterized by extreme clinical variability which is poorly understood, as are genotype–phenotype correlations. Intra- and interfamilial evaluation of the NF1 phenotype suggests that genetic modifiers which are not linked to the NF1 locus contribute to the variable expressivity of the disease (52, 53). Differently skewed expression of the NF1 alleles as well as somatic “second hit” variants or loss of heterozygosity may account in part for the phenotypic variability (54, 55). In addition to NF1, atypical manifestations, such as familial spinal neurofibromatosis, multiple spinal ganglioneuromas, optic gliomas, or Lentigines, Electrocardiographic abnormalities, Ocular hypertelorism, Pulmonary stenosis, Abnormalities of genitalia, Retardation of growth and Deafness (LEOPARD) syndrome, have been associated with NF1 mutations. Finally, incidental occurrence of NF1 mutations together with mutations in other genes may account for atypical phenotypic associations.
NS with Multiple Lentigines (syn. LEOPARD Syndrome, Multiple Lentigines Syndrome, Lentiginosis profusa and Progressive Cardiomyopathic Lentiginosis; MIM 151100)
Clinical Features
Noonan syndrome with multiple lentigines is a rare RASopathy that manifests in childhood. The incidence is unknown; so far, more than 200 cases were published. The characteristic cutaneous appearance is described well by the acronym LEOPARD: the skin appears spotted due to thousands of dark brown lentigines of 1–5 mm size which are distributed on the entire body (including sun-protected areas), cooccurring with café-au-lait macules (hence sometimes confused with NF1), hypomelanotic macules, and sometimes axillary freckling. Apart from LEOPARD are defining features (56). CAKUT, including horseshoe kidneys, occur in 11% of affected individuals (57). NS with multiple lentigines is sometimes difficult to distinguish from NF1 and the allelic NS (58), especially in early childhood when pigmentation anomalies are not yet pronounced (59). The prognosis is generally good, but can be limited by hypertrophic cardiomyopathy, arrhythmias, and sudden cardiac death. Annual cardiologic check-up should be performed life-long, and hearing assessment should be undertaken until adulthood. If auxologic follow-up indicates small statue, growth hormone therapy should be considered (56). Intense pulsed light has been used for cosmetic treatment of lentigines (60).
Genetics and Molecular Pathology
Noonan syndrome with multiple lentigines is allelic with NS and with the cardio-facio-cutaneous syndrome. The genetic basis of NS with multiple lentigines is heterogeneous including heterozygous pathogenic variants in one of four genes PTPN11 (90% of cases), RAF1 (less than 5% of cases), BRAF and MAP2K1 (both in single cases) (61). One or more additional, as-yet undefined genes are probably associated with about 5% of cases in whom no pathogenic variant has been identified (61). Genotype–phenotype correlations are not well established (62).
All involved genes code for components of the Ras/MAPK pathway:
• PTPN11 encodes the protein tyrosine phosphatase non-receptor type 11 that in its active form increases downstream Ras activity
• RAF1 encodes a serine–threonine kinase that activates MEK1 and MEK2
• BRAF encodes the B-Raf proto-oncogene serine/threonine kinase that activates MEK1 and/or MEK2 by phosphorylation
• MAP2K1 encodes the mitogen-activated protein kinase kinase 1 that activates ERK1 and/or ERK2 by phosphorylation (41).
Somatic mutations in all these genes are present in various types of cancers. Indeed, individuals with NS have a threefold increased risk of malignancies, such as juvenile myelomonocytic leukemia, acute lymphoblastic leukemia, rhabdomyosarcoma, and neuroblastoma (63, 64).
Genetic Tumor Predisposition Syndromes Affecting both Skin and Kidney
This is a large group of disorders characterized by both numerous hamartomas (benign tumors that can develop in basically all tissues) and premature development of malignant tumors during childhood. The molecular pathology of these conditions is highly heterogeneous. The most common conditions are described below or in Table 4. The tumors in these syndromes can occur in both cutaneous and extracutaneous locations, including the kidney (Table 4). Other tumor predisposition syndromes which usually manifest in adult age are only briefly mentioned.
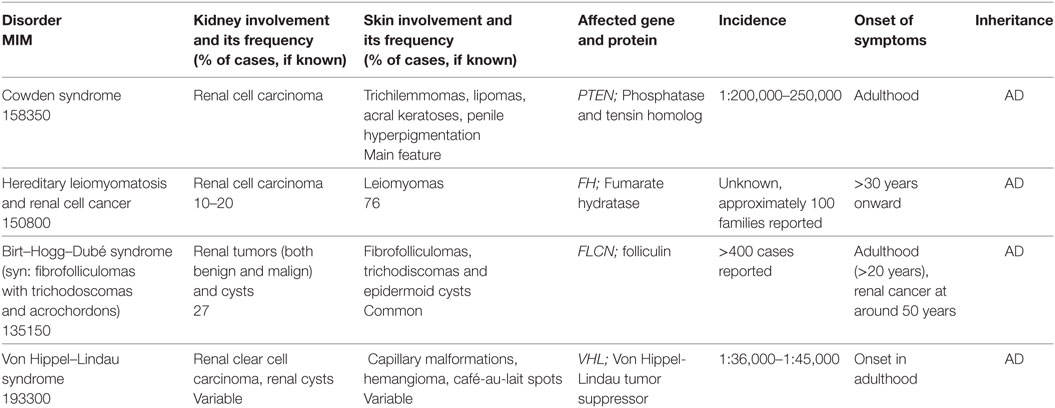
Table 4. Genetic tumor predisposition syndromes with cutaneous and reno-urinary involvement.
Tuberous Sclerosis Complex (TSC, TSC1 and TSC2, syn. Bourneville Disease; MIM 191100)
Clinical Features
Tuberous sclerosis complex occurs with an estimated incidence of 1:5,800–1:10,000 (65). It is mostly diagnosed in infancy when it manifests with skin findings and seizures due to cerebral hamartomas and giant cell astrocytomas. The diagnosis of TSC is made according to clinical criteria (66) (Table 5, either two major features are required or, alternatively, one major and two or more minor features). Typical cutaneous features are hypopigmented macules (best seen in Wood’s light), angiofibromas (mostly facial), periungual fibromas, and connective tissue nevi (shagreen patches). The frequency of cutaneous findings increases with age, but polygonal hypomelanotic macules, known as “ash-leaf spots,” are the earliest manifestation and are invariably present at birth. Renal involvement is also common, with angiomyolipomas and cysts as the most frequent renal manifestations found in 17% of children with TSC by age 2 years and 65% of 14 years old children with TSC (67). Renal cell carcinoma is more common in TSC than in the overall population (68).
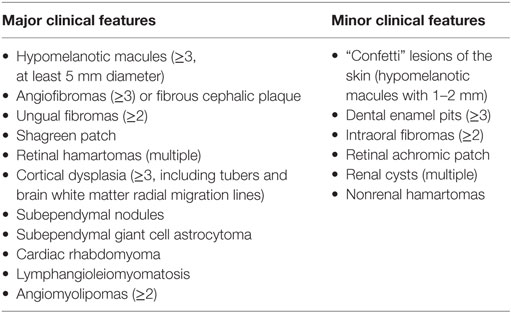
Table 5. Diagnostic criteria of tuberous sclerosis complex [adapted from Ref. (66)].
There is large variability in the clinical course, neurological development, and life expectancy in TSC, mostly depending on the number and location of hamartomas. While cutaneous features are crucial for clinical diagnosis, central nervous system tumors are the main cause of morbidity and mortality, while renal disease is the second leading cause of early death (69).
As TSC involves multiple organ systems, interdisciplinary care is necessary. Skin examinations should be performed annually. Diagnostic work-up of the kidney should include annual assessment of renal function and blood pressure and imaging (preferably with MRI) every 1–3 years (70). Since 2005, mTOR inhibitors have been evaluated for the use in TSC. Everolimus (Votubia®) is approved as a system therapeutic for use in children of 3 years and older with subependymal giant cell astrocytomas and for adults with complicated renal angiomyolipomas (71). Cutaneous lesions can be treated surgically, using laser (CO2/Erbium:YAG/Dye laser combination, or CO2, or Nd:YAG, or pulsed-dye laser) (72–74) and pharmacologically using topical mTOR inhibitors (75, 76). Surgical intervention can be considered as a therapeutic option for painful hemorrhagic renal angiomyolipomas and cerebral lesions.
Genetics and Molecular Pathology
Tuberous sclerosis complex is caused by monoallelic mutations in TSC1 (about 20% of cases) or TSC2 (about 70% of cases) (69) (Leiden open variation database). Two-thirds of TSC cases result from de novo pathogenic variants, and in about 10% no mutation can be detected (69). Large gene rearrangements, intronic pathogenic variants, and somatic or germ line mosaicism may explain the failure to detect mutations (77, 78). Specialized methods, such as targeted-deep sequencing of introns and exons and high-resolution SNP arrays improved the mutation detection rate to 94% (79). Genotype–phenotype correlations revealed that TSC2 mutations lead to earlier onset and more severe phenotype, as compared with TSC1 mutations (80). The occurrence of autosomal dominant polycystic kidney disease in TSC may be due to a contiguous deletion of TSC2 and PKD1 (81).
TSC1 and TSC2 code for hamartin and tuberin which form heterodimers within the TSC protein complex. Loss-of-function mutations in either TSC1 or TSC2 lead to constitutive activation of the mammalian target of rapamycin complex 1 (mTORC1) that is uncoupled from inhibitory mechanisms. Thus tumor cells in TSC have increased activation of mTORC1 signaling, resulting in increased protein synthesis and cell growth, and reduced autophagy (82). In fact, somatic inactivation of normal alleles is expected to drive mTOR activation, but second hit mutations are not always observed. The pathogenesis of angiofibromas involves UV-induced mutations suggesting that sun exposure is the initiating event (83). In angiomyolipomas, about 70% of the second-hit events are loss-of-heterozygosity mutations (84). A recent study showed that in TSC, somatic mutation rates were lower than most malignant tumors, while whole or arm-level chromosome gains and losses were the most remarkable finding in over 10% of patients (79).
Basal Cell Nevus Syndrome (syn. Gorlin Syndrome, Gorlin–Goltz Syndrome, Nevoid Basal Cell Carcinoma Syndrome; MIM 109400)
Clinical Features
The basal cell nevus syndrome is a rare autosomal dominant condition, occurring with an estimated incidence of 1:30,000 (85). It formally belongs to the group of hamartoses, but is mainly ranked among the tumor predisposition syndromes. Its characteristic feature is the occurrence of multiple basal cell carcinomas from young adulthood onward. Development of basal cell carcinoma in infancy has also been described (86). Other skin manifestations include palmar and plantar punctate dyskeratotic pits and facial milia. Skeletal abnormalities (e.g., polydactily), jaw cysts, and medulloblastoma occurring in 5% of patients are early features that can hint toward the diagnosis of basal cell nevus syndrome. Renal anomalies, such as renal agenesis (87) or Wilms tumors (88, 89), were reported in single cases. Diagnosis can be difficult in childhood due to few or unspecific findings. In suspected basal cell nevus syndrome, a systematic work-up including examinations by a dermatologist, a radiologist, a dentist, a gynecologist, a cardiologist, and a geneticist is recommended (90). After the occurrence of the first basal cell carcinoma, dermatologic examinations should be performed every 6–12 months. A baseline cerebral MRI with yearly controls until the age of 8 years is recommended. Echocardiography should be performed at baseline to rule out cardiac fibromas. X-ray of the jaw should be repeated yearly until a first jaw cyst is detected, after that every 6 months or according to symptoms. For scoliosis detection, an X-ray at the age of 1 year or at time of diagnosis is recommended. If normal, it is only repeated in case of symptoms. If scoliosis is present, regular follow-ups are appointed. Other baseline evaluations include pelvic ultrasound and ophthalmologic assessments. Psychological evaluation and support is advisable (90).
For many years, excision of basal carcinoma was the main treatment option for this condition. Understanding of the molecular pathology has recently led to development and approval of vismodegib (Erivedge®) as an effective therapy. Vismodegib targets the sonic Hh pathway and leads to regression of existing and inhibits the development of new tumors (91). Radiation should be avoided as it will trigger the eruption of multiple new tumors (92).
Genetics and Molecular Pathology
The genetic basis of the basal cell nevus syndrome is heterogeneous. The main cause is represented by monoallelic germline pathogenic variants in PTCH1 responsible for approximately 85% of the cases. SUFU pathogenic variants reside in about 5% of the cases (93). Rare causes are PTCH2 and SMO mutations: a missense mutation in PTCH2 was disclosed in a Chinese family (94), and a SMO mutation in a single case with a segmental basal cell nevus syndrome (95). In about 15–27% of cases, the genetic basis remains unclear (93). Low level of postzygotic mosaicism may explain some of the genetically unsolved cases (96). PTCH1 pathogenic missense variants have also been associated with holoprosencephaly (97).
All these genes encode key players in the Hh signaling pathway, which is essential for development of vertebrates and drives proliferation, migration, and differentiation of progenitor cells (98):
• PTCH1 encodes the patched homolog 1, the receptor for sonic Hh
• SUFU encodes the suppressor of fused homolog, a negative regulator of the Hh signaling pathway
• PTCH2 encodes patched 2
• SMO encodes smoothened frizzled class receptor, a G protein-coupled receptor that interacts with the patched protein.
Activation of the Hh pathway is initiated by the Hh ligand binding and inhibition of the transmembrane receptor patched 1, allowing the signal transducer smoothened to activate Gli transcription factors and amplify the expression of Hh target genes (98). Somatic mutations that activate the Hh signaling pathway drive growth of various cancers including basal cell carcinomas, medulloblastomas, pancreatic, prostate, and small cell lung cancer, that account for up to 25% of all human cancer deaths (99).
Birt–Hogg–Dubé Syndrome
Clinical Features
The Birt–Hogg–Dubé syndrome is an autosomal dominant disorder which manifests with cutaneous lesions, pulmonary cysts and/or history of pneumothorax, and various types of renal tumors (100). Skin involvement occurs during the second, third, or fourth decade of life and progresses with age. It includes various benign tumors such as fibrofolliculomas, trichodiscomas/angiofibromas, perifollicular fibromas, and acrochordons. Fibrofolliculomas are the most common phenotypic features of the Birt–Hogg–Dubé syndrome, occurring in more than 85% of the patients over the age of 25 years (101). They appear as multiple, small, skin-colored papules disseminated on the face, neck, and upper trunk. Treatment by laser ablation results in temporary improvement, but relapse usually occurs.
Individuals with Birt–Hogg–Dubé syndrome have a sevenfold increased risk to develop renal tumors, that are typically bilateral and multifocal (102, 103). They are usually diagnosed in adults (median of diagnosis is 48 years, but have been described as early as 20 years of age) and have a slow progression (103). The histologic types of renal tumors found in individuals with Birt–Hogg–Dubé syndrome are: by far predominant are chromophobe renal cell carcinomas, followed by hybrid oncocytic tumors and oncocytomas, while clear cell renal cell carcinomas are uncommon. Yearly screening by renal MRI is indicated in individuals with Birt–Hogg–Dubé syndrome age 18 years or older. In some families, renal tumors and/or spontaneous pneumothorax occur without cutaneous manifestations.
Genetics and Molecular Pathology
The Birt–Hogg–Dubé syndrome is caused by monoallelic pathogenic variants in FLCN, encoding folliculin. Mutation analysis detects disease-causing variants in 88% of the affected families; the deletion or duplication of a cytosine at position c.1285 is a mutational hot spot. Partial- or whole-gene deletions account for 3–5% of the cases and must be identified with specific methods. About 7–9% of the cases remain genetically unsolved. The protein folliculin forms a complex with folliculin-interacting protein 1 or 2 and binds to the 5′ AMP-activated protein kinase suppressing tumorigenesis (104). Moreover, it plays a role in mTOR activation (105–107).
Hereditary Leiomyomatosis and Renal Cell Cancer (HLRCC)
Clinical Features
Hereditary leiomyomatosis and renal cell cancer is characterized by the occurrence of cutaneous and uterine leiomyomata, and/or a single, unilateral, and aggressive renal tumor (108). Cutaneous leiomyomata may be multiple or single, appear in adults (mean age of 25 years), and increase in size and number with age. They manifest as skin-colored papules or nodules, disseminated on the trunk, extremities, and face. The treatment consists of surgical or laser excision, or cryoablation. Renal tumors occur in about 10–16% of individuals with HLRCC at a median age of 44 years and cause hematuria and lower back pain. Histologically they are classically classified as type 2 papillary (108).
Genetics and Molecular Pathology
Hereditary leiomyomatosis and renal cell cancer is caused by monoallelic FH mutations that lead to reduced activity of the enzyme fumarate hydratase (109). In tumor tissue, somatic variants and loss of heterozygosity are found. No genotype–phenotype correlations are known, and there is significant intrafamilial variability. Biallelic mutations resulting in fumarase deficiency cause an inborn error of metabolism characterized by rapidly progressive neurologic impairment including hypotonia, seizures, and cerebral atrophy (110).
Disorders Due to Postzygotic Mosaicism
The disorders in this group are caused by mutations that are mostly lethal if occurring as a germline mutation affecting all cells. However, if these mutations occur postzygotic in early embryogenesis, disorders with unilateral or segmental manifestations result, as proposed by Happle (111, 112). His hypothesis is supported by the elucidation of the molecular basis of several segmental disorders since the implementation of next-generation sequencing technologies.
Linear Sebaceous Nevus Sequence [Schimmelpenning–Feuerstein–Mims Syndrome, Nevus Sebaceous of Jadassohn; MIM 163200]
Clinical Features
This syndrome belongs to the group of epidermal nevus syndromes. More than 100 sporadic cases have been described, the incidence is not known. While solitary sebaceous nevi are a reasonably common finding in infants, the sebaceous nevi in this syndrome are associated with seizures, mental retardation, skeletal and ophthalmic anomalies, and asymmetric growth. At birth, one (or multiple) sebaceous nevi is/are found mostly in the mid-face. Involvement of the head/neck area is possible, as are locations on trunk or extremities. The sebaceous nevus shows a linear configuration along the lines of Blaschko. While it is mostly flat and wax-like in infancy, verrucous changes, hyperpigmentation, hyperkeratosis, and hypertrophy are seen toward puberty. In adulthood, development of (mostly benign) tumors within the sebaceous nevus is noted. Skeletal, ophthalmic, and renal involvements occur occasionally, the latter comprising CAKUT, such as double urinary collecting system and horseshoe kidneys, and renal hamartoma and nephroblastoma.
Surgical treatment of sebaceous nevi can be offered for cosmetic or psychological reasons. Excision is generally not indicated because of cancer prophylaxis, as the risk for malignant tumors is very low (113). Therapeutic options include excision and laser ablation (114). Regarding the main complications, seizures, neurological retardation, and rickets, interdisciplinary care for children with linear sebaceous nevus sequence should be offered.
Genetics and Molecular Pathology
This syndrome can be considered a mosaic RASopathy because it is caused by postzygotic pathogenic variants in HRAS (HRas Proto-Oncogene, GTPase), KRAS (KRAS Proto-Oncogene, GTPase), or the NRAS (NRas Proto-Oncogene, GTPase) genes (115, 116). The recurrent activating mutations induce constitutive activation of the MAPK and PI3K/AKT signaling pathways. Somatic mutations in the HRAS and/or KRAS genes were also found in isolated sebaceous nevi. These mutations are only present in keratinocytes which give rise to the cutaneous lesions.
The proteins encoded by the Ras oncogene family have intrinsic GTPase activity and function in signal transduction pathways important for cell growth, proliferation, and survival. Defects in these genes are present in various cancers.
Neurocutaneous Melanocytosis (syn. Neurocutaneous Melanosis Sequence, Neuromelanosis; MIM 249400)
Clinical Features
The landmark lesion of this syndrome, a giant pigmented nevus, is seen at birth. It is usually located in the posterior head or trunk (117). Sometimes, three or more large congenital nevi are found rather than a single giant nevus. Numerous disseminated (“satellite”) nevi can be present at birth and more will develop in the course of disease. The presence and proliferation of melanin-producing cells within cranium and spine leads to increased intracranial pressure, seizures, mental deterioration, and death in early childhood (117). Leptomeningeal and intracranial melanoma occur in a significant portion of patients. Occasional abnormalities found in neurocutaneous melanocytosis are cerebral malformations such as syringomyelia and Dandy–Walker malformation, CAKUT, and unilateral renal cysts. Other tumors occurring in the syndrome include rhabdomyosarcoma, liposarcoma, and malignant peripheral nerve sheath tumors.
There is a risk of development of cutaneous malignant melanoma within the congenital nevi. They develop in the depth of the lesion and can be felt earlier than they can be seen. This has been suggested to be as high as 15% (118) in giant congenital melanocytic nevi, although others have reported incidences of 0.7% (119). The amount of patients with the full picture of neurocutaneous melanocytosis developing malignant melanoma is not known, probably as most of these patients die before developing melanomas.
They clinical course is mostly determined by neurologic symptoms. If these occur, there is no effective therapeutic approach. If the child shows normal psychomotoric development, excision for the giant melanocytic nevi is recommended. Such surgical procedures may require several steps and the use of tissue expanders. Dermabrasio is not considered as a therapeutic option any more. To date, a causal therapy is not available, but a recent in vitro assessment of inhibitors of the NRAS-signaling pathway (drugs also successfully used in the therapy of malignant melanoma) showed promising results (120).
Genetics and Molecular Pathology
Somatic oncogenic missense mutations affecting codon 61 of the NRAS gene were identified in affected cutaneous (melanocytes) and nervous tissues from patients with congenital melanocytic nevus syndrome and/or neurocutaneous melanosis (118).
CHILD Syndrome (Congenital Hemidysplasia with Ichthyosiform Erythroderma and Limb Defects; MIM 308050)
Clinical Features
This epidermal nevus syndrome was coined with the acronyme “CHILD” by Happle and colleagues in 1980 (121) to sum up the main findings in children with this condition: a characteristic, mostly unilateral epidermal nevus in combination with ipsilateral congenital hemidysplasia of bones (affecting any part of the body, mainly limbs). The epidermal nevus is usually present at birth but can also develop in the first weeks of life. Spontaneous involution is sometimes witnessed (113). The CHILD nevus is red and scaly. It can show strict lateralization (right side more frequently than left side, 3:2) and midline demarcation, but it can also follow lines of Blaschko, and both patterns may be present in an affected individual (113) (Figure 2). Next to cardiovascular anomalies, renal findings comprise CAKUT, such as renal agenesis (122) or hypoplasia (123), to unilateral hydronephrosis, but their frequency is unknown (124).
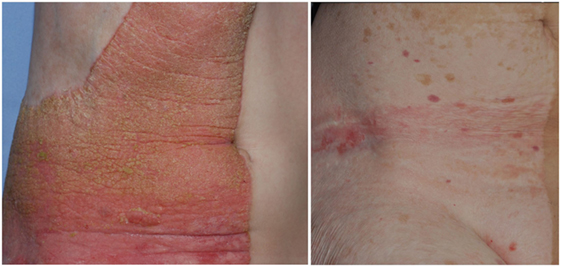
Figure 2. Unilateral epidermal nevus in a patient with CHILD syndrome, before (left panel) and after 5 years topical application of a simvastatin/cholesterol cream (right panel).
Addressing the molecular pathology of CHILD, a therapeutic approach for treating the epidermal nevus combining simvastatin and cholesterol for topical use proved to be effective (125, 126) (Figure 2).
Genetics, Molecular Pathology
CHILD syndrome is caused by monoallelic loss-of-function pathogenic variants in the NSDHL gene encoding the NAD(P)H steroid dehydrogenase-like protein, which is a C4 demethylase involved in postsqualene cholesterol biosynthesis (127). The enzyme is located within the membranes of the endoplasmic reticulum. Its deficiency leads to impaired cholesterol processing, causing abnormal sonic Hh signaling, which affects spatial patterning during embryogenesis (128). The cutaneous features may result from a dual mechanism: accumulation of cholesterol precursors and cholesterol deficiency (128).
This X-linked dominant disorder is lethal in male during gestation and thus predominantly affects females. The CK syndrome [initials of the original proband (129)] is an X-linked recessive disorder that affects males being also caused by pathogenic NSDHL variants (130). In CHILD syndrome, mosaicism results from inactivation of an X-chromosome in females. Inter-individual differences in the pattern of X inactivation explain the phenotypic variations.
Focal Dermal Hypoplasia (Goltz syndrome; MIM 305600)
Clinical Features
Focal dermal hypoplasia is rare; more than 175 cases have been reported. The focal dermal hypoplasia is mostly encountered in females (90%), as its X-linked dominant inheritance leads to lethality in male fetuses. Affected males usually show a mosaic form of focal dermal hypoplasia. This syndrome is evident at birth, when skin and skeletal symptoms are predominant. Children with focal dermal hypoplasia show skin atrophy with Blaschko linear arrangement, appearing as depressed or slightly raised red macules. This finding explains the original name “focal dermal hypoplasia” (131). Over the course of disease, fatty tissue can herniate through gaps in the underdeveloped connective tissue forming lipomatous papules. Papillomas and angiofibroma occur on the face and in the urogenitoanal region (132). Additional findings are patchy alopecia and thin hair. Affected children show facial dysmorphies and asymmetric skeletal deformities (e.g., syndactyly, polydactyly, amelia, scoliosis). Renal anomalies occur occasionally and include horseshoes kidneys and hydronephrosis.
No specific therapy for focal dermal hypoplasia exists. Papillomas can be surgically removed, but may reoccur.
Genetics and Molecular Pathology
Focal dermal hypoplasia is an X-linked dominant disorder which reflects mosaicism resulting from inactivation of an X-chromosome in females. The pathogenic variants affect PORCN (133). PORCN is a gene of the porcupine family, which code for endoplasmic reticulum proteins with multiple transmembrane domains involved in the processing of Wnt (wingless and int homolog) proteins. Mutations in different players of the Wnt signaling pathway have been described before to cause CAKUT (134), explaining the pathogenesis of CAKUT in focal dermal hypoplasia. The disease is lethal in males; live-born affected males are rare and nearly always have somatic mosaicism for a de novo postzygotic pathogenic variant. Postzygotic mutations may also cause mild disease in females (135).
Chromosomal Aberrations
In case of chromosomal aberrations, e.g., deletions or trisomies, a large number of genes are affected by the defect. Therefore, the resulting clinical picture is broad and includes renal and cutaneous anomalies in some syndromes (Table 6). However, these are not defining for the clinical picture.
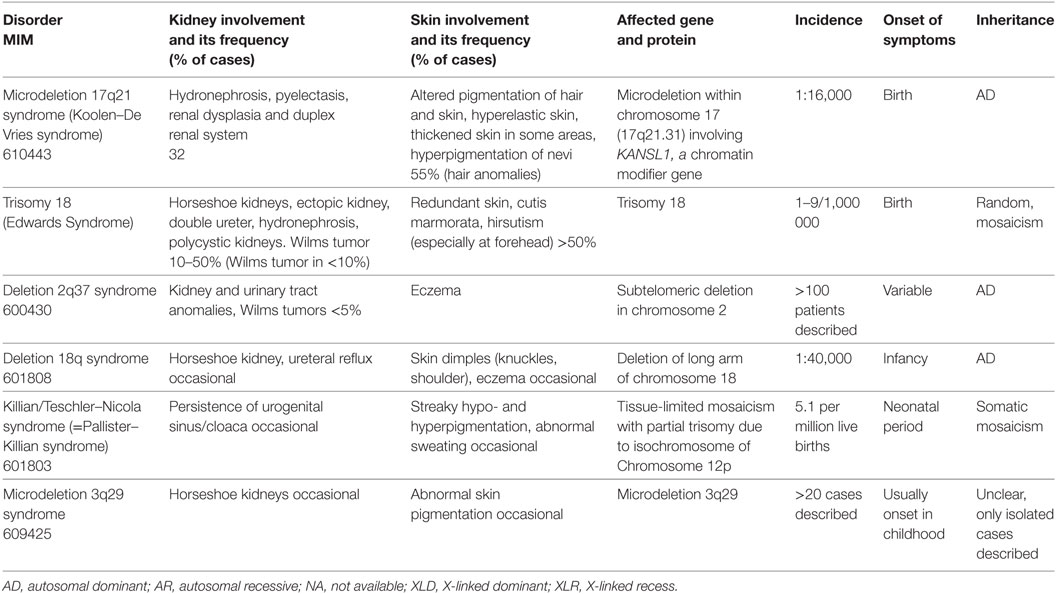
Table 6. Chromosome anomalies with cutaneous and reno-urinary involvement.
Differential Diagnosis in Newborns
Several acquired conditions affect both skin and kidney either pre- or postnatally. For example, maternal intake of valproate leads to fetal valproate syndrome commonly showing hemangiomas, altered pigmentation, and occasional renal malformations. Intake of phenytoin during pregnancy causes fetal hydantoin syndrome, in which hirsutism and coarse hair are common and renal malformations can occur. The oligohydramnios sequence (Potter syndrome) arises from lack of amniotic fluid. This anhydramnion or oligohydramnion can either be caused by primary renal problems such as agenesis, severe polycystic kidney deformation or obstruction of the urinary tract, or by chronic leakage from the amniotic sac. Fetal development, especially of the lungs, and life expectancy are severely limited.
Conclusion
Genetic disorders affecting the skin and the kidneys cover a broad range of phenotypes and molecular mechanisms, which have been largely uncovered in the last decades. Many of these conditions comprise involvement of multiple organs and systems. Although, in many cases, the cutaneous findings (e.g., café-au-lait spots, angiofibromas, nevi) have no significant impact on the prognosis, they represent precious signs for the clinical diagnosis and should alert pediatricians to carefully evaluate the patients.
• Because of the clinical complexity these patients require an interdisciplinary care, comprising geneticists, dermatologists, nephrologists, cardiologists, etc., in which the pediatrician has a central coordinating role.
• The rarity of these disorders renders their diagnosis sometimes difficult, underrecognized and delayed. In addition, the cutaneous lesions have an esthetic impact (e.g., angiofibromas of the face, neurofibromas, nevi) and the tumors have an unpredictable, mostly progressive course. All these factors have a high psychological impact on the patients and their families, who require psychological aid and support from patients’ organizations.
• Illumination of the molecular pathomechanisms of some rare disorders (e.g., Ras/MAPK, mTOR, cholesterol biosynthesis) opened new opportunities to repurpose known drugs and use them to slow down disease progression. Such evolving therapies, either in the clinical practice or in clinical trials, have been briefly outlined in this review.
Author Contributions
AR wrote most of the clinical part (clinical features) and the tables. YH prepared the figures and reviewed the manuscript. CH drafted the manuscript, wrote the genetics and molecular part, and reviewed the entire manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
CH was supported by the Deutsche Forschungsgemeinschaft (DFG) CRC/SFB 1140 and AR by the Berta-Ottenstein Programme of the Faculty of Medicine, University of Freiburg. YH was a fellow of the Else-Kröner-Fresenius Foundation.
Abbreviations
CAKUT, congenital abnormalities of the kidney and urinary tract; EB, epidermolysis bullosa; Hh, hedgehog; HLRCC, hereditary leiomyomatosis and renal cell cancer; LEOPARD, Lentigines, Electrocardiographic abnormalities, Ocular hypertelorism, Pulmonary stenosis, Abnormalities of genitalia, Retardation of growth and Deafness; NF1, neurofibromatosis type 1; NS, Noonan syndrome; TSC, tuberous sclerosis complex.
References
1. Haller M, Mo Q, Imamoto A, Lamb DJ. Murine model indicates 22q11.2 signaling adaptor CRKL is a dosage-sensitive regulator of genitourinary development. Proc Natl Acad Sci U S A (2017) 114:4981–6. doi:10.1073/pnas.1619523114
2. Has C, Bruckner-Tuderman L. The genetics of skin fragility. Annu Rev Genomics Hum Genet (2014) 15:245–68. doi:10.1146/annurev-genom-090413-025540
3. Fine J-D, Bruckner-Tuderman L, Eady RAJ, Bauer EA, Bauer JW, Has C, et al. Inherited epidermolysis bullosa: updated recommendations on diagnosis and classification. J Am Acad Dermatol (2014) 70:1103–26. doi:10.1016/j.jaad.2014.01.903
4. Fine JD, Johnson LB, Weiner M, Stein A, Cash S, DeLeoz J, et al. Genitourinary complications of inherited epidermolysis bullosa: experience of the national epidermylosis bullosa registry and review of the literature. J Urol (2004) 172:2040–4. doi:10.1097/01.ju.0000143200.86683.2c
5. Has C, He Y. Renal-skin syndromes. Cell Tissue Res (2017) 369:63–73. doi:10.1007/s00441-017-2623-y
6. Fine JD, Mellerio JE. Extracutaneous manifestations and complications of inherited epidermolysis bullosa: part I. Epithelial associated tissues. J Am Acad Dermatol (2009) 61:367–84. doi:10.1016/j.jaad.2009.03.052
7. Has C, Spartà G, Kiritsi D, Weibel L, Moeller A, Vega-Warner V, et al. Integrin α3 mutations with kidney, lung, and skin disease. N Engl J Med (2012) 366:1508–14. doi:10.1056/NEJMoa1110813
8. Nicolaou N, Margadant C, Kevelam SH, Lilien MR, Oosterveld MJ, Kreft M, et al. Gain of glycosylation in integrin alpha3 causes lung disease and nephrotic syndrome. J Clin Invest (2012) 122:4375–87. doi:10.1172/JCI64100
9. Yalcin EG, He Y, Orhan D, Pazzagli C, Emiralioglu N, Has C. Crucial role of posttranslational modifications of integrin α3 in interstitial lung disease and nephrotic syndrome. Hum Mol Genet (2015) 24:3679–88. doi:10.1093/hmg/ddv111
10. He Y, Balasubramanian M, Humphreys N, Waruiru C, Brauner M, Kohlhase J, et al. Intronic ITGA3 mutation impacts splicing regulation and causes interstitial lung disease, nephrotic syndrome, and epidermolysis bullosa. J Invest Dermatol (2016) 136:1056–9. doi:10.1016/j.jid.2015.11.031
11. Lovric S, Fang H, Vega-Warner V, Sadowski CE, Gee HY, Halbritter J, et al. Rapid detection of monogenic causes of childhood-onset steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol (2014) 9:1109–16. doi:10.2215/CJN.09010813
12. Shukrun R, Vivante A, Pleniceanu O, Vax E, Anikster Y, Dekel B, et al. A human integrin-alpha3 mutation confers major renal developmental defects. PLoS One (2014) 9:e90879. doi:10.1371/journal.pone.0090879
13. Colombo EA, Spaccini L, Volpi L, Negri G, Cittaro D, Lazarevic D, et al. Viable phenotype of ILNEB syndrome without nephrotic impairment in siblings heterozygous for unreported integrin alpha3 mutations. Orphanet J Rare Dis (2016) 11:136. doi:10.1186/s13023-016-0514-z
14. Yamada M, Sekiguchi K. Disease-associated single amino acid mutation in the calf-1 domain of integrin alpha3 leads to defects in its processing and cell surface expression. Biochem Biophys Res Commun (2013) 441:988–93. doi:10.1016/j.bbrc.2013.11.003
15. Borza CM, Chen X, Zent R, Pozzi A. Cell receptor-basement membrane interactions in health and disease: a kidney-centric view. Curr Top Membr (2015) 76:231–53. doi:10.1016/bs.ctm.2015.07.003
16. Pozzi A, Zent R. Integrins in kidney disease. J Am Soc Nephrol (2013) 24:1034–9. doi:10.1681/ASN.2013010012
17. Longmate WM, Dipersio CM. Integrin regulation of epidermal functions in wounds. Adv Wound Care (2014) 3:229–46. doi:10.1089/wound.2013.0516
18. Campbell ID, Humphries MJ. Integrin structure, activation, and interactions. Cold Spring Harb Perspect Biol (2011) 3:a004994. doi:10.1101/cshperspect.a004994
19. Nishiuchi R, Takagi J, Hayashi M, Ido H, Yagi Y, Sanzen N, et al. Ligand-binding specificities of laminin-binding integrins: a comprehensive survey of laminin-integrin interactions using recombinant alpha3beta1, alpha6beta1, alpha7beta1 and alpha6beta4 integrins. Matrix Biol (2006) 25:189–97. doi:10.1016/j.matbio.2005.12.001
20. Sawada K, Toyoda M, Kaneyama N, Shiraiwa S, Moriya H, Miyatake H, et al. Upregulation of α3β1-integrin in podocytes in early-stage diabetic nephropathy. J Diabetes Res (2016) 2016:9265074. doi:10.1155/2016/9265074
21. Chen HC, Chen CA, Guh JY, Chang JM, Shin SJ, Lai YH. Altering expression of alpha3beta1 integrin on podocytes of human and rats with diabetes. Life Sci (2000) 67:2345–53. doi:10.1016/S0024-3205(00)00815-8
22. Chen C-A, Hwang J-C, Guh J-Y, Chang J-M, Lai Y-H, Chen H-C. Reduced podocyte expression of alpha3beta1 integrins and podocyte depletion in patients with primary focal segmental glomerulosclerosis and chronic PAN-treated rats. J Lab Clin Med (2006) 147:74–82. doi:10.1016/j.lab.2005.08.011
23. Reiser J, Oh J, Shirato I, Asanuma K, Hug A, Mundel TM, et al. Podocyte migration during nephrotic syndrome requires a coordinated interplay between cathepsin L and alpha3 integrin. J Biol Chem (2004) 279:34827–32. doi:10.1074/jbc.M401973200
24. Kagan A, Feld S, Chemke J, Bar-Khayim Y. Occurrence of hereditary nephritis, pretibial epidermolysis bullosa and beta-thalassemia minor in two siblings with end-stage renal disease. Nephron (1988) 49:331–2. doi:10.1159/000185086
25. Karamatic Crew V, Burton N, Kagan A, Green CA, Levene C, Flinter F, et al. CD151, the first member of the tetraspanin (TM4) superfamily detected on erythrocytes, is essential for the correct assembly of human basement membranes in kidney and skin. Blood (2004) 104:2217–23. doi:10.1182/blood-2004-04-1512
26. Vahidnezhad H, Youssefian L, Saeidian AH, Mahmoudi HR, Touati A, Abiri M, et al. Recessive mutation in tetraspanin CD151 causes Kindler syndrome-like epidermolysis bullosa with multi-systemic manifestations including nephropathy. Matrix Biol (2017). doi:10.1016/j.matbio.2017.11.003
27. Zoller M. Tetraspanins: push and pull in suppressing and promoting metastasis. Nat Rev Cancer (2009) 9:40–55. doi:10.1038/nrc2543
28. Sterk LM, Geuijen CA, van den Berg JG, Claessen N, Weening JJ, Sonnenberg A. Association of the tetraspanin CD151 with the laminin-binding integrins alpha3beta1, alpha6beta1, alpha6beta4 and alpha7beta1 in cells in culture and in vivo. J Cell Sci (2002) 115:1161–73.
29. Serru V, Le Naour F, Billard M, Azorsa DO, Lanza F, Boucheix C, et al. Selective tetraspan-integrin complexes (CD81/alpha4beta1, CD151/alpha3beta1, CD151/alpha6beta1) under conditions disrupting tetraspan interactions. Biochem J (1999) 340(Pt 1):103–11. doi:10.1042/bj3400103
30. Berditchevski F, Gilbert E, Griffiths MR, Fitter S, Ashman L, Jenner SJ. Analysis of the CD151-alpha3beta1 integrin and CD151-tetraspanin interactions by mutagenesis. J Biol Chem (2001) 276:41165–74. doi:10.1074/jbc.M104041200
31. Baldwin G, Novitskaya V, Sadej R, Pochec E, Litynska A, Hartmann C, et al. Tetraspanin CD151 regulates glycosylation of (alpha)3(beta)1 integrin. J Biol Chem (2008) 283:35445–54. doi:10.1074/jbc.M806394200
32. Baleato RM, Guthrie PL, Gubler M-C, Ashman LK, Roselli S. Deletion of CD151 results in a strain-dependent glomerular disease due to severe alterations of the glomerular basement membrane. Am J Pathol (2008) 173:927–37. doi:10.2353/ajpath.2008.071149
33. Burgu B, Duffy PG, Wilcox DT. Single-centre experience of genitourinary complications of epidermolysis bullosa. J Pediatr Urol (2006) 2:583–6. doi:10.1016/j.jpurol.2006.01.007
34. Schumann H, Kiritsi D, Pigors M, Hausser I, Kohlhase J, Peters J, et al. Phenotypic spectrum of epidermolysis bullosa associated with alpha6beta4 integrin mutations. Br J Dermatol (2013) 169:115–24. doi:10.1111/bjd.12317
35. Borza CM, Pozzi A. The role of cell-extracellular matrix interactions in glomerular injury. Exp Cell Res (2012) 318:1001–10. doi:10.1016/j.yexcr.2012.02.033
36. Yazlovitskaya EM, Tseng H-Y, Viquez O, Tu T, Mernaugh G, McKee KK, et al. Integrin α3β1 regulates kidney collecting duct development via TRAF6-dependent K63-linked polyubiquitination of Akt. Mol Biol Cell (2015) 26:1857–74. doi:10.1091/mbc.E14-07-1203
37. Viquez OM, Yazlovitskaya EM, Tu T, Mernaugh G, Secades P, McKee KK, et al. Integrin alpha6 maintains the structural integrity of the kidney collecting system. Matrix Biol (2017) 57–58:244–57. doi:10.1016/j.matbio.2016.12.003
38. Hammersen J, Has C, Naumann-Bartsch N, Stachel D, Kiritsi D, Söder S, et al. Genotype, clinical course, and therapeutic decision making in 76 infants with severe generalized junctional epidermolysis bullosa. J Invest Dermatol (2016) 136:2150–7. doi:10.1016/j.jid.2016.06.609
39. Hata D, Miyazaki M, Seto S, Kadota E, Muso E, Takasu K, et al. Nephrotic syndrome and aberrant expression of laminin isoforms in glomerular basement membranes for an infant with Herlitz junctional epidermolysis bullosa. Pediatrics (2005) 116:e601–7. doi:10.1542/peds.2005-0160
40. Tidyman WE, Rauen KA. Expansion of the RASopathies. Curr Genet Med Rep (2016) 4:57–64. doi:10.1007/s40142-016-0100-7
41. Tidyman WE, Rauen KA. Pathogenetics of the RASopathies. Hum Mol Genet (2016) 25:R123–32. doi:10.1093/hmg/ddw191
42. Ferner RE, Huson SM, Thomas N, Moss C, Willshaw H, Evans DG, et al. Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. J Med Genet (2007) 44:81–8. doi:10.1136/jmg.2006.045906
43. Neurofibromatosis. Conference statement. National Institutes of Health Consensus Development Conference. Arch Neurol (1988) 45:575–8.
44. Stay EJ, Vawter G. The relationship between nephroblastoma and neurofibromatosis (Von Recklinghausen’s disease). Cancer (1977) 39:2550–5. doi:10.1002/1097-0142(197706)39:6<2550::AID-CNCR2820390636>3.0.CO;2-Y
45. Walden PA, Johnson AG, Bagshawe KD. Wilms’s tumour and neurofibromatosis. Br Med J (1977) 1:813. doi:10.1136/bmj.1.6064.813
46. Korf BR. Malignancy in neurofibromatosis type 1. Oncologist (2000) 5:477–85. doi:10.1634/theoncologist.5-6-477
47. Evans DGR, O’Hara C, Wilding A, Ingham SL, Howard E, Dawson J, et al. Mortality in neurofibromatosis 1: in North West England: an assessment of actuarial survival in a region of the UK since 1989. Eur J Hum Genet EJHG (2011) 19:1187–91. doi:10.1038/ejhg.2011.113
48. Méni C, Sbidian E, Moreno JC, Lafaye S, Buffard V, Goldzal S, et al. Treatment of neurofibromas with a carbon dioxide laser: a retrospective cross-sectional study of 106 patients. Dermatol Basel Switz (2015) 230:263–8. doi:10.1159/000368078
49. Kriechbaumer LK, Susani M, Kircher SG, Distelmaier K, Happak W. Comparative study of CO2- and Er:YAG laser ablation of multiple cutaneous neurofibromas in von Recklinghausen’s disease. Lasers Med Sci (2014) 29:1083–91. doi:10.1007/s10103-013-1469-0
50. Rad E, Tee AR. Neurofibromatosis type 1: fundamental insights into cell signalling and cancer. Semin Cell Dev Biol (2016) 52:39–46. doi:10.1016/j.semcdb.2016.02.007
51. Friedman JM. Neurofibromatosis 1. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Mefford HC, Stephens K, Amemiya A, Ledbetter N, editors. GeneReviews(®). Seattle, WA: University of Washington, Seattle (2017). Available from: http://www.ncbi.nlm.nih.gov/books/NBK1109/
52. Sabbagh A, Pasmant E, Laurendeau I, Parfait B, Barbarot S, Guillot B, et al. Unravelling the genetic basis of variable clinical expression in neurofibromatosis 1. Hum Mol Genet (2009) 18:2768–78. doi:10.1093/hmg/ddp212
53. Sabbagh A, Pasmant E, Imbard A, Luscan A, Soares M, Blanché H, et al. NF1 molecular characterization and neurofibromatosis type I genotype-phenotype correlation: the French experience. Hum Mutat (2013) 34:1510–8. doi:10.1002/humu.22392
54. Jentarra GM, Rice SG, Olfers S, Rajan C, Saffen DM, Narayanan V. Skewed allele-specific expression of the NF1 gene in normal subjects: a possible mechanism for phenotypic variability in neurofibromatosis type 1. J Child Neurol (2012) 27:695–702. doi:10.1177/0883073811423439
55. Upadhyaya M, Spurlock G, Kluwe L, Chuzhanova N, Bennett E, Thomas N, et al. The spectrum of somatic and germline NF1 mutations in NF1 patients with spinal neurofibromas. Neurogenetics (2009) 10:251–63. doi:10.1007/s10048-009-0178-0
56. Sarkozy A, Digilio MC, Dallapiccola B. LEOPARD syndrome. Orphanet J Rare Dis (2008) 3:13. doi:10.1186/1750-1172-3-13
57. George CD, Patton MA, el Sawi M, Sharland M, Adam EJ. Abdominal ultrasound in Noonan syndrome: a study of 44 patients. Pediatr Radiol (1993) 23:316–8. doi:10.1007/BF02010926
58. Santoro C, Pacileo G, Limongelli G, Scianguetta S, Giugliano T, Piluso G, et al. LEOPARD syndrome: clinical dilemmas in differential diagnosis of RASopathies. BMC Med Genet (2014) 15:44. doi:10.1186/1471-2350-15-44
59. Digilio MC, Sarkozy A, de Zorzi A, Pacileo G, Limongelli G, Mingarelli R, et al. LEOPARD syndrome: clinical diagnosis in the first year of life. Am J Med Genet A (2006) 140:740–6. doi:10.1002/ajmg.a.31156
60. Kontoes PP, Vlachos SP, Marayiannis KV. Intense pulsed light for the treatment of lentigines in LEOPARD syndrome. Br J Plast Surg (2003) 56:607–10. doi:10.1016/S0007-1226(03)00218-2
61. Gelb BD, Tartaglia M. Noonan syndrome with multiple lentigines. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Mefford HC, Stephens K, Amemiya A, Ledbetter N, editors. GeneReviews(®). Seattle, WA: University of Washington, Seattle (2017). Available from: http://www.ncbi.nlm.nih.gov/books/NBK1383/
62. Pandit B, Sarkozy A, Pennacchio LA, Carta C, Oishi K, Martinelli S, et al. Gain-of-function RAF1 mutations cause Noonan and LEOPARD syndromes with hypertrophic cardiomyopathy. Nat Genet (2007) 39:1007–12. doi:10.1038/ng2073
63. Jongmans MCJ, van der Burgt I, Hoogerbrugge PM, Noordam K, Yntema HG, Nillesen WM, et al. Cancer risk in patients with Noonan syndrome carrying a PTPN11 mutation. Eur J Hum Genet EJHG (2011) 19:870–4. doi:10.1038/ejhg.2011.37
64. Denayer E, Devriendt K, de Ravel T, Van Buggenhout G, Smeets E, Francois I, et al. Tumor spectrum in children with Noonan syndrome and SOS1 or RAF1 mutations. Genes Chromosomes Cancer (2010) 49:242–52. doi:10.1002/gcc.20735
65. Jones K, Jones MC, del Campo M. Smith’s Recognizable Patterns of Human Malformation: Expert Consult – Online and Print, 7e. Philadelphia: Saunders (2013).
66. Northrup H, Krueger DA; International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 Iinternational Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol (2013) 49:243–54. doi:10.1016/j.pediatrneurol.2013.08.001
67. Józwiak S, Schwartz RA, Janniger CK, Bielicka-Cymerman J. Usefulness of diagnostic criteria of tuberous sclerosis complex in pediatric patients. J Child Neurol (2000) 15:652–9. doi:10.1177/088307380001501003
68. Bjornsson J, Short MP, Kwiatkowski DJ, Henske EP. Tuberous sclerosis-associated renal cell carcinoma. Clinical, pathological, and genetic features. Am J Pathol (1996) 149:1201–8.
69. Northrup H, Koenig MK, Pearson DA, Au K-S. Tuberous sclerosis complex. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Mefford HC, Stephens K, Amemiya A, Ledbetter N, editors. GeneReviews(®). Seattle, WA: University of Washington, Seattle (2017). Available from: http://www.ncbi.nlm.nih.gov/books/NBK1220/
70. Krueger DA, Northrup H; International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex surveillance and management: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol (2013) 49:255–65. doi:10.1016/j.pediatrneurol.2013.08.002
71. Palavra F, Robalo C, Reis F. Recent advances and challenges of mTOR inhibitors use in the treatment of patients with tuberous sclerosis complex. Oxid Med Cell Longev (2017) 2017:9820181. doi:10.1155/2017/9820181
72. Fioramonti P, De Santo L, Ruggieri M, Carella S, Federico LT, Onesti MG, et al. Co2/Erbium:YAG/Dye laser combination: an effective and successful treatment for angiofibromas in tuberous sclerosis. Aesthetic Plast Surg (2014) 38:192–8. doi:10.1007/s00266-013-0252-8
73. Ma G, Wu P, Lin X, Chen H, Li W, Hu X, et al. Nd:YAG laser for “fractional” treatment of angiofibromas. Int J Dermatol (2014) 53:638–42. doi:10.1111/ijd.12384
74. Weiss ET, Geronemus RG. New technique using combined pulsed dye laser and fractional resurfacing for treating facial angiofibromas in tuberous sclerosis. Lasers Surg Med (2010) 42:357–60. doi:10.1002/lsm.20939
75. Cardis MA, DeKlotz CMC. Cutaneous manifestations of tuberous sclerosis complex and the paediatrician’s role. Arch Dis Child (2017) 102:858–63. doi:10.1136/archdischild-2016-312001
76. Jóźwiak S, Sadowski K, Kotulska K, Schwartz RA. Topical use of mammalian target of rapamycin (mTOR) inhibitors in tuberous sclerosis complex – a comprehensive review of the literature. Pediatr Neurol (2016) 61:21–7. doi:10.1016/j.pediatrneurol.2016.04.003
77. Rosset C, Netto CBO, Ashton-Prolla P. TSC1 and TSC2 gene mutations and their implications for treatment in tuberous sclerosis complex: a review. Genet Mol Biol (2017) 40:69–79. doi:10.1590/1678-4685-GMB-2015-0321
78. Tyburczy ME, Dies KA, Glass J, Camposano S, Chekaluk Y, Thorner AR, et al. Mosaic and intronic mutations in TSC1/TSC2 explain the majority of TSC patients with no mutation identified by conventional testing. PLoS Genet (2015) 11:e1005637. doi:10.1371/journal.pgen.1005637
79. Martin KR, Zhou W, Bowman MJ, Shih J, Au KS, Dittenhafer-Reed KE, et al. The genomic landscape of tuberous sclerosis complex. Nat Commun (2017) 8:15816. doi:10.1038/ncomms15816
80. Kothare SV, Singh K, Chalifoux JR, Staley BA, Weiner HL, Menzer K, et al. Severity of manifestations in tuberous sclerosis complex in relation to genotype. Epilepsia (2014) 55:1025–9. doi:10.1111/epi.12680
81. Boronat S, Caruso P, Auladell M, Van Eeghen A, Thiele EA. Arachnoid cysts in tuberous sclerosis complex. Brain Dev (2014) 36:801–6. doi:10.1016/j.braindev.2013.11.003
82. Lam HC, Nijmeh J, Henske EP. New developments in the genetics and pathogenesis of tumours in tuberous sclerosis complex. J Pathol (2017) 241:219–25. doi:10.1002/path.4827
83. Tyburczy ME, Wang J-A, Li S, Thangapazham R, Chekaluk Y, Moss J, et al. Sun exposure causes somatic second-hit mutations and angiofibroma development in tuberous sclerosis complex. Hum Mol Genet (2014) 23:2023–9. doi:10.1093/hmg/ddt597
84. Henske EP, Wessner LL, Golden J, Scheithauer BW, Vortmeyer AO, Zhuang Z, et al. Loss of tuberin in both subependymal giant cell astrocytomas and angiomyolipomas supports a two-hit model for the pathogenesis of tuberous sclerosis tumors. Am J Pathol (1997) 151:1639–47.
85. Evans DG, Howard E, Giblin C, Clancy T, Spencer H, Huson SM, et al. Birth incidence and prevalence of tumor-prone syndromes: estimates from a UK family genetic register service. Am J Med Genet A (2010) 152A:327–32. doi:10.1002/ajmg.a.33139
86. Mendes-Bastos P, Vale-Fernandes P, Brás S, Carvalho R, João A, Amaro C. Acral basal cell carcinomas in an infant with Gorlin syndrome: expanding the phenotype? J Dtsch Dermatol Ges J Ger Soc Dermatol JDDG (2017) 15:89–90. doi:10.1111/ddg.12934
87. Bacanli A, Ciftcioglu MA, Savas B, Alpsoy E. Nevoid basal cell carcinoma syndrome associated with unilateral renal agenesis: acceleration of basal cell carcinomas following radiotherapy. J Eur Acad Dermatol Venereol JEADV (2005) 19:510–1. doi:10.1111/j.1468-3083.2004.01169.x
88. Cajaiba MM, Bale AE, Alvarez-Franco M, McNamara J, Reyes-Múgica M. Rhabdomyosarcoma, Wilms tumor, and deletion of the patched gene in Gorlin syndrome. Nat Clin Pract Oncol (2006) 3:575–80. doi:10.1038/ncponc0608
89. Garavelli L, Piemontese MR, Cavazza A, Rosato S, Wischmeijer A, Gelmini C, et al. Multiple tumor types including leiomyoma and Wilms tumor in a patient with Gorlin syndrome due to 9q22.3 microdeletion encompassing the PTCH1 and FANC-C loci. Am J Med Genet A (2013) 161A:2894–901. doi:10.1002/ajmg.a.36259
90. Bree AF, Shah MR, BCNS Colloquium Group. Consensus statement from the first international colloquium on basal cell nevus syndrome (BCNS). Am J Med Genet A (2011) 155A:2091–7. doi:10.1002/ajmg.a.34128
91. Bakshi A, Chaudhary SC, Rana M, Elmets CA, Athar M. Basal cell carcinoma pathogenesis and therapy involving hedgehog signaling and beyond. Mol Carcinog (2017) 56(12):2543–57. doi:10.1002/mc.22690
92. Solis DC, Kwon GP, Ransohoff KJ, Li S, Chahal HS, Ally MS, et al. Risk factors for basal cell carcinoma among patients with basal cell nevus syndrome: development of a basal cell nevus syndrome patient registry. JAMA Dermatol (2016). doi:10.1001/jamadermatol.2016.4347
93. Evans DG, Farndon PA. Nevoid basal cell carcinoma syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Mefford HC, Stephens K, Amemiya A, Ledbetter N, editors. GeneReviews(®). Seattle, WA: University of Washington, Seattle (2017). Available from: http://www.ncbi.nlm.nih.gov/books/NBK1151/
94. Fan Z, Li J, Du J, Zhang H, Shen Y, Wang C-Y, et al. A missense mutation in PTCH2 underlies dominantly inherited NBCCS in a Chinese family. J Med Genet (2008) 45:303–8. doi:10.1136/jmg.2007.055343
95. Khamaysi Z, Bochner R, Indelman M, Magal L, Avitan-Hersh E, Sarig O, et al. Segmental basal cell naevus syndrome caused by an activating mutation in smoothened. Br J Dermatol (2016) 175:178–81. doi:10.1111/bjd.14425
96. Reinders MGHC, Boersma HJ, Leter EM, Vreeburg M, Paulussen ADC, Arits AHMM, et al. Postzygotic mosaicism in basal cell naevus syndrome. Br J Dermatol (2017) 177:249–52. doi:10.1111/bjd.15082
97. Ribeiro LA, Murray JC, Richieri-Costa A. PTCH mutations in four Brazilian patients with holoprosencephaly and in one with holoprosencephaly-like features and normal MRI. Am J Med Genet A (2006) 140:2584–6. doi:10.1002/ajmg.a.31369
98. Atwood SX, Chang ALS, Oro AE. Hedgehog pathway inhibition and the race against tumor evolution. J Cell Biol (2012) 199:193–7. doi:10.1083/jcb.201207140
99. Epstein EH. Basal cell carcinomas: attack of the hedgehog. Nat Rev Cancer (2008) 8:743–54. doi:10.1038/nrc2503
100. Toro JR. Birt-Hogg-Dubé syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Mefford HC, Stephens K, Amemiya A, Ledbetter N, editors. GeneReviews®. Seattle, WA: University of Washington, Seattle (2017). Available from: http://www.ncbi.nlm.nih.gov/books/NBK1522/
101. Schmidt LS, Linehan WM. Molecular genetics and clinical features of Birt-Hogg-Dubé syndrome. Nat Rev Urol (2015) 12:558–69. doi:10.1038/nrurol.2015.206
102. Zbar B, Alvord WG, Glenn G, Turner M, Pavlovich CP, Schmidt L, et al. Risk of renal and colonic neoplasms and spontaneous pneumothorax in the Birt-Hogg-Dubé syndrome. Cancer Epidemiol Biomark Prev (2002) 11:393–400.
103. Schmidt LS, Nickerson ML, Warren MB, Glenn GM, Toro JR. Germline BHD-mutation spectrum and phenotype analysis of a large cohort of families with Birt-Hogg-Dubé syndrome. Am J Hum Genet (2005) 76(6):1023–33. doi:10.1086/430842
104. Yan M, Gingras MC, Dunlop EA, Nouet Y, Dupuy F, Jalali Z, et al. The tumor suppressor folliculin regulates AMPK-dependent metabolic transformation. J Clin Invest (2014) 124:2640–50. doi:10.1172/JCI71749
105. Baba M, Hong S-B, Sharma N, Warren MB, Nickerson ML, Iwamatsu A, et al. Folliculin encoded by the BHD gene interacts with a binding protein, FNIP1, and AMPK, and is involved in AMPK and mTOR signaling. Proc Natl Acad Sci U S A (2006) 103:15552–7. doi:10.1073/pnas.0603781103
106. Hasumi H, Baba M, Hong S-B, Hasumi Y, Huang Y, Yao M, et al. Identification and characterization of a novel folliculin-interacting protein FNIP2. Gene (2008) 415:60–7. doi:10.1016/j.gene.2008.02.022
107. Takagi Y, Kobayashi T, Shiono M, Wang L, Piao X, Sun G, et al. Interaction of folliculin (Birt-Hogg-Dubé gene product) with a novel Fnip1-like (FnipL/Fnip2) protein. Oncogene (2008) 27:5339–47. doi:10.1038/onc.2008.261
108. Pithukpakorn M, Toro JR. Hereditary leiomyomatosis and renal cell cancer. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Mefford HC, Stephens K, Amemiya A, Ledbetter N, editors. GeneReviews®. Seattle, WA: University of Washington, Seattle (2017). Available from: http://www.ncbi.nlm.nih.gov/books/NBK1252/
109. Tomlinson IPM, Alam NA, Rowan AJ, Barclay E, Jaeger EEM, Kelsell D, et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet (2002) 30:406–10. doi:10.1038/ng849
110. Coughlin EM, Christensen E, Kunz PL, Krishnamoorthy KS, Walker V, Dennis NR, et al. Molecular analysis and prenatal diagnosis of human fumarase deficiency. Mol Genet Metab (1998) 63:254–62. doi:10.1006/mgme.1998.2684
111. Happle R. Cutaneous manifestation of lethal genes. Hum Genet (1986) 72:280. doi:10.1007/BF00291899
112. Happle R. Lethal genes surviving by mosaicism: a possible explanation for sporadic birth defects involving the skin. J Am Acad Dermatol (1987) 16:899–906. doi:10.1016/S0190-9622(87)80249-9
113. Happle R. The group of epidermal nevus syndromes Part I. Well defined phenotypes. J Am Acad Dermatol (2010) 63:1–22–24. doi:10.1016/j.jaad.2010.01.017
114. Kiedrowicz M, Kacalak-Rzepka A, Królicki A, Maleszka R, Bielecka-Grzela S. Therapeutic effects of CO2 laser therapy of linear nevus sebaceous in the course of the Schimmelpenning-Feuerstein-Mims syndrome. Postepy Dermatol Alergol (2013) 30:320–3. doi:10.5114/pdia.2013.38363
115. Groesser L, Herschberger E, Ruetten A, Ruivenkamp C, Lopriore E, Zutt M, et al. Postzygotic HRAS and KRAS mutations cause nevus sebaceous and Schimmelpenning syndrome. Nat Genet (2012) 44:783–7. doi:10.1038/ng.2316
116. Sun BK, Saggini A, Sarin KY, Kim J, Benjamin L, LeBoit PE, et al. Mosaic activating RAS mutations in nevus sebaceus and nevus sebaceus syndrome. J Invest Dermatol (2013) 133:824–7. doi:10.1038/jid.2012.377
117. DeDavid M, Orlow SJ, Provost N, Marghoob AA, Rao BK, Wasti Q, et al. Neurocutaneous melanosis: clinical features of large congenital melanocytic nevi in patients with manifest central nervous system melanosis. J Am Acad Dermatol (1996) 35:529–38. doi:10.1016/S0190-9622(96)90674-X
118. Kinsler VA, Thomas AC, Ishida M, Bulstrode NW, Loughlin S, Hing S, et al. Multiple congenital melanocytic nevi and neurocutaneous melanosis are caused by postzygotic mutations in codon 61 of NRAS. J Invest Dermatol (2013) 133:2229–36. doi:10.1038/jid.2013.70
119. Krengel S, Hauschild A, Schäfer T. Melanoma risk in congenital melanocytic naevi: a systematic review. Br J Dermatol (2006) 155:1–8. doi:10.1111/j.1365-2133.2006.07218.x
120. Basu D, Salgado CM, Bauer BS, Johnson D, Rundell V, Nikiforova M, et al. Nevospheres from neurocutaneous melanocytosis cells show reduced viability when treated with specific inhibitors of NRAS signaling pathway. Neuro Oncol (2016) 18:528–37. doi:10.1093/neuonc/nov184
121. Happle R, Koch H, Lenz W. The CHILD syndrome. Congenital hemidysplasia with ichthyosiform erythroderma and limb defects. Eur J Pediatr (1980) 134:27–33. doi:10.1007/BF00442399
122. Happle R, Karlić D, Steijlen PM. [CHILD syndrome in a mother and daughter]. Hautarzt Z Dermatol Venerol Verwandte Geb (1990) 41:105–8.
123. Schmidt-Sidor B, Obersztyn E, Szymańska K, Wychowski J, Mierzewska H, Wierzba-Bobrowicz T, et al. Brain and cerebellar hemidysplasia in a case with ipsilateral body dysplasia and suspicion of CHILD syndrome. Folia Neuropathol (2008) 46:232–7.
124. du Souich C, Raymond FL, Grzeschik K-H, Boerkoel CF. NSDHL-related disorders. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Mefford HC, Stephens K, Amemiya A, Ledbetter N, editors. GeneReviews(®). Seattle, WA: University of Washington, Seattle (2017). Available from: http://www.ncbi.nlm.nih.gov/books/NBK51754/
125. Kiritsi D, Schauer F, Wolfle U, Valari M, Bruckner-Tuderman L, Has C, et al. Targeting epidermal lipids for treatment of Mendelian disorders of cornification. Orphanet J Rare Dis (2014) 9:33. doi:10.1186/1750-1172-9-33
126. Paller AS, van Steensel MAM, Rodriguez-Martín M, Sorrell J, Heath C, Crumrine D, et al. Pathogenesis-based therapy reverses cutaneous abnormalities in an inherited disorder of distal cholesterol metabolism. J Invest Dermatol (2011) 131:2242–8. doi:10.1038/jid.2011.189
127. König A, Happle R, Bornholdt D, Engel H, Grzeschik KH. Mutations in the NSDHL gene, encoding a 3beta-hydroxysteroid dehydrogenase, cause CHILD syndrome. Am J Med Genet (2000) 90:339–46. doi:10.1002/(SICI)1096-8628(20000214)90:4<339::AID-AJMG15>3.0.CO;2-5
128. Seeger MA, Paller AS. The role of abnormalities in the distal pathway of cholesterol synthesis in the Congenital Hemidysplasia with Ichthyosiform erythroderma and Limb Defects (CHILD) syndrome. Biochim Biophys Acta (2014) 1841:345–52. doi:10.1016/j.bbalip.2013.09.006
129. du Souich C, Chou A, Yin J, Oh T, Nelson TN, Hurlburt J, et al. Characterization of a new X-linked mental retardation syndrome with microcephaly, cortical malformation, and thin habitus. Am J Med Genet A (2009) 149A:2469–78. doi:10.1002/ajmg.a.33071
130. McLarren KW, Severson TM, du Souich C, Stockton DW, Kratz LE, Cunningham D, et al. Hypomorphic temperature-sensitive alleles of NSDHL cause CK syndrome. Am J Hum Genet (2010) 87:905–14. doi:10.1016/j.ajhg.2010.11.004
131. Goltz RW, Peterson WC, Gorlin RJ, Ravits HG. Focal dermal hypoplasia. Arch Dermatol (1962) 86:708–17. doi:10.1001/archderm.1962.01590120006002
132. Bree AF, Grange DK, Hicks MJ, Goltz RW. Dermatologic findings of focal dermal hypoplasia (Goltz syndrome). Am J Med Genet C Semin Med Genet (2016) 172C:44–51. doi:10.1002/ajmg.c.31472
133. Grzeschik K-H, Bornholdt D, Oeffner F, König A, del Carmen Boente M, Enders H, et al. Deficiency of PORCN, a regulator of Wnt signaling, is associated with focal dermal hypoplasia. Nat Genet (2007) 39:833–5. doi:10.1038/ng2052
134. Sanna-Cherchi S, Westland R, Ghiggeri GM, Gharavi AG. Genetic basis of human congenital anomalies of the kidney and urinary tract. J Clin Invest (2018) 128:4–15. doi:10.1172/JCI95300
Keywords: epidermolysis bullosa, mosaicism, genodermatosis, kidney, mutation, RASopathy, nevus, renal anomaly
Citation: Reimer A, He Y and Has C (2018) Update on Genetic Conditions Affecting the Skin and the Kidneys. Front. Pediatr. 6:43. doi: 10.3389/fped.2018.00043
Received: 26 October 2017; Accepted: 14 February 2018;
Published: 02 March 2018
Edited by:
Miriam Schmidts, Radboud University Nijmegen, NetherlandsReviewed by:
Katja Höpker, University Hospital Cologne, GermanyRoman-Ulrich Mueller, University Hospital Cologne, Germany
Copyright: © 2018 Reimer, He and Has. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cristina Has, cristina.has@uniklinik-freiburg.de