Autoimmune Polyendocrinopathy-Candidiasis-Ectodermal Dystrophy
 Elise M. N. Ferré
Elise M. N. Ferré Monica M. Schmitt
Monica M. Schmitt Michail S. Lionakis
Michail S. Lionakis- Fungal Pathogenesis Section, Laboratory of Clinical Immunology and Microbiology, National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health (NIH), Bethesda, MD, United States
Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED), also known as autoimmune polyglandular syndrome type-1 (APS-1), is a rare monogenic autoimmune disease caused by loss-of-function mutations in the autoimmune regulator (AIRE) gene. AIRE deficiency impairs immune tolerance in the thymus and results in the peripheral escape of self-reactive T lymphocytes and the generation of several cytokine- and tissue antigen-targeted autoantibodies. APECED features a classic triad of characteristic clinical manifestations consisting of chronic mucocutaneous candidiasis (CMC), hypoparathyroidism, and primary adrenal insufficiency (Addison's disease). In addition, APECED patients develop several non-endocrine autoimmune manifestations with variable frequencies, whose recognition by pediatricians should facilitate an earlier diagnosis and allow for the prompt implementation of targeted screening, preventive, and therapeutic strategies. This review summarizes our current understanding of the genetic, immunological, clinical, diagnostic, and treatment features of APECED.
Introduction
APECED is a rare monogenic autoimmune disease (OMIM#, 240300) caused by loss-of-function AIRE mutations that impair central immune tolerance and result in the peripheral escape of self-reactive T lymphocytes, which infiltrate various endocrine (e.g., parathyroids, adrenals, gonads, thyroid, pancreas) and non-endocrine (e.g., enamel, stomach, small intestine, lungs, liver, salivary glands, kidneys, spleen, skin) organs and cause autoimmune tissue destruction. Since the discovery of mutations in the AIRE gene as the cause of APECED by positional cloning in 1997 (1, 2) and the initial characterization of the immunological functions of AIRE in the thymus in 2002 (3), significant progress has been made in our fundamental understanding of the genetic and immunological basis of AIRE deficiency via the study of APECED patients and of Aire-deficient mice, which develop a multisystem autoimmune disease that closely resembles human APECED particularly on the NOD genetic background, featuring endocrine, non-endocrine, and fungal infection disease manifestations (3–6). In addition, recent patient cohort studies have uncovered an expanded clinical spectrum of APECED (7, 8), which has led to novel observations that may help (a) accelerate diagnosis via earlier recognition of certain disease manifestations and (b) devise effective screening, preventive, and treatment strategies for affected patients. In this review, we present our current knowledge of the genetic and immunological underpinnings of AIRE deficiency and discuss the clinical presentation, diagnostic criteria, and management of APECED patients.
Genetics of APECED
Bi-allelic loss-of-function mutations in AIRE typically underlie APECED with >100 mutations and/or deletions being described throughout the AIRE gene (9). Certain APECED cohorts harbor characteristic founder mutations. For example, Finnish, Sardinian, Persian Jew, and Sicilian APECED patients typically carry homozygous p.R257X, p.R139X, p.Y85C, and p.R203X mutations, respectively (9, 10). In the genetically diverse American APECED cohort, compound heterozygous AIRE mutations were observed often, with the two most common mutations being p.L323SfsX51 followed by p.R257X (7). In that cohort, p.L323SfsX51, was associated with the development of certain non-endocrine autoimmune manifestations such as pneumonitis and hepatitis (11, 12). Notably, a remarkable variability in the spectrum and severity of the clinical phenotype is seen among APECED patients who carry the same AIRE mutations, including among siblings (13–16). This suggests that yet-unknown genetic modifiers may affect individual patient susceptibility to various organ-specific manifestations of the syndrome. To that end, the peripheral tolerance immune checkpoint molecules Cbl-b and Lyn were shown to cooperate with AIRE in modulating the development of autoimmune retinitis and exocrine pancreatitis in mice, respectively (17, 18). In addition, when deficiency in the IL-2/STAT5 response regulatory element CNS0 was combined with AIRE deficiency in mice the result was exacerbation of autoimmune destruction in multiple organs, including in tissues (e.g., adipose tissue) that did not exhibit autoimmunity in isolated CNS0 or AIRE deficiencies (19).
Moreover, several dominant-negative AIRE mutations have now been recognized within the SAND and PHD1 domains of the gene and exhibit varying degrees of dominant negative effects in vitro and in mice (20–25). Affected patients typically develop milder APECED with fewer autoimmune manifestations compared to patients with classical APECED, while some patients remain unaffected without autoimmunity, indicative of incomplete clinical penetrance (21). Moreover, patients with dominant-negative AIRE mutations do not always harbor the cytokine- and tissue antigen-targeted autoantibodies of classical APECED patients (21). Because the minor allelic frequency of some of these mutations (e.g., p.V301M, p.R303Q) is relatively high in the general population, genetic variation in AIRE may contribute to the development of organ-specific autoimmune diseases with a greater frequency than previously anticipated. Of interest, a recent genome-wide association study (GWAS) identified two protein-coding AIRE variants (rs74203920; rs2075876) associated with autoimmune Addison's disease, one of which (rs74203920, p.R471C) was also found to be associated with pernicious anemia in an independent GWAS (26, 27).
In recent years, the cis-regulatory element CNS1, which promotes AIRE expression in medullary thymic epithelial cells (mTECs) (28, 29) and several regulatory molecules that affect the expression and/or transcriptional activity of AIRE (i.e., HIPK2, FBXO3, JMJD6, SIRT1, DGCR8) have been characterized (30–34). In the subset of patients with a clinical diagnosis of APECED who have wild-type AIRE genotype (7), it will be important to examine whether mutations in these or other AIRE regulators and/or partners may underlie their autoimmune disease.
Pathogenesis of Aire Deficiency
AIRE-Deficient T Lymphocytes
AIRE is a transcriptional regulator that is highly expressed in a subset of mTECs where it promotes the expression of a large number of -but not all- tissue-specific antigens (3). This process facilitates the negative selection of self-reactive T lymphocytes and the differentiation of self-antigen–specific regulatory T lymphocytes, which collectively shape and maintain central immune tolerance, reviewed elsewhere (13, 14, 35–40). In AIRE deficiency, self-reactive CD4+ T lymphocytes escape from the thymus into the periphery and are both necessary and sufficient to cause autoimmune tissue infiltration and destruction, as shown by adoptive transfer experiments of Aire−/− CD4+ T lymphocytes in immunodeficient mice, by CD4+ T lymphocyte depletion experiments in Aire−/− mice, and by experiments in Aire−/−Tcra−/− mice (3, 4, 41, 42). The recent discovery and characterization of the post-Aire expressing mTEC subset and of extrathymic Aire-expressing cells (eTACs) in secondary lymphoid tissues require additional functional studies to precisely decipher their contributions in maintaining immune tolerance (43–48). The use of tetramer reagents, epitope mapping, and T cell receptor (TCR) sequencing has helped characterize self-reactive T lymphocyte populations and TCR repertoires in AIRE-deficient mice and/or humans (49–56). As mentioned earlier, AIRE also participates in the positive selection of thymic regulatory T cells (57) and mouse studies have shown that defective neonatal output of thymic regulatory T cells in AIRE deficiency contributes to the development of organ-specific autoimmunity (58). Beyond CD4+ T lymphocytes, AIRE-deficient CD8+ T lymphocytes and γδ T lymphocytes have also been implicated in the development of certain -but not all- organ-specific autoimmune manifestations in Aire−/− mice (i.e., oral candidiasis, pneumonitis, retinitis, peripheral neuropathy) and more work is needed to further define their roles in the breakdown of organ-specific tolerance in cooperation with or independent of Aire−/− CD4+ T lymphocytes (6, 59, 60).
Tissue Antigen-Specific Autoantibodies
Furthermore, B lymphocytes are dysregulated in AIRE deficiency associated with expansion of autoreactive naïve B lymphocytes, increased frequency of the CD21loCD38− B lymphocyte subset, and production of a broad array of autoantibodies directed against cytokines and tissue antigens (7, 13, 56). Studies in Aire−/− mice have shown that deletion of mature B lymphocytes and their autoantibody-producing potential ameliorates certain organ-specific autoimmune manifestations (41, 42). Whether AIRE-deficient B lymphocytes contribute to autoimmunity via direct priming of AIRE-deficient T lymphocytes and/or via autoantibody production remains unclear (42). Of note, experimental transfer of autoantibody-containing AIRE-deficient serum in mice is not sufficient to promote autoimmunity (41, 42). Serum transfer experiments in the recently developed AIRE-deficient rat model, which harbors a broader spectrum of autoantibodies relative to AIRE-deficient mice, may help further elucidate the potential direct role of autoantibodies in autoimmune tissue destruction (61, 62).
In APECED patients, the detection of several tissue antigen-directed autoantibodies has been associated with the presence of corresponding organ-specific autoimmune manifestations (13, 52, 63–75). New experimental approaches such as phage/bacterial peptide display (PhiP-Seq) and yeast surface display (REAP) have been recently used to uncover novel autoantigen specificities such as KHDC3L, associated with primary ovarian failure, RFX6, associated with intestinal dysfunction, and colipase, associated with exocrine pancreatic insufficiency (76, 77). However, for most of the tissue antigen-targeted autoantibodies detected in APECED patients, it is difficult to reliably predict the development of the corresponding autoimmune manifestation at the individual patient level as their sensitivity and specificity is not very high. Indeed, patients may harbor an autoantibody without featuring the corresponding clinical manifestation while other patients may have a clinical manifestation without harboring the corresponding tissue antigen-directed autoantibody.
Three examples of tissue antigen-directed autoantibodies that can be helpful in the clinical management of APECED patients are worthwhile briefly mentioning. The detection of autoantibodies to 21-hydroxylase in an APECED patient without Addison's disease who did not previously harbor these autoantibodies is often a herald for the forthcoming development of primary adrenal insufficiency (71). Intensified screening with ACTH stimulation testing can help prevent acute adrenal crises in such patients. Moreover, the lung-targeted autoantibodies KCNRG and BPIFB1 have very high specificity (>90%) for autoimmune pneumonitis, although their sensitivity is ~30–60% and, thus, a negative result does not rule out the presence of pneumonitis (11, 52, 74, 78, 79). APECED patients carrying KCNRG- and/or BPIFB1-directed autoantibodies should undergo chest imaging with computed tomography and pulmonary function testing to evaluate for the presence of autoimmune pneumonitis, even if alternative diagnoses (e.g., asthma, bronchitis) were previously made (11). Furthermore, autoantibodies observed in classical autoimmune hepatitis (e.g., anti-LKM, anti-SLA, anti-ASMA) are not typically detected in patients with APECED–associated autoimmune hepatitis (12). Thus, a diagnostic liver biopsy should be performed in APECED patients with persistent transaminase elevation even when classical autoimmune hepatitis-associated biomarkers are negative.
Autoantibodies Against Type-I Interferons (IFNs)
Beyond tissue antigen-directed autoantibodies, APECED patients harbor neutralizing autoantibodies against certain cytokines, primarily type-I IFNs and type-17 cytokines (80–83). Neutralizing autoantibodies against type-I IFNs, predominantly directed to IFN-ω and the majority of the 13 subtypes of IFN-α, are present in >95% of APECED patients, whereas autoantibodies against IFN-β are detected in ~20% of patients (81, 82), and autoantibodies against IFN-ε are infrequently detected (84). Because IFN-ω-directed autoantibodies are detectable with high sensitivity during infancy before the development of clinical manifestations, and because at that early age these autoantibodies are highly specific for APECED, their early detection carries significant diagnostic utility in children with suspected APECED (81, 82). Of note, IFN-α autoantibodies were proposed to act as ameliorating factors for the development of type-1 diabetes in APECED patients who carry GAD65-directed autoantibodies, indicating that these autoantibodies may also have therapeutic utility (73).
Despite the presence of autoantibodies against type-I IFNs, APECED patients do not develop the severe viral infections that are seen in patients with inherited complete deficiencies of IFNAR1 and IFNAR2 including herpes simplex encephalitis and live attenuated measles-mumps-rubella vaccine-associated disease (85, 86), although ~10–20% of APECED patients have been reported to develop prolonged and/or severe manifestations of cutaneous varicella zoster and/or mucosal herpes simplex infections (87). These clinical observations suggest that APECED patients retain residual compensatory activity of some of the type-I IFNs and/or that alternative type-I IFN-independent immune pathways may provide protection against these viral diseases in the setting of neutralizing autoantibodies against type-I IFNs (88–90). The recent report of severe live attenuated yellow fever 17D vaccine-associated disease in three individuals with neutralizing autoantibodies to type-I IFNs without APECED suggests that this vaccine should be avoided in APECED patients (91).
COVID-19 Infection and Vaccination in APECED Patients
Neutralizing autoantibodies against type-I IFNs, particularly to the 13 IFN-α subtypes and IFN-ω, were recently identified in ~10% of patients suffering from critical COVID-19 pneumonia (92) and were shown to delay SARS-CoV-2 clearance (93). This observation was confirmed in independent patient cohorts (94–96). In addition, inborn errors of type-I IFN immunity were reported in some -but not all- examined cohorts of patients with life-threatening COVID-19 (97–99). Given these observations, and the life-threatening pneumonia requiring mechanical ventilation in the first three reported APECED patients with COVID-19 (92, 100), we performed a follow-up international observational study of 22 SARS-CoV-2–infected APECED patients (84). We found that most APECED patients developed severe, hypoxemic COVID-19 pneumonia requiring hospitalization and intensive care unit admission, and four patients (18%) succumbed to the infection (84). These data suggest that the presence of neutralizing autoantibodies to type-I IFNs and the inflammation-prone lung tissue of APECED patients may heighten their risk for life-threatening COVID-19 complications (11, 84, 101). Of interest, another recent study reported four APECED patients who developed mild COVID-19 infection despite the presence of neutralizing autoantibodies to type-I IFNs, consistent with a model of incomplete clinical penetrance (102). These patients were <26-year-old, female, and did not have pre-existing autoimmune pneumonitis (102). Collectively, these reports indicate that APECED patients, particularly adults and/or those with underlying pneumonitis, can be at risk for severe COVID-19. Therefore, APECED patients should be prioritized for vaccination against SARS-CoV-2, which they appear to tolerate without unusual adverse events; however, it is important to note that not all APECED patients develop robust humoral responses to the SARS-CoV-2 vaccine, especially those receiving immunomodulatory therapy (103). In our experience with SARS-CoV-2–infected APECED patients at the NIH Clinical Center, we proceed with prophylactic hospital admission upon diagnosis for close clinical monitoring. In the early ambulatory non-hypoxemic phase of the infection, we consider administration of anti-spike SARS-CoV-2 monoclonal antibodies (104), which was shown to decrease the risk of hospitalization, severe infection, and death from COVID-19 in high-risk individuals without APECED (105). Administration of IFN-β and/or plasmapheresis could also be considered in this setting (84, 106). In the late hypoxemic phase of infection, prompt initiation of corticosteroids is critical to ameliorate lung injury (84, 107), while remdesivir may curtail viral proliferation (108). Caution should be exercised with the use of anti-spike SARS-CoV-2 monoclonal antibodies or IFN-β during the hypoxemic phase of COVID-19, as these modalities could worsen lung inflammation and hypoxemia (109, 110).
Autoantibodies Against Type-17 Cytokines, IFN-γ-Driven Defects in Oral Epithelial Barrier, and CMC
The presence of neutralizing autoantibodies against type-17 cytokines is associated with CMC, the “signature” infectious disease in APECED patients (80, 83). The majority of APECED patients carry neutralizing autoantibodies against IL-22 (frequency, ~70–90%), whereas neutralizing autoantibodies against IL-17F (frequency ranging from ~20% to ~80% depending on the patient cohort) and IL-17A (frequency, ~35%) are detected less often and neutralizing autoantibodies against IL-17B and IL-17C are not detected (7, 8, 80, 83, 111, 112). However, the association between CMC and the presence of autoantibodies against type-17 cytokines is incompletely penetrant as some patients carry autoantibodies but do not manifest CMC and some others develop CMC without harboring these autoantibodies (7, 8, 80, 83, 113). In fact, in the Russian and American patient cohorts, the frequencies of IL-17 autoantibodies were similar in APECED patients with or without CMC (7, 8), indicating that additional factors must also contribute to CMC susceptibility. Moreover, patients who receive IL-17–targeted monoclonal antibodies (e.g., for psoriasis or inflammatory bowel disease) infrequently develop mild, treatment-responsive oropharyngeal candidiasis (OPC; mean frequency, <10%), as opposed to the ~80–90% frequency of CMC in APECED patients (114, 115). These clinical observations are consistent with the incomplete blockade of IL-17 receptor signaling conferred by these monoclonal antibodies at the mucocutaneous barrier (116, 117), as opposed to the complete abrogation of IL-17 receptor signaling in patients with inherited complete deficiencies of the IL-17 receptors IL-17RA and IL-17RC and of the IL-17 receptor adaptor ACT1 who develop CMC with complete penetrance (118–120). Furthermore, patients with inherited IL-10RB deficiency whose cells do not respond to IL-22 (nor to IL-10, IL-26, IL-28, and IFNL1) do not develop CMC; a single case of treatment-responsive OPC in the absence of iatrogenic immunosuppression has been reported in IL-10RB–deficient patients (OPC frequency, ~3%) who develop very severe early-onset inflammatory bowel disease requiring treatment with corticosteroids and/or TNF-α inhibitors (121–123). Taken together, although APECED patients harbor neutralizing autoantibodies against type-17 cytokines, their presence is unlikely to be the sole factor that might contribute to CMC in the APECED population (124).
This led us examine potential additional mechanisms of CMC susceptibility in AIRE deficiency. Aire-deficient mice, which infrequently harbor neutralizing autoantibodies against type-17 cytokines, were susceptible to OPC despite mounting intact type-17 mucosal responses, indicating that impaired type-17 immunity is not the primary driver of mucosal fungal susceptibility in the model (6). Instead, Aire−/− T lymphocytes were both necessary and sufficient to drive OPC susceptibility (6), in agreement with their previously established necessary and sufficient roles in driving susceptibility to all endocrine and non-endocrine autoimmune manifestations of AIRE deficiency in the model (3, 4, 41). Mechanistically, excessive production of IFN-γ by mucosal Aire−/− CD4+ and CD8+ T lymphocytes impaired the integrity of the oral epithelial barrier and promoted OPC, which were both ameliorated by inhibition of IFN-γ and/or JAK/STAT (6). IFN-γ was similarly toxic to human oral epithelial cells in vitro and evaluation of mucosal responses in a large cohort of APECED patients, including with RNA-sequencing of oral mucosal tissue in five adult individuals with a history of CMC, showed clear corroborative evidence of exaggerated type-1 responses, while type-17 mucosal responses were intact (6). These findings are consistent with residual compensatory activity of type-17 cytokines in the oral mucosa of the examined patients. Future work will be required to evaluate type-1 and type-17 mucosal responses in infants with APECED before the development of CMC and at the onset of CMC, and to longitudinally study oral mucosal immune responses in patients during acute and quiescent phases of CMC. These studies will help further define the relative contributions of excessive IFN-γ vs. type-17 impairment to the initiation, severity and/or relapse frequency of CMC in APECED patients; it is conceivable that the two mechanisms converge in a subset of APECED patients at different times. Moreover, a clinical trial is being deployed at the NIH Clinical Center to evaluate the safety and efficacy of JAK/STAT inhibition in the management of CMC in APECED patients. Of note, although CMC is the “signature” infection of APECED, these patients are not at risk for invasive candidiasis or other invasive fungal infections, which rely on myeloid phagocytes for effective host defense (125).
Taken together, these findings show that, in certain defined settings, mucosal fungal susceptibility may be driven by aberrant T lymphocyte-mediated immunopathology, not only by impaired type-17 immunity, and support a novel conceptual framework for classifying distinct molecular subtypes of CMC based on the balance between impaired type-17 immunity and/or immunopathology-promoting excessive type-1 inflammation (124).
Clinical Presentation and Diagnosis of APECED
APECED is clinically defined by the classic triad manifestations of CMC, hypoparathyroidism, and adrenal insufficiency. Developing any dyad among these classic triad manifestations establishes a clinical diagnosis of APECED. Developing a single classic triad manifestation in a patient whose sibling has APECED also establishes a clinical diagnosis. The development of a classic diagnostic dyad raises suspicion for APECED, which then leads to sequencing of the AIRE gene and/or testing for autoantibodies against IFN-ω. Several APECED cohorts have been described worldwide with varying disease prevalence. The highest prevalence has been reported in Finnish, Sardinian, and Persian Jew populations (~1:9,000–1:25,000), whereas in the United States, the prevalence of APECED is estimated between 1:100,000–1:300,000 (126).
APECED is a multisystem autoimmune disease that involves several endocrine and non-endocrine organs. More than 30 different autoimmune manifestations have been reported over the past decades with variable, cohort-specific representation of some of these disease components (Figures 1–3); among these manifestations, over 25 involve non-endocrine tissues (7, 8, 13, 112, 126–140). In a prospective natural history study of APECED at the NIH Clinical Center, where we have thus far enrolled >150 patients and evaluated them in a uniform, systematic manner with a multidisciplinary team of clinicians regardless of their underlying clinical manifestations, we have observed a dramatic enrichment of certain non-endocrine autoimmune manifestations relative to other APECED cohorts (7). Specifically, American APECED patients develop a hexad of non-endocrine disease manifestations consisting of urticarial eruption (“APECED rash”), autoimmune gastritis, intestinal malabsorption, autoimmune pneumonitis, autoimmune hepatitis, and Sjögren's-like syndrome with much greater frequency (~40–80%) compared to previously reported European APECED cohorts (<5–20%) (7) (Figures 2, 3). In the American and Russian APECED cohorts that collectively follow >250 patients, several uncommon disease manifestations have also been described, which had not been apparent in previously reported smaller cohorts (8, 141–144).
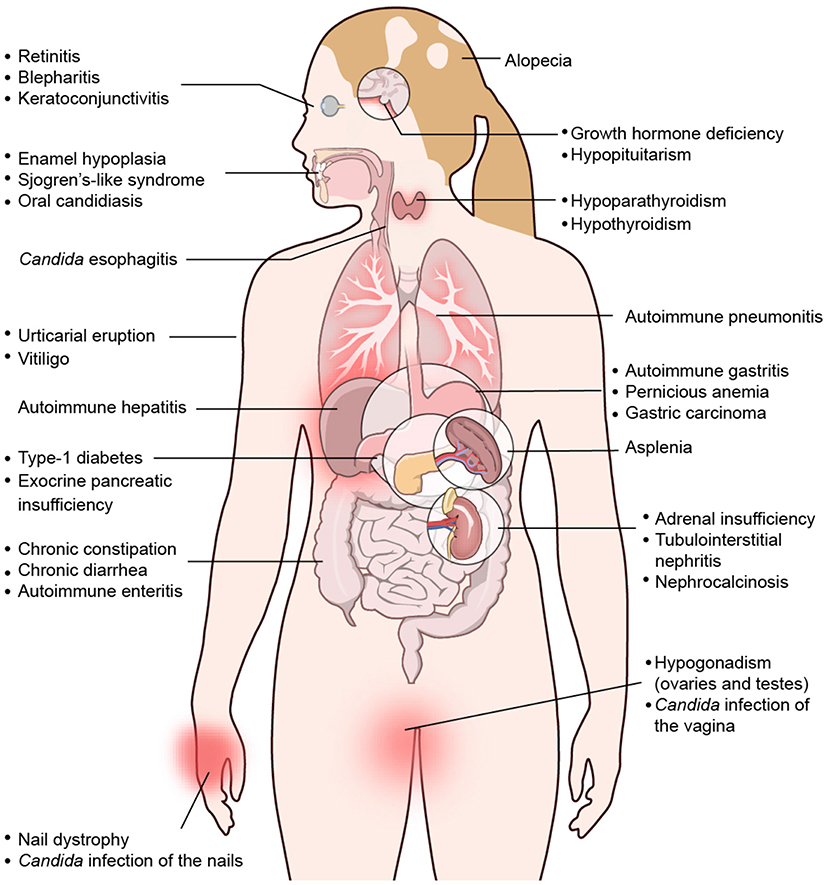
Figure 1. Spectrum of clinical manifestations in APECED patients. Depiction of organ-specific autoimmune manifestations observed with variable frequencies in patients with APECED. Derived from Constantine and Lionakis (13) with permission from Wiley.
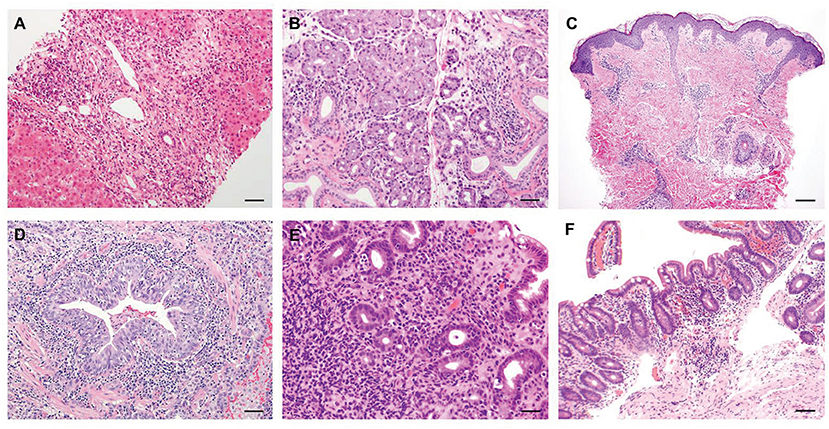
Figure 2. Representative histological features of common non-endocrine manifestations in APECED patients. (A) A liver biopsy displaying severe chronic hepatitis with expansion of portal areas by inflammation and fibrosis and extensive interface hepatitis with numerous plasma cells infiltrating into the hepatic parenchyma (H&E; scale bar: 50 μm; original magnification, ×200). (B) A minor salivary gland biopsy depicting lymphocytic and plasma cell infiltration in and around the ducts of the gland (H&E; scale bar: 50 μm; original magnification, ×200). (C) Skin biopsy of a patient with APECED rash demonstrates perivascular and periadnexal inflammation in the superficial and deep dermis with pallor of the papillary dermis (H&E; scale bar: 2 mm; original magnification, ×40) (D) An open lung biopsy revealing chronic bronchiolitis with lymphocytic infiltration within and around the bronchiolar mucosa and lymphoid aggregates in the interstitium nearby (H&E; scale bar: 50 μm; original magnification, ×200). (E) A stomach biopsy exhibiting chronic antral inflammation with lymphoplasmacytic infiltrates in the lamina propria and occasionally on glands (H&E; scale bar: 50 μm; original magnification, ×200). (F) A jejunal biopsy depicting mild villus blunting and focal acute inflammation (H&E; scale bar: 50 μm; original magnification, ×200). Images in panels A-E are derived from Ferré et al., (7).
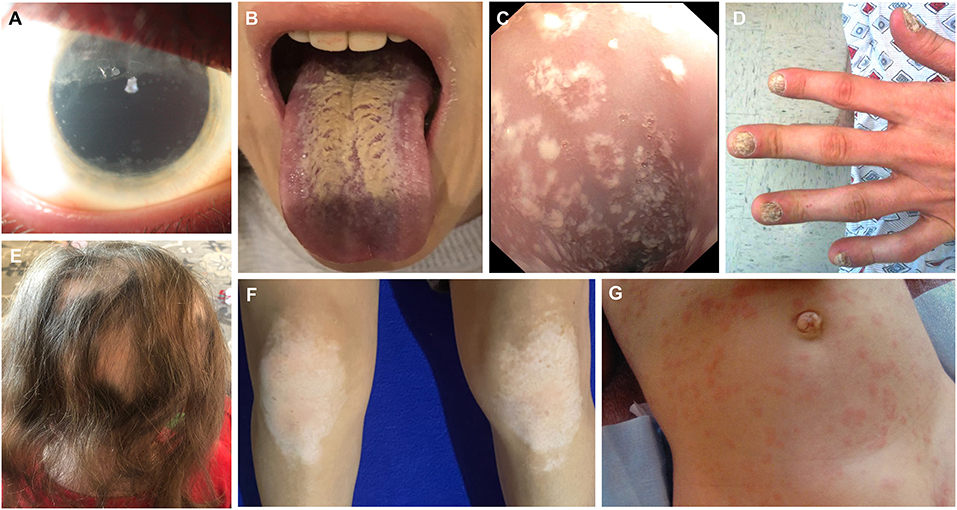
Figure 3. Clinical images depicting ophthalmologic, mucocutaneous and dermatologic manifestations in APECED patients. (A) Extensive keratopathy secondary to chronic keratoconjunctivitis. (B) Depiction of Candida chelitis and thrush affecting the tongue. (C) Endoscopic visualization of the esophagus demonstrating Candida esophagitis. (D) Nail dystrophy. (E) Alopecia areata. (F) Vitiligo affecting the knees. (G) APECED rash in an infant with APECED. Image 3G is derived from Ferré et al., (7).
In the American APECED cohort, we found the mean age of reaching a classic diagnostic dyad to be ~7.5 years; this delay in developing a classic diagnostic dyad is consistent with prior reports (145). Notably, only ~20% of the patients developed their first two consecutive manifestations among the classic triad manifestations. In contrast, the remaining ~80% of the patients developed a median of three non-triad manifestations before eventually reaching a classic diagnostic dyad, therefore resulting in significant delays in establishing a clinical diagnosis (7). Among the non-triad manifestations that occurred early before the development of a classic diagnostic dyad in American APECED patients, three were more prominent: (a) an urticarial eruption (“APECED rash”), which together with CMC were the most common initial disease manifestations, and presented typically as a self-limited, non-pruritic, and often recurrent maculopapular rash with a characteristic histological appearance of combined neutrophilic and lymphocytic dermatosis associated with NLRP3 inflammasome activation but without eosinophilic infiltration or vasculitis (7, 146) (Figure 2); (b) enamel hypoplasia, often featuring early tooth cavity formation, which underscores the importance of close cooperation between dental and medical professionals in the management of APECED patients (147–149); and (c) intestinal malabsorption, which is associated with increased fecal fat, loss of small intestinal enteroendocrine and/or Paneth cells, and gut dysbiosis (150–152). Less frequently, keratoconjunctivitis, autoimmune hepatitis, and autoimmune pneumonitis were observed early in the course of the disease before the development of a classic diagnostic dyad (7, 11, 12) (Figures 2, 3).
These findings led us to propose expanded diagnostic criteria that incorporate the adjunct triad manifestations of APECED rash, enamel hypoplasia, and intestinal malabsorption with the classic triad manifestations (7). With these expanded diagnostic criteria, the development of a diagnostic dyad among the classic and adjunct triad manifestations would be reached ~4 years earlier compared to the development of a diagnostic dyad among the classic triad manifestations, thereby decreasing the time to clinical diagnosis by half (7). In that dataset, accelerated APECED diagnosis by applying the expanded diagnostic criteria would have led to the prevention of life-threatening hypocalcemic seizures and/or adrenal crises in about half of the patients. We have now validated the diagnostic utility of the expanded diagnostic criteria in independent patient cohorts that have been evaluated in our prospective natural history study from both the Americas and non-Nordic European countries (Ferré & Schmitt et al., submitted). In addition, independent re-analysis of previously published APECED cohorts from Finland, Turkey, Sardinia, India, and Brazil indicated that the implementation of our proposed expanded diagnostic criteria would have also resulted in accelerated clinical diagnosis and earlier recognition of APECED in those populations (136–138), attesting to the broader applicability and usefulness of these expanded diagnostic criteria. Future prospective evaluation of patients from other countries in a similar uniform, systematic, multidisciplinary manner will help further define the diagnostic utility of the expanded diagnostic criteria in APECED. An earlier recognition of APECED via the expanded diagnostic criteria could have major clinical implications including (a) early screening for life-threatening endocrinopathies (hypoparathyroidism, adrenal insufficiency), (b) early initiation of treatment for life-threatening non-endocrine autoimmune manifestations (hepatitis, pneumonitis), and, possibly, (c) early administration of prophylactic immunomodulation with the goal to prevent the development of autoimmunity in children.
Taken together, these findings show that children affected by APECED in the Americas and other geographic regions are likely to be evaluated by dermatologists, dentists, allergists, immunologists, gastroenterologists as well as hepatologists, pulmonologists, and ophthalmologists early in the course of their disease, not only by endocrinologists who are traditionally more familiar with APECED. The presence of any of the manifestations within the adjunct triad of APECED rash, enamel hypoplasia, and intestinal malabsorption in a child with or without CMC or endocrinopathies or other organ-specific autoimmune manifestations (e.g., hepatitis, pneumonitis, keratoconjunctivitis, other) should raise suspicion for APECED. Such children should undergo a) sequencing of the AIRE gene with copy number variation analysis to evaluate for AIRE mutations and/or deletions, and b) testing for the presence of autoantibodies against IFN-ω (13, 67, 126) (Figure 4).
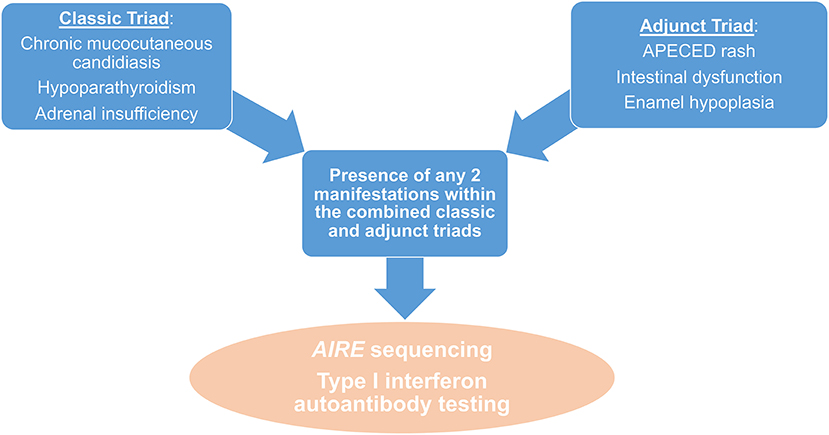
Figure 4. Diagnostic algorithm in patients with suspected APECED. The presence of any 2 manifestations amongst the combined classic and adjunct triads should raise suspicion for APECED and should prompt further work-up with AIRE sequencing and measurement of IFNω autoantibodies.
Clinical Management of APECED Patients
The management of APECED patients can be challenging due to the complex medical conditions that they develop, which are associated with poor quality of life and substantial psychosocial burden to both patients and their families (153). Mortality may exceed 30% even with best available medical treatment, driven by adrenal or hypocalcemic crises, end-organ failure (e.g., fulminant autoimmune hepatitis, pneumonitis-associated respiratory failure), malignancies (i.e., oral, esophageal and/or gastric), infections, or suicide (7, 128, 154). Therefore, a coordinated multidisciplinary approach that incorporates several medical and dental specialties is required to provide the best clinical care for the individual patient. Herein, we briefly outline a roadmap of general principles for the management of APECED patients.
Surveillance for New Manifestations
A critical component of the management of APECED patients is the systematic surveillance for the early detection of new endocrine and non-endocrine manifestations. In patients without hypoparathyroidism, periodic measurement of calcium and intact parathyroid hormone levels can help prevent unsuspected acute hypocalcemic seizures and/or tetany. In patients without Addison's disease, periodic ACTH stimulation testing can help prevent unsuspected acute adrenal crises. Screening for subclinical hypoparathyroidism and/or Addison's disease is particularly important before performing invasive procedures (e.g., esophagogastroduodenoscopy, bronchoscopy) as these can precipitate acute hypocalcemic and adrenal crises in APECED patients with subclinical hypoparathyroidism and Addison's disease. Periodic measurement of thyroid stimulating hormone, glucose, hemoglobin A1c, follicle stimulating hormone, and luteinizing hormone levels can help with early detection of subclinical hypothyroidism, type-1 diabetes, and hypogonadism. Periodic monitoring of transaminases, creatinine, and vitamin B12 levels, chest imaging with computed tomography, and bone density (DEXA) scan can help with early detection of subclinical autoimmune hepatitis, tubulointerstitial nephritis (TIN), pernicious anemia, pneumonitis, and osteopenia, respectively (11, 12, 155). Periodic examination of the oral mucosa by dental specialists can help with early detection of pre-cancerous or cancerous lesions (156). Periodic screening for asplenia, which develops in early adolescence in ~10–20% of patients, includes monitoring for new-onset leukocytosis and thrombocytosis, examination of peripheral blood smears for Howell-Jolly bodies, and/or nuclear liver-spleen scans.
Management of CMC
Untreated candidiasis can lead to esophageal strictures and may contribute to the development of squamous cell carcinomas of the oral mucosa and/or esophagus; therefore, it is important to treat APECED patients with acute mucosal fungal infection (156, 157). Acute episodes of mucosal candidiasis respond well to induction therapy for four weeks -to reduce the rate of infection relapse following treatment discontinuation- with a triazole such as fluconazole. An echinocandin (e.g., caspofungin, micafungin) is effective when fungal cultures reveal azole-resistant Candida strains, which are observed often in patients with APECED and other CMC syndromes (158, 159). In patients with frequent infection relapses (i.e., >3–4/year) in the absence of antifungal prophylaxis, we opt to transition from induction therapy to secondary prophylaxis with swish and swallow amphotericin B solution; in patients with infrequent infection relapses (i.e., <1–2/year), we opt to discontinue antifungal agents at the end of induction therapy and repeat induction therapy when candidiasis recurs. JAK/STAT inhibition has successfully remitted CMC in some patients with STAT1 gain-of-function (160) and it may have a role in the treatment of APECED-associated CMC given the exaggerated mucosal IFN-γ responses that we recently reported in the setting of AIRE deficiency (6); a formal clinical trial has been prepared at the NIH to investigate the efficacy of this treatment modality.
Management of Endocrine Manifestations
Close monitoring of serum and 24-h urine levels of calcium and phosphorus are needed to optimize the management of hypoparathyroidism. The goals of replacement treatment include the prevention of hypocalcemic crises but also avoidance of the long-term development of nephrocalcinosis, nephrolithiasis, and renal failure resulting from excessive replacement. To that end, maintaining serum calcium levels within the lower range of normal or just below the normal range is favored (161). The recent introduction of recombinant PTH in clinical practice for the treatment of adult patients with hypoparathyroidism (162) may help a subset of APECED patients with hypoparathyroidism to achieve optimal calcium homeostasis, particularly in the setting of pre-existing renal impairment and/or intestinal malabsorption. We recently reported that the peri-procedural use of recombinant PTH in APECED patients was safe and successfully maintained serum calcium levels without the need for intravenous calcium supplementation that can worsen nephrocalcinosis (163). Close monitoring of electrolytes and blood pressure are needed to optimize the management of adrenal insufficiency. The goals of replacement treatment include the prevention of adrenal crises but also avoidance of the long-term development of osteopenia resulting from excessive replacement. All patients with adrenal insufficiency should have access to parenteral hydrocortisone for acute stress dosing and a medical alert bracelet and/or Addison's disease emergency card and should be educated by their treating physicians on the appropriate clinical indications that warrant increasing their hydrocortisone dose (e.g., febrile illness). Furthermore, prompt initiation of hormone replacement is critical to achieve optimal pubertal development and growth in patients with hypogonadism; sperm and oocyte cryopreservation could be considered, when feasible, in adolescent patients before the development of gonadal failure (164).
Management of Non-endocrine Manifestations
Early diagnosis of certain non-endocrine autoimmune manifestations and initiation of immunomodulatory treatment is critical to prevent the development of irreversible end-organ damage. Autoimmune hepatitis develops in up to 40% of APECED patients, with presentations that may range between asymptomatic laboratory abnormalities to life-threatening fulminant failure requiring liver transplantation (12, 165, 166). A liver biopsy should be performed in APECED patients with persistent elevation in transaminases and/or bilirubin levels, even when classical serological biomarkers for autoimmune hepatitis are negative (e.g., anti-LKM, anti-SLA, anti-ASMA). Histological analysis shows lymphoplasmacytic infiltration and varying degrees of, typically mild, fibrosis in those with autoimmune hepatitis or may reveal an alternative diagnosis (e.g., fatty liver disease) (12) (Figure 2). APECED-associated autoimmune hepatitis responds clinically and biochemically to immunomodulatory treatment. Azathioprine-based or 6-mercapropurine–based therapy is most often used in clinical practice similar to classical autoimmune hepatitis in patients without APECED. In patients with mutations in the thiopurine S-methyltransferase (TPMT) gene that alter the metabolism of azathioprine and result in greater drug exposures and higher risk of azathioprine-induced toxicity, other T-lymphocyte–modulating agents such as mycophenolate, sirolimus, cyclosporine, and tacrolimus have been reported to remit liver inflammation (12). We favor the use mycophenolate or sirolimus in APECED patients due to the renal impairment that cyclosporine or tacrolimus may cause. Of note, we have observed a higher frequency of biopsy-proven mycophenolate-induced colitis in this patient population (~40%) relative to that observed in solid organ transplant recipients (<10%), which requires additional observational studies (167).
Autoimmune pneumonitis develops in up to 40% of APECED patients, typically presenting with chronic cough that is often misdiagnosed as asthma or bronchitis. Chronically untreated pneumonitis progresses to cause severe bronchiectatic structural lung disease with development of secondary pulmonary infections with bacteria and non-tuberculous mycobacteria (11, 168). We perform periodic chest imaging with computed tomography in all APECED patients because some patients can be asymptomatic in the early stages of autoimmune pneumonitis. A bronchoscopy should be performed in patients with radiographic evidence of bronchiectasis and/or ground glass or nodular opacities and/or in patients with abnormal pulmonary function testing, even when BPIFB1 and/or KCNRG autoantibodies are negative. Bronchoscopic findings consistent with the diagnosis of autoimmune pneumonitis include (a) the presence of increased numbers of activated neutrophils in the airways and (b) a thickened basal membrane with increased intraepithelial (CD8+ T lymphocytes > CD4+ T lymphocytes) and submucosal (CD4+ T lymphocytes > CD8+ T lymphocytes > B lymphocytes) inflammation in endobronchial tissue biopsies (11). Peribronchial lymphocytic inflammation (CD4+ T lymphocytes and CD8+ T lymphocytes > B lymphocytes) with B lymphocyte aggregates deeper in the lung parenchyma are typically observed when patients undergo transbronchial tissue biopsies (Figure 2). We recently reported that treatment with the combination of azathioprine (or mycophenolate) and rituximab results in remission of pneumonitis with improvement in clinical symptoms and in radiographic and pulmonary function abnormalities (11). Future studies will be aimed at examining whether targeting the IFN-γ/JAK-STAT axis may ameliorate non-endocrine autoimmune manifestations (including pneumonitis) in APECED patients.
TIN is uncommon (<5–10%) and presents with rapidly increasing creatinine levels that can lead to renal failure requiring kidney transplantation, as opposed to the slower progression of renal impairment that is seen in APECED patients with nephrocalcinosis who do not suffer from TIN (13, 169). Thus, a kidney biopsy should be performed in patients with rapidly evolving renal disease. Azathioprine or mycophenolate, when initiated early, may halt the progression of TIN in some -but not all- patients (13, 169). It is important to note that patients require T lymphocyte-directed immunomodulatory treatment following kidney or liver transplantation because TIN or autoimmune hepatitis will recur in the transplanted organs without immunosuppressive treatment (170, 171). Intestinal malabsorption develops in up to 80% of APECED patients and treatment can be challenging. It is important to rule out exocrine pancreatic insufficiency with measurement of fecal pancreatic elastase-1 levels as these patients respond clinically to pancreatic enzyme replacement therapy. Autoimmune enteritis features lymphocytic infiltration (T > B lymphocytes) in the small intestine and may respond clinically to T lymphocyte-directed immunomodulatory treatment, with sirolimus providing better results (unpublished observations) (Figure 2). Lactobacillus-based probiotic therapy was recently reported to ameliorate some gastrointestinal symptoms in a subset of APECED patients based on the presence of gut dysbiosis that is seen in intestinal malabsorption (151). Untreated keratoconjunctivitis can cause blindness and topical application of ophthalmic cyclosporine solution is effective. Patients with asplenia require vaccinations and prophylactic antibiotics to prevent the development of life-threatening sepsis by encapsulated bacteria (172). Anemia is a common laboratory abnormality in APECED patients and may be caused by (a) iron deficiency, which is often associated with intestinal malabsorption and may thus require parenteral supplementation, and/or (b) anemia of chronic disease, and/or (c) B12 deficiency, which requires supplementation to prevent the development of irreversible neurological sequelae, and/or, (d) rarely, autoimmune pure red cell aplasia, which is often refractory to T-lymphocyte–modulating therapy (141, 173).
Conclusions
APECED is a multisystem autoimmune disease caused by AIRE deficiency, which impairs the negative selection of T lymphocytes in the thymus. In recent years, the characterization of genetically diverse APECED patient cohorts and of dominant-negative AIRE mutations that cause milder autoimmune manifestations with incomplete clinical penetrance, and basic studies in Aire-deficient mice have shed more light into the genetics, immunology, clinical presentation, diagnosis, and treatment of AIRE deficiency. Moving forward, improved awareness of APECED among clinicians and a better understanding of the pathogenesis of AIRE deficiency should help devise enhanced strategies for earlier diagnosis and for effective preventive and therapeutic interventions, which should collectively improve the clinical outcomes of affected patients.
Author Contributions
EF, MS, and ML wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This research was supported by the Division of Intramural Research of the National Institute of Allergy and Infectious Diseases, NIH.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Finnish-German AC. An autoimmune disease, APECED, caused by mutations in a novel gene featuring two PHD-type zinc-finger domains. Nat Genet. (1997) 17:399–403. doi: 10.1038/ng1297-399
2. Nagamine K, Peterson P, Scott HS, Kudoh J, Minoshima S, Heino M, et al. Positional cloning of the APECED gene. Nat Genet. (1997) 17:393–8. doi: 10.1038/ng1297-393
3. Anderson MS, Venanzi ES, Klein L, Chen Z, Berzins SP, Turley SJ, et al. Projection of an immunological self shadow within the thymus by the aire protein. Science. (2002) 298:1395–401. doi: 10.1126/science.1075958
4. Niki S, Oshikawa K, Mouri Y, Hirota F, Matsushima A, Yano M, et al. Alteration of intra-pancreatic target-organ specificity by abrogation of Aire in NOD mice. J Clin Invest. (2006) 116:1292–301. doi: 10.1172/JCI26971
5. Jiang W, Anderson MS, Bronson R, Mathis D, Benoist C. Modifier loci condition autoimmunity provoked by Aire deficiency. J Exp Med. (2005) 202:805–15. doi: 10.1084/jem.20050693
6. Break TJ, Oikonomou V, Dutzan N, Desai JV, Swidergall M, Freiwald T, et al. Aberrant type 1 immunity drives susceptibility to mucosal fungal infections. Science. (2021) 371:eaay5731. doi: 10.1126/science.aay5731
7. Ferre EM, Rose SR, Rosenzweig SD, Burbelo PD, Romito KR, Niemela JE, et al. Redefined clinical features and diagnostic criteria in autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. JCI Insight. (2016) 1:e88782. doi: 10.1172/jci.insight.88782
8. Orlova EM, Sozaeva LS, Kareva MA, Oftedal BE, Wolff ASB, Breivik L, et al. Expanding the phenotypic and genotypic landscape of autoimmune polyendocrine syndrome type 1. J Clin Endocrinol Metab. (2017) 102:3546–56. doi: 10.1210/jc.2017-00139
9. Bruserud O, Oftedal BE, Wolff AB, Husebye ES. AIRE-mutations and autoimmune disease. Curr Opin Immunol. (2016) 43:8–15. doi: 10.1016/j.coi.2016.07.003
10. Scott HS, Heino M, Peterson P, Mittaz L, Lalioti MD, Betterle C, et al. Common mutations in autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy patients of different origins. Mol Endocrinol. (1998) 12:1112–9. doi: 10.1210/mend.12.8.0143
11. Ferre EMN, Break TJ, Burbelo PD, Allgauer M, Kleiner DE, Jin D, et al. Lymphocyte-driven regional immunopathology in pneumonitis caused by impaired central immune tolerance. Sci Transl Med. (2019) 11:eaav5597. doi: 10.1126/scitranslmed.aav5597
12. Chascsa DM, Ferre EMN, Hadjiyannis Y, Alao H, Natarajan M, Quinones M, et al. APECED-associated hepatitis: clinical, biochemical, histological and treatment data from a large, predominantly american cohort. Hepatology. (2021) 73:1088–104. doi: 10.1002/hep.31421
13. Constantine GM, Lionakis MS. Lessons from primary immunodeficiencies: autoimmune regulator and autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. Immunol Rev. (2019) 287:103–20. doi: 10.1111/imr.12714
14. Proekt I, Miller CN, Lionakis MS, Anderson MS. Insights into immune tolerance from AIRE deficiency. Curr Opin Immunol. (2017) 49:71–8. doi: 10.1016/j.coi.2017.10.003
15. Kogawa K, Kudoh J, Nagafuchi S, Ohga S, Katsuta H, Ishibashi H, et al. Distinct clinical phenotype and immunoreactivity in Japanese siblings with autoimmune polyglandular syndrome type 1 (APS-1) associated with compound heterozygous novel AIRE gene mutations. Clin Immunol. (2002) 103:277–83. doi: 10.1006/clim.2002.5208
16. Carpino A, Buganza R, Matarazzo P, Tuli G, Pinon M, Calvo PL, et al. Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy in two siblings: same mutations but very different phenotypes. Genes (Basel). (2021) 12. doi: 10.3390/genes12020169
17. Proekt I, Miller CN, Jeanne M, Fasano KJ, Moon JJ, Lowell CA, et al. LYN- and AIRE-mediated tolerance checkpoint defects synergize to trigger organ-specific autoimmunity. J Clin Invest. (2016) 126:3758–71. doi: 10.1172/JCI84440
18. Teh CE, Daley SR, Enders A, Goodnow CC. T-cell regulation by casitas B-lineage lymphoma (Cblb) is a critical failsafe against autoimmune disease due to autoimmune regulator (Aire) deficiency. Proc Natl Acad Sci U S A. (2010) 107:14709–14. doi: 10.1073/pnas.1009209107
19. Dikiy S, Li J, Bai L, Jiang M, Janke L, Zong X, et al. A distal Foxp3 enhancer enables interleukin-2 dependent thymic Treg cell lineage commitment for robust immune tolerance. Immunity. (2021) 54:931–46 e911. doi: 10.1016/j.immuni.2021.03.020
20. Ilmarinen T, Eskelin P, Halonen M, Ruppell T, Kilpikari R, Torres GD, et al. Functional analysis of SAND mutations in AIRE supports dominant inheritance of the G228W mutation. Hum Mutat. (2005) 26:322–31. doi: 10.1002/humu.20224
21. Oftedal BE, Hellesen A, Erichsen MM, Bratland E, Vardi A, Perheentupa J, et al. Dominant mutations in the autoimmune regulator AIRE are associated with common organ-specific autoimmune diseases. Immunity. (2015) 42:1185–96. doi: 10.1016/j.immuni.2015.04.021
22. Abbott JK, Huoh YS, Reynolds PR, Yu L, Rewers M, Reddy M, et al. Dominant-negative loss of function arises from a second, more frequent variant within the SAND domain of autoimmune regulator (AIRE). J Autoimmun. (2018) 88:114–20. doi: 10.1016/j.jaut.2017.10.010
23. Su MA, Giang K, Zumer K, Jiang H, Oven I, Rinn JL, et al. Mechanisms of an autoimmunity syndrome in mice caused by a dominant mutation in Aire. J Clin Invest. (2008) 118:1712–26. doi: 10.1172/JCI34523
24. Cetani F, Barbesino G, Borsari S, Pardi E, Cianferotti L, Pinchera A, et al. A novel mutation of the autoimmune regulator gene in an Italian kindred with autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy, acting in a dominant fashion and strongly cosegregating with hypothyroid autoimmune thyroiditis. J Clin Endocrinol Metab. (2001) 86:4747–52. doi: 10.1210/jcem.86.10.7884
25. Goldfarb Y, Givony T, Kadouri N, Dobes J, Peligero-Cruz C, Zalayat I, et al. Mechanistic dissection of dominant AIRE mutations in mouse models reveals AIRE autoregulation. J Exp Med. (2021) 218:e20201076. doi: 10.1084/jem.20201076
26. Eriksson D, Royrvik EC, Aranda-Guillen M, Berger AH, Landegren N, Artaza H, et al. GWAS for autoimmune Addison's disease identifies multiple risk loci and highlights AIRE in disease susceptibility. Nat Commun. (2021) 12:959. doi: 10.1038/s41467-021-21015-8
27. Laisk T, Lepamets M, Koel M, Abner E, Estonian Biobank Research T, Magi R. Genome-wide association study identifies five risk loci for pernicious anemia. Nat Commun. (2021) 12:3761. doi: 10.1038/s41467-021-24051-6
28. Laflam TN, Seumois G, Miller CN, Lwin W, Fasano KJ, Waterfield M, et al. Identification of a novel cis-regulatory element essential for immune tolerance. J Exp Med. (2015) 212:1993–2002. doi: 10.1084/jem.20151069
29. Haljasorg U, Bichele R, Saare M, Guha M, Maslovskaja J, Kond K, et al. A highly conserved NF-kappaB-responsive enhancer is critical for thymic expression of Aire in mice. Eur J Immunol. (2015) 45:3246–56. doi: 10.1002/eji.201545928
30. Yanagihara T, Sanematsu F, Sato T, Uruno T, Duan X, Tomino T, et al. Intronic regulation of Aire expression by Jmjd6 for self-tolerance induction in the thymus. Nat Commun. (2015) 6:8820. doi: 10.1038/ncomms9820
31. Rattay K, Claude J, Rezavandy E, Matt S, Hofmann TG, Kyewski B, et al. Homeodomain-interacting protein kinase 2, a novel autoimmune regulator interaction partner, modulates promiscuous gene expression in medullary thymic epithelial cells. J Immunol. (2015) 194:921–8. doi: 10.4049/jimmunol.1402694
32. Chuprin A, Avin A, Goldfarb Y, Herzig Y, Levi B, Jacob A, et al. The deacetylase Sirt1 is an essential regulator of Aire-mediated induction of central immunological tolerance. Nat Immunol. (2015) 16:737–45. doi: 10.1038/ni.3194
33. Shao W, Zumer K, Fujinaga K, Peterlin BM. FBXO3 protein promotes ubiquitylation and transcriptional activity of AIRE (Autoimmune Regulator). J Biol Chem. (2016) 291:17953–63. doi: 10.1074/jbc.M116.724401
34. Khan IS Taniguchi RT Fasano KJ Anderson MS Jeker LT. Canonical microRNAs in thymic epithelial cells promote central tolerance. Eur J Immunol. (2014) 44:1313–9. doi: 10.1002/eji.201344079
35. Anderson MS, Su MA. AIRE expands: new roles in immune tolerance and beyond. Nat Rev Immunol. (2016) 16:247–58. doi: 10.1038/nri.2016.9
36. Mathis D, Benoist C. Aire. Annu Rev Immunol. (2009) 27:287–312. doi: 10.1146/annurev.immunol.25.022106.141532
37. Klein L, Kyewski B, Allen PM, Hogquist KA. Positive and negative selection of the T cell repertoire: what thymocytes see (and don't see). Nat Rev Immunol. (2014) 14:377–91. doi: 10.1038/nri3667
38. Abramson J, Anderson G. Thymic epithelial cells. Annu Rev Immunol. (2017) 35:85–118. doi: 10.1146/annurev-immunol-051116-052320
39. Bansal K, Yoshida H, Benoist C, Mathis D. The transcriptional regulator Aire binds to and activates super-enhancers. Nat Immunol. (2017) 18:263–73. doi: 10.1038/ni.3675
40. Sansom SN, Shikama-Dorn N, Zhanybekova S, Nusspaumer G, Macaulay IC, Deadman ME, et al. Population and single-cell genomics reveal the Aire dependency, relief from Polycomb silencing, and distribution of self-antigen expression in thymic epithelia. Genome Res. (2014) 24:1918–31. doi: 10.1101/gr.171645.113
41. Devoss JJ, Shum AK, Johannes KP, Lu W, Krawisz AK, Wang P, et al. Effector mechanisms of the autoimmune syndrome in the murine model of autoimmune polyglandular syndrome type 1. J Immunol. (2008) 181:4072–9. doi: 10.4049/jimmunol.181.6.4072
42. Gavanescu I, Benoist C, Mathis D. B cells are required for Aire-deficient mice to develop multi-organ autoinflammation: a therapeutic approach for APECED patients. Proc Natl Acad Sci U S A. (2008) 105:13009–14. doi: 10.1073/pnas.0806874105
43. Fergusson JR, Morgan MD, Bruchard M, Huitema L, Heesters BA, Van Unen V, et al. Maturing human CD127+ CCR7+ PDL1+ dendritic cells express AIRE in the absence of tissue restricted antigens. Front Immunol. (2018) 9:2902. doi: 10.3389/fimmu.2018.02902
44. Gardner JM, Devoss JJ, Friedman RS, Wong DJ, Tan YX, Zhou X, et al. Deletional tolerance mediated by extrathymic Aire-expressing cells. Science. (2008) 321:843–7. doi: 10.1126/science.1159407
45. Gardner JM, Metzger TC, Mcmahon EJ, Au-Yeung BB, Krawisz AK, Lu W, et al. Extrathymic Aire-expressing cells are a distinct bone marrow-derived population that induce functional inactivation of CD4(+) T cells. Immunity. (2013) 39:560–72. doi: 10.1016/j.immuni.2013.08.005
46. Metzger TC Khan IS Gardner JM Mouchess ML Johannes KP Krawisz AK . Lineage tracing and cell ablation identify a post-Aire-expressing thymic epithelial cell population. Cell Rep. (2013) 5:166–79. doi: 10.1016/j.celrep.2013.08.038
47. Wang X, Laan M, Bichele R, Kisand K, Scott HS, Peterson P. Post-Aire maturation of thymic medullary epithelial cells involves selective expression of keratinocyte-specific autoantigens. Front Immunol. (2012) 3:19. doi: 10.3389/fimmu.2012.00019
48. Yamano T, Dobes J, Voboril M, Steinert M, Brabec T, Zietara N, et al. Aire-expressing ILC3-like cells in the lymph node display potent APC features. J Exp Med. (2019) 216:1027–37. doi: 10.1084/jem.20181430
49. Devoss J, Hou Y, Johannes K, Lu W, Liou GI, Rinn J, et al. Spontaneous autoimmunity prevented by thymic expression of a single self-antigen. J Exp Med. (2006) 203:2727–35. doi: 10.1084/jem.20061864
50. Hou Y, Devoss J, Dao V, Kwek S, Simko JP, Mcneel DG, et al. An aberrant prostate antigen-specific immune response causes prostatitis in mice and is associated with chronic prostatitis in humans. J Clin Invest. (2009) 119:2031–41. doi: 10.1172/JCI38332
51. Oftedal BE, Ardesjo Lundgren B, Hamm D, Gan PY, Holdsworth SR, Hahn CN, et al. T cell receptor assessment in autoimmune disease requires access to the most adjacent immunologically active organ. J Autoimmun. (2017) 81:24–33. doi: 10.1016/j.jaut.2017.03.002
52. Shum AK, Devoss J, Tan CL, Hou Y, Johannes K, O'gorman CS, et al. Identification of an autoantigen demonstrates a link between interstitial lung disease and a defect in central tolerance. Sci Transl Med. (2009) 1:9ra20. doi: 10.1126/scitranslmed.3000284
53. Taniguchi RT, Devoss JJ, Moon JJ, Sidney J, Sette A, Jenkins MK, et al. Detection of an autoreactive T-cell population within the polyclonal repertoire that undergoes distinct autoimmune regulator (Aire)-mediated selection. Proc Natl Acad Sci U S A. (2012) 109:7847–52. doi: 10.1073/pnas.1120607109
54. Wirasinha RC, Davies AR, Srivastava M, Sheridan JM, Sng XYX, Delmonte OM, et al. Nfkb2 variants reveal a p100-degradation threshold that defines autoimmune susceptibility. J Exp Med. (2021) 218:e20200476. doi: 10.1084/jem.20200476
55. Niemi HJ, Laakso S, Salminen JT, Arstila TP, Tuulasvaara A. A normal T cell receptor beta CDR3 length distribution in patients with APECED. Cell Immunol. (2015) 295:99–104. doi: 10.1016/j.cellimm.2015.03.005
56. Sng J, Ayoglu B, Chen JW, Schickel JN, Ferre EMN, Glauzy S, et al. AIRE expression controls the peripheral selection of autoreactive B cells. Sci Immunol. (2019) 4:eaav6778. doi: 10.1126/sciimmunol.aav6778
57. Malchow S, Leventhal DS, Lee V, Nishi S, Socci ND, Savage PA. Aire enforces immune tolerance by directing autoreactive T cells into the regulatory T cell lineage. Immunity. (2016) 44:1102–13. doi: 10.1016/j.immuni.2016.02.009
58. Yang S, Fujikado N, Kolodin D, Benoist C, Mathis D. Immune tolerance. Regulatory T cells generated early in life play a distinct role in maintaining self-tolerance. Science. (2015) 348:589–94. doi: 10.1126/science.aaa7017
59. Fujikado N, Mann AO, Bansal K, Romito KR, Ferre EMN, Rosenzweig SD, et al. Aire inhibits the generation of a perinatal population of interleukin-17A-producing gammadelta T cells to promote immunologic tolerance. Immunity. (2016) 45:999–1012. doi: 10.1016/j.immuni.2016.10.023
60. Su MA, Davini D, Cheng P, Giang K, Fan U, Devoss JJ, et al. Defective autoimmune regulator-dependent central tolerance to myelin protein zero is linked to autoimmune peripheral neuropathy. J Immunol. (2012) 188:4906–12. doi: 10.4049/jimmunol.1200493
61. Ossart J, Moreau A, Autrusseau E, Menoret S, Martin JC, Besnard M, et al. Breakdown of immune tolerance in AIRE-deficient rats induces a severe autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy-like autoimmune disease. J Immunol. (2018) 201:874–87. doi: 10.4049/jimmunol.1701318
62. Besnard M, Padonou F, Provin N, Giraud M, Guillonneau C. AIRE deficiency, from preclinical models to human APECED disease. Dis Model Mech. (2021) 14:dmm046359. doi: 10.1242/dmm.046359
63. Alimohammadi M, Bjorklund P, Hallgren A, Pontynen N, Szinnai G, Shikama N, et al. Autoimmune polyendocrine syndrome type 1 and NALP5, a parathyroid autoantigen. N Engl J Med. (2008) 358:1018–28. doi: 10.1056/NEJMoa0706487
64. Bensing S, Fetissov SO, Mulder J, Perheentupa J, Gustafsson J, Husebye ES, et al. Pituitary autoantibodies in autoimmune polyendocrine syndrome type 1. Proc Natl Acad Sci U S A. (2007) 104:949–54. doi: 10.1073/pnas.0610070104
65. Betterle C, Dal Pra C, Mantero F, Zanchetta R. Autoimmune adrenal insufficiency and autoimmune polyendocrine syndromes: autoantibodies, autoantigens, and their applicability in diagnosis and disease prediction. Endocr Rev. (2002) 23:327–64. doi: 10.1210/edrv.23.3.0466
66. Ekwall O, Hedstrand H, Grimelius L, Haavik J, Perheentupa J, Gustafsson J, et al. Identification of tryptophan hydroxylase as an intestinal autoantigen. Lancet. (1998) 352:279–83. doi: 10.1016/S0140-6736(97)11050-9
67. Kisand K, Peterson P. Autoimmune polyendocrinopathy candidiasis ectodermal dystrophy. J Clin Immunol. (2015) 35:463–78. doi: 10.1007/s10875-015-0176-y
68. Landegren N, Sharon D, Freyhult E, Hallgren A, Eriksson D, Edqvist PH, et al. Proteome-wide survey of the autoimmune target repertoire in autoimmune polyendocrine syndrome type 1. Sci Rep. (2016) 6:20104. doi: 10.1038/srep20104
69. Landegren N Sharon D Shum AK Khan IS Fasano KJ Hallgren A . Transglutaminase 4 as a prostate autoantigen in male subfertility. Sci Transl Med. (2015) 7:292ra101. doi: 10.1126/scitranslmed.aaa9186
70. Winqvist O, Gustafsson J, Rorsman F, Karlsson FA, Kampe O. Two different cytochrome P450 enzymes are the adrenal antigens in autoimmune polyendocrine syndrome type I and Addison's disease. J Clin Invest. (1993) 92:2377–85. doi: 10.1172/JCI116843
71. Winqvist O, Karlsson FA, Kampe O. 21-Hydroxylase, a major autoantigen in idiopathic Addison's disease. Lancet. (1992) 339:1559–62. doi: 10.1016/0140-6736(92)91829-W
72. Burbelo PD, Ferre EMN, Chaturvedi A, Chiorini JA, Alevizos I, Lionakis MS, et al. Profiling autoantibodies against salivary proteins in sicca conditions. J Dent Res. (2019) 98:772–8. doi: 10.1177/0022034519850564
73. Meyer S, Woodward M, Hertel C, Vlaicu P, Haque Y, Karner J, et al. AIRE-deficient patients harbor unique high-affinity disease-ameliorating autoantibodies. Cell. (2016) 166:582–95. doi: 10.1016/j.cell.2016.06.024
74. Alimohammadi M, Dubois N, Skoldberg F, Hallgren A, Tardivel I, Hedstrand H, et al. Pulmonary autoimmunity as a feature of autoimmune polyendocrine syndrome type 1 and identification of KCNRG as a bronchial autoantigen. Proc Natl Acad Sci U S A. (2009) 106:4396–401. doi: 10.1073/pnas.0809986106
75. Fishman D, Kisand K, Hertel C, Rothe M, Remm A, Pihlap M, et al. Autoantibody repertoire in APECED patients targets two distinct subgroups of proteins. Front Immunol. (2017) 8:976. doi: 10.3389/fimmu.2017.00976
76. Vazquez SE, Ferre EM, Scheel DW, Sunshine S, Miao B, Mandel-Brehm C, et al. Identification of novel, clinically correlated autoantigens in the monogenic autoimmune syndrome APS1 by proteome-wide PhIP-Seq. Elife. (2020) 9:e55053. doi: 10.7554/eLife.55053
77. Wang EY, Dai Y, Rosen CE, Schmitt MM, Dong MX, Ferré EM, et al. REAP: A platform to identify autoantibodies that target the human exoproteome. biorxiv. (2021). doi: 10.1101/2021.02.11.430703
78. Shum AK, Alimohammadi M, Tan CL, Cheng MH, Metzger TC, Law CS, et al. BPIFB1 is a lung-specific autoantigen associated with interstitial lung disease. Sci Transl Med. (2013) 5:206ra139. doi: 10.1126/scitranslmed.3006998
79. Popler J, Alimohammadi M, Kampe O, Dalin F, Dishop MK, Barker JM, et al. Autoimmune polyendocrine syndrome type 1: Utility of KCNRG autoantibodies as a marker of active pulmonary disease and successful treatment with rituximab. Pediatr Pulmonol. (2012) 47:84–7. doi: 10.1002/ppul.21520
80. Kisand K, Boe Wolff AS, Podkrajsek KT, Tserel L, Link M, Kisand KV, et al. Chronic mucocutaneous candidiasis in APECED or thymoma patients correlates with autoimmunity to Th17-associated cytokines. J Exp Med. (2010) 207:299–308. doi: 10.1084/jem.20091669
81. Meager A, Visvalingam K, Peterson P, Moll K, Murumagi A, Krohn K, et al. Anti-interferon autoantibodies in autoimmune polyendocrinopathy syndrome type 1. PLoS Med. (2006) 3:e289. doi: 10.1371/journal.pmed.0030289
82. Meloni A, Furcas M, Cetani F, Marcocci C, Falorni A, Perniola R, et al. Autoantibodies against type I interferons as an additional diagnostic criterion for autoimmune polyendocrine syndrome type I. J Clin Endocrinol Metab. (2008) 93:4389–97. doi: 10.1210/jc.2008-0935
83. Puel A, Doffinger R, Natividad A, Chrabieh M, Barcenas-Morales G, Picard C, et al. Autoantibodies against IL-17A, IL-17F, and IL-22 in patients with chronic mucocutaneous candidiasis and autoimmune polyendocrine syndrome type I. J Exp Med. (2010) 207:291–7. doi: 10.1084/jem.20091983
84. Bastard P, Orlova E, Sozaeva L, Levy R, James A, Schmitt MM, et al. Preexisting autoantibodies to type I IFNs underlie critical COVID-19 pneumonia in patients with APS-1. J Exp Med. (2021) 218:e20210554. doi: 10.1084/jem.20210554
85. Bastard P, Manry J, Chen J, Rosain J, Seeleuthner Y, Abuzaitun O, et al. Herpes simplex encephalitis in a patient with a distinctive form of inherited IFNAR1 deficiency. J Clin Invest. (2021) 131:e139980. doi: 10.1172/JCI139980
86. Hernandez N, Bucciol G, Moens L, Le Pen J, Shahrooei M, Goudouris E, et al. Inherited IFNAR1 deficiency in otherwise healthy patients with adverse reaction to measles and yellow fever live vaccines. J Exp Med. (2019) 216:2057–70. doi: 10.1084/jem.20182295
87. Hetemaki I, Laakso S, Valimaa H, Kleino I, Kekalainen E, Makitie O, et al. Patients with autoimmune polyendocrine syndrome type 1 have an increased susceptibility to severe herpesvirus infections. Clin Immunol. (2021) 231:108851. doi: 10.1016/j.clim.2021.108851
88. Wu J, Dobbs N, Yang K, Yan N. Interferon-independent activities of mammalian STING mediate antiviral response and tumor immune evasion. Immunity. (2020) 53 115–26 e115. doi: 10.1016/j.immuni.2020.06.009
89. Yamashiro LH, Wilson SC, Morrison HM, Karalis V, Chung JJ, Chen KJ, et al. Interferon-independent STING signaling promotes resistance to HSV-1 in vivo. Nat Commun. (2020) 11:3382. doi: 10.1038/s41467-020-17156-x
90. Yum S, Li M, Fang Y, Chen ZJ. TBK1 recruitment to STING activates both IRF3 and NF-kappaB that mediate immune defense against tumors and viral infections. Proc Natl Acad Sci U S A. (2021) 118:e2100225118. doi: 10.1073/pnas.2100225118
91. Bastard P, Michailidis E, Hoffmann HH, Chbihi M, Le Voyer T, Rosain J, et al. Auto-antibodies to type I IFNs can underlie adverse reactions to yellow fever live attenuated vaccine. J Exp Med. (2021) 218:e20202486. doi: 10.1084/jem.20202486
92. Bastard P, Rosen LB, Zhang Q, Michailidis E, Hoffmann HH, Zhang Y, et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science. (2020) 370:eabd4585. doi: 10.1126/science.abd4585
93. Abers MS, Rosen LB, Delmonte OM, Shaw E, Bastard P, Imberti L, et al. Neutralizing type-I interferon autoantibodies are associated with delayed viral clearance and intensive care unit admission in patients with COVID-19. Immunol Cell Biol. (2021) 99:917–21. doi: 10.1111/imcb.12495
94. Koning R, Bastard P, Casanova JL, Brouwer MC, van de Beek D. Autoantibodies against type I interferons are associated with multi-organ failure in COVID-19 patients. Intensive Care Med. (2021) 47:704–6 doi: 10.1007/s00134-021-06392-4
95. Troya J, Bastard P, Planas-Serra L, Ryan P, Ruiz M, De Carranza M, et al. Neutralizing autoantibodies to type I IFNs in >10% of patients with severe COVID-19 pneumonia hospitalized in Madrid, Spain. J Clin Immunol. (2021) 13:1–9 doi: 10.1007/s10875-021-01036-0
96. Van Der Wijst MGP, Vazquez SE, Hartoularos GC, Bastard P, Grant T, Bueno R, et al. Type I interferon autoantibodies are associated with systemic immune alterations in patients with COVID-19. Sci Transl Med. (2021) 13:eabh2624 doi: 10.1126/scitranslmed.abh2624
97. Povysil G, Butler-Laporte G, Shang N, Wang C, Khan A, Alaamery M, et al. Rare loss-of-function variants in type I IFN immunity genes are not associated with severe COVID-19. J Clin Invest. (2021) 131:e147834. doi: 10.1172/JCI147834
98. Van Der Made CI, Simons A, Schuurs-Hoeijmakers J, Van Den Heuvel G, Mantere T, Kersten S, et al. Presence of Genetic Variants Among Young Men With Severe COVID-19. JAMA. (2020) 324:663–73. doi: 10.1001/jama.2020.13719
99. Zhang Q, Bastard P, Liu Z, Le Pen J, Moncada-Velez M, Chen J, et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science. (2020) 370:eabd4570. doi: 10.1126/science.abd4570
100. Beccuti G, Ghizzoni L, Cambria V, Codullo V, Sacchi P, Lovati E, et al. A COVID-19 pneumonia case report of autoimmune polyendocrine syndrome type 1 in Lombardy, Italy: letter to the editor. J Endocrinol Invest. (2020) 43:1175–7. doi: 10.1007/s40618-020-01323-4
101. Ferre EMN, Lionakis MS. An AIREless Breath: Pneumonitis Caused by Impaired Central Immune Tolerance. Front Immunol. (2020) 11:609253. doi: 10.3389/fimmu.2020.609253
102. Meisel C, Akbil B, Meyer T, Lankes E, Corman VM, Staudacher O, et al. Mild COVID-19 despite autoantibodies to type I IFNs in Autoimmune-Polyendocrine-Syndrome Type 1 (APS-1). J Clin Invest. (2021) 131:e150867. doi: 10.1172/JCI150867
103. Delmonte OM, Bergerson JRE, Burbelo PD, Durkee-Shock JR, Dobbs K, Bosticardo M, et al. Antibody responses to the SARS-CoV-2 vaccine in individuals with various Inborn Errors of Immunity. J Allergy Clin Immunol. (2021) S0091-6749(21)01356-7. doi: 10.1016/j.jaci.2021.08.016
104. Ferre EMN, Schmitt MM, Ochoa S, Rosen LB, Shaw ER, Burbelo PD, et al. SARS-CoV-2 spike protein-directed monoclonal antibodies may ameliorate COVID-19 complications in APECED patients. Front Immunol. (2021) 12:720205. doi: 10.3389/fimmu.2021.720205
105. Gottlieb RL, Nirula A, Chen P, Boscia J, Heller B, Morris J, et al. Effect of bamlanivimab as monotherapy or in combination with etesevimab on viral load in patients with mild to moderate COVID-19: a randomized clinical trial. JAMA. (2021) 325:632–44. doi: 10.1001/jama.2021.0202
106. Lemarquis A, Campbell T, Aranda-Guillen M, Hennings V, Brodin P, Kampe O, et al. Severe COVID-19 in an APS1 patient with interferon autoantibodies treated with plasmapheresis. J Allergy Clin Immunol. (2021) 148:96–8. doi: 10.1016/j.jaci.2021.03.034
107. Group RC, Horby P, Lim WS, Emberson JR, Mafham M, Bell JL, et al. Dexamethasone in hospitalized patients with Covid-19. N Engl J Med. (2021) 384:693–704. doi: 10.1056/NEJMoa2021436
108. Beigel JH, Tomashek KM, Dodd LE, Mehta AK, Zingman BS, Kalil AC, et al. Remdesivir for the treatment of Covid-19 - final report. N Engl J Med. (2020) 383:1813–26. doi: 10.1056/NEJMc2022236
109. Consortium WHOST, Pan H, Peto R, Henao-Restrepo AM, Preziosi MP, Sathiyamoorthy V, et al. Repurposed antiviral drugs for Covid-19 - interim WHO solidarity trial results. N Engl J Med. (2021) 384:497–511. doi: 10.1056/NEJMoa2023184
110. Hung IF, Lung KC, Tso EY, Liu R, Chung TW, Chu MY, et al. Triple combination of interferon beta-1b, lopinavir-ritonavir, and ribavirin in the treatment of patients admitted to hospital with COVID-19: an open-label, randomised, phase 2 trial. Lancet. (2020) 395:1695–704. doi: 10.1016/S0140-6736(20)31042-4
111. Ahlgren KM, Moretti S, Lundgren BA, Karlsson I, Ahlin E, Norling A, et al. Increased IL-17A secretion in response to Candida albicans in autoimmune polyendocrine syndrome type 1 and its animal model. Eur J Immunol. (2011) 41:235–45. doi: 10.1002/eji.200939883
112. Bruserud O, Oftedal BE, Landegren N, Erichsen MM, Bratland E, Lima K, et al. A longitudinal follow-up of autoimmune polyendocrine syndrome type 1. J Clin Endocrinol Metab. (2016) 101:2975–83. doi: 10.1210/jc.2016-1821
113. Sarkadi AK, Tasko S, Csorba G, Toth B, Erdos M, Marodi L. Autoantibodies to IL-17A may be correlated with the severity of mucocutaneous candidiasis in APECED patients. J Clin Immunol. (2014) 34:181–93. doi: 10.1007/s10875-014-9987-5
114. Ritchlin CT, Kavanaugh A, Merola JF, Schett G, Scher JU, Warren RB, et al. Bimekizumab in patients with active psoriatic arthritis: results from a 48-week, randomised, double-blind, placebo-controlled, dose-ranging phase 2b trial. Lancet. (2020) 395:427–40. doi: 10.1016/S0140-6736(19)33161-7
115. Saunte DM, Mrowietz U, Puig L, Zachariae C. Candida infections in patients with psoriasis and psoriatic arthritis treated with interleukin-17 inhibitors and their practical management. Br J Dermatol. (2017) 177:47–62. doi: 10.1111/bjd.15015
116. Krueger JG, Fretzin S, Suarez-Farinas M, Haslett PA, Phipps KM, Cameron GS, et al. IL-17A is essential for cell activation and inflammatory gene circuits in subjects with psoriasis. J Allergy Clin Immunol. (2012) 130:145–54e149. doi: 10.1016/j.jaci.2012.04.024
117. Krueger JG, Wharton KA, Schlitt T, Suprun M, Torene RI, Jiang X, et al. IL-17A inhibition by secukinumab induces early clinical, histopathologic, and molecular resolution of psoriasis. J Allergy Clin Immunol. (2019) 144:750–63. doi: 10.1016/j.jaci.2019.04.029
118. Puel A, Cypowyj S, Bustamante J, Wright JF, Liu L, Lim HK, et al. Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science. (2011) 332:65–8. doi: 10.1126/science.1200439
119. Boisson B, Wang C, Pedergnana V, Wu L, Cypowyj S, Rybojad M, et al. An ACT1 mutation selectively abolishes interleukin-17 responses in humans with chronic mucocutaneous candidiasis. Immunity. (2013) 39:676–86. doi: 10.1016/j.immuni.2013.09.002
120. Levy R, Okada S, Beziat V, Moriya K, Liu C, Chai LY, et al. Genetic, immunological, and clinical features of patients with bacterial and fungal infections due to inherited IL-17RA deficiency. Proc Natl Acad Sci U S A. (2016) 113:E8277–85. doi: 10.1073/pnas.1618300114
121. Forbester JL, Lees EA, Goulding D, Forrest S, Yeung A, Speak A, et al. Interleukin-22 promotes phagolysosomal fusion to induce protection against Salmonella enterica Typhimurium in human epithelial cells. Proc Natl Acad Sci U S A. (2018) 115:10118–23. doi: 10.1073/pnas.1811866115
122. Glocker EO, Kotlarz D, Boztug K, Gertz EM, Schaffer AA, Noyan F, et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N Engl J Med. (2009) 361:2033–45. doi: 10.1056/NEJMoa0907206
123. Yazdani R, Moazzami B, Madani SP, Behniafard N, Azizi G, Aflatoonian M, et al. Candidiasis associated with very early onset inflammatory bowel disease: first IL10RB deficient case from the National Iranian Registry and review of the literature. Clin Immunol. (2019) 205:35–42. doi: 10.1016/j.clim.2019.05.007
124. Oikonomou V, Break TJ, Gaffen SL, Moutsopoulos NM, Lionakis MS. Infections in the monogenic autoimmune syndrome APECED. Curr Opin Immunol. (2021) 72:286–97. doi: 10.1016/j.coi.2021.07.011
125. Lionakis MS, Levitz SM. Host control of fungal infections: lessons from basic studies and human cohorts. Annu Rev Immunol. (2018) 36:157–91. doi: 10.1146/annurev-immunol-042617-053318
126. Husebye ES, Perheentupa J, Rautemaa R, Kampe O. Clinical manifestations and management of patients with autoimmune polyendocrine syndrome type I. J Intern Med. (2009) 265:514–29. doi: 10.1111/j.1365-2796.2009.02090.x
127. Ahonen P, Myllarniemi S, Sipila I, Perheentupa J. Clinical variation of autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) in a series of 68 patients. N Engl J Med. (1990) 322:1829–36. doi: 10.1056/NEJM199006283222601
128. Garelli S, Dalla Costa M, Sabbadin C, Barollo S, Rubin B, Scarpa R, et al. Autoimmune polyendocrine syndrome type 1: an Italian survey on 158 patients. J Endocrinol Invest. (2021). 44:2493–510 doi: 10.1007/s40618-021-01585-6
129. Sharifinejad N, Zaki-Dizaji M, Tebyanian S, Zainaldain H, Jamee M, Rizvi FS, et al. Clinical, immunological, and genetic features in 938 patients with autoimmune polyendocrinopathy candidiasis ectodermal dystrophy (APECED): a systematic review. Expert Rev Clin Immunol. (2021) 17:807–17. doi: 10.1080/1744666X.2021.1925543
130. Dominguez M, Crushell E, Ilmarinen T, Mcgovern E, Collins S, Chang B, et al. Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) in the Irish population. J Pediatr Endocrinol Metab. (2006) 19:1343–52. doi: 10.1515/JPEM.2006.19.11.1343
131. Meloni A, Willcox N, Meager A, Atzeni M, Wolff AS, Husebye ES, et al. Autoimmune polyendocrine syndrome type 1: an extensive longitudinal study in Sardinian patients. J Clin Endocrinol Metab. (2012) 97:1114–24. doi: 10.1210/jc.2011-2461
132. Ng'weina FM, Pura M, Anette SB. Autoimmune polyendocrine syndrome type I in Slovakia: relevance of screening patients with autoimmune Addison's disease. Eur J Endocrinol. (2008) 158:705–9. doi: 10.1530/EJE-07-0843
133. Perheentupa J. Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. J Clin Endocrinol Metab. (2006) 91:2843–50. doi: 10.1210/jc.2005-2611
134. Zlotogora J, Shapiro MS. Polyglandular autoimmune syndrome type I among Iranian Jews. J Med Genet. (1992) 29:824–6. doi: 10.1136/jmg.29.11.824
135. Pearce SH, Cheetham T, Imrie H, Vaidya B, Barnes ND, Bilous RW, et al. A common and recurrent 13-bp deletion in the autoimmune regulator gene in British kindreds with autoimmune polyendocrinopathy type 1. Am J Hum Genet. (1998) 63:1675–84. doi: 10.1086/302145
136. Fierabracci A, Arena A, Toto F, Gallo N, Puel A, Migaud M, et al. Autoimmune polyendocrine syndrome type 1 (APECED) in the Indian population: case report and review of a series of 45 patients. J Endocrinol Invest. (2021) 44:661–77. doi: 10.1007/s40618-020-01376-5
137. Fierabracci A, Pellegrino M, Frasca F, Kilic SS, Betterle C. APECED in Turkey: A case report and insights on genetic and phenotypic variability. Clin Immunol. (2018) 194:60–6. doi: 10.1016/j.clim.2018.06.012
138. Weiler FG, Peterson P, Costa-Carvalho BT, De Barros Dorna M, Correia-Deur JE, Sader SL, et al. The heterogeneity of autoimmune polyendocrine syndrome type 1: Clinical features, new mutations and cytokine autoantibodies in a Brazilian cohort from tertiary care centers. Clin Immunol. (2018) 197:231–8. doi: 10.1016/j.clim.2018.09.012
139. Friedman TC, Thomas PM, Fleisher TA, Feuillan P, Parker RI, Cassorla F, et al. Frequent occurrence of asplenism and cholelithiasis in patients with autoimmune polyglandular disease type I. Am J Med. (1991) 91:625–30. doi: 10.1016/0002-9343(91)90215-J
140. Stolarski B, Pronicka E, Korniszewski L, Pollak A, Kostrzewa G, Rowinska E, et al. Molecular background of polyendocrinopathy-candidiasis-ectodermal dystrophy syndrome in a Polish population: novel AIRE mutations and an estimate of disease prevalence. Clin Genet. (2006) 70:348–54. doi: 10.1111/j.1399-0004.2006.00690.x
141. Orlova EM, Kareva MA, Melikyan MA, Boyakova E, Peterkova VA, Maschan AA. Response of pure red cell aplasia to cyclophosphamide after failure of mycofenolate mofetil in a patient with polyglandular syndrome type I. J Pediatr Hematol Oncol. (2013) 35:e338–340. doi: 10.1097/MPH.0b013e3182755c52
142. Mazerkina N, Trunin Y, Gorelyshev S, Golanov A, Kadashev B, Shishkina L, et al. Thyrotropin-secreting pituitary adenoma in an 11-year-old boy with type 1 autoimmune polyglandular syndrome. J Pediatr Endocrinol Metab. (2016) 29:237–40. doi: 10.1515/jpem-2015-0018
143. Ferre EMN, Heller T, Lionakis MS. Progressive Small Bowel Obstruction in a Patient With the Autoimmune Syndrome APECED. Gastroenterology. (2020) 159:e4–5. doi: 10.1053/j.gastro.2019.12.040
144. Hadjiyannis Y, Ferre EMN, Schmitt MM, Davis JL, Heller T, Lionakis MS. Peritonitis on fasting-refeeding in APECED patients: a case series. Am J Gastroenterol. (2020) 115:790–5. doi: 10.14309/ajg.0000000000000599
145. Mazza C, Buzi F, Ortolani F, Vitali A, Notarangelo LD, Weber G, et al. Clinical heterogeneity and diagnostic delay of autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy syndrome. Clin Immunol. (2011) 139:6–11. doi: 10.1016/j.clim.2010.12.021
146. Zhu F, Willette-Brown J, Zhang J, Ferre EMN, Sun Z, Wu X, et al. NLRP3 inhibition ameliorates severe cutaneous autoimmune manifestations in a mouse model of autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy-like disease. J Invest Dermatol. (2021) 141:1404–15. doi: 10.1016/j.jid.2020.10.016
147. Brenchley L, Ferre E, Schmitt MM, Gardner PJ, Lionakis MS, Moutsopoulos N. Case report: dental findings can aid in early diagnosis of APECED syndrome. Front Dent Med. (2021) 2:670624. doi: 10.3389/fdmed.2021.670624
148. Mcgovern E, Fleming P, Costigan C, Dominguez M, Coleman DC, Nunn J. Oral health in autoimmune polyendocrinopathy candidiasis ectodermal dystrophy (APECED). Eur Arch Paediatr Dent. (2008) 9:236–44. doi: 10.1007/BF03262642
149. Perniola R, Tamborrino G, Marsigliante S, De Rinaldis C. Assessment of enamel hypoplasia in autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED). J Oral Pathol Med. (1998) 27:278–82. doi: 10.1111/j.1600-0714.1998.tb01956.x
150. Dobes J, Neuwirth A, Dobesova M, Voboril M, Balounova J, Ballek O, et al. Gastrointestinal Autoimmunity Associated With Loss of Central Tolerance to Enteric alpha-Defensins. Gastroenterology. (2015) 149:139–50. doi: 10.1053/j.gastro.2015.05.009
151. Petersen AØ, Jokinen M, Plichta DR, Liebisch G, Gronwald W, Dettmer K, et al. Cytokine-specific autoantibodies shape the gut microbiome in autoimmune polyendocrine syndrome type 1. J Allergy Clin Immunol. (2021) 148:876–88. doi: 10.1016/j.jaci.2021.03.025
152. Scarpa R, Alaggio R, Norberto L, Furmaniak J, Chen S, Smith BR, et al. Tryptophan hydroxylase autoantibodies as markers of a distinct autoimmune gastrointestinal component of autoimmune polyendocrine syndrome type 1. J Clin Endocrinol Metab. (2013) 98:704–12. doi: 10.1210/jc.2012-2734
153. Kluger N, Jokinen M, Krohn K, Ranki A. What is the burden of living with autoimmune polyendocrinopathy candidiasis ectodermal dystrophy (APECED) in 2012? A health-related quality-of-life assessment in Finnish patients. Clin Endocrinol (Oxf). (2013) 79:134–41. doi: 10.1111/cen.12087
154. Borchers J, Pukkala E, Makitie O, Laakso S. Patients With APECED Have Increased Early Mortality Due to Endocrine Causes, Malignancies and infections. J Clin Endocrinol Metab. (2020) 105:e2207-e2213. doi: 10.1530/endoabs.70.OC5.6
155. Laakso S, Borchers J, Toiviainen-Salo S, Pekkinen M, Makitie O. Severe Phenotype of APECED (APS1) Increases Risk for Structural Bone Alterations. Front Endocrinol (Lausanne). (2020) 11:109. doi: 10.3389/fendo.2020.00109
156. Bruserud O, Costea DE, Laakso S, Garty BZ, Mathisen E, Makitie A, et al. Oral Tongue Malignancies in Autoimmune Polyendocrine Syndrome Type 1. Front Endocrinol (Lausanne). (2018) 9:463. doi: 10.3389/fendo.2018.00463
157. Zhu F, Willette-Brown J, Song NY, Lomada D, Song Y, Xue L, et al. Autoreactive T cells and chronic fungal infection drive esophageal carcinogenesis. Cell Host Microbe. (2017) 21:478–93 e477. doi: 10.1016/j.chom.2017.03.006
158. Break TJ, Desai JV, Healey KR, Natarajan M, Ferre EMN, Henderson C, et al. VT-1598 inhibits the in vitro growth of mucosal Candida strains and protects against fluconazole-susceptible and -resistant oral candidiasis in IL-17 signalling-deficient mice. J Antimicrob Chemother. (2018) 73:2089–94. doi: 10.1093/jac/dky170
159. Siikala E, Rautemaa R, Richardson M, Saxen H, Bowyer P, Sanglard D. Persistent Candida albicans colonization and molecular mechanisms of azole resistance in autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) patients. J Antimicrob Chemother. (2010) 65:2505–13. doi: 10.1093/jac/dkq354
160. Higgins E, Al Shehri T, Mcaleer MA, Conlon N, Feighery C, Lilic D, et al. Use of ruxolitinib to successfully treat chronic mucocutaneous candidiasis caused by gain-of-function signal transducer and activator of transcription 1 (STAT1) mutation. J Allergy Clin Immunol. (2015) 135:551–3. doi: 10.1016/j.jaci.2014.12.1867
161. Winer KK, Ye S, Ferre EMN, Schmitt MM, Zhang B, Cutler GB, et al. Therapy with PTH 1-34 or calcitriol and calcium in diverse etiologies of hypoparathyroidism over 27 years at a single tertiary care center. Bone. (2021) 149:115977. doi: 10.1016/j.bone.2021.115977
162. Mannstadt M, Clarke BL, Vokes T, Brandi ML, Ranganath L, Fraser WD, et al. ya. Lancet Diabetes Endocrinol. (2013) 1:275–83. doi: 10.1016/S2213-8587(13)70106-2
163. Winer KK, Schmitt MM, Ferre EMN, Fennelly KP, Olivier KN, Heller T, et al. Impact of periprocedural subcutaneous parathyroid hormone on control of hypocalcaemia in APS-1/APECED patients undergoing invasive procedures. Clin Endocrinol (Oxf). (2021) 94:377–83. doi: 10.1111/cen.14335
164. Saari V, Holopainen E, Makitie O, Laakso S. Pubertal development and premature ovarian insufficiency in patients with APECED. Eur J Endocrinol. (2020) 183:513–20. doi: 10.1530/EJE-20-0516
165. Sinha R, Chapman AR, Reid GT, Hayes PC. Fulminant hepatic failure in autoimmune polyendocrine syndrome type-1. J R Coll Physicians Edinb. (2015) 45:136–40. doi: 10.4997/JRCPE.2015.210
166. Smith D, Stringer MD, Wyatt J, O'Meara M, Davison S, Cheetham TD, et al. Orthotopic liver transplantation for acute liver failure secondary to autoimmune hepatitis in a child with autoimmune polyglandular syndrome type 1. Pediatr Transplant. (2002) 6:166–70. doi: 10.1034/j.1399-3046.2002.01063.x
167. Schmitt M, Ferre EMN, Sampaio De Melo M, Cooper MA, Quezado MA, Heller T, et al. Mycophenolate-induced colitis in autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy patients. JPGN Rep in press. (2021). doi: 10.1097/PG9.0000000000000131
168. Kubala SA, Do HM, Ferre EMN, Schrump DS, Olivier KN, Walls JG, et al. Fatal autoimmune pneumonitis requiring bilobectomy and omental flap repair in a patient with autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED). Respir Med Case Rep. (2021) 33:101476. doi: 10.1016/j.rmcr.2021.101476
169. Kluger N, Kataja J, Aho H, Ronn AM, Krohn K, Ranki A. Kidney involvement in autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy in a Finnish cohort. Nephrol Dial Transplant. (2014) 29:1750–7. doi: 10.1093/ndt/gfu064
170. Gwertzman R, Corey H, Roberti I. Autoimmune polyglandular syndrome type I can have significant kidney disease in children including recurrence in renal allograft - a report of two cases. Clin Nephrol. (2016) 85:358–62. doi: 10.5414/CN108782
171. Ulinski T, Perrin L, Morris M, Houang M, Cabrol S, Grapin C, et al. Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy syndrome with renal failure: impact of posttransplant immunosuppression on disease activity. J Clin Endocrinol Metab. (2006) 91:192–5. doi: 10.1210/jc.2005-1538
172. Schmitt MM, Lionakis MS. Fulminant pneumococcal sepsis due to APECED-associated autoimmune asplenia. Blood. (2020) 135:298. doi: 10.1182/blood.2019003900
173. Chinello M, Mauro M, Cantalupo G, Balter R, De Bortoli M, Vitale V, et al. Pure red cell aplasia (PRCA) and cerebellar hypoplasia as atypical features of polyglandular autoimmune syndrome type I (APS-1): two sisters with the same AIRE mutation but different phenotypes. Front Pediatr. (2019) 7:51. doi: 10.3389/fped.2019.00051
Keywords: APECED syndrome, APS-1, AIRE, self-reactive T cells, autoantibodies, type-I interferons, IL-17, IFN-γ
Citation: Ferré EMN, Schmitt MM and Lionakis MS (2021) Autoimmune Polyendocrinopathy-Candidiasis-Ectodermal Dystrophy. Front. Pediatr. 9:723532. doi: 10.3389/fped.2021.723532
Received: 10 June 2021; Accepted: 07 October 2021;
Published: 01 November 2021.
Edited by:
Andrew R. Gennery, Newcastle University, United KingdomReviewed by:
Pattraporn Satitsuksanoa, University of Zurich, SwitzerlandRicardo U. Sorensen, Louisiana State University, United States
Copyright © 2021 Ferré, Schmitt and Lionakis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michail S. Lionakis, lionakism@mail.nih.gov
†These authors have contributed equally to this work