
- 1 Centre for Translational Medicine and Therapeutics, William Harvey Research Institute, Barts and The London, Queen Mary’s School of Medicine and Dentistry, London, UK
- 2 Centre for Neuroscience, Division of Experimental Medicine, Imperial College London, London, UK
Contrary to early views, we now know that systemic inflammatory/immune responses transmit to the brain. The microglia, the resident “macrophages” of the brain’s innate immune system, are most responsive, and increasing evidence suggests that they enter a hyper-reactive state in neurodegenerative conditions and aging. As sustained over-production of microglial pro-inflammatory mediators is neurotoxic, this raises great concern that systemic inflammation (that also escalates with aging) exacerbates or possibly triggers, neurological diseases (Alzheimer’s, prion, motoneuron disease). It is known that inflammation has an essential role in the progression of Alzheimer’s disease (AD), since amyloid-β (Aβ) is able to activate microglia, initiating an inflammatory response, which could have different consequences for neuronal survival. On one hand, microglia may delay the progression of AD by contributing to the clearance of Aβ, since they phagocyte Aβ and release enzymes responsible for Aβ degradation. Microglia also secrete growth factors and anti-inflammatory cytokines, which are neuroprotective. In addition, microglia removal of damaged cells is a very important step in the restoration of the normal brain environment, as if left such cells can become potent inflammatory stimuli, resulting in yet further tissue damage. On the other hand, as we age microglia become steadily less efficient at these processes, tending to become over-activated in response to stimulation and instigating too potent a reaction, which may cause neuronal damage in its own right. Therefore, it is critical to understand the state of activation of microglia in different AD stages to be able to determine the effect of potential anti-inflammatory therapies. We discuss here recent evidence supporting both the beneficial or detrimental performance of microglia in AD, and the attempt to find molecules/biomarkers for early diagnosis or therapeutic interventions.
Introduction
For many years, the central nervous system (CNS) was considered to be immune privileged, neither susceptible nor contributing to infection/inflammation. It is now evident that CNS infection and neurological diseases trigger local inflammation and consequently activation of the immune response. In particular, the response to aggression is driven by the resident immune cells, the microglia distributed throughout the normal adult brain (Perry and Andersson, 1992; Nimmerjahn et al., 2005). Specifically, Alzheimer’s disease (AD) is characterized by an inflammatory response to Amyloid-β (Aβ), inducing the activation of microglia and the recruitment of astrocytes to the sites where Aβ deposits occur (Sastre et al., 2006a).
It is nowadays accepted that there is a dynamic microglia turnover in the brain and that microglia phenotype may change depending on aging, stage of the disease, and/or the presence of peripheral inflammation. In the brain there are also infiltrated macrophages, which play an essential role in the immune response.
The purpose of this review is first, to describe microglia as a cell of the immune system and the effects of peripheral inflammation on their activation. Secondly, our aim is to describe how this system is altered in a neurodegenerative disease, such as AD. Therefore, targeting microglia could serve as a potential therapy to treat AD patients.
Microglia the Innate Immunity Cell Component
Role of Microglia and Macrophages in the Brain
– Microglia represent around 10% of the cells in the nervous system. Although there are many theories concerning the origin of microglia, the general consensus today is its hematopoietic origin, derived from myeloid precursor cells, which enter the developing CNS during embryogenesis. Many questions remain about the recruitment and the life of the resident microglial in adult and aging brain (Chan et al., 2007).
Microglia constitute the first line of defense against invading pathogens or other types of brain tissue injury. The general agreement is that microglia are the “sentinels” of the CNS. Their fundamental role is sensing both pathogen- and host-derived ligands within the CNS. By detecting the type of insult and consequently directing the innate to the adaptive immune response (e.g., removal of pathogen) they are fundamental to the resolution of inflammation. Under pathological situations, such as neurodegenerative disease, stroke, and tumor invasion, microglia become activated, surround damaged and dead cells, and clear cellular debris from the area, in analogy to phagocytic macrophages of the immune system (Fetler and Amigorena, 2005). This process plays a fundamental part in the reorganization of neural circuitry and repair mechanisms that arise following injury (Neumann et al., 2009; Neher et al., 2011). As part of a beneficial role microglial phagocytosis is a highly regulated process, with activated microglia expressing a wide, and redundant, variety of distinct receptors for the removal of pathogenic organisms, e.g., Toll-like receptors (TLRs; Neumann et al., 2009), or of apoptotic cell debris, e.g., CD36 and integrins (Napoli and Neumann, 2009; Lue et al., 2010). The microglial phagocytic response is thus a central part of the brain’s defense mechanisms, and is a powerful contributor to the systems in place that ensure healthy neural function.
Activated microglia also up-regulate other cell-surface receptors, including the major histocompatibility complex and complement receptors (Liu and Hong, 2003). Microglia experience dramatic morphological changes from ramified cells to activated amoeboid microglia (Kreutzberg, 1996). In addition, when microglia become activated they generate inflammatory mediators like cytokines, chemokines, prostaglandins, inducible nitric oxide synthase (iNOS), cyclooxygenase-2 (COX-2), free radicals, and stimulating an adaptive immune response (Nimmerjahn et al., 2005; Ransohoff and Perry, 2009). The principal goals of such reactions include the repair and the restoration of the homeostasis, but complications often arise, resulting in detrimental effects and actual exacerbation of the occurring damage (Lehnardt, 2010).
– Apart from resident microglia, in the brain there are monocyte-derived macrophages (Schwartz and Shechter, 2010). Perivascular macrophages have a phagocytic role and are also implicated in the presentation of antigens to T cells that have been activated in the periphery, thereby facilitating the recognition of CNS antigens (Perry et al., 2010). The macrophage and microglia phenotype has been defined as M1 (classically activated via TLRs or interferon γ) and M2 (alternatively activated by interleukin 4 or interleukin 13), although it is assumed that a mixed population of both phenotypes exists (Cameron and Landreth, 2010; Perry et al., 2010). Because most techniques are unable to differentiate between both populations of microglia and macrophages in the brain, they are collectively referred as microglia. However, both microglia and infiltrated monocytes are not functionally redundant and have different properties, so they are both necessary to display functions such as brain repair.
Impact of Peripheral Inflammation in the Brain
Inflammation is a response mounted by the innate immune system in response to injury and infections in order to promote recovery. It was long thought that the brain was protected from systemic inflammation. However, growing evidence shows that systemic inflammatory stimuli, such as infection, also trigger a central response through microglia with consequent release of pro-inflammatory mediators (cytokines, lipid metabolites, free radicals). As part of the host-defense process, this stimulates autonomic, neuroendocrine, and behavioral responses that promote recovery. Under normal conditions the neuroinflammatory response resolves and microglia resume their “resting” state and role of monitoring the microenvironment. However, increasing experimental and clinical evidence indicates that systemic inflammation can worsen, or possibly trigger, neurological diseases (Weller et al., 2005; Ransohoff and Perry, 2009). These include stroke (McColl et al., 2007), Alzheimer’s, prion, and motoneuron diseases (Nguyen et al., 2004; Perry, 2004; Rogers et al., 2007). Although the underlying mechanisms are not clear, mounting evidence suggests that microglia enter a primed/hyper-sensitive state in neurodegenerative conditions. This pre-disposes to an exaggerated production of toxic pro-inflammatory mediators in response to systemic inflammogens, thereby exacerbating neuronal loss (Streit et al., 2004; Godbout et al., 2005; Perry et al., 2007).
Understanding the routes of communication between peripheral immune responses and the brain has not been an easy challenge (Perry et al., 2007). The general belief is that immune messages are passed to the brain mainly through three different pathways: first, peripherally derived signals (mainly pro-inflammatory cytokines like IL-1β, TNF-α, and IL-6) and even pathogen-associated molecular patterns (PAMPs; for example the lipopolysaccharide (LPS), the main cell-wall component of Gram-negative bacteria) can access the nervous system through brain sites that lack a proper blood brain barrier (BBB), or through fenestrated capillaries (Rivest, 2003); secondly, on-going peripheral reactions can be sensed and transmitted to the brain via neural afferent pathways, mainly through the vagus nerve (Gao et al., 2008); lastly, the BBB itself, through the role of its numerous cellular components like endothelial cells and perivascular macrophages can sense circulating signals and respond to them, affecting behavior of neurons, astrocytes, and especially resident microglia population (Rivest, 2009; Figure 1).
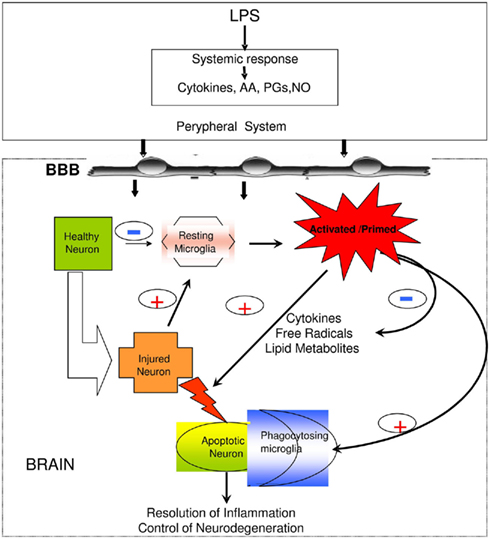
Figure 1. Peripheral infection/inflammation causes the release of pro-inflammatory mediators, including cytokines (TNF-α, IL-1β, IL-6), arachidonic acid (AA), prostaglandins (PGs), and nitric oxide (NO) synthesis. The brain also mounts an inflammatory response to systemic inflammation, as well as to local injury (neurodegeneration, trauma, stroke), with the microglial cells responding soonest and with production of the greatest amounts of pro-inflammatory mediators. However, the central response appears to be under tighter control than the peripheral response in that it is delayed and more modest, probably in order to avoid the dire consequences of a full-blown inflammatory response within the confines of the skull. Modified after Solito et al. (2008).
Microglia in Aging
Despite the similarities with the innate immune response in the peripheral system, there are important differences with brain environment, where the microglia activity could be sometimes deleterious for the brain. This in-balance phenotype is determined by the fact that neuroinflammation may be chronic or acute (Colton, 2009). Both chronic low-grade peripheral inflammation (Ouchi et al., 2005) and microglial priming/hypersensitivity are associated with aging.
It is well known that microglia exhibit significant phenotypic changes during normal aging. Microglia cells from both aged humans and rodents show profoundly altered morphology, characterized by dystrophic processes, and abnormal clustering (Perry et al., 1996; Streit et al., 2008). These changes in morphology are accompanied by increased expression of activation markers such as MHCII and RAGE (Perry et al., 1993), raised basal production of the pro-inflammatory cytokines TNFα, IL-6, and IL-1β (Ye and Johnson, 1999; Xie et al., 2003; Sierra et al., 2007), and hyper-responsiveness to inflammatory stimulation (Njie et al., 2012), together suggesting that microglia become progressively dysfunctional with age. A likely explanation for this fact lies in the loss of endogenous factors which would normally control/drive prevent excessive microglial activation, and promote the beneficial, anti-inflammatory, phenotype. We could further hypothesize that microglia turnover with aging is reduced and that newly attracted monistic cells may acquire a wrong phenotype once enriched the brain environment (Kofler and Wiley, 2011).
In summary, microglial activation presents a double-edged sword: from one side we can define it as neuroprotective, forming the first line of defense in the CNS; from the other, it can become a neurodegenerative force, when its power is excessive and to represent risk factors for developing age-associated neurodegenerative disease (AD), contributing to the worsening of symptoms that occur after systemic infection (Perry et al., 2007). Understanding the shift between these two opposite and the changes that occurring with aging will allow us to minimize the harmful and capitalize the beneficial effects and consequently the treatment of neurodegenerative diseases (Yong and Rivest, 2009).
Microglia in Alzheimer’s Disease
Implications of AD Pathogenesis in Microglia Activation
Inflammation has been implicated in neuronal damage, increased Aβ generation, increased phosphorylation of tau, and cognitive impairment in AD. Cause or consequence of disease progression is still not clear. Clinical work and studies in animal models suggest that microglial activation precede amyloid plaques and tangles formation (Griffin et al., 1989; Heneka et al., 2005a) while PET studies have reported inflammatory changes in one-third of amnestic mild cognitive impairment (MCI; Cagnin et al., 2001; Okello et al., 2009).
Many of the cytokines and chemokines secreted by microglia such as IL-1β, IL-6, TNF-α, IL-8, TGF-β, and macrophage inflammatory protein-1α (MIP-1α) have been found to have altered expression in AD patients compared to control individuals (Sastre et al., 2006a). Animal models of AD, including the APP transgenic line Tg2567 carrying the Swedish mutation, also show elevated levels for TNF-α, IL-1β, IL-1α, chemoattractant protein-1, COX-2, and complement component 1q (Benzing et al., 2000; Matsuoka et al., 2001). In addition, an increased risk of AD has been associated with several polymorphisms of pro-inflammatory genes, including IL-1 (Nicoll et al., 2000), IL-6 (Capurso et al., 2004), TNF-α (McCusker et al., 2001; Perry et al., 2001), and α1-antichymotrypsin (Kamboh et al., 1995).
Amyloid peptides and their precursor protein (APP) are strong glial activators (Barger and Harmon, 1997) and knockdown of APP gene and its proteolytic products delay and decrease microglial activation (DeGiorgio et al., 2002). The extent of astrocytosis and microglial activation is directly dependent on the amyloid load, and treatment with β-sheet breaker peptides leads to reduced brain inflammation (Permanne et al., 2002). Aβ is able to activate a NFκB-dependent pathway that is required for cytokine production (Combs et al., 2001). In addition, the C-terminal (CT) 100 amino acids of βAPP, which is also present in senile plaques, can induce gliosis and neuronal death. CT100 exposure results in activation of mitogen-activated protein kinase (MAPK) pathways as well as NFκB (Bach et al., 2001). At the same time, inflammation may increase Aβ generation by affecting the transcription of the β-secretase (BACE1), the main enzyme responsible for Aβ generation (Sastre et al., 2006b, 2008), therefore creating a feed-forward cycle.
In addition, neuroinflammation participates in tau-mediated neurodegeneration (Jaworski et al., 2011). Animal models of tauopathy such as the P301S tau transgenic mice exhibit accumulation of activated microglial cells around tau-positive nerve cells (Yoshiyama et al., 2007). Eventually, pro-inflammatory cytokines are also able to modify the activity of kinases involved in Tau phosphorylation (Arnaud et al., 2006). Products of inflammation might change the substrate specificity of kinases/phosphatases leading to tau phosphorylation at pathological sites. It was shown recently that inflammation induced by infection increased GSK3 activity in the triple-transgenic mouse model of AD, associated with a shift of tau from the detergent-soluble to the detergent-insoluble fraction (Sy et al., 2011).
On the other hand, other proteins involved in AD, such as presenilin, have been implicated in inflammation. Presenilin conditional knockout mice present differential up-regulation of inflammatory markers in the cerebral cortex, such as strong microglial activation, and elevated levels of glial fibrillary acidic protein (GFAP), complement component C1q, and cathepsin S (Beglopoulos et al., 2004). In fact, γ-secretase inhibitors have been reported to impair microglial activity as measured in gene expression, protein levels, and migration ability, which resulted in a reduction of soluble β-amyloid phagocytosis. Moreover, microglia deficient in presenilin 1 and 2 showed impairment in phagocytosis of soluble β-amyloid (Farfara et al., 2011).
Mechanisms of Aβ-Induced Microglial Activation
As indicated above, Aβ is able to bind and activate microglia. The mechanism of action for this is through interaction with pattern recognition receptors (PRRS). Microglia express many PRRs (Farina et al., 2007; Falsig et al., 2008), which recognize and bind to both PAMPs, or danger-associated molecular patterns (DAMPs), such as Aβ (Salminen et al., 2009). Interaction of microglia with Aβ, via PRRs provokes their inflammatory actions.
Toll-like receptors
Toll-like receptors are a type-1 integral glycoproteins (Pancer and Cooper, 2006; Miyake, 2007). Among the cell-surface TLRs, TLR2 and 4 can recognize Aβ (Carty and Bowie, 2011). Several studies have confirmed that TLR4 mediates microglial-induced neurotoxicity both in vivo and in vitro (Lehnardt et al., 2003; Walter et al., 2007).
Toll-like receptor activation is regulated by co-receptors, including MD-2, CD14, and CD36 (Akashi-Takamura and Miyake, 2006). Research using knockout mice for TLR4 or TLR2 demonstrated an increase in Aβ deposition and acceleration in cognitive decline (Tahara et al., 2006; Richard et al., 2008). These results suggest that TLR2 and TLR4 may be involved in Aβ clearance in vivo and hence provide neuroprotection in AD. In fact, it was shown that response of microglial cells to fibrillar forms of Aβ requires the participation of TLRs and the co-receptor CD14 (Reed-Geaghan et al., 2009). However, microglia internalize soluble Aβ through a non-saturable, fluid phase macropinocytic mechanism that is distinct from phagocytosis and receptor-mediated endocytosis (Mandrekar et al., 2009).
Receptor for advanced end glycation products (RAGE)
RAGE is a member of the immunoglobulin superfamily of cell-surface proteins (Schmidt et al., 2001; Chavakis et al., 2003; Bierhaus et al., 2005). It is a multiligand receptor, which recognizes Aβ peptides and fibrils (Knapp and Prince, 2007). Interestingly, RAGE-expressing microglia are upregulated in AD, and microglial RAGE is reported to mediate the pro-inflammatory effects of Aβ (Yan et al., 1996; Lue et al., 2001; Arancio et al., 2004). This is supported by recent work whereby it was demonstrated in transgenic AD models that the interaction of microglial RAGE with Aβ activates signal transduction cascades (MAP kinase, p38, and ERK1/2), enhances cytokines production (IL-β and TNF-α), and accelerates or amplifies the inflammatory response, leading to recruitment or activation of microglia and astrocytes (Fang et al., 2010).
Scavenger receptors
Scavenger receptor (SR) type-A (SR-A), type B1 (SR-B1), CD36, and CD40 are established receptors for insoluble fibrillar Aβ aggregates, and are expressed by activated microglia, mediating the endocytosis of oligomeric and fibrillar Aβ (El Khoury et al., 1996; Paresce et al., 1996; Coraci et al., 2002; Husemann et al., 2002). Microglial adherence via SR-A binding to fibrillar Aβ leads to microglial immobilization, production of ROS, secretion of cytokines such as TNF-α and complement proteins (El Khoury et al., 1996).
Formyl peptide receptors
Aβ can also bind to members of the seven-transmembrane G protein coupled receptors known as formyl peptide receptors (FPRs; Le et al., 2002). FPR, FPR-like 1 (FPRL-1), and FPR-like 2 (FPRL2) have been characterized as series of receptors, for which the main endogenous ligand is Annexin A1 (ANXA1; Solito et al., 2008). These receptors bind with high affinity to N-formylated bacterial peptides. FPRs are expressed on several immune cells including leukocytes, monocytes, and microglia. Among them the FPRL-1 mediates the chemotactic activity of Aβ42 for mononuclear phagocytes and therefore appear to be pathophysiologically relevant in the AD (Iribarren et al., 2005). In addition, Aβ bound to FPRL-1 is rapidly internalized into the cytoplasmic region as ligand/receptor complexes in mononuclear phagocytes. This process may represent responses of host-defense aiming at the clearance of abnormally elevated, pathogenic Aβ. However, the Aβ interaction with FPRL-1 is clearly associated with cell activation (Cui et al., 2002) and the release of pro-inflammatory and neurotoxic mediators (Pan et al., 2011). Interestingly FPRL-1 is highly express in mononuclear phagocytes surrounding and infiltrating Congo red-positive plaques in AD patients’ brain tissue (Le et al., 2001).
Complement receptors
Complement receptors are one of the categories of cell-surface molecules on microglia that are upregulated in response to the activation of these cells (Liu and Hong, 2003). Aβ-induced complement activation leads to generation of C1q, C4, and C3 activation fragments around the plaques. Here microglia express complement proteins C1q, C3, and receptors C1qR, CR3, CR4, and C5aR, which support phagocytic uptake (Keene et al., 2011). Inhibition of the complement system results in an increase of Aβ plaque formation and neurodegeneration in AD transgenic mice (Shen and Meri, 2003).
In contrast, lack of C1q in mice models of AD results in decrease pathology (Hafer-Macko et al., 2000). This indicates that one mechanism by which microglia could recruit further reactive cells to the site of a plaque and cause neurotoxic damage is by activating the classic complement pathway and the inflammatory machinery associated with it (pro-inflammatory cytokines, oxidative products) through production of C1q (McGeer and McGeer, 1998; Bonifati and Kishore, 2007).
The demonstration that the peripheral benzodiazepine receptor is upregulated in activated microglia led to the development of a ligand, [11C](R)-PK11195, which binds to this receptor also known as the 18-kDa translocator protein (TSPO). Extensive amyloid deposition and microglial activation can be demonstrated in the same group of AD patients in vivo by PET using [11C]PK11195 and a negative correlation between microglial activation and levels of cognition has been reported. Both amyloid deposition and microglial activation can be detected in vivo with PET in around 50% of patients with MCI. However, amyloid deposition and microglial activation are not necessarily correlated in MCI suggesting both can occur in the absence of the other (Okello et al., 2009; Sastre et al., 2011). On the other hand, a significant age-dependent increase in specific [3H](R)-PK11195 binding was also demonstrated in a transgenic mouse model of AD (TASTPM: APPswxPS1M146V; Roberts et al., 2009). This was consistent with immunohistochemical data showing age-dependent increases in CD68 immunoreactivity co-localized with Aβ deposits. CD68 is a 110-kDa transmembrane glycoprotein, expressed by monocyte/macrophage lineages and serves as a marker for microglia. Interestingly, an antibody to human TSPO revealed induction of TSPO-positive microgliosis by tau fibrils in tauopathy brains. In addition, in transgenic PS19 mice, carrying the P301S Tau mutation, radiolabeling of TSPO with [11C]AC-5216 was linearly proportional to the amount of phospho-tau immunolabeling (Maeda et al., 2011). The results of that study indicated that TSPO immunoreactivities are associated with NFTs, neuropil threads, and plaque neuritis rather than Aβ deposits. All together, the analysis of microglia by PET in AD and MCI patients plus the studies of microglial activation over time in animal models suggest that microglia activation occurs before Aβ deposition and correlates better with cognitive deficits and tau phosphorylation.
Microglial Activation in Different Stages of AD
It has been hypothesized that early microglial activation in AD delays disease progression by promoting clearance of Aβ before formation of senile plaques. It is conceivable that glial activation is protective early in the disease (Wyss-Coray et al., 2003; Maragakis and Rothstein, 2006; Wyss-Coray, 2006). In fact, studies have shown that blood derived macrophages (BMDM) are able to efficiently eliminate amyloid and confer neuroprotection by secretion of growth factors such as the glia-derived neurotrophic factor (GDNF), which are potentially beneficial to the survival of neurons (Liu and Hong, 2003). Activated microglia in early stages of AD can reduce Aβ accumulation by increasing its phagocytosis, clearance, and degradation (Frautschy et al., 1998; Qiu et al., 1998). The mechanism by which Aβ is phagocytosed depends on the physical properties of Aβ and whether it is soluble or fibrillar. Secreted Aβ1-40 and Aβ1-42 peptides are constitutively degraded by neprilysin and the insulin degrading enzyme (IDE), a metalloprotease released by microglia and other neural cells, whose enzymatic activity is enhanced by inflammatory events, such as LPS stimulation (Qiu et al., 1997).
In later stages, with persistent production of pro-inflammatory cytokines, microglia lose their protective effect (Hickman et al., 2008; Jimenez et al., 2008) and may become detrimental through the release of cytokines and chemokines (Hickman et al., 2008). These inflammatory mediators modulate immune and inflammatory function and may also alter neuronal function. In addition, microglia from old transgenic mice have a decrease in the expression of the Aβ-binding SR-A, CD36 and RAGE, and the Aβ degrading enzymes IDE, neprilysin, and matrix metalloprotease 9 (MMP9), compared with wild-type controls (Hickman et al., 2008). Therefore, all the evidences support the idea that over-activated microglia could cause uncontrolled inflammation that may drive the chronic progression of AD by exacerbating Aβ deposition and stimulating neuronal death (Mrak and Griffin, 2005; Gao and Hong, 2008). This concept constitutes the “Neuroinflammatory hypothesis.”
By comparison, the “Microglial dysfunction hypothesis” stipulates that rather than an increase of inflammatory function there is a loss of the microglial neuroprotective function in AD (Polazzi and Monti, 2010). Research has shown that the phagocytic abilities of microglia are altered in aging and impaired in neurodegenerative diseases. Therefore this “senescent” or dystrophic microglia can also contribute to the onset of sporadic AD (Streit et al., 2004, 2009).
In addition, other studies have shown that inadequate recruitment of blood monocytes with aging might be a critical event that leads to disease onset. Because the dynamics of the local and systemic inflammatory response may vary with aging and stage of the disease, this would important for the outcome of immunosuppressive treatments (Schwartz and Shechter, 2010).
Studies with animal models have provided controversial data regarding the role of microglia in AD. Experiments crossing APP animal models with Iba1-TK mice, leading to nearly complete ablation of microglia, did not display differences in plaque formation (Grathwohl et al., 2009). These results suggest that microglia may not have a direct role on Aβ deposition, but affect neuronal function. These results also reinforce the role of blood monocytes, which may support the phagocytic function of microglia. However, another study, using two-photon microscopy, performed in triple-transgenic mice crossed with the microglial chemokine receptor CX3CR1 knockout mouse, revealed that microglia is involved in neuron elimination, indicated by locally increased number and migration velocity of microglia around lost neurons (Fuhrmann et al., 2010). Microglia were recruited to the neuron before and not after the elimination of the neuron. Furthermore, CX3CR1 knockout prevented neuronal loss, indicating that neuronal loss depends on the communication between neurons and microglia (Fuhrmann et al., 2010).
Microglia as Target for AD Therapy
Anti-inflammatory drugs
Microglia associated with the senile plaques is thought to be a potential target of non-steroidal anti-inflammatory drugs (NSAIDs). A study by Mackenzie and Muñoz (1998) carried out in non-demented patients showed that those treated with NSAIDs had three times less activated microglia as non-treated controls. These data have been confirmed by in vivo treatment with NSAIDs such as ibuprofen in mouse models of AD, which have shown decreases in microglial activation and in inflammatory mediators such as iNOS, cyclooxygenase (COX), and cytokines (Lim et al., 2000; Heneka et al., 2005b). Experiments performed using cultured microglia have revealed that incubation with NSAIDs decreased the secretion of pro-inflammatory cytokines and may increase Aβ phagocytosis (Lleo et al., 2007). However, the reduction of activated microglia and astroglia by NSAIDs was not significant in AD patients, indicating an age or stage dependent difference in the glial response, i.e., in their activation rate (Alafuzoff et al., 2000). Microglia in aged or diseased brains are primed and usually behave differently to those in younger individuals (Gao and Hong, 2008). Thus, it is likely that microglia do not respond equally to anti-inflammatory therapy in old age and therefore, treatment of patients with NSAIDs in advanced stages of the disease may not produce any benefit. In this regard, NSAIDs have been shown to have beneficial effects in young individuals with robust immune systems. In aged patients, these drugs may affect the weak systemic immune response of the patients, exacerbating the local damage by eliminating the capacity of the immune system to introduce disease-modifying factors to the inflamed area (Sastre and Gentleman, 2010) (Figure 2).
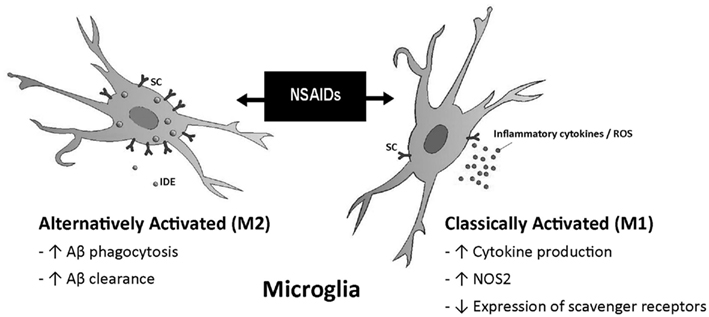
Figure 2. Different effects of NSAIDs on microglia. The response to NSAIDs may differ depending on whether they are used in early stages of disease, in which microglia present an alternatively activated phenotype compared with late stages which is associated with a classical microglia phenotype. (Adapted from Sastre and Gentleman, 2010). Abbreviations: ROS, reactive oxygen species; NOS2, same as iNOS; IDE, insulin degrading enzyme; SC, scavenger receptors.
A potential downstream target of some NSAIDs such as ibuprofen, indomethacin, and naproxen is the peroxisome proliferator-activated receptor-γ (PPAR-γ; Lehmann et al., 1997; Willson et al., 2000). Several PPAR-γ activators including NSAIDs, drugs of the thiazolidinedione class, and the natural ligand prostaglandin J2 (15d-PGJ2) have been shown to be able to inhibit the β-amyloid-stimulated secretion of pro-inflammatory products by microglia and monocytes responsible for neurotoxicity and astrocyte activation (Combs et al., 2000). Furthermore 15d-PGJ(2) caused microglial death, which terminates brain inflammation (Yang et al., 2006).
Interestingly, anti-TNFα treatment reduced Aβ and Tau phosphorylation in transgenic mice. Treatment with the antibody against TNF-α Infliximab increased the number of CD11c-positive dendritic-like cells and the expression of CD11c. These data suggested that the CD11c-positive dendritic-like cells might contribute to the Infliximab-induced reduction of AD-like pathology (Shi et al., 2011).
Therapeutic vaccination with Aβ antibodies in mice evidenced the Fc-mediated uptake and clearance of Aβ antibody complexes by local activated microglia (Bard et al., 2000; Weiner and Selkoe, 2002). Therefore, it was proposed that microglial activation by active immunization might be a valid mechanism for clearance of senile plaques (Gelinas et al., 2004).
Endogenous molecule for the control of microglia detrimental action
The major breakthrough in the therapy of neurodegenerative disease would be controlling the switch between the beneficial versus the detrimental microglia phenotype in order to control inflammation.
Because Aβ stimulates microglia phagocytosis with consequent release of toxic factors, many studies have reported possible mechanism of action implicating receptors on microglia surface. In this regard, therapeutic agents that are able to disrupt the interaction between Aβ42 and FPR may prove beneficial in the treatment of AD.
We have recently published data providing strong indication for a protective role of a protein called annexin A1 (ANXA1), a glucocorticoid anti-inflammatory mediator in the peripheral system (Perretti and D’Acquisto, 2009). ANXA1 plays a key role in ensuring the effective and selective removal of apoptotic neuron-like cells under inflammatory and non-inflammatory conditions which is a ligand for FPRL-1 receptor (McArthur et al., 2010).
Our studies have shown that ANXA1 is upregulated in human microglia in AD, supporting a possible role for the protein in regulating the microglial response to amyloid plaques and inflammatory response in neurodegeneration. This view is strongly supported by further findings in which recombinant ANXA1 administration in vitro suppress microglial activation following an inflammatory challenge (McArthur et al., 2010).
The identification of microglial FPRL-1 as a receptor for ANXA1, together with our identification of strong expression of ANXA1 in neuritic plaque-associated microglia in AD, suggests a fascinating connection with published data indicating a link between Aβ and FPRL-1 (Heurtaux et al., 2010). Microglia have clearly been shown to phagocytose Aβ through this receptor, but they appear unable to digest this protein, leading to persistent internalization of Aβ/FPRL-1 complexes and culminating in intracellular fibril formation and apoptosis (Pan et al., 2011). The binding of ANXA1 to FPRL-1 in microglia may thus be able to disrupt the interaction of the receptor with Aβ, potentially being of significant benefit in the treatment of AD (Figure 3).
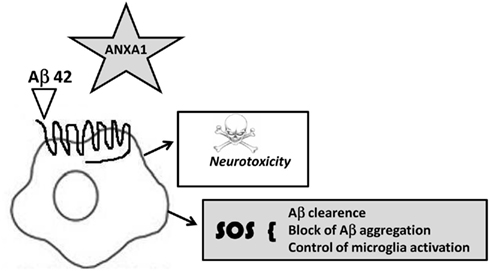
Figure 3. Hypothetical annexin A1- FPRLI mechanism of action in microglia. Annexin A1 binds to FPRL-1 triggering an SOS signaling limiting microglia response to Aβ and preventing a constant uptake and apoptosis, altering microglia pro-inflammatory phenotype.
Conclusion
Microglia effects on AD seem to have double component. On one hand their activation seems to be neuroprotective at early stages of the disease but at older ages and in severely ill patients the effects could be counterproductive. There is therefore the need to investigate the changes in phenotype of resident microglia and how they react to anti-inflammatory therapy over age. In addition, it would be relevant to determine the role of monocyte-derived macrophages along different stages of the disease. One could speculate that the use of BMDM, which are more effective at degrading Aβ could compensate the defective functions of senescent resident microglia (Davoust et al., 2008).
In the future, the combined use of amyloid and microglial imaging should allow one to determine how different anti-amyloid strategies exert their therapeutic effects. Follow-up and further imaging of patients at risk of developing AD with radiotracers that bind to the TSPO/peripheral benzodiazepine receptor will enable a better understanding of both the pattern and the time course of microglial activation and will also provide the opportunity to monitor the pathophysiological effects of anti-inflammatory agents in these patients in vivo.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Prof. Glenda Gillies (Imperial College London) for critical reading of the manuscript and the Alzheimer’s Research UK for their support. We also thank The Wellcome Trust.
References
Akashi-Takamura, S., and Miyake, K. (2006). Toll-like receptors (TLRs) and immune disorders. J. Infect. Chemother. 12, 233–240.
Alafuzoff, I., Overmyer, M., Helisalmi, S., and Soininen, H. (2000). Lower counts of astroglia and activated microglia in patients with Alzheimer’s disease with regular use of nonsteroidal anti-inflammatory drugs. J. Alzheimers Dis. 2, 37–46.
Arancio, O., Zhang, H. P., Chen, X., Lin, C., Trinchese, F., Puzzo, D., Liu, S., Hegde, A., Yan, S. F., Stern, A., Luddy, J. S., Lue, L. F., Walker, D. G., Roher, A., Buttini, M., Mucke, L., Li, W., Schmidt, A. M., Kindy, M., Hyslop, P. A., Stern, D. M., and Du Yan, S. S. (2004). RAGE potentiates Abeta-induced perturbation of neuronal function in transgenic mice. EMBO J. 23, 4096–4105.
Arnaud, L., Robakis, N. K., and Figueiredo-Pereira, M. E. (2006). It may take inflammation, phosphorylation and ubiquitination to ‘tangle’ in Alzheimer’s disease. Neurodegener. Dis. 3, 313–319.
Bach, J. H., Chae, H. S., Rah, J. C., Lee, M. W., Park, C. H., Choi, S. H., Choi, J. K., Lee, S. H., Kim, Y. S., Kim, K. Y., Lee, W. B., Suh, Y. H., and Kim, S. S. (2001). C-terminal fragment of amyloid precursor protein induces astrocytosis. J. Neurochem. 78, 109–120.
Bard, F., Cannon, C., Barbour, R., Burke, R. L., Games, D., Grajeda, H., Guido, T., Hu, K., Huang, J., Johnson-Wood, K., Khan, K., Kholodenko, D., Lee, M., Lieberburg, I., Motter, R., Nguyen, M., Soriano, F., Vasquez, N., Weiss, K., Welch, B., Seubert, P., Schenk, D., and Yednock, T. (2000). Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat. Med. 6, 916–919.
Barger, S. W., and Harmon, A. D. (1997). Microglial activation by Alzheimer amyloid precursor protein and modulation by apolipoprotein E. Nature 388, 878–881.
Beglopoulos, V., Sun, X., Saura, C. A., Lemere, C. A., Kim, R. D., and Shen, J. (2004). Reduced b-amyloid production and increased inflammatory responses in presenilin conditional knock-out mice. J. Biol. Chem. 279, 46907–46914.
Benzing, W. C., Wujek, J. R., Ward, E. K., Shaffer, D., Ashe, K. H., Younkin, S. G., and Brunden, K. R. (2000). Evidence for glial-mediated inflammation in aged APPSw transgenic mice. Neurobiol. Aging 16, 523–530.
Bierhaus, A., Humpert, P. M., Morcos, M., Wendt, T., Chavakis, T., Arnold, B., Stern, D. M., and Nawroth, P. P. (2005). Understanding RAGE, the receptor for advanced glycation end products. J. Mol. Med. (Berl.) 83, 876–886.
Bonifati, D. M., and Kishore, U. (2007). Role of complement in neurodegeneration and neuroinflammation. Mol. Immunol. 44, 999–1010.
Cagnin, A., Brooks, D. J., Kennedy, A. M., Gunn, R. N., Myers, R., Turkheimer, F. E., Jones, T., and Banati, R. B. (2001). In-vivo measurement of activated microglia in dementia. Lancet 358, 461–467.
Cameron, B., and Landreth, G. E. (2010). Inflammation, microglia, and Alzheimer’s disease. Neurobiol. Dis. 37, 503–509.
Capurso, C., Solfrizzi, V., D’Introno, A., Colacicco, A. M., Capurso, S. A;., Capurso, A., and Panza, F. (2004). Interleukin 6-174 G/C promoter gene polymorphism and sporadic Alzheimer’s disease: geographic allele and genotype variations in Europe. Exp. Gerontol. 39, 1567–1573.
Carty, M., and Bowie, A. G. (2011). Evaluating the role of Toll-like receptors in diseases of the central nervous system. Biochem. Pharmacol. 81, 825–837.
Chan, W. Y., Kohsaka, S., and Rezaie, P. (2007). The origin, and cell lineage of microglia: new concepts. Brain Res. Rev. 53, 344–354.
Chavakis, T., Bierhaus, A., Al-Fakhri, N., Schneider, D., Witte, S., Linn, T., Nagashima, M., Morser, J., Arnold, B., Preissner, K. T., and Nawroth, P. P. (2003). The pattern recognition receptor (RAGE) is a counterreceptor for leukocyte integrins: a novel pathway for inflammatory cell recruitment. J. Exp. Med. 198, 1507–1515.
Colton, C. A. (2009). Heterogeneity of microglial activation in the innate immune response in the brain. J. Neuroimmune Pharmacol. 4, 399–418.
Combs, C. K., Johnson, D. E., Karlo, J. C., Cannady, S. B., and Landreth, G. E. (2000). Inflammatory mechanisms in Alzheimer’s disease: inhibition of b-amyloid stimulated proinflammatory responses and neurotoxicity by PPARg agonists. J. Neurosci. 20, 558–567.
Combs, C. K., Karlo, J. C., Kao, S. C., and Landreth, G. E. (2001). b-Amyloid stimulation of microglia and monocytes results in TNFa-dependent expression of inducible nitric oxide synthase and neuronal apoptosis. J. Neurosci. 21, 1179–1188.
Coraci, I. S., Husemann, J., Berman, J. W., Hulette, C., Dufour, J. H., Campanella, G. K., Luster, A. D., Silverstein, S. C., and El-Khoury, J. B. (2002). CD36, a class B scavenger receptor, is expressed on microglia in Alzheimer’s disease brains and can mediate production of reactive oxygen species in response to beta-amyloid fibrils. Am. J. Pathol. 160, 101–112.
Cui, Y., Le, Y., Yazawa, H., Gong, W., and Wang, J. M. (2002). Potential role of the formyl peptide receptor-like 1 (FPRL1) in inflammatory aspects of Alzheimer’s disease. J. Leukoc. Biol. 72, 628–635.
Davoust, N., Vuaillat, C., Androdias, G., and Nataf, S. (2008). From bone marrow to microglia: barriers and avenues. Trends Immunol. 29, 227–234.
DeGiorgio, L. A., Shimizu, Y., Chun, H. S., Kim, Y. S., Sugama, S., Son, J. H., Joh, T. H., and Volpe, B. T. (2002). Amyloid precursor protein gene disruption attenuates degeneration of substantia nigra compacta neurons following axotomy. Brain Res. 938, 38–44.
El Khoury, J., Hickman, S. E., Thomas, C. A., Cao, L., Silverstein, S. C., and Loike, J. D. (1996). Scavenger receptor-mediated adhesion of microglia to beta-amyloid fibrils. Nature 382, 716–719.
Falsig, J., van Beek, J., Hermann, C., and Leist, M. (2008). Molecular basis for detection of invading pathogens in the brain. J. Neurosci. Res. 86, 1434–1447.
Fang, F., Lue, L. F., Yan, S., Xu, H., Luddy, J. S., Chen, D., Walker, D. G., Stern, D. M., Yan, S., Schmidt, A. M., Chen, J. X., and Yan, S. S. (2010). RAGE-dependent signaling in microglia contributes to neuroinflammation, Abeta accumulation, and impaired learning/memory in a mouse model of Alzheimer’s disease. FASEB J. 24, 1043–1055.
Farfara, D., Trudler, D., Segev-Amzaleg, N., Galron, R., Stein, R., and Frenkel, D. (2011). γ-Secretase component presenilin is important for microglia β-amyloid clearance. Ann. Neurol. 69, 170–180.
Farina, C., Aloisi, F., and Meinl, E. (2007). Astrocytes are active players in cerebral innate immunity. Trends Immunol. 28, 138–145.
Fetler, L., and Amigorena, S. (2005). Neuroscience. Brain under surveillance: the microglia patrol. Science 309, 392–393.
Frautschy, S. A., Yang, F., Irrizarry, M., Hyman, B., Saido, T. C., Hsiao, K., and Cole, G. M. (1998). Microglial response to amyloid plaques in APPsw transgenic mice. Am. J. Pathol. 152, 307–317.
Fuhrmann, M., Bittner, T., Jung, C. K., Burgold, S., Page, R. M., Mitteregger, G., Haass, C., Laferla, F. M., Kretzschmar, H., and Herms, J. (2010). Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer’s disease. Nat. Neurosci. 13, 411–413.
Gao, H. M., and Hong, J. S. (2008). Why neurodegenerative diseases are progressive: uncontrolled inflammation drives disease progression. Trends Immunol. 29, 357–365.
Gao, H. M., Kotzbauer, P. T., Uryu, K., Leight, S., Trojanowski, J. Q., and Lee, V. M. (2008). Neuroinflammation and oxidation/nitration of alpha-synuclein linked to dopaminergic neurodegeneration. J. Neurosci. 28, 7687–7698.
Gelinas, D. S., DaSilva, K., Fenili, D., St George-Hyslop, P., and McLaurin, J. (2004). Immunotherapy for Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 101(Suppl. 2), 14657–14662.
Godbout, J. P., Chen, J., Abraham, J., Richwine, A. F., Berg, B. M., Kelley, K. W., and Johnson, R. W. (2005). Exaggerated neuroinflammation and sickness behavior in aged mice following activation of the peripheral innate immune system. FASEB J. 19, 1329–1331.
Grathwohl, S. A., Kälin, R. E., Bolmont, T., Prokop, S., Winkelmann, G., Kaeser, S. A., Odenthal, J., Radde, R., Eldh, T., Gandy, S., Aguzzi, A., Staufenbiel, M., Mathews, P. M., Wolburg, H., Heppner, F. L., and Jucker, M. (2009). Formation and maintenance of Alzheimer’s disease beta-amyloid plaques in the absence of microglia. Nat. Neurosci. 12, 1361–1363.
Griffin, W. S., Stanley, L. C., Ling, C., White, L., MacLeod, V., Perrot, L. J., White, C. L. III, and Araoz, C. (1989). Brain interleukin 1 and S-100 immunoreactivity are elevated in down syndrome and Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 86, 7611–7615.
Hafer-Macko, C. E., Dyck, P. J., and Koski, C. L. (2000). Complement activation in acquired and hereditary amyloid neuropathy. J. Peripher. Nerv. Syst. 5, 131–139.
Heneka, M. T., Sastre, M., Dumitrescu-Ozimek, L., Dewachter, I., Walter, J., Klockgether, T., and Van Leuven, F. (2005a). Focal glial activation coincides with increased BACE1 activation and precedes amyloid plaque deposition in APP(V717I) transgenic mice. J. Neuroinflammation 2, 22.
Heneka, M. T., Sastre, M., Dumitrescu-Ozimek, L., Kreutz, A., Dewachter, I., Kuiperi, C., Klockgether, T., Van Leuven, F., and Landreth, G. (2005b). The PPARγ agonist pioglitazone reduces inflammation and Aβ1-42 levels in APP V717I transgenic mice. Brain 128, 1442–1453.
Heurtaux, T., Michelucci, A., Losciuto, S., Gallotti, C., Felten, P., Dorban, G., Grandbarbe, L., Morga, E., and Heuschling, P. (2010). Microglial activation depends on beta-amyloid conformation: role of the formylpeptide receptor 2. J. Neurochem. 114, 576–586.
Hickman, S. E., Allison, E. K., and El Khoury, J. (2008). Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J. Neurosci. 28, 8354–8360.
Husemann, J., Loike, J. D., Anankov, R., Febbraio, M., and Silverstein, S. C. (2002). Scavenger receptors in neurobiology and neuropathology: their role on microglia and other cells of the nervous system. Glia 40, 195–205.
Iribarren, P., Zhou, Y., Hu, J., Le, Y., and Wang, J. M. (2005). Role of formyl peptide receptor-like 1 (FPRL1/FPR2) in mononuclear phagocyte responses in Alzheimer disease. Immunol. Res. 31, 165–176.
Jaworski, T., Lechat, B., Demedts, D., Gielis, L., Devijver, H., Borghgraef, P., Duimel, H., Verheyen, F., Kügler, S., and Van Leuven, F. (2011). Dendritic degeneration, neurovascular defects, and inflammation precede neuronal loss in a mouse model for tau-mediated neurodegeneration. Am. J. Pathol. 179, 2001–2015.
Jimenez, S., Baglietto-Vargas, D., Caballero, C., Moreno-Gonzalez, I., Torres, M., Sanchez-Varo, R., Ruano, D., Vizuete, M., Gutierrez, A., and Vitorica, J. (2008). Inflammatory response in the hippocampus of PS1M146L/APP751SL mouse model of Alzheimer’s disease: age-dependent switch in the microglial phenotype from alternative to classic. J. Neurosci. 28, 11650–11661.
Kamboh, M. I., Sanghera, D. K., Ferrell, R. E., and DeKosky, S. T. (1995). APOE*4-associated Alzheimer’s disease risk is modified by alpha 1-antichymotrypsin polymorphism. Nat. Genet. 10, 486–488.
Keene, C. D., Cudaback, E., Li, X., Montine, K. S., and Montine, T. J. (2011). Apolipoprotein E isoforms and regulation of the innate immune response in brain of patients with Alzheimer’s disease. Curr. Opin. Neurobiol. 21, 920–928.
Kofler, J., and Wiley, C. A. (2011). Microglia: key innate immune cells of the brain. Toxicol. Pathol. 39, 103–114.
Kreutzberg, G. W. (1996). Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 19, 312–318.
Le, Y., Gong, W., Tiffany, H. L., Tumanov, A., Nedospasov, S., Shen, W., Dunlop, N. M., Gao, J. L., Murphy, P. M., Oppenheim, J. J., and Wang, J. M. (2001). Amyloid (beta)42 activates a G-protein-coupled chemoattractant receptor, FPR-like-1. J. Neurosci. 21, RC123.
Le, Y., Murphy, P. M., and Wang, J. M. (2002). Formyl-peptide receptors revisited. Trends Immunol. 23, 541–548.
Lehmann, J. M., Lenhard, J. M., Oliver, B. B., Ringold, G. M., and Kliewer, S. A. (1997). Peroxisome proliferator-activated receptors α and γ are activated by indomethacin and other non-steroidal anti-inflammatory drugs. J. Biol. Chem. 272, 3406–3410.
Lehnardt, S. (2010). Innate immunity and neuroinflammation in the CNS: the role of microglia in Toll-like receptor-mediated neuronal injury. Glia 58, 253–263.
Lehnardt, S., Massillon, L., Follett, P., Jensen, F. E., Ratan, R., Rosenberg, P. A., Volpe, J. J., and Vartanian, T. (2003). Activation of innate immunity in the CNS triggers neurodegeneration through a Toll-like receptor 4-dependent pathway. Proc. Natl. Acad. Sci. U.S.A. 100, 8514–8519.
Lim, G. P., Yang, F., Chu, T., Chen, P., Beech, W., Teter, B., Tran, T., Ubeda, O., Ashe, K. H., Frautschy, S. A., and Cole, G. M. (2000). Ibuprofen suppresses plaque pathology and inflammation in a mouse model for Alzheimer’s disease. J. Neurosci. 20, 5709–5714.
Liu, B., and Hong, J.-S. (2003). Role of microglia in inflammation-mediated neurodegenerative diseases: mechanisms and strategies for therapeutic intervention. J. Pharmacol. Exp. Ther. 304, 1–7.
Lleo, A., Galea, E., and Sastre, M. (2007). Molecular targets of non-steroidal anti-inflammatory drugs in neurodegenerative diseases. Cell. Mol. Life Sci. 64, 1403–1418.
Lue, L. F., Kuo, Y. M., Beach, T., and Walker, D. G. (2010). Microglia activation and anti-inflammatory regulation in Alzheimer’s disease. Mol. Neurobiol. 41, 115–128.
Lue, L. F., Walker, D. G., Brachova, L., Beach, T. G., Rogers, J., Schmidt, A. M., Stern, D. M., and Yan, S. D. (2001). Involvement of microglial receptor for advanced glycation endproducts (RAGE) in Alzheimer’s disease: identification of a cellular activation mechanism. Exp. Neurol. 171, 29–45.
Mackenzie, I. R., and Muñoz, D. G. (1998). Nonsteroidal anti-inflammatory drug use and Alzheimer-type pathology in aging. Neurology 50, 986–990.
Maeda, J., Zhang, M. R., Okauchi, T., Ji, B., Ono, M., Hattori, S., Kumata, K., Iwata, N., Saido, T. C., Trojanowski, J. Q., Lee, V. M., Staufenbiel, M., Tomiyama, T., Mori, H., Fukumura, T., Suhara, T., and Higuchi, M. (2011). In vivo positron emission tomographic imaging of glial responses to amyloid-beta and tau pathologies in mouse models of Alzheimer’s disease and related disorders. J. Neurosci. 31, 4720–4730.
Mandrekar, S., Jiang, Q., Lee, C. Y., Koenigsknecht-Talboo, J., Holtzman, D. M., and Landreth, G. E. (2009). Microglia mediate the clearance of soluble Abeta through fluid phase macropinocytosis. J. Neurosci. 29, 4252–4262.
Maragakis, N. J., and Rothstein, J. D. (2006). Mechanisms of disease: astrocytes in neurodegenerative disease. Nat. Clin. Pract. Neurol. 2, 679–689.
Matsuoka, Y., Picciano, M., Malester, B., LaFrancois, J., Zehr, C., Daeschner, J. M., Olschowka, J. A., Fonseca, M. I., O’Banion, M. K., Tenner, A. J., Lemer, C. A., and Duff, K. (2001). Inflammatory responses to amyloidosis in a transgenic mouse model of Alzheimer’s disease. Am. J. Pathol. 158, 1345–1354.
McArthur, S., Cristante, E., Paterno, M., Christian, H., Roncaroli, F., Gillies, G. E., and Solito, E. (2010). Annexin A1: a central player in the anti-inflammatory and neuroprotective role of microglia. J. Immunol. 185, 6317–6328.
McColl, B. W., Rothwell, N. J., and Allan, S. M. (2007). Systemic inflammatory stimulus potentiates the acute phase and CXC chemokine responses to experimental stroke and exacerbates brain damage via interleukin-1- and neutrophil-dependent mechanisms. J. Neurosci. 27, 4403–4412.
McCusker, S. M., Curran, M. D., Dynan, K. B., McCullagh, C. D., Urquhart, D. D., Middleton, D., Patterson, C. C., McIlroy, S. P., and Passmore, A. P. (2001). Association between polymorphism in regulatory region of gene encoding tumour necrosis factor alpha and risk of Alzheimer’s disease and vascular dementia: a case-control study. Lancet 357, 436–439.
McGeer, P. L., and McGeer, E. G. (1998). Mechanisms of cell death in Alzheimer disease – immunopathology. J. Neural Transm. Suppl. 54, 159–166.
Miyake, K. (2007). Innate immune sensing of pathogens and danger signals by cell surface toll-like receptors. Semin. Immunol. 19, 3–10.
Mrak, R. E., and Griffin, W. S. T. (2005). Glia and their cytokines in progression of neurodegeneration. Neurobiol. Aging 26, 349–354.
Napoli, I., and Neumann, H. (2009). Microglial clearance function in health and disease. Neuroscience 158, 1030–1038.
Neher, J. J., Neniskyte, U., Zhao, J. W., Bal-Price, A., Tolkovsky, A. M., and Brown, G. C. (2011). Inhibition of microglial phagocytosis is sufficient to prevent inflammatory neuronal death. J. Immunol. 186, 4973–4983.
Neumann, H., Kotter, M. R., and Franklin, R. J. (2009). Debris clearance by microglia: an essential link between degeneration and regeneration. Brain 132, 288–295.
Nguyen, M. D., D’Aigle, T., Gowing, G., Julien, J. P., and Rivest, S. (2004). Exacerbation of motor neuron disease by chronic stimulation of innate immunity in a mouse model of amyotrophic lateral sclerosis. J. Neurosci. 24, 1340–1349.
Nicoll, J. A., Mrak, R. E., Graham, D. I., Stewart, J., Wilcock, G., MacGowan, S., Esiri, M. M., Murray, L. S., Dewar, D., Love, S., Moss, T., and Griffin, W. S. (2000). Association of interleukin-1 gene polymorphisms with Alzheimer’s disease. Ann. Neurol. 47, 365–368.
Nimmerjahn, A., Kirchhoff, F., and Helmchen, F. (2005). Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308, 1314–1318.
Njie, E. G., Boelen, E., Stassen, F. R., Steinbusch, H. W., Borchelt, D. R., and Streit, W. J. (2012). Ex vivo cultures of microglia from young and aged rodent brain reveal age-related changes in microglial function. Neurobiol. Aging 33, 195.e1–195.e12.
Okello, A., Edison, P., Archer, H. A., Turkheimer, F. E., Kennedy, J., Bullock, R., Walker, Z., Kennedy, A., Fox, N., Rossor, M., and Brooks, D. J. (2009). Microglial activation and amyloid deposition in mild cognitive impairment: a PET study. Neurology 72, 56–62.
Ouchi, Y., Yoshikawa, E., Sekine, Y., Futatsubashi, M., Kanno, T., Ogusu, T., and Torizuka, T. (2005). Microglial activation and dopamine terminal loss in early Parkinson’s disease. Ann. Neurol. 57, 168–175.
Pan, X. D., Zhu, Y. G., Lin, N., Zhang, J., Ye, Q. Y., Huang, H. P., and Chen, X. C. (2011). Microglial phagocytosis induced by fibrillar beta-amyloid is attenuated by oligomeric beta-amyloid: implications for Alzheimer’s disease. Mol. Neurodegener. 6, 45.
Pancer, Z., and Cooper, M. D. (2006). The evolution of adaptive immunity. Annu. Rev. Immunol. 24, 497–518.
Paresce, D. M., Ghosh, R. N., and Maxfield, F. R. (1996). Microglial cells internalize aggregates of the Alzheimer’s disease amyloid beta-protein via a scavenger receptor. Neuron 17, 553–565.
Permanne, B., Adessi, C., Saborio, G. P., Fraga, S., Frossard, M. J., Van Dorpe, J., Dewachter, I., Banks, W. A., Van Leuven, F., and Soto, C. (2002). Reduction of amyloid load and cerebral damage in a transgenic mouse model of Alzheimer’s disease by treatment with a b-sheet breaker peptide. FASEB J. 16, 860–862.
Perretti, M., and D’Acquisto, F. (2009). Annexin A1 and glucocorticoids as effectors of the resolution of inflammation. Nat. Rev. Immunol. 9, 62–70.
Perry, R. T., Collins, J. S., Wiener, H., Acton, R., and Go, R. C. P. (2001). The role of TNF and its receptors in Alzheimer’s disease. Neurobiol. Aging 22, 873–883.
Perry, V. H. (2004). The influence of systemic inflammation on inflammation in the brain: implications for chronic neurodegenerative disease. Brain Behav. Immun. 18, 407–413.
Perry, V. H., and Andersson, P. B. (1992). The inflammatory response in the CNS. Neuropathol. Appl. Neurobiol. 18, 454–459.
Perry, V. H., Anthony, D. C., Bell, M. D., Lawson, L. J., Reid, D. M., and Gordon, S. (1996). Microglia activation and inflammation in the CNS. J. Neurochem. 66, S70.
Perry, V. H., Cunningham, C., and Holmes, C. (2007). Systemic infections and inflammation affect chronic neurodegeneration. Nat. Rev. Immunol. 7, 161–167.
Perry, V. H., Matyszak, M. K., and Fearn, S. (1993). Altered antigen expression of microglia in the aged rodent CNS. Glia 7, 60–67.
Perry, V. H., Nicoll, J. A., and Holmes, C. (2010). Microglia in neurodegenerative disease. Nat. Rev. Neurol. 6, 193–201.
Polazzi, E., and Monti, B. (2010). Microglia and neuroprotection: from in vitro studies to therapeutic applications. Prog. Neurobiol. 92, 293–315.
Qiu, W. Q., Walsh, D. M., Ye, Z., Vekrellis, K., Zhang, J., Podlisny, M. B., Rosner, M. R., Safavi, A., Hersh, L. B., and Selkoe, D. J. (1998). Insulin-degrading enzyme regulates extracellular levels of amyloid b-protein by degradation. J. Biol. Chem. 273, 32730–32738.
Qiu, W. Q., Ye, Z., Kholodenko, D., Seubert, P., and Selkoe, D. J. (1997). Degradation of amyloid b-protein by a metalloprotease secreted by microglia and other neural and non-neural cells. J. Biol. Chem. 272, 6641–6646.
Ransohoff, R. M., and Perry, V. H. (2009). Microglial physiology: unique stimuli, specialized responses. Annu. Rev. Immunol. 27, 119–145.
Reed-Geaghan, E. G., Savage, J. C., Hise, A. G., and Landreth, G. E. (2009). CD14 and toll-like receptors 2 and 4 are required for fibrillar A{beta}-stimulated microglial activation. J. Neurosci. 29, 11982–11992.
Richard, K. L., Filali, M., Préfontaine, P., and Rivest, S. (2008). Toll-like receptor 2 acts as a natural innate immune receptor to clear amyloid beta 1-42 and delay the cognitive decline in a mouse model of Alzheimer’s disease. J. Neurosci. 28, 5784–5793.
Rivest, S. (2003). Molecular insights on the cerebral innate immune system. Brain Behav. Immun. 17, 13–19.
Rivest, S. (2009). Regulation of innate immune responses in the brain. Nat. Rev. Immunol. 9, 429–439.
Roberts, J. C., Friel, S. L., Roman, S., Perren, M., Harper, A., Davis, J. B., Richardson, J. C., Virley, D., and Medhurst, A. D. (2009). Autoradiographical imaging of PPARgamma agonist effects on PBR/TSPO binding in TASTPM mice. Exp. Neurol. 216, 459–470.
Rogers, J., Mastroeni, D., Leonard, B., Joyce, J., and Grover, A. (2007). Neuroinflammation in Alzheimer’s disease and Parkinson’s disease: are microglia pathogenic in either disorder? Int. Rev. Neurobiol. 82, 235–246.
Salminen, A., Ojala, J., Kauppinen, A., Kaarniranta, K., and Suuronen, T. (2009). Inflammation in Alzheimer’s disease: amyloid-beta oligomers trigger innate immunity defence via pattern recognition receptors. Prog. Neurobiol. 87, 181–194.
Sastre, M., and Gentleman, S. M. (2010). NSAIDs: how they work and their prospects as therapeutics in Alzheimer’s disease. Front. Aging Neurosci. 2:20. doi:10.3389/fnagi.2010.00020
Sastre, M., Klockgether, T., and Heneka, M. T. (2006a). Contribution of inflammatory processes to Alzheimer’s disease: molecular mechanisms. Int. J. Dev. Neurosci. 24, 167–176.
Sastre, M., Roßner, S., Bogdanovic, N., Rosen, E., Dewachter, I., Thal, D., Evert, B., Klockgether, T., van Leuven, F., and Heneka, M. T. (2006b). Non-steroidal anti-inflammatory drugs repress beta-secretase gene promoter activity by the activation of PPARgamma. Proc. Natl. Acad. Sci. U.S.A. 103, 443–448.
Sastre, M., Richardson, J., Gentleman, S. M., and Brooks, D. (2011). Inflammatory risk factors and pathologies associated with Alzheimer’s disease. Curr. Alzheimer Res. 8, 132–141.
Sastre, M., Walter, J., and Gentleman, S. M. (2008). Interactions between APP secretases and inflammatory mediators. J. Neuroinflammation 5, 25.
Schmidt, A. M., Yan, S. D., Yan, S. F., and Stern, D. M. (2001). The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J. Clin. Invest. 108, 949–955.
Schwartz, M., and Shechter, R. (2010). Systemic inflammatory cells fight off neurodegenerative disease. Nat. Rev. Neurol. 6, 405–410.
Shen, Y., and Meri, S. (2003). Yin and Yang: complement activation and regulation in Alzheimer’s disease. Prog. Neurobiol. 70, 463–472.
Shi, J. Q., Shen, W., Chen, J., Wang, B. R., Zhong, L. L., Zhu, Y. W., Zhu, H. Q., Zhang, Q. Q., Zhang, Y. D., and Xu, J. (2011). Anti-TNF-α reduces amyloid plaques and tau phosphorylation and induces CD11c-positive dendritic-like cell in the APP/PS1 transgenic mouse brains. Brain Res. 1368, 239–247.
Sierra, A., Gottfried-Blackmore, A. C., McEwen, B. S., and Bulloch, K. (2007). Microglia derived from aging mice exhibit an altered inflammatory profile. Glia 55, 412–424.
Solito, E., McArthur, S., Christian, H., Gavins, F., Buckingham, J. C., and Gillies, G. E. (2008). Annexin A1 in the brain – undiscovered roles? Trends Pharmacol. Sci. 29, 135–142.
Streit, W., Braak, H., Xue, Q. S., and Bechmann, I. (2009). Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer’s disease. Acta Neuropathol. 118, 475–485.
Streit, W. J., Miller, K. R., Lopes, K. O., and Njie, E. (2008). Microglial degeneration in the aging brain – bad news for neurons? Front. Biosci. 13, 3423–3438.
Streit, W. J., Sammons, N. W., Kuhns, A. J., and Sparks, D. L. (2004). Dystrophic microglia in the aging human brain. Glia 45, 208–212.
Sy, M., Kitazawa, M., Medeiros, R., Whitman, L., Cheng, D., Lane, T. E., and Laferla, F. M. (2011). Inflammation induced by infection potentiates tau pathological features in transgenic mice. Am. J. Pathol. 178, 2811–2822.
Tahara, K., Kim, H. D., Jin, J. J., Maxwell, J. A., Li, L., and Fukuchi, K. (2006). Role of toll-like receptor signalling in Abeta uptake and clearance. Brain 129, 3006–3019.
Walter, S., Letiembre, M., Liu, Y., Heine, H., Penke, B., Hao, W., Bode, B., Manietta, N., Walter, J., Schulz-Schuffer, W., and Fassbender, K. (2007). Role of the toll-like receptor 4 in neuroinflammation in Alzheimer’s disease. Cell. Physiol. Biochem. 20, 947–956.
Weiner, H. L., and Selkoe, D. J. (2002). Inflammation and therapeutic vaccination in CNS diseases. Nature 420, 879–884.
Weller, C., Oxlade, N., Dobbs, S. M., Dobbs, R. J., Charlett, A., and Bjarnason, I. T. (2005). Role of inflammation in gastrointestinal tract in aetiology and pathogenesis of idiopathic parkinsonism. FEMS Immunol. Med. Microbiol. 44, 129–135.
Willson, T. M., Brown, P. J., Sternbach, D. D., and Henke, B. R. (2000). The PPARs: from orphan receptors to drug discovery. J. Med. Chem. 43, 527–550.
Wyss-Coray, T. (2006). Inflammation in Alzheimer disease: driving force, bystander or beneficial response? Nat. Med. 12, 1005–1015.
Wyss-Coray, T., Loike, J. D., Brionne, T. C., Lu, E., Anankov, R., Yan, F., Silverstein, S. C., and Husemann, J. (2003). Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nat. Med. 9, 453–457.
Xie, Z., Morgan, T. E., Rozovsky, I., and Finch, C. E. (2003). Aging and glial responses to lipopolysaccharide in vitro: greater induction of IL-1 and IL-6, but smaller induction of neurotoxicity. Exp. Neurol. 182, 135–141.
Yan, S. D., Chen, X., Fu, J., Chen, M., Zhu, H., Roher, A., Slattery, T., Zhao, L., Nagashima, M., Morser, J., Migheli, A., Nawroth, P., Stern, D., and Schmidt, A. M. (1996). RAGE and amyloid-beta peptide neurotoxicity in Alzheimer’s disease. Nature 382, 685–691.
Yang, M. S., Ji, K. A., Jeon, S. B., Jin, B. K., Kim, S. U., Jou, I., and Joe, E. (2006). Interleukin-13 enhances cyclooxygenase-2 expression in activated rat brain microglia: implications for death of activated microglia. J. Immunol. 177, 1323–1329.
Ye, S. M., and Johnson, R. W. (1999). Increased interleukin-6 expression by microglia from brain of aged mice. J. Neuroimmunol. 93, 139–148.
Yong, V. W., and Rivest, S. (2009). Taking advantage of the systemic immune system to cure brain diseases. Neuron 64, 55–60.
Keywords: microglia, amyloid-β, Alzheimer’s disease, inflammation, NSAIDs, annexin A1, immunity
Citation: Solito E and Sastre M (2012) Microglia function in Alzheimer’s disease. Front. Pharmacol. 3:14. doi: 10.3389/fphar.2012.00014
Received: 14 November 2011;
Accepted: 21 January 2012;
Published online: 10 February 2012.
Edited by:
Roger A. Barker, University of Cambridge and Addenbrooke’ Hospital, UKReviewed by:
Emmanuel Planel, Centre Hospitalier de l’Université Laval, CanadaMariela Fermanda Perez, Universidad Nacional de Cordoba, Argentina
Copyright: © 2012 Solito and Sastre. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Magdalena Sastre, Centre for Neuroscience, Division of Experimental Medicine, Imperial College London, Hammersmith Hospital, Du Cane Road, London W12 0NN, UK. e-mail: m.sastre@imperial.ac.uk