 Dora Brites1,2*
Dora Brites1,2*- 1 Neuron Glia Biology in Health and Disease Unit, Research Institute for Medicines and Pharmaceutical Sciences, Faculty of Pharmacy, University of Lisbon, Lisbon, Portugal
- 2 Department of Biochemistry and Human Biology, Faculty of Pharmacy, University of Lisbon, Lisbon, Portugal
Unconjugated hyperbilirubinemia is a common condition in the first week of postnatal life. Although generally harmless, some neonates may develop very high levels of unconjugated bilirubin (UCB), which may surpass the protective mechanisms of the brain in preventing UCB accumulation. In this case, both short-term and long-term neurodevelopmental disabilities, such as acute and chronic UCB encephalopathy, known as kernicterus, or more subtle alterations defined as bilirubin-induced neurological dysfunction (BIND) may be produced. There is a tremendous variability in babies’ vulnerability toward UCB for reasons not yet explained, but preterm birth, sepsis, hypoxia, and hemolytic disease are comprised as risk factors. Therefore, UCB levels and neurological abnormalities are not strictly correlated. Even nowadays, the mechanisms of UCB neurotoxicity are still unclear, as are specific biomarkers, and little is known about lasting sequelae attributable to hyperbilirubinemia. On autopsy, UCB was shown to be within neurons, neuronal processes, and microglia, and to produce loss of neurons, demyelination, and gliosis. In isolated cell cultures, UCB was shown to impair neuronal arborization and to induce the release of pro-inflammatory cytokines from microglia and astrocytes. However, cell dependent sensitivity to UCB toxicity and the role of each nerve cell type remains not fully understood. This review provides a comprehensive insight into cell susceptibilities and molecular targets of UCB in neurons, astrocytes, and oligodendrocytes, and on phenotypic and functional responses of microglia to UCB. Interplay among glia elements and cross-talk with neurons, with a special emphasis in the UCB-induced immunostimulation, and the role of sepsis in BIND pathogenesis are highlighted. New and interesting data on the anti-inflammatory and antioxidant activities of different pharmacological agents are also presented, as novel and promising additional therapeutic approaches to BIND.
Introduction
Jaundice occurs during the first week of life in most infants due to elevated levels of unconjugated bilirubin (UCB) by increased breakdown of fetal erythrocytes, deficient human serum albumin (HSA) transport to the liver, and inefficient conjugation. Although the outcome for the majority is benign, untreated neonates, or newborns with very high UCB levels can develop acute encephalopathy or kernicterus. The most disabling neurological findings in kernicterus are auditory and motor sequelae. At autopsy of kernicteric infants, UCB is macroscopically visible within globus pallidus, hippocampus, lateral ventricular walls, cerebellum, and subthalamic nuclei, reflecting a preferential deposition of UCB in specific brain areas. Unfortunately, no established criteria recognize whether neurological or developmental problems result from hyperbilirubinemia. Nevertheless, patient history, physical examination, and laboratory findings allow the categorization of kernicterus as mild, moderate, or severe. Recently, Shapiro (2010) has suggested the term of subtle kernicterus for the less severe injuries, including minor neurological abnormalities. This milder form of bilirubin induced neurological dysfunction (BIND) in the central nervous system (CNS) needs to be distinguished from other known causes of neurodevelopmental disabilities. Further studies looking at potential alterations produced by UCB on neurodevelopmental programs and lasting neurological deficits that may involve vulnerability to aging and neurodegenerative diseases are needed.
To reduce BIND incidence, normograms, and guidelines were proposed (Bhutani et al., 2008) for jaundiced newborn infants of 35 or more weeks of gestation. However, in sick and preterm infants, the absence of precise data on hyperbilirubinemia prevalence and the lack of proven predictive indices has made impossible to establish such guidelines. Therefore, the problem is even more worrying for premature babies and low birth weight infants, mainly when infection/sepsis is associated, but also because they are particularly vulnerable to BIND (Bhutani et al., 2004; Okumura et al., 2009; Moll et al., 2011), probably due to neurodevelopmental maturity differences. In fact, temporal windows of CNS vulnerability to UCB toxicity have been suggested and the neurodevelopmental age at the time of UCB brain injury influences the location of the selective neuropathological damage (Keino and Kashiwamata, 1989; Conlee and Shapiro, 1997). While auditory predominant kernicterus subtype prevails in infants with peak levels at earlier gestational ages of exposure to total serum bilirubin (TSB), motor kernicterus subtype usually develops in infants with more than 34 weeks of gestation (Shapiro, 2010). TSB is the sum of UCB plus the fraction of the pigment that has already been conjugated in the liver. Interestingly, if we look into two different mice models with equivalent TSB levels, results indicate that when hyperbilirubinemia develops in the first week of life, such as in the Ugt1−/− model (Nguyen et al., 2008), all the animals succumb, while if occurring in the second week of life as in the Tg(UGT1A1*28)Ugt1−/− model (Fujiwara et al., 2010), only 10% of the mice dye. These findings suggest an acquired resistance with the postnatal age. Therefore, high levels of UCB during early neurodevelopment may determine injuries that can be diverse in nature, severity, and enduring effects from those produced a week later.
Neonatal jaundice, if prolonged or severe, could have an impact on the infant’s learning and memory, and even moderate levels of UCB have been associated with developmental delay, attention-deficit disorder, autism, and isolated neural hearing loss (Gkoltsiou et al., 2008; Jangaard et al., 2008; Shapiro, 2010). However, the establishment of these casual relationships was never demonstrated with certainty because of the two decades that separate neonatal jaundice from signs of mental disorders at puberty. Moreover, neonatal hyperbilirubinemia, mainly in prematures, could be one of the multifactorial risk factors leading to long-lasting deficits as recently evidenced for moderate systemic inflammation (Favrais et al., 2011). Thus, elucidation of structural–functional relationship might help in understanding the pathological processes and developmental difficulties experienced by jaundiced babies.
In vitro experiments have been elucidating the mechanisms of UCB neurotoxicity (Brites et al., 2009; Brites, 2011), while in vivo studies have almost exclusively focused on homozygous (jj) Gunn rats (Rice and Shapiro, 2008; Daood et al., 2009), the kernicterus classic model. Hyperbilirubinemic Gunn rats have a congenital deficiency of the bilirubin liver conjugating enzyme uridine-diphosphate-glucuronosyltransferase 1A1 (UGT1A1) and present neurological abnormalities similar to kernicterus (Billing, 1972) when acute bilirubin encephalopathy is induced by the administration of phenylhydrazine (Rice and Shapiro, 2008) or sulfadimethoxine (Daood et al., 2009). Indeed the model, although indicated as mimicking the Crigler–Najjar I syndrome due to a defect in the UGT1A1 isoform of the UDPGT gene (Johnson et al., 1959), evidence less severe hyperbilirubinemia, and the natural course of the disease is milder in the Gunn rat than in human Crigler–Najjar I syndrome (Shapiro, 2003). Interestingly, Gunn rats with mild concentrations of UCB at 2 months of age (∼3 mg/dL) have exhibited behavioral alterations and neuropathological changes similar to those found in schizophrenia (Hayashida et al., 2009).
Hence, clarification of the molecular mechanisms of UCB neurotoxicity is an emerging area of research as it may open new perspectives for therapeutic approaches at circumventing potential enduring injurious effects of neonatal brain insults by UCB. The aim of this review is to highlight the most recent and relevant information on the determinants and widespread effects of UCB neurotoxicity, the potential molecular mechanisms involved and the associated neurodevelopmental susceptibilities, as well as the glia immune-inflammatory responses and the most promising neuropharmacological agents based on in vitro experimental models.
Determinants of Brain Bilirubin Entrance and Load
Several factors in the first week of postnatal life may concur to increase the amount of UCB in the circulation, which when surpassing the albumin-binding capacity for UCB lead to an increase of the unbound (free) fraction of UCB (Bf). Although UCB entrance into the brain is prevented by the brain microvascular endothelial cells tightly jointed by the elaborated junctional complexes that form the blood–brain barrier (BBB; Cardoso et al., 2010; Figure 1A), Bf diffuses into the brain (Ostrow et al., 2003b), and its accumulation in the CNS will depend on the total amount of Bf in the systemic circulation, on the efficacy of BBB transporters, and on the presence of acidosis, which increases binding of Bf to the brain parenchyma and the risk of kernicterus (Cashore et al., 1983; Meisel et al., 1988), due to its decreased solubility in acidic aqueous solutions (less than 1 nM at pH = 7 and about 0.1 μM at pH = 8; Ostrow and Celic, 1984).
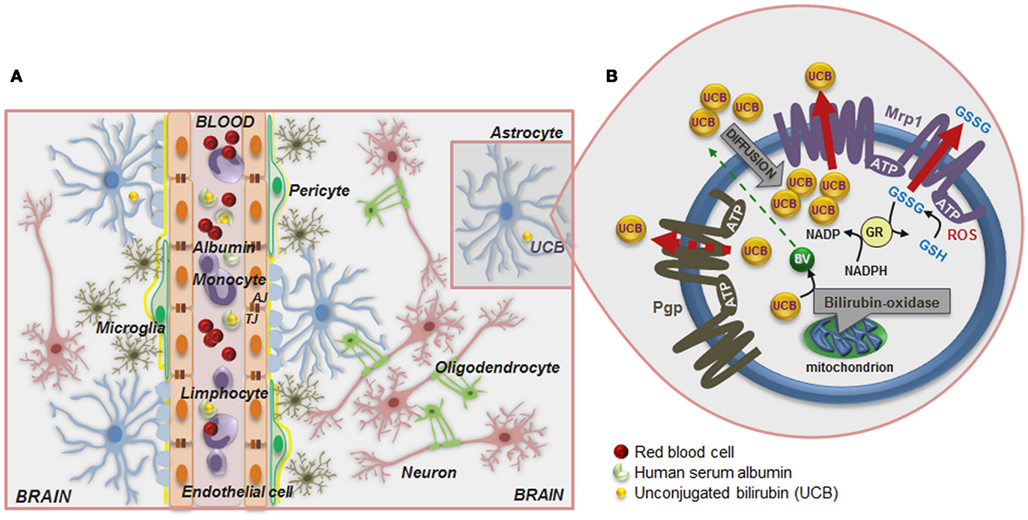
Figure 1. Schematic of the blood–brain barrier (BBB) in a condition of mild hyperbilirubinemia (A) and overview of the mechanisms that prevent the accumulation of unconjugated bilirubin (UCB) in astrocytes (B). (A) The normally functioning BBB maintains the central nervous system homeostasis by separating the brain from the systemic blood circulation and regulating the exchange of various compounds. The BBB is composed by endothelial cells (orange) and their tight junctions (TJ), located more apically, and adherens junctions (AJ), located underneath, as well as by astrocyte end-feet (light blue spots), perivascular microglia (greenish-brown), pericytes (light-green), and the basement membrane (yellow). Integrity of all BBB elements is essential for neuron (pink) function and for oligodendrocytes (solid-green) to exert protection of axon function and promotion of neuronal survival. Circulation of UCB in the blood is provided by its binding to human serum albumin. Thus, decreased levels of albumin, the lower capacity for UCB binding, and the high UCB concentrations favor the entrance of unbound (free) UCB (Bf) fraction by passive or facilitated diffusion into the brain. In a condition of mild hyperbilirubinemia the Bf levels in the plasma rarely surpass 100 nM and in the brain are usually lower than 10 nM; entrance restriction to UCB is afforded by the P-glycoprotein (Pgp) at the BBB, which is an ATP-dependent cellular export protein of UCB. (B) At the brain cells, UCB is a substrate for the efflux pump multidrug resistance-associated protein 1 (Mrp1) and together with Pgp limit the intracellular accumulation of UCB, mainly in astrocytes. Reduced expression of Pgp and Mrp1 in young astrocytes and neurons, as well as in Gunn rats’ pups, probably account to the vulnerability of premature babies to UCB. A high intracellular concentration of glutathione (GSH) protects against reactive oxygen species (ROS). GSH is regenerated from glutathione disulfide (GSSG) in a reaction catalyzed by glutathione reductase (GR) using NADPH as a second substrate. Astrocytes release GSSG under oxidative stress via Mrp1. Another protective mechanism against UCB toxicity is the mitochondrial membrane enzyme bilirubin oxidase which in the presence of molecular oxygen converts UCB to biliverdin (BV) and other products that are much less toxic than UCB. Changes in BBB integrity will lead to UCB entrance into the brain, and failure of cell defensive mechanisms against UCB toxicity will favor brain injury.
ATP-dependent cellular export of UCB by P-glycoprotein (Pgp) and multidrug resistance-associated protein 1 (MRP1/Mrp1), which are highly expressed in BBB and in CSF (Gazzin et al., 2011), respectively, limit Bf passage across the BBB (the first one) and its intracellular accumulation (mainly the last one; Sequeira et al., 2007; Daood et al., 2008; Bellarosa et al., 2009). However, the lower expression of Pgp in early-life (Daood et al., 2008; Gazzin et al., 2011) may be critical in preventing UCB entrance into the CNS. Similarly, Mrp1 down regulation by long exposure to elevated concentrations of UCB, recently observed at the blood–CSF barrier in the Gunn rats, might favor BIND (Gazzin et al., 2011). UCB can even compromise the integrity of BBB lining by disrupting glutathione (GSH) homeostasis and increasing endothelial nitric oxide synthase (NOS) expression followed by nitrite (NO) production and cytokine release (Palmela et al., 2011), thus favoring UCB brain uptake in prolonged hyperbilirubinemia. Formation of NO, originally described as the endothelial-derived relaxing factor (Bellefontaine et al., 2011), requires NOS, in which there are three isoforms: two constitutive (i) neural-type NOS I (nNOS) and (ii) endothelial-type III (eNOS), and one inducible NOS II (iNOS) isoform. Interestingly, upregulation of eNOS expression was more pronounced in the first 4 h incubation and showed to increase with the concentration of UCB (Palmela et al., 2011). The elevated levels of NO generated may increase microvascular permeability (Kim and Jung, 2011), thus favoring passage of UCB into the brain.
Once in the brain, preferential deposition and neurotoxicity of UCB may result from cell specific susceptibilities and CNS regional vulnerabilities, while time of exposure will be critical in extending UCB load and toxicity (Wennberg et al., 2006). Localization of Mrp1 in astrocytes and of Pgp in those associated to BBB may help in restricting UCB injury to cells located at BBB proximity and to maintain BBB properties in mild hyperbilirubinemia (Watchko et al., 2001). Rigato et al. (2004) have demonstrated that Mrp1 mediates ATP-dependent cellular export of UCB by astrocytes (Figure 1B). GSH protects astrocytes from reactive oxygen species (ROS) by directly reacting with radicals and by reducing peroxides generating oxidized glutathione (GSSG). In order to maintain a reduced thiol reduction potential under oxidative stress, Mrp1 mediates GSSG release from astrocytes (Hirrlinger et al., 2001). Mrp1 expression was also shown in neurons (Falcão et al., 2007a). Moreover, it was observed that immature neurons and astrocytes exhibit low levels of the protein as compared with mature cells, in line with what was observed for Pgp, and that inhibition of Mrp1 expression with MK571 increases UCB toxicity to neurons and astrocytes (Falcão et al., 2007a), as well as to human neuroblastoma SH-SY5Y cells when RNA interference technology was used (Corich et al., 2009).
Another important defense mechanism resides on the ability of UCB oxidation by brain mitochondrial membranes (Figure 1B), at a rate of 100–300 pM UCB/mg protein/min (Hansen, 2000a). The enzyme activity showed to be cytochrome c dependent, but it is not influenced either by GSH or GSSG (Hansen et al., 1999). Once it is less expressed in immature brains, as well as in undifferentiated cells, and in neurons than in glia (Hansen and Allen, 1997), it is hypothesized that it could be related to the increased vulnerability of the newborn to UCB, mainly if premature.
Microscopic evaluation of icteric stained brain sections revealed that UCB interacts with neurons, neuronal processes, and glia elements (Martich-Kriss et al., 1995), but the contribution of each cell type to BIND is not clear, mainly when considering the UCB-induced nerve cell pathological alterations as an integrated phenomena, i.e., by using organotypic slice cultures. If neurons are highly specialized for the processing and transmission of cellular signals, glial cells, which include astrocytes, microglia, and oligodendrocytes (OLGs), are essential in the maintenance of the neuronal network, in neuronal migration during development and in the generation of myelin, respectively. Therefore, once such cells are important players in neuronal dysfunction they should be considered as promising therapeutic targets in BIND. Moreover, one must consider glial damage not as a marginal event or a secondary consequence of neuronal injury, but rather as an intrinsic participant and determinant of the UCB-induced neuropathological processes.
Biomarkers and Predictors of Bilirubin Neurotoxicity
Classical and subtypes of kernicterus associated with kernicterus and BIND, respectively, have been related to auditory impairment (Shapiro and Popelka, 2011). Intriguingly, it was observed that from a total of 13 neonates with severe hyperbilirubinemia, six infants had audiologic findings of acute auditory neuropathy spectrum disorder, while only two from the six had clinical signs and symptoms of acute UCB encephalopathy (Saluja et al., 2010). Therefore, abnormal neurological function with auditory brainstem responses (ABRs), also known as brainstem auditory evoked potentials (BAEPs), or responses (BAERs), is considered a very objective and sensitive test for the diagnosis of kernicterus and UCB neurotoxicity, once these auditory regions are too small to be seen on conventional magnetic resonance imaging (MRI) scans (Gupta and Mann, 1998; Shapiro, 2010). Actually, audiological diagnostic work-up in neonatal hyperbilirubinemia above 20 mg/dL, and even for UCB at <20 mg/dL, has been recommended (Sheykholeslami and Kaga, 2000; Nickisch et al., 2009).
Aberrant ABRs have been consistently used as a non-invasive method to identify UCB toxicity in Gunn rats. However, UCB by itself is not able to cause hearing impairment in jaundiced Gunn rats (Levi et al., 1981), unless combined with induced asphyxia (Silver et al., 1995a). Other sensitive markers for the massive entry of UCB into the nervous system include the somatosensory evoked potential (Silver et al., 1996) and visual evoked potential abnormalities (Silver et al., 1995b), both observed in the Gunn rats, in the absence or in the presence of sulfadimethoxine, respectively.
Nevertheless, in what concerns biomarkers to evaluate the risk of BIND, Bf is considered a more sensitive predictor (Ahlfors et al., 2009b; Lee et al., 2009) than either bilirubin–albumin molar ratio (UCB/HSA) or abnormal ABR maturation (Amin et al., 2001) and, in contrast to TSB levels, revealed to be associated with ABRs results (Ahlfors and Parker, 2008). In sum, Bf or TSB plus Bf, instead of TSB alone, are more rational indicators (Wennberg et al., 2006). However, due to the difficulties of Bf assessment in the clinical practice it has been referred the use of TSB/HSA instead of UCB alone (Hulzebos et al., 2008). Additionally, the HSA (Ahlfors et al., 2009a) and the “reserve HSA binding capacity” (McDonagh, 2010) determinations are indicated as complementary, and a Bf of 20 nM proposed as the risk threshold for UCB toxicity in term infants (Wennberg et al., 2006). In fact, while the values of TSB only varied between 116 and 615 μM in infants with birth weight higher or equal to 2.5 kg, greater variations in Bf concentrations (from 0.9 to 130 nM) were encountered, reinforcing the need of individualized jaundice management (Ahlfors et al., 2009b). Finally, the Bf/TSB ratio is even believed to be a best displayer of abnormal BAEPs than Bf alone (Ahlfors et al., 2009a). However, when the challenge resides in relating Bf or TSB with abnormal neurodevelopmental and lasting neurological changes, alterations in ABRs are considered the top clinical approach.
Recently, it was suggested that serum tau and S100B protein levels are equally promising biomarkers, which besides being correlated with TSB, were found to be more elevated in infants with auditory neuropathy, neurologic defects, or electroencephalogram abnormalities (Okumus et al., 2008), and consequently with UCB-induced brain sequelae.
How Does Bilirubin Make Neurons Sick?
Most published studies on the neurotoxicity of UCB have been performed either at too high UCB/HSA molar ratio (>3) or at UCB values in the absence of serum or albumin, Bf, vastly higher (>1 μM) than those seen in jaundiced neonates at risk of kernicterus or BIND (between 100 and 500 nM), and thus are of questionable relevance to the clinical manifestations of neurotoxicity (Ostrow et al., 2003a). In fact, those Bf concentrations in the absence of HSA form microsuspensions, followed by coarser aggregates that precipitate spontaneously, mainly at the level of the cellular membranes. Some data suggest that metastable aggregates begin to occur at concentrations of Bf as low as 1–2 μM and probably not at 500 nM (Mukerjee et al., 2002; Ostrow et al., 2003a). Once bilirubin has been referred as a neuroprotective compound, as well, it deserves to be noted that only Bf concentrations below 100 nM are able to protect CNS against oxidative damage (Doré and Snyder, 1999; Dora Brites lab, unpublished results). Therefore, we have privileged in this section the studies addressing the neurotoxic effects produced by moderately supersaturated Bf levels (between 100 nM and 1 μM) and/or UCB/HSA molar ratios ≤3.
Neurons are more susceptible than astrocytes to UCB-induced demise, probably as a result of lower GSH content (Brito et al., 2008b). Indeed, neurons generally show higher levels of ROS, protein oxidation, and lipid peroxidation upon UCB exposure than astrocytes, despite the different vulnerability of neuronal subpopulations (Conforti et al., 2007). One of the most challenging issues is to define the earliest steps of injury once it can differ on time, severity, and site between one cell and another (Muramatsu et al., 2009). Years ago, Chen et al. (1971) suggested, based in their experimental kernicterus model, that after absorption of UCB from astrocytes at the dendritic level, UCB is transported from the distorted microtubules to the Golgi complex, before the enlarged vesicles coming from the Golgi reach the granular endoplasmic reticulum (ER), which in turn show vesiculation, vacuolation, and dilated cisternae.
Interaction of UCB with the cells occurs first at the cell membrane (Figure 2A), and although UCB rapidly diffuses through membranes (Zucker et al., 1999), it was shown that it causes increased polarity and fluidity at carbon numbers 5 and 7 (Rodrigues et al., 2002c), ultimately perturbing the neuronal cell membrane structure. These features showed to be associated with oxidative injury to membrane lipids and were not sensed in the more hydrophobic regions. Similar effects were observed on membranes of astrocytes (Rodrigues et al., 2002c) and erythrocytes (Brito et al., 2001), as well as on mitochondrial membrane (Rodrigues et al., 2002b). This increased membrane fluidity favors the externalization of phosphatidylserine to the outer leaflet, which allows phagocytic recognition. This finding, also observed in erythrocyte membranes (Brito et al., 2002) and synaptosomes (Brito et al., 2004) after exposure to UCB, was accompanied by reduced activities of Mg2+-ATPase aminophospholipid translocase (flippase) and Na+, K+-ATPase, together with enhanced intracellular levels of calcium (Ca2+; Figure 2B). These effects can lead to cell destruction, as evidenced by the hemolysis of erythrocytes (Brites et al., 1997; Khan and Poduval, 2011). Ca2+ influx or increased release of Ca2+ from ER or other stores may contribute to the intracellular increase of Ca2+. However, Zhang et al. (2010) demonstrated that UCB mainly induces the opening of the Ca2+ channel. Ca2+ influx into cortical neurons was shown to regulate the expression of nNOS mediated by a cAMP response element-binding (CREB) family transcription factor-dependent mechanism (Sasaki et al., 2000). Later it was shown that UCB-induced phosphorylation of CREB, induction of nNOS, and generation of NO, was indeed dependent of extracellular Ca2+ (Mancuso et al., 2008). Both extracellular signal regulated kinases (ERK1/2) (Mancuso et al., 2008) and c-Jun N-terminal kinases (JNK1/2) (Vaz et al., 2011b) phosphorylation is mediated through the NOS/NO pathway, but at least ERK activation seems to not depend from extracellular Ca2+ (Mancuso et al., 2008).
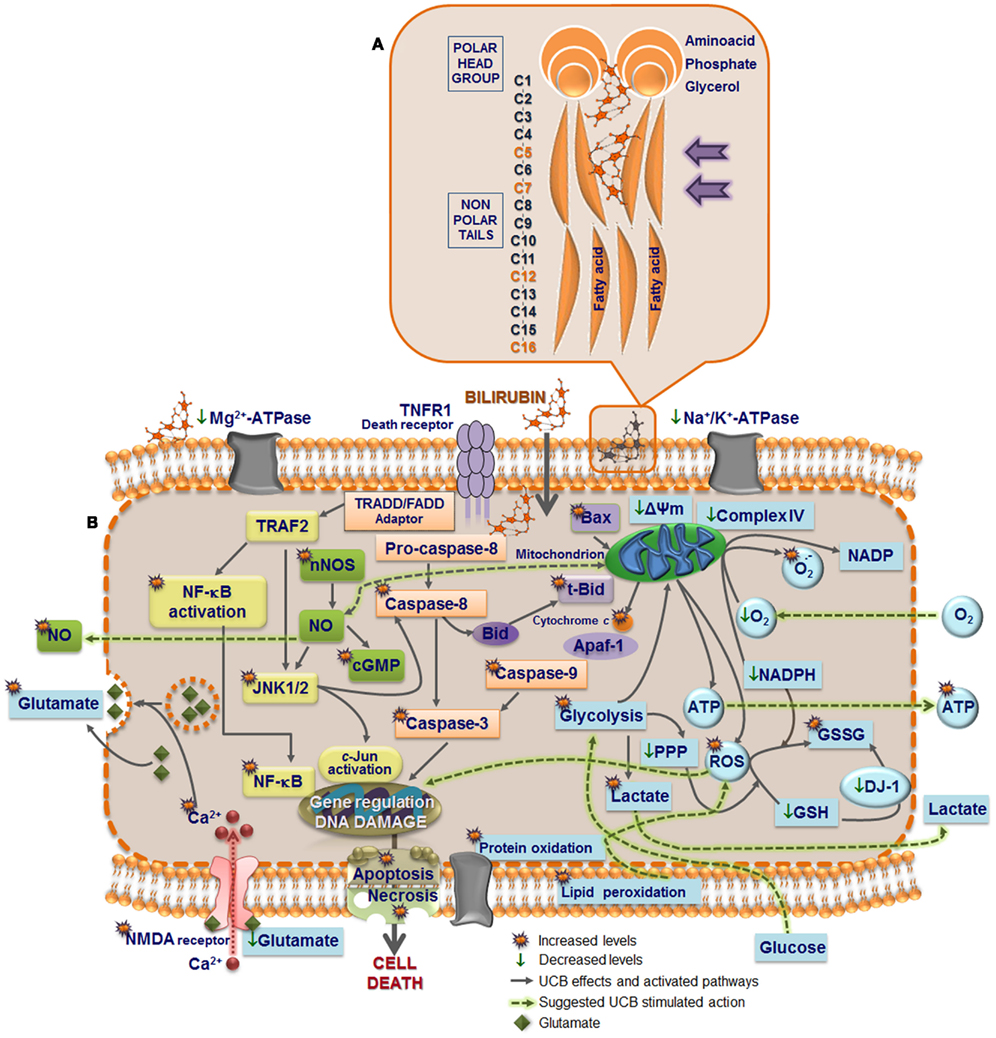
Figure 2. Model of UCB interaction with neural plasma membrane (A) and pathways by which UCB may affect neuronal function (B). Cell membrane includes a bilayer of packed phospholipids, cholesterol, and integral proteins. (A) The inset represents a phospholipid of the outer membrane containing a phosphatidyl-glycerol polar (hydrophilic head) and two fatty acids (hydrophobic tails). The phosphate group is attached to the glycerol and form ester links with a variety of other molecules. UCB decreases phospholipid mobility and the polarity at carbon (C) 5, while increased it at C7, and less markedly at C12 and C16 (orange). The intercalation at C5 may trigger the release of phospholipids and cholesterol, as well as the externalization of the phosphatidylserine, a recognition signal for phagocytosis. (B) UCB-induced protein oxidation and lipid peroxidation may arise from increased reactive oxygen species (ROS) production. UCB impairs Mg2+-ATPase activity, responsible for the maintenance of phosphatidylserine in the inner leaflet, and Na+, K+-ATPase, which may trigger the intracellular intrusion of calcium (Ca2+). Opening of Ca2+channels, as well as the activation of NMDA receptors by UCB, recognized to modulate the exocytosis of glutamate, can also account to it. Changes in membrane polarity and fluidity, equally produced at the mitochondrial membrane by UCB, lead to the collapse in the mitochondrial transmembrane potential (ΔΨm), translocation of proapoptic Bax to the mitochondria, inhibition of cytochrome c oxidase activity (complex IV of the respiratory chain), and efflux of cytochrome c. Then, activation of caspases 3 and 9 (intrinsic apoptotic cascade) takes place, as well as UCB-induced activation of caspase-8 (extrinsic apoptotic cascade) and the cleavage of Bid into truncated Bid (tBid). Oxidation of GSH to GSSG and inability to be restored is associated with the increased ROS generation, which may determine DNA damage. DJ-1 that is early up-regulated, to diminish UCB-induced oxidative stress, is latter on oxidized and proteolitically degraded due to the reduced levels of GSH. Increased superoxide anion radical production and decreased NADPH upon exposure to UCB also account to the oxidative stress. Glycolysis upregulation by UCB lead to increased intracellular lactate content and inhibition of the pentose phosphate pathway (PPP), providing ATP to support the bioenergetic crisis. UCB-asscociated nitrosative stress derives from the increased expression of nNOS and generation of NO and cyclic guanosine 3′,5′-monophosphate (cGMP). Actually, NO signaling, c-Jun N-terminal kinases (JNK1/2) stimulation, activation of nuclear factor-kappaB (NF-κB) with translocation to the nucleus, ROS generation, and activation of caspases are key players in the induction of genes involved in UCB-induced neuronal demise by necrosis and apoptosis.
Developing neurons are particularly susceptible to UCB interaction which causes neurogenesis impairment, neuritic atrophy, and cell death by both necrosis and apoptosis (Figures 3A,B; Brito et al., 2002; Falcão et al., 2007c) that is enhanced by associated hypoxia (Vert et al., 2001). Impact results in sudden and lasting decrease in neuronal arborization, as well as in reduced number of dendritic spines and synapses (Fernandes et al., 2009b), and cells stayed sensitized and increasingly vulnerable to a secondary toxic stimulus (Falcão et al., 2007c). The observed neurite outgrowth impairment is consistent with UCB-induced neurodevelopmental abnormalities, by interfering with neural plasticity, and hippocampal susceptibility corroborates the brain regional specificity toward UCB in bilirubin encephalopathy (Jangaard et al., 2008). It deserves to be noted that even in more differentiated neurons (10 days in vitro) impairment in neurite outgrowth by UCB is still observed after 12–24 h of exposure (Zhang et al., 2010). However, it was reported that cells are able to reverse some of the UCB-mediated effects when it is used a shorter (6 h), instead of a longer exposure time (12 or 24 h; Hankø et al., 2006b).
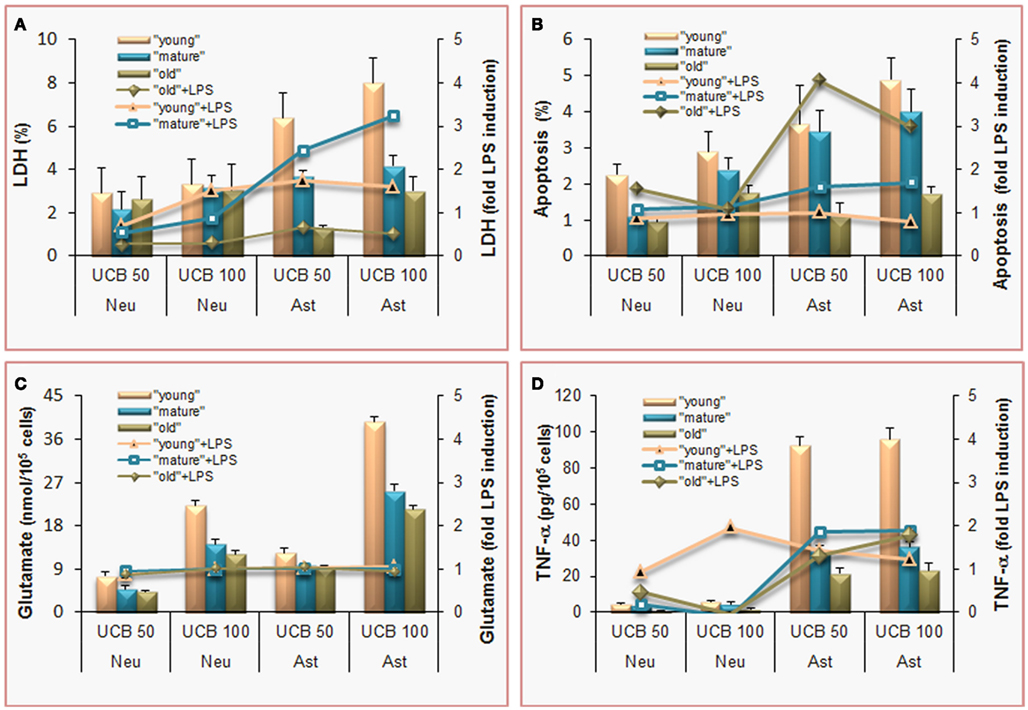
Figure 3. Effects of unconjugated bilirubin (UCB) are concentration-dependent, more pronounced in astrocytes (Ast) than in neurons (Neu), and most sensed by undifferentiated (young) cells, which also show increased demise by co-incubation with lipopolysaccharide (LPS). To mimic young (immature), mature, and old cells, and to achieve the same stage of differentiation in both, rat cortical astrocytes and neurons were cultured for 5, 10, and 20 days in vitro (DIV) or 4, 8, and 18 DIV, respectively. Cells were incubated with 50 or 100 μM purified UCB, or no addition (control), in the presence of 100 μM human serum albumin, at pH 7.4, for 4 h, at 37°C. In sister experiments, cells were co-incubated with 1 ng/L LPS and results were expressed as fold LPS induction. (A) Results of lactate dehydrogenase (LDH) release by non-viable cells are expressed as percentage of cell death relatively to total cell lysis. Astrocytes are the most susceptible cells, and LPS effects are similarly produced in both type of cells, although mature astrocytes are the most vulnerable. (B) Apoptosis was estimated by analysis of nuclear morphology following staining with Hoechst 33258 dye. Immature and mature cells reveal enhanced vulnerability to UCB and old astrocytes greater apoptosis by associated LPS. (C) Release of glutamate to the incubation medium proportionally increases with UCB concentration and it is not modified by LPS co-incubation. (D) The pro-inflammatory cytokine tumor necrosis factor (TNF)-α is released to the culture medium mainly by astrocytes and always increase by LPS addition. Mean values ± SEM are differences from the respective control, for all the parameters except for the fold increase by LPS, which data are means. Data derived from Fernandes et al. (2004) and Falcão et al. (2005, 2006, 2007c).
Alterations of UCB at neuronal arborization derive, at least in part, from cytoskeleton changes elicited in microtubule associated proteins (MAP) in immature neurons. Extended MAP2 entry in the axonal shaft (Figures 4A,B) and increased Tau1 expression by UCB, promotes Tau1 binding to microtubules and its release into the cytoplasm (Fernandes et al., 2009a, 2010; Figures 4C,D). Tau is localized in the axon and MAP2 is somato-dendritically compartmentalized. Mislocalization of Tau to dendritic spines and subsequent synaptic impairment appears to precede neuronal loss (Hoover et al., 2010) and its release into the extracellular space, from where it can reach the CSF and the plasma (Qiang et al., 2006). Therefore, the increased Tau levels observed in serum are considered to be a predictive indicator of neurodegeneration (Wang and Liu, 2008; Noguchi-Shinohara et al., 2011) and Okumus et al. (2008) have proposed that it should be considered a biomarker of BIND. Further studies should elucidate on this and other promising biomarkers of early neuronal dysfunction by UCB, as well as on the temporal window of reversibility, so that clinical detection and rescue intervention treatments can be achieved.
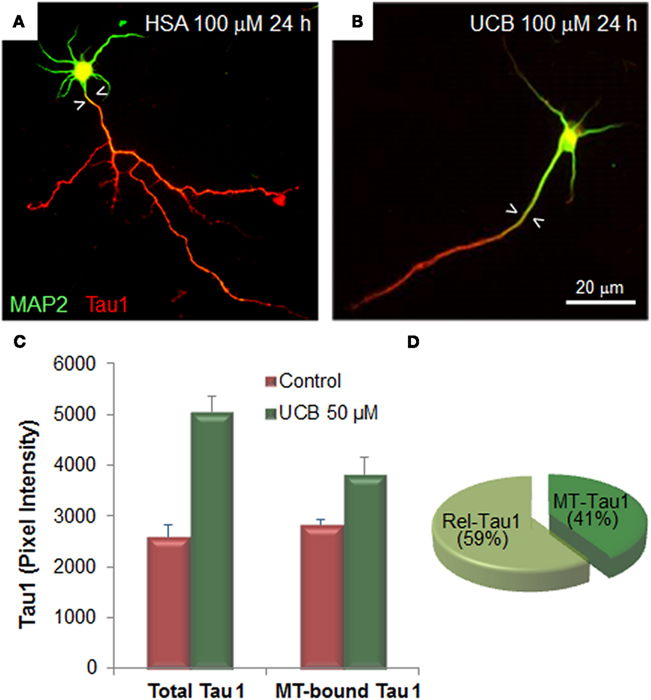
Figure 4. Exposure of immature hippocampal neurons to unconjugated bilirubin (UCB) impairs neuronal arborization by reducing axonal ramification and dendritic output, while increasing microtubule stability by changing the expression and localization patterns of the microtubule (MT) associated protein 2 (MAP2) and Tau1. Embryonic hippocampal neurons were treated with vehicle (human serum albumin, HSA) alone (control) or with UCB 100 μM (UCB/HSA = 1) for 24 h at 1 day in vitro (DIV) and fixed at 3 DIV. Representative images of hippocampal neurons immunostained with MAP2 (green) to identify the cell body and dendrites, and Tau1 (red) to identify the axon, are shown in (A) for control and (B) for UCB. UCB induces MAP2 axonal entry, evidenced by the white arrowheads pointing to the portion of axon with MAP2 entry, and a decrease in the number of axonal and dendritic branches when compared with HSA alone. Graph bars representing total and MT-bound Tau1 are shown in (C) illustrating that UCB induces axonal expression of Tau1. Percentage of Rel-Tau1 and MT-Tau1 in the pie chart in (D) depicts that part of Tau1 will be increasingly bound to microtubules (MT-Tau) (C,D), although the major portion is released (Rel-Tau), as illustrated in (D). Data derived from Fernandes et al. (2009a, 2010).
In addition, UCB also causes synaptotoxicity that has been related with potentiation of inhibitory synaptic transmission (Shi et al., 2006), as well as with impairment of the induction of long-term potentiation (LTP) and long-term depression (LTD; Chang et al., 2009). Interestingly, UCB was shown to decrease the expression of synaptophysin and SNAP-25 (Silva et al., 2012), proteins that participate in synapse establishment and neurotransmitter release (Wang and Tang, 2006). Accordingly, UCB was demonstrated to cause presynaptic degeneration in the Gunn rat, while preserving postsynaptic neurons (Haustein et al., 2010).
Mitochondria swelling by UCB (Solá et al., 2002), as well as Bax translocation, mitochondrial depolarization, release of cytochrome c, activation of caspase-3, and degradation of poly(ADP)ribose polymerase (Rodrigues et al., 2002a) were shown to be implicated in neuronal apoptosis by UCB (Figure 2B). Inhibition of cytochrome c oxidase in mitochondria isolated from mice brain (Malik et al., 2010) and in immature cortical neurons obtained from rats, in which UCB evidenced to affect cell respiration due to the low oxygen consumption, account for the decrease in mitochondrial functional activity by UCB (Vaz et al., 2010). In addition, while low and moderate concentrations of UCB may favor a delayed apoptosis in cultured human NT2-N neurons, moderate and high levels preferentially trigger early necrosis (Hankø et al., 2005). Cell death prevalence in immature (3–4 days in vitro) neurons, mostly by apoptosis (Falcão et al., 2006; Figure 3B), involves a bioenergetic and oxidative crisis (Vaz et al., 2010). Oxidative stress derives from the increased superoxide anion radical production () (Figure 2B) together with an elevation of the GSSG relatively to the total glutathione (GSH plus two times GSSG), as well as from a decrease in NADPH concentration. It is conceivable that the upregulation of glycolysis by UCB will be an attempt to provide ATP to support the bioenergetic crisis, while ATP release resulting from the NO production and oxidative stress should be associated with apoptosis.
The N-methyl-D-aspartate (NMDA) receptor-mediated excitotoxic mechanism is another pathway leading to UCB-induced neuronal injury, providing the necessary increase in intracellular Ca2+ required for activation of nNOS (Bellefontaine et al., 2011). In fact, UCB was demonstrated to raise the extracellular concentration of glutamate, by enhancing its release mainly from immature neurons (Falcão et al., 2006; Figures 2B and 3C), thus engendering overstimulation of NMDA receptors (Ostrow et al., 2004; Falcão et al., 2006; Brites et al., 2009). In addition, prolonged UCB exposure led to a decrease in presynaptic transmitter release but also impaired the postsynaptic NMDA receptor function (Chang et al., 2009). Therefore, the use of NMDA receptor antagonist MK-801 was shown to prevent the expression of nNOS, the consequent release of NO, the production of cyclic guanosine 3′,5′-monophosphate (cGMP), as well as cell dysfunction and demise (Hankø et al., 2006a; Brito et al., 2010), but not the extracellular accumulation of glutamate in primary cultures of cortical neurons (Brito et al., 2010). Conflicting results were observed for MK-801 in hippocampal neurons treated with high UCB levels (Shapiro et al., 2007), not considered clinically relevant by surpassing UCB aqueous solubility (Ostrow et al., 2003b; Zhou et al., 2010), and in Gunn rats pups, where no protection against BAEPs abnormalities was observed (Shapiro et al., 2007).
Increased expression of nNOS by upregulation at both the mRNA and the protein level and the generation of NO were shown to depend from neurotrophins (Mancuso et al., 2008). These Authors have shown that 10 μM Bf reduces the formation of H2O2-induced ROS, contributing to the inhibition of the nerve growth factor and the brain-derived neurotrophic factor signaling to Akt and ERK1/2 decreased phosphorylation. Intriguingly, in the absence of endogenous growth factors, UCB evidenced to activate an NO-dependent cascade leading to ERK activation, actions that the same Authors hypothesize to result from cell membrane perturbation and calcium influx in PC12 cells. Therefore, UCB neurotoxicity would derive in the perspective of these Authors from the presence of neurotrophins which predominate in hippocampus and cerebellum on the perinatal life in order to promote neuronal development and differentiation. Recently, it was observed an increased expression of nNOS by UCB in hippocampal neurons, as compared to cerebellar or cortical neurons (Vaz et al., 2011a), indicating a particular susceptibility of hippocampus to nitrosative stress. Most important, acute hyperbilirubinemia by administration of sulfadimethoxine to the Gunn rat was evidenced to lead to a presynaptic failure of synaptic transmission, considered to be in the basis of UCB-induced deafness and related to the upregulation of nNOS and generation of NO. In fact, the administration of a nNOS antagonist has shown to protect the loss of auditory function (Haustein et al., 2010), highlighting the role of NO in the neurotoxic mechanisms of UCB. Additional studies are necessary to further explore the UCB-dependent neurotoxicity from growth factor availability, as proposed by Mancuso et al. (2008).
Both glutamate (Silva et al., 2012) and oxidative stress (Vaz et al., 2011a) were shown to mediate the disruption of neuronal dynamics by UCB. Interestingly this effect, similarly to nNOS induction, was also more evident in neurons from the hippocampus than in those from the cortex and the cerebellum. To that should account the decreased levels of DJ-1, a protein with a protective role against oxidative damage (Lev et al., 2008; Kahle et al., 2009), the increased nitrosative stress, ROS production and GSSG evidenced by hippocampal neurons when treated with UCB (Vaz et al., 2011a). Similarly to other studies pointing to DJ-1 over-expression as a mechanism to protect cells from UCB-induced cell death (Deganuto et al., 2010), our data also evidenced an early upregulation effect (Vaz et al., 2011a), probably as an initial effort to diminish UCB-induced oxidative stress. However, in this last study it only lasted 4 h instead of the 24 h observed by Deganuto et al. (2010). These different results may be caused by the use of different models, in the case the human neuroblastoma cell line SH-SY5Y and not hippocampal neurons from Wistar rats as we used. The low levels of DJ-1 later observed (24 h) may result from its oxidation due to the decreased levels of GSH (Miyama et al., 2011) and degradation through proteolysis (Saeed et al., 2010). Inability to restore GSH from GSSG is related to the increased production of ROS and oxidative damage of proteins and lipids (green broken line, Figure 2), leading to DNA damage (Wiseman and Halliwell, 1996), which was evidenced to be produced by UCB in the neuroblastoma cell line (Deganuto et al., 2010). However, it was recently shown that a subpopulation of SH-SY5Y cells appear to survive to the initial damage of 140 nM UCB by increasing GSH content, thus becoming more resistant to oxidative stress (Giraudi et al., 2011).
Finally, even though that microglia and astrocytes are the predominant sources of pro-inflammatory cyto/chemokines, neurons can also express these factors in disease settings (Ohtori et al., 2004; Janelsins et al., 2008; Nazmi et al., 2011). Nevertheless, far smaller concentrations of tumor necrosis factor (TNF)-α than the ones produced by astrocytes were secreted from UCB-treated neurons (Falcão et al., 2006; Figure 3D).
Once glial cells degeneration has direct deleterious consequences on the function and survival of neurons, which will be deprived of their optimal microenvironment, UCB-induced glial damage cannot be simply considered as marginal events or secondary reactions to neuronal injury, but rather should be jointly included in the BIND processes. Therefore, in the following sections we will describe how glial cells sense and respond to UCB as important players in neuronal dysfunction and promising therapeutic targets.
Glial Reactivity and Dysfunction by Bilirubin
The survival and proper function of neurons is ensured by the large number of glial cells (Streit, 2002). They have acquired a special relevance in brain injuries and include OLGs and astrocytes, as well as microglial cells which share features with immune cells. Microglia and astrocytes communicate each other through the cytokine IL-1 and ATP and are recognized as active participants in various pathological conditions, reason why their activation is associated with the development and severity of diseases. Once microglia are activated earlier than astrocytes, it is believed that they promote the activation of astrocytes (Liu et al., 2011a).
Interestingly, both astrocytes and microglia have lately been referred to be involved in “synaptic stripping” after axon lesion (Cullheim and Thams, 2007). Recently, it was observed that microglia make contacts with neuronal synapses at a rate of about one per hour with a lasting time of 5 min (Wake et al., 2009), being considered vital members of the “quadripartite synapse,” which also includes presynaptic and postsynaptic neurons, astrocytes, and extracellular matrix (Sykova and Nicholson, 2008). Therefore, we should consider that microglia–neuronal cross-talk is a key point in guiding microglial activation and that the cytokines released by activated microglia influence not only neighboring neurons but also astrocytes.
Interaction of all these cell types thus fulfills brain function. In vitro cell models have been used to unravel specific cell dysfunction mechanisms by UCB and the intricate signaling processes comprised, as well to screen target-driven therapeutic agents, in an attempt to incorporate this heterogeneity to better understand the basis of BIND pathogenesis. In the following sections, we will describe the research that is being undertaken using specific cell populations.
Astrocytes are Key Players in Bilirubin Injury
Astrocytes that are the most abundant type of glia (Nair et al., 2008), outnumbering neurons by over fivefold (Sofroniew and Vinters, 2010), have multiple homeostatic properties (Orellana et al., 2009). They are closely associated to neurons, and most synapses are intimately ensheathed by astrocytes processes. Astrocytes, like microglia, may have either a neuroprotective or a neurotoxic role, where the release of pro-inflammatory cytokines, glutamate, ATP, and free radicals may decrease neuronal survival and increase their susceptibility to neurotoxins. In fact, injuries in astrocyte function have been recognized as highly contributing to neuronal dysfunction (Allaman et al., 2011).
First reports on the UCB toxicity to astroglia came out in the 1960s, either in rat cerebellum cultures (Silberberg and Schutta, 1967) or in experimental kernicterus (Chen et al., 1969), in which the Authors observed swelling of the perivascular astrocyte foot and evidence of impending cell damage. Access of UCB to neurons was later proposed on the basis of a first absorption from astrocytes foot processes followed by diffusion from presynaptic astrocytes processes into the dendrites.
In primary cultures of fetal rat glial cells (Amit and Brenner, 1993), as well as in astrocytes (Falcão et al., 2006), toxic effects of UCB were noticed on cell morphology, cell viability, and mitochondrial function (Figure 5A). Astrocytes, similarly to neurons exhibited an age-dependent sensitivity to UCB toxic effects. Decreased expression of Mrp1 and Pgp, observed in immature astrocytes when compared to more differentiated cells (Watchko et al., 2001; Falcão et al., 2007a; Sequeira et al., 2007), surely contributes for the increased susceptibility of immature astrocytes to UCB. Moreover, induction of necrotic-like cell death by UCB was higher in astrocytes than in neurons, in immature than in mature cells, and more elevated than its own apoptosis (Figures 3A,B). In addition, both hypoxia and combined oxygen–glucose deprivation (OGD) followed by reoxygenation demonstrated to increase astrocyte susceptibility to UCB injury (Falcão et al., 2007b), accounting as risk factors of BIND.
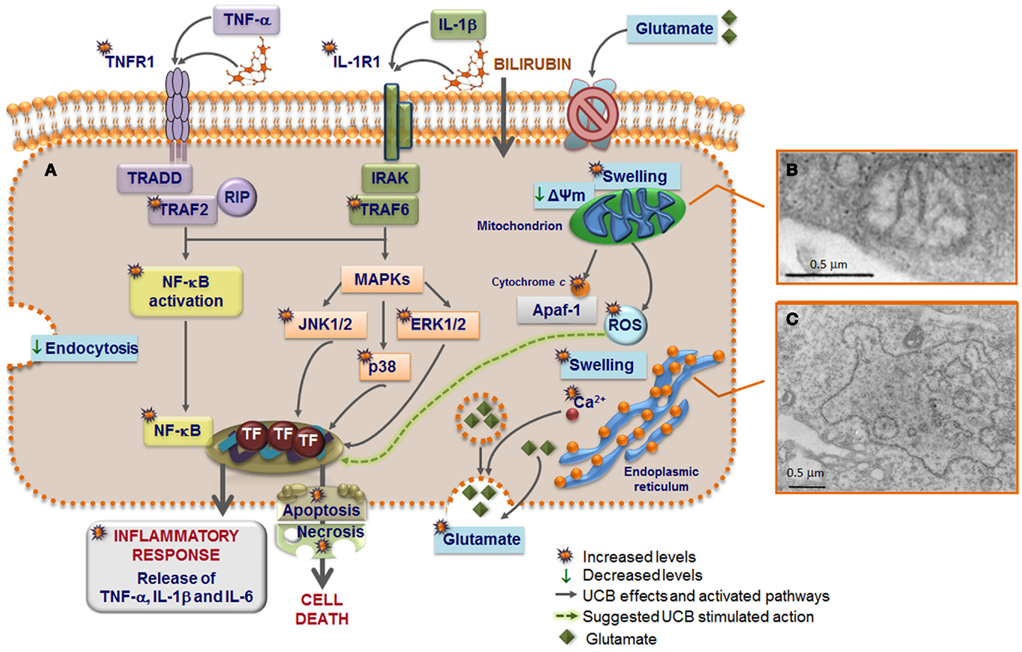
Figure 5. Model of astrocytic reactivity to unconjugated bilirubin (UCB) evidencing the pathways involved in the release of pro-inflammatory cytokines and in cell demise (A) mediated by mitochondrial (B) and rough endoplasmic reticulum (ER) (C) abnormalities. UCB causes inhibition of glutamate uptake and elevation of intracellular calcium (Ca2+) levels that modulate the exocytosis of glutamate, thus increasing the extracellular content of glutamate. High concentrations of UCB disrupt endocytosis in astrocytes and produce mitochondria swelling, mediated by the opening of the permeability transition megapore, and ER enlargement probably preceded by ER stress response and release of Ca2+. UCB-induced apoptosis include mitochondrial permeabilization and cytochrome c efflux. UCB induces the secretion of both tumor necrosis factor (TNF)-α and interleukin (IL)-1β, which stimulate the synthesis of IL-6 after the first 4 h of interaction. The primary inflammatory stimulus causes a rapid and transient activation of the mitogen-activated protein kinase (MAPK) pathways that include p38, c-Jun N-terminal kinases (JNK) 1/2, and extracellular signal regulated kinases (ERK) 1/2, as well as of the nuclear factor-kappaB (NF-κB) that is translocated to the nucleus. Then, downstream transcription factors (TF) are activated and the expression of genes involved in cytokine release and cell death is induced. UCB interacts with the cytoplasmatic membrane of astrocytes inducing a rapid and sustained upregulation of both TNF-α receptor (TNFR)1 and IL-1β receptor (IL-1R)1. TNFR1 activation triggers the recruitment of the receptor-associated factor (TRAF)2, TNF receptor-associated death domain (TRADD), and receptor-interacting protein (RIP). Formation of an active IL-1R1 signaling complex mediate the recruitment of IL-1 receptor-associated kinases (IRAK) and leads to the recruitment of TRAF6 that is required for NF-κB mobilization to the nucleus and to the phosphorylation of the three MAPKs. Activation of this intracellular cascade, upon direct UCB challenge or indirectly by the increased secretion of TNF-α and IL-1β, lead to a number of astrocytic death paradigms, which cover a spectrum from apoptosis to necrosis.
Similarly to neurons, UCB determines a fast increase in the extracellular glutamate content (Fernandes et al., 2004), mainly in immature astrocytes (Figure 3C; Falcão et al., 2005) that appears to result from the inhibition of its uptake by UCB (Silva et al., 1999), more effective in these glial cells than in neurons (Brito et al., 2002), but also from enhanced reactive astrocyte secretion (Falcão et al., 2006), which may either occur directly from the cytosol or via regulated exocytosis (Figure 5A). Abnormally prolonged exposure to glutamate causes neuronal injury, and such “excitotoxicity” can be deleterious to neurons and OLGs and cause disease. Interestingly, this glutamate elicits a fast communication between astrocytes and adjacent cells in the brain (De Keyser et al., 2008) and although glutamate-induced Ca2+ influx can trigger DNA damage by a mitochondrial ROS-mediated mechanism, the Ca2+was indicated to simultaneously enhance DNA repair capability, thereby protecting neurons against injury and disease (Yang et al., 2011).
To the best of our knowledge, our lab provided the first evidence that elevated concentrations of UCB trigger the secretion of TNF-α (Figure 6A) and IL-1β by astrocytes (Figure 6B; Fernandes et al., 2004) through the increase of TNF-α and IL-1β mRNA expression and activation of TNF-α-converting enzyme (TACE) and IL-1β-converting enzyme (ICE), known as caspase-1, raising the conversion of the cytokine pro-forms into active forms (Fernandes et al., 2006). Again, immature astrocytes were the main producers of TNF-α (Figure 3D) and IL-1β (Falcão et al., 2006).
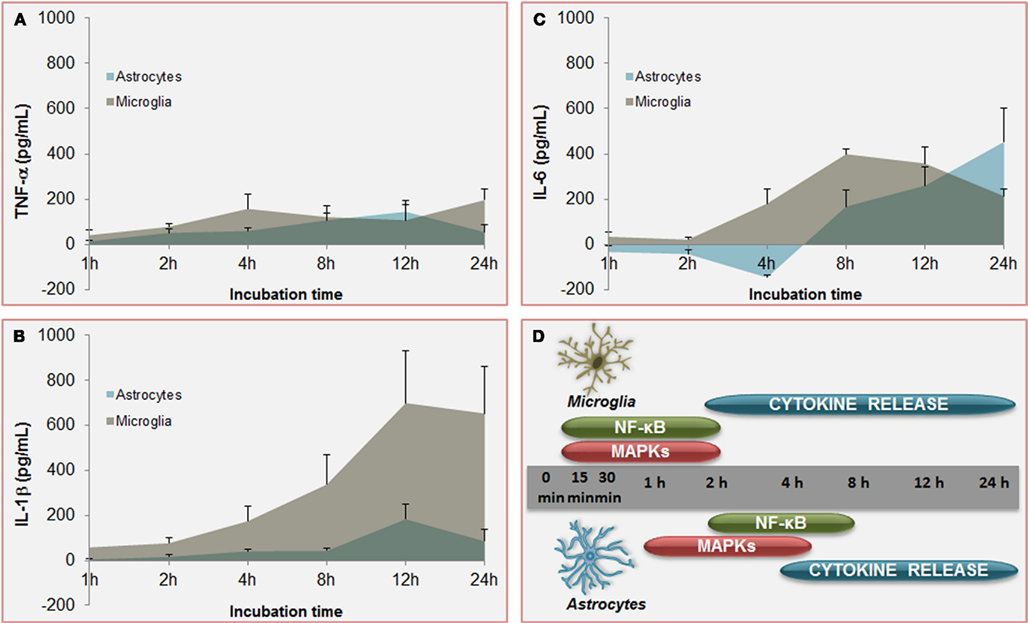
Figure 6. Temporal profile of unconjugated bilirubin-induced (UCB) activation of inflammatory pathways: a comparison between microglia and astrocytes. Increased and equivalent release of tumor necrosis factor (TNF)-α (A) is determined by UCB on both astrocytes and microglia. However, secretion of interleukin (IL)-1β (B) by astrocytes is much higher in microglia and earlier produced upon UCB challenge than in astrocytes. Initial effects of UCB at the level of IL-6 release (C) are of opposite signals. While IL-6 production by astrocytes is depressed in the first 4 h of incubation with UCB before being enhanced, IL-6 secretion by microglia is almost immediately induced by UCB. These timely differences in the secretion of pro-inflammatory cytokines between astrocytes and microglia should reside in the former activation of nuclear factor (NF)-κB and mitogen-activated protein kinases (MAPKs) (D) in microglia than is astrocytes. These features corroborate the belief that microglia is activated earlier than astrocytes, thus promoting astrocytic activation. Data, presented as mean ± SEM, derived from Fernandes et al. (2006) and Silva et al. (2010).
Effects of UCB at the level of the membrane of astrocytes also include the inhibition of cell endocytosis (Silva et al., 2001a) and the upregulation of both TNF-α receptor (TNFR)1 and IL-1β receptor (IL-1R)1 (Fernandes et al., 2011), triggering the recruitment of receptors’ molecular adaptors TRAF2 and TRAF6, respectively (Figure 5). Activation of the three mitogen-activated protein kinases (MAPK) pathways, p38, JNK1/2, and ERK1/2 is subsequently achieved, inducing the pro-inflammatory cytokine gene expression and the release of TNF-α and IL-1β. Although IL-6 expression was initially down-regulated by UCB, it substantially increased after 6 h of incubation with UCB (Figure 6C; Fernandes et al., 2006). Similarly, UCB activated nuclear factor-kappaB (NF-κB) signaling pathway (Figures 5A and 6D) via activation of TNF-α/TNFR1 and IL-1β/IL-1R1 cascades (Fernandes et al., 2006, 2007a).
Injury of astrocytes by high levels of UCB determines ER and mitochondria morphological changes (Figures 5B,C; Silva et al., 2001a), which have been related with oxidative stress and cell death in other neurological diseases (Higgins et al., 2010). Even considering the higher GSH levels and the elevated resistance to UCB-induced oxidative stress in astrocytes, as compared to neurons, depletion of GSH was still observed after incubation with Bf concentrations of 0.1 and 1 μM (Brito et al., 2008b).
Because all these aspects may contribute to the degeneration of astrocytes in brain parenchyma, compromising neuronal survival, and functional recovery after BIND, we propose that astrocytes should be considered one of the targets when searching for novel therapeutics for preventing neurological sequelae by UCB in conditions where the most used clinical approaches evidence to fail.
Microglia in the Process of Bilirubin-Induced Inflammation
Microglia reside within the CNS parenchyma, in both gray and white matter (WM), and constitute about 12% of total glial cell population (Aloisi, 2001; Ladeby et al., 2005). In the surveillant/vigilating state, previously designed as resting state, microglia through their fine processes have an elevated motility, higher than that of astrocytes, allowing them to constantly monitor their microenvironment (Davalos et al., 2005; Nimmerjahn et al., 2005). Therefore, microglia were shown to be involved in neurogenesis, postlesional tissue repair, and “synaptic stripping” (Graeber, 2010). Microglia should be considered an additional target to protect in hyperbilirubinemia because they play a major role in synaptic pruning during postnatal development, as suggested by data obtained in mice during normal brain development (Paolicelli et al., 2011). Moreover, phagocytosis of tissue debris by microglia is essential for tissue homeostasis and is considered to be associated to a neuroprotective outcome. The first reaction of microglia toward UCB insult is the increase in phagocytosis (Figure 7), probably as an attempt to constrain the lesion extent, although only transiently exerted once increased levels of apoptosis were shown to follow until the 12 h of incubation (Silva et al., 2010).
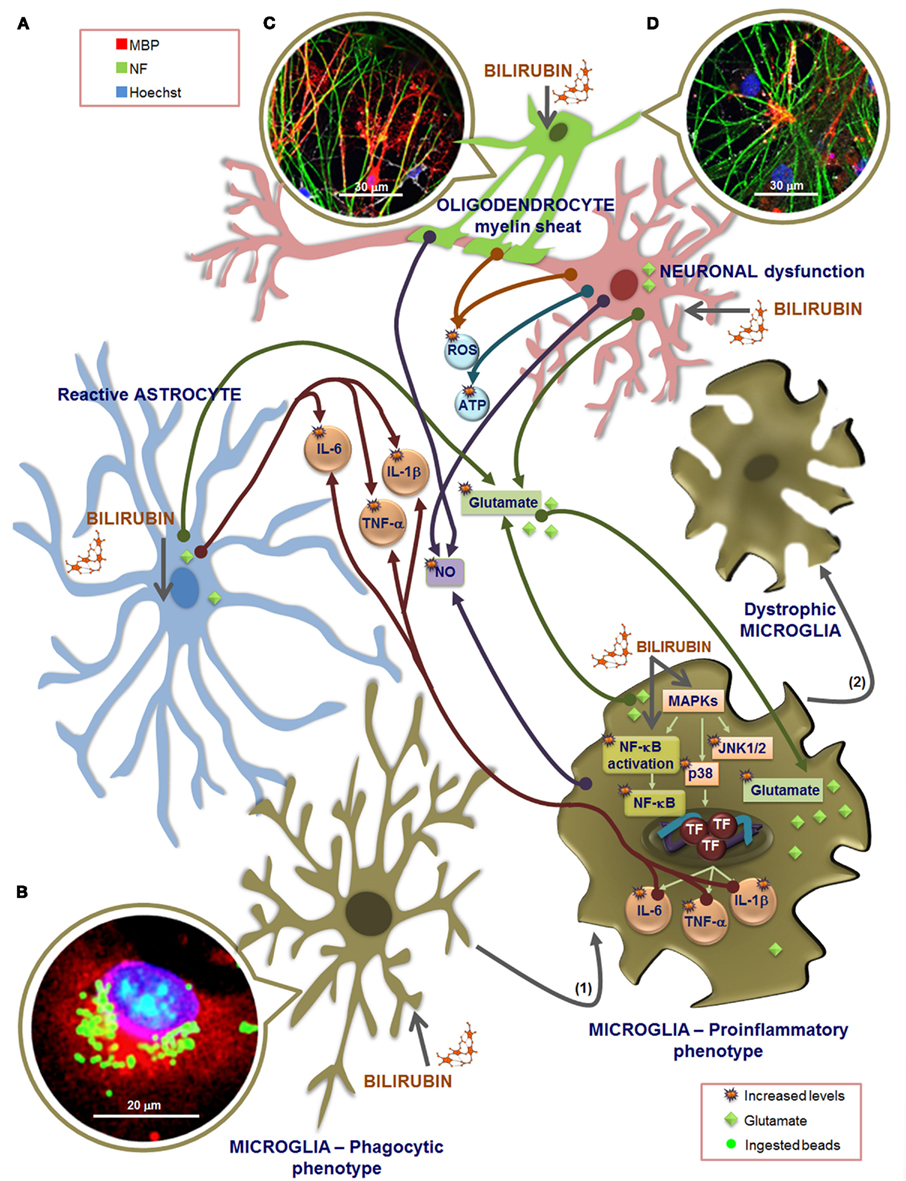
Figure 7. Overview of the effectors that may be involved in microglia dialog with other glial cells and neurons, upon exposure to unconjugated bilirubin (UCB). Extracellular increased levels of glutamate by UCB-induced secretion from astrocytes, microglia, and neurons, as well as by astrocytic uptake inhibition, together with the release of inflammatory mediators, such as interleukin (IL)-6, tumor necrosis factor (TNF)-α, and IL-1β from astrocytes and microglia (A), are included in cross-talk effectors. Also comprised are members of the oxidative stress, including the reactive oxygen species (ROS) produced by neurons and oligodendrocytes (OLGs), as well as nitrosative stress by nitric oxide (NO) generated by microglia, neurons, and OLGs, and elevated release of ATP from neurons, during exposure to UCB. (B) Phagocytosis upon short stimulation with UCB is visualized in microglia immunostained with an antibody against Iba1 (red) and using Hoechst 33258 stain to visualize the nucleus (blue), by counting the number of ingested fluorescent latex beads (green). Microglia intervenes in glutamate tissue homeostasis by both releasing and retaining glutamate when exposed to UCB. But microglia also reacts to UCB stimulus, very rapidly, through the activation of mitogen-activated protein kinases (MAPKs), mainly the c-Jun N-terminal kinases (JNK)1/2 and p38, and of the nuclear factor-kappaB (NF-κB) that is translocated to the nucleus, determining the activation of transcription factors (TF) and further release of cytokines. These pathways may mediate both the phagocytic and the inflammatory phenotypes and are related with a shift from an elongated morphology to a large and amoeboid shape (1). Longer exposure to UCB leas to fragmented and condensed cytoplasm, a feature of dystrophic microglia (2) indicative of cell degeneration and senescence. The multifaceted profile of microglia is determined by the stimulus type and duration, and dialog between cells. Although microglia activation may promote astrocytic reactivity, the release of pro-inflammatory cytokines, glutamate, and NO from these cells may exacerbate microglia reactivity, which by propagating inflammation will sensitize neurons and decrease their survival upon UCB exposure. Moreover, astrocytic deregulation and degeneration by UCB determine inaccurate neuron–astrocyte interactions, causing neuritic arborization and synaptic transmission impairment, favoring brain damage. OLGs do not play a role in promoting inflammation although, like neurons, are damaged by UCB and associated inflammatory processes.(C) OLGs and dorsal root ganglion neurons co-cultures incubated for 24 h with 100 μM human serum albumin (HSA) at 7 days in vitro evidence an increased number of myelinating OLGs. (D) Incubation with 50 μM UCB plus 100 μM HSA reveal a delayed myelination once a number of neurofilaments are still not myelinated when compared to the image in (C). To evaluate myelination, OLGs were fixed at 21 days in vitro and immunolabeled for myelin basic protein (MBP, red) and neurofilament (NF, green). Nuclei were counterstained with Hoechst 33258 dye (blue).
However, microglia can also be activated or overactivated, depending on the stimuli, thus becoming neurotoxic (Carson et al., 2007). When activated by UCB microglia change from an elongated morphology to an amoeboid-appearance (Gordo et al., 2006) and even can dye by several pathways (Silva et al., 2010). Maximal levels for caspase-8 (extrinsic pathway), caspase-9 (intrinsic) and the effector caspase-3 were obtained at 6 h of treatment. Moreover, the cells release pro-inflammatory cytokines such as TNF-α (Figure 6A), IL-1β (Figure 6B), and IL-6 (Figure 6C), which parallels the alterations in cell morphology. Similarly to astrocytes, UCB activates MAPKs and NF-κB in microglia. MAPKs involvement and NF-κB engagement in UCB-challenged microglia occurs at an early time point (Figure 6D), and appear to underlie the phagocytic activity observed at 4 h incubation and the inflammatory phenotypes initiated at 2 h with peak levels at 4 and 24 h for TNF-α, at 8 h for IL-6, and at 12 h IL-1β (Figure 6D). TNF-α has shown to have a critical role in several neuropathologic disorders. Indeed, TNF-α lies at the beginning of a signal cascade that may lead to neuronal cell death, potentiates glutamate neurotoxicity, and can stimulate the production of IL-1β, IL-6, and other cytotoxic cytokines (Lee et al., 1993; Chao and Hu, 1994). Moderate upregulation of cyclooxygenase (COX)-2 and of matrix metalloproteinase (MMP)-2 and -9 activities were induced by UCB only after 12 and 24 h incubation. The most marked effect of UCB on microglia was shown to consist in the elevated levels of IL-1β what was curiously related with increased Tau phosphorylation (Li et al., 2003) and loss of neuronal integrity (Depino et al., 2005).
Microglia, similarly to neurons and astrocytes (Falcão et al., 2006), also release the cytotoxic molecule glutamate (Piani et al., 1991) when exposed to UCB (Gordo et al., 2006). The levels were found to be threefold enhanced when compared to those in astrocytes and sixfold to those in neurons (Brites et al., 2009). Release of glutamate by microglia was observed by other neurotoxins, is related to disorders of synaptic plasticity (Patrizio and Levi, 1994; Maezawa and Jin, 2010) and indicated to participate in microglial neurotoxicity (Barger and Basile, 2001; Liang et al., 2008).
What triggers increased microglial activation is still unclear but oxidative stress, resulting from increased ROS, appears to have a determinant role (Floyd and Hensley, 2002; Lu et al., 2004). As it can be seen in Figure 7, UCB is implicated in ROS generation by neurons. After CNS injury, microglia can rapidly populate the site of injury (Morgese et al., 1983) and migration of these cells is triggered by compounds released by the damaged tissue (Kurpius et al., 2007), such as ATP and NO (Duan et al., 2009). Actually, ATP was shown to be released from neurons treated with UCB (Vaz et al., 2010), and NO generated by OLGs (Genc et al., 2003), neurons (Vaz et al., 2011b), and microglia (Silva et al., 2012) upon exposure to UCB (Figure 7). Therefore, since microglial cells themselves can release ATP, it is possible that a positive feedback mechanism perpetuates migration and a gradient of NO will direct migration to the site of injury, as the stopping location for microglial cells reparative functions (Samuels et al., 2010). These include phagocytosis of cellular debris leading to the axonal sprouting and reestablishment of synaptic contacts (Morgese et al., 1983; Ngu et al., 2007). Nevertheless, microglial phagocytosis of stressed neurons also contributes to their loss. Moreover, excessive microglial production of NO induced by UCB in hippocampal slices (Silva et al., 2012) may lead to increased cell death. In fact, and in contrast to the “protective” effects exerted by microglia in preventing excessive glutamate, microglia additionally contribute to the generation of NO, an important mediator in microglia-induced neuron death (Golde et al., 2002; Graber et al., 2012). Generation of NO may derive from iNOS activation, which once activated produces moderate levels of NO chronically without any requirement for further activation (Brown, 2010), or from constitutive NOS, probably originating in the case of UCB an overproduction that may inhibit phagocytosis (Kopec and Carroll, 2000), as we observed following the 4 h of incubation (Silva et al., 2010).
Furthermore, microglia exhibit partial or complete fragmentation of cytoplasm, as well as nuclear condensation, when exposed to UCB for longer periods of time (Silva et al., 2010), thus compromising any beneficial effect that they could have in BIND protection. This feature is designed as dystrophy of microglia (Streit et al., 2008), or cytorrhexis (Fendrick et al., 2007; Hasegawa-Ishii et al., 2011) and is an indication of microglia degeneration and senescence (Streit et al., 2004).
Collectively, we may assume that the release of cytokines and, eventually other soluble products from microglia activated by damaged neurons, contributes to the initiation and maintenance of astrogliosis observed in kernicterus (Steinborn et al., 1999), and an underlying component of a diverse range of diseases and associated neuropathologies. Thus, development of targeted anti-microglial activation therapies might help to attenuate reactive astrogliosis and generate a more beneficial environment to protect brain from UCB-induced neurotoxicity.
Bilirubin Damages Myelinating Glia
In the CNS, WM is primarily comprised of axonal bundles ensheathed with myelin, which reduces axonal loss. The cells forming these sheaths are the OLGs that spirally wrap segments of axons during late fetal and early postnatal stages. Just before and after birth, oligodendrocyte precursor cells (OPCs) multiply rapidly, differentiate into myelinating OLGs, and develop processes, which are then involved in the formation of myelin (Arai and Lo, 2009; Piaton et al., 2010). Reciprocal communication between neurons and OLGs are essential for myelin biogenesis and myelin repair (Piaton et al., 2010). When OPCs and OLGs are damaged there is a loss of myelin synthesis and interruption of proper axonal function and, consequently, of neuronal connectivity and function (Arai et al., 2009). However, compared to the mechanisms of neuronal injury in gray matter, WM pathophysiology remains relatively understudied and poorly understood, probably accounting for the scarce information about its influence on BIND and kernicterus.
Moderate perinatal systemic inflammation and hypoxia–ischemia, and perhaps most important, combinations of these, are related to deficient OPCs maturation, with reduced density of myelinating OLGs and disruption of the expression of several transcription factors important in OPCs maturation, thus causing long-lasting effects (Favrais et al., 2011; Volpe et al., 2011). In these circumstances, susceptible microglia during early postnatal development may modify their functional capacity, similarly to a “primed” state, and become more susceptible to a secondary reaction by injury or to the effects of aging (Harry and Kraft, 2012).
Several years ago, it was reported that UCB adversely affects myelination in tissue cultures of rat cerebellum (Silberberg and Schutta, 1967). Binding of UCB to myelin basic protein (MBP) was later on evidenced (Gurba and Zand, 1974) and suggested to provide a mechanism for retention of UCB in the brain, while causing the inhibition of cerebellar protein synthesis by an undefined mechanism. Curiously elevated concentrations of UCB were found in the CNS myelin fraction after injection of UCB into rats (Hansen et al., 2001). Recently, by using cranial ultrasound and MRI scans, Gkoltsiou et al. (2008) observed abnormal WM signal intensity in 10/11 later scans in infants that have shown to be in risk of kernicterus, as well as delayed myelination in some of them. These authors suggested that reaction to UCB neurotoxicity in extremely immature brains may be delayed as compared to term or near term infants, determining the observation of changes only in late scans. Impairments in myelination and WM involvement in UCB encephalopathy were also observed at autopsy (Ahdab-Barmada and Moossy, 1984; Koo and Roessmann, 1988). Furthermore, separation of the myelin lamellae was visualized in a model of experimental kernicterus (Chen et al., 1971), and myelin figures surrounding vacuoles, bits of cytoplasm, and other intracytoplasmic debris were identified in the Gunn rat (Jew and Williams, 1977). In addition, a decreased density of myelinated fibers was recently evidenced in the cerebellum of a preterm infant who died from kernicterus (Brito et al., 2012).
In vitro studies have indicated that UCB is responsible for decreased OPCs (Barateiro et al., 2010) and OLGs viability (Genc et al., 2003). OPC displayed increased apoptosis (Barateiro et al., 2010), and necrosis-like cell death upon UCB exposure, mediated by early signals of ER stress followed by mitochondrial dysfunction (Figures 8A,B). Indeed, UCB activates the unfolded protein response in OPCs by up-regulating the expression of GRP78 and GRP94. This is not without precedent once recent reports have shown that both UCB and Bf up-regulate many genes involved in ER stress both in SH-SY5Y (Calligaris et al., 2009) and Hepa 1c1c7 cells (Oakes and Bend, 2010). Other effects produced by UCB on OPCs include intracellular Ca2+ overload, ROS generation and JNK activation (Barateiro et al., 2010), while NO production via UCB-induced iNOS mRNA expression was observed in OLGs (Genc et al., 2003). Interestingly, no release of TNF-α, IL-1β, or glutamate could be noticed in OPCs cultured with UCB (Barateiro et al., 2010). A consequence of the sustained high concentration of intracellular Ca2+ is the activation of calpains by UCB, compromising cellular specific functions, and promoting cell death (Figure 8C). Moreover, UCB also delays differentiation of OPCs enhancing the number of immature (NG2+) cells and reducing that of OLGs (MBP+) when compared to vehicle-treated OPCs. These findings indicate that UCB, besides compromising OPCs proliferation, may also delay myelination (Figures 7C,D). Therefore, hyperbilirubinemia during the early phase of developmental myelination, where an unusual synthesis rate of myelin structural proteins are necessary (Nave, 2010), may impair OPCs differentiation into OLGs triggering defective myelination and neurological damage.
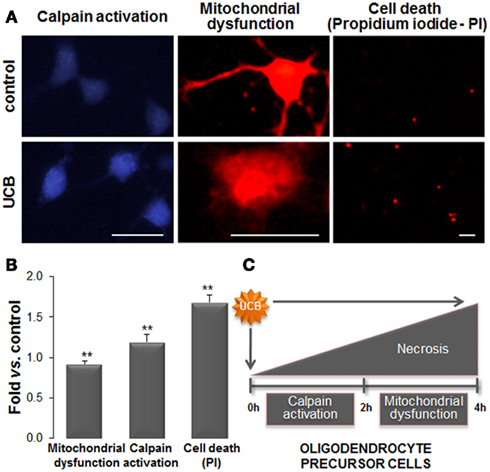
Figure 8. Exposure of oligodendrocyte precursor cells (OPCs) to unconjugated bilirubin (UCB) leads to mitochondrial dysfunction, followed by calpain activation, and cell death. Primary cultures of OPCs were isolated from mixed glial cultures and stimulated to differentiate. OPCs were treated with 100 μM of human serum albumin (HSA; control) or 50 μM UCB plus 100 μM HSA for 24 h (UCB). Mitochondrial functionality was evaluated by Mitotracker Red. Endoplasmic reticulum stress was assessed by the activity of calcium-dependent calpain using a specific calpain substrate. Necrotic-like cell death was evaluated by propidium iodide (PI) through the emission of red fluorescence. (A) Representative results of one experiment. (B) Graph bars represent the fold increase in staining intensity comparatively to control (mean ± SEM) in labeled active mitochondria (red), in calpain activity (blue), and in PI+ cells (red) from at least three independent experiments performed in duplicate. (C) Effects produced by UCB in OPCs start by mitochondrial dysfunction that induces the increase in intracellular calcium levels and activation of calpains. This cascade of events culminates in OPCs demise. Impairment of OPCs differentiation into oligodendrocytes by UCB will determine defective myelination. Scale bar represents 20 μm. **p < 0.01 vs. control. Data derived from Barateiro et al. (2010).
Collectively, these findings indicate that UCB compromises myelinogenesis and determines degeneration and eventual loss of functional myelin sheaths, features that should be taken in consideration for BIND neuroprotective strategies, mainly in premature babies. Indeed, neuroprotection cannot be truly attained without effective oligoprotection (Arai and Lo, 2009). Thus, OPCs count among the most vulnerable cells of the CNS due to their complex differentiation program (Bradl and Lassmann, 2010), and neuroinflammation, and OLGs dysfunction were shown as a main cause of axonal loss (Edgar et al., 2010). Dysregulation of myelination pathways by UCB surely deserves further investigation.
Role of Neuroinflammation in Bind
Some past studies have evidenced that UCB inhibits the function of the polymorphonuclear leukocytes and lymphocytes (Table 1; Thong and Rencis, 1977; Thong et al., 1979; Iwanaga et al., 1987; Haga et al., 1996a,b). Inflammatory properties by UCB have lately been observed in a mice model of intracerebral hemorrhage (Loftspring et al., 2011) and immunotoxic effects on both bone marrow and spleen cell suspensions obtained from mice (Khan and Poduval, 2011). As previously commented for neural cells, UCB has shown to trigger intrinsic and extrinsic pathways activation, GSH depletion, p38MAPK activation, and oxidative stress in splenocytes. Production of systemic pro-inflammatory mediators ultimately activates microglia, as well as it causes neuronal apoptosis and disruption of BBB and neuroinflammation (Morandi et al., 2011; Daulatzai, 2012). This alteration disrupts the delicate balance between neuro-glial interactive components resulting in deficient neuritic arborization and synaptic transmission (Rossi and Volterra, 2009), as well as in impaired memory, neural plasticity, and neurogenesis (Yirmiya and Goshen, 2011).
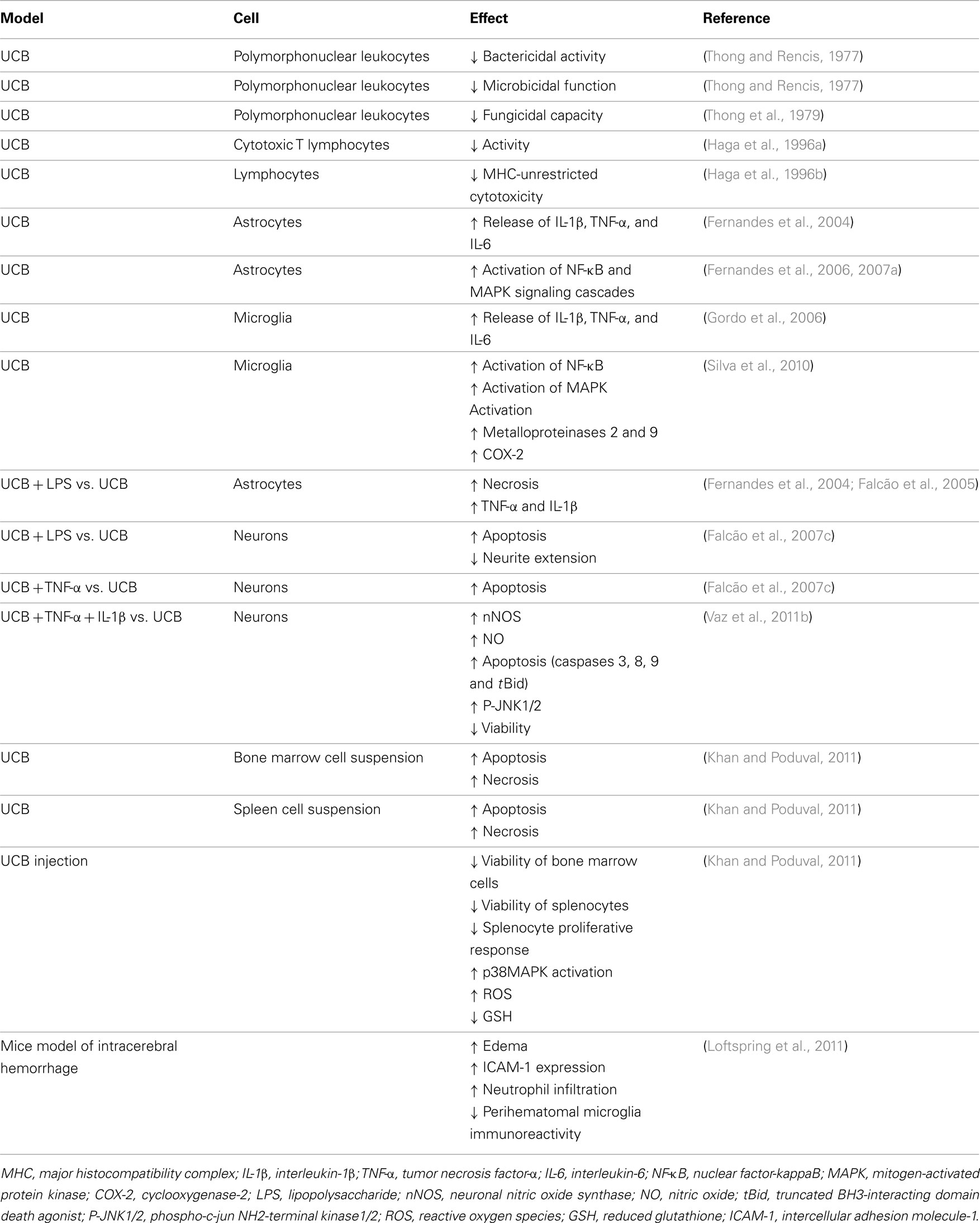
Table 1. Immunostimulant and immunotoxic effects of unconjugated bilirubin (UCB) in cell primary cultures, cell suspensions, and animal models.
As commented before, UCB is able to cause the reactivity of rat cortical astrocytes and microglia (Table 1), which in turn exacerbate inflammation (Sofroniew and Vinters, 2010) and increase BBB permeability by acting on endothelial cells and tight junctions (Nair et al., 2008). While the secreted levels of TNF-α (Figure 6A) are at the same order either in astrocytes or in microglia exposed to UCB, those of IL-1β and IL-6 are increasingly produced by microglia (Figures 6B,C) and earlier secreted (Figure 6D), suggesting that microglia participate in astrocyte reactivity by UCB and corroborating microglia faster response to stimuli (Liu et al., 2011a). Release of pro-inflammatory cytokines, as well as of ROS and NOS, can disrupt nerve terminals activity causing dysfunction and loss of synapses by UCB (Haustein et al., 2010). Therefore, UCB-induced inflammatory processes have emerged as a critical concept to understand the neurotoxic effects of UCB.
Unconjugated bilirubin-induced immunostimulation involves the activation of NF-κB signal transduction pathway in both astrocytes (Fernandes et al., 2006, 2007a, 2011) and microglia (Silva et al., 2010), one of the most important determinants of inflammatory gene expression and cytokine synthesis in glia (Table 1; Figure 7). Activation of NF-κB was mainly evidenced in immature astrocytes and increase by risk factors common in neonatal life (Falcão et al., 2007b), pointing to an added determinant for the susceptibility of preterm infants. Interestingly, activation of this transcription factor evidenced to precede MAPK activation and to be directly associated with TNF-α and IL-1β secretion, as well as with the loss of cell membrane integrity, but not the release of glutamate by astrocytes (Fernandes et al., 2006, 2007a; Brites et al., 2009). As already noticed for MAPKs activation and cytokine release, activation of NF-κB was faster in microglia than in astrocytes, as well (Figure 6D). It should be emphasized that activation of NF-κB, although not as markedly as in astrocytes and microglia, was also induced in neurons cultured with UCB (Figure 2), mainly in the immature ones (Falcão et al., 2006). However, only mild levels of IL-6, even less of TNF-α, and undetectable of IL-1β secretion were obtained under UCB exposure. Actually, NF-κB has diverse functions in the CNS, depending on the cellular context. Activation of NF-κB in neurons is implicated in axon growth, which is important for long-term memory (Kaltschmidt and Kaltschmidt, 2009; Teng and Tang, 2010). In this context, NF-κB in the synapse can be activated and transported retrogradely to the cell nucleus to regulate neural development, plasticity, and even neurogenesis (Gutierrez and Davies, 2011; Yakovleva et al., 2011). Thus, it is recognized as a major regulator of the growth and morphology of process initiation to stages when neurons are establishing functional connections and dendritic spines (Gutierrez and Davies, 2011). Collectively, these findings indicate that while NF-κB inhibition in glia might ameliorate disease, induced activation in neurons might enhance memory.
Associated conditions, such as sepsis, during, or following moderate to severe hyperbilirubinemia, are believed to contribute to BIND (Connolly and Volpe, 1990; Kaplan and Hammerman, 2005) and bacterial infection is considered a risk factor in the development of kernicterus (Pearlman et al., 1980; Yeung and Ngai, 2001; Figure 9). Moreover, in a Nigerian study, sepsis was the sole cause of hyperbilirubinaemia in 5% of the cases with severe neonatal jaundice (Dawodu et al., 1984). In in-vitro experimental conditions, addition of lipopolyssacharide (LPS) to cultures of astrocytes or neurons was shown to increase UCB-induced cell death (Figures 3A,B), as well as the release of TNF-α (Figure 3D) and IL-1β by astrocytes (Fernandes et al., 2004).
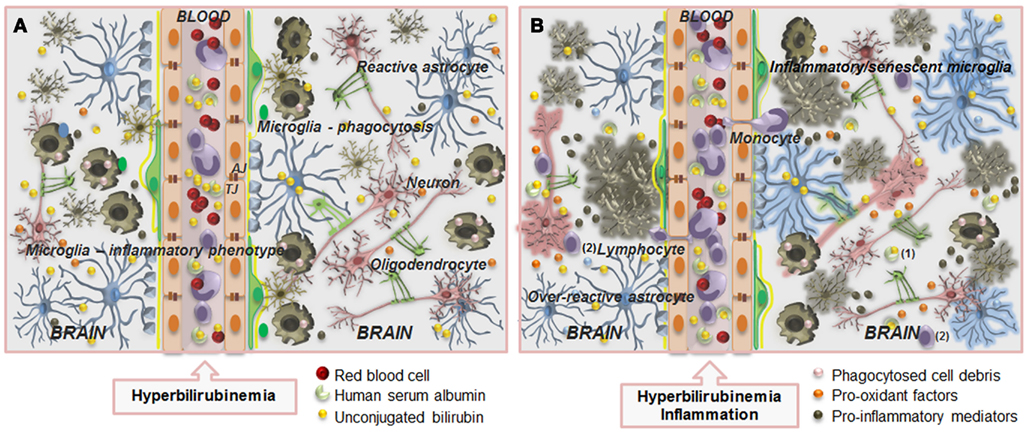
Figure 9. Overview of the main alterations occurring at the blood–brain barrier (BBB) and in the interplay between neurons and glial cells during severe hyperbilirubinemia (A) and associated infection (B). The BBB is composed by endothelial cells (orange) and their tight (TJ) and adherens junctions (AJ), astrocyte end-feet (light blue spots), perivascular microglia (greenish-brown), pericytes (light-green), and the basement membrane (yellow). Circulation of unconjugated bilirubin (UCB) in the blood (yellow bullets) is provided by its binding to human serum albumin, but when the UCB levels greatly increase, interaction of unbound (free) fraction of UCB (Bf) with the BBB is enhanced, as well as its passage into the brain (A), by passive or facilitated diffusion. Entrance will be also facilitated by the increased number of caveolae and caveolin-1 upregulation. Then, mainly after long-term exposure, UCB decreases zonula occludens-1 and β-catenin levels, and thus TJ strands and cell-to-cell contacts, favoring UCB passage across the BBB. Pericytes will be damaged and glutathione homeostasis disruption, together with increased nitric oxide synthase expression, nitrite production, and cytokine release will cause endothelial cell degeneration. In the brain parenchyma, elevated levels of UCB will surpass protective mechanisms (see Figure 1) and will cause neuron (pink) degeneration and release of pro-oxidant factors (orange bullets), microglia activation that will first phagocyte debris (pink bullets), and together with astrocytes (blue) reactivity will secrete pro-inflammatory mediators (greenish-brown bullets). In addition, decreased number of myelinating oligodendrocytes (solid-green) is also observed. (B) When sepsis is associated to hyperbilirubinemia, increased alterations at structural and functional levels are produced. Endothelial cell and astrocyte end-feet degeneration, modified basement membrane composition, tight junction, and transporter system impairment, vascular protein leakage, extensive extracellular edema, and activation of metalloproteinases are hallmarks of BBB impairment. Facilitated entrance of blood-borne harmful substances may accelerate neuron degeneration and activate microglia and reactive astrocytes leading to the release of cytokines further damaging neurons and detrimentally affecting vascular endothelium. Cross-talk between monocytes and endothelial cells in this inflammatory state leads to reciprocal activation of the two cell types, allowing the diapedesis of monocytes and lymphocytes. Leukocytes extravasation across the activated endothelium includes its arrest and adhesion, as well as alterations in their morphology and transmigration. Alterations in BBB dynamic properties may favor the presence of albumin carrying UCB (1) and increased passage of lymphocytes (2) into the brain parenchyma. In this condition, astrocytes become over-reactive, microglia will acquire inflammatory/senescent phenotypes and both neurons and oligodendrocytes will degenerate. Thus, therapeutic strategies directed at controlling the activation of microglia and astrocytes, and the excessive production of pro-inflammatory mediators and pro-oxidant factors may add on the prevention and recovery of neurologic sequelae resulting from unconjugated hyperbilirubinemia.
Addition of LPS to astrocytes has revealed to increase UCB-induced necrosis mainly in mature cells (Figure 3A), apoptosis in old ones (Figure 3B; Falcão et al., 2005), and the release of IL-1β (Fernandes et al., 2004) and TNF-α at all cell ages (Falcão et al., 2005). Interestingly, increased secretion of TNF-α by LPS was also evidenced in immature neurons when co-incubated with UCB (Figure 3D). Although LPS has previously shown to inhibit glutamate uptake (Parry et al., 2010), elevation of extracellular glutamate when neurons, astrocytes, and microglia were simultaneously treated with LPS and UCB was not sensed (Figure 3C; Fernandes et al., 2004; Falcão et al., 2005). The same was observed in astrocytes when co-incubated with UCB and TNF-α (Fernandes et al., 2004). However, when neurons were treated with UCB plus TNF-α + IL-1β, these cytokines were shown to have cumulative effects on the generation of NO and activation of nNOS, JNK1/2, and caspase cascades (Vaz et al., 2011b). Increased cleavage of Bid into truncated Bid (tBid), as well as cytotoxic potential, was also observed (Figure 2; Table 1). These events provide a reason for the risk of sepsis in BIND and point it as a potential target for therapeutic intervention in the BIND prevention. Also remarkable was the observation that neurons previously exposed to UCB evidence an increased susceptibility to a subsequent inflammatory insult with LPS or TNF-α (Falcão et al., 2007c), suggesting that priming of neurons by UCB may result in long-lasting neurological dysfunction, as observed for early-life seizures through enhanced and persistent glial activation in adult life (Somera-Molina et al., 2007). Inflammatory priming was also shown to influence dopaminergic neurons sensitivity to subsequent environmental toxins and suggested to contribute to neurodegenerative diseases (Mangano and Hayley, 2009). Moreover, microglial priming by acute or chronic stress was shown to predispose the brain to neurodegeneration and to affect disease progression as well (Frank et al., 2012; Ramaglia et al., 2012). Microglia activation was observed in the Gunn rats at 8 weeks of age and suggested to contribute to chronic neuronal inflammation (Liaury et al., 2012) and behavioral abnormalities similar to schizophrenia (Hayashida et al., 2009). These findings may have a high relevance in the evaluation of potential lasting sequelae by unconjugated hyperbilirubinemia and surely deserve further investigation.
Prostaglandin E (PGE), also considered one mediator in neuroinflammatory processes, originates from the degradation of arachidonic acid through the sequential actions of COX-1 and COX-2, and three terminal PGE synthases (PGES) one cytosolic and two membrane associated PGES, the mPGES-1 and the mPGES-2. COX-1 is expressed constitutively at very low levels in the brain, while COX-2, the inducible isoform, is acutely expressed in several cell types after brain injury (Zhuang et al., 2003). COX-2 is induced by pro-inflammatory challenges (Font-Nieves et al., 2012) and pointed as a defensive response to the excess of glutamate, at least in neurons (Bidmon et al., 2000). LPS was shown to significantly induce PGES-1 in microglia (de Oliveira et al., 2008), as well as PGE2 release from astrocytes by the stimulation of both the constitutive and inducible COX isoforms (Pistritto et al., 1998, 1999). Intriguingly, studies in COX-deficient cells and using COX inhibitors evidenced that COX-2 mediated the production of PGE2, and COX-1 deficiency further increased PGE2 production after LPS treatment (Font-Nieves et al., 2012). In fact, the contribution of these enzymes varies accordingly with the stimuli and cell type. While UCB/HSA = 0.5 revealed to moderately increase COX-2 in microglia (Silva et al., 2010) no effects on basal PGE2 release were produced by 10 μM Bf on primary cultures of rat hypothalamic astrocytes (Mancuso et al., 1997). Nevertheless, in rat cortical astrocytes the same Bf amount was able to increase PGE2 production (Vairano et al., 2001). Since no other studies on PGE2 increased production by UCB in the presence or in the absence of HSA can be found in the literature, further studies should attest on the effects of UCB on the PGE2 production, on whether it should be taken or not as an index of inflammation, and on its invariable coupling to COX-2 activation, since LPS-induced mPGES-1 synthesis showed to not strictly be coupled to the synthesis of COX-2 in activated primary rat microglia (de Oliveira et al., 2008).
Neuronal and glial interplay may reinforce the cascade of inflammatory signaling in astrocytes and microglia interfering with the reactivity of each other and enhancing detrimental outcomes in neurons (Figure 7). Conditioned media obtained from either UCB-treated neurons (NCM) or UCB-treated astrocytes (ACM) revealed to be able to modulate microglia response, leading to an enhanced secretion of IL-6 while decreasing that IL-1β (Silva et al., 2011). NCM also showed to down-regulate TNF-α production by microglia, and to increase NO production and MMPs activities, thus accounting to microglia demise. In contrast, ACM protected microglia from loss of viability by UCB, in line with the property of reactive astrocytes in facilitating activation of distant microglia, while inhibiting microglial activities (Liu et al., 2011a) and preventing overactivation (Renault-Mihara et al., 2008). These results emphasize the ability of neurons and astrocytes to modulate the pro-inflammatory properties of microglia. However, evaluation of neuroprotective or neurotoxic responsiveness of cells to a certain stimulus is only possible if unrestricted dialog between neurons and glial cells is achieved, as the one allowed by organotypic-cultured slices.
Brain slice models offer unique advantages over other in vitro platforms in that interactions between neurons or between neurons and glial cells are fundamentally preserved replicating many aspects of the in vivo context (Cho et al., 2007; Lossi et al., 2009). In hippocampal slices incubated with UCB microglia showed to contribute to the release of NO, thus potentiating nitrosative stress. However, and alike astrocytes (Matute et al., 2007), microglia seem to participate in the homeostasis of glutamate by promoting its uptake (Silva et al., 2012), as a way of contributing to restoration of homeostasis (Shaked et al., 2005). In fact, microglia express the glutamate transporter 1 (Nakajima et al., 2001), thus reinforcing the neuroprotective role of microglia (see Role of Neuroinflammation in BIND). By using organotypic-cultured hippocampal slices, depleted, or not-depleted in microglia (Silva et al., 2012), the highest extracellular levels of glutamate were obtained in microglia-depleted slices, thus corroborating the participation of microglia in the homeostasis of glutamate, protecting neurons against excitotoxicity. Indeed, it was recently considered that depending on the stimuli, microglia can adopt a phenotype that facilitates rather than impairs glutamate clearance, as a way of contributing to restoration of homeostasis (Shaked et al., 2005). Nevertheless, other studies underscore glutamate’s key role in microglial neurotoxicity (Barger and Basile, 2001; Liang et al., 2008) that may be related to a more “silent” microglia resulting from detrimental effects over the course of the injury upon various regulatory processes (Sargsyan et al., 2011; Harry and Kraft, 2012).
Once degeneration of glial cells has direct deleterious effects on neuronal function and survival, it will be then important to look for UCB-induced pathophysiological alterations as integrated phenomena (Figure 7), only achieved with organotypic slice cultures. These cultures will be ideal for looking at the mechanisms of neuroinflammation and the death of neurons. Their potential for drug discovery is substantial once microglial and astrocytic activation could be specifically targeted to preserve cross-talk between neurons and glia, constituting new strategic therapeutics to prevent BIND.
Bind: in Search of Potential Neurotherapeutics
Incidence of kernicterus varies from 1:30 000 to 1:100 000 births (Manning et al., 2007), but can be higher in developing countries. Excessive hyperbilirubinemia in human neonates may cause permanent dysfunction of the auditory system, as assessed with BAEPs (Rice et al., 2011). Phototherapy has been a widely and successfully used therapy to reduce the levels of UCB by transforming UCB in the skin into water-soluble isomers that are cleared without the need of liver conjugation (Stokowski, 2011). Guidelines for the management of hyperbilirubinemia in newborn infants and recommendations to use phototherapy were published by the American Academy of Pediatrics (2004) and Bhutani (2011), respectively. If phototherapy fails in reducing UCB levels, or at the readmission of very ill infants, aggressive management, as exchange transfusion (ET), is usually recommended. Administration of albumin 1 h prior to ET was shown to significantly reduce the tissue-bound UCB and to increase ET efficacy (Shahian and Moslehi, 2010; Mitra et al., 2011). However, ET is not always successful in preventing acute bilirubin encephalopathy and adverse outcome (Mukhopadhyay et al. 2010), and complications such as thrombocytopenia and seizure were indicated to be present in 21.5% of jaundiced neonates performing such intervention (Davutoglu et al., 2010). Since kernicterus continues to occur whereas it ought to be avoidable (Hansen, 2011), recognition of new therapeutic modulation capacities as adjunctive therapies may contribute to a more consistent regimen for treatment and prevention of kernicterus.
Furthermore, once the threshold for UCB neurotoxicity is unknown, and kernicterus was only evidenced in 5% of yellow brains ensuing from jaundiced infants at autopsy, as described by Schmorl and cited by Hansen (2000b), these other cases probably resultant from moderate levels of UCB may also have harmful consequences. We speculate that neonatal hyperbilirubinemia, like perinatal infection/inflammation (Favrais et al., 2011), may disrupt the neurodevelopmental program and lead to potential lifetime effects, mainly in preterm infants, warranting an intensified search for neuroprotective actions.
Minocycline can be an effective treatment in neurological diseases associated with both oxidative stress (Zhong and Lee, 2007) and inflammation (Chu et al., 2007), by inhibiting microglia activation (Tikka et al., 2001; Suzuki et al., 2010), caspase-1 expression (Kim and Suh, 2009), and both caspase-dependent and -independent programmed cell death (Ossola et al., 2012; Figure 10). It also induces autophagic cell death (Liu et al., 2011b) and decreases glutamate excitotoxicity (Maier et al., 2007), NO production (Levkovitz et al., 2007), activation of NF-κB (Cai et al., 2011), formation of ROS (Schildknecht et al., 2011), MMP-9 expression (Guo et al., 2011), and the delayed responses of microglia (Yamada and Jinno, 2011). Its efficacy has been tested in Gunn rats, where it was shown to almost completely block the loss of Purkinje and granule neurons, probably by inhibiting p38 activation (Lin et al., 2005). Minocycline has a graded neuroprotective capability in acute UCB neurotoxicity induced by sulfadimethoxine in Gunn rat pups; while minocycline administration 30 min post-sulfadimethoxine totally prevented alterations in BAEPs, the minocycline 120 min post-sulfadimethoxine did not work anymore (Rice et al., 2011). In addition, it deserves to be noticed that minocycline has several side effects such as gastrointestinal symptoms, pediatric tooth discoloration, and dizziness, among many others (Smith and Leyden, 2005). Thus, applications in clinical settings dealing with pregnant women and children are restricted and contentious.
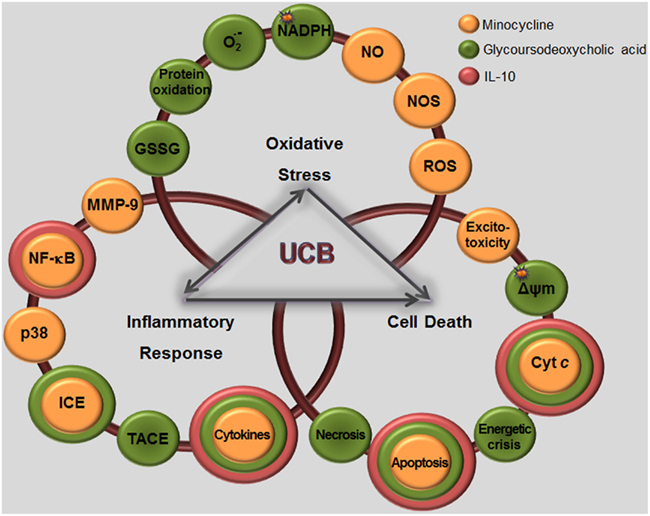
Figure 10. Shematic representation of some potential neuroprotective, antioxidant, and anti-inflammatory adjunctive therapies for preventing bilirubin-induced neurological dysfunction. Given the marked oxidative stress, neuroinflammatory response, and cell death elicited by unconjugated bilirubin (UCB) in neurons and glial cells, it will be interesting to revise the potentialities of some particular antioxidants and immunomodulatory agents such as the interleukin (IL)-10, the bile acid glycoursodeoxycholic acid (GUDCA), and the antibiotic minocycline. IL-10 revealed to reduce the UCB-induced release of cytokines such as tumor necrosis factor (TNF)-α and IL-1β, as well as nuclear factor-kappaB (NF-κB) activation and translocation to the nucleus. In other conditions, IL-10 prevented apoptosis through the inhibition of the cytochrome c (Cytc) release and of the caspase-3 cleavage, pointing to its antiapoptic properties. Neuroprotective effects of the synthetic tetracycline minocycline include: (i) cytoprotective, by decreasing glutamate and excitotoxicity; (ii) anti-apoptotic, by reducing the release of Cytc and therefore apoptosis; (iii) anti-inflammatory by decreasing the activity of the metalloproteinase-9 (MMP-9), of caspase-1 or IL-1β converting enzyme (ICE) and of NF-κB, and specifically directed to UCB immunostimulation by preventing p38 phosphorylation, cytokine release, and cell death; (iv) antioxidant by reducing reactive oxygen species (ROS) formation, as well as nitric oxide synthase (NOS) expression and NO production. Concerning GUDCA benefits, it prevents the inflammatory effects of UCB by reducing both TNF-α and IL-1β release through the inhibition of TNF-α converting enzyme (TACE) and ICE inhibition. GUDCA also prevented UCB-induced energetic crisis by counter acting cytochrome c oxidase or complex IV inhibition, oxygen consumption, extracellular ATP increase, intracellular lactate, fructose-2,6-bisphosphate, oxidized glutathione (GSSG) increase, protein oxidation, production of superoxide anion radical () and NADPH reduction, as well as necrosis and apoptosis by avoiding the collapse in the mitochondrial transmembrane potential (ΔΨm) and caspase activity. Taken together, minocycline and GUDCA may prove to have additional benefits over IL-10 in preserving neural cells functionality in cases of lasting unconjugated hyperbilirubinemia, by abolishing and/or decreasing the dangerous determinants of UCB brain injury, and reestablishing the compromised ones, marked with stars. However, in what concerns minocycline, applications in clinical settings dealing with pregnant women and children are restricted and contentious.
Ursodeoxycholic acid (UDCA) is an endogenous bile acid used for the treatment of hepatobiliary disorders (Lazaridis et al., 2001). Following oral administration, it is conjugated originating tauroursodeoxycholic acid and, mostly, glycoursodeoxycholic acid (GUDCA), which acquires the highest clinical relevance (Rudolph et al., 2002). Recent data indicate that unconjugated and conjugated UDCA species have anti-apoptotic, antioxidant, and anti-inflammatory effects in nerve cells (Fernandes et al., 2007b; Solá et al., 2007; Brito et al., 2008a), pointing to their therapeutic potential for CNS disorders (Parry et al., 2010; Fonseca et al., 2012). UDCA has also shown to have beneficial effects on itching and in preventing liver failure in children with progressive familial intrahepatic cholestasis (Davit-Spraul et al., 2010), as well as in recovering liver function in patients with intrahepatic cholestasis of pregnancy, a condition with fetal poor prognosis resulting from increased transfer of bile acids from mother to fetus (Brites et al., 1998c). In this disorder, UDCA is administered until labor, decreasing the content of bile acids in the maternal serum, amniotic fluid, cord blood serum, meconium and colostrum, improving fetal prognosis and showing to be safe for the mother and babies (Brites and Rodrigues, 1998; Brites et al., 1998a,b; Brites, 2002). Thus, no secondary effects were observed in newborns from UDCA-treated mothers, which showed a better liver function than the non-treated babies (Mazzella et al., 2001). Interestingly, UDCA has shown to decrease unconjugated hyperbilirubinemia in the Gunn rat model, supporting its application in preventing BIND (Cuperus et al., 2009).
In the neurotoxic effects produced by UCB in vitro, UDCA revealed ability to prevent both necrosis and apoptosis in cultured rat astrocytes and neurons (Figure 10; Silva et al., 2001b). Experiments with GUDCA produced similar results in astrocytes (Fernandes et al., 2007b). Interestingly, GUDCA also prevented the UCB-induced release of TNF-α and IL-1β, but only reduced the TNF-α mRNA induction. Curiously, it was observed that UCB increases TACE and ICE activities, which were counteracted in the presence of GUDCA, leading to the increase of the cytokine pro-forms at the cytosol. Thus, whereas UCB promotes the maturation of the cytokine pro-forms and their consequent release, GUDCA inhibits their activation. Nevertheless, GUDCA did not modulate either glutamate release or NF-κB activation. In addition, it was observed that it prevents the decrease of GSH produced by UCB in neurons together with the oxidation of proteins (Brito et al., 2008a). GUDCA also completely precludes the inhibition of the cytochrome c oxidase activity by UCB, the increase of the production and the GSSG formation, as well as the decrease in NADPH concentration and in oxygen consumption (Vaz et al., 2010). The ability of GUDCA to prevent UCB-induced energetic crisis and mitochondria dysfunction further supports the efficacy of this compound as a potential preventive therapy for BIND.
However, despite the beneficial effects that UDCA may have in early-life as a neuroprotectant, fundamental aspects of investigation are still outstanding. Harnessing the potential of UDCA for use in neonates clearly requires further investigation to identify definitively the window of opportunity for UDCA intervention once shortcoming in that most of the studies performed have been by administration to the mother in the course of the intrahepatic cholestasis of pregnancy.
IL-10 is another anti-inflammatory compound that was tested in in vitro conditions mimicking BIND. It did not produce any effect in both apoptotic and necrotic cell death of astrocytes, neither in glutamate release (Fernandes et al., 2007b). In contrast, it prevented UCB-induced release of TNF-α and IL-1β by down-regulating their expression, as well as abrogated NF-κB translocation to the nucleus. In a recent study of spinal cord injury it was used a herpes simplex virus-based vector to express IL-10 in spinal cord in vivo (Zhou et al., 2009). In this case, the cytokine was able to inhibit cytochrome c release and caspase-3 cleavage, pointing to the possibility that IL-10 may present separate effects from the anti-inflammatory properties. However, the anti-apoptotic effect may derive from the suppression of glial activation as previously observed by Pang et al. (2005).
The aminoacid taurine is not only a neurotransmitter, but also a trophic factor in CNS development. It maintains the structural integrity of the membrane and regulates calcium transport and homeostasis (Wu and Prentice, 2010). Taurine was initially indicated to have benefits in stroke together with anti-inflammatory effects (Yamori et al., 2010; Sun et al., 2012). In 2010, taurine has shown to prevent the loss of neuronal viability induced by UCB, the changes in neurite outgrowth and the influx of Ca2+from external microenvironment, but with limited efficacy on the release of Ca2+ from ER (Zhang et al., 2010). Later, Gao et al. (2011) observed the same properties in a jaundiced baby mice model established by intraperitoneal injection with UCB. When these mice were pretreated with taurine for 4 h, intracellular levels of Ca2+ were inhibited and down regulation of caspase-3 activity was achieved. Besides this anti-apoptotic effect, the new perspective for the use of taurine for preventing and/or treating neural damage in neonatal jaundice comes from its anti-inflammatory potential.
Conclusion
Although we have still a long way to envisage the complex and diverse reactivity of astrocytes and microglia to UCB, and their cross-talk with neurons in BIND conditions, in the present review we attempted to provide information on how individual cell response depends on the time of exposure to UCB, on the associated stimuli and on the presence of each type of neural cell. Receptor activation by UCB and signaling cascades followed by injury due to lipid peroxidation and protein oxidation starts at the cell membrane level. Targeted subcellular compartments by UCB are ER, mitochondria and nucleus that orchestrate cell death by apoptosis and necrosis. Alterations induced by UCB on neurogenesis, neuritogenesis, spinogenesis, and on axonal cytoskeleton dynamics are suggestive of synaptic plasticity abnormalities and lasting neurodevelopmental disabilities that should be further investigated for their relevance and priming effects. The extracellular increase in glutamate, in pro-oxidant factors, and the presence of sepsis or inflammation that further enhances the susceptibility of neurons to UCB, even more in immature cells explain, at least in part, the vulnerability of premature infants to BIND. How temporal windows of neurodevelopment will influence and determine increased brain damage by BIND and different type or sequelae at early ages surely deserve additional studies, but preliminary data suggest that infants in the first postnatal week are more susceptible to BIND than in the second week of life. UCB-induced reactivity of microglia, although initially triggering a neuroprotective phagocytosis of damaged neurons, decreasing their number but restricting the area of damage, later it leads to the release of pro-inflammatory mediators that activate TNF-α and IL-1β receptor signaling pathways in astrocytes, culminating in an additional production of pro-inflammatory cytokines mediated by MAPKs and NF-κB activation pathways. How far is the microglia reactivity modulated by the other glial cells and neurons during unconjugated hyperbilirubinemia is just beginning to be studied. By interfering with OPCs differentiation and proliferation, UCB can directly damage myelinogenesis, although activated microglia can similarly produce the same effects, thus probably contributing to enhance the damage of UCB to OPCs. Therefore, therapeutic strategies such as GUDCA, IL-10, taurine, and minocycline directed at controlling the activation of microglia and astrocytes, and the excessive production of pro-inflammatory and pro-oxidant factors, may be valuable to control BIND and prevent long-lasting vulnerabilities to elderly and adult neurodegenerative diseases resultant from glial sensitization or priming. Further studies using the organotypic slice culture model will contribute to better identify risks, targets, and determinants of BIND and to improve our knowledge on nerve cell pathological alterations as integrated phenomena. It also will have an important role in the pharmacological assessment of novel preventive and curative approaches, as well as strategies based on new therapeutic indications. In closing, dysregulation of “neurodevelopmental programming” by moderate and high levels of UCB, enduring impacts on brain microglial function, and risk for cognitive aging disorders warrant further research that should lead to innovative treatments.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The author wishes to express her appreciation to Rui Silva, Maria Alexandra Brito, Adelaide Fernandes, Ana Sofia Falcão, Ana Rita Vaz, and Sandra Silva who participated in the realization of the publications in which the findings are described in this review. The authors’s experimental work and the studies from the author’s Research Unit were funded by the PTDC/SAU-NEU/64385/2006 grant (to Dora Brites) and by the strategic project PEst-OE/SAU/UI4013/2011 (to iMed.UL and to author’s Neuron Glia Biology in Health and Disease Research Unit), from Fundação para a Ciência e a Tecnologia (FCT), Lisbon, Portugal.
References
Ahdab-Barmada, M., and Moossy, J. (1984). The neuropathology of kernicterus in the premature neonate: diagnostic problems. J. Neuropathol. Exp. Neurol. 43, 45–56.
Ahlfors, C. E., Amin, S. B., and Parker, A. E. (2009a). Unbound bilirubin predicts abnormal automated auditory brainstem response in a diverse newborn population. J. Perinatol. 29, 305–309.
Ahlfors, C. E., Wennberg, R. P., Ostrow, J. D., and Tiribelli, C. (2009b). Unbound (free) bilirubin: improving the paradigm for evaluating neonatal jaundice. Clin. Chem. 55, 1288–1299.
Ahlfors, C. E., and Parker, A. E. (2008). Unbound bilirubin concentration is associated with abnormal automated auditory brainstem response for jaundiced newborns. Pediatrics 121, 976–978.
Allaman, I., Belanger, M., and Magistretti, P. J. (2011). Astrocyte-neuron metabolic relationships: for better and for worse. Trends Neurosci. 34, 76–87.
American Academy of Pediatrics. (2004). Management of hyperbilirubinemia in the newborn infant 35 or more weeks of gestation. Pediatrics 114, 297–316.
Amin, S. B., Ahlfors, C., Orlando, M. S., Dalzell, L. E., Merle, K. S., and Guillet, R. (2001). Bilirubin and serial auditory brainstem responses in premature infants. Pediatrics 107, 664–670.
Amit, Y., and Brenner, T. (1993). Age-dependent sensitivity of cultured rat glial cells to bilirubin toxicity. Exp. Neurol. 121, 248–255.
Arai, K., Jin, G., Navaratna, D., and Lo, E. H. (2009). Brain angiogenesis in developmental and pathological processes: neurovascular injury and angiogenic recovery after stroke. FEBS J. 276, 4644–4652.
Arai, K., and Lo, E. H. (2009). Experimental models for analysis of oligodendrocyte pathophysiology in stroke. Exp. Transl. Stroke Med. 1, 6.
Barateiro, A., Vaz, A. R., Silva, S. L., Fernandes, A., and Brites, D. (2010). Unconjugated bilirubin induces oligodendrocyte progenitor cell death following a programmed course of intracellular events. J. Neuroimmunol. 228, 27.
Barger, S. W., and Basile, A. S. (2001). Activation of microglia by secreted amyloid precursor protein evokes release of glutamate by cystine exchange and attenuates synaptic function. J. Neurochem. 76, 846–854.
Bellarosa, C., Bortolussi, G., and Tiribelli, C. (2009). The role of ABC transporters in protecting cells from bilirubin toxicity. Curr. Pharm. Des. 15, 2884–2892.
Bellefontaine, N., Hanchate, N. K., Parkash, J., Campagne, C., de Seranno, S., Clasadonte, J., d’Anglemont de Tassigny, X., and Prevot, V. (2011). Nitric oxide as key mediator of neuron-to-neuron and endothelia-to-glia communication involved in the neuroendocrine control of reproduction. Neuroendocrinology 93, 74–89.
Bhutani, V. K. (2011). Phototherapy to prevent severe neonatal hyperbilirubinemia in the newborn infant 35 or more weeks of gestation. Pediatrics 128, e1046–e1052.
Bhutani, V. K., Johnson, L. H., and Shapiro, S. M. (2004). Kernicterus in sick and preterm infants (1999–2002): a need for an effective preventive approach. Semin. Perinatol. 28, 319–325.
Bhutani, V. K., Maisels, M. J., Stark, A. R., and Buonocore, G. (2008). Management of jaundice and prevention of severe neonatal hyperbilirubinemia in infants >or=35 weeks gestation. Neonatology 94, 63–67.
Bidmon, H. J., Oermann, E., Schiene, K., Schmitt, M., Kato, K., Asayama, K., Witte, O. W., and Zilles, K. (2000). Unilateral upregulation of cyclooxygenase-2 following cerebral, cortical photothrombosis in the rat: suppression by MK-801 and co-distribution with enzymes involved in the oxidative stress cascade. J. Chem. Neuroanat. 20, 163–176.
Billing, B. (1972). The Gunn rat: an animal model for the study of jaundice. Proc. R. Soc. Med. 65, 350–351.
Bradl, M., and Lassmann, H. (2010). Oligodendrocytes: biology and pathology. Acta Neuropathol. 119, 37–53.
Brites, D. (2002). Intrahepatic cholestasis of pregnancy: changes in maternal-fetal bile acid balance and improvement by ursodeoxycholic acid. Ann. Hepatol. 1, 20–28.
Brites, D. (2011). Bilirubin injury to neurons and glial cells: new players, novel targets, and newer insights. Semin. Perinatol. 35, 114–120.
Brites, D., Fernandes, A., Falcão, A. S., Gordo, A. C., Silva, R. F., and Brito, M. A. (2009). Biological risks for neurological abnormalities associated with hyperbilirubinemia. J. Perinatol. 29(Suppl. 1), S8–S13.
Brites, D., and Rodrigues, C. M. (1998). Elevated levels of bile acids in colostrum of patients with cholestasis of pregnancy are decreased following ursodeoxycholic acid therapy [see comments]. J. Hepatol. 29, 743–751.
Brites, D., Rodrigues, C. M., Cardoso Mda, C., and Graca, L. M. (1998a). Unusual case of severe cholestasis of pregnancy with early onset, improved by ursodeoxycholic acid administration. Eur. J. Obstet. Gynecol. Reprod. Biol. 76, 165–168.
Brites, D., Rodrigues, C. M., Oliveira, N., Cardoso, M., and Graca, L. M. (1998b). Correction of maternal serum bile acid profile during ursodeoxycholic acid therapy in cholestasis of pregnancy. J. Hepatol. 28, 91–98.
Brites, D., Rodrigues, C. M., van-Zeller, H., Brito, A., and Silva, R. (1998c). Relevance of serum bile acid profile in the diagnosis of intrahepatic cholestasis of pregnancy in an high incidence area: Portugal. Eur. J. Obstet. Gynecol. Reprod. Biol. 80, 31–38.
Brites, D., Silva, R., and Brito, A. (1997). Effect of bilirubin on erythrocyte shape and haemolysis, under hypotonic, aggregating or non-aggregating conditions, and correlation with cell age. Scand. J. Clin. Lab. Invest. 57, 337–349.
Brito, M. A., Brites, D., and Butterfield, D. A. (2004). A link between hyperbilirubinemia, oxidative stress and injury to neocortical synaptosomes. Brain Res. 1026, 33–43.
Brito, M. A., Brondino, C. D., Moura, J. J., and Brites, D. (2001). Effects of bilirubin molecular species on membrane dynamic properties of human erythrocyte membranes: a spin label electron paramagnetic resonance spectroscopy study. Arch. Biochem. Biophys. 387, 57–65.
Brito, M. A., Lima, S., Fernandes, A., Falcão, A. S., Silva, R. F., Butterfield, D. A., and Brites, D. (2008a). Bilirubin injury to neurons: contribution of oxidative stress and rescue by glycoursodeoxycholic acid. Neurotoxicology 29, 259–269.
Brito, M. A., Rosa, A. I., Falcão, A. S., Fernandes, A., Silva, R. F., Butterfield, D. A., and Brites, D. (2008b). Unconjugated bilirubin differentially affects the redox status of neuronal and astroglial cells. Neurobiol. Dis. 29, 30–40.
Brito, M. A., Silva, R. F., and Brites, D. (2002). Bilirubin induces loss of membrane lipids and exposure of phosphatidylserine in human erythrocytes. Cell Biol. Toxicol. 18, 181–192.
Brito, M. A., Vaz, A. R., Silva, S. L., Falcão, A. S., Fernandes, A., Silva, R. F., and Brites, D. (2010). N-methyl-aspartate receptor and neuronal nitric oxide synthase activation mediate bilirubin-induced neurotoxicity. Mol. Med. 16, 372–380.
Brito, M. A., Zurolo, E., Pereira, P., Barroso, C., Aronica, E., and Brites, D. (2012). Cerebellar Axon/Myelin Loss, angiogenic sprouting, and neuronal increase of vascular endothelial growth factor in a preterm infant with kernicterus. J. Child Neurol. 27, 615–624.
Cai, Z., Zhao, Y., Yao, S., and Bin Zhao, B. (2011). Increases in beta-amyloid protein in the hippocampus caused by diabetic metabolic disorder are blocked by minocycline through inhibition of NF-kappaB pathway activation. Pharmacol. Rep. 63, 381–391.
Calligaris, R., Bellarosa, C., Foti, R., Roncaglia, P., Giraudi, P., Krmac, H., Tiribelli, C., and Gustincich, S. (2009). A transcriptome analysis identifies molecular effectors of unconjugated bilirubin in human neuroblastoma SH-SY5Y cells. BMC Genomics 10, 543.
Cardoso, F. L., Brites, D., and Brito, M. A. (2010). Looking at the blood-brain barrier: molecular anatomy and possible investigation approaches. Brain Res. Rev. 64, 328–363.
Carson, M. J., Bilousova, T. V., Puntambekar, S. S., Melchior, B., Doose, J. M., and Ethell, I. M. (2007). A rose by any other name? The potential consequences of microglial heterogeneity during CNS health and disease. Neurotherapeutics 4, 571–579.
Cashore, W. J., Oh, W., and Brodersen, R. (1983). Reserve albumin and bilirubin toxicity index in infant serum. Acta Paediatr. Scand. 72, 415–419.
Chang, F. Y., Lee, C. C., Huang, C. C., and Hsu, K. S. (2009). Unconjugated bilirubin exposure impairs hippocampal long-term synaptic plasticity. PLoS ONE 4, e5876.
Chao, C. C., and Hu, S. (1994). Tumor necrosis factor-alpha potentiates glutamate neurotoxicity in human fetal brain cell cultures. Dev. Neurosci. 16, 172–179.
Chen, H. C., Tsai, D. J., Wang, C. H., and Chen, Y. C. (1969). An electron microscopic and radioautographic study on experimental kernicterus. I. Bilirubin transport via astroglia. Am. J. Pathol. 56, 31–58.
Chen, H. C., Wang, C. H., Tsan, K. W., and Chen, Y. C. (1971). An electron microscopic and radioautographic study on experimental kernicterus. II. Bilirubin movement within neurons and release of waste products via astroglia. Am. J. Pathol. 64, 45–66.
Cho, S., Wood, A., and Bowlby, M. R. (2007). Brain slices as models for neurodegenerative disease and screening platforms to identify novel therapeutics. Curr. Neuropharmacol. 5, 19–33.
Chu, L. S., Fang, S. H., Zhou, Y., Yu, G. L., Wang, M. L., Zhang, W. P., and Wei, E. Q. (2007). Minocycline inhibits 5-lipoxygenase activation and brain inflammation after focal cerebral ischemia in rats. Acta Pharmacol. Sin. 28, 763–772.
Conforti, L., Adalbert, R., and Coleman, M. P. (2007). Neuronal death: where does the end begin? Trends Neurosci. 30, 159–166.
Conlee, J. W., and Shapiro, S. M. (1997). Development of cerebellar hypoplasia in jaundiced Gunn rats: a quantitative light microscopic analysis. Acta Neuropathol. 93, 450–460.
Connolly, A. M., and Volpe, J. J. (1990). Clinical features of bilirubin encephalopathy. Clin. Perinatol. 17, 371–379.
Corich, L., Aranda, A., Carrassa, L., Bellarosa, C., Ostrow, J. D., and Tiribelli, C. (2009). The cytotoxic effect of unconjugated bilirubin in human neuroblastoma SH-SY5Y cells is modulated by the expression level of MRP1 but not MDR1. Biochem. J. 417, 305–312.
Cullheim, S., and Thams, S. (2007). The microglial networks of the brain and their role in neuronal network plasticity after lesion. Brain Res. Rev. 55, 89–96.
Cuperus, F. J., Hafkamp, A. M., Havinga, R., Vitek, L., Zelenka, J., Tiribelli, C., Ostrow, J. D., and Verkade, H. J. (2009). Effective treatment of unconjugated hyperbilirubinemia with oral bile salts in Gunn rats. Gastroenterology 136, 673–682 e671.
Daood, M., Tsai, C., Ahdab-Barmada, M., and Watchko, J. F. (2008). ABC transporter (P-gp/ABCB1, MRP1/ABCC1, BCRP/ABCG2) expression in the developing human CNS. Neuropediatrics 39, 211–218.
Daood, M. J., McDonagh, A. F., and Watchko, J. F. (2009). Calculated free bilirubin levels and neurotoxicity. J. Perinatol. 29(Suppl. 1), S14–S19.
Daulatzai, M. A. (2012). Dysfunctional nucleus tractus solitarius: its crucial role in promoting neuropathogentic cascade of Alzheimer’s dementia-a novel hypothesis. Neurochem. Res. 37, 846–868.
Davalos, D., Grutzendler, J., Yang, G., Kim, J. V., Zuo, Y., Jung, S., Littman, D. R., Dustin, M. L., and Gan, W. B. (2005). ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 8, 752–758.
Davit-Spraul, A., Fabre, M., Branchereau, S., Baussan, C., Gonzales, E., Stieger, B., Bernard, O., and Jacquemin, E. (2010). ATP8B1 and ABCB11 analysis in 62 children with normal gamma-glutamyl transferase progressive familial intrahepatic cholestasis (PFIC): phenotypic differences between PFIC1 and PFIC2 and natural history. Hepatology 51, 1645–1655.
Davutoglu, M., Garipardic, M., Guler, E., Karabiber, H., and Erhan, D. (2010). The etiology of severe neonatal hyperbilirubinemia and complications of exchange transfusion. Turk. J. Pediatr. 52, 163–166.
Dawodu, A. H., Owa, J. A., and Familusi, J. B. (1984). A prospective study of the role of bacterial infection and G6PD deficiency in severe neonatal jaundice in Nigeria. Trop. Geogr. Med. 36, 127–132.
De Keyser, J., Mostert, J. P., and Koch, M. W. (2008). Dysfunctional astrocytes as key players in the pathogenesis of central nervous system disorders. J. Neurol. Sci. 267, 3–16.
de Oliveira, A. C., Candelario-Jalil, E., Bhatia, H. S., Lieb, K., Hull, M., and Fiebich, B. L. (2008). Regulation of prostaglandin E2 synthase expression in activated primary rat microglia: evidence for uncoupled regulation of mPGES-1 and COX-2. Glia 56, 844–855.
Deganuto, M., Cesaratto, L., Bellarosa, C., Calligaris, R., Vilotti, S., Renzone, G., Foti, R., Scaloni, A., Gustincich, S., Quadrifoglio, F., Tiribelli, C., and Tell, G. (2010). A proteomic approach to the bilirubin-induced toxicity in neuronal cells reveals a protective function of DJ-1 protein. Proteomics 10, 1645–1657.
Depino, A., Ferrari, C., Pott Godoy, M. C., Tarelli, R., and Pitossi, F. J. (2005). Differential effects of interleukin-1beta on neurotoxicity, cytokine induction and glial reaction in specific brain regions. J. Neuroimmunol. 168, 96–110.
Doré, S., and Snyder, S. H. (1999). Neuroprotective action of bilirubin against oxidative stress in primary hippocampal cultures. Ann. N. Y. Acad. Sci. 890, 167–172.
Doré, S., Takahashi, M., Ferris, C. D., Zakhary, R., Hester, L. D., Guastella, D., and Snyder, S. H. (1999). Bilirubin, formed by activation of heme oxygenase-2, protects neurons against oxidative stress injury. Proc. Natl. Acad. Sci. U. S. A. 96, 2445–2450.
Duan, Y., Sahley, C. L., and Muller, K. J. (2009). ATP and NO dually control migration of microglia to nerve lesions. Dev. Neurobiol. 69, 60–72.
Edgar, J. M., McCulloch, M. C., Montague, P., Brown, A. M., Thilemann, S., Pratola, L., Gruenenfelder, F. I., Griffiths, I. R., and Nave, K. A. (2010). Demyelination and axonal preservation in a transgenic mouse model of Pelizaeus-Merzbacher disease. EMBO Mol. Med. 2, 42–50.
Falcão, A. S., Bellarosa, C., Fernandes, A., Brito, M. A., Silva, R. F., Tiribelli, C., and Brites, D. (2007a). Role of multidrug resistance-associated protein 1 expression in the in vitro susceptibility of rat nerve cell to unconjugated bilirubin. Neuroscience 144, 878–888.
Falcão, A. S., Silva, R. F., Fernandes, A., Brito, M. A., and Brites, D. (2007b). Influence of hypoxia and ischemia preconditioning on bilirubin damage to astrocytes. Brain Res. 1149, 191–199.
Falcão, A. S., Silva, R. F., Pancadas, S., Fernandes, A., Brito, M. A., and Brites, D. (2007c). Apoptosis and impairment of neurite network by short exposure of immature rat cortical neurons to unconjugated bilirubin increase with cell differentiation and are additionally enhanced by an inflammatory stimulus. J. Neurosci. Res. 85, 1229–1239.
Falcão, A. S., Fernandes, A., Brito, M. A., Silva, R. F., and Brites, D. (2005). Bilirubin-induced inflammatory response, glutamate release, and cell death in rat cortical astrocytes are enhanced in younger cells. Neurobiol. Dis. 20, 199–206.
Falcão, A. S., Fernandes, A., Brito, M. A., Silva, R. F., and Brites, D. (2006). Bilirubin-induced immunostimulant effects and toxicity vary with neural cell type and maturation state. Acta Neuropathol. 112, 95–105.
Favrais, G., van de Looij, Y., Fleiss, B., Ramanantsoa, N., Bonnin, P., Stoltenburg-Didinger, G., Lacaud, A., Saliba, E., Dammann, O., Gallego, J., Sizonenko, S., Hagberg, H., Lelievre, V., and Gressens, P. (2011). Systemic inflammation disrupts the developmental program of white matter. Ann. Neurol. 70, 550–565.
Fendrick, S. E., Xue, Q. S., and Streit, W. J. (2007). Formation of multinucleated giant cells and microglial degeneration in rats expressing a mutant Cu/Zn superoxide dismutase gene. J. Neuroinflammation 4, 9.
Fernandes, A., Barateiro, A., Falcão, A. S., Silva, S. L., Vaz, A. R., Brito, M. A., Silva, R. F., and Brites, D. (2011). Astrocyte reactivity to unconjugated bilirubin requires TNF-alpha and IL-1beta receptor signaling pathways. Glia 59, 14–25.
Fernandes, A., Coutinho, E., Lanier, L. M., and Brites, D. (2009a). Bilirubin-induced changes at neuronal cytoskeletal dynamics: novel cues for reduced axonal arborization. Glia 57, S102–S102.
Fernandes, A., Falcão, A. S., Abranches, E., Bekman, E., Henrique, D., Lanier, L. M., and Brites, D. (2009b). Bilirubin as a determinant for altered neurogenesis, neuritogenesis, and synaptogenesis. Dev. Neurobiol. 69, 568–582.
Fernandes, A., Coutinho, E., Lanier, L. M., and Brites, D. (2010). Exploring neuronal cytoskeleton defects by unconjugated bilirubin. Int. J. Dev. Neurosci. 28, 715–716.
Fernandes, A., Falcão, A. S., Silva, R. F., Brito, M. A., and Brites, D. (2007a). MAPKs are key players in mediating cytokine release and cell death induced by unconjugated bilirubin in cultured rat cortical astrocytes. Eur. J. Neurosci. 25, 1058–1068.
Fernandes, A., Vaz, A. R., Falcão, A. S., Silva, R. F., Brito, M. A., and Brites, D. (2007b). Glycoursodeoxycholic acid and interleukin-10 modulate the reactivity of rat cortical astrocytes to unconjugated bilirubin. J. Neuropathol. Exp. Neurol. 66, 789–798.
Fernandes, A., Falcão, A. S., Silva, R. F., Gordo, A. C., Gama, M. J., Brito, M. A., and Brites, D. (2006). Inflammatory signalling pathways involved in astroglial activation by unconjugated bilirubin. J. Neurochem. 96, 1667–1679.
Fernandes, A., Silva, R. F., Falcão, A. S., Brito, M. A., and Brites, D. (2004). Cytokine production, glutamate release and cell death in rat cultured astrocytes treated with unconjugated bilirubin and LPS. J. Neuroimmunol. 153, 64–75.
Floyd, R. A., and Hensley, K. (2002). Oxidative stress in brain aging. Implications for therapeutics of neurodegenerative diseases. Neurobiol. Aging 23, 795–807.
Fonseca, M. B., Nunes, A. F., and Rodrigues, C. M. (2012). c-Jun regulates the stability of anti-apoptotic DeltaNp63 in amyloid-beta-induced apoptosis. J. Alzheimers Dis. 28, 685–694.
Font-Nieves, M., Sans-Fons, M. G., Gorina, R., Bonfill-Teixidor, E., Salas-Perdomo, A., Marquez-Kisinousky, L., Santalucia, T., and Planas, A. M. (2012). Induction of COX-2 enzyme and down-regulation of COX-1 expression by lipopolysaccharide (LPS) control prostaglandin E2 production in astrocytes. J. Biol. Chem. 287, 6454–6468.
Frank, M. G., Thompson, B. M., Watkins, L. R., and Maier, S. F. (2012). Glucocorticoids mediate stress-induced priming of microglial pro-inflammatory responses. Brain Behav. Immun. 26, 337–345.
Fujiwara, R., Nguyen, N., Chen, S., and Tukey, R. H. (2010). Developmental hyperbilirubinemia and CNS toxicity in mice humanized with the UDP glucuronosyltransferase 1 (UGT1) locus. Proc. Natl. Acad. Sci. U.S.A. 107, 5024–5029.
Gao, X., Yang, X., and Zhang, B. (2011). Neuroprotection of taurine against bilirubin-induced elevation of apoptosis and intracellular free calcium ion in vivo. Toxicol. Mech. Methods 21, 383–387.
Gazzin, S., Berengeno, A. L., Strazielle, N., Fazzari, F., Raseni, A., Ostrow, J. D., Wennberg, R., Ghersi-Egea, J. F., and Tiribelli, C. (2011). Modulation of Mrp1 (ABCc1) and Pgp (ABCb1) by bilirubin at the blood-CSF and blood-brain barriers in the Gunn rat. PLoS ONE 6, e16165.
Genc, S., Genc, K., Kumral, A., Baskin, H., and Ozkan, H. (2003). Bilirubin is cytotoxic to rat oligodendrocytes in vitro. Brain Res. 985, 135–141.
Giraudi, P. J., Bellarosa, C., Coda-Zabetta, C. D., Peruzzo, P., and Tiribelli, C. (2011). Functional induction of the cystine-glutamate exchanger system Xc(-) activity in SH-SY5Y cells by unconjugated bilirubin. PLoS ONE 6, e29078.
Gkoltsiou, K., Tzoufi, M., Counsell, S., Rutherford, M., and Cowan, F. (2008). Serial brain MRI and ultrasound findings: relation to gestational age, bilirubin level, neonatal neurologic status and neurodevelopmental outcome in infants at risk of kernicterus. Early Hum. Dev. 84, 829–838.
Golde, S., Chandran, S., Brown, G. C., and Compston, A. (2002). Different pathways for iNOS-mediated toxicity in vitro dependent on neuronal maturation and NMDA receptor expression. J. Neurochem. 82, 269–282.
Gordo, A. C., Falcão, A. S., Fernandes, A., Brito, M. A., Silva, R. F., and Brites, D. (2006). Unconjugated bilirubin activates and damages microglia. J. Neurosci. Res. 84, 194–201.
Graber, D. J., Snyder-Keller, A., Lawrence, D. A., and Turner, J. N. (2012). Neurodegeneration by activated microglia across a nanofiltration membrane. J. Biochem. Mol. Toxicol. 26, 45–53.
Guo, Z. D., Wu, H. T., Sun, X. C., Zhang, X. D., and Zhang, J. H. (2011). Protection of minocycline on early brain injury after subarachnoid hemorrhage in rats. Acta Neurochir. Suppl. 110, 71–74.
Gupta, A. K., and Mann, S. B. (1998). Is auditory brainstem response a bilirubin neurotoxicity marker? Am. J. Otolaryngol. 19, 232–236.
Gurba, P. E., and Zand, R. (1974). Bilirubin binding to myelin basic protein, histones and its inhibition in vitro of cerebellar protein synthesis. Biochem. Biophys. Res. Commun. 58, 1142–1147.
Gutierrez, H., and Davies, A. M. (2011). Regulation of neural process growth, elaboration and structural plasticity by NF-kappaB. Trends Neurosci. 34, 316–325.
Haga, Y., Tempero, M. A., and Zetterman, R. K. (1996a). Unconjugated bilirubin inhibits in vitro cytotoxic T lymphocyte activity of human lymphocytes. Biochim. Biophys. Acta 1317, 65–70.
Haga, Y., Tempero, M. A., and Zetterman, R. K. (1996b). Unconjugated bilirubin inhibits in vitro major histocompatibility complex-unrestricted cytotoxicity of human lymphocytes. Biochim. Biophys. Acta 1316, 29–34.
Hankø, E., Hansen, T. W., Almaas, R., Lindstad, J., and Rootwelt, T. (2005). Bilirubin induces apoptosis and necrosis in human NT2-N neurons. Pediatr. Res. 57, 179–184.
Hankø, E., Hansen, T. W., Almaas, R., Paulsen, R., and Rootwelt, T. (2006a). Synergistic protection of a general caspase inhibitor and MK-801 in bilirubin-induced cell death in human NT2-N neurons. Pediatr. Res. 59, 72–77.
Hankø, E., Hansen, T. W., Almaas, R., and Rootwelt, T. (2006b). Recovery after short-term bilirubin exposure in human NT2-N neurons. Brain Res. 1103, 56–64.
Hansen, T., Tommarello, S., and Allen, J. (2001). Subcellular localization of bilirubin in rat brain after in vivo i.v. administration of [3H]bilirubin. Pediatr. Res. 49, 203–207.
Hansen, T. W. (2000b). Pioneers in the scientific study of neonatal jaundice and kernicterus. Pediatrics 106, E15.
Hansen, T. W. (2011). Prevention of neurodevelopmental sequelae of jaundice in the newborn. Dev. Med. Child Neurol. 53(Suppl. 4), 24–28.
Hansen, T. W., and Allen, J. W. (1997). Oxidation of bilirubin by brain mitochondrial membranes – dependence on cell type and postnatal age. Biochem. Mol. Med. 60, 155–160.
Hansen, T. W., Allen, J. W., and Tommarello, S. (1999). Oxidation of bilirubin in the brain-further characterization of a potentially protective mechanism. Mol. Genet. Metab. 68, 404–409.
Harry, G. J., and Kraft, A. D. (2012). Microglia in the developing brain: a potential target with lifetime effects. Neurotoxicology 33, 191–206.
Hasegawa-Ishii, S., Takei, S., Chiba, Y., Furukawa, A., Umegaki, H., Iguchi, A., Kawamura, N., Yoshikawa, K., Hosokawa, M., and Shimada, A. (2011). Morphological impairments in microglia precede age-related neuronal degeneration in senescence-accelerated mice. Neuropathology 31, 20–28.
Haustein, M. D., Read, D. J., Steinert, J. R., Pilati, N., Dinsdale, D., and Forsythe, I. D. (2010). Acute hyperbilirubinaemia induces presynaptic neurodegeneration at a central glutamatergic synapse. J. Physiol. (Lond.) 588, 4683–4693.
Hayashida, M., Miyaoka, T., Tsuchie, K., Yasuda, H., Wake, R., Nishida, A., Inagaki, T., Toga, T., Nagami, H., Oda, T., and Horiguchi, J. (2009). Hyperbilirubinemia-related behavioral and neuropathological changes in rats: a possible schizophrenia animal model. Prog. Neuropsychopharmacol. Biol. Psychiatry 33, 581–588.
Higgins, G. C., Beart, P. M., Shin, Y. S., Chen, M. J., Cheung, N. S., and Nagley, P. (2010). Oxidative stress: emerging mitochondrial and cellular themes and variations in neuronal injury. J. Alzheimers Dis. 20(Suppl. 2), S453–S473.
Hirrlinger, J., Konig, J., Keppler, D., Lindenau, J., Schulz, J. B., and Dringen, R. (2001). The multidrug resistance protein MRP1 mediates the release of glutathione disulfide from rat astrocytes during oxidative stress. J. Neurochem. 76, 627–636.
Hoover, B. R., Reed, M. N., Su, J., Penrod, R. D., Kotilinek, L. A., Grant, M. K., Pitstick, R., Carlson, G. A., Lanier, L. M., Yuan, L. L., Ashe, K. H., and Liao, D. (2010). Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron 68, 1067–1081.
Hulzebos, C. V., van Imhoff, D. E., Bos, A. F., Ahlfors, C. E., Verkade, H. J., and Dijk, P. H. (2008). Usefulness of the bilirubin/albumin ratio for predicting bilirubin-induced neurotoxicity in premature infants. Arch. Dis. Child. Fetal Neonatal Ed. 93, F384–F388.
Iwanaga, M., Nakagawara, A., Matsuo, S., and Ikeda, K. (1987). Impaired polymorphonuclear leukocyte function in biliary atresia: role of bilirubin and bile acids. J. Pediatr. Surg. 22, 967–972.
Janelsins, M. C., Mastrangelo, M. A., Park, K. M., Sudol, K. L., Narrow, W. C., Oddo, S., LaFerla, F. M., Callahan, L. M., Federoff, H. J., and Bowers, W. J. (2008). Chronic neuron-specific tumor necrosis factor-alpha expression enhances the local inflammatory environment ultimately leading to neuronal death in 3xTg-AD mice. Am. J. Pathol. 173, 1768–1782.
Jangaard, K. A., Fell, D. B., Dodds, L., and Allen, A. C. (2008). Outcomes in a population of healthy term and near-term infants with serum bilirubin levels of >or=325 micromol/L (>or=19 mg/dL) who were born in Nova Scotia, Canada, between 1994 and 2000. Pediatrics 122, 119–124.
Jew, J. Y., and Williams, T. H. (1977). Ultrastructural aspects of bilirubin encephalopathy in cochlear nuclei of the Gunn rat. J. Anat. 124, 599–614.
Johnson, L., Sarmiento, F., Blanc, W. A., and Day, R. (1959). Kernicterus in rats with an inherited deficiency of glucuronyl transferase. AMA J. Dis. Child. 97, 591–608.
Kahle, P. J., Waak, J., and Gasser, T. (2009). DJ-1 and prevention of oxidative stress in Parkinson’s disease and other age-related disorders. Free Radic. Biol. Med. 47, 1354–1361.
Kaltschmidt, B., and Kaltschmidt, C. (2009). NF-kappaB in the nervous system. Cold Spring Harb. Perspect. Biol. 1, a001271.
Kaplan, M., and Hammerman, C. (2005). Understanding severe hyperbilirubinemia and preventing kernicterus: adjuncts in the interpretation of neonatal serum bilirubin. Clin. Chim. Acta 356, 9–21.
Keino, H., and Kashiwamata, S. (1989). Critical period of bilirubin-induced cerebellar hypoplasia in a new Sprague-Dawley strain of jaundiced Gunn rats. Neurosci. Res. 6, 209–215.
Khan, N. M., and Poduval, T. B. (2011). Immunomodulatory and immunotoxic effects of bilirubin: molecular mechanisms. J. Leukoc. Biol. 90, 997–1015.
Kim, H. S., and Suh, Y. H. (2009). Minocycline and neurodegenerative diseases. Behav. Brain Res. 196, 168–179.
Kim, J., and Jung, Y. (2011). Different expressions of AQP1, AQP4, eNOS, and VEGF proteins in ischemic versus non-ischemic cerebropathy in rats: potential roles of AQP1 and eNOS in hydrocephalic and vasogenic edema formation. Anat. Cell Biol. 44, 295–303.
Koo, H., and Roessmann, U. (1988). Astrocyte response to perinatal liver disease, hyperammonemia, and hyperbilirubinemia: an immunohistochemical study. Pediatr. Pathol. 8, 301–311.
Kopec, K. K., and Carroll, R. T. (2000). Phagocytosis is regulated by nitric oxide in murine microglia. Nitric Oxide 4, 103–111.
Kurpius, D., Nolley, E. P., and Dailey, M. E. (2007). Purines induce directed migration and rapid homing of microglia to injured pyramidal neurons in developing hippocampus. Glia 55, 873–884.
Ladeby, R., Wirenfeldt, M., Garcia-Ovejero, D., Fenger, C., Dissing-Olesen, L., Dalmau, I., and Finsen, B. (2005). Microglial cell population dynamics in the injured adult central nervous system. Brain Res. Brain Res. Rev. 48, 196–206.
Lazaridis, K. N., Gores, G. J., and Lindor, K. D. (2001). Ursodeoxycholic acid “mechanisms of action and clinical use in hepatobiliary disorders.” J. Hepatol. 35, 134–146.
Lee, S. C., Liu, W., Dickson, D. W., Brosnan, C. F., and Berman, J. W. (1993). Cytokine production by human fetal microglia and astrocytes. Differential induction by lipopolysaccharide and IL-1 beta. J. Immunol. 150, 2659–2667.
Lee, Y. K., Daito, Y., Katayama, Y., Minami, H., and Negishi, H. (2009). The significance of measurement of serum unbound bilirubin concentrations in high-risk infants. Pediatr. Int. 51, 795–799.
Lev, N., Ickowicz, D., Melamed, E., and Offen, D. (2008). Oxidative insults induce DJ-1 upregulation and redistribution: implications for neuroprotection. Neurotoxicology 29, 397–405.
Levi, G., Sohmer, H., and Kapitulnik, J. (1981). Auditory nerve and brain stem responses in homozygous jaundiced Gunn rats. Arch. Otorhinolaryngol. 232, 139–143.
Levkovitz, Y., Levi, U., Braw, Y., and Cohen, H. (2007). Minocycline, a second-generation tetracycline, as a neuroprotective agent in an animal model of schizophrenia. Brain Res. 1154, 154–162.
Li, Y., Liu, L., Barger, S. W., and Griffin, W. S. (2003). Interleukin-1 mediates pathological effects of microglia on tau phosphorylation and on synaptophysin synthesis in cortical neurons through a p38-MAPK pathway. J. Neurosci. 23, 1605–1611.
Liang, J., Takeuchi, H., Doi, Y., Kawanokuchi, J., Sonobe, Y., Jin, S., Yawata, I., Li, H., Yasuoka, S., Mizuno, T., and Suzumura, A. (2008). Excitatory amino acid transporter expression by astrocytes is neuroprotective against microglial excitotoxicity. Brain Res. 1210, 11–19.
Liaury, K., Miyaoka, T., Tsumori, T., Furuya, M., Wake, R., Ieda, M., Tsuchie, K., Taki, M., Ishihara, K., Tanra, A. J., and Horiguchi, J. (2012). Morphological features of microglial cells in the hippocampal dentate gyrus of Gunn rat: a possible schizophrenia animal model. J. Neuroinflammation 9, 56.
Lin, S., Wei, X., Bales, K. R., Paul, A. B., Ma, Z., Yan, G., Paul, S. M., and Du, Y. (2005). Minocycline blocks bilirubin neurotoxicity and prevents hyperbilirubinemia-induced cerebellar hypoplasia in the Gunn rat. Eur. J. Neurosci. 22, 21–27.
Liu, W., Tang, Y., and Feng, J. (2011a). Cross talk between activation of microglia and astrocytes in pathological conditions in the central nervous system. Life Sci. 89, 141–146.
Liu, W. T., Lin, C. H., Hsiao, M., and Gean, P. W. (2011b). Minocycline inhibits the growth of glioma by inducing autophagy. Autophagy 7, 166–175.
Loftspring, M. C., Johnson, H. L., Feng, R., Johnson, A. J., and Clark, J. F. (2011). Unconjugated bilirubin contributes to early inflammation and edema after intracerebral hemorrhage. J. Cereb. Blood Flow Metab. 31, 1133–1142.
Lossi, L., Alasia, S., Salio, C., and Merighi, A. (2009). Cell death and proliferation in acute slices and organotypic cultures of mammalian CNS. Prog. Neurobiol. 88, 221–245.
Lu, T., Pan, Y., Kao, S. Y., Li, C., Kohane, I., Chan, J., and Yankner, B. A. (2004). Gene regulation and DNA damage in the ageing human brain. Nature 429, 883–891.
Maezawa, I., and Jin, L. W. (2010). Rett syndrome microglia damage dendrites and synapses by the elevated release of glutamate. J. Neurosci. 30, 5346–5356.
Maier, K., Merkler, D., Gerber, J., Taheri, N., Kuhnert, A. V., Williams, S. K., Neusch, C., Bahr, M., and Diem, R. (2007). Multiple neuroprotective mechanisms of minocycline in autoimmune CNS inflammation. Neurobiol. Dis. 25, 514–525.
Malik, S. G., Irwanto, K. A., Ostrow, J. D., and Tiribelli, C. (2010). Effect of bilirubin on cytochrome c oxidase activity of mitochondria from mouse brain and liver. BMC Res Notes 3, 162.
Mancuso, C., Capone, C., Ranieri, S. C., Fusco, S., Calabrese, V., Eboli, M. L., Preziosi, P., Galeotti, T., and Pani, G. (2008). Bilirubin as an endogenous modulator of neurotrophin redox signaling. J. Neurosci. Res. 86, 2235–2249.
Mancuso, C., Pistritto, G., Tringali, G., Grossman, A. B., Preziosi, P., and Navarra, P. (1997). Evidence that carbon monoxide stimulates prostaglandin endoperoxide synthase activity in rat hypothalamic explants and in primary cultures of rat hypothalamic astrocytes. Brain Res. Mol. Brain Res. 45, 294–300.
Mangano, E. N., and Hayley, S. (2009). Inflammatory priming of the substantia nigra influences the impact of later paraquat exposure: neuroimmune sensitization of neurodegeneration. Neurobiol. Aging 30, 1361–1378.
Manning, D., Todd, P., Maxwell, M., and Jane Platt, M. (2007). Prospective surveillance study of severe hyperbilirubinaemia in the newborn in the UK and Ireland. Arch. Dis. Child. Fetal Neonatal Ed. 92, F342–F346.
Martich-Kriss, V., Kollias, S. S., and Ball, W. S. Jr. (1995). MR findings in kernicterus. AJNR Am. J. Neuroradiol. 16, 819–821.
Matute, C., Alberdi, E., Domercq, M., Sanchez-Gomez, M. V., Perez-Samartin, A., Rodriguez-Antiguedad, A., and Perez-Cerda, F. (2007). Excitotoxic damage to white matter. J. Anat. 210, 693–702.
Mazzella, G., Rizzo, N., Azzaroli, F., Simoni, P., Bovicelli, L., Miracolo, A., Simonazzi, G., Colecchia, A., Nigro, G., Mwangemi, C., Festi, D., and Roda, E. (2001). Ursodeoxycholic acid administration in patients with cholestasis of pregnancy: effects on primary bile acids in babies and mothers. Hepatology 33, 504–508.
McDonagh, A. F. (2010). Controversies in bilirubin biochemistry and their clinical relevance. Semin. Fetal Neonatal Med. 15, 141–147.
Meisel, P., Jahrig, D., Beyersdorff, E., and Jahrig, K. (1988). Bilirubin binding and acid-base equilibrium in newborn infants with low birthweight. Acta Paediatr. Scand. 77, 496–501.
Mitra, S., Samanta, M., Sarkar, M., De, A. K., and Chatterjee, S. (2011). Pre-exchange 5% albumin infusion in low birth weight neonates with intensive phototherapy failure – a randomized controlled trial. J. Trop. Pediatr. 57, 217–221.
Miyama, A., Saito, Y., Yamanaka, K., Hayashi, K., Hamakubo, T., and Noguchi, N. (2011). Oxidation of DJ-1 induced by 6-hydroxydopamine decreasing intracellular glutathione. PLoS ONE 6, e27883.
Moll, M., Goelz, R., Naegele, T., Wilke, M., and Poets, C. F. (2011). Are recommended phototherapy thresholds safe enough for extremely low birth weight (ELBW) infants? A report on 2 ELBW infants with kernicterus despite only moderate hyperbilirubinemia. Neonatology 99, 90–94.
Morandi, A., Hughes, C. G., Girard, T. D., McAuley, D. F., Ely, E. W., and Pandharipande, P. P. (2011). Statins and brain dysfunction: a hypothesis to reduce the burden of cognitive impairment in patients who are critically ill. Chest 140, 580–585.
Morgese, V. J., Elliott, E. J., and Muller, K. J. (1983). Microglial movement to sites of nerve lesion in the leech CNS. Brain Res. 272, 166–170.
Mukerjee, P., Ostrow, J. D., and Tiribelli, C. (2002). Low solubility of unconjugated bilirubin in dimethylsulfoxide – water systems: implications for pKa determinations. BMC Biochem. 3, 17.
Mukhopadhyay, K., Chowdhary, G., Singh, P., Kumar, P., and Narang, A. (2010). Neurodevelopmental outcome of acute bilirubin encephalopathy. J. Trop. Pediatr. 56, 333–336.
Muramatsu, R., Ueno, M., and Yamashita, T. (2009). Intrinsic regenerative mechanisms of central nervous system neurons. Biosci. Trends 3, 179–183.
Nair, A., Frederick, T. J., and Miller, S. D. (2008). Astrocytes in multiple sclerosis: a product of their environment. Cell. Mol. Life Sci. 65, 2702–2720.
Nakajima, K., Tohyama, Y., Kohsaka, S., and Kurihara, T. (2001). Ability of rat microglia to uptake extracellular glutamate. Neurosci. Lett. 307, 171–174.
Nave, K. A. (2010). Oligodendrocytes and the “micro brake” of progenitor cell proliferation. Neuron 65, 577–579.
Nazmi, A., Dutta, K., and Basu, A. (2011). RIG-I mediates innate immune response in mouse neurons following Japanese encephalitis virus infection. PLoS ONE 6, e21761.
Ngu, E. M., Sahley, C. L., and Muller, K. J. (2007). Reduced axon sprouting after treatment that diminishes microglia accumulation at lesions in the leech CNS. J. Comp. Neurol. 503, 101–109.
Nguyen, N., Bonzo, J. A., Chen, S., Chouinard, S., Kelner, M. J., Hardiman, G., Belanger, A., and Tukey, R. H. (2008). Disruption of the ugt1 locus in mice resembles human Crigler-Najjar type I disease. J. Biol. Chem. 283, 7901–7911.
Nickisch, A., Massinger, C., Ertl-Wagner, B., and von Voss, H. (2009). Pedaudiologic findings after severe neonatal hyperbilirubinemia. Eur. Arch. Otorhinolaryngol. 266, 207–212.
Nimmerjahn, A., Kirchhoff, F., and Helmchen, F. (2005). Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308, 1314–1318.
Noguchi-Shinohara, M., Hamaguchi, T., Nozaki, I., Sakai, K., and Yamada, M. (2011). Serum tau protein as a marker for the diagnosis of Creutzfeldt-Jakob disease. J. Neurol. 258, 1464–1468.
Oakes, G. H., and Bend, J. R. (2010). Global changes in gene regulation demonstrate that unconjugated bilirubin is able to upregulate and activate select components of the endoplasmic reticulum stress response pathway. J. Biochem. Mol. Toxicol. 24, 73–88.
Ohtori, S., Takahashi, K., Moriya, H., and Myers, R. R. (2004). TNF-alpha and TNF-alpha receptor type 1 upregulation in glia and neurons after peripheral nerve injury: studies in murine DRG and spinal cord. Spine 29, 1082–1088.
Okumura, A., Kidokoro, H., Shoji, H., Nakazawa, T., Mimaki, M., Fujii, K., Oba, H., and Shimizu, T. (2009). Kernicterus in preterm infants. Pediatrics 123, e1052–e1058.
Okumus, N., Turkyilmaz, C., Onal, E. E., Atalay, Y., Serdaroglu, A., Elbeg, S., Koc, E., Deda, G., Cansu, A., and Gunduz, B. (2008). Tau and S100B proteins as biochemical markers of bilirubin-induced neurotoxicity in term neonates. Pediatr. Neurol. 39, 245–252.
Orellana, J. A., Saez, P. J., Shoji, K. F., Schalper, K. A., Palacios-Prado, N., Velarde, V., Giaume, C., Bennett, M. V., and Saez, J. C. (2009). Modulation of brain hemichannels and gap junction channels by pro-inflammatory agents and their possible role in neurodegeneration. Antioxid. Redox Signal. 11, 369–399.
Ossola, B., Lantto, T. A., Puttonen, K. A., Tuominen, R. K., Raasmaja, A., and Mannisto, P. T. (2012). Minocycline protects SH-SY5Y cells from 6-hydroxydopamine by inhibiting both caspase-dependent and -independent programmed cell death. J. Neurosci. Res. 90, 682–690.
Ostrow, J. D., and Celic, L. (1984). Bilirubin chemistry, ionization and solubilization by bile salts. Hepatology 4, 38S–45S.
Ostrow, J. D., Pascolo, L., Brites, D., and Tiribelli, C. (2004). Molecular basis of bilirubin-induced neurotoxicity. Trends. Mol. Med. 10, 65–70.
Ostrow, J. D., Pascolo, L., and Tiribelli, C. (2003a). Reassessment of the unbound concentrations of unconjugated bilirubin in relation to neurotoxicity in vitro. Pediatr. Res. 54, 98–104.
Ostrow, J. D., Pascolo, L., Shapiro, S. M., and Tiribelli, C. (2003b). New concepts in bilirubin encephalopathy. Eur. J. Clin. Invest. 33, 988–997.
Palmela, I., Cardoso, F. L., Bernas, M., Correia, L., Vaz, A. R., Silva, R. F., Fernandes, A., Kim, K. S., Brites, D., and Brito, M. A. (2011). Elevated levels of bilirubin and long-term exposure impair human brain microvascular endothelial cell integrity. Curr. Neurovasc. Res. 8, 153–169.
Pang, Y., Rodts-Palenik, S., Cai, Z., Bennett, W. A., and Rhodes, P. G. (2005). Suppression of glial activation is involved in the protection of IL-10 on maternal E. coli induced neonatal white matter injury. Brain Res. Dev. Brain Res. 157, 141–149.
Paolicelli, R. C., Bolasco, G., Pagani, F., Maggi, L., Scianni, M., Panzanelli, P., Giustetto, M., Ferreira, T. A., Guiducci, E., Dumas, L., Ragozzino, D., and Gross, C. T. (2011). Synaptic pruning by microglia is necessary for normal brain development. Science 333, 1456–1458.
Parry, G. J., Rodrigues, C. M., Aranha, M. M., Hilbert, S. J., Davey, C., Kelkar, P., Low, W. C., and Steer, C. J. (2010). Safety, tolerability, and cerebrospinal fluid penetration of ursodeoxycholic acid in patients with amyotrophic lateral sclerosis. Clin. Neuropharmacol. 33, 17–21.
Patrizio, M., and Levi, G. (1994). Glutamate production by cultured microglia: differences between rat and mouse, enhancement by lipopolysaccharide and lack effect of HIV coat protein gp120 and depolarizing agents. Neurosci. Lett. 178, 184–189.
Pearlman, M. A., Gartner, L. M., Lee, K., Eidelman, A. I., Morecki, R., and Horoupian, D. S. (1980). The association of kernicterus with bacterial infection in the newborn. Pediatrics 65, 26–29.
Piani, D., Frei, K., Do, K. Q., Cuenod, M., and Fontana, A. (1991). Murine brain macrophages induced NMDA receptor mediated neurotoxicity in vitro by secreting glutamate. Neurosci. Lett. 133, 159–162.
Piaton, G., Gould, R. M., and Lubetzki, C. (2010). Axon-oligodendrocyte interactions during developmental myelination, demyelination and repair. J. Neurochem. 114, 1243–1260.
Pistritto, G., Franzese, O., Pozzoli, G., Mancuso, C., Tringali, G., Preziosi, P., and Navarra, P. (1999). Bacterial lipopolysaccharide increases prostaglandin production by rat astrocytes via inducible cyclo-oxygenase: evidence for the involvement of nuclear factor kappaB. Biochem. Biophys. Res. Commun. 263, 570–574.
Pistritto, G., Mancuso, C., Tringali, G., Perretti, M., Preziosi, P., and Navarra, P. (1998). The relative contribution of constitutive and inducible cyclooxygenase activity to lipopolysaccharide-induced prostaglandin production by primary cultures of rat hypothalamic astrocytes. Neurosci. Lett. 246, 45–48.
Qiang, L., Yu, W., Andreadis, A., Luo, M., and Baas, P. W. (2006). Tau protects microtubules in the axon from severing by katanin. J. Neurosci. 26, 3120–3129.
Ramaglia, V., Hughes, T. R., Donev, R. M., Ruseva, M. M., Wu, X., Huitinga, I., Baas, F., Neal, J. W., and Morgan, B. P. (2012). C3-dependent mechanism of microglial priming relevant to multiple sclerosis. Proc. Natl. Acad. Sci. U.S.A. 109, 965–970.
Renault-Mihara, F., Okada, S., Shibata, S., Nakamura, M., Toyama, Y., and Okano, H. (2008). Spinal cord injury: emerging beneficial role of reactive astrocytes’ migration. Int. J. Biochem. Cell Biol. 40, 1649–1653.
Rice, A. C., Chiou, V. L., Zuckoff, S. B., and Shapiro, S. M. (2011). Profile of minocycline neuroprotection in bilirubin-induced auditory system dysfunction. Brain Res. 1368, 290–298.
Rice, A. C., and Shapiro, S. M. (2008). A new animal model of hemolytic hyperbilirubinemia-induced bilirubin encephalopathy (kernicterus). Pediatr. Res. 64, 265–269.
Rigato, I., Pascolo, L., Fernetti, C., Ostrow, J. D., and Tiribelli, C. (2004). The human multidrug-resistance-associated protein MRP1 mediates ATP-dependent transport of unconjugated bilirubin. Biochem. J. 383, 335–341.
Rodrigues, C. M., Solá, S., and Brites, D. (2002a). Bilirubin induces apoptosis via the mitochondrial pathway in developing rat brain neurons. Hepatology 35, 1186–1195.
Rodrigues, C. M., Solá, S., Brito, M. A., Brites, D., and Moura, J. J. (2002b). Bilirubin directly disrupts membrane lipid polarity and fluidity, protein order, and redox status in rat mitochondria. J. Hepatol. 36, 335–341.
Rodrigues, C. M., Solá, S., Castro, R. E., Laires, P. A., Brites, D., and Moura, J. J. (2002c). Perturbation of membrane dynamics in nerve cells as an early event during bilirubin-induced apoptosis. J. Lipid Res. 43, 885–894.
Rossi, D., and Volterra, A. (2009). Astrocytic dysfunction: insights on the role in neurodegeneration. Brain Res. Bull. 80, 224–232.
Rudolph, G., Kloeters-Plachky, P., Sauer, P., and Stiehl, A. (2002). Intestinal absorption and biliary secretion of ursodeoxycholic acid and its taurine conjugate. Eur. J. Clin. Invest. 32, 575–580.
Saeed, U., Ray, A., Valli, R. K., Kumar, A. M., and Ravindranath, V. (2010). DJ-1 loss by glutaredoxin but not glutathione depletion triggers Daxx translocation and cell death. Antioxid. Redox Signal. 13, 127–144.
Saluja, S., Agarwal, A., Kler, N., and Amin, S. (2010). Auditory neuropathy spectrum disorder in late preterm and term infants with severe jaundice. Int. J. Pediatr. Otorhinolaryngol. 74, 1292–1297.
Samuels, S. E., Lipitz, J. B., Dahl, G., and Muller, K. J. (2010). Neuroglial ATP release through innexin channels controls microglial cell movement to a nerve injury. J. Gen. Physiol. 136, 425–442.
Sargsyan, S. A., Blackburn, D. J., Barber, S. C., Grosskreutz, J., De Vos, K. J., Monk, P. N., and Shaw, P. J. (2011). A comparison of in vitro properties of resting SOD1 transgenic microglia reveals evidence of reduced neuroprotective function. BMC Neurosci. 12, 91.
Sasaki, M., Gonzalez-Zulueta, M., Huang, H., Herring, W. J., Ahn, S., Ginty, D. D., Dawson, V. L., and Dawson, T. M. (2000). Dynamic regulation of neuronal NO synthase transcription by calcium influx through a CREB family transcription factor-dependent mechanism. Proc. Natl. Acad. Sci. U.S.A. 97, 8617–8622.
Schildknecht, S., Pape, R., Muller, N., Robotta, M., Marquardt, A., Burkle, A., Drescher, M., and Leist, M. (2011). Neuroprotection by minocycline caused by direct and specific scavenging of peroxynitrite. J. Biol. Chem. 286, 4991–5002.
Sequeira, D., Watchko, J. F., Daood, M. J., O’Day, T. L., and Mahmood, B. (2007). Unconjugated bilirubin efflux by bovine brain microvascular endothelial cells in vitro. Pediatr. Crit. Care Med. 8, 570–575.
Shahian, M., and Moslehi, M. A. (2010). Effect of albumin administration prior to exchange transfusion in term neonates with hyperbilirubinemia – a randomized controlled trial. Indian Pediatr. 47, 241–244.
Shaked, I., Tchoresh, D., Gersner, R., Meiri, G., Mordechai, S., Xiao, X., Hart, R. P., and Schwartz, M. (2005). Protective autoimmunity: interferon-gamma enables microglia to remove glutamate without evoking inflammatory mediators. J. Neurochem. 92, 997–1009.
Shapiro, S. M. (2003). Bilirubin toxicity in the developing nervous system. Pediatr. Neurol. 29, 410–421.
Shapiro, S. M. (2010). Chronic bilirubin encephalopathy: diagnosis and outcome. Semin. Fetal Neonatal Med. 15, 157–163.
Shapiro, S. M., and Popelka, G. R. (2011). Auditory impairment in infants at risk for bilirubin-induced neurologic dysfunction. Semin. Perinatol. 35, 162–170.
Shapiro, S. M., Sombati, S., Geiger, A., and Rice, A. C. (2007). NMDA channel antagonist MK-801 does not protect against bilirubin neurotoxicity. Neonatology 92, 248–257.
Sheykholeslami, K., and Kaga, K. (2000). Otoacoustic emissions and auditory brainstem responses after neonatal hyperbilirubinemia. Int. J. Pediatr. Otorhinolaryngol. 52, 65–73.
Shi, H. B., Kakazu, Y., Shibata, S., Matsumoto, N., Nakagawa, T., and Komune, S. (2006). Bilirubin potentiates inhibitory synaptic transmission in lateral superior olive neurons of the rat. Neurosci. Res. 55, 161–170.
Silberberg, D. H., and Schutta, H. S. (1967). The effects of unconjugated bilirubin and related pigments on cultures of rat cerebellum. J. Neuropathol. Exp. Neurol. 26, 572–583.
Silva, R., Mata, L. R., Gulbenkian, S., Brito, M. A., Tiribelli, C., and Brites, D. (1999). Inhibition of glutamate uptake by unconjugated bilirubin in cultured cortical rat astrocytes: role of concentration and pH. Biochem. Biophys. Res. Commun. 265, 67–72.
Silva, R. F., Mata, L. M., Gulbenkian, S., and Brites, D. (2001a). Endocytosis in rat cultured astrocytes is inhibited by unconjugated bilirubin. Neurochem. Res. 26, 793–800.
Silva, R. F., Rodrigues, C. M., and Brites, D. (2001b). Bilirubin-induced apoptosis in cultured rat neural cells is aggravated by chenodeoxycholic acid but prevented by ursodeoxycholic acid. J. Hepatol. 34, 402–408.
Silva, S. L., Osório, C., Vaz, A. R., Barateiro, A., Falcão, A. S., Silva, R. F., and Brites, D. (2011). Dynamics of neuron-glia interplay upon exposure to unconjugated bilirubin. J. Neurochem. 117, 412–424.
Silva, S. L., Vaz, A. R., Barateiro, A., Falcão, A. S., Fernandes, A., Brito, M. A., Silva, R. F., and Brites, D. (2010). Features of bilirubin-induced reactive microglia: from phagocytosis to inflammation. Neurobiol. Dis. 40, 663–675.
Silva, S. L., Vaz, A. R., Diógenes, M. J., van Rooijen, N., Sebastião, A. M., Fernandes, A., Silva, R. F. M., and Brites, D. (2012). Impairment of neurite sprouting by unconjugated bilirubin is mediated by NO and glutamate, modulated by microglia, and prevented by glycoursodeoxycholic acid and interleukin-10. Neuropharmacology 62, 2397–2407.
Silver, S., Kapitulnik, J., and Sohmer, H. (1995a). Contribution of asphyxia to the induction of hearing impairment in jaundiced Gunn rats. Pediatrics 95, 579–583.
Silver, S., Sohmer, H., and Kapitulnik, J. (1995b). Visual evoked potential abnormalities in jaundiced Gunn rats treated with sulfadimethoxine. Pediatr. Res. 38, 258–261.
Silver, S., Sohmer, H., and Kapitulnik, J. (1996). Postnatal development of somatosensory evoked potential in jaundiced Gunn rats and effects of sulfadimethoxine administration. Pediatr. Res. 40, 209–214.
Smith, K., and Leyden, J. J. (2005). Safety of doxycycline and minocycline: a systematic review. Clin. Ther. 27, 1329–1342.
Sofroniew, M. V., and Vinters, H. V. (2010). Astrocytes: biology and pathology. Acta Neuropathol. 119, 7–35.
Solá, S., Aranha, M. M., Steer, C. J., and Rodrigues, C. M. (2007). Game and players: mitochondrial apoptosis and the therapeutic potential of ursodeoxycholic acid. Curr. Issues Mol. Biol. 9, 123–138.
Solá, S., Diógenes, M. J., Brites, D., and Rodrigues, C. M. (2002). Release of cytochrome c with the interaction of bilirubin, amyloid beta-peptide and glycochenodeoxycholate from isolated mitochondria. Acta Med. Port. 15, 269–275.
Somera-Molina, K. C., Robin, B., Somera, C. A., Anderson, C., Stine, C., Koh, S., Behanna, H. A., Van Eldik, L. J., Watterson, D. M., and Wainwright, M. S. (2007). Glial activation links early-life seizures and long-term neurologic dysfunction: evidence using a small molecule inhibitor of proinflammatory cytokine upregulation. Epilepsia 48, 1785–1800.
Steinborn, M., Seelos, K. C., Heuck, A., von Voss, H., and Reiser, M. (1999). MR findings in a patient with Kernicterus. Eur. Radiol. 9, 1913–1915.
Stokowski, L. A. (2011). Fundamentals of phototherapy for neonatal jaundice. Adv. Neonatal Care 11, S10–S21.
Streit, W. J. (2002). Microglia as neuroprotective, immunocompetent cells of the CNS. Glia 40, 133–139.
Streit, W. J., Miller, K. R., Lopes, K. O., and Njie, E. (2008). Microglial degeneration in the aging brain-bad news for neurons? Front. Biosci. 13, 3423–3438.
Streit, W. J., Sammons, N. W., Kuhns, A. J., and Sparks, D. L. (2004). Dystrophic microglia in the aging human brain. Glia 45, 208–212.
Sun, M., Zhao, Y., Gu, Y., and Xu, C. (2012). Anti-inflammatory mechanism of taurine against ischemic stroke is related to down-regulation of PARP and NF-kappaB. Amino Acids. 42, 1735–1747.
Suzuki, H., Sugimura, Y., Iwama, S., Nobuaki, O., Nagasaki, H., Arima, H., Sawada, M., and Oiso, Y. (2010). Minocycline prevents osmotic demyelination syndrome by inhibiting the activation of microglia. J. Am. Soc. Nephrol. 21, 2090–2098.
Sykova, E., and Nicholson, C. (2008). Diffusion in brain extracellular space. Physiol. Rev. 88, 1277–1340.
Teng, F. Y., and Tang, B. L. (2010). NF-kappaB signaling in neurite growth and neuronal survival. Rev. Neurosci. 21, 299–313.
Thong, Y. H., Ness, D., and Ferrante, A. (1979). Effect of bilirubin on the fungicidal capacity of human neutrophils. Sabouraudia 17, 125–129.
Thong, Y. H., and Rencis, V. (1977). Bilirubin inhibits hexose-monophosphate shunt activity of phagocytosing neutrophils. Acta Paediatr. Scand. 66, 757–759.
Tikka, T., Fiebich, B. L., Goldsteins, G., Keinanen, R., and Koistinaho, J. (2001). Minocycline, a tetracycline derivative, is neuroprotective against excitotoxicity by inhibiting activation and proliferation of microglia. J. Neurosci. 21, 2580–2588.
Vairano, M., Dello Russo, C., Pozzoli, G., Tringali, G., Preziosi, P., and Navarra, P. (2001). A functional link between heme oxygenase and cyclo-oxygenase activities in cortical rat astrocytes. Biochem. Pharmacol. 61, 437–441.
Vaz, A. R., Delgado-Esteban, M., Brito, M. A., Bolanos, J. P., Brites, D., and Almeida, A. (2010). Bilirubin selectively inhibits cytochrome c oxidase activity and induces apoptosis in immature cortical neurons: assessment of the protective effects of glycoursodeoxycholic acid. J. Neurochem. 112, 56–65.
Vaz, A. R., Silva, S. L., Barateiro, A., Falcão, A. S., Fernandes, A., Brito, M. A., and Brites, D. (2011a). Selective vulnerability of rat brain regions to unconjugated bilirubin. Mol. Cell. Neurosci. 48, 82–93.
Vaz, A. R., Silva, S. L., Barateiro, A., Fernandes, A., Falcão, A. S., Brito, M. A., and Brites, D. (2011b). Pro-inflammatory cytokines intensify the activation of NO/NOS, JNK1/2 and caspase cascades in immature neurons exposed to elevated levels of unconjugated bilirubin. Exp. Neurol. 229, 381–390.
Vert, P., Grojean, S., and Daval, J. L. (2001). Combined neuronal toxicity of bilirubin and hypoxia. Study of cultured rat neurons. Bull. Acad. Natl. Med. 185, 1417–1426; discussion 1427–1418.
Volpe, J. J., Kinney, H. C., Jensen, F. E., and Rosenberg, P. A. (2011). The developing oligodendrocyte: key cellular target in brain injury in the premature infant. Int. J. Dev. Neurosci. 29, 423–440.
Wake, H., Moorhouse, A. J., Jinno, S., Kohsaka, S., and Nabekura, J. (2009). Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J. Neurosci. 29, 3974–3980.
Wang, J. Z., and Liu, F. (2008). Microtubule-associated protein tau in development, degeneration and protection of neurons. Prog. Neurobiol. 85, 148–175.
Wang, Y., and Tang, B. L. (2006). SNAREs in neurons – beyond synaptic vesicle exocytosis (Review). Mol. Membr. Biol. 23, 377–384.
Watchko, J. F., Daood, M. J., Mahmood, B., Vats, K., Hart, C., and Ahdab-Barmada, M. (2001). P-glycoprotein and bilirubin disposition. J. Perinatol. 21(Suppl. 1), S43–S47; discussion S59–S62.
Wennberg, R. P., Ahlfors, C. E., Bhutani, V. K., Johnson, L. H., and Shapiro, S. M. (2006). Toward understanding kernicterus: a challenge to improve the management of jaundiced newborns. Pediatrics 117, 474–485.
Wiseman, H., and Halliwell, B. (1996). Damage to DNA by reactive oxygen and nitrogen species: role in inflammatory disease and progression to cancer. Biochem. J. 313(Pt 1), 17–29.
Wu, J. Y., and Prentice, H. (2010). Role of taurine in the central nervous system. J. Biomed. Sci. 17(Suppl. 1), :S1.
Yakovleva, T., Bazov, I., Watanabe, H., Hauser, K. F., and Bakalkin, G. (2011). Transcriptional control of maladaptive and protective responses in alcoholics: a role of the NF-kappaB system. Brain Behav. Immun. 25(Suppl. 1), S29–S38.
Yamada, J., and Jinno, S. (2011). Alterations in neuronal survival and glial reactions after axotomy by ceftriaxone and minocycline in the mouse hypoglossal nucleus. Neurosci. Lett. 504, 295–300.
Yamori, Y., Taguchi, T., Hamada, A., Kunimasa, K., Mori, H., and Mori, M. (2010). Taurine in health and diseases: consistent evidence from experimental and epidemiological studies. J. Biomed. Sci. 17(Suppl. 1), S6.
Yang, J. L., Sykora, P., Wilson, D. M. III, Mattson, M. P., and Bohr, V. A. (2011). The excitatory neurotransmitter glutamate stimulates DNA repair to increase neuronal resiliency. Mech. Ageing Dev. 132, 405–411.
Yeung, C. Y., and Ngai, K. C. (2001). Cytokine- and endotoxin-enhanced bilirubin cytotoxicity. J. Perinatol. 21(Suppl. 1), S56–S58; discussion S59–S62.
Yirmiya, R., and Goshen, I. (2011). Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav. Immun. 25, 181–213.
Zhang, B., Yang, X., and Gao, X. (2010). Taurine protects against bilirubin-induced neurotoxicity in vitro. Brain Res. 1320, 159–167.
Zhong, J., and Lee, W. H. (2007). Hydrogen peroxide attenuates insulin-like growth factor-1 neuroprotective effect, prevented by minocycline. Neurochem. Int. 51, 398–404.
Zhou, Y., Qin, H., Zhang, M., Shen, T., Chen, H., Ma, Y., Chu, Z., Zhang, P., and Liu, Z. (2010). Lactobacillus plantarum inhibits intestinal epithelial barrier dysfunction induced by unconjugated bilirubin. Br. J. Nutr. 104, 390–401.
Zhou, Z., Peng, X., Insolera, R., Fink, D. J., and Mata, M. (2009). IL-10 promotes neuronal survival following spinal cord injury. Exp. Neurol. 220, 183–190.
Zhuang, H., Pin, S., Li, X., and Dore, S. (2003). Regulation of heme oxygenase expression by cyclopentenone prostaglandins. Exp. Biol. Med. (Maywood) 228, 499–505.
Keywords: unconjugated hyperbilirubinemia, neurotoxicity, reactive astrocytes, activated microglia, sepsis, pro-inflammatory cytokines, oxidative stress, myelin
Citation: Brites D (2012) The evolving landscape of neurotoxicity by unconjugated bilirubin: role of glial cells and inflammation. Front. Pharmacol. 3:88. doi: 10.3389/fphar.2012.00088
Received: 23 January 2012; Paper pending published: 21 February 2012;
Accepted: 23 April 2012; Published online: 29 May 2012.
Edited by:
Jaime Kapitulnik, The Hebrew University of Jerusalem, IsraelReviewed by:
Jaime Kapitulnik, The Hebrew University of Jerusalem, IsraelCesare Mancuso, Catholic University School of Medicine, Italy
Andreas Daiber, University Medical Center Mainz, Germany
Copyright: © 2012 Brites. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Dora Brites, Faculdade de Farmácia da Universidade de Lisboa, Avenida Professor Gama Pinto, 1649-003 Lisbon, Portugal. e-mail: dbrites@ff.ul.pt