
- 1 Italian Liver Foundation, AREA Science Park Basovizza, Trieste, Italy
- 2 Brain-i, Lyon, France
- 3 Neurooncology and Neuroinflammation Team, INSERM U1028, CNRS UMR5292, Lyon Neuroscience Research Center, Lyon-1 University, Lyon, France
Bilirubin, the end-product of heme catabolism, circulates in non-pathological plasma mostly as a protein-bound species. When bilirubin concentration builds up, the free fraction of the molecule increases. Unbound bilirubin then diffuses across blood–brain interfaces (BBIs) into the brain, where it accumulates and exerts neurotoxic effects. In this classical view of bilirubin neurotoxicity, BBIs act merely as structural barriers impeding the penetration of the pigment-bound carrier protein, and neural cells are considered as passive targets of its toxicity. Yet, the role of BBIs in the occurrence of bilirubin encephalopathy appears more complex than being simple barriers to the diffusion of bilirubin, and neural cells such as astrocytes and neurons can play an active role in controlling the balance between the neuroprotective and neurotoxic effects of bilirubin. This article reviews the emerging in vivo and in vitro data showing that transport and metabolic detoxification mechanisms at the blood–brain and blood–cerebrospinal fluid barriers may modulate bilirubin flux across both cellular interfaces, and that these protective functions can be affected in chronic unconjugated hyperbilirubinemia. Then the in vivo and in vitro arguments in favor of the physiological antioxidant function of intracerebral bilirubin are presented, as well as the potential role of transporters such as ABCC1 and metabolizing enzymes such as cytochromes P-450 in setting the cerebral cell- and structure-specific toxicity of bilirubin following hyperbilirubinemia. The relevance of these data to the pathophysiology of bilirubin-induced neurological diseases is discussed.
Introduction
Unconjugated bilirubin (UCB) is the end-product of heme catabolism, and is eliminated from the body following hepatic conjugation to glucuronic acid. When UCB clearance is inefficient, plasma UCB concentration increases markedly, and brain damage may ensue. This most frequently occurs in severe jaundice of the neonate, or in cases of severe congenital impairment of bilirubin conjugation, such as in the Crigler–Najjar syndrome. In adults, the average total plasma UCB concentration is <20 μM. This value is higher in newborns, due to the immaturity of transport and metabolic processes in the liver, which is faced with a higher production rate. This typically results in a mild “physiological” neonatal jaundice (UCB concentrations up to 170 μM), which is not harmful to newborns and may even be beneficial to the organism owing to the antioxidant properties of the pigment (for review, see Jansen, 1999; Wennberg et al., 2009). When serum concentrations increase further, UCB may become neurotoxic, leading to clinical manifestations of encephalopathy, sometimes resulting in irreversible brain damage when jaundice is especially severe or prolonged (Jansen, 1999; Wennberg et al., 2009).
The prevalent explanation for the existence of a sharp threshold plasma concentration of UCB setting its neurotoxicity is based on the ability of UCB to bind with a high affinity to plasma proteins, mostly to albumin. The slow dissociation rate of the albumin/bilirubin complex and the short transit time of plasma in the brain do not favor the cerebral penetration of bound UCB (Robinson and Rapoport, 1986). Numerous early works (reviewed in Wennberg, 2000) demonstrated experimentally that the pigment accumulates into the brain and neurotoxicity occurs when the circulating free (unbound) UCB increases abnormally. This happens especially when plasma protein binding to UCB approaches saturation, or when UCB is dissociated from albumin by drugs competing for albumin binding sites. UCB is a dicarboxylic acid which has remarkably high pKa values (8.1 and 8.4), and is thus mostly non-ionized at the physiological plasma pH of 7.4. The relative hydrophobicity of UCB allows it to readily permeate the cell layers that form the tight blood–brain interfaces (BBIs) by passive diffusion (Zucker et al., 1999; Vitek and Ostrow, 2009). Within the brain some regions (mainly the basal ganglia, selected brain stem areas, hippocampus, and cerebellum) show a greater sensitivity to UCB toxicity (Shapiro, 2010). UCB alters neuronal cell functions and causes cell death by mechanisms that seem to involve synaptic dysfunction, but also oxidative stress, impairment of mitochondrial oxidative phosphorylation and apoptosis (reviewed in Watchko, 2006). In this pathophysiological scheme, BBIs merely acts as structural barriers limiting the penetration of the carrier protein, while allowing unrestricted passive diffusion of free UCB into the brain, and neural cells are considered as passive targets of UCB toxicity.
Yet, our current knowledge on transport and metabolic processes in the brain supports the concepts that (1) the implication of BBIs in the occurrence of bilirubin encephalopathy is more complex than a simple role as a diffusion barrier to carrier proteins, and (2) neural cells such as astrocytes and neurons appear to play an active role in controlling the balance between the neuroprotective and neurotoxic properties of UCB.
Relevant to the first point, BBIs may actively participate in the regulation of the cerebral bioavailability of UCB. First, the binding affinity of UCB to human serum albumin has been shown to be strongly influenced by both albumin and chloride concentrations and free UCB concentration is actually much higher than previously thought (Weisiger et al., 2001). This suggests that parameters other than the rate of diffusional influx of UCB exist at the BBIs to account for the very low physiological UCB intracerebral concentrations (Daood and Watchko, 2006; Zelenka et al., 2008) and the absence of neurotoxicity. Furthermore, the numerous transporters and detoxifying enzymes that have been described in BBIs may confer an active role to the barriers in limiting the entry of free UCB into the CNS, and/or in increasing its elimination from the CNS. In line with this latter possibility, Levine et al. (1985) reported that bilirubin efflux from brain and cerebrospinal fluid (CSF) was an important factor of UCB cerebral homeostasis. An alteration of the neuroprotective functions of the BBIs is another aspect that may have been overlooked in bilirubin-induced encephalopathy. The BBIs are key elements in establishing the cerebral homeostasis required for proper brain development and neuronal function throughout life. In addition to supplying the brain with nutrients and micronutrients (Davson and Segal, 1996; Redzic and Segal, 2004), the BBIs protect the cerebral tissue by decreasing brain exposition to numerous circulating drug and toxic substances, and by their antioxidant properties. The cells forming the BBIs are the first cerebral cells exposed to plasma free UCB, so that any impairment of their antioxidant and neuroprotective functions, especially during the postnatal period when the developing neuropil is highly sensitive to toxic insults, may contribute to the occurrence of bilirubin encephalopathy.
Relevant to the second point, UCB is present in the brain under normal conditions at nanomolar concentrations (40–50 nM in normobilirubinemic rats). UCB confers neuroprotection against oxidative stress to cultured neuronal cells at similar (20–50 nM) concentrations (Doré and Snyder, 1999), and become toxic for neuronal cells at extracellular concentrations only slightly higher (Ostrow et al., 1994). Physiologically, the main fraction of cerebral UCB derives from the peripheral degradation of hemoglobin, but the pigment is also produced within the brain by catabolism of other heme-containing proteins. Neural cells seem to possess a biochemical machinery to maintain the intracellular concentration of UCB within a range compatible with the beneficial effect of the pigment, and damage occurs only when these mechanisms are overrun by elevated UCB brain extracellular concentrations such as during chronic unconjugated hyperbilirubinemia. In this situation, the brain UCB content increases to micromolar levels throughout the brain as shown in animal models (Zelenka et al., 2008; Gazzin et al., 2012) while neuronal damage occurs with a selective topography (Shapiro, 2010; Gazzin et al., 2012). This suggests that factors other than intracerebral UCB concentration are involved in the regional selectivity of UCB toxicity.
The following sections review these complex facets of bilirubin interactions with the BBIs as well as with glial and neuronal cells (Figure 1). They raise some questions that still need to be solved in view to optimize the development of efficient prophylactic and therapeutic treatments to protect the brain against the accumulation of excessive levels of UCB and prevent the neurotoxic effect of the pigment.
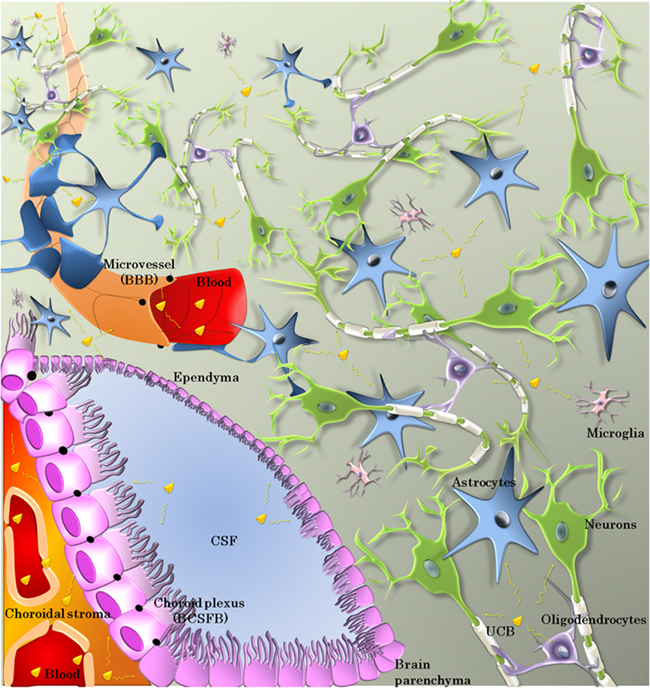
Figure 1. Cellular effectors involved in the bioavailability and toxicity of UCB. Bilirubin exchanges between blood and brain occur mainly across the microvessel walls forming the blood–brain barrier (BBB) or the choroid plexus epithelium forming the blood–cerebrospinal fluid barrier (BCSFB). The cells forming these barriers are sealed by tight junctions (black dots and lines). The fenestrated choroidal vessels allow extensive exchanges between the blood and the choroidal stroma. CSF–brain exchanges take place across the ependyma, or the pia–glia limitans (not shown here). Within the neuropil, neurons are the terminal target of bilirubin toxicity. Astrocytes, microglia, and oligodendrocytes all play a role in controlling bilirubin toxicity-over-benefit balance.
Bilirubin Interactions with Metabolic/Transport Processes at the Blood–Brain Interfaces
Specific Properties of the Blood–Brain and Blood–Cerebrospinal Fluid Barriers
The BBIs, together with the CSF circulatory system, are responsible for the homeostasis of the cerebral extracellular fluid necessary to neuronal functions. The interface between the blood and the brain parenchyma proper, referred to as the blood–brain barrier (BBB), is located at the endothelium of the cerebral microvessels. Pericytes ensheathed in the endothelial basal membrane and astrocytic end-feet apposed to this membrane are thought to be essential for the induction of the barrier phenotype characteristic of the brain endothelium. Occasional macrophages are also associated with the vasculature. Another barrier component is the blood–CSF barrier (BCSFB), formed by the epithelium of the four choroid plexuses (CPs; Figure 1). Located in brain ventricles, the CPs display the highest local blood flow rate among all cerebral structures, and are responsible for the secretion of CSF. The CP–CSF system adds a degree of complexity to the mechanisms that set the cerebral bioavailability of both endogenous and exogenous bioactive compounds. The CSF circulatory pathways, the precise histological organization of these barriers and the interplay between the BBB, BCSFB, and CSF have been described elsewhere (Strazielle and Ghersi-Egea, 2000a; Ghersi-Egea et al., 2009a,b).
The molecular mechanisms that support the neuroprotective functions of BBIs are multiple. The tight junctions efficiently restrict the entry of blood-borne polar compounds into the brain, with the exception of nutrients, polypeptides, and hormones required for brain function, that are carried by selective influx transport systems. The penetration of more lipid-soluble molecules capable of diffusing across the cell membranes is limited by binding to serum albumin and influenced by transport proteins present in both barriers. These transport systems include P-glycoprotein, Breast Cancer Resistance Protein, or Multidrug-associated Resistance Proteins (MRPs)1, all belonging to several subfamilies of ATP Binding Cassette (ABC) efflux transporters. They are also organic anion and cation transporters of the Solute Carrier (SLC) superfamily. All these systems display broad substrate specificities, and a pattern of expression specific to each barrier. Being distributed between the blood-facing and brain-facing membranes in both endothelial and epithelial cells, they work in concert to achieve highly efficient neuroprotection (Strazielle et al., 2004; Leslie et al., 2005; Strazielle and Ghersi-Egea, 2005; Gazzin et al., 2008). Finally, detoxifying enzymes present at BBIs can influence the cerebral availability of toxic compounds (Ghersi-Egea et al., 1994; Dauchy et al., 2008). In particular, CPs possess a high conjugation capacity based on phase II drug metabolizing enzymes such as glutathione-S-transferases, as well as a high epoxide hydrolase activity and antioxidant capacity, hence fulfilling “liver-like” functions within the brain (Ghersi-Egea et al., 1995, 2006; Strazielle et al., 2004).
Within the CNS, diffusion between the CSF and the parenchyma occurs more freely across the ependyma bordering the ventricles, or the pia–glia limitans separating the neurons from the subarachnoid and cisternal spaces as the cells forming these interfaces are not linked by tight junctions. However, exchanges between the different CNS compartments may be influenced by glutathione-S-transferases and ABC transporters, which have been described in the ependymal and glial cells (Strazielle and Ghersi-Egea, 2000b; Mercier et al., 2004; see infra).
The structural features of the barriers are established early on during brain development. The paracellular pathway is already impeded in the fetal brain, pointing to the efficiency of the BBIs in protecting the developing brain. In addition the BBIs may fulfill fence and transport functions which are specific to the immature brain (Ek et al., 2006; Johansson et al., 2008; Daneman et al., 2010). Less is known about the developmental regulation of the efflux transporters and enzymes involved in detoxification processes (see infra).
It is well established that in the liver a complex interplay between ABC efflux pumps, detoxifying enzymes, and possibly SLC transporters (Table 1) allows controlling the plasma UCB level. Similar interactions between UCB and these families of proteins may occur at the BBIs, hence influence UCB cerebral bioavailability.

Table 1. Main potential mechanisms involved in bilirubin detoxification and elimination.
Mechanisms Controlling Bilirubin Permeability at BBIs
Transporters
Different studies (reviewed in Ghersi-Egea et al., 2009a) pointed out UCB as a substrate for various transporters. These include the ATP-dependent ABCC1 transporter which is a member of the multidrug-resistance associated protein family also called MRP1, the ATP-dependent multidrug-resistance protein ABCB1 (human)/Abcb1a/b(rodents; see text footnote 1) also called P-glycoprotein or MDR1, which displays a lower affinity than MRP1, and finally transporters of the SLC21/Organic Anion Transport Polypeptide (OATP) family2, such as OATP1B3 (also called OATP8, SLC21A8), or OATP1B1 (OATP-C, OATP2, SLC21A6) in human.
P-glycoprotein and MRP1 are two major efflux transporters involved in neuroprotection. They prevent access to, or increase elimination from the brain of various endo- and xenobiotics (Schinkel et al., 1996; Wijnholds et al., 2000; Schinkel and Jonker, 2003). P-glycoprotein is almost exclusively located at the blood-facing luminal membrane of the endothelium forming the BBB, while MRP1 is mainly located at the basolateral membrane of the choroidal epithelium. This mirror image of the relative abundance of P-glycoprotein and MRP1 between the two interfaces was confirmed in human (Gazzin et al., 2008). Different members of the OATP family have also been localized at the BBB and BCSFB in both laboratory animals and human. For most of these OATPs, the precise cellular localization, directionality of transport and functional significance remain to be assessed (for details, see Strazielle et al., 2004; Strazielle and Ghersi-Egea, 2005; Ghersi-Egea et al., 2009a). OATP1A2 can transport opioid peptides across the human BBB (Gao et al., 2000; Lee et al., 2005), and at the BCSFB, Oatp1a5 (Oatp3, Slc21a7) located at the apical membranes of the rodent choroidal epithelium, transports organic anions from the CSF into the choroidal epithelial cells, for further efflux into the blood. All P-glycoprotein, MRP1, and those of the OATP transporters located at brain-facing membranes, are ideally localized to prevent the entry of blood-borne substrates, including UCB into the brain, or to allow their elimination from the central nervous system.
In line with this hypothesis, a role of P-glycoprotein in decreasing UCB penetration into the brain has been highlighted in Abcb1a/b knockout mice which display a cerebral UCB concentration 1.8-fold higher than wild type animals after a 5-min intravenous UCB infusion. Importantly, this treatment resulted in a high plasma total UCB that was not different between the two types of animals (Watchko et al., 1998). Brain-to-plasma bilirubin ratios were also increased in wild type rats treated with P-glycoprotein inhibitors (Hanko et al., 2003). The significance of a P-glycoprotein-dependent efflux process limiting free UCB diffusion from plasma into the neuropil remains to be assessed at lower clinically relevant UCB concentrations and in the developing brain. Indeed, a role for P-glycoprotein in the canalicular efflux of UCB in the liver could not be established, and the amount of P-glycoprotein associated to microvessels isolated from 9-day-old animals is only one-fifth of that measured in capillaries from adult animals (Gazzin et al., 2008). No study has specifically investigated the involvement of MRP1 in UCB transport across the BCSFB. A considerable uptake of UCB by isolated rabbit CP has been described, which is inhibited by iodipamide, a substrate of organic anion transport systems (Jakobson, 1991). Which OATP or other transport proteins located at the brain-facing membranes of the BCSFB are involved in the clearance of UCB from the CNS remains an open question. It is however tempting to hypothesize that the carrier-mediated uptake of UCB at the apical membrane of the choroidal epithelium, coupled to active efflux into the blood via the basolateral MRP1 pump forms a pathway accelerating the clearance of UCB from the CNS. Such mechanism would be of special interest during early postnatal life, as the CP is a brain structure maturing early during development, and already expresses high levels of MRP1 at birth (Gazzin et al., 2008; Johansson et al., 2008).
Metabolizing enzymes
In the liver, UCB interacts mainly with three families of detoxifying enzymes, cytochrome P-450-dependent mixed function oxygenases (CYPs), glutathione-S-transferases (GSTs), and UDP-glucuronosyltransferases (UGTs).
Unconjugated bilirubin taken up by hepatocytes binds to cytosolic isoforms of GST, which act as a sink that maintains the gradient of free UCB and limits its backflux into plasma. There are several GST classes, those best characterized in mammals being the alpha, mu, and pi classes (see Mannervik et al., 2005 for GST nomenclature in human, rat, and mouse). GSTs are homo- or heterodimer proteins. The GSTA (alpha class) subunits have the highest affinity for free UCB. Dimers of GSTA1 and/or GSTA2 form ligandin, the main GST isoform involved in hepatic UCB binding. This binding is independent of the enzymatic activity (bilirubin is not conjugated to glutathione), and contributes to protect the cells by maintaining the intracellular pool of free UCB at low level. It may also facilitate UCB transfer to the smooth endoplasmic reticulum for further catabolism. The three main classes of cytosolic GSTs of all three alpha, mu, and pi classes are present in BBIs, although GST pi rather than GST alpha seems to be the major GST proteins in adult human brain capillaries (Shawahna et al., 2011). GST-dependent activities are especially high in CPs isolated from both animal and human developing brain (reviewed in Ghersi-Egea et al., 2006, 2009a). Thus, in the course of UCB transport across BBI cells, binding of UCB to GST subunits, especially of the alpha class, may contribute to an overall barrier effect. The developmental profile of GST proteins at the interfaces differs between the pi, mu, and alpha classes. In rat, transcripts of mu and pi subunits are higher in CPs from newborn compared to adult animals, while those of the alpha subunits which display the highest affinity for UCB are absent or down regulated in newborn pups (unpublished data). The alpha class subunits were detectable by immunohistochemistry only 10 days after birth in mouse choroidal cells (Beiswanger et al., 1995), or in adult but not fetal CP in human (Carder et al., 1990). Hence, at CSF-contacting interfaces, UCB binding to GST subunits is likely to be less efficient during development than at the adult stage.
Bilirubin metabolic pathways include oxidation by CYPs, namely CYP1A1 and CYP1A2 (Kapitulnik and Gonzalez, 1993), as well as CYP2A5 (Abu-Bakar et al., 2005; Cyp2a3 in rodents). Activities of CYP isoenzymes appear modest in cells forming the BBIs (Strazielle and Ghersi-Egea, 2000b; Miksys and Tyndale, 2002). Total cytochrome P-450 has been detected by spectrophotometry in microvessels isolated from both rat and human brains (Ghersi-Egea et al., 1993). Gene and protein expression studies conducted in human microvessels showed that CYP1B1 was the main endothelial CYP, while CYP1A1 and 2 expression levels were low or undetected (Dauchy et al., 2008; Shawahna et al., 2011). Cyp1a1 protein has been detected in cultured mouse brain endothelial cells only after treatment with 2,3,7,8-tetrachlorodibenzo-p-dioxin, an inducer acting via the Ah receptor (Filbrandt et al., 2004). Cyp1a1 mRNA was detected in isolated rat capillaries only following in vivo treatment with Ah receptor-dependent inducers but the Cyp1a1 protein was not detected (Jacob et al., 2011). Another study performed by Western blot detected the protein in brain microvessels following Ah receptor activation (Wang et al., 2011). This apparent discrepancy may have come from differences in the composition of the microvessels preparations between the two studies. Indeed previous in vivo works had shown that Cyp1a1 was detected in mouse cerebral veins rather than capillaries following treatment with Ah receptor-dependent inducers. Induced Cyp1a1 was also detected in rat and mouse choroidal sinusoidal vessels (Brittebo, 1994; Morse et al., 1998; Dey et al., 1999; Granberg et al., 2003). No information is available concerning CYP2A5 in brain barriers. Overall, CYP activities likely play only a minor role in UCB metabolism at BBIs, unless specific inductive mechanisms occur in the course of hyperbilirubinemia (see infra).
The main pathway of bilirubin detoxification in the liver is conjugation with glucuronic acid. It is specifically catalyzed by the UGT1A1 isoenzyme (Kadakol et al., 2000). This pathway leads to the production of the more water soluble mono and di-glucuronide metabolites which are mainly transported from the liver into the bile by ABCC1, 2, and 3 proteins (Keppler, 2011). In the rat brain, Ugt activity toward planar compounds is several times higher in microvessels than in the cerebral parenchyma, and is notably comparable to the hepatic activity in the choroidal epithelium, even in developing animals (Ghersi-Egea et al., 1994; Strazielle and Ghersi-Egea, 1999). In both barriers these activities increase following in vivo treatment of rats with exogenous inducers (Ghersi-Egea et al., 1988; Leininger-Muller et al., 1994). The UGT isoenzyme responsible in rat BBIs for the conjugation of planar compounds is likely to be Ugt1a6 (Gradinaru et al., 2009). Ugt1a1 mRNA has been reported in whole rat brain extract (Shelby et al., 2003), but it is not known whether Ugt1a1 is associated with BBIs in rodents. Neither UGT1A6 nor UGT1A1 mRNA was detected in human brain capillaries (Shawahna et al., 2011). As the CPs fulfill “liver-like” functions for the brain, and as the conjugation to glucuronic acid within the epithelial cells, coupled to a basolateral efflux of the resulting glucuronides forms a functional metabolic barrier preventing the entry of selected substrates into the CSF (Strazielle and Ghersi-Egea, 1999), the choroidal tissue might prove to be an important site of UCB detoxification in the brain. This and the presence of different ABCC proteins located in both brains barriers (reviewed in Ghersi-Egea et al., 2009a), call for more investigations on UCB and bilirubin conjugate disposition at the BBIs.
CSF proteins
The protein concentration is of 3.2 g/l in CSF at birth in rat and decreases thereafter to reach adult values as low as 200 mg/l. This is not due to an immaturity of the BBIs allowing non-specific leakage of plasma proteins into the brain, but rather to a regulated mechanism involving a specialized transport function of the BCSFB during development (Johansson et al., 2008). This opens new fields of investigation to appreciate the relevance of CSF and ICF proteins in sequestering UCB within the extracellular space in the brain during early postnatal development, and to decipher the choroidal mechanisms that control protein CSF concentration in normal and jaundiced brain.
Photoisomers
Finally, the BBIs may play a significant role in the mechanisms behind the efficacy of phototherapy, a treatment degrading the light sensitive plasma UCB in jaundiced children. During phototherapy, both photooxidation and photoisomerization of UCB occur. While the former is a slow process, photoisomers are detected in blood within minutes after starting the treatment, and although the reaction is reversible, the major 4Z, 15E photoisomer represents 20–30% of total bilirubin after 2 h. Total bilirubin concentration is not affected as rapidly in cases of phototherapy in jaundiced newborn, and does not decrease in blood during the 4-h following treatment initiation (Mreihil et al., 2010). Yet, because the isomers have lower lipid solubility relative to the parent UCB species, and should be restricted to a greater extent by the BBIs, an immediate phototherapy benefit should be achieved before UCB plasma levels are normalized. This hypothesis remains to be proven experimentally (McDonagh, 2010).
Alteration of the Neuroprotective Functions of BBIs by Hyperbilirubinemia
At low concentration UCB behaves as an antioxidant agent and may represent a transitional antioxidative mechanism during physiological jaundice (see infra). However, higher concentrations of bilirubin are harmful. In vitro studies have shown that a prolonged exposure of cells to free bilirubin at concentrations that were within the range of aqueous solubility, i.e., similar to plasma free bilirubin concentrations reached in moderate jaundice, elicited cytotoxic effects toward different cell types (Chuniaud et al., 1996 and reviewed in Ostrow et al., 2002). The molecular mechanisms of UCB cytotoxicity, assessed mostly in vitro, are not fully unraveled. Besides neuron-specific mechanisms, key events in UCB-induced toxicity would involve both oxidative injury and mitochondrial damage. The latter is probably triggered by bilirubin-membrane interactions and leads to an impairment of oxidative phosphorylation (reviewed in Watchko, 2006; Ghersi-Egea et al., 2009a).
This mechanism is especially relevant when evaluating the role of BBIs in bilirubin cerebral toxicity, because the cells forming the BBIs are the first cerebral cells exposed to blood-borne free bilirubin, and many of the key functions of the barriers are energy-dependent. Mitochondria are numerous in cells forming the BBIs, especially the choroidal epithelial cells (Schlosshauer, 1993; Cornford et al., 1997). Tight junction protein complexes undergo continuous molecular remodeling, which is linked to the energy status of the cells (Eckert and Fleming, 2008; Shen et al., 2008). ABC efflux transporters involved in neuroprotection are unidirectional, and require ATP-dependent energy to transport substrates against the concentration gradient. Several other organic anion and cation transporters are secondarily dependent on inorganic anion gradients generated by the Na+K+ATPase, as demonstrated at the BCSFB (Pritchard et al., 1999). Finally, at the CP, CSF secretion is also a tightly controlled process linked to Na+K+ATPase.
These different BBI-related neuroprotective mechanisms have hardly been evaluated in the context of bilirubin encephalopathy. Experimental hyperbilirubinemia obtained by short-term continuous infusion of bilirubin does not lead to any prominent alteration of the barrier integrity (Roger et al., 1993, 1995). Alterations secondary to mitochondrial dysfunction are likely to develop only after sustained exposure to elevated levels of UCB. Accordingly, a decrease in the expression of occludin, one of the proteins forming the tight junctions in BBB and BCSFB, was observed in microvessel fractions prepared from brains of rats subjected to obstructive jaundice for several days. The direct involvement of bilirubin in this process was however not evaluated (Faropoulos et al., 2010). Exposition of bovine brain endothelial cells in culture to UCB concentrations generating high levels of extracellular free bilirubin-induced cell apoptosis (Akin et al., 2002). A similar increase in apoptotic/necrotic processes was more recently described in an immortalized cell line and in primary cultures of human brain endothelial cells exposed for several hours to lower and more physiopathologically relevant free bilirubin concentrations (Palmela et al., 2011). In this study, the cell loss in bilirubin-treated cultures was twice that in untreated cells after 24 h. The relevance of these in vitro data to in vivo UCB–induced brain endothelial cell lethality remains to be established. In these cells, bilirubin only marginally affected inflammatory mediators, and induced an oxidative stress reflected by a large increase in nitrite production by comparison with untreated cells. The effect of such UCB-induced alteration of the redox status on the barrier properties of the endothelial cell monolayers was not assessed. The potential influence of bilirubin-activated perivascular astrocytes and macrophages on BBB properties would also deserve attention.
The effect of UCB on the BCSFB has been investigated using monolayers of rat choroidal epithelial cells cultured in bicameral devices that maintain the barrier and transport properties of this barrier (Strazielle and Ghersi-Egea, 1999; Strazielle and Preston, 2003; Ghersi-Egea et al., 2006). The cells were exposed at their basolateral (blood-side) membrane to 40 and 140 nM free bilirubin, concentrations respectively below and above the aqueous solubility of the pigment and which mimic levels evaluated in physiological and pathological jaundices, respectively (Rigato et al., 2005; Ahlfors et al., 2009). Treatment was applied for six consecutive days to approach the in vivo prolonged postnatal exposure. This treatment did not alter the integrity of the barrier, as assessed by the measurement of the paracellular permeability of the monolayers to sucrose (Gazzin et al., 2011). The high antioxidant capacity of the choroidal tissue (Tayarani et al., 1989; Ghersi-Egea et al., 2006), in concert with efflux transporters, may allow maintaining the integrity of the barrier under pathophysiologically relevant UCB-challenge.
By contrast, ABC transporters are affected by sustained UCB exposure. The homozygous hyperbilirubinemic jj Gunn rat bears a congenital inherited deficiency of the hepatic UDP-glucuronosyltransferase enzyme and is used as an animal model of Crigler–Najjar type I syndrome and neonatal jaundice. P-glycoprotein expression is increased up to twofold in brain microvessels from developing jj Gunn rats as compared to heterozygous littermates; however, it does not reach the high levels observed in microvessels of adult animals. More strikingly, the level of MRP1 protein, already high in CPs of newborn animals, is decreased by 50% in jj Gunn rats compared to heterozygous littermates. This decrease in protein content was a direct effect of bilirubin, because it could be reproduced by treating choroidal epithelial cells for 6 days with UCB at a dose generating a 140-nM free bilirubin concentration in the medium (Gazzin et al., 2011), the latter concentration mimicking the levels measured in pathological jaundice in Gunn rats 9 days after birth (Gazzin et al., personal communication). Mrp1 is involved in regulating the cerebral availability of endogenous biologically active compounds and of circulating potentially toxic xenobiotics. Hence the UCB-induced decrease in the efflux transport activity of choroidal MRP1 may critically impact on normal brain maturation.
Finally, the CPs strongly participate to thyroid hormone homeostasis which is crucial for brain maturation (reviewed in Zheng, 2001; Ghersi-Egea et al., 2009a). UCB competitively inhibits cellular uptake of thyroid hormones in hepatocytes (Lim et al., 1993; Hennemann et al., 2001). Whether hyperbilirubinemia also disturbs cerebral thyroid hormone transport and homeostasis needs to be assessed in jaundiced neonates.
Control of Bilirubin Intracellular Concentration and Toxicity in the Neuropil
Antioxidant Function of Bilirubin: The Biliverdin/Bilirubin Redox Cycle in Neural Cells
Among all body organs, the brain shows the highest rate of oxygen metabolism, consuming 20% of the whole body oxygen while weighting only 2% of the total body (Clarke and Sokoloff, 1999). Oxygen reactive species (ROS) are continuously generated at a high rate, and oxidative stress does occur to a certain degree in the absence of any pathology. ROS may damage sugars, proteins, DNA, and lipids. The brain is rich in unsaturated fatty acids which are main targets for lipid peroxidation. Relevant to perinatal diseases, birth is accompanied by a sudden exposition to oxygen, leading to an increased oxidative stress. In premature newborns, antioxidant defenses are not fully efficient because their maturation occurs during the late gestation period (Saugstad, 1989; Friel et al., 2004). Thus newborns and especially premature infants exhibit a unique sensitivity to oxidant injury.
The brain possesses only moderate antioxidant superoxide dismutase and glutathione peroxidase activities compared to the liver (Cooper et al., 1997; Ho et al., 1997), with the exception of the CP (Tayarani et al., 1989). Besides the reduced/oxidized glutathione redox cycle functioning as a cell antioxidant mechanism, the bilirubin-dependent redox cycle also seems to play a role in cell protection against oxidative stress in brain, for the following reasons.
Bilirubin is present in brain tissue under normal conditions in nanomolar concentrations (20–50 nM; Gazzin et al., 2012). The dynamic of bilirubin movement between extracellular and intracellular compartment is complex, and may involve both transporter-mediated processes and diffusion. Hence data on UCB concentration in tissue do not inform on the actual intracellular concentration in different neural cells. Bilirubin concentrations in the overall brain represent less than 0.1% of the level of the antioxidant molecule glutathione (Baranano et al., 2002). Nevertheless the pigment ability to protect against oxidative stress is well acknowledged (Dennery et al., 1995; Dohi et al., 2005; Vitek, 2005a,b), and evidence that this holds true for brain cells has been documented. Bilirubin is the reduction product of biliverdin. The biliverdin/bilirubin and glutathione redox cycles share some similarities. Biliverdin synthesis results from the degradation of heme-containing proteins by heme oxygenases (HO). The inducible HO-1 is expressed in glial cells (Dwyer et al., 1995) and is induced by oxidative stress (Calabrese et al., 2006). The constitutive HO-2 accounts for the main portion of this enzymatic activity in brain, where it seems to be expressed in neuronal populations in several regions (Ewing and Maines, 1997; Mancuso, 2004). HO-2 impairment results in a loss of bilirubin in cells and a higher susceptibility to different CNS damages (Chen et al., 2005). Doré et al. (1999) described a correlation between an activation of HO-2 by protein kinase C-mediated phosphorylation, increased cellular bilirubin levels (sixfolds after 24 h), and resistance to H2O2 induced stress in cultured hippocampal and cortex neurons.
Biliverdin is reduced to bilirubin by the cytosolic enzyme biliverdin reductase (BVR), the depletion of which strongly induces (20–40%) the apoptosis of cells cultured from hippocampal/cortical structures (Sedlak et al., 2009). BVR activity requires free SH groups and NADH or NADPH (at pH of 6.8 and 8.7, respectively; Maines and Trakshel, 1993). BVR pattern of expression in normal brain coincides with HO gene expression (Ewing et al., 1993). BVR is also involved in cell signaling (Kapitulnik and Maines, 2009), and can transport the transcription factor hematin from the cytoplasm to the nucleus, thereby allowing hematin-dependent HO-1 gene transcription (Tudor et al., 2008). Silencing BVR leads to a depletion of cellular bilirubin, increases cellular ROS and promotes apoptotic death in neuronal cultures (Baranano et al., 2002). Finally BVR can increase bilirubin production from heme degradation during oxidative stress (Miralem et al., 2005). In turn, the increase in UCB inhibits BVR and HO activity, maintaining a balanced intracellular biliverdin/bilirubin ratio (Maines, 2005). When produced intracellularly, bilirubin may act as ROS scavenger by quenching reactive radicals (Nag et al., 2009) before being reoxidized to biliverdin (Baranano et al., 2002). In brain homogenate, biliverdin, and bilirubin have been reported to inhibit lipid peroxidation induced by hemoglobin. This was not observed in vivo after intrastriatal injection of hemoglobin, probably because of its conversion into biliverdin and bilirubin by the endogenous enzymatic machinery (Van Bergen et al., 1999).
Finally, approaches using bilirubin antioxidant properties for therapeutic purposes have been evaluated experimentally. The administration of exogenous biliverdin (immediately converting into bilirubin) to rat following transient cerebral artery occlusion, significantly reduced the volume of infarct tissue as well as superoxide production and lipid peroxidation (Deguchi et al., 2008). In autoimmune encephalomyelitis, an animal model for multiple sclerosis, treatment with BVR (10 μg/day) ameliorates the outcome of the pathology more efficiently than optimal treatments with catalase or HO-1, while treatments with superoxide dismutase and glutathione reductase have no significant effect (Liu et al., 2006).
Overall, these sets of data indicate that biliverdin production and the biliverdin/bilirubin cycle are effective in the brain, and can play a role in the regulation of oxidative stress. The huge capacity of bilirubin to protect against oxidative stress (10 nM UCB protects against 75 μM H2O2 in cell culture, Doré and Snyder, 1999), seems to result from the high turn-over rate of the biliverdin/bilirubin cycle (Baranano et al., 2002) by comparison to the glutathione redox system (Hollensworth et al., 2000). The two redox systems appear complementary. Unlike glutathione, bilirubin binds avidly to albumin and to cell membranes, especially myelin rich membranes (Mustafa and King, 1970; Brodersen, 1981), where it may prevent lipid peroxidation and protect the proteins inside the bilayer (Van Bergen et al., 1999). Reduced glutathione and bilirubin have thus been hypothesized to be complementary in their effects against oxidative stress. Reduced glutathione may serve as a protectant for cytoplasmic ROS targets, while bilirubin acts preferentially against lipid peroxidation. This was demonstrated in cultured HEK293 cells (Sedlak et al., 2009). The sixfold increase in protein oxidation in H2O2 treated cells compared to control cells was reversed by addition of reduced glutathione but not bilirubin, while a 2.5- to 3-fold increase in lipid peroxidation was obtained by silencing BVR activity, but not by BSO-induced glutathione depletion. A confirmation of this hypothesis was obtained in vivo by comparing wild type and HO-2 deleted mice (Sedlak et al., 2009).
Besides complementing each other, the glutathione redox cycle and the biliverdin/bilirubin cycle undergo complex interactions. On one hand, the reduced/oxidized glutathione equilibrium acts as a sensor for redox stress. Reduced glutathione depletion activates HO-1 gene transcription, probably by mitogen-activated protein kinases, and thereby could favor the bilirubin/biliverdin cycle. On the other hand, molecules bearing free thiol groups are necessary to BVR activity. Thus a decrease in the concentration of reduced glutathione and other molecules bearing SH groups might impact on the efficacy of the bilirubin/biliverdin cycle.
A threshold UCB concentration sets the switch between anti- and pro-oxidant effects of bilirubin. Doré and Snyder (1999) reported that its maximal neuroprotective effects in hippocampal cultures was reached at nanomolar concentrations (10–50 nM), while at higher pigment concentrations the pro-oxidant effects of bilirubin started to dominate. A similar dual effect was reported in primary cultures of oligodendrocytes (Liu et al., 2003). The exact concentration thresholds between anti- and pro-oxidant effects of bilirubin in different brain structures and cells in vivo are however not well estimated, and need further investigation.
Metabolic/Transport Processes Controlling Bilirubin Concentration and Toxicity in Glial and Neuronal Cells
The pro-oxidant activity of UCB is probably the most studied aspect of bilirubin encephalopathy and kernicterus in which UCB content in tissue increases from protective nanomolar to toxic micromolar concentrations, mainly as a result of UCB influx from blood (Zelenka et al., 2008; Gazzin et al., 2012). Neural cells possess regulatory mechanisms that allow controlling UCB intracellular concentrations to some extent. Even if expressed at lower levels than in BBI (see supra), transporters, and detoxifying enzymes such as those described in Table 1 might act also at the brain parenchyma level.
Among the two ABC proteins that transport bilirubin, P-glycoprotein level of expression is at least 20 times lower in whole brain parenchyma compared to cerebral capillaries, indicating that P-glycoprotein is almost exclusively located at the BBB (Gazzin et al., 2008) and is unlikely to play a role in bilirubin transport in neural cells. Of note, P-glycoprotein has been identified in cultured microglia (Lee et al., 2001) and astrocytes (Ronaldson et al., 2004). The high affinity bilirubin transporter Mrp1 (Rigato et al., 2004) is present in rat brain parenchyma, where the protein content represents 6% of that measured in the BCSFB (Gazzin et al., 2008). Mrp1 is not enriched in the capillary fraction compared to parenchyma, indicating its presence in neural cells. Accordingly, it has been identified on brain tissue sections in rat astrocytes and in the glia limitans (Mercier et al., 2004). In vitro, the expression of MRP1 was shown in microglia primary cultures or cell lines (Dallas et al., 2003), in oligodendrocytes (Hirrlinger et al., 2002), and in rat, mice, and human primary cultures of astrocytes (Decleves et al., 2000; Hirrlinger et al., 2002, 2005; Spiegl-Kreinecker et al., 2002; Gennuso et al., 2004; Falcão et al., 2007). The simultaneous up-regulation and redistribution of the transporter from the perinuclear region to the plasma membrane after exposure to UCB was interpreted as a mechanism to confer cellular protection by allocating the UCB transporter in the most adequate subcellular position to face pigment entry (Gennuso et al., 2004). Mrp1 was also detected in primary cultures of embryonic rat neurons, even if in amounts lower than in astrocytes (Falcão et al., 2007). An inverse correlation between Mrp1 expression and sensibility to UCB, was reported in neurons and astrocytes, possibly explaining the higher UCB toxicity observed in cultured neurons which display the lowest levels of Mrp1 (Falcão et al., 2007). In agreement with a protective function of MRP1, mouse embryonic fibroblast cells derived from Mrp1 KO mice (Calligaris et al., 2006), and SH-SY5Y cells in which the transporter activity was silenced (Corich et al., 2009), displayed a higher mortality after bilirubin treatment compared to their respective controls. Cell culture conditions substantially modulate the expression of several ABCs transporters, however, and the observations obtained from in vitro experiments may not be fully applicable to the in vivo scenario. Accordingly, the actual levels of expression and functional relevance of UCB transporters in brain parenchyma in vivo are still debated.
The potential relationship between GST isoforms present in astrocytes and bilirubin toxicity has not been investigated, but in one study that showed higher cytosolic GST activity and level of alpha class GST subunits in cerebellum of hyperbilirubinemic jj Gunn rats compared to the Jj heterozygous animals. Within this distinct structure, the alterations in GST levels were region-specific, developmentally regulated, and inversely related to region-specific cerebellar hypoplasia (Johnson et al., 1993). This calls for additional investigation on the potential implication of GSTs in the control of long-term hyperbilirubinemic cerebral intoxication.
Cytochrome P-450 monooxygenases are involved in controlling bilirubin concentration in tissues and cells, as originally shown by Kapitulnik and colleagues. Selected hepatic Cyps isoforms (Cyp1a1 and 1a2) are able to compensate for the lack of hepatic glucuronidation in Gunn rat (Kapitulnik et al., 1987; Kapitulnik and Gonzalez, 1993), by increasing UCB body clearance following its oxidation (Zaccaro et al., 2001; De Matteis et al., 2006). CYP isoenzymes display a highly heterogeneous expression among different regions or cellular types in the CNS. For instance it is proposed that certain cellular subsets might express CYPs at levels as high as in the liver (Chinta et al., 2005b). Thus, it is conceivable that local CYP-dependent metabolic pathways have considerable effects on brain pathology and physiology. CYP 1A1 and 1A2 seems to be largely expressed in brain, predominantly in cerebral cortical neurons, Purkinje, and granule cells of hippocampus (Chinta et al., 2005a). CYP1A2 pattern of expression in human brain seems to be diffuse (Farin and Omiecinski, 1993). The role played by CYPs in brain resistance to bilirubin has just started to be investigated.
The expression of CYPs was investigated in the context of typical kernicterus which occurs only when an acute shift of bilirubin from blood to tissues is generated. It was reproduced experimentally in homozygous Gunn rats exposed to sulfadimethoxine to displace bilirubin from albumin. In this model, ABC proteins appear inefficient to control the sudden increase in tissue free bilirubin concentrations (Gazzin et al., 2012). By contrast, a strong correlation was observed between the time-course and extent of induction of the Cyp genes involved in bilirubin oxidation on one hand, and the kinetic of tissue UCB disappearance on the other hand, in the different brain regions. Cyp mRNAs increased up to 70-fold in some regions, but returned to basal levels within 48–72 h, indicating that these changes are transitory. Overall, this set of data points to the possible role of UCB-mediated CYP induction in limiting UCB accumulation in selected brain regions, and suggests that these enzymes may protect selected brain areas from bilirubin neurotoxicity (Gazzin et al., 2012).
Understanding Cellular and Regional Selectivity of Bilirubin Toxicity
A decade ago, based on the observation that yellow staining of the basal nuclei is an extremely rare event, even in newborns that die from severe neonatal jaundice, Hansen challenged the concept that hyperbilirubinemia always leads to kernicterus by UCB accumulation in distinctive brain areas. In fact, only few cases of “real kernicterus” strictly describing bilirubin accumulation in specific brain regions have been reported in humans (Hansen, 2000). This conclusion was recently corroborated experimentally using the Gunn rat as an animal model for this disease. Bilirubin concentrations measured in four different brain regions in hyperbilirubinemic animals were found identical (Gazzin et al., 2012). Along the same line, the local cerebral blood flow that could influence the net blood-to-brain UCB flux is more homogeneous among regions in both human and rodent during early postnatal development than in adult, and is not correlated to the regional susceptibility to UCB toxicity (reviewed in Ghersi-Egea et al., 2009a). Similarly P-glycoprotein-dependent efflux processes which could marginally influence the net UCB flux in brain structures (see supra) is rather homogeneous among brain regions at least in the human adult (Eyal et al., 2010).
Despite the absence of a typical kernicterus, no doubt exists, however, as to the localization of damage to specific brain structures in both the human pathology and its experimental models (Shapiro, 2010). One hypothesis to explain this selective topography of bilirubin-induced injury would be based on a superimposable selective regionalization of the threshold concentrations between the anti- and pro-oxidant effects of the pigment.
Evidence for such a heterogenous sensitivity to bilirubin toxicity has already been reported between organs, regions within a given organ, or cells types in the same region. In hyperbilirubinemic Gunn rats, the bilirubin content in organs involved in heme metabolism such as the liver and spleen is several folds higher than that determined in brain (Zelenka et al., 2008; Gazzin et al., 2012), yet UCB-induced tissue damage has never been reported in the formers. In the brain, UCB-induced toxicity leads mainly to neuronal loss (especially Purkinje and granular cells from cerebellum; Keino et al., 1985; Conlee and Shapiro, 1997) and myelination defects (Brito et al., 2012). In line with this, astrocytes displayed a greater resistance than neurons to UCB toxicity (Brito et al., 2008), and the myelin-forming oligodendrocytes and their precursors have also emerged as possible sensitive targets in bilirubin encephalopathy (Kapitulnik, 2004). The latter hypothesis is corroborated by in vitro studies showing that oligodendrocyte viability decreases after exposure to UCB (Genc et al., 2003). It is in agreement with both the myelin alterations described in hyperbilirubinemic Gunn rats (O’Callaghan and Miller, 1985) and the loss of myelin fibers described in a preterm infant with kernicterus (Brito et al., 2012). With respect to regional differences, when primary cultures of neurons isolated from the cerebral cortex, the hippocampus or the cerebellum were exposed to UCB, hippocampal cells presented more oxidative damage associated with lower reduced glutathione levels, impairment of neurite outgrowth, and maximal cell death compared with cerebellar and cortical neurons (Vaz et al., 2011). This points to differences in the intrinsic sensitivity to UCB between neuronal sub-populations.
As bilirubin detoxification mechanisms are inducible/repressible processes, a differential modulation of transporters and enzymes following exposure to UCB must also be considered when searching for factors involved in the cell- and tissue-specific toxicity. The initial data described above such as those obtained for ABC transporters in the BBIs and neural cells, or for CYPs and GSTs in the neuropil, support a link between the expression level of these genes and bilirubin-induced toxicity, which needs to be further investigated.
Finally, the vulnerability of the brain to UCB intoxication is dependent on the timing of UCB exposure. The maximal sensitivity is observed during early postnatal development (Rice and Barone, 2000), at a time when the CNS undergoes extensive remodeling and maturation with temporal differences between the various cerebral structures. Thus the developmental age at the time of insult may determine the neural cell sensitivity to UCB intoxication, and possibly the efficiency of neuroprotective pathways in the different brain regions, and thereby influence the regional pattern of damage.
Future Challenges
An in-depth characterization of the detoxification and transport processes at BBIs and in the different neural cells of the developing brain are probably the most important basic knowledge we need to acquire in order to better appreciate the functional basis of selective brain structure damage following exposure to elevated UCB levels. The phenotype, the magnitude of symptoms, and the outcome of the disease vary with the developmental age at the time of bilirubin exposition. This heterogeneity thus calls for studies defining the developmental profile of the various effector molecules. These include all transporters and enzymes involved in UCB detoxification or in the regulation of CSF protein level and those exerting neuroprotective functions (antioxidants). Furthermore, these key actors should be investigated in the context of the pathology to evaluate the possible UCB-induced alteration in their expression or activity. Due to the complexity of the brain architecture and the developmental pattern specific to each cerebral structure, it is reasonable to speculate that several “sensible targets” are yet to be identified, and that those targets are developmentally regulated.
From a therapeutic point of view, conventional treatments include phototherapy and blood exchanges for hyperbilirubinemic infants, and liver transplantation or gene therapy for patients with the Crigler–Najjar syndrome. While waiting for new therapeutic targets to be identified, neuroprotective therapies that complement the conventional treatments in their initiation phase would be highly beneficial to the final outcome of the disease. A current approach is to target the perturbations of the neural cell redox status. Minocycline, a drug with properties similar to Vitamin E (Kraus et al., 2005), and already in use in several other central nervous system disorders, inhibits the cerebellar hypoplasia in Gunn rats (Lin et al., 2005). Unfortunately, its use in bilirubin-induced neurological disorders is impeded by side effects in pediatric patients (Yong et al., 2004). This highlights the difficulty of developing therapeutic strategies for this category of patients and stresses out the real need to fully understand the differences between adult and children in the factors setting the cerebral bioavailability of drugs, including drug transporters at BBIs. Among milder therapeutic approaches, essential fatty acid supply may be considered. Polyunsaturated fatty acids are major components of brain membranes and especially of myelin, and fatty acid depletion impairs nervous system functions leading to learning, motor, vision, and auditory abnormalities (Yehuda et al., 2005). Omega-3 and -6 lipid supplementation by oral administration has proved beneficial in several neurodegenerative pathologies that involve oxidative stress (review in Kim et al., 2010). Since myelin is a target for UCB, this therapeutic approach could be relevant to the prevention of UCB-mediated toxicity in children. In addition oral administration is possible, and side effects are not expected. To the best of our knowledge, this approach has never been tested.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the European Union (HEALTH-F2-2009-241778) to Nathalie Strazielle and Jean-François Ghersi-Egea, and by the Regione Friuli Venezia Giulia (Fondo Regionale FVG LR26/2005) to Silvia Gazzin and Claudio Tiribelli.
Footnotes
- ^Capitalized characters are used when referring to human transporters and enzymes or to transporters and enzymes listed in a general context. Minor characters are used when specifically referring to rat or mouse transporters.
- ^See Hagenbuch and Meier (2004) for review on new and old protein and gene OATP nomenclatures.
References
Abu-Bakar, A., Moore, M. R., and Lang, M. A. (2005). Evidence for induced microsomal bilirubin degradation by cytochrome P450 2A5. Biochem. Pharmacol. 70, 1527–1535.
Ahlfors, C. E., Wennberg, R. P., Ostrow, J. D., and Tiribelli, C. (2009). Unbound (free) bilirubin: improving the paradigm for evaluating neonatal jaundice. Clin. Chem. 55, 1288–1299.
Akin, E., Clower, B., Tibbs, R., Tang, J., and Zhang, J. (2002). Bilirubin produces apoptosis in cultured bovine brain endothelial cells. Brain Res. 931, 168–175.
Baranano, D. E., Rao, M., Ferris, C. D., and Snyder, S. H. (2002). Biliverdin reductase: a major physiologic cytoprotectant. Proc. Natl. Acad. Sci. U.S.A. 99, 16093–16098.
Beiswanger, C. M., Diegmann, M. H., Novak, R. F., Philbert, M. A., Graessle, T. L., Reuhl, K. R., and Lowndes, H. E. (1995). Developmental changes in the cellular distribution of glutathione and glutathione S-transferases in the murine nervous system. Neurotoxicology 16, 425–440.
Brito, M. A., Rosa, A. I., Falcao, A. S., Fernandes, A., Silva, R. F., Butterfield, D. A., and Brites, D. (2008). Unconjugated bilirubin differentially affects the redox status of neuronal and astroglial cells. Neurobiol. Dis. 29, 30–40.
Brito, M. A., Zurolo, E., Pereira, P., Barroso, C., Aronica, E., and Brites, D. (2012). Cerebellar axon/myelin loss, angiogenic sprouting, and neuronal increase of vascular endothelial growth factor in a preterm infant with kernicterus. J. Child Neurol. 27, 615–624.
Brittebo, E. B. (1994). Metabolism-dependent binding of the heterocyclic amine Trp-P-1 in endothelial cells of choroid plexus and in large cerebral veins of cytochrome P450-induced mice. Brain Res. 659, 91–98.
Brodersen, R. (1981). “Binding of bilirubin to albumin and tissues,” in Physiological and Biochemical Basis for Perinatal Medicine, eds M. Monset-Couchard and A. E. B. S. K. Minkowski Basel, Kargel, 144–152.
Calabrese, V., Butterfield, D. A., Scapagnini, G., Stella, A. M., and Maines, M. D. (2006). Redox regulation of heat shock protein expression by signaling involving nitric oxide and carbon monoxide: relevance to brain aging, neurodegenerative disorders, and longevity. Antioxid. Redox Signal. 8, 444–477.
Calligaris, S., Cekic, D., Roca-Burgos, L., Gerin, F., Mazzone, G., Ostrow, J. D., and Tiribelli, C. (2006). Multidrug resistance associated protein 1 protects against bilirubin-induced cytotoxicity. FEBS Lett. 580, 1355–1359.
Carder, P. J., Hume, R., Fryer, A. A., Strange, R. C., Lauder, J., and Bell, J. E. (1990). Glutathione S-transferase in human brain. Neuropathol. Appl. Neurobiol. 16, 293–303.
Chen, J., Tu, Y., Connolly, E. C., and Ronnett, G. V. (2005). Heme oxygenase-2 protects against glutathione depletion-induced neuronal apoptosis mediated by bilirubin and cyclic GMP. Curr. Neurovasc. Res. 2, 121–131.
Chinta, S. J., Kommaddi, R. P., Turman, C. M., Strobel, H. W., and Ravindranath, V. (2005a). Constitutive expression and localization of cytochrome P-450 1A1 in rat and human brain: presence of a splice variant form in human brain. J. Neurochem. 93, 724–736.
Chinta, S. J., Pai, H. V., and Ravindranath, V. (2005b). Presence of splice variant forms of cytochrome P4502D1 in rat brain but not in liver. Brain Res. Mol. Brain Res. 135, 81–92.
Chuniaud, L., Dessante, M., Chantoux, F., Blondeau, J. P., Francon, J., and Trivin, F. (1996). Cytotoxicity of bilirubin for human fibroblasts and rat astrocytes in culture. Effect of the ratio of bilirubin to serum albumin. Clin. Chim. Acta 256, 103–114.
Clarke, D. D., and Sokoloff, L. (1999) “Circulation and energy metabolism of the brain,” in Basic Neurochemistry: Molecular, Cellular and Medical Aspects. eds G. J. Siegl, B. W. Agranoff, R. W. Albers, S. K. Fisher, and M. D. Uhler (Philadelphia: Lippincott-Raven), 637–669.
Conlee, J. W., and Shapiro, S. M. (1997). Development of cerebellar hypoplasia in jaundiced Gunn rats: a quantitative light microscopic analysis. Acta Neuropathol. 93, 450–460.
Cooper, A. J. L., Rosemberg, R. N., Prusiner, S. B., DiMauro, S., Barchi, R. L., and Kunk, L. M. (1997). “Glutathione in the brain: disorders of glutathione metabolism,” in The Molecular and Genetic Basis of Neurological Disease. eds R. N. Rosemberg, S. B. Prusiner, S. DiMauro, R. L. Barchi, and L. M. Kunk (Boston: Butterworth-Heinemann), 1195–1230.
Corich, L., Aranda, A., Carrassa, L., Bellarosa, C., Ostrow, J. D., and Tiribelli, C. (2009). The cytotoxic effect of unconjugated bilirubin in human neuroblastoma SH-SY5Y cells is modulated by the expression level of MRP1 but not MDR1. Biochem. J. 417, 305–312.
Cornford, E. M., Varesi, J. B., Hyman, S., Damian, R. T., and Raleigh, M. J. (1997). Mitochondrial content of choroid plexus epithelium. Exp. Brain Res. 116, 399–405.
Dallas, S., Zhu, X., Baruchel, S., Schlichter, L., and Bendayan, R. (2003). Functional expression of the multidrug resistance protein 1 in microglia. J. Pharmacol. Exp. Ther. 307, 282–290.
Daneman, R., Zhou, L., Kebede, A. A., and Barres, B. A. (2010). Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature 468, 562–566.
Daood, M. J., and Watchko, J. F. (2006). Calculated in vivo free bilirubin levels in the central nervous system of Gunn rat pups. Pediatr. Res. 60, 44–49.
Dauchy, S., Dutheil, F., Weaver, R. J., Chassoux, F., Daumas-Duport, C., Couraud, P. O., Scherrmann, J. M., De Waziers, I., and Decleves, X. (2008). ABC transporters, cytochromes P450 and their main transcription factors: expression at the human blood-brain barrier. J. Neurochem. 107, 1518–1528.
Davson, H., and Segal, M. B. (1996). Physiology of the CSF and the Blood-Brain Barriers. Boca Raton: CRC Press.
De Matteis, F., Lord, G. A., Kee, L. C., and Pons, N. (2006). Bilirubin degradation by uncoupled cytochrome P450. Comparison with a chemical oxidation system and characterization of the products by high-performance liquid chromatography/electrospray ionization mass spectrometry. Rapid Commun. Mass Spectrom. 20, 1209–1217.
Decleves, X., Regina, A., Laplanche, J. L., Roux, F., Boval, B., Launay, J. M., and Scherrmann, J. M. (2000). Functional expression of P-glycoprotein and multidrug resistance-associated protein (Mrp1) in primary cultures of rat astrocytes. J. Neurosci. Res. 60, 594–601.
Deguchi, K., Hayashi, T., Nagotani, S., Sehara, Y., Zhang, H., Tsuchiya, A., Ohta, Y., Tomiyama, K., Morimoto, N., Miyazaki, M., Huh, N. H., Nakao, A., Kamiya, T., and Abe, K. (2008). Reduction of cerebral infarction in rats by biliverdin associated with amelioration of oxidative stress. Brain Res. 1188, 1–8.
Dennery, P. A., McDonagh, A. F., Spitz, D. R., Rodgers, P. A., and Stevenson, D. K. (1995). Hyperbilirubinemia results in reduced oxidative injury in neonatal Gunn rats exposed to hyperoxia. Free Radic. Biol. Med. 19, 395–404.
Dey, A., Jones, J. E., and Nebert, D. W. (1999). Tissue- and cell type-specific expression of cytochrome P450 1A1 and cytochrome P450 1A2 mRNA in the mouse localized in situ hybridization. Biochem. Pharmacol. 58, 525–537.
Dohi, K., Satoh, K., Ohtaki, H., Shioda, S., Miyake, Y., Shindo, M., and Aruga, T. (2005). Elevated plasma levels of bilirubin in patients with neurotrauma reflect its pathophysiological role in free radical scavenging. In vivo 19, 855–860.
Doré, S., and Snyder, S. H. (1999). Neuroprotective action of bilirubin against oxidative stress in primary hippocampal cultures. Ann. N. Y. Acad. Sci. 890, 167–172.
Doré, S., Takahashi, M., Ferris, C. D., Zakhary, R., Hester, L. D., Guastella, D., and Snyder, S. H. (1999). Bilirubin, formed by activation of heme oxygenase-2, protects neurons against oxidative stress injury. Proc. Natl. Acad. Sci. U.S.A. 96, 2445–2450.
Dwyer, B. E., Nishimura, R. N., and Lu, S. Y. (1995). Differential expression of heme oxygenase-1 in cultured cortical neurons and astrocytes determined by the aid of a new heme oxygenase antibody. Response to oxidative stress. Brain Res. Mol. Brain Res. 30, 37–47.
Eckert, J. J., and Fleming, T. P. (2008). Tight junction biogenesis during early development. Biochim. Biophys. Acta 1778, 717–728.
Ek, C. J., Dziegielewska, K. M., Stolp, H., and Saunders, N. R. (2006). Functional effectiveness of the blood-brain barrier to small water-soluble molecules in developing and adult opossum (Monodelphis domestica). J. Comp. Neurol. 496, 13–26.
Ewing, J. F., and Maines, M. D. (1997). Histochemical localization of heme oxygenase-2 protein and mRNA expression in rat brain. Brain Res. Brain Res. Protoc. 1, 165–174.
Ewing, J. F., Weber, C. M., and Maines, M. D. (1993). Biliverdin reductase is heat resistant and coexpressed with constitutive and heat shock forms of heme oxygenase in brain. J. Neurochem. 61, 1015–1023.
Eyal, S., Ke, B., Muzi, M., Link, J. M., Mankoff, D. A., Collier, A. C., and Unadkat, J. D. (2010). Regional P-glycoprotein activity and inhibition at the human blood-brain barrier as imaged by positron emission tomography. Clin. Pharmacol. Ther. 87, 579–585.
Falcão, A. S., Bellarosa, C., Fernandes, A., Brito, M. A., Silva, R. F., Tiribelli, C., and Brites, D. (2007). Role of multidrug resistance-associated protein 1 expression in the in vitro susceptibility of rat nerve cell to unconjugated bilirubin. Neuroscience 144, 878–888.
Farin, F. M., and Omiecinski, C. J. (1993). Regiospecific expression of cytochrome P-450s and microsomal epoxide hydrolase in human brain tissue. J. Toxicol. Environ. Health 40, 317–335.
Faropoulos, K., Chroni, E., Assimakopoulos, S. F., Mavrakis, A., Stamatopoulou, V., Toumpeki, C., Drainas, D., Grintzalis, K., Papapostolou, I., Georgiou, C. D., and Konstantinou, D. (2010). Altered occludin expression in brain capillaries induced by obstructive jaundice in rats. Brain Res. 1325, 121–127.
Filbrandt, C. R., Wu, Z., Zlokovic, B., Opanashuk, L., and Gasiewicz, T. A. (2004). Presence and functional activity of the aryl hydrocarbon receptor in isolated murine cerebral vascular endothelial cells and astrocytes. Neurotoxicology 25, 605–616.
Friel, J. K., Friesen, R. W., Harding, S. V., and Roberts, L. J. (2004). Evidence of oxidative stress in full-term healthy infants. Pediatr. Res. 56, 878–882.
Gao, B., Hagenbuch, B., Kullak-Ublick, G. A., Benke, D., Aguzzi, A., and Meier, P. J. (2000). Organic anion-transporting polypeptides mediate transport of opioid peptides across blood-brain barrier. J. Pharmacol. Exp. Ther. 294, 73–79.
Gazzin, S., Berengeno, A. L., Strazielle, N., Fazzari, F., Raseni, A., Ostrow, J. D., Wennberg, R., Ghersi-Egea, J. F., and Tiribelli, C. (2011). Modulation of Mrp1 (ABCc1) and Pgp (ABCb1) by bilirubin at the blood-CSF and blood-brain barriers in the Gunn rat. PLoS ONE 6, e16165. doi:10.1371/journal.pone.0016165
Gazzin, S., Strazielle, N., Schmitt, C., Fevre-Montange, M., Ostrow, J. D., Tiribelli, C., and Ghersi-Egea, J. F. (2008). Differential expression of the multidrug resistance-related proteins ABCb1 and ABCc1 between blood-brain interfaces. J. Comp. Neurol. 510, 497–507.
Gazzin, S., Zelenka, J., Adralova, L., Konickova, R., Coda Zabetta, C., Giraudi, P., Berengeno, A. L., Raseni, A., Robert, M. C., Vitek, L., and Tiribelli, C. (2012). Postnatal changes of bilirubin accumulation in selective brain regions of hyperbilirubinemic Gunn rat. Pediatr. Res. doi: 10.1038/pr2012.23
Genc, S., Genc, K., Kumral, A., Baskin, H., and Ozkan, H. (2003). Bilirubin is cytotoxic to rat oligodendrocytes in vitro. Brain Res. 985, 135–141.
Gennuso, F., Fernetti, C., Tirolo, C., Testa, N., L’Episcopo, F., Caniglia, S., Morale, M. C., Ostrow, J. D., Pascolo, L., Tiribelli, C., and Marchetti, B. (2004). Bilirubin protects astrocytes from its own toxicity by inducing up-regulation and translocation of multidrug resistance-associated protein 1 (Mrp1). Proc. Natl. Acad. Sci. U.S.A. 101, 2470–2475.
Ghersi-Egea, J. F., Gazzin, S., and Strazielle, N. (2009a). Blood-brain interfaces and bilirubin-induced neurological diseases. Curr. Pharm. Des. 15, 2893–2907.
Ghersi-Egea, J. F., Monkkonen, K. S., Schmitt, C., Honnorat, J., Fevre-Montange, M., and Strazielle, N. (2009b). Blood-brain interfaces and cerebral drug bioavailability. Rev. Neurol. (Paris) 165, 1029–1038.
Ghersi-Egea, J. F., Leininger-Muller, B., Cecchelli, R., and Fenstermacher, J. D. (1995). Blood-brain interfaces: relevance to cerebral drug metabolism. Toxicol. Lett. 82–83, 645–653.
Ghersi-Egea, J. F., Leninger-Muller, B., Suleman, G., Siest, G., and Minn, A. (1994). Localization of drug-metabolizing enzyme activities to blood-brain interfaces and circumventricular organs. J. Neurochem. 62, 1089–1096.
Ghersi-Egea, J. F., Minn, A., and Siest, G. (1988). A new aspect of the protective functions of the blood-brain barrier: activities of four drug-metabolizing enzymes in isolated rat brain microvessels. Life Sci. 42, 2515–2523.
Ghersi-Egea, J. F., Perrin, R., Leininger-Muller, B., Grassiot, M. C., Jeandel, C., Floquet, J., Cuny, G., Siest, G., and Minn, A. (1993). Subcellular localization of cytochrome P450, and activities of several enzymes responsible for drug metabolism in the human brain. Biochem. Pharmacol. 45, 647–658.
Ghersi-Egea, J. F., Strazielle, N., Murat, A., Jouvet, A., Buenerd, A., and Belin, M. F. (2006). Brain protection at the blood-cerebrospinal fluid interface involves a glutathione-dependent metabolic barrier mechanism. J. Cereb. Blood Flow Metab. 26, 1165–1175.
Gradinaru, D., Minn, A. L., Artur, Y., Minn, A., and Heydel, J. M. (2009). Drug metabolizing enzyme expression in rat choroid plexus: effects of in vivo xenobiotics treatment. Arch. Toxicol. 83, 581–586.
Granberg, L., Ostergren, A., Brandt, I., and Brittebo, E. B. (2003). CYP1A1 and CYP1B1 in blood-brain interfaces: CYP1A1-dependent bioactivation of 7,12-dimethylbenz(a)anthracene in endothelial cells. Drug Metab. Dispos. 31, 259–265.
Hagenbuch, B., and Meier, P. J. (2004). Organic anion transporting polypeptides of the OATP/SLC21 family: phylogenetic classification as OATP/SLCO superfamily, new nomenclature and molecular/functional properties. Pflugers Arch. 447, 653–665.
Hanko, E., Tommarello, S., Watchko, J. F., and Hansen, T. W. (2003). Administration of drugs known to inhibit P-glycoprotein increases brain bilirubin and alters the regional distribution of bilirubin in rat brain. Pediatr. Res. 54, 441–445.
Hansen, T. W. (2000). Pioneers in the scientific study of neonatal jaundice and kernicterus. Pediatrics 106, E15.
Hennemann, G., Docter, R., Friesema, E. C., de Jong, M., Krenning, E. P., and Visser, T. J. (2001). Plasma membrane transport of thyroid hormones and its role in thyroid hormone metabolism and bioavailability. Endocr. Rev. 22, 451–476.
Hirrlinger, J., Konig, J., and Dringen, R. (2002). Expression of mRNAs of multidrug resistance proteins (Mrps) in cultured rat astrocytes, oligodendrocytes, microglial cells and neurones. J. Neurochem. 82, 716–719.
Hirrlinger, J., Moeller, H., Kirchhoff, F., and Dringen, R. (2005). Expression of multidrug resistance proteins (Mrps) in astrocytes of the mouse brain: a single cell RT-PCR study. Neurochem. Res. 30, 1237–1244.
Ho, Y. S., Magnenat, J. L., Bronson, R. T., Cao, J., Gargano, M., Sugawara, M., and Funk, C. D. (1997). Mice deficient in cellular glutathione peroxidase develop normally and show no increased sensitivity to hyperoxia. J Biol. Chem. 272, 16644–16651.
Hollensworth, S. B., Shen, C., Sim, J. E., Spitz, D. R., Wilson, G. L., and LeDoux, S. P. (2000). Glial cell type-specific responses to menadione-induced oxidative stress. Free Radic. Biol. Med. 28, 1161–1174.
Jacob, A., Hartz, A. M., Potin, S., Coumoul, X., Yousif, S., Scherrmann, J. M., Bauer, B., and Decleves, X. (2011). Aryl hydrocarbon receptor-dependent upregulation of Cyp1b1 by TCDD and diesel exhaust particles in rat brain microvessels. Fluids Barriers CNS 8, 23.
Jakobson, A. M. (1991). Bilirubin accumulation by the rabbit choroid plexus in vitro. Biol. Neonate 60, 221–229.
Jansen, P. L. (1999). Diagnosis and management of Crigler-Najjar syndrome. Eur. J. Pediatr. 158(Suppl. 2), S89–S94.
Johansson, P. A., Dziegielewska, K. M., Liddelow, S. A., and Saunders, N. R. (2008). The blood-CSF barrier explained: when development is not immaturity. Bioessays 30, 237–248.
Johnson, J. A., Hayward, J. J., Kornguth, S. E., and Siegel, F. L. (1993). Effects of hyperbilirubinaemia on glutathione S-transferase isoenzymes in cerebellar cortex of the Gunn rat. Biochem. J. 291, 453–461.
Kadakol, A., Ghosh, S. S., Sappal, B. S., Sharma, G., Chowdhury, J. R., and Chowdhury, N. R. (2000). Genetic lesions of bilirubin uridine-diphosphoglucuronate glucuronosyltransferase (UGT1A1) causing Crigler-Najjar and Gilbert syndromes: correlation of genotype to phenotype. Hum. Mutat. 16, 297–306.
Kapitulnik, J. (2004). Bilirubin: an endogenous product of heme degradation with both cytotoxic and cytoprotective properties. Mol. Pharmacol. 66, 773–779.
Kapitulnik, J., and Gonzalez, F. J. (1993). Marked endogenous activation of the CYP1A1 and CYP1A2 genes in the congenitally jaundiced Gunn rat. Mol. Pharmacol. 43, 722–725.
Kapitulnik, J., Hardwick, J. P., Ostrow, J. D., Webster, C. C., Park, S. S., and Gelboin, H. V. (1987). Increase in a specific cytochrome P-450 isoenzyme in the liver of congenitally jaundiced Gunn rats. Biochem. J. 242, 297–300.
Kapitulnik, J., and Maines, M. D. (2009). Pleiotropic functions of biliverdin reductase: cellular signaling and generation of cytoprotective and cytotoxic bilirubin. Trends Pharmacol. Sci. 30, 129–137.
Keino, H., Sato, H., Semba, R., Aono, S., Aoki, E., and Kashiwamata, S. (1985). Mode of prevention by phototherapy of cerebellar hypoplasia in a new Sprague-Dawley strain of jaundiced Gunn rats. Pediatr. Neurosci. 12, 145–150.
Keppler, D. (2011). Multidrug resistance proteins (MRPs, ABCCs): importance for pathophysiology and drug therapy. Handb. Exp. Pharmacol. 201, 299–323.
Kim, S. J., Zhang, Z., Saha, A., Sarkar, C., Zhao, Z., Xu, Y., and Mukherjee, A. B. (2010). Omega-3 and omega-6 fatty acids suppress ER- and oxidative stress in cultured neurons and neuronal progenitor cells from mice lacking PPT1. Neurosci. Lett. 479, 292–296.
Kraus, R. L., Pasieczny, R., Lariosa-Willingham, K., Turner, M. S., Jiang, A., and Trauger, J. W. (2005). Antioxidant properties of minocycline: neuroprotection in an oxidative stress assay and direct radical-scavenging activity. J. Neurochem. 94, 819–827.
Lee, G., Schlichter, L., Bendayan, M., and Bendayan, R. (2001). Functional expression of P-glycoprotein in rat brain microglia. J. Pharmacol. Exp. Ther. 299, 204–212.
Lee, W., Glaeser, H., Smith, L. H., Roberts, R. L., Moeckel, G. W., Gervasini, G., Leake, B. F., and Kim, R. B. (2005). Polymorphisms in human organic anion-transporting polypeptide 1A2 (OATP1A2): implications for altered drug disposition and central nervous system drug entry. J. Biol. Chem. 280, 9610–9617.
Leininger-Muller, B., Ghersi-Egea, J. F., Siest, G., and Minn, A. (1994). Induction and immunological characterization of the uridine diphosphate-glucuronosyltransferase conjugating 1-naphthol in the rat choroid plexus. Neurosci. Lett. 175, 37–40.
Leslie, E. M., Deeley, R. G., and Cole, S. P. (2005). Multidrug resistance proteins: role of P-glycoprotein, MRP1, MRP2, and BCRP (ABCG2) in tissue defense. Toxicol. Appl. Pharmacol. 204, 216–237.
Levine, R. L., Fredericks, W. R., and Rapoport, S. I. (1985). Clearance of bilirubin from rat brain after reversible osmotic opening of the blood-brain barrier. Pediatr. Res. 19, 1040–1043.
Lim, C. F., Docter, R., Visser, T. J., Krenning, E. P., Bernard, B., van Toor, H., de Jong, M., and Hennemann, G. (1993). Inhibition of thyroxine transport into cultured rat hepatocytes by serum of nonuremic critically ill patients: effects of bilirubin and nonesterified fatty acids. J. Clin. Endocrinol. Metab. 76, 1165–1172.
Lin, S., Wei, X., Bales, K. R., Paul, A. B., Ma, Z., Yan, G., Paul, S. M., and Du, Y. (2005). Minocycline blocks bilirubin neurotoxicity and prevents hyperbilirubinemia-induced cerebellar hypoplasia in the Gunn rat. Eur. J. Neurosci. 22, 21–27.
Liu, Y., Liu, J., Tetzlaff, W., Paty, D. W., and Cynader, M. S. (2006). Biliverdin reductase, a major physiologic cytoprotectant, suppresses experimental autoimmune encephalomyelitis. Free Radic. Biol. Med. 40, 960–967.
Liu, Y., Zhu, B., Wang, X., Luo, L., Li, P., Paty, D. W., and Cynader, M. S. (2003). Bilirubin as a potent antioxidant suppresses experimental autoimmune encephalomyelitis: implications for the role of oxidative stress in the development of multiple sclerosis. J. Neuroimmunol. 139, 27–35.
Maines, M. D. (2005). New insights into biliverdin reductase functions: linking heme metabolism to cell signaling. Physiology (Bethesda) 20, 382–389.
Maines, M. D., and Trakshel, G. M. (1993). Purification and characterization of human biliverdin reductase. Arch. Biochem. Biophys. 300, 320–326.
Mancuso, C. (2004). Heme oxygenase and its products in the nervous system. Antioxid. Redox Signal. 6, 878–887.
Mannervik, B., Board, P. G., Hayes, J. D., Listowsky, I., and Pearson, W. R. (2005). Nomenclature for mammalian soluble glutathione transferases. Meth. Enzymol. 401, 1–8.
McDonagh, A. F. (2010). Controversies in bilirubin biochemistry and their clinical relevance. Semin. Fetal Neonatal Med. 15, 141–147.
Mercier, C., Masseguin, C., Roux, F., Gabrion, J., and Scherrmann, J. M. (2004). Expression of P-glycoprotein (ABCB1) and Mrp1 (ABCC1) in adult rat brain: focus on astrocytes. Brain Res. 1021, 32–40.
Miksys, S. L., and Tyndale, R. F. (2002). Drug-metabolizing cytochrome P450s in the brain. J. Psychiatry Neurosci. 27, 406–415.
Miralem, T., Hu, Z., Torno, M. D., Lelli, K. M., and Maines, M. D. (2005). Small interference RNA-mediated gene silencing of human biliverdin reductase, but not that of heme oxygenase-1, attenuates arsenite-mediated induction of the oxygenase and increases apoptosis in 293A kidney cells. J. Biol. Chem. 280, 17084–17092.
Morse, D. C., Stein, A. P., Thomas, P. E., and Lowndes, H. E. (1998). Distribution and induction of cytochrome P450 1A1 and 1A2 in rat brain. Toxicol. Appl. Pharmacol. 152, 232–239.
Mreihil, K., McDonagh, A. F., Nakstad, B., and Hansen, T. W. (2010). Early isomerization of bilirubin in phototherapy of neonatal jaundice. Pediatr. Res. 67, 656–659.
Mustafa, M. G., and King, T. E. (1970). Binding of bilirubin with lipid. A possible mechanism of its toxic reactions in mitochondria. J. Biol. Chem. 245, 1084–1089.
Nag, N., Halder, S., Chaudhuri, R., Adhikary, S., and Mazumder, S. (2009). Role of bilirubin as antioxidant in neonatal jaundice and effect of ethanolic extract of sweet lime peel on experimentally induced jaundice in rat. Indian J. Biochem. Biophys. 46, 73–78.
O’Callaghan, J. P., and Miller, D. B. (1985). Cerebellar hypoplasia in the Gunn rat is associated with quantitative changes in neurotypic and gliotypic proteins. J. Pharmacol. Exp. Ther. 234, 522–533.
Ostrow, J. D., Mukerjee, P., and Tiribelli, C. (1994). Structure and binding of unconjugated bilirubin: relevance for physiological and pathophysiological function. J. Lipid Res. 35, 1715–1737.
Ostrow, J. D., Pascolo, L., and Tiribelli, C. (2002). Mechanisms of bilirubin neurotoxicity. Hepatology 35, 1277–1280.
Palmela, I., Cardoso, F. L., Bernas, M., Correia, L., Vaz, A. R., Silva, R. F., Fernandes, A., Kim, K. S., Brites, D., and Brito, M. A. (2011). Elevated levels of bilirubin and long-term exposure impair human brain microvascular endothelial cell integrity. Curr. Neurovasc. Res. 8, 153–169.
Pritchard, J. B., Sweet, D. H., Miller, D. S., and Walden, R. (1999). Mechanism of organic anion transport across the apical membrane of choroid plexus. J. Biol. Chem. 274, 33382–33387.
Redzic, Z. B., and Segal, M. B. (2004). The structure of the choroid plexus and the physiology of the choroid plexus epithelium. Adv. Drug Deliv. Rev. 56, 1695–1716.
Rice, D., and Barone, S. Jr. (2000). Critical periods of vulnerability for the developing nervous system: evidence from humans and animal models. Environ. Health Perspect. 108(Suppl. 3), 511–533.
Rigato, I., Ostrow, J. D., and Tiribelli, C. (2005). Bilirubin and the risk of common non-hepatic diseases. Trends. Mol. Med. 11, 277–283.
Rigato, I., Pascolo, L., Fernetti, C., Ostrow, J. D., and Tiribelli, C. (2004). The human multidrug-resistance-associated protein MRP1 mediates ATP-dependent transport of unconjugated bilirubin. Biochem. J. 383, 335–341.
Robinson, P. J., and Rapoport, S. I. (1986). Kinetics of protein binding determine rates of uptake of drugs by brain. Am. J. Physiol. 251, R1212–R1220.
Roger, C., Koziel, V., Vert, P., and Nehlig, A. (1993). Effects of bilirubin infusion on local cerebral glucose utilization in the immature rat. Brain Res. Dev. Brain Res. 76, 115–130.
Roger, C., Koziel, V., Vert, P., and Nehlig, A. (1995). Mapping of the consequences of bilirubin exposure in the immature rat: local cerebral metabolic rates for glucose during moderate and severe hyperbilirubinemia. Early Hum. Dev. 43, 133–144.
Ronaldson, P. T., Bendayan, M., Gingras, D., Piquette-Miller, M., and Bendayan, R. (2004). Cellular localization and functional expression of P-glycoprotein in rat astrocyte cultures. J. Neurochem. 89, 788–800.
Schinkel, A. H., and Jonker, J. W. (2003). Mammalian drug efflux transporters of the ATP binding cassette (ABC) family: an overview. Adv. Drug Deliv. Rev. 55, 3–29.
Schinkel, A. H., Wagenaar, E., Mol, C. A., and van Deemter, L. (1996). P-glycoprotein in the blood-brain barrier of mice influences the brain penetration and pharmacological activity of many drugs. J. Clin. Invest. 97, 2517–2524.
Schlosshauer, B. (1993). The blood-brain barrier: morphology, molecules, and neurothelin. Bioessays 15, 341–346.
Sedlak, T. W., Saleh, M., Higginson, D. S., Paul, B. D., Juluri, K. R., and Snyder, S. H. (2009). Bilirubin and glutathione have complementary antioxidant and cytoprotective roles. Proc. Natl. Acad. Sci. U.S.A. 106, 5171–5176.
Shapiro, S. M. (2010). Chronic bilirubin encephalopathy: diagnosis and outcome. Semin. Fetal Neonatal Med. 15, 157–163.
Shawahna, R., Uchida, Y., Decleves, X., Ohtsuki, S., Yousif, S., Dauchy, S., Jacob, A., Chassoux, F., Daumas-Duport, C., Couraud, P. O., Terasaki, T., and Scherrmann, J. M. (2011). Transcriptomic and quantitative proteomic analysis of transporters and drug metabolizing enzymes in freshly isolated human brain microvessels. Mol. Pharm. 8, 1332–1341.
Shelby, M. K., Cherrington, N. J., Vansell, N. R., and Klaassen, C. D. (2003). Tissue mRNA expression of the rat UDP-glucuronosyltransferase gene family. Drug Metab. Dispos. 31, 326–333.
Shen, L., Weber, C. R., and Turner, J. R. (2008). The tight junction protein complex undergoes rapid and continuous molecular remodeling at steady state. J. Cell Biol. 181, 683–695.
Spiegl-Kreinecker, S., Buchroithner, J., Elbling, L., Steiner, E., Wurm, G., Bodenteich, A., Fischer, J., Micksche, M., and Berger, W. (2002). Expression and functional activity of the ABC-transporter proteins P-glycoprotein and multidrug-resistance protein 1 in human brain tumor cells and astrocytes. J. Neurooncol. 57, 27–36.
Strazielle, N., and Ghersi-Egea, J. F. (1999). Demonstration of a coupled metabolism-efflux process at the choroid plexus as a mechanism of brain protection toward xenobiotics. J. Neurosci. 19, 6275–6289.
Strazielle, N., and Ghersi-Egea, J. F. (2000a). Choroid plexus in the central nervous system: biology and physiopathology. J. Neuropathol. Exp. Neurol. 59, 561–574.
Strazielle, N., and Ghersi-Egea, J. F. (2000b). “Implication of blood-brain interfaces in cerebral drug metabolism and drug metabolite disposition,” in Molecular Drug Metabolism and Toxicology, eds G. Williams and O. I. Aruoma (Saint Lucia: OICA International), 181–200.
Strazielle, N., and Ghersi-Egea, J. F. (2005). Factors affecting delivery of antiviral drugs to the brain. Rev. Med. Virol. 15, 105–133.
Strazielle, N., Khuth, S. T., and Ghersi-Egea, J. F. (2004). Detoxification systems, passive and specific transport for drugs at the blood-CSF barrier in normal and pathological situations. Adv. Drug Deliv. Rev. 56, 1717–1740.
Strazielle, N., and Preston, J. E. (2003). Transport across the choroid plexuses in vivo and in vitro. Methods Mol. Med. 89, 291–304.
Tayarani, I., Cloez, I., Clement, M., and Bourre, J. M. (1989). Antioxidant enzymes and related trace elements in aging brain capillaries and choroid plexus. J. Neurochem. 53, 817–824.
Tudor, C., Lerner-Marmarosh, N., Engelborghs, Y., Gibbs, P. E., and Maines, M. D. (2008). Biliverdin reductase is a transporter of haem into the nucleus and is essential for regulation of HO-1 gene expression by haematin. Biochem. J. 413, 405–416.
Van Bergen, P., Rauhala, P., Spooner, C. M., and Chiueh, C. C. (1999). Hemoglobin and iron-evoked oxidative stress in the brain: protection by bile pigments, manganese and S-nitrosoglutathione. Free Radic. Res. 31, 631–640.
Vaz, A. R., Silva, S. L., Barateiro, A., Falcao, A. S., Fernandes, A., Brito, M. A., and Brites, D. (2011). Selective vulnerability of rat brain regions to unconjugated bilirubin. Mol. Cell Neurosci. 48, 82–93.
Vitek, L., and Ostrow, J. D. (2009). Bilirubin chemistry and metabolism; harmful and protective aspects. Curr. Pharm. Des. 15, 2869–2883.
Wang, X., Hawkins, B. T., and Miller, D. S. (2011). Aryl hydrocarbon receptor-mediated up-regulation of ATP-driven xenobiotic efflux transporters at the blood-brain barrier. FASEB J. 25, 644–652.
Watchko, J. F. (2006). Kernicterus and the molecular mechanisms of bilirubin-induced CNS injury in newborns. Neuromolecular Med. 8, 513–529.
Watchko, J. F., Daood, M. J., and Hansen, T. W. (1998). Brain bilirubin content is increased in P-glycoprotein-deficient transgenic null mutant mice. Pediatr. Res. 44, 763–766.
Weisiger, R. A., Ostrow, J. D., Koehler, R. K., Webster, C. C., Mukerjee, P., Pascolo, L., and Tiribelli, C. (2001). Affinity of human serum albumin for bilirubin varies with albumin concentration and buffer composition: results of a novel ultrafiltration method. J. Biol. Chem. 276, 29953–29960.
Wennberg, R. P. (2000). The blood-brain barrier and bilirubin encephalopathy. Cell. Mol. Neurobiol. 20, 97–109.
Wennberg, R. P., Ahlfors, C. E., and Aravkin, A. Y. (2009). Intervention guidelines for neonatal hyperbilirubinemia: an evidence based quagmire. Curr. Pharm. Des. 15, 2939–2945.
Wijnholds, J., deLange, E. C., Scheffer, G. L., van den Berg, D. J., Mol, C. A., van der Valk, M., Schinkel, A. H., Scheper, R. J., Breimer, D. D., and Borst, P. (2000). Multidrug resistance protein 1 protects the choroid plexus epithelium and contributes to the blood-cerebrospinal fluid barrier. J. Clin. Invest. 105, 279–285.
Yehuda, S., Rabinovitz, S., and Mostofsky, D. I. (2005). Essential fatty acids and the brain: from infancy to aging. Neurobiol. Aging 26(Suppl. 1), 98–102.
Yong, V. W., Wells, J., Giuliani, F., Casha, S., Power, C., and Metz, L. M. (2004). The promise of minocycline in neurology. Lancet Neurol. 3, 744–751.
Zaccaro, C., Sweitzer, S., Pipino, S., Gorman, N., Sinclair, P. R., Sinclair, J. F., Nebert, D. W., and De, M. F. (2001). Role of cytochrome P450 1A2 in bilirubin degradation Studies in Cyp1a2 (-/-) mutant mice. Biochem. Pharmacol. 61, 843–849.
Zelenka, J., Lenicek, M., Muchova, L., Jirsa, M., Kudla, M., Balaz, P., Zadinova, M., Ostrow, J. D., Wong, R. J., and Vitek, L. (2008). Highly sensitive method for quantitative determination of bilirubin in biological fluids and tissues. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 867, 37–42.
Zheng, W. (2001). Toxicology of choroid plexus: special reference to metal-induced neurotoxicities. Microsc. Res. Tech. 52, 89–103.
Keywords: ABC transporters, astrocyte, biliverdin, blood–brain barrier, choroid plexus, glutathione-S-transferase, OATP, UDP-glucuronosyltransferase
Citation: Gazzin S, Strazielle N, Tiribelli C and Ghersi-Egea J (2012) Transport and metabolism at blood–brain interfaces and in neural cells: relevance to bilirubin-induced encephalopathy. Front. Pharmacol. 3:89. doi: 10.3389/fphar.2012.00089
Received: 03 February 2012; Accepted: 25 April 2012;
Published online: 18 May 2012.
Edited by:
Jaime Kapitulnik, The Hebrew University of Jerusalem, IsraelReviewed by:
Dora Brites, University of Lisbon, PortugalXavier Decleves, University Paris Descartes, France
Copyright: © 2012 Gazzin, Strazielle, Tiribelli and Ghersi-Egea. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Jean-François Ghersi-Egea, Faculté de Médecine Laennec, Neurooncology and Neuroinflammation Team, INSERM U1028, Lyon Neuroscience Research Center, 7 rue G. Paradin, 69372 Lyon Cedex 08, France. e-mail: jean-francois.ghersi-egea@inserm.fr