 Raghda Ramadan1,2
Raghda Ramadan1,2 Els Vromans3
Els Vromans3 Dornatien Chuo Anang4Ines Goetschalckx5Delphine Hoorelbeke2Elke Decrock2Sarah Baatout1,6
Dornatien Chuo Anang4Ines Goetschalckx5Delphine Hoorelbeke2Elke Decrock2Sarah Baatout1,6 Luc Leybaert2†An Aerts1*†
Luc Leybaert2†An Aerts1*†
- 1Radiobiology Unit, Belgian Nuclear Research Centre (SCK•CEN), Mol, Belgium
- 2Department of Fundamental and Basic Medical Sciences, Physiology Group, Ghent University, Ghent, Belgium
- 3Centre for Environmental Health Sciences, Hasselt University, Hasselt, Belgium
- 4Biomedical Research Institute and Transnational University of Limburg, Hasselt University, Hasselt, Belgium
- 5Protein Chemistry, Proteomics and Epigenetic Signaling Group, Faculty of Pharmaceutical, Biomedical and Veterinary Sciences, University of Antwerp, Antwerp, Belgium
- 6Department of Molecular Biotechnology, Ghent University, Ghent, Belgium
Background: Emerging evidence indicates an excess risk of late occurring cardiovascular diseases, especially atherosclerosis, after thoracic cancer radiotherapy. Ionizing radiation (IR) induces cellular effects which may induce endothelial cell dysfunction, an early marker for atherosclerosis. In addition, intercellular communication through channels composed of transmembrane connexin proteins (Cxs), i.e. Gap junctions (direct cell-cell coupling) and hemichannels (paracrine release/uptake pathway) can modulate radiation-induced responses and therefore the atherosclerotic process. However, the role of endothelial hemichannel in IR-induced atherosclerosis has never been described before.
Materials and Methods: Telomerase-immortalized human Coronary Artery/Microvascular Endothelial cells (TICAE/TIME) were exposed to X-rays (0.1 and 5 Gy). Production of reactive oxygen species (ROS), DNA damage, cell death, inflammatory responses, and senescence were assessed with or without applying a Cx43 hemichannel blocker (TAT-Gap19).
Results: We report here that IR induces an increase in oxidative stress, cell death, inflammatory responses (IL-8, IL-1β, VCAM-1, MCP-1, and Endothelin-1) and premature cellular senescence in TICAE and TIME cells. These effects are significantly reduced in the presence of the Cx43 hemichannel-targeting peptide TAT-Gap19.
Conclusion: Our findings suggest that endothelial Cx43 hemichannels contribute to various IR-induced processes, such as ROS, cell death, inflammation, and senescence, resulting in an increase in endothelial cell damage, which could be protected by blocking these hemichannels. Thus, targeting Cx43 hemichannels may potentially exert radioprotective effects.
Introduction
Adjuvant radiotherapy is a standard therapy for breast cancer treatment after conservative surgery and mastectomy (Yang and Ho, 2013). However, exposure of healthy tissue, in particular, the heart, to ionizing radiation (IR) often increases the risk for the development of cardiovascular diseases (CVD), especially atherosclerosis (Darby et al., 2003; Marks et al., 2005; Baker et al., 2011; Cutter et al., 2011; Yusuf et al., 2011; Darby et al., 2013; Aleman et al., 2014; Van Nimwegen et al., 2015; Baselet et al., 2016). Although modern radiotherapy techniques reduce the volume of the heart and major coronary vessels exposed to high doses of IR, some exposure is often unavoidable, especially in the case of left-sided breast cancer, in which case the accumulative dose received by the heart area is in the order of ∼6.6 Gy (compared to 2.9 Gy for right-sided breast cancer) (Darby et al., 2013; Eftekhari et al., 2015; Al-Kindi and Oliveira, 2016; Cho et al., 2018). A population-based case-control study in women who underwent radiotherapy for breast cancer indicated a significant increase of 7.4% in the rates of major coronary events (i.e. myocardial infarction, coronary revascularization, or death from ischemic heart disease) per increase of 1 Gy in the cardiac exposure dose, without apparent threshold (Darby et al., 2013). In addition to breast cancer, exposure of the cardiovascular system to IR may occur during thoracic cancer radiotherapy such as head-and-neck cancer, Hodgkin’s lymphoma, and esophageal cancer (Borghini et al., 2013). Indeed, growing evidence indicates a link between IR exposure at high and medium doses (>0.5 Gy) and atherosclerosis development (Stewart et al., 2006; Hoving et al., 2008; Shimizu et al., 2010; Icrp et al., 2012; Darby et al., 2013; Kreuzer et al., 2015; Little, 2016; Cheng et al., 2017). However, the underlying molecular mechanisms remain not entirely understood. In addition, there is growing epidemiological evidence nowadays that indicates an excess risk for CVD at much lower IR doses than previously thought (<0.5 Gy) (Yamada et al., 2005; Kreuzer et al., 2015; Baselet et al., 2016; Little, 2016). However, these data are suggestive rather than persuasive due to a lack of statistical power and the fairly limited knowledge on the underlying molecular mechanisms. Hence, effects of radiation exposure on the cardiovascular system, especially at low dose exposure, are still not fully characterized, possibly resulting in improper radiation protection.
Exposure of endothelial cells to IR induces DNA damage and ROS which in turn induces oxidative stress, cellular Ca2+ overload, inflammation, senescence and apoptosis which may cause endothelial cell dysfunction, an early marker for atherosclerosis (Baker et al., 2011; Yusuf et al., 2011; Borghini et al., 2013; Madan et al., 2015; Baselet et al., 2016; Tapio, 2016). Intercellular communication mediated by gap junctions and hemichannels, may modulate endothelial response to IR, and therefore, the atherosclerotic process (Azzam et al., 2001; Little, 2006; Ohshima et al., 2012). Gap junctions permit passive diffusion of small hydrophilic molecules and ions of below ∼1.5 kDa molecular weight between adjacent cells. Gap junctions are usually open under physiological conditions, and they are important both for slow physiological processes (e.g. cell growth and proliferation) and fast activities such as electrical impulse conduction in the heart or communication between vascular endothelial cells to transmit upstream vasodilatory messages (De Wit and Griffith, 2010). In contrast to gap junctions, plasma membrane hemichannels are typically closed under physiological conditions (Orellana et al., 2011), and can open in response to several stress signals (e.g. mechanical stimulation, stress-associated stimuli such as ischemia, oxidative stress, a modest increase in intracellular Ca2+ or pro-inflammatory conditions) (Ramachandran et al., 2007; De Vuyst et al., 2009; Orellana et al., 2009; Johansen et al., 2011; Meunier et al., 2017). When open, hemichannels facilitate bidirectional passage over the plasma membrane of ions and signaling or metabolic molecules of below ∼1.5 kDa molecular weight (e.g. Ca2+, prostaglandin E2, glutathione, glutamate, ATP, NAD+, and others) (Wang et al., 2013a). Uncontrolled opening of hemichannels can result in loss of cell-essential metabolites, excessive entry of Na+ and Ca2+ and ATP leakage which in turn can lead to activation of apoptotic caspases, nitric oxide production, and inflammation (Decrock et al., 2009a, b; Saez et al., 2010; Ohshima et al., 2012; Decrock et al., 2017; Leybaert et al., 2017). These events may potentially trigger significant changes in cell homeostasis and cause cell dysfunction.
Gap junctions and hemichannels are composed of transmembrane connexin (Cx) proteins. Endothelial cells, the interior lining of blood vessels and cardiac valves, express three main Cx isotypes, namely Cx37, Cx40, and Cx43. Increasing evidence indicates that Cxs play a role in atherosclerosis progression (27, 29–31). Cx37 and Cx40 are suggested to have atheroprotective effects as it was reported that healthy endothelial cells have a widely distributed Cx37 and Cx40 expression pattern, while both Cxs are lost in the endothelium covering the advanced atherosclerotic plaques (Kwak et al., 2002; Morel, 2014). In contrast, Cx43 has been associated with atherogenesis as it is highly expressed in atherosclerotic plaques compared to control (Kwak et al., 2002; Morel, 2014). Cx43 also has atherogenic properties as decreasing Cx43 expression reduces the formation of atherosclerotic lesions in vivo (Kwak et al., 2003; Wong et al., 2003; Wang et al., 2013b) while in vivo Cx43 upregulation increased the expression of cell adhesion proteins such as VCAM-1, thereby enhancing monocyte-endothelial adhesion, a key event in initiating atherosclerosis (Yuan et al., 2015). Furthermore, Cx43 has been implicated in endothelial cellular stiffness that is associated with cardiovascular disease and atherosclerosis (Okamoto et al., 2017).
Besides the role of Cx in the development of atherosclerosis, it was previously reported that Cx expression is sensitive to ionizing radiation. Cx43 expression was increased in human skin fibroblast after exposure to 10 mGy of α-particles (Azzam et al., 2003). In addition, Cx43 expression is elevated in response to low dose gamma-ray exposure (137Cs source) in human neonatal foreskin fibroblast (Glover et al., 2003), in response to 5 Gy X-rays in mouse brain endothelial cell line bend3 (Banaz-Yasar et al., 2005) and in cardiac myocytes in an in vivo animal model upon exposure to high-LET radiation (Amino et al., 2010). Finally, it was reported that 0.5 Gy of gamma-rays exposure induced Cx43 hemichannels opening in B16 melanoma cells (Ohshima et al., 2012).
Although Cxs and their channels have been reported to be involved in the pathogenesis of atherosclerosis and to be sensitive to IR exposure, their role in radiation-induced endothelial cell response were never investigated (Azzam et al., 2003; Pfenniger et al., 2013). We previously demonstrated that single and fractionated IR induces acute and persistent upregulation of Cx43 gene and protein expression in the coronary artery and microvascular endothelial cells (Ramadan et al., 2019). In addition, we demonstrated that IR induces acute and long-lived Cx43 hemichannel opening in a dose-dependent manner (Ramadan et al., 2019). Here, we aimed at investigating the involvement of Cx43 hemichannels in endothelial cell responses induced by IR exposure, by making use of the Cx43-targeting peptide TAT-Gap19 (Abudara et al., 2014). TAT-Gap19 was previously reported to specifically block Cx43 hemichannels in different in vitro experimental models, as observed by ATP release and dye uptake assays (Abudara et al., 2014; Willebrords et al., 2017; Maatouk et al., 2018; Saez et al., 2018; Walrave et al., 2018). These findings were supported by electrophysiological measurements of Cx43 hemichannel unitary currents demonstrating that Gap19 inhibits Cx43 hemichannels in HeLa cells overexpressing Cx43 (Wang et al., 2013b; Gadicherla et al., 2017), in acutely isolated pig ventricular cardiomyocytes (Wang et al., 2013b) and in primary astrocytes (Freitas-Andrade et al., 2019). In depth mechanistic investigations based on surface plasmon resonance studies revealed a direct binding of Gap19 to the C-terminal tail of Cx43, thereby preventing the CL loop/CT tail interaction which is essential for Cx43 hemichannel activity, whereas closure of gap junctions is prevented (Ponsaerts et al., 2010; Iyyathurai et al., 2018). Linking Gap19 to the HIV-derived TAT internalization sequence promotes its membrane permeability and reduces the concentration needed for half-maximal Cx43 hemichannel inhibition by 5-folds. We found that TAT-Gap19 reduced intracellular ROS generation, cell death, inflammation, and premature cell senescence induced by IR in the immortalized human coronary artery and microvascular endothelial cells. Collectively, these results indicate a functional linkage between hemichannel opening and post-irradiation ROS, inflammation, and cell death/senescence responses.
Materials and Methods
Cell Culture
Two human endothelial cell lines: Telomerase Immortalized human Coronary Artery Endothelial cells (TICAE) from the European Collection of Authenticated Cell Cultures (ECACC), and Telomerase Immortalized human Microvascular Endothelial cells (TIME) from the American Type Cell Culture (ATTC), were grown in MesoEndo Cell Growth Medium (Sigma-Aldrich Co., LCC, Diegem, Belgium) at 37°C in a humidified incubator supplemented with 5% CO2. Cells were split every 3/4 days, with 0.05% trypsin supplemented with 0.02% ethylenediaminetetraacetic acid (EDTA). The passage number 28 until passage 36 was used in all the experiments. Moxi Z Mini Automated Cell Counter (ORFLO Technologies, Ketchum, ID, United States) was used to count the cells. Cells were not passaged after irradiation for all experiments.
Irradiation
X-irradiation was performed at the Laboratory for Nuclear Calibrations (LNK) of the Belgian Nuclear Research Centre (SCK•CEN), in accordance to ISO 4037 and under ISO 17025 accreditation of LNK. TICAE and TIME cells were irradiated when reached 100% confluence with 0.1 and 5 Gy of single X-rays doses, at a dose rate of 0.5 Gy/minute with a vertical, point source X-ray beam using a Xstrahl 320 kV tube (tube voltage: 250 kV; filtration: 3.8 mmAl+ 1.4 mm Cu+ Dose Area Product (DAP) monitor ionizing chamber; tube current 12 ma) (Camberley, United Kingdom).
Intracellular ROS Detection
In order to assess intracellular ROS production, IncuCyte live cell imaging was used to have fast imaging in a large number of replicates over multiple time points (Jeong et al., 2014). TICAE and TIME cells were seeded in 96-well plate in 16 replicates at a density of 10 000 cells/well. Three days later, cells reached 100% confluence. At 30 min before irradiation, a fresh MesoEndo Cell Growth Medium without phenol red (Sigma-Aldrich Co., LCC, Diegem, Belgium) supplemented with 10 μmol/L of the CM-H2DCFDA dye (Sigma-Aldrich Co., LCC, Diegem, Belgium) was added to the cells, with or without 100 μM TAT-Gap19 (YGRKKRRQRRRKQIEIKKFK) (Genosphere Biotechnologies, Paris, France). Fluorescence signals were measured at 45 min, 2 h and 3 h after irradiation [CM-H2DCFDA dye became saturated that might be due to prolonged laser illumination-induced cellular ROS production, or the dye leaked outside the cells after this time (Koopman et al., 2006)] using the Incucyte ZOOM® system (Essen Bioscience, Ann Arbor, MI, United States), 10x objective and FITC filter. Phase contrast imaging was performed to count the cells. Positive control of 10 μmol/L tert-butyl hydroxyperoxide (tBHP) was added to the cells directly before imaging. Images were analyzed, making use of the software package provided by the manufacturer; we used a training set of images from different doses to define the image processing procedure. The so-called Top-Hat background subtraction method was applied by measuring the radius of the fluorescence object (30 μm), and adjust a threshold of 1.5 (above the local background fluorescent intensity level) (Kapellos et al., 2016; Gupta et al., 2018), as described in Incucyte Background Fluorescence Technical Note. These process definitions were applied for all conditions in TICAE and TIME cells.
Cell Death Assessment
Annexin V and Caspase 3/7
TICAE and TIME cells were seeded in 96-well plates at a density of 10 000 cells/well in eight biological replicates. After 3 days, cells were at 100% confluence. At 30 min before X-ray exposure, cells were refreshed with 150 μl medium supplemented with Caspase 3/7 reagent (1:1000 dilution) and Annexin V reagent (1:200 dilution) (Essen Bioscience, United Kingdom) with or without 100 μM TAT-Gap19 (Genosphere Biotechnologies, Paris, France). Fluorescence signals were measured from 4 h until 100 h after irradiation by imaging every 2 h using the Incucyte ZOOM® system (Essen Bioscience, Ann Arbor, MI, United States). Different channels (TRITC, FITC and phase contrast) and a 10x objective were used. Spectral unmixing set as 8% of red removed from green was applied. The Incucyte software’s processing definition was set to recognize red (Annexin V) and green (Caspase 3/7) stained cells, making use of the Top-Hat background subtraction method. The fluorescence signal was normalized to cell count.
Dextran Fluorescein Dye Uptake Assay
Dextran fluorescein, staining late apoptotic and necrotic cells, was used to validate apoptosis in TIME cells after irradiation. Cells were seeded in a 24-well plate at a density of 1.5 × 105 cells/well in 6 biological replicates. Three days later, cells reached 100% confluence. Before X-ray exposure with 30 min, cells were refreshed with medium with or without 100 μM TAT-Gap19 (Genosphere Biotechnologies, Paris, France). At 6 and 72 h after irradiation, cells were washed twice with HBSS complemented with 25 mm HEPES (Sigma-Aldrich Co., LCC, Diegem, Belgium) (HBSS-HEPES). Afterward, the cells were incubated with 200 μM dextran fluorescein (10 kDa) (Life Technologies, Merelbeke, Belgium), dissolved in HBSS-HEPES. After 6 min, the staining was removed by washing six times with HBSS-HEPES. Finally, the fluorescence was measured using the IncuCyte ZOOM® system (Essen Bioscience, Ann Arbor, MI, United States) by using FITC channel and phase contrast to count the cells using a 10x objective. The fluorescence signal was normalized to cell count.
Cytokine Detection
TICAE and TIME cells were seeded in a 6-well plate at a density of 2.5 × 105 cells/well in 5 to 6 biological replicates. Two to 3 days later, cells reached 100% confluence. At 30 min before X-ray exposure, cells were refreshed with medium with or without 100 μM TAT-Gap19 (Genosphere Biotechnologies, Paris, France). At 24, 48, 72 h, and 7 days after irradiation, the supernatant was collected. The medium was changed only for the 7 days time point experiment on the fourth day with fresh medium alone or with medium supplemented with 100 μM TAT-Gap19. For the simultaneous detection of multiple cytokines in the supernatants, the Magnetic Luminex® Assay (R&D Systems, Minneapolis, Canada) was used following the manufacturer’s instructions. Briefly, supernatants, standards, and microparticles were incubated into a 96-well plate which was pre-coated with cytokine-specific antibodies. The immobilized antibodies were able to bind the cytokines of interest after 2 h incubation, then the plate was washed, and incubation with a biotinylated antibody cocktail specific to the cytokines of interest was performed for 1 h. A second wash was performed in order to remove the unbound biotinylated antibodies. Further, a streptavidin-phycoerythrin conjugate was added to each well to bind to the biotinylated antibodies. After a final wash, the microparticles were resuspended in buffer and read using the Luminex® MAGPIX Analyzer (R&D systems, Minneapolis, MN, Canada).
In order to normalize to cell number, DAPI staining was performed for the 48 h after irradiation time point. Briefly, cells were washed with phosphate buffered saline (PBS) and fixed with 4% paraformaldehyde (PFA) for 15 min at room temperature. Afterward, cells were washed with PBS and stained with 1 μg/ml 4′,6-Diamidine-2′-phenylindole (DAPI) dissolved in 1x TBST, 0.005 g/v% TSA blocking powder (PerkinElmer, FP1012) (TNB). After 1 h, cells were washed with PBS and DAPI signals were visualized using the Nikon Eclipse Ti inverted wide-field epifluorescence microscope equipped with a 5× magnification dry objective (Plan Fluor, numerical aperture 0.6) and TE2000-E Nikon camera, controlled by the NIS Elements software. The intensity of the DAPI signals was determined with the FIJI software. Cell count for 24 h after irradiation experiment was performed by Moxi Z Mini Automated Cell Counter (ORFLO Technologies, Ketchum, ID, United States) after trypsinization. For the 72 h, and 7 days post-irradiation cell counts, we used Incucyte ZOOMTM phase-contrast imaging based on 49 images taken per well with the 10x objective.
Senescence Detection by CPRG Assay
TICAE and TIME cells were seeded in 96 well plates at a density of 10 000 cells/well in 16 replicates. Three days after, cells were at 100% confluent. At 30 min before X-ray exposure, the cells were refreshed with 150 μl medium with or without with 100 μM TAT-Gap19 (Genosphere Biotechnologies, Paris, France). For 7 and 9 days post irradiation experiments, the medium was refreshed once, while for 14 days post irradiation experiment the medium was refreshed twice with 200 μl medium alone or with medium supplemented with 100 μM TAT-Gap19. At 7, 9, and 14 days post irradiation, cell number was determined using an Incucyte ZOOM® live cell analysis system and related software (Essen Bioscience, Hertfordshire, United Kingdom). Afterward, the senescence-associated β-galactosidase activity in the cells was determined using the chlorophenol red β-D-galactopyranoside (CPRG) assay, as described previously (Baselet et al., 2017). The cells were washed with PBS and lysed using M-PERTM buffer (Thermo Fisher Scientific, Asse, Belgium). Subsequently, 1 × CPRG substrate (2 mM Chlorophenol Red β-D-galactopyranoside in CPRG assay buffer containing 50 mm KPO4, 1 mM MgCl2, pH 6) (Sigma-Aldrich, Overijse, Belgium) was added to each well and the plates incubated for 18 h at 37°C without CO2. A Clariostar® microplate reader (BMG Labtech, Temse, Belgium) was used to measure the absorbance at 570 nm.
DNA Damage Detection
To detect DNA double strand breaks after IR, TICAE and TIME cells were seeded in NuncTM Lab-TekTM Chamber Slide (Thermo Fisher Scientific, Asse, Belgium) in eight replicates, at a seeding density of 50 000 cells/well. Two days later, cells reached 100% confluence. At 30 min before IR, a fresh medium was added to the cells with or without 100 μm TAT-Gap19. At 1 h post irradiation, cells were fixed with 2% PFA (Sigma-Aldrich Co., LCC, Diegem, Belgium) and permeabilized with 0.25% Triton X-100 (Sigma-Aldrich Co., LCC, Diegem, Belgium) in PBS. Cells were blocked for 1 h in 1% normal goat serum (Thermo Fisher Scientific, Asse, Belgium) in Tris-NaCl (Perkin Elmer, Brussels, Belgium). Thereafter, staining with 1/300 primary anti-gamma H2AX antibody (Merck-Millipore #05-636) and 1/1000 anti-TP53BP1 antibody (Novus Biologicals #NB100-304) was performed for 1 h at 37°C. After washing three times with PBS, secondary Alexa Fluor 488 and 568 antibodies (Life Technologies, Merelbeke, Belgium) were applied for 1 h at 37°C. Finally, the slides were mounted using prolong Diamond Antifade Mountant with DAPI (Life Technologies, Merelbeke, Belgium), and cells were visualized with an Eclipse Ti inverted wide-field epifluorescence microscope (Nikon, Brussels, Belgium) with a 20 × Plan Fluor objective (NA 0.6) and an Andor Ixon EMCCD camera, controlled by the NIS Elements software. A z-stack of nine planes axially separated by 1 μm was applied, and 16 fields were captured for each replicate. The analysis was performed with FIJI software using the CellBlocks.ijm script (De Vos et al., 2010; Schindelin et al., 2012).
Statistical Analysis
Non-parametric two-tailed Mann–Whitney T-test was performed for the analysis of all the experiments, except for cell death experiment using live cell imaging, where two-way ANOVA analysis followed by Tukey test was used. Data are presented as mean ± standard error of the mean. Statistical significance was considered when P < 0.05. GraphPad Prism 5.01 (GraphPad Software Inc., La Jolla, CA, United States) was used in all the analysis and occasional exclusion of outlier data points were performed using Grubbs’ test.
Results
Radiation-Induced Oxidative Stress Is Suppressed by TAT-Gap19
Intracellular ROS production was measured in TICAE and TIME cells in response to exposure to 0.1 and 5 Gy X-rays, making use of Incucyte live cell imaging of CM-H2DCFDA fluorescence at 45 min, 2 h, and 3 h after IR exposure. In TICAE and TIME cells, a radiation-induced dose-dependent increase in intracellular ROS production was observed from 45 min until 3 h post irradiation (p.i.) (significant for 0.1 and 5 Gy), with the highest response at 45 min p.i. (Figures 1A–D). TIME cells showed to be more sensitive, as the extent of ROS production after 45 min of IR was 5-fold larger for the 0.1 Gy dose, and two times higher for the 5 Gy dose than in TICAE cells (Figures 1A–D). TAT-Gap19 significantly reduced ROS production at both doses (0.1 and 5 Gy) in TICAE cells at 45 min p.i., while in TIME cells, it significantly reduced ROS production only for the 0.1 Gy dose at 45 min p.i. (Figures 1A,B). Tert-Butyl hydroperoxide (tBHP) was used in the experiment as a positive control (Figure 1E).
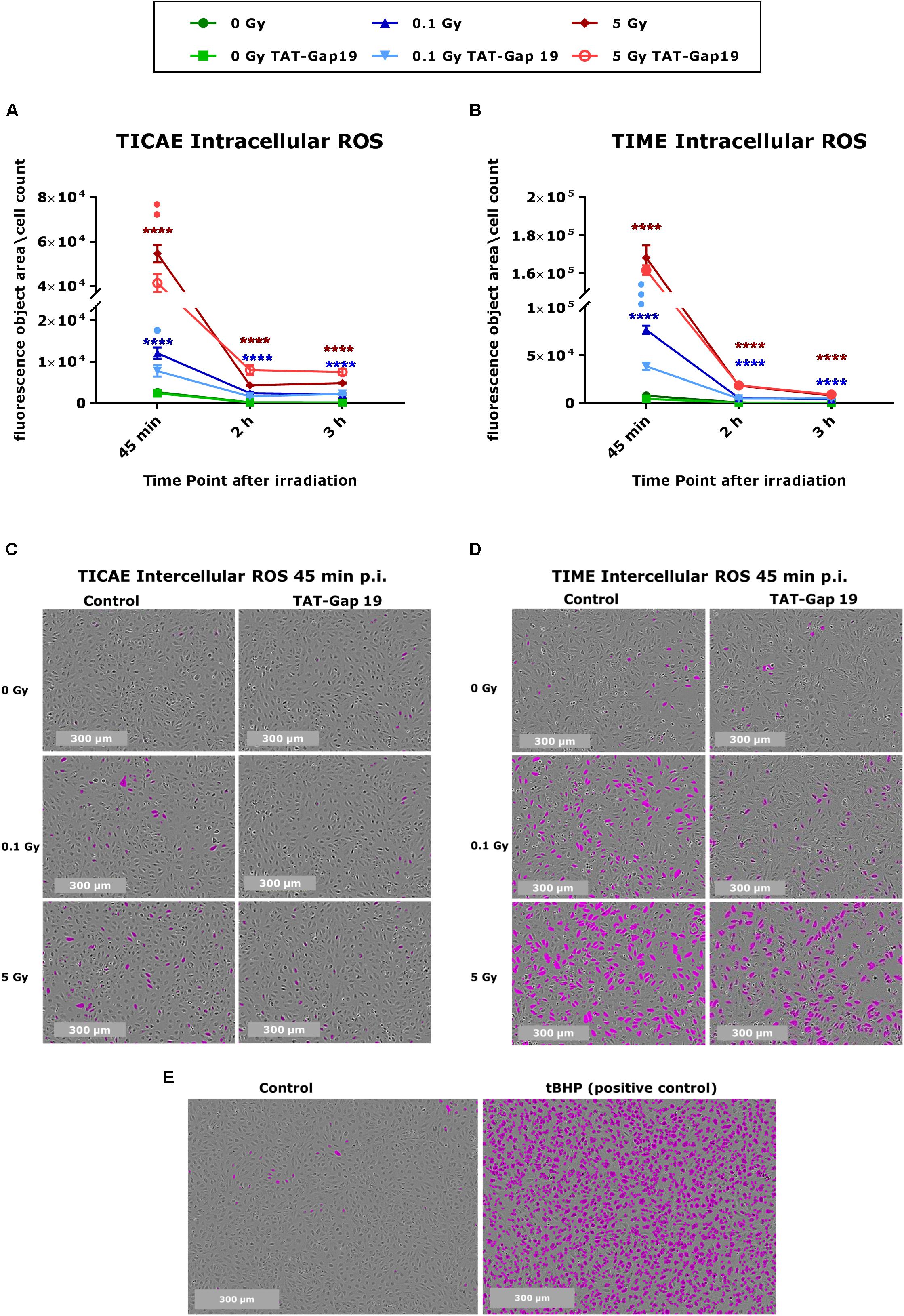
Figure 1. Radiation-induced ROS production and the effect of TAT-Gap19 in TICAE and TIME cells. Intracellular ROS was assessed using CM-H2DCFDA combined with Incucyte live cell imaging at three time points (45 min, 2 and 3 h) after irradiation (A,B). (C,D) Representative images for ROS production at 45 min p.i. in TICAE and TIME cells, with purple colored cells indicating above threshold ROS signal. (E) Representative images for ROS production in TICAE cells in response to tert-Butyl hydroperoxide (tBHP) as a positive control condition; purple colored cells indicate above threshold ROS signal. The values represent the average ± SEM of 8–16 biological replicates for TICAE and TIME cells. Statistical analysis was done with a non-parametric Mann–Whitney T-test. *Indicates statistically significant differences compared to the respective 0 Gy controls. •Indicate statistically significant differences compared to the respective control condition (not treated with TAT-Gap19). */•p < 0.05; **/••p < 0.01; ***/•••p < 0.0001, ****/••••p < 0.00001.
TAT-Gap19 Protects Against Radiation-Induced Cell Death
In order to investigate cell death after IR exposure, Annexin V (early and late apoptotic cells) and Caspase 3/7 activity (late apoptotic cells) were assessed in TICAE and TIME cells from 4 h until 100 h after IR using Incucyte live cell imaging. We furthermore tested the effect of hemichannel inhibition with TAT-Gap19.
In TICAE cells, a significant increase in Caspase 3/7 activity and Annexin V was observed for the 5 Gy dose starting from 4 h and persistent until 100 h p.i., the last point of the recording (Figures 2A,B). TAT-Gap19 significantly reduced Caspase 3/7 and Annexin V for the 5 Gy dose starting from 12 h p.i. and persistent until the end of the recording (Figures 2A,B). In addition, TAT-Gap19 significantly reduced Caspase 3/7 activity for 0 and 0.1 Gy conditions starting from 16 h p.i, and persisting until the end (Figure 2A). TAT-Gap19 also significantly reduced Annexin V for the 0 Gy and 0.1 Gy conditions starting from 4 to 8 h on, respectively (Figure 2B).
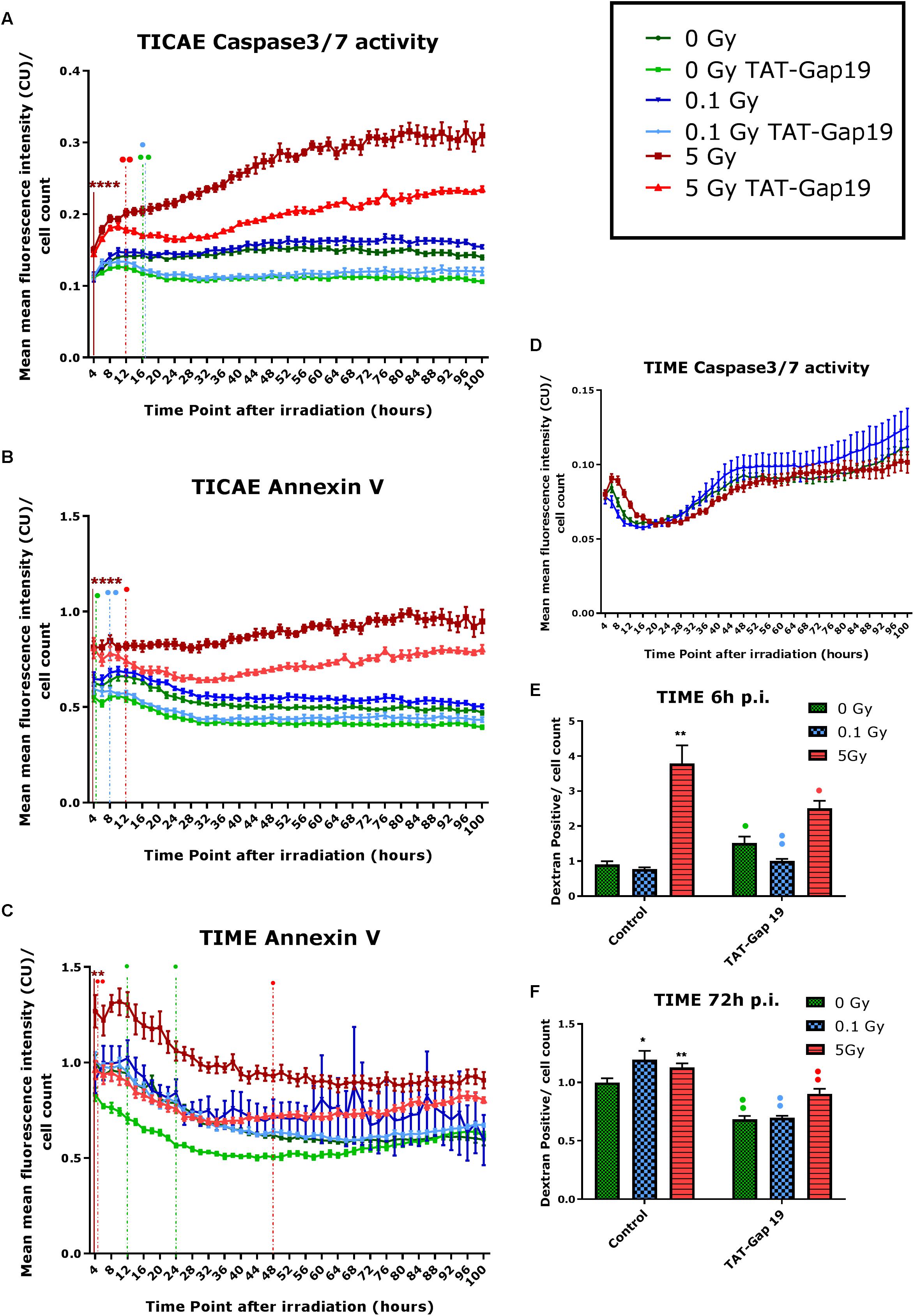
Figure 2. Radiation-induced cell death as assessed by Caspase 3/7 activity, Annexin V and 10 kDa dextran fluorescein staining, and effect of TAT-Gap19. (A,D) Caspase 3/7 activity in TICAE and TIME cells from 4 to 100 h p.i., (B,C) Annexin V level in TICAE and TIME cells from 4 to 100 h p.i. assessed by Incucyte live cell imaging of the mean fluorescence intensity measured in Calibrated Unit (CU) that takes into account acquisition time and fluorescent intensity. (E,F) Dextran fluorescein stained TIME cells at 6 and 72 h p.i. The values represent the average ± SEM of 6–8 biological replicates; statistical analysis was done with non-parametric two-way ANOVA followed by a Tukey test for Annexin V and Caspase 3/7 activity, and with a non-parametric Mann–Whitney T-test for the dextran fluorescein assay. *Indicates statistically significant differences compared to the respective 0 Gy controls. •Indicates statistically significant differences between the TAT-Gap19 group compared to the corresponding respective control conditions (not treated with TAT-Gap19). */•p < 0.05; **/••p < 0.01; ***/•••p < 0.0001, ****/••••p < 0.00001.
In TIME cells, there was no significant change in Caspase 3/7 activity after 0.1 and 5 Gy of IR until 100 h p.i. (Figure 2D). However, a significant persistent increase in Annexin V level was observed at 5 Gy starting from 4 h p.i. until the end of the recording (Figure 2C). TAT-Gap19 significantly reduced Annexin V elevation for the 5 Gy dose in the 4–48 h p.i. time window. TAT-Gap19 also transiently reduced Annexin V for the 0 Gy condition (12–24 h p.i. time window; Figure 2C).
To further validate cell death in TIME cells, 10 kDa dextran fluorescein dye, which is known to enter and stain cells during necrosis or late apoptosis, was assessed at 6 and 72 h p.i. A radiation-induced increase in dextran fluorescein was observed for 5 Gy at 6 h p.i. and for both 0.1 and 5 Gy doses at 72 h p.i. (Figures 2E,F). These responses (including the control response) were inhibited by TAT-Gap19 (Figures 2E,F). An unexpected small increase in dextran fluorescein staining was observed with TAT-Gap19 for the control and 0.1 Gy conditions at 6 h p.i. (Figure 2E). We also evaluated the effect of TAT peptide alone (100 μm) on cell death in TICAE and TIME cells using Incucyte live cell imaging (Supplementary Figure 1). In TICAE cells, no significant changes in Caspase 3/7 activity and Annexin V were observed for TAT peptide from 4 h until 100 h after treatment (Supplementary Figures 1A,B). In TIME cells, it slightly increased Annexin V between 24 and 30 h after exposure (Supplementary Figure 1C).
TAT-Gap19 Mitigates Radiation-Induced Inflammatory Responses
Different atherosclerosis inflammatory markers, selected based on a survey of available literature, were assessed in TICAE and TIME cells after 0.1 and 5 Gy of IR exposure at 24 h, 48 h, 72 h and 7 days p.i. We subsequently tested whether TAT-Gap19 affected these responses. The various inflammatory markers used are summarized in Supplementary Table 1. In total, we considered 12 different markers.
In general, IR exposure at a 5 Gy dose induced an increase in the majority of the markers (IL-6, MCP-1, PECAM-1, IL-1β, TNF-α, CRP, VCAM-1, E-Selectin, Endothelin-1, IL-8, and PAI-1) in both TICAE and TIME cells. TAT-Gap19 displayed variable effects, in some conditions decreasing while in others increasing the responses, especially in TICAE cells (Supplementary Table 1). In order to test the possibility that the viral TAT membrane translocation sequence could perhaps by itself elicit an effect, we selected several markers and evaluated their response to TAT peptide exposure at the 24 h time point, that corresponds to the time at which TAT-Gap 19 increased all tested markers in TICAE cells. We found that TAT (100 μm) significantly increased IL-6, MCP-1, IL-1β, and CRP in TICAE cells (Supplementary Figure 2). Notwithstanding these early (24 h) TAT side-effects on a subset of the markers in TICAE cells, TAT-Gap19 significantly reduced the responses of several markers in TICAE and TIME cells mostly at later time points. Figure 3 summarizes these responses for selected markers and time points, which are considered in the account that follows. In TICAE cells, MCP-1, VCAM-1, and IL-8 were significantly increased after 5 Gy IR exposure at 72 h and 7 days p.i., and TAT-Gap 19 significantly decreased these responses (Figures 3A–F). Radiation induced an increase in Endothelin-1 and IL-1β at 7 days p.i. in TICAE cells, and TAT-Gap 19 again significantly reduced these responses (Figures 3G,H).
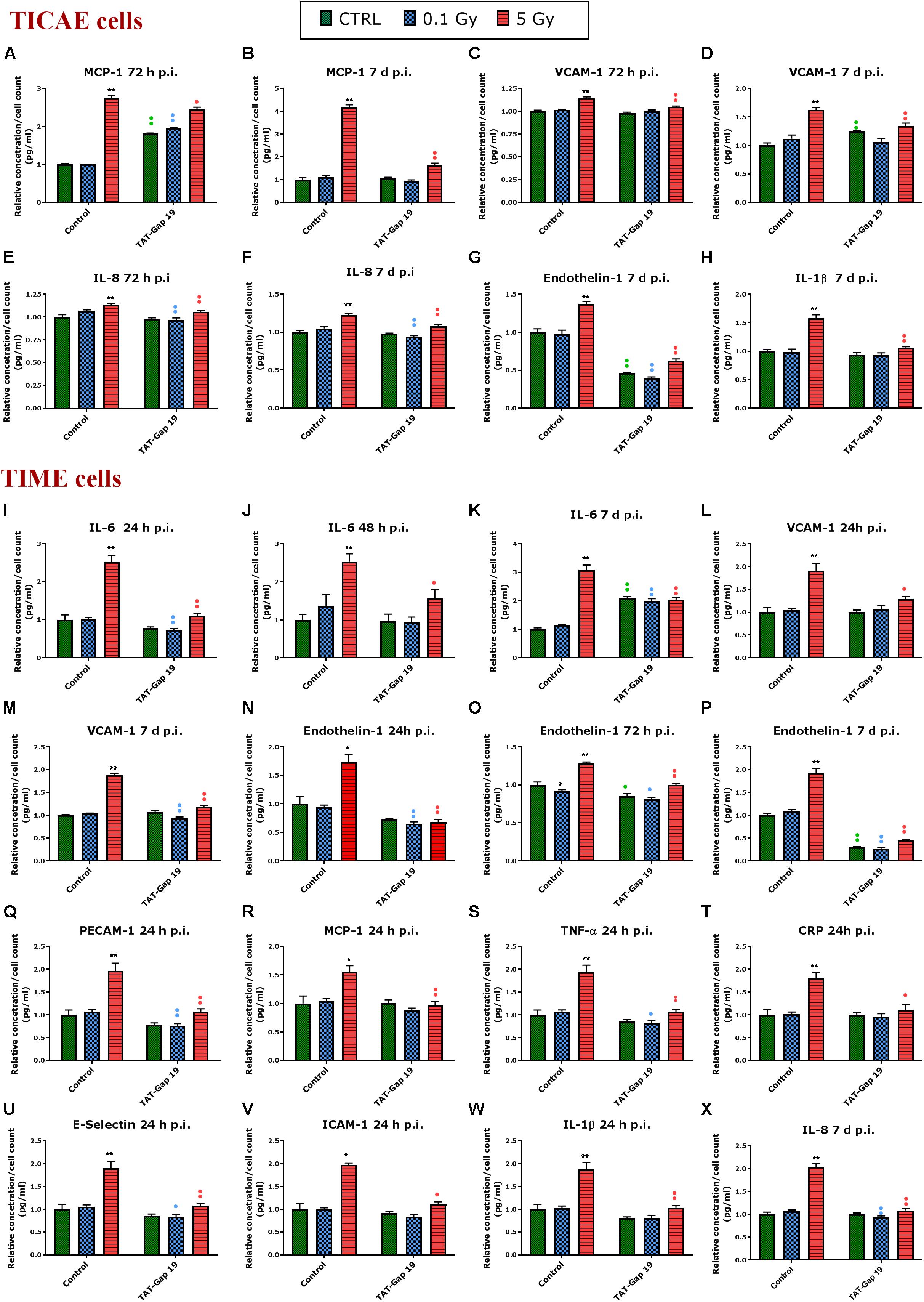
Figure 3. Radiation-induced inflammatory responses and the effect of TAT-Gap19. The response of various inflammatory markers in (A–H) TICAE cells and (I–X) TIME cells to 0.1 and 5 Gy irradiation conditions. The values represent the average ± SEM of 5–6 biological replicates; data were analyzed with a non-parametric Mann–Whitney T-test. *Indicates statistical differences compared to the respective 0 Gy controls. •Indicates statistically significant differences between the TAT-Gap19 group compared to the corresponding responses in the control group (not-treated with TAT-Gap19). */•p < 0.05; **/••p < 0.01. IL-1β, Interleukin 1 beta; IL-6, Interleukin 6; IL-8, Interleukin 8; ICAM-1, Intracellular adhesion molecule 1; VCAM-1, Vascular cell adhesion protein 1; PECAM-1, Platelet endothelial cell adhesion molecule-1; MCP-1, Monocyte chemotactic protein-1; TNF- α, Tumor necrosis factor alpha; CRP, C-reactive protein.
In TIME cells, a radiation-induced significant increase in IL-6 at 5 Gy was observed at 24 h, 48 h, and 7 days p.i., and TAT-Gap19 significantly reduced these responses (Figures 3I–K). Next to that, radiation induced a significant increase in VCAM-1 for 5 Gy at 24 h and 7 days p.i. in TIME cells, and TAT-Gap19 again significantly reduced these responses (Figures 3L,M). Likewise, Endothelin-1 was significantly increased after 5 Gy irradiation in TIME cells at 24 h, 72 h and 7 days p.i., and TAT-Gap19 significantly decreased these responses (Figures 3N–P). Furthermore, TAT-Gap19 significantly reduced the increase of PECAM-1, MCP-1, TNF-α, CRP, E-Selectin, ICAM-1, and IL-1β at 24 h after 5 Gy irradiation in TIME cells (Figures 3Q–W). TAT-Gap19 also significantly decreased IL-8 levels after 5 Gy IR exposure at 7 days p.i. (Figure 3X) in TIME cells. In some of the experiments described above, TAT-Gap19 also had effects on the 0 Gy control and 0.1 Gy conditions in both cell types: it increased the MCP-1 control readout as well as the 0.1 Gy at 72 h p.i. measurement in TICAE cells (Figure 3A), and also the IL-6 control and 0.1 Gy at 7 days p.i. readouts in TIME cells (Figure 3K). By contrast, it decreased the control and the 0.1 Gy readouts for Endothelin-1 in TICAE and TIME cells (Figures 3G,O,P), and the 0.1 Gy readouts for, IL-6, VCAM-1, PECAM-1, TNF-α, E-Selectin and IL-8 in TIME cells (Figures 3I,M,Q,S,U,X), suggesting baseline/low dose effect of TAT-Gap19. We further evaluated the effect of TAT peptide exposure (100 μm) at later time points and conditions where TAT-Gap 19 demonstrated inhibitory effects in both TICAE and TIME cells (Supplementary Figure 3). TAT peptide did not influence IL-1β, IL-8, MCP-1, VCAM-1, and Endothelin-1 levels at 72 h and 7 days post exposure in TICAE cells. In TIME cells, TAT peptide only increased MCP-1 and VCAM-1 at 72 h, and IL-8 at 7 days post exposure (Supplementary Figure 3).
TAT-Gap19 Protects From Radiation-Induced Premature Endothelial Senescence
In order to assess premature senescence in TICAE and TIME cells after 0.1 and 5 Gy irradiation and to evaluate the effect of TAT-Gap19, senescence-associated β-galactosidase (SA β-gal) activity was measured at 7, 9, and 14 days after exposure.
In TICAE cells, a radiation-induced increase in SA β-gal activity was observed for 5 Gy at 7 and 9 days p.i. By contrast, at the 0.1 Gy 7 days p.i. time point, SA β-gal activity was significantly decreased (Figure 4A). TAT-Gap19 significantly reduced SA β-gal activity at control, 0.1 and 5 Gy conditions for 7 days and at 0 and 5 Gy irradiated conditions for 9 days p.i. (Figure 4A). In TIME cells, a radiation-induced increase in SA β-gal activity was observed for 5 Gy at 7 days p.i. and for 0.1 Gy at 9 days p.i. (Figure 4B). TAT-Gap19 significantly reduced SA β-gal activity in all conditions (0 Gy, 0.1 and 5 Gy) at the 7 days p.i. time point and at the 9 days p.i. time point for 5 Gy; although it unexpectedly increased SA β-gal activity in the control condition in TIME cells at 9 days p.i. (Figure 4B). At 14 days p.i., SA β-gal activity was decreased after 5 Gy exposure but significantly increased after 0.1 Gy exposure in both TICAE and TIME cells, and TAT-Gap19 significantly inhibited these responses (Figures 4A,B).
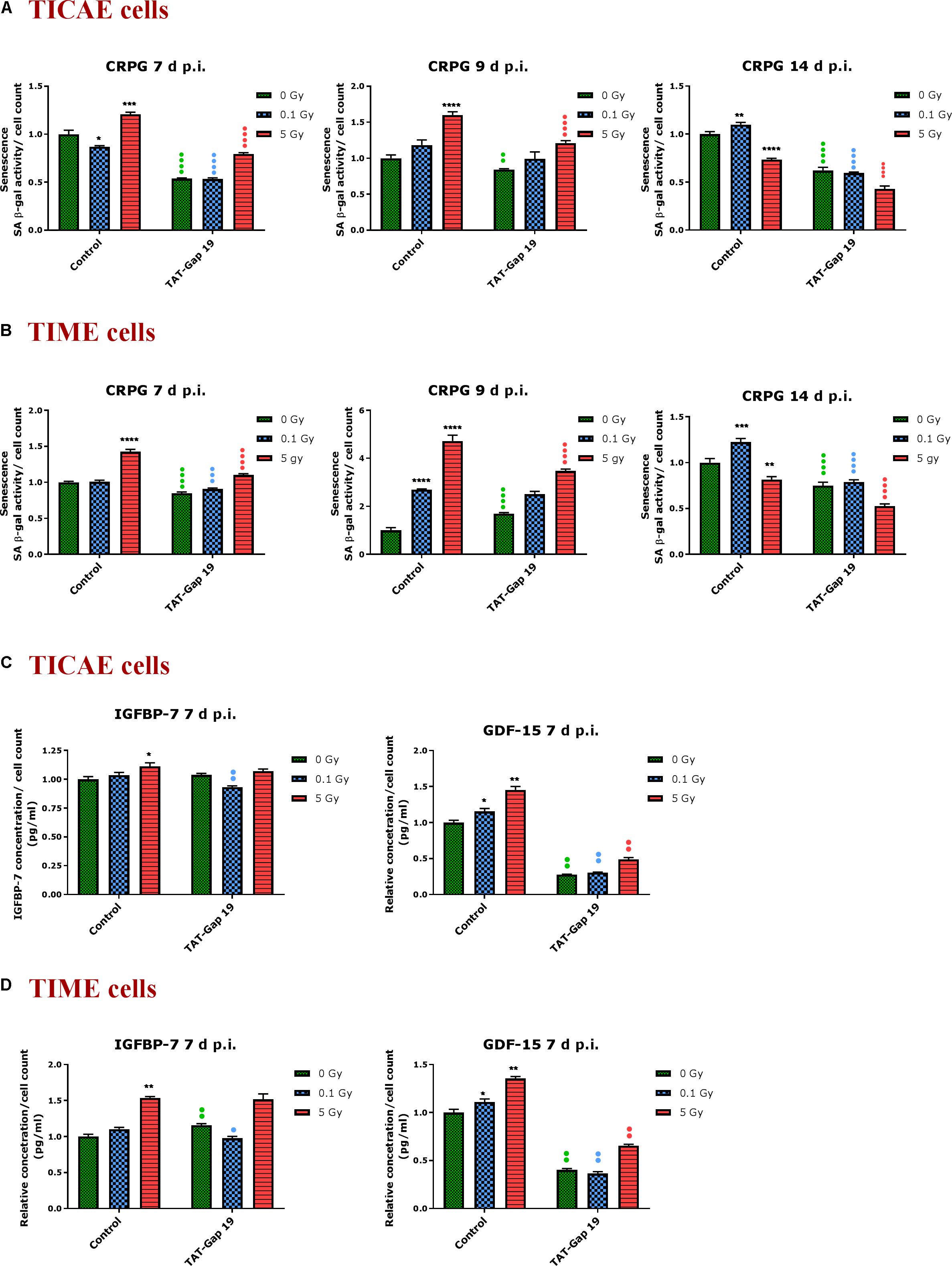
Figure 4. Radiation-induced premature endothelial senescence and effect of TAT-Gap19. Senescence-associated β-galactosidase activity was measured in a CPRG assay at 7, 9, and 14 days after radiation exposure (0.1 and 5 Gy) in (A) TICAE cells and (B) TIME cells. IGFBP-7 and GDF-15 were assessed in (C) TICAE cells and (D) TIME cells at 7 days p.i. The values represent the average ± SEM of 16–24 biological replicates in the CPRG assay and of six biological replicates in multiplex-based assays. Statistical significance was analyzed with a non-parametric Mann–Whitney T-test. *Indicates statistically significant differences compared to the respective 0 Gy controls. •Indicates statistically significant differences between the TAT-Gap19 group compared to the respective control condition (not treated with TAT-Gap19). */•p < 0.05; **/••p < 0.01., ***/•••P < 0.001, ****/••••P < 0.0001.
In order to further assess premature senescence, levels of the Insulin-like Growth Factor-Binding Protein-7 (IGFBP-7) and Growth Differentiation Factor 15 (GDF-15), known to be involved in senescence, were assessed at 7 days after IR exposure using multiplex-based assays. A radiation-induced increase in IGFBP-7 was observed at 5 Gy for TICAE and TIME cells, but this response was not inhibited by TAT-Gap19 (Figures 4C,D). By contrast, 0.1 Gy did not significantly increase IGFBP-7 at 7 days p.i. but TAT-Gap19 acted in a significantly inhibitory way in this condition (Figures 4C,D). The control readout of IGFBP-7 in TIME cells was increased by TAT-Gap19 (Figure 4D). GDF-15 appeared more sensitive to irradiation and was significantly increased in TICAE and TIME cells at both 0.1 and 5 Gy doses at 7 days p.i. TAT-Gap19 significantly inhibited these responses in all treated conditions including the 0 Gy condition (Figures 4C,D).
Radiation-Induced DNA Damage Is Not Affected by TAT-Gap19
DNA damage was assessed by immunocytochemical staining for gamma H2AX and TP53BP1 (Figure 5C), DNA double strand break markers (Wang et al., 2014; Kleiner et al., 2015; Baselet et al., 2017), in TICAE and TIME cells after 1 h of X-ray exposure (0.1 and 5 Gy). We furthermore tested the effect of hemichannel inhibition with TAT-Gap19.
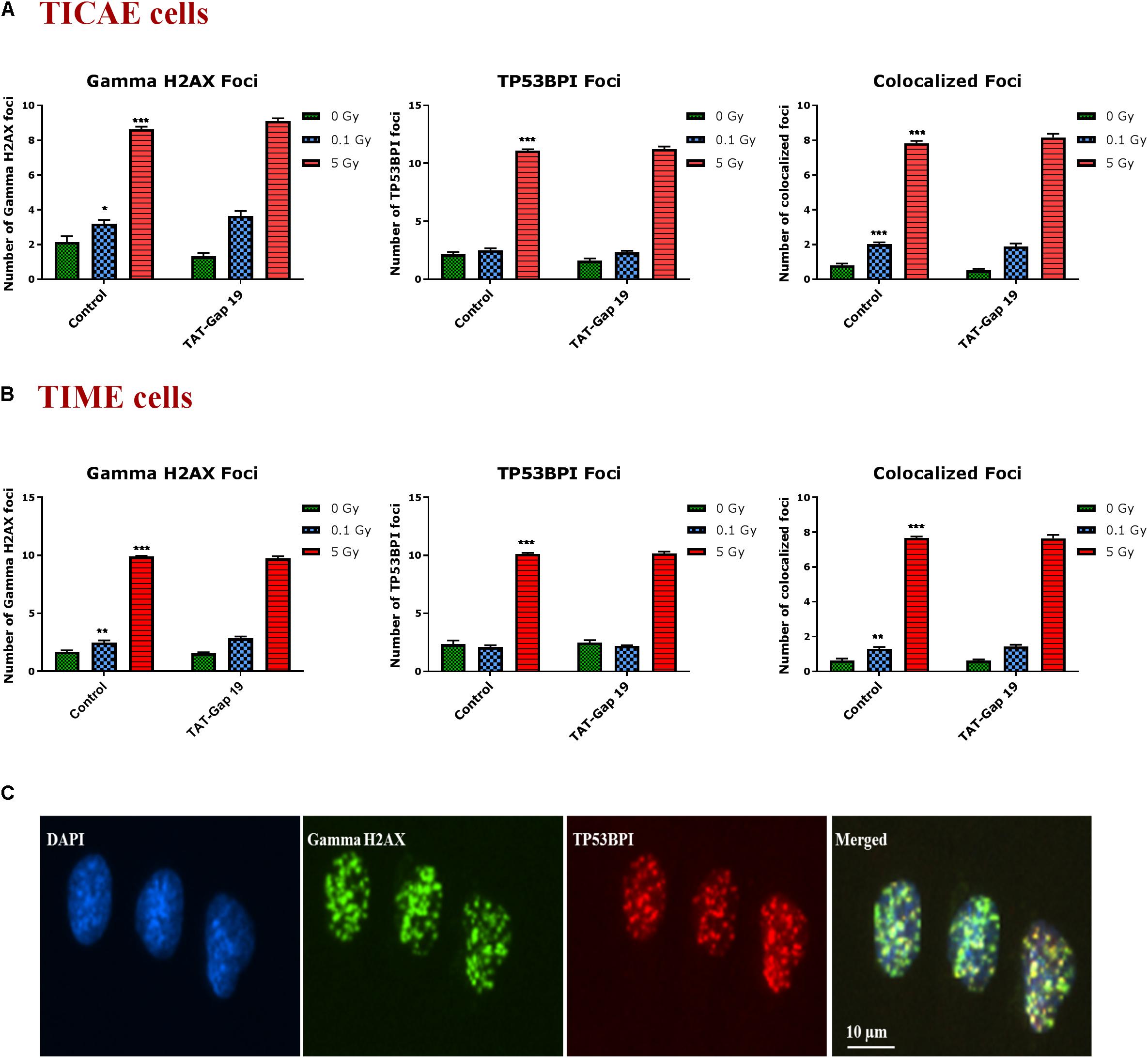
Figure 5. Radiation-induced endothelial DNA damage and effect of TAT-Gap19. Gamma H2AX foci, TP53BPI foci and the colocalized Gamma H2AX/TP53BPI foci were assessed in (A) TICAE cells and (B) TIME cells at 1 h after 0.1 and 5 Gy of X-ray exposure with or without applying TAT-Gap19. (C) Representative images showing gamma H2AX foci (green), TP53BPI foci (red) and colocalized gamma H2AX/TP53BPI foci (yellow) in DAPI stained nuclei of TICAE cells at 5 Gy dose. The values represent the average ± SEM of eight biological replicates; statistical significance was analyzed with a non-parametric Mann–Whitney T-test. *Indicates statistically significant differences compared to the respective 0 Gy controls. •Indicates statistically significant differences in the TAT-Gap19 group compared to the corresponding responses in the control group (not treated with TAT-Gap19). */•p < 0.05; **/••p < 0.01., ***/•••p < 0.001.
In TICAE and TIME cells, radiation induced a significant dose-dependent increase in gamma H2AX and TP53BP1 foci at the 5 Gy dose (Figures 5A,B). The colocalized gamma H2AX/TP53BPI foci also significantly increased in a dose-dependent manner in the irradiated TICAE and TIME cells (Figures 5A,B). These responses were not affected by TAT-Gap19.
Discussion
Patients treated with thoracic radiotherapy have an increased risk of cardiovascular disease, but the underlying pathophysiology is complex and not fully understood (Yamada et al., 2005; Stewart et al., 2006; Hoving et al., 2008; Shimizu et al., 2010; Icrp et al., 2012; Darby et al., 2013; Kreuzer et al., 2015; Baselet et al., 2016). Here, we aimed to investigate the role of Cx43-based hemichannels in radiation-induced endothelial cell damage, an early marker of atherosclerosis, making use of the peptide inhibitor TAT-Gap19 (Wang et al., 2013b; Delvaeye et al., 2018). We found that this peptide significantly reduced oxidative stress, cell death, key inflammatory cytokines and premature cell senescence induced by X-ray exposure of endothelial cell lines derived from coronary artery and microvascular endothelial cells (Figure 6). Below we discuss these findings in more detail.
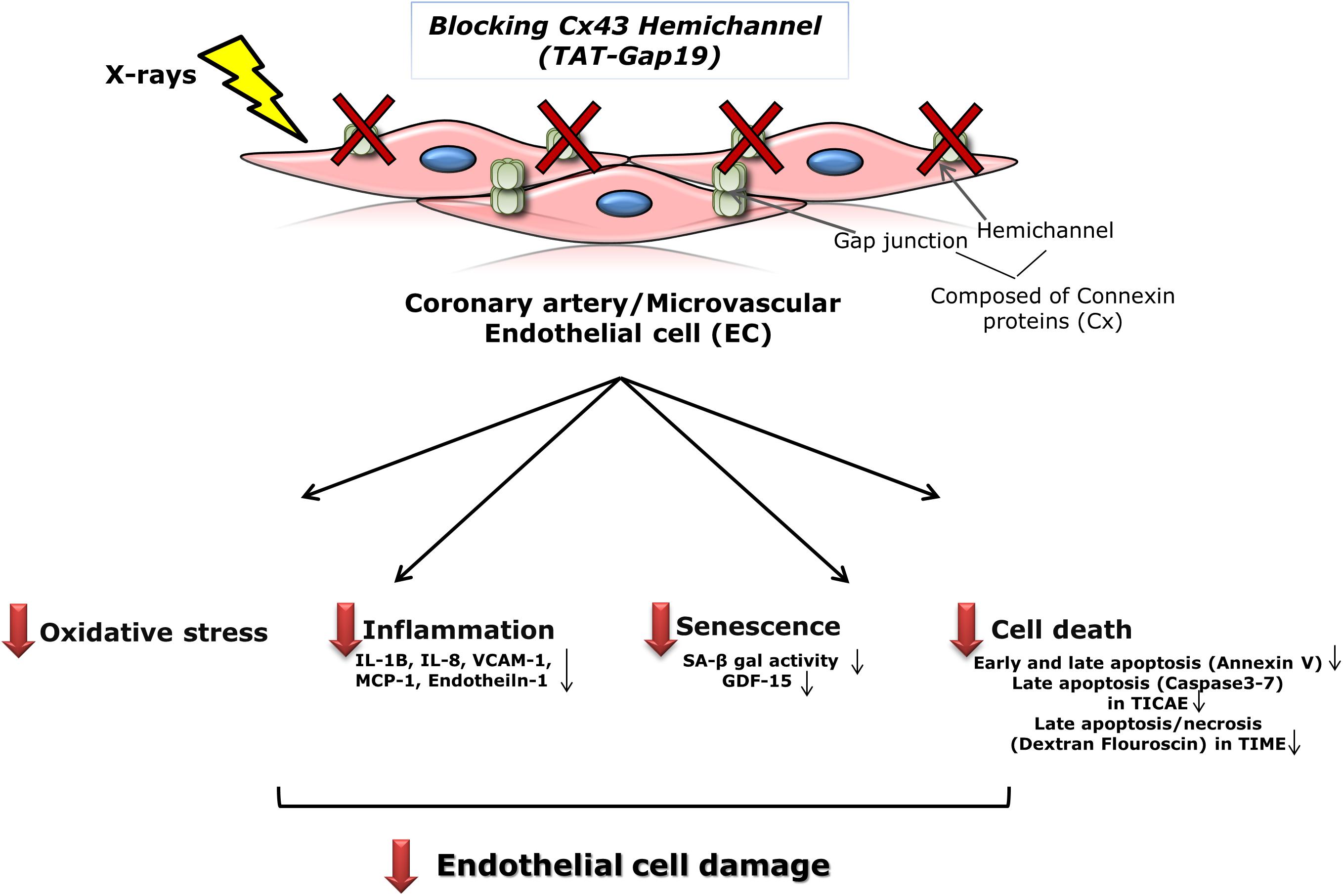
Figure 6. Schematic overview of the experimental findings. Blocking Cx43 hemichannel with TAT-Gap19 significantly reduced oxidative stress, inflammation, senescence and cell death in the irradiated coronary artery and microvascular (TICAE and TIME) endothelial cells, and these responses were mainly observed at the high dose (5 Gy).
A predominant effect of IR exposure is ROS production and oxidative stress (Robbins and Zhao, 2004; Azzam et al., 2012; Yamamori et al., 2012). Accordingly, in TICAE (coronary artery-derived) and TIME cells (microvascular endothelial cells TIME), we found a dose-dependent increase in oxidative stress after 0.1 and 5 Gy of X-ray exposure as measured by CM-H2DCFDA-based live cell imaging approache. The highest oxidative stress response was observed at 45 min p.i. in both cell types, followed by a decline to the 2 and 3 h time points; the latter were, however, still significantly above non-irradiated controls. The decline at 2–3 h may be linked to antioxidant defense reactions of enzymes such as superoxide dismutases (SOD), glutathione, glutathione peroxidases, catalase and others (Robbins and Zhao, 2004; Azzam et al., 2012; Szumiel, 2015). A similar timeline has been observed in primary microvascular endothelial cells exposed to gamma-rays (Laurent et al., 2006). The observed differences between TICAE and TIME cells in the extent of ROS production is possibly related to different radiosensitivity and distinct endothelial properties at different sites of the vascular tree (Aird, 2012; Park et al., 2012). Interestingly, we found that blocking Cx43 hemichannels with TAT-Gap19 significantly reduced ROS production in both cell types at 45 min p.i. Cx43 hemichannel opening has been demonstrated previously in osteocytes in response to oxidative stress, which involved changes of intracellular Ca2+ concentration (Kar et al., 2013; Riquelme and Jiang, 2013). Oxidative stress was also reported to induce Cx43 hemichannels opening in fibroblastoid rat mammary tumor cell line (Marshall Cells) which express Cx43 only (Ramachandran et al., 2007). Further downstream, hemichannel opening has been linked to an extracellular depletion of the major antioxidant glutathione, thereby exaggerating oxidative stress in renal tubular epithelial cells (Chi et al., 2016). Along the same line, Cx43 hemichannel inhibition with TAT-Gap 19 increased SOD activity in mice liver treated with thioacetamide to induce liver fibrosis (Crespo Yanguas et al., 2018). We hypothesize that IR exposure induces ROS production in endothelial cells, which opens Cx43 hemichannels and leads to either loss of hemichannel-permeable antioxidants such as glutathione or activates other signaling pathways that lead to the loss of larger antioxidants such as SOD. TAT-Gap19 attenuated this oxidative stress response, but given the important bidirectional relation between [Ca2+]i elevation and ROS generation (Yan et al., 2006; Gorlach et al., 2015), it is also possible that hemichannel inhibition directly reduces ROS production by inhibiting Ca2+ entry through hemichannels, though further investigations are needed to clarify the involvement of Cx43 hemichannels in intracellular oxidative stress induced by ionizing radiation exposure.
It is well known that oxidative stress is associated with the activation of various cascades including the DNA damage response, apoptosis and inflammatory pathways, thereby amplifying endothelial dysfunction (Elahi et al., 2009; Frey et al., 2009; Soucy et al., 2011; Schramm et al., 2012; Tapio, 2016; Kattoor et al., 2017; Incalza et al., 2018). We observed radiation-induced DNA damage in the two cell types at 1 h p.i., as evidenced by gamma-H2AX and TP53BP1 foci analysis. These findings are consistent with previous studies where a dose-dependent increase in DNA damage was observed in EA.hy926 endothelial hybrid cells, in umbilical vein endothelial cells, as well as in coronary artery endothelial cells in response to X-ray doses in the range of 0.05 to 2 Gy (Rombouts et al., 2013; Baselet et al., 2017), and in microvascular endothelial cells after 3 and 10 Gy of gamma-rays (Laurent et al., 2006). Our findings showed that TAT-Gap19 did not alter this DNA damage response in the irradiated TICAE and TIME cells.
We found that cell death in the two cell types mainly occurred after 5 Gy irradiation, with TICAE cells responding with Annexin V and Caspase 3/7 elevation while TIME cells responded with Annexin V elevation and membrane leakage as indicated by cellular dextran fluorescein positivity. The absence of Caspase 3/7 elevation in TIME cells confirms the findings in primary human microvascular endothelial cells exposed to X-rays in the 2 to 12 Gy dose range (Muschter et al., 2018). Overall, the cell death observed in our study is in line with previous findings in umbilical vein endothelial cells (Rombouts et al., 2013) and microvascular endothelial cells (Langley et al., 1997; Rousseau et al., 2011). Interestingly, we show that TAT-Gap19 significantly reduced cell death in irradiated TICAE and TIME cells. Cx43 hemichannels are known to mediate cell death in various model systems [reviewed in Decrock et al. (2009b)] as well as in cardiomyocytes exposed to hypoxia-reoxygenation or ischemia-reperfusion (Wang et al., 2013b; Gadicherla et al., 2017), by facilitating the passage of soluble factors and molecules such as ATP, ROS and Ca2+ (Leist et al., 1997; Li et al., 2001; Hur et al., 2003; Kalvelyte et al., 2003; Rodriguez-Sinovas et al., 2007; Decrock et al., 2009a, b; Saez et al., 2010; Belousov et al., 2017). We (in a previous study) and others reported that IR induces hemichannel-related ATP release (Kang et al., 2008; Ohshima et al., 2012; Ramadan et al., 2019) which may cause intracellular ATP depletion, thereby activating cell death process through necrosis or apoptosis (Leist et al., 1997; Kalvelyte et al., 2003; Decrock et al., 2009a). Hemichannel opening has furthermore been suggested to facilitate ROS entry into the cells eventually leading to cell death (Ramachandran et al., 2007). Moreover, Cx43 hemichannel inhibition with TAT-Gap19 was previously reported to reduce cell death in various model systems (Wang et al., 2013b; Gadicherla et al., 2017), and to have anti-apoptotic activity by increasing the expression of Bcl-2 and decreasing that of Bax (Chen et al., 2019). Our experiment further showed that TAT-Gap19 decreased cell death in the control non-irradiated condition, as observed by an increase in Annexin V and Caspase 3/7 detection in TICAE cells and by an increase in Annexin V and Dextran fluorescein detection at 72 h in TIME cells. This could be explained by Cx43 hemichannels opening in response to several stress stimuli (Ramachandran et al., 2007; De Vuyst et al., 2009; Orellana et al., 2009; Johansen et al., 2011; Meunier et al., 2017), which could be associated with manipulation of cells during experiments or confluent cells in culture, resulting in a basel level of ATP leakage and cell death, counteracted by TAT-Gap19 protection.
Inflammation plays a key role in atherosclerosis development and progression (Libby, 2012). In this study, we showed that radiation induced an increase in atherogenic inflammatory markers (IL-6, MCP-1, PECAM-1, IL-1β, TNF-α, CRP, VCAM-1, E-Selectin, Endothelin-1, IL-8, and PAI-1) mainly at 5 Gy, in both TICAE and TIME cells. Observations in various endothelial cells confirm an elevation of these inflammatory markers mainly at higher irradiation doses (>2 Gy) (Hallahan et al., 1995; Van Der Meeren et al., 1999; Lanza et al., 2007; Scharpfenecker et al., 2009; Kiyohara et al., 2011; Abderrahmani et al., 2012; Haubner et al., 2013; Sievert et al., 2015; Himburg et al., 2016; Baselet et al., 2017). We found that TAT-Gap19 significantly reduced the elevated IL-1β, IL-8, VCAM-1, MCP-1, and Endothelin-1 levels in the two cell lines, at late time points (72 h and 7 days), while it also had early (24 h) inhibitory effects in TIME cells mainly at 5 Gy. In addition, TAT-Gap19 displayed early (24 h) inhibition of TNF-α, E-Selectin, PECAM-1, CRP, and ICAM-1 mainly at 5 Gy but only in TIME cells, indicating that Cx43 hemichannel involvement in the inflammatory response is time and cell line dependent. All cytokines listed are known to be strongly involved in the pathogenesis of radiation-induced atherosclerosis. The cytokine response is triggered by IR/oxidative stress-induced NF-κb signaling and results in endothelial cell activation with subsequent expression of adhesion molecules (Kim et al., 2001; Roedel et al., 2002; Elahi et al., 2009; Liao, 2013; Sievert et al., 2015). IL-6 has been shown to be responsible for propagating downstream inflammatory responses, and expression of adhesion molecules, such as VCAM-1, ICAM-1, PECAM-1, and E-Selectin, on the vascular wall which is known to initiate the recruitment of macrophages and leukocyte from the vasculature, that acts together with MCP-1 and IL-8 to induce infiltration of macrophages into the subendothelial cell layer, leading to the formation of atherosclerotic lesions (Harrington, 2000; Libby et al., 2002; Schuett et al., 2009; Kiyohara et al., 2011; Libby, 2012). Moreover, increased the production of the potent vasoconstrictor peptide Endothelin-1 was linked to endothelial cell dysfunction, as it may decrease endothelial nitric oxide synthase (eNOS) expression, thereby reducing nitric oxide (NO) vasodilatory signaling and it was also reported to activate macrophages (Kinlay et al., 2001; Bousette and Giaid, 2003; Loomis et al., 2005; Napoli et al., 2006; Bohm and Pernow, 2007). Importantly, it is thought that Cx43 hemichannel opening may continue and propagate the inflammatory scene (Retamal et al., 2007; Guo et al., 2016; Li et al., 2018; Saez et al., 2018). In addition, upregulated Cx43 expression often correlates with increased inflammatory responses, while reduced Cx43 expression inhibits inflammation (Mori et al., 2006; Retamal et al., 2007; Cronin et al., 2008; Danesh-Meyer et al., 2008; Tsuchida et al., 2013; Mugisho et al., 2018). TAT-Gap19 has been demonstrated to significantly decrease IL-1β and TNF-α in non-alcoholic fatty liver mouse model (Willebrords et al., 2017). Peptide5, another mimetic peptide-based hemichannel blocker, was reported to reduce TNF-α and IL-1β in rat model of spinal cord injury (O’Carroll et al., 2013). This peptide also reduced inflammation in a rat model of light-induced retinal damage, as assessed by glial fibrillary acidic protein (GFAP) and leukocyte common antigen (CD45) immunohistochemistry (Guo et al., 2016; Mat Nor et al., 2018). A possible mechanism by which Cx43 hemichannels contribute to inflammation is through their regulatory effect on high-mobility group box 1 (HMGB1), which serves as a damage-associated molecular pattern molecule (DAMP) (Li et al., 2018). HMGB1, released by apoptotic cells, induces IL-6, IL-8, and MCP-1 secretion and increases the expression of ICAM-1 and VCAM-1 in endothelial cells (Sun et al., 2013). Furthermore, by releasing ATP, Cx43 hemichannels enhance inflammation through downstream activation of the NLRP3 inflammasome (Mugisho et al., 2018; Mugisho et al., 2019). Cx43 hemichannels may thus act to amplify and perpetuate an initial inflammatory condition. Interestingly, IR induces a rapid and persisting increase in Cx43 gene and protein expression in TICAE and TIME cells (Ramadan et al., 2019), thereby creating pro-inflammatory substrate.
In contrast to the inflammation mitigating effect of TAT-Gap19 on irradiated endothelial cells, the peptide transiently increased some other inflammatory markers at various time points (Supplementary Table 1). TAT-Gap19 is composed of the nonapeptide Gap19, linked to a HIV-derived TAT sequence to promote its membrane permeability (Abudara et al., 2014). It is possible that this HIV-derived membrane translocation sequence may induce pro-inflammatory side effects in the cells. In line with this, alpha-carboxy terminus 1-peptide (αCT1), another Cx43 based peptide containing the antennapedia translocation motif, triggered a transient increase in TNF−α upon topical corneal application (Moore et al., 2014). Here, we report that the TAT translocation motif by itself induces a transient increase in IL-6, MCP-1, IL-1β and CRP in TICAE cells at 24 h p.i. (the time point at which TAT-Gap19 significantly increased all assessed cytokines in TICAE cells). In addition, TAT peptide increased MCP-1 and VCAM-1 only in TIME cells at 72 h post exposure (of note; TAT-Gap19 significantly increased the 0 Gy control and some of the irradiated conditions in these cytokines at 72 h p.i. in TIME cells). Taken this into account, our results indicate that TAT-Gap19 exerts a late inflammation mitigating effect, by reducing the atherogenic inflammatory markers IL-1β, IL-8, VCAM-1, MCP-1, and Endothelin-1, in both TICAE and TIME cells after IR exposure.
Interestingly, inflammation has been reported to contribute to cellular senescence (Freund et al., 2010; Tian and Li, 2014). Premature cell senescence is known to contribute to endothelial dysfunction by stimulation a complex pro-inflammatory response, increasing ROS levelsand it is known to trigger the initiation of cell death by reducing cell repair ability (Collado et al., 2007; Xue et al., 2007; Donato et al., 2015). Our experiments showed an increase in SA-β gal activity in TICAE and TIME cells mainly at 7 and 9 days p.i. This increase in SA-β gal activity was persistent until 14 days p.i. only at 0.1 Gy. For 5 Gy, an unexpected decrease in SA-β gal activity was observed, which may be caused by the frequent medium change for this time point, resulting in washout of senescent and/or apoptotic cells, which in our experiment mainly occurred at the high dose, and replacement by proliferating cells. Premature senescence in TICAE and TIME cells was also apparent from the increase in IGFBP-7 (at 5 Gy) and GDF-15 (at 0.1 and 5 Gy) 7 days p.i. Our results are in line with previous studies, where cellular senescence was observed after low and high doses of IR exposure in different types of endothelial cells, including human coronary artery (Oh et al., 2001; Ungvari et al., 2013; Yentrapalli et al., 2013a, b; Kim et al., 2014; Lowe and Raj, 2014; Baselet et al., 2017). Interestingly, TAT-Gap19 significantly suppressed the radiation-induced increases in SA-β gal activity, mainly at 5 Gy at 7 and 9 days and from 0.1 Gy at 14 days, and GDF-15 levels from 0.1 Gy in TICAE and TIME cells suggesting a role for Cx43 hemichannels. Chronic Cx43 hemichannel opening, as reported in our previous study at 72 h after irradiation in TICAE and TIME cells (Ramadan et al., 2019), may result in ATP leakage that in turn can activate downstream cellular processes including propagation of intercellular Ca2+ waves, ROS production, activation of the NLRP3 inflammasome pathway and inflammation (De Bock et al., 2012; Mugisho et al., 2018; Tonkin et al., 2018), which are all known to stimulate premature cellular senescence (Kurz et al., 2004; Acosta et al., 2013; Tchkonia et al., 2013). In addition, senescence can be communicated to neighboring cells in a paracrine manner involving secretion of the senescence-associated secretory phenotype (SASP) and inflammasome activation (Acosta et al., 2013). Presumably, Cx43 hemichannels may perhaps facilitate paracrine senescence communication between cells via its effects on pro-inflammatory parameters like IL-8, MCP-1, and IL-6 which are inhibited by TAT-Gap19 in our study. We further showed that TAT-Gap19 decreased senescence in some of the control non-irradiated conditions. Several stress stimuli might be associated with confluent cells in culture for longer times (7 to 14 days), which may result in a basal level of senescence that can be protected by TAT-Gap19, as shown in our experiment.
In summary, this study demonstrates that TAT-Gap19 mitigates radiation-induced endothelial cell damage by reducing oxidative stress, cell death, pro-inflammatory and pathological factors like IL-1β, IL-8, VCAM-1, MCP-1, and Endothelin-1 and premature senescence. This effect is observed mainly for the 5 Gy dose. Further research is needed to investigate the underlying pathways and molecular mechanisms leading to radiation-induced Cx43 hemichannel opening, including changes in membrane potential and intracellular Ca2+, redox status, the surface amount of Cx43 hemichannels and the channel opening probability. Investigating the amount of functional connexin hemichannels at the cell surface may also help in understanding the variable response of TAT-Gap19 in different cell types. In addition, it will be interesting to gather information regarding the downstream effects of Cx43 hemichannel opening that trigger major changes in endothelial function and survival such as unveiling the role of purinergic signaling, intracellular Ca2+, ionic balance and intracellular redox status. Other open question that needs to be resolved is what is the effect of other Cx43 hemichannel targeting approaches, such as Gap19 without HIV-derived TAT, siRNA/shRNA or other mimetic peptides, on endothelial cell response after irradiation. This may open perspectives for the establishment of novel therapeutic strategies to protect from secondary radiotherapy side effects in thoracic cancer patients.
Conclusion
In conclusion, to the best of our knowledge, this is the first study to show the possible involvement of endothelial Cx43 hemichannels in contributing to various radiation-induced processes, such as oxidative stress, cell death, inflammation and premature cell senescence that lead to endothelial activation and dysfunction, which is known as an early marker for atherosclerosis. Therefore, targeting Cx43 hemichannels holds potential to therapeutically protect against radiation-induced endothelial cell damage.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation, to any qualified researcher.
Author Contributions
RR conducted all the experiments and wrote the manuscript text. EV did a MSc project contributing to inflammatory marker measurement at 48 h and Dextran fluorescein experiments. DA did a MSc project contributing to ROS experiment using Flow Cytometry, inflammatory markers measurements at 24 and 72 h, and apoptosis live cell imaging experiments. IG performed a MSc project contributing to live cell imaging of ROS and DNA damage experiment. DH participated in Calcium measurement experiment which directed us in the discussion. ED, SB, LL, and AA contributed in designing the experiments and supervision of the work. All authors, except the MSc students EV, DA, and IG contributed equally in reviewing the manuscript. All authors read and approved the final version of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
RR is supported by a doctoral SCK•CEN/Ghent University Grant.
Acknowledgments
TICAE cell line was kindly donated by Dr. Kenneth Raj, Centre for Radiation, Chemical and Environmental Hazards, Public Health England, Didcot, United Kingdom. The authors thank Prof. Dr. Winnok de Vos, Laboratory of Cell Biology and Histology, Department of Veterinary Sciences, University of Antwerp, Antwerp, Belgium for the CellBlocks.ijm script used in FIJI. The authors also thank Nele Weyens and Koen Hasaers, Business Development & Support unit (BD&S) at the Belgian Nuclear Research Centre (SCK•CEN) for filing a patent for this work.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2020.00212/full#supplementary-material
References
Abderrahmani, R., Francois, A., Buard, V., Tarlet, G., Blirando, K., Hneino, M., et al. (2012). PAI-1-dependent endothelial cell death determines severity of radiation-induced intestinal injury. PLoS ONE 7:e35740. doi: 10.1371/journal.pone.0035740
Abudara, V., Bechberger, J., Freitas-Andrade, M., De Bock, M., Wang, N., Bultynck, G., et al. (2014). The connexin43 mimetic peptide Gap19 inhibits hemichannels without altering gap junctional communication in astrocytes. Front. Cell. Neurosci. 8:306. doi: 10.3389/fncel.2014.00306
Acosta, J. C., Banito, A., Wuestefeld, T., Georgilis, A., Janich, P., Morton, J. P., et al. (2013). A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 15, 978–990. doi: 10.1038/ncb2784
Aleman, B. M., Moser, E. C., Nuver, J., Suter, T. M., Maraldo, M. V., Specht, L., et al. (2014). Cardiovascular disease after cancer therapy. EJC Suppl. 12, 18–28.
Al-Kindi, S. G., and Oliveira, G. H. (2016). Incidence and trends of cardiovascular mortality after common cancers in young adults: analysis of surveillance, epidemiology and end-results program. World J. Cardiol. 8, 368–374.
Amino, M., Yoshioka, K., Fujibayashi, D., Hashida, T., Furusawa, Y., Zareba, W., et al. (2010). Year-long upregulation of connexin43 in rabbit hearts by heavy ion irradiation. Am. J. Physiol. Heart Circ. Physiol. 298, H1014–H1021.
Azzam, E. I., De Toledo, S. M., and Little, J. B. (2001). Direct evidence for the participation of gap junction-mediated intercellular communication in the transmission of damage signals from alpha-particle irradiated to nonirradiated cells. Proc. Natl. Acad. Sci. U.S.A. 98, 473–478. doi: 10.1073/pnas.011417098
Azzam, E. I., De Toledo, S. M., and Little, J. B. (2003). Expression of CONNEXIN43 is highly sensitive to ionizing radiation and other environmental stresses. Cancer Res. 63, 7128–7135.
Azzam, E. I., Jay-Gerin, J. P., and Pain, D. (2012). Ionizing radiation-induced metabolic oxidative stress and prolonged cell injury. Cancer Lett. 327, 48–60. doi: 10.1016/j.canlet.2011.12.012
Baker, J. E., Moulder, J. E., and Hopewell, J. W. (2011). Radiation as a risk factor for cardiovascular disease. Antioxid. Redox. Signal. 15, 1945–1956.
Banaz-Yasar, F., Tischka, R., Iliakis, G., Winterhager, E., and Gellhaus, A. (2005). Cell line specific modulation of connexin43 expression after exposure to ionizing radiation. Cell Commun. Adhes. 12, 249–259. doi: 10.1080/15419060500514101
Baselet, B., Belmans, N., Coninx, E., Lowe, D., Janssen, A., Michaux, A., et al. (2017). Functional gene analysis reveals cell cycle changes and inflammation in endothelial cells irradiated with a single X-ray dose. Front. Pharmacol. 8:213. doi: 10.3389/fphar.2017.00213
Baselet, B., Rombouts, C., Benotmane, A. M., Baatout, S., and Aerts, A. (2016). Cardiovascular diseases related to ionizing radiation: the risk of low-dose exposure (Review). Int. J. Mol. Med. 38, 1623–1641. doi: 10.3892/ijmm.2016.2777
Belousov, A. B., Fontes, J. D., Freitas-Andrade, M., and Naus, C. C. (2017). Gap junctions and hemichannels: communicating cell death in neurodevelopment and disease. BMC Cell Biol. 18:4. doi: 10.1186/s12860-016-0120-x
Bohm, F., and Pernow, J. (2007). The importance of endothelin-1 for vascular dysfunction in cardiovascular disease. Cardiovasc. Res. 76, 8–18. doi: 10.1016/j.cardiores.2007.06.004
Borghini, A., Gianicolo, E. A., Picano, E., and Andreassi, M. G. (2013). Ionizing radiation and atherosclerosis: current knowledge and future challenges. Atherosclerosis 230, 40–47. doi: 10.1016/j.atherosclerosis.2013.06.010
Bousette, N., and Giaid, A. (2003). Endothelin-1 in atherosclerosis and other vasculopathies. Can. J. Physiol. Pharmacol. 81, 578–587. doi: 10.1139/y03-010
Chen, B., Yang, L., Chen, J., Chen, Y., Zhang, L., Wang, L., et al. (2019). Inhibition of Connexin43 hemichannels with Gap19 protects cerebral ischemia/reperfusion injury via the JAK2/STAT3 pathway in mice. Brain Res. Bull. 146, 124–135. doi: 10.1016/j.brainresbull.2018.12.009
Cheng, Y. J., Nie, X. Y., Ji, C. C., Lin, X. X., Liu, L. J., Chen, X. M., et al. (2017). Long-term cardiovascular risk after radiotherapy in women with breast cancer. J. Am. Heart Assoc. 6:e005633.
Chi, Y., Zhang, X., Zhang, Z., Mitsui, T., Kamiyama, M., Takeda, M., et al. (2016). Connexin43 hemichannels contributes to the disassembly of cell junctions through modulation of intracellular oxidative status. Redox Biol. 9, 198–209. doi: 10.1016/j.redox.2016.08.008
Cho, W. K., Park, W., Choi, D. H., Cha, H., Nam, S. J., Kim, S. W., et al. (2018). Which patients with left breast cancer should be candidates for heart-sparing radiotherapy? J. Breast Cancer 21, 206–212.
Collado, M., Blasco, M. A., and Serrano, M. (2007). Cellular senescence in cancer and aging. Cell 130, 223–233.
Crespo Yanguas, S., Da Silva, T. C., Pereira, I. V. A., Willebrords, J., Maes, M., Sayuri Nogueira, M., et al. (2018). TAT-Gap19 and carbenoxolone alleviate liver fibrosis in mice. Int. J. Mol. Sci. 19:E817.
Cronin, M., Anderson, P. N., Cook, J. E., Green, C. R., and Becker, D. L. (2008). Blocking connexin43 expression reduces inflammation and improves functional recovery after spinal cord injury. Mol. Cell. Neurosci. 39, 152–160. doi: 10.1016/j.mcn.2008.06.005
Cutter, D. J., Darby, S. C., and Yusuf, S. W. (2011). Risks of heart disease after radiotherapy. Tex. Heart Inst. J. 38, 257–258.
Danesh-Meyer, H. V., Huang, R., Nicholson, L. F., and Green, C. R. (2008). Connexin43 antisense oligodeoxynucleotide treatment down-regulates the inflammatory response in an in vitro interphase organotypic culture model of optic nerve ischaemia. J. Clin. Neurosci. 15, 1253–1263. doi: 10.1016/j.jocn.2008.08.002
Darby, S., Mcgale, P., Peto, R., Granath, F., Hall, P., and Ekbom, A. (2003). Mortality from cardiovascular disease more than 10 years after radiotherapy for breast cancer: nationwide cohort study of 90 000 Swedish women. BMJ 326, 256–257. doi: 10.1136/bmj.326.7383.256
Darby, S. C., Ewertz, M., Mcgale, P., Bennet, A. M., Blom-Goldman, U., Bronnum, D., et al. (2013). Risk of ischemic heart disease in women after radiotherapy for breast cancer. N. Engl. J. Med. 368, 987–998.
De Bock, M., Wang, N., Bol, M., Decrock, E., Ponsaerts, R., Bultynck, G., et al. (2012). Connexin 43 hemichannels contribute to cytoplasmic Ca2+ oscillations by providing a bimodal Ca2+-dependent Ca2+ entry pathway. J. Biol. Chem. 287, 12250–12266. doi: 10.1074/jbc.m111.299610
De Vos, W. H., Van Neste, L., Dieriks, B., Joss, G. H., and Van Oostveldt, P. (2010). High content image cytometry in the context of subnuclear organization. Cytometry A 77, 64–75.
De Vuyst, E., Wang, N., Decrock, E., De Bock, M., Vinken, M., Van Moorhem, M., et al. (2009). Ca(2+) regulation of connexin 43 hemichannels in C6 glioma and glial cells. Cell Calcium 46, 176–187. doi: 10.1016/j.ceca.2009.07.002
De Wit, C., and Griffith, T. M. (2010). Connexins and gap junctions in the EDHF phenomenon and conducted vasomotor responses. Pflugers. Arch. 459, 897–914. doi: 10.1007/s00424-010-0830-4
Decrock, E., De Vuyst, E., Vinken, M., Van Moorhem, M., Vranckx, K., Wang, N., et al. (2009a). Connexin 43 hemichannels contribute to the propagation of apoptotic cell death in a rat C6 glioma cell model. Cell Death Differ. 16, 151–163. doi: 10.1038/cdd.2008.138
Decrock, E., Hoorelbeke, D., Ramadan, R., Delvaeye, T., De Bock, M., Wang, N., et al. (2017). Calcium, oxidative stress and connexin channels, a harmonious orchestra directing the response to radiotherapy treatment? Biochim. Biophys. Acta 1864, 1099–1120. doi: 10.1016/j.bbamcr.2017.02.007
Decrock, E., Vinken, M., De Vuyst, E., Krysko, D. V., D’herde, K., Vanhaecke, T., et al. (2009b). Connexin-related signaling in cell death: to live or let die? Cell Death Differ. 16, 524–536. doi: 10.1038/cdd.2008.196
Delvaeye, T., Vandenabeele, P., Bultynck, G., Leybaert, L., and Krysko, D. V. (2018). Therapeutic targeting of connexin channels: new views and challenges. Trends Mol. Med. 24, 1036–1053. doi: 10.1016/j.molmed.2018.10.005
Donato, A. J., Morgan, R. G., Walker, A. E., and Lesniewski, L. A. (2015). Cellular and molecular biology of aging endothelial cells. J. Mol. Cell Cardiol. 89, 122–135. doi: 10.1016/j.yjmcc.2015.01.021
Eftekhari, M., Anbiaei, R., Zamani, H., Fallahi, B., Beiki, D., Ameri, A., et al. (2015). Radiation-induced myocardial perfusion abnormalities in breast cancer patients following external beam radiation therapy. Asia Ocean J. Nucl. Med. Biol. 3, 3–9.
Elahi, M. M., Kong, Y. X., and Matata, B. M. (2009). Oxidative stress as a mediator of cardiovascular disease. Oxid. Med. Cell. Longevity 2, 259–269. doi: 10.4161/oxim.2.5.9441
Freitas-Andrade, M., Wang, N., Bechberger, J. F., De Bock, M., Lampe, P. D., Leybaert, L., et al. (2019). Targeting MAPK phosphorylation of Connexin43 provides neuroprotection in stroke. J. Exp. Med. 216, 916–935. doi: 10.1084/jem.20171452
Freund, A., Orjalo, A. V., Desprez, P. Y., and Campisi, J. (2010). Inflammatory networks during cellular senescence: causes and consequences. Trends Mol. Med. 16, 238–246. doi: 10.1016/j.molmed.2010.03.003
Frey, R. S., Ushio-Fukai, M., and Malik, A. B. (2009). NADPH oxidase-dependent signaling in endothelial cells: role in physiology and pathophysiology. Antioxid. Redox. Signal. 11, 791–810. doi: 10.1089/ars.2008.2220
Gadicherla, A. K., Wang, N., Bulic, M., Agullo-Pascual, E., Lissoni, A., De Smet, M., et al. (2017). Mitochondrial Cx43 hemichannels contribute to mitochondrial calcium entry and cell death in the heart. Basic Res. Cardiol. 112:27.
Glover, D., Little, J. B., Lavin, M. F., and Gueven, N. (2003). Low dose ionizing radiation-induced activation of connexin 43 expression. Int. J. Radiat. Biol. 79, 955–964. doi: 10.1080/09553000310001632895
Gorlach, A., Bertram, K., Hudecova, S., and Krizanova, O. (2015). Calcium and ROS: a mutual interplay. Redox Biol. 6, 260–271. doi: 10.1016/j.redox.2015.08.010
Guo, C. X., Mat Nor, M. N., Danesh-Meyer, H. V., Vessey, K. A., Fletcher, E. L., O’Carroll, S. J., et al. (2016). Connexin43 mimetic peptide improves retinal function and reduces inflammation in a light-damaged albino rat model. Invest. Ophthalmol. Vis. Sci. 57, 3961–3973.
Gupta, S., Chan, D. W., Zaal, K. J., and Kaplan, M. J. (2018). A high-throughput real-time imaging technique to quantify NETosis and distinguish mechanisms of cell death in human neutrophils. J. Immunol. 200, 869–879. doi: 10.4049/jimmunol.1700905
Hallahan, D., Clark, E. T., Kuchibhotla, J., Gewertz, B. L., and Collins, T. (1995). E-selectin gene induction by ionizing radiation is independent of cytokine induction. Biochem. Biophys. Res. Commun. 217, 784–795. doi: 10.1006/bbrc.1995.2841
Harrington, J. R. (2000). The role of MCP-1 in atherosclerosis. Stem Cells 18, 65–66. doi: 10.1634/stemcells.18-1-65
Haubner, F., Leyh, M., Ohmann, E., Pohl, F., Prantl, L., and Gassner, H. G. (2013). Effects of external radiation in a co-culture model of endothelial cells and adipose-derived stem cells. Radiat. Oncol. 8:66.
Himburg, H. A., Sasine, J., Yan, X., Kan, J., Dressman, H., and Chute, J. P. (2016). A molecular profile of the endothelial cell response to ionizing radiation. Radiat. Res. 186, 141–152.
Hoving, S., Heeneman, S., Gijbels, M. J., Te Poele, J. A., Russell, N. S., Daemen, M. J., et al. (2008). Single-dose and fractionated irradiation promote initiation and progression of atherosclerosis and induce an inflammatory plaque phenotype in ApoE(-/-) mice. Int. J. Radiat. Oncol. Biol. Phys. 71, 848–857. doi: 10.1016/j.ijrobp.2008.02.031
Hur, K. C., Shim, J. E., and Johnson, R. G. (2003). A potential role for cx43-hemichannels in staurosporin-induced apoptosis. Cell Commun. Adhes. 10, 271–277. doi: 10.1080/cac.10.4-6.271.277
Icrp, Stewart, F. A., Akleyev, A. V., Hauer-Jensen, M., Hendry, J. H., Kleiman, N. J., et al. (2012). ICRP publication 118: ICRP statement on tissue reactions and early and late effects of radiation in normal tissues and organs–threshold doses for tissue reactions in a radiation protection context. Ann. ICRP 41, 1–322. doi: 10.1016/j.icrp.2012.02.001
Incalza, M. A., D’oria, R., Natalicchio, A., Perrini, S., Laviola, L., and Giorgino, F. (2018). Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vascul. Pharmacol. 100, 1–19. doi: 10.1016/j.vph.2017.05.005
Iyyathurai, J., Wang, N., D’hondt, C., Jiang, J. X., Leybaert, L., and Bultynck, G. (2018). The SH3-binding domain of Cx43 participates in loop/tail interactions critical for Cx43-hemichannel activity. Cell Mol. Life. Sci. 75, 2059–2073. doi: 10.1007/s00018-017-2722-7
Jeong, Y. I., Kim, Y. J., Ju, J. W., Hong, S. H., Lee, M. R., Cho, S. H., et al. (2014). Identification of anti-allergic effect of Clonorchis sinensis-derived protein venom allergen-like proteins (CsVAL). Biochem. Biophys. Res. Commun. 445, 549–555. doi: 10.1016/j.bbrc.2014.01.189
Johansen, D., Cruciani, V., Sundset, R., Ytrehus, K., and Mikalsen, S. O. (2011). Ischemia induces closure of gap junctional channels and opening of hemichannels in heart-derived cells and tissue. Cell. Physiol. Biochem. 28, 103–114. doi: 10.1159/000331719
Kalvelyte, A., Imbrasaite, A., Bukauskiene, A., Verselis, V. K., and Bukauskas, F. F. (2003). Connexins and apoptotic transformation. Biochem. Pharmacol. 66, 1661–1672. doi: 10.1016/s0006-2952(03)00540-9
Kang, J., Kang, N., Lovatt, D., Torres, A., Zhao, Z., Lin, J., et al. (2008). Connexin 43 hemichannels are permeable to ATP. J. Neurosci. 28, 4702–4711. doi: 10.1523/jneurosci.5048-07.2008
Kapellos, T. S., Taylor, L., Lee, H., Cowley, S. A., James, W. S., Iqbal, A. J., et al. (2016). A novel real time imaging platform to quantify macrophage phagocytosis. Biochem. Pharmacol. 116, 107–119. doi: 10.1016/j.bcp.2016.07.011
Kar, R., Riquelme, M. A., Werner, S., and Jiang, J. X. (2013). Connexin 43 channels protect osteocytes against oxidative stress-induced cell death. J. Bone. Miner. Res. 28, 1611–1621. doi: 10.1002/jbmr.1917
Kattoor, A. J., Pothineni, N. V. K., Palagiri, D., and Mehta, J. L. (2017). Oxidative stress in atherosclerosis. Curr. Atheroscler. Rep. 19:42.
Kim, I., Moon, S. O., Kim, S. H., Kim, H. J., Koh, Y. S., and Koh, G. Y. (2001). Vascular endothelial growth factor expression of intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1), and E-selectin through nuclear factor-kappa B activation in endothelial cells. J. Biol. Chem. 276, 7614–7620. doi: 10.1074/jbc.m009705200
Kim, K. S., Kim, J. E., Choi, K. J., Bae, S., and Kim, D. H. (2014). Characterization of DNA damage-induced cellular senescence by ionizing radiation in endothelial cells. Int. J. Radiat. Biol. 90, 71–80. doi: 10.3109/09553002.2014.859763
Kinlay, S., Behrendt, D., Wainstein, M., Beltrame, J., Fang, J. C., Creager, M. A., et al. (2001). Role of endothelin-1 in the active constriction of human atherosclerotic coronary arteries. Circulation 104, 1114–1118. doi: 10.1161/hc3501.095707
Kiyohara, H., Ishizaki, Y., Suzuki, Y., Katoh, H., Hamada, N., Ohno, T., et al. (2011). Radiation-induced ICAM-1 expression via TGF-β1 pathway on human umbilical vein endothelial cells, comparison between x-ray and carbon-ion beam irradiation. J. Radiat. Res. 52, 287–292. doi: 10.1269/jrr.10061
Kleiner, R. E., Verma, P., Molloy, K. R., Chait, B. T., and Kapoor, T. M. (2015). Chemical proteomics reveals a gammaH2AX-53BP1 interaction in the DNA damage response. Nat. Chem. Biol. 11, 807–814. doi: 10.1038/nchembio.1908
Koopman, W. J., Verkaart, S., Van Emst-De Vries, S. E., Grefte, S., Smeitink, J. A., and Willems, P. H. (2006). Simultaneous quantification of oxidative stress and cell spreading using 5-(and-6)-chloromethyl-2′,7′-dichlorofluorescein. Cytometry A 69, 1184–1192. doi: 10.1002/cyto.a.20348
Kreuzer, M., Auvinen, A., Cardis, E., Hall, J., Jourdain, J. R., Laurier, D., et al. (2015). Low-dose ionising radiation and cardiovascular diseases–Strategies for molecular epidemiological studies in Europe. Mutat. Res. Rev. Mutat. Res. 764, 90–100. doi: 10.1016/j.mrrev.2015.03.002
Kurz, D. J., Decary, S., Hong, Y., Trivier, E., Akhmedov, A., and Erusalimsky, J. D. (2004). Chronic oxidative stress compromises telomere integrity and accelerates the onset of senescence in human endothelial cells. J. Cell Sci. 117, 2417–2426. doi: 10.1242/jcs.01097
Kwak, B. R., Mulhaupt, F., Veillard, N., Gros, D. B., and Mach, F. (2002). Altered pattern of vascular connexin expression in atherosclerotic plaques. Arterioscler. Thromb. Vasc. Biol. 22, 225–230. doi: 10.1161/hq0102.104125
Kwak, B. R., Veillard, N., Pelli, G., Mulhaupt, F., James, R. W., Chanson, M., et al. (2003). Reduced connexin43 expression inhibits atherosclerotic lesion formation in low-density lipoprotein receptor-deficient mice. Circulation 107, 1033–1039. doi: 10.1161/01.cir.0000051364.70064.d1
Langley, R. E., Bump, E. A., Quartuccio, S. G., Medeiros, D., and Braunhut, S. J. (1997). Radiation-induced apoptosis in microvascular endothelial cells. Br. J. Cancer 75, 666–672. doi: 10.1038/bjc.1997.119
Lanza, V., Fadda, P., Iannone, C., and Negri, R. (2007). Low-dose ionizing radiation stimulates transcription and production of endothelin by human vein endothelial cells. Radiat. Res. 168, 193–198. doi: 10.1667/rr0780.1
Laurent, C., Voisin, P., and Pouget, J. P. (2006). DNA damage in cultured skin microvascular endothelial cells exposed to gamma rays and treated by the combination pentoxifylline and alpha-tocopherol. Int. J. Radiat. Biol. 82, 309–321. doi: 10.1080/09553000600733150
Leist, M., Single, B., Castoldi, A. F., Kuhnle, S., and Nicotera, P. (1997). Intracellular adenosine triphosphate (ATP) concentration: a switch in the decision between apoptosis and necrosis. J. Exp. Med. 185, 1481–1486. doi: 10.1084/jem.185.8.1481
Leybaert, L., Lampe, P. D., Dhein, S., Kwak, B. R., Ferdinandy, P., Beyer, E. C., et al. (2017). Connexins in cardiovascular and neurovascular health and disease: pharmacological implications. Pharmacol. Rev. 69, 396–478. doi: 10.1124/pr.115.012062
Li, F., Sugishita, K., Su, Z., Ueda, I., and Barry, W. H. (2001). Activation of connexin-43 hemichannels can elevate [Ca(2+)]i and [Na(+)]i in rabbit ventricular myocytes during metabolic inhibition. J. Mol. Cell Cardiol. 33, 2145–2155. doi: 10.1006/jmcc.2001.1477
Li, W., Bao, G., Chen, W., Qiang, X., Zhu, S., Wang, S., et al. (2018). Connexin 43 hemichannel as a novel mediator of sterile and infectious inflammatory diseases. Sci. Rep. 8:166.
Liao, J. K. (2013). Linking endothelial dysfunction with endothelial cell activation. J. Clin. Invest. 123, 540–541. doi: 10.1172/jci66843
Libby, P., Ridker, P. M., and Maseri, A. (2002). Inflammation and atherosclerosis. Circulation 105, 1135–1143.
Little, J. B. (2006). Cellular radiation effects and the bystander response. Mutat. Res. 597, 113–118. doi: 10.1016/j.mrfmmm.2005.12.001
Little, M. P. (2016). Radiation and circulatory disease. Mutat. Res. 770, 299–318. doi: 10.1016/j.mrrev.2016.07.008
Loomis, E. D., Sullivan, J. C., Osmond, D. A., Pollock, D. M., and Pollock, J. S. (2005). Endothelin mediates superoxide production and vasoconstriction through activation of NADPH oxidase and uncoupled nitric-oxide synthase in the rat aorta. J. Pharmacol. Exp. Ther. 315, 1058–1064. doi: 10.1124/jpet.105.091728
Lowe, D., and Raj, K. (2014). Premature aging induced by radiation exhibits pro-atherosclerotic effects mediated by epigenetic activation of CD44 expression. Aging Cell 13, 900–910. doi: 10.1111/acel.12253
Maatouk, L., Yi, C., Carrillo-De Sauvage, M. A., Compagnion, A. C., Hunot, S., Ezan, P., et al. (2018). Glucocorticoid receptor in astrocytes regulates midbrain dopamine neurodegeneration through connexin hemichannel activity. Cell Death Differ. 26, 580–596. doi: 10.1038/s41418-018-0150-3
Madan, R., Benson, R., Sharma, D. N., Julka, P. K., and Rath, G. K. (2015). Radiation induced heart disease: pathogenesis, management and review literature. J. Egpt. Natl. Cancer Inst. 27, 187–193. doi: 10.1016/j.jnci.2015.07.005
Marks, L. B., Yu, X., Prosnitz, R. G., Zhou, S. M., Hardenbergh, P. H., Blazing, M., et al. (2005). The incidence and functional consequences of RT-associated cardiac perfusion defects. Int. J. Radiat. Oncol. Biol. Phys. 63, 214–223. doi: 10.1016/j.ijrobp.2005.01.029
Mat Nor, N., Guo, C. X., Rupenthal, I. D., Chen, Y. S., Green, C. R., and Acosta, M. L. (2018). Sustained Connexin43 mimetic peptide release from loaded nanoparticles reduces retinal and choroidal photodamage. Invest. Ophthalmol. Vis. Sci. 59, 3682–3693.
Meunier, C., Wang, N., Yi, C., Dallerac, G., Ezan, P., Koulakoff, A., et al. (2017). Contribution of astroglial Cx43 hemichannels to the modulation of glutamatergic currents by d-serine in the mouse prefrontal cortex. J. Neurosci. 37, 9064–9075. doi: 10.1523/jneurosci.2204-16.2017
Moore, K., Ghatnekar, G., Gourdie, R. G., and Potts, J. D. (2014). Impact of the controlled release of a connexin 43 peptide on corneal wound closure in an STZ model of type I diabetes. PLoS ONE 9:e86570. doi: 10.1371/journal.pone.0086570
Morel, S. (2014). Multiple roles of connexins in atherosclerosis- and restenosis-induced vascular remodelling. J. Vasc. Res. 51, 149–161. doi: 10.1159/000362122
Mori, R., Power, K. T., Wang, C. M., Martin, P., and Becker, D. L. (2006). Acute downregulation of connexin43 at wound sites leads to a reduced inflammatory response, enhanced keratinocyte proliferation and wound fibroblast migration. J. Cell Sci. 119, 5193–5203. doi: 10.1242/jcs.03320
Mugisho, O. O., Green, C. R., Kho, D. T., Zhang, J., Graham, E. S., Acosta, M. L., et al. (2018). The inflammasome pathway is amplified and perpetuated in an autocrine manner through connexin43 hemichannel mediated ATP release. Biochim. Biophys. Acta 1862, 385–393. doi: 10.1016/j.bbagen.2017.11.015
Mugisho, O. O., Green, C. R., Squirrell, D. M., Bould, S., Danesh-Meyer, H. V., Zhang, J., et al. (2019). Connexin43 hemichannel block protects against the development of diabetic retinopathy signs in a mouse model of the disease. J. Mol. Med. (Berl.) 97, 215–229. doi: 10.1007/s00109-018-1727-5
Muschter, D., Geyer, F., Bauer, R., Ettl, T., Schreml, S., and Haubner, F. (2018). A comparison of cell survival and heat shock protein expression after radiation in normal dermal fibroblasts, microvascular endothelial cells, and different head and neck squamous carcinoma cell lines. Clin. Oral Investig. 22, 2251–2262. doi: 10.1007/s00784-017-2323-8
Napoli, C., De Nigris, F., Williams-Ignarro, S., Pignalosa, O., Sica, V., and Ignarro, L. J. (2006). Nitric oxide and atherosclerosis: an update. Nitric Oxide 15, 265–279. doi: 10.1016/j.niox.2006.03.011
O’Carroll, S. J., Gorrie, C. A., Velamoor, S., Green, C. R., and Nicholson, L. F. (2013). Connexin43 mimetic peptide is neuroprotective and improves function following spinal cord injury. Neurosci. Res. 75, 256–267. doi: 10.1016/j.neures.2013.01.004
Oh, C. W., Bump, E. A., Kim, J. S., Janigro, D., and Mayberg, M. R. (2001). Induction of a senescence-like phenotype in bovine aortic endothelial cells by ionizing radiation. Radiat. Res. 156, 232–240. doi: 10.1667/0033-7587(2001)156[0232:ioaslp]2.0.co;2
Ohshima, Y., Tsukimoto, M., Harada, H., and Kojima, S. (2012). Involvement of connexin43 hemichannel in ATP release after γ-irradiation. J. Radiat. Res. 53, 551–557. doi: 10.1093/jrr/rrs014
Okamoto, T., Kawamoto, E., Takagi, Y., Akita, N., Hayashi, T., Park, E. J., et al. (2017). Gap junction-mediated regulation of endothelial cellular stiffness. Sci. Rep. 7:6134.
Orellana, J. A., Diaz, E., Schalper, K. A., Vargas, A. A., Bennett, M. V., and Saez, J. C. (2011). Cation permeation through connexin 43 hemichannels is cooperative, competitive and saturable with parameters depending on the permeant species. Biochem. Biophys. Res. Commun. 409, 603–609. doi: 10.1016/j.bbrc.2011.05.031
Orellana, J. A., Saez, P. J., Shoji, K. F., Schalper, K. A., Palacios-Prado, N., Velarde, V., et al. (2009). Modulation of brain hemichannels and gap junction channels by pro-inflammatory agents and their possible role in neurodegeneration. Antioxid. Redox. Signal. 11, 369–399. doi: 10.1089/ars.2008.2130
Park, M. T., Oh, E. T., Song, M. J., Lee, H., and Park, H. J. (2012). Radio-sensitivities and angiogenic signaling pathways of irradiated normal endothelial cells derived from diverse human organs. J. Radiat. Res. 53, 570–580. doi: 10.1093/jrr/rrs011
Pfenniger, A., Chanson, M., and Kwak, B. R. (2013). Connexins in atherosclerosis. Biochim. Biophys. Acta 1828, 157–166.
Ponsaerts, R., De Vuyst, E., Retamal, M., D’hondt, C., Vermeire, D., Wang, N., et al. (2010). Intramolecular loop/tail interactions are essential for connexin 43-hemichannel activity. FASEB J. 24, 4378–4395. doi: 10.1096/fj.09-153007
Ramachandran, S., Xie, L. H., John, S. A., Subramaniam, S., and Lal, R. (2007). A novel role for connexin hemichannel in oxidative stress and smoking-induced cell injury. PLoS ONE 2:e712. doi: 10.1371/journal.pone.0000712
Ramadan, R., Vromans, E., Anang, D. C., Decrock, E., Mysara, M., Monsieurs, P., et al. (2019). Single and fractionated ionizing radiation induce alterations in endothelial connexin expression and channel function. Sci. Rep. 9:4643.
Retamal, M. A., Froger, N., Palacios-Prado, N., Ezan, P., Saez, P. J., Saez, J. C., et al. (2007). Cx43 hemichannels and gap junction channels in astrocytes are regulated oppositely by proinflammatory cytokines released from activated microglia. J. Neurosci. 27, 13781–13792. doi: 10.1523/jneurosci.2042-07.2007
Riquelme, M. A., and Jiang, J. X. (2013). Elevated intracellular Ca(2+) signals by oxidative stress activate connexin 43 hemichannels in osteocytes. Bone Res. 1, 355–361. doi: 10.4248/br201304006
Robbins, M. E., and Zhao, W. (2004). Chronic oxidative stress and radiation-induced late normal tissue injury: a review. Int. J. Radiat. Biol. 80, 251–259. doi: 10.1080/09553000410001692726
Rodriguez-Sinovas, A., Cabestrero, A., Lopez, D., Torre, I., Morente, M., Abellan, A., et al. (2007). The modulatory effects of connexin 43 on cell death/survival beyond cell coupling. Prog. Biophys. Mol. Biol. 94, 219–232. doi: 10.1016/j.pbiomolbio.2007.03.003
Roedel, F., Kley, N., Beuscher, H. U., Hildebrandt, G., Keilholz, L., Kern, P., et al. (2002). Anti-inflammatory effect of low-dose X-irradiation and the involvement of a TGF-beta1-induced down-regulation of leukocyte/endothelial cell adhesion. Int. J. Radiat. Biol. 78, 711–719. doi: 10.1080/09553000210137671
Rombouts, C., Aerts, A., Beck, M., De Vos, W. H., Van Oostveldt, P., Benotmane, M. A., et al. (2013). Differential response to acute low dose radiation in primary and immortalized endothelial cells. Int. J. Radiat. Biol. 89, 841–850. doi: 10.3109/09553002.2013.806831
Rousseau, M., Gaugler, M. H., Rodallec, A., Bonnaud, S., Paris, F., and Corre, I. (2011). RhoA GTPase regulates radiation-induced alterations in endothelial cell adhesion and migration. Biochem. Biophys. Res. Commun. 414, 750–755. doi: 10.1016/j.bbrc.2011.09.150
Saez, J. C., Contreras-Duarte, S., Gomez, G. I., Labra, V. C., Santibanez, C. A., Gajardo-Gomez, R., et al. (2018). Connexin 43 hemichannel activity promoted by pro-inflammatory cytokines and high glucose alters endothelial cell function. Front. Immunol. 9:1899. doi: 10.3389/fimmu.2018.01899
Saez, J. C., Schalper, K. A., Retamal, M. A., Orellana, J. A., Shoji, K. F., and Bennett, M. V. (2010). Cell membrane permeabilization via connexin hemichannels in living and dying cells. Exp. Cell Res. 316, 2377–2389. doi: 10.1016/j.yexcr.2010.05.026
Scharpfenecker, M., Kruse, J. J., Sprong, D., Russell, N. S., Ten Dijke, P., and Stewart, F. A. (2009). Ionizing radiation shifts the PAI-1/ID-1 balance and activates notch signaling in endothelial cells. Int. J. Radiat. Oncol. Biol. Phys. 73, 506–513. doi: 10.1016/j.ijrobp.2008.09.052
Schindelin, J., Arganda-Carreras, I., Frise, E., Kaynig, V., Longair, M., Pietzsch, T., et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. doi: 10.1038/nmeth.2019
Schramm, A., Matusik, P., Osmenda, G., and Guzik, T. J. (2012). Targeting NADPH oxidases in vascular pharmacology. Vascul. Pharmacol. 56, 216–231. doi: 10.1016/j.vph.2012.02.012
Schuett, H., Luchtefeld, M., Grothusen, C., Grote, K., and Schieffer, B. (2009). How much is too much? Interleukin-6 and its signalling in atherosclerosis. Thromb. Haemost. 102, 215–222. doi: 10.1160/th09-05-0297
Shimizu, Y., Kodama, K., Nishi, N., Kasagi, F., Suyama, A., Soda, M., et al. (2010). Radiation exposure and circulatory disease risk: hiroshima and nagasaki atomic bomb survivor data, 1950-2003. BMJ 340:b5349. doi: 10.1136/bmj.b5349
Sievert, W., Trott, K. R., Azimzadeh, O., Tapio, S., Zitzelsberger, H., and Multhoff, G. (2015). Late proliferating and inflammatory effects on murine microvascular heart and lung endothelial cells after irradiation. Radiother. Oncol. 117, 376–381. doi: 10.1016/j.radonc.2015.07.029
Soucy, K. G., Lim, H. K., Kim, J. H., Oh, Y., Attarzadeh, D. O., Sevinc, B., et al. (2011). HZE (5)(6)Fe-ion irradiation induces endothelial dysfunction in rat aorta: role of xanthine oxidase. Radiat. Res. 176, 474–485. doi: 10.1667/rr2598.1
Stewart, F. A., Heeneman, S., Te Poele, J., Kruse, J., Russell, N. S., Gijbels, M., et al. (2006). Ionizing radiation accelerates the development of atherosclerotic lesions in ApoE-/- mice and predisposes to an inflammatory plaque phenotype prone to hemorrhage. Am. J. Pathol. 168, 649–658. doi: 10.2353/ajpath.2006.050409
Sun, W., Jiao, Y., Cui, B., Gao, X., Xia, Y., and Zhao, Y. (2013). Immune complexes activate human endothelium involving the cell-signaling HMGB1-RAGE axis in the pathogenesis of lupus vasculitis. Lab. Invest. 93, 626–638. doi: 10.1038/labinvest.2013.61
Szumiel, I. (2015). Ionizing radiation-induced oxidative stress, epigenetic changes and genomic instability: the pivotal role of mitochondria. Int. J. Radiat. Biol. 91, 1–12. doi: 10.3109/09553002.2014.934929
Tapio, S. (2016). Pathology and biology of radiation-induced cardiac disease. J. Radiat. Res. 57, 439–448. doi: 10.1093/jrr/rrw064
Tchkonia, T., Zhu, Y., Van Deursen, J., Campisi, J., and Kirkland, J. L. (2013). Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J. Clin. Invest. 123, 966–972. doi: 10.1172/jci64098
Tian, X. L., and Li, Y. (2014). Endothelial cell senescence and age-related vascular diseases. J. Genet. Genomics 41, 485–495. doi: 10.1016/j.jgg.2014.08.001
Tonkin, R. S., Bowles, C., Perera, C. J., Keating, B. A., Makker, P. G. S., Duffy, S. S., et al. (2018). Attenuation of mechanical pain hypersensitivity by treatment with Peptide5, a connexin-43 mimetic peptide, involves inhibition of NLRP3 inflammasome in nerve-injured mice. Exp. Neurol. 300, 1–12. doi: 10.1016/j.expneurol.2017.10.016
Tsuchida, S., Arai, Y., Kishida, T., Takahashi, K. A., Honjo, K., Terauchi, R., et al. (2013). Silencing the expression of connexin 43 decreases inflammation and joint destruction in experimental arthritis. J. Orthop. Res. 31, 525–530. doi: 10.1002/jor.22263
Ungvari, Z., Podlutsky, A., Sosnowska, D., Tucsek, Z., Toth, P., Deak, F., et al. (2013). Ionizing radiation promotes the acquisition of a senescence-associated secretory phenotype and impairs angiogenic capacity in cerebromicrovascular endothelial cells: role of increased DNA damage and decreased DNA repair capacity in microvascular radiosensitivity. J. Gerontol. A Biol. Sci. Med. Sci. 68, 1443–1457. doi: 10.1093/gerona/glt057
Van Der Meeren, A., Squiban, C., Gourmelon, P., Lafont, H., and Gaugler, M. H. (1999). Differential regulation by IL-4 and IL-10 of radiation-induced IL-6 and IL-8 production and ICAM-1 expression by human endothelial cells. Cytokine 11, 831–838. doi: 10.1006/cyto.1999.0497
Van Nimwegen, F. A., Schaapveld, M., Janus, C. P., Krol, A. D., Petersen, E. J., Raemaekers, J. M., et al. (2015). Cardiovascular disease after Hodgkin lymphoma treatment: 40-year disease risk. JAMA Int. Med. 175, 1007–1017.
Walrave, L., Pierre, A., Albertini, G., Aourz, N., De Bundel, D., Van Eeckhaut, A., et al. (2018). Inhibition of astroglial connexin43 hemichannels with TAT-Gap19 exerts anticonvulsant effects in rodents. Glia 66, 1788–1804. doi: 10.1002/glia.23341
Wang, H., Adhikari, S., Butler, B. E., Pandita, T. K., Mitra, S., and Hegde, M. L. (2014). A perspective on chromosomal double strand break markers in mammalian cells. Jacobs J. Radiat. Oncol. 1:003.
Wang, N., De Bock, M., Decrock, E., Bol, M., Gadicherla, A., Vinken, M., et al. (2013a). Paracrine signaling through plasma membrane hemichannels. Biochim. Biophys. Acta 1828, 35–50. doi: 10.1016/j.bbamem.2012.07.002
Wang, N., De Vuyst, E., Ponsaerts, R., Boengler, K., Palacios-Prado, N., Wauman, J., et al. (2013b). Selective inhibition of Cx43 hemichannels by Gap19 and its impact on myocardial ischemia/reperfusion injury. Basic Res. Cardiol. 108:309.
Willebrords, J., Cogliati, B., Pereira, I. V. A., Da Silva, T. C., Crespo Yanguas, S., Maes, M., et al. (2017). Inhibition of connexin hemichannels alleviates non-alcoholic steatohepatitis in mice. Sci. Rep. 7:8268.
Wong, C. W., Burger, F., Pelli, G., Mach, F., and Kwak, B. R. (2003). Dual benefit of reduced Cx43 on atherosclerosis in LDL receptor-deficient mice. Cell Commun. Adhes. 10, 395–400. doi: 10.1080/714040458
Xue, W., Zender, L., Miething, C., Dickins, R. A., Hernando, E., Krizhanovsky, V., et al. (2007). Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 445, 656–660. doi: 10.1038/nature05529
Yamada, M., Naito, K., Kasagi, F., Masunari, N., and Suzuki, G. (2005). Prevalence of atherosclerosis in relation to atomic bomb radiation exposure: an RERF Adult Health Study. Int. J. Radiat. Biol. 81, 821–826. doi: 10.1080/09553000600555504
Yamamori, T., Yasui, H., Yamazumi, M., Wada, Y., Nakamura, Y., Nakamura, H., et al. (2012). Ionizing radiation induces mitochondrial reactive oxygen species production accompanied by upregulation of mitochondrial electron transport chain function and mitochondrial content under control of the cell cycle checkpoint. Free Radic. Biol. Med. 53, 260–270. doi: 10.1016/j.freeradbiomed.2012.04.033
Yan, Y., Wei, C. L., Zhang, W. R., Cheng, H. P., and Liu, J. (2006). Cross-talk between calcium and reactive oxygen species signaling. Acta Pharmacol. Sin. 27, 821–826. doi: 10.1111/j.1745-7254.2006.00390.x
Yang, T. J., and Ho, A. Y. (2013). Radiation therapy in the management of breast cancer. Surg. Clin. N. Am. 93, 455–471.
Yentrapalli, R., Azimzadeh, O., Barjaktarovic, Z., Sarioglu, H., Wojcik, A., Harms-Ringdahl, M., et al. (2013a). Quantitative proteomic analysis reveals induction of premature senescence in human umbilical vein endothelial cells exposed to chronic low-dose rate gamma radiation. Proteomics 13, 1096–1107. doi: 10.1002/pmic.201200463
Yentrapalli, R., Azimzadeh, O., Sriharshan, A., Malinowsky, K., Merl, J., Wojcik, A., et al. (2013b). The PI3K/Akt/mTOR pathway is implicated in the premature senescence of primary human endothelial cells exposed to chronic radiation. PLoS ONE 8:e70024. doi: 10.1371/journal.pone.0070024
Yuan, D., Sun, G., Zhang, R., Luo, C., Ge, M., Luo, G., et al. (2015). Connexin 43 expressed in endothelial cells modulates monocyte-endothelial adhesion by regulating cell adhesion proteins. Mol. Med. Rep. 12, 7146–7152. doi: 10.3892/mmr.2015.4273
Keywords: atherosclerosis, endothelial damage, ionizing radiation, connexin43 hemichannels, TAT-Gap19
Citation: Ramadan R, Vromans E, Anang DC, Goetschalckx I, Hoorelbeke D, Decrock E, Baatout S, Leybaert L and Aerts A (2020) Connexin43 Hemichannel Targeting With TAT-Gap19 Alleviates Radiation-Induced Endothelial Cell Damage. Front. Pharmacol. 11:212. doi: 10.3389/fphar.2020.00212
Received: 10 December 2019; Accepted: 14 February 2020;
Published: 05 March 2020.
Edited by:
Aida Salameh, Leipzig University, GermanyReviewed by:
Juan Andrés Orellana, Pontifical Catholic University of Chile, ChileManuel Antonio Riquelme, The University of Texas Health Science Center at San Antonio, United States
Copyright © 2020 Ramadan, Vromans, Anang, Goetschalckx, Hoorelbeke, Decrock, Baatout, Leybaert and Aerts. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: An Aerts, an.aerts@sckcen.be
†These authors share senior authorship