 Gemma Vilahur
Gemma Vilahur Lina Badimon
Lina Badimon- 1Cardiovascular Research Center, CSIC-ICCC, Hospital de la Santa Creu i Sant Pau, Institut d'Investigació Biomèdica Sant Pau, Barcelona, Spain
- 2Cardiovascular Research Chair, Universitat Autònoma de Barcelona, Barcelona, Spain
Recent data have indicated that the myocardium may act as an immune organ initiating cardiac innate immune response and inflammation. It has been suggested that activation of the immune system occurs upon the interaction of damage-associated molecular patterns (DAMPs) generated and released during ischemic damage with pattern recognition receptors (Toll like receptors; TLR) present in cardiac cells. Among TLRs, TLR4, and TLR2 are the ones mostly expressed in cardiac tissue. Whereas TLR4 has shown to play a detrimental role in myocardial ischemia/reperfusion (I/R) injury, the effect elicited by TLR2 activation remains controversial. Once activated, TLR signaling may occur via the Myd88- and Trif- dependent pathways leading to NFκB and IFN-3 activation, respectively, and subsequent stimulation of pro-inflammatory and immunomodulatory cytokine gene expression. Cytokine release contributes to neutrophils activation, recruitment, adhesion and infiltration to the site of cardiac injury further perpetuating the inflammatory process. This mini-review will focus on the current knowledge regarding the role of the heart in inducing and coordinating the innate inflammatory response via the TLR signaling pathway in myocardial I/R injury.
Activation of Myocardial TLR in Ischemia/Reperfusion
Activation of the innate immunity is an essential component of the acute inflammatory response in the setting of ischemia/reperfusion (I/R). It is widely recognized the key role of neutrophils, monocytes, macrophages and dendritic cells in innate immunity. However, recent experimental lines of evidence have suggested that ischemic cardiomyocytes can also contribute to the innate immune response besides being a target organ. Activation of the immune response mainly occurs when “danger” signals released from the damaged cardiomyocytes [the so called damage-associated molecular patterns (DAMPs)] interact with pattern recognition receptors (PRRs). Among PRRs, Toll-Like Receptors (TLR), originally identified 20 years ago for its role in the embryonic development of the fruit fly Drosophila melanogaster (Anderson et al., 1985), have been later recognized as a key elements of the immune system (Lemaitre et al., 1996). Ten TLR have been identified so far in humans six of which are located in the cell surface and are activated by a variety of exogenous/endogenous ligands (TLR1, TLR 2, TLR4, TLR5, TLR6, and TLR10) and four are expressed intracellularly (TLR3, TLR7, TLR8, and TLR9) and are generally recognized by nucleic acid structures (Wang et al., 2010; Mann, 2011). Almost all cells of the heart have shown to express TLR (TLR 2, 3, 4, and 6 are found in cardiomyocytes, and TLR1 through 6 are found in the smooth muscle and endothelial cells) (Frantz et al., 2007). The discovery of the notably contribution of TLR2 and TLR4 in the development of atherosclerotic lesions and vascular remodeling (Kiechl et al., 2002; Lin et al., 2009) and their abundance in adult human hearts (Lin and Knowlton, 2014) frosted the interest in deciphering their potential role in the setting of myocardial I/R (Frantz et al., 1999). Although the mechanisms involved in TLR 2 and 4 activation in myocardial I/R remain to be fully described, a variety of endogenous molecules have been identified to act as potential endogenous DAMPS. In one hand, myocardial cell necrosis triggered during ischemia results in the release of necrotic products and cellular constituents such as heat shock proteins (HSP) and nuclear DNA-binding protein high mobility group box (HMGB-1) capable of activating the TLR2 and TLR4 signaling. Dynamic alterations of extra cellular matrix (ECM) resulting from I/R have also shown to generate DAMPs molecules. In this regard, either breakdown products of the cardiac ECM or de novo synthesized matrix molecules may function as endogenous ligands (Lin and Knowlton, 2014). Reactive oxygen species have also demonstrated to activate cytokine production through TLR4-dependent mechanisms, although the molecules involved remain unknown (Flohe et al., 1997; Ha et al., 2011). Finally, physical alterations of the receptor-plasma membrane environment are thought to activate TLRs in the absence of any cellular-/ECM-derived ligand (Figure 1) (Scaffidi et al., 2002; Scheibner et al., 2006). Once activated, signal transduction of TLR2 and/or 4 may take place via two main pathways, the Myd88- and TRIF- dependent pathway, eventually leading to cytokine production.
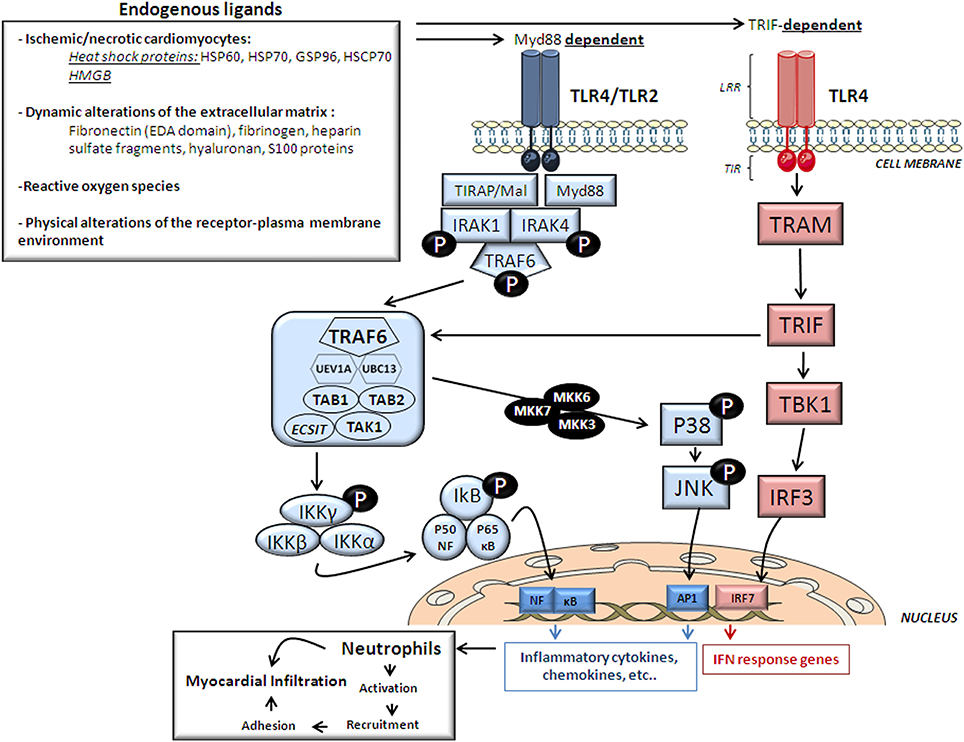
Figure 1. Cardiac TLR2/4 structure, endogenous ligands and signaling. This figure depicts the endogenous ligands released upon ischemia and reperfusion that trigger signaling pathways activated by the two most important cardiac TLR receptors, TLR2 and TLR4 and TLR structure. HMGB, high mobility group box; HSP, heat-shock protein; HSCP, heat shock cognate protein. LRR, extracellular C-terminal leucine-rich repeat (ligand recognition domain); TIR-domain, N-terminal cytoplasmic toll/interleukin receptor homologous signaling domain; IRAK, interleukin -1 receptor-associated kinase; TRAF6, tumor necrosis factor-receptor-associated factor-6; TAK1, TGF-β-Activated Kinase-1; TAB1, TAK1-Binding Protein-1; TAB2, TAK1-Binding Protein-2; UBC13, ubiquitin-conjugating enzyme-13; UEV1A, ubiquitin-conjugating enzyme E2-variant-1; IKK, Inhibitor of κ Light Polypeptide Gene Enhancer in B-Cells Kinase; NF-κB, nuclear transcription factor- κB; MKK, mitogen-activated protein kinase; TRIF, TIR domain-containing adaptor protein inducing type 1 interferons; TRAM, Trif-related adaptor molecule; TBK1, TANK Binding Kinase-1; IRF, interferon regulatory factor. AP-1, transcription factor complex-1.
TLR Signal Transduction
TLR are type-I transmembrane proteins composed of 3 structural domains: an extracellular C-terminal leucine-rich repeat (LRR) as ligand recognition domain, a central transmembrane domain, and a cytoplasmic domain similar to the IL-1 receptor (the toll/IL-1 receptor; TIR). Whereas TLR4 acts as homodimer, TLR2 forms heterodimers with TLR1 and TLR6 (Wang et al., 2010). TLR dimerization leads to changes in the conformation of the cytoplasmic TIR domain which subsequently facilitates the recruitment of adaptors that initiate TLRs downstream signaling. Depending on the usage of different adaptor molecules, TLRs signaling if divided in two major pathways, a MyD88-dependent pathway and a TRIF-dependent pathway.
MyD88-Dependent Pathway
Myd88 adaptor protein requires the recruitment of a TIR domain-containing adaptor protein (TIRAP/Mal) to initiate TLR4/2-related signaling. As shown in Figure 1 once stimulated, the MyD88/TIRAP complex recruits and activates the downstream interleukin -1 receptor-associated kinase 4 (IRAK4) which induces IRAK1 phosphorylation and in turn recruits TNF-Receptor-Associated Factor-6 (TRAF6) by associating with phosphorylated IRAK1. Hereafter, phosphorylated TRAF6 then dissociates from the receptor and forms a complex with the linker molecules transforming growth factor β-activated kinase (TAK)-1, TAK1-Binding Protein-1 (TAB1) and TAB2. TRAF6, TAK1, TAB1, and TAB2 associate with the Ubiquitin ligases UBC13 (Ubiquitin-Conjugating Enzyme-13) and UEV1A (Ubiquitin-conjugating Enzyme E2-Variant-1). TAK1 is then activated and phosphorylates the IKK complex (Inhibitor of κ Light Polypeptide Gene Enhancer in B-Cells Kinase; IKK-α, IKK-β, and IKK-γ) and p38 Kinases by activating MKK3 (Mitogen-Activated Protein Kinase Kinase-3), MKK6 and MKK7. The IKK complex then phosphorylates I-κB which leads to its ubiquitylation and subsequent degradation. This allows NF-κB to translocate to the nucleus which brings about the production of pro-inflammatory cytokines and expression of co-stimulatory molecules (Ha et al., 2011). On the other hand, p38 Kinases activates c-Jun Kinase (JNKs) which then enter the nucleus and activates the transcription factor complex (AP1) which also stimulates the expression of pro-inflammatory cytokine genes (Figure 1) (Chao, 2009).
TRIF-Dependent Pathway
TLR4 can also signal through a TRIF-dependent pathway (also known as the MyD88-independent pathway; Figure 1). In the TRIF-dependent pathway TLR4 may signal via TRIF-related adaptor molecule (TRAM) and TRIF which activates TBK1 (TANK Binding Kinase-1) eventually activating interferon-regulatory factor-3 (IRF3). This pathway may also signal through NF-κB signaling through TRAF6 recruitment. Activation of the TRIF-dependent pathway induces IFNβ and IFN-inducible genes including a variety of cytokines (Figure 1) (Chao, 2009).
All TLRs exert their action through the Myd88-dependent pathway except for TLR3 and partially for TLR4 which exert their action through the TRIF-dependent pathway. In fact, TRIF-knock-out mice has shown impaired cytokine production despite IRAK-1 phosphorylation and NF-κB activation suggesting that TLR4 requires both Myd88- and TRIF-dependent pathways to exert its action.
Myocardial TLR4 a New Player in I/R Injury
Oyama et al. were the first to report a detrimental role for TLR4 in myocardial I/R injury (Oyama et al., 2004). Since then, several investigations have corroborated that TLR4 deficient mice show suppressed inflammatory response (lower NF-κB activation and reduced TNFα and IL1β levels), decreased cardiac inflammatory infiltration and smaller infarctions as compared with their wild-type littermates (Chong et al., 2004; Oyama et al., 2004). Moreover, administration of a specific TLR4 antagonist has shown to decrease NF-κB activation and subsequent pro-inflammatory cytokine release limiting the size of infarction as compared to non-treated animals (Kim et al., 2007a). However, inconsistent results have been reported regarding the role of TLR4 in cardiac recovery after I/R, some studies showing improvement (Kim et al., 2007b) and others no effect (Fallach et al., 2010).
A short-period of coronary ischemia (5 min) has not shown to trigger an inflammatory reaction (Bolli and Marban, 1999). On the contrary, brief ischemic periods implemented before an ischemic insult have shown to confer protection against prolonged ischemia (i.e., ischemic pre-conditioning) (Murry et al., 1986). However, sustained ischemia has shown to result in a detrimental inflammatory response. In this regard, we have reported in a preclinical model of closed-chest coronary balloon occlusion that 30 min of ischemia suffice to enhance the inflammatory potential of circulating leukocytes and increase cytokines release (Vilahur et al., 2009). Interestingly, such systemic inflammatory response was associated with a marked upregulation of both myocardial TLR-4- MyD88 -dependent and -TRIF-dependent signaling pathways in the ischemic myocardium leading to NF-κB nucleus translocation and IRF3 activation, respectively (Vilahur et al., 2011). These observations support a relatively prompt activation of the cardiac innate immune response upon an ischemic insult that rapidly translates into a pro-inflammatory systemic response through TLR4/cytokine networks. In addition, these data also suggests that timely interception of TLR4 might be critical to prevent adverse myocardial remodeling post-I/R (Vilahur et al., 2011). Once activated, myocardial TLR4-related inflammatory response stimulates the homing and recruitment of neutrophils to the site of cardiac injury (Oyama et al., 2004). Neutrophils recruitment and further infiltration may then contribute to the detected raise in cardiac TLR-4 expression detected up to 1 week after reperfusion (Vilahur et al., 2011). Although the relative role of neutrophils TLR4 and myocardial TLR4 in I/R-induced neutrophils cardiac infiltration has yet to be clearly defined, an interesting study in Langendorff perfused heart supported that functional TLR4 in the myocardium is required for neutrophils infiltration after global I/R. Besides, it has been suggested that myocardial TLR4 mediates cardiac chemokine response through a mechanisms involving HSC70, released during ischemia (Ao et al., 2009). Neutrophils are the major leukocytes found in I/R injury and not only are target cells for the cytokines released by the ischemic-reperfused myocardium but also contribute to the progression of I/R tissue damage. Released pro-inflammatory cytokines from the ischemic reperfused myocardium activates both neutrophils and cardiomyocytes leading to MAC-1 and intercellular adhesion molecule-1 (ICAM1) expression, respectively. MAC-1/ICAM1 interaction favors neutrophils adherence to cardiomyocytes which, in turn, accelerates and perpetuates tissue injury via reactive oxygen species (ROS) generation and pro-inflammatory cytokine release by both cell types triggering a positive feedback loop (Arslan et al., 2008).
The Controversial Role of TLR2 in Myocardial I/R Injury
The role of myocardial TLR2 in the setting of I/R has been poorly investigated. In fact, it still remains uncertain whether it has a detrimental role in I/R injury similar to that of TLR4 or exerts beneficial effects. Shishido et al. (2003) reported that infarct size and the degree of inflammatory cell infiltration in the infarcted area were similar between wild type and TLR-2 knock-out mice. However, longer time-period analysis (up to 4 weeks) revealed that TLR-2 deficient mice showed lower fibrosis in the non-infarcted myocardium and improved left-ventricular remodeling parameters, as assessed by echocardiography, suggesting a key role for TLR2 in the tissue repair process after reperfusion (i.e., remodeling phase). Yet, it is unknown the relative contribution of TLR2 expressing inflammatory cells and myocardial-related TLR2 in myocardial I/R injury. On the other hand, studies in isolated hearts and infarcted mice have shown, by using specific synthetic TLR2 ligands, that TLR2 is required for ischemic pre-conditioning-induced left ventricle functional improvement and infarct size reduction and cardiac recovery, respectively (Dong et al., 2010; Mersmann et al., 2010).
Concluding Remarks
Innate immunity triggers the initial inflammatory response upon myocardial ischemia in part by the TLR signaling. Several lines of evidence suggest the active involvement of the heart in inducing and regulating the innate inflammatory response in the progression of myocardial I/R injury. Cardiac myocytes exhibit properties associated with the innate immune response, they are able to generate DAMPs and express TLRs. Although the mechanisms by which TLRs exert their detrimental cardiovascular effects in the setting of I/R are incompletely understood, during the last years emerging evidence has indicated that cardiac TLR4 contribute to cytokine release and neutrophils activation and recruitment to the site of injury (Aikawa et al., 2001). In contrast, the role of TLR2 still remains unclear. Further research is needed to explore the pathways and mechanisms by which the cardiac innate response is activated following tissue injury. Development of selective compounds capable to inhibit the TLR signaling may provide important insights in this regard. Yet, it is important to consider that although excessive pro-inflammatory cytokine production exerts myocardial detrimental effects in the ischemic reperfused myocardium, cytokines also drive the tissue repair process within the heart.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The work presented in this review has been partially financed by PNS 2012-40208 (to Gemma Vilahur) and PNS 2013-42962-R (to Lina Badimon) from the Spanish Ministry of Science and Innovation and Red Investigación Cardiovascular RD12/0042/0027 (Spanish Cardiovascular Network) from Instituto Salud Carlos III (to Lina Badimon). We thank Fundacion Jesus Serra, Barcelona for continuous support. Gemma Vilahur is a “Ramon y Cajal” scientist funded by the Spanish Ministry of Science and Innovation (MICINN).
References
Aikawa, R., Nagai, T., Tanaka, M., Zou, Y., Ishihara, T., Takano, H., et al. (2001). Reactive oxygen species in mechanical stress-induced cardiac hypertrophy. Biochem. Biophys. Res. Commun. 289, 901–907. doi: 10.1006/bbrc.2001.6068
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Anderson, K. V., Bokla, L., and Nusslein-Volhard, C. (1985). Establishment of dorsal-ventral polarity in the Drosophila embryo: the induction of polarity by the Toll gene product. Cell 42, 791–798. doi: 10.1016/0092-8674(85)90275-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ao, L., Zou, N., Cleveland, J. C. Jr., Fullerton, D. A., and Meng, X. (2009). Myocardial TLR4 is a determinant of neutrophil infiltration after global myocardial ischemia: mediating KC and MCP-1 expression induced by extracellular HSC70. Am. J. Physiol. Heart Circ. Physiol. 297, H21–H28. doi: 10.1152/ajpheart.00292.2009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Arslan, F., de Kleijn, D. P., Timmers, L., Doevendans, P. A., and Pasterkamp, G. (2008). Bridging innate immunity and myocardial ischemia/reperfusion injury: the search for therapeutic targets. Curr. Pharm. Des. 14, 1205–1216. doi: 10.2174/138161208784246090
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bolli, R., and Marban, E. (1999). Molecular and cellular mechanisms of myocardial stunning. Physiol. Rev. 79, 609–634.
Chao, W. (2009). Toll-like receptor signaling: a critical modulator of cell survival and ischemic injury in the heart. Am. J. Physiol. Heart Circ. Physiol. 296, H1–H12. doi: 10.1152/ajpheart.00995.2008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chong, A. J., Shimamoto, A., Hampton, C. R., Takayama, H., Spring, D. J., Rothnie, C. L., et al. (2004). Toll-like receptor 4 mediates ischemia/reperfusion injury of the heart. J. Thorac. Cardiovasc. Surg. 128, 170–179. doi: 10.1016/j.jtcvs.2003.11.036
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dong, J. W., Vallejo, J. G., Tzeng, H. P., Thomas, J. A., Mann, D. L. (2010). Innate immunity mediates myocardial preconditioning through Toll-like receptor 2 and TIRAP-dependent signaling pathways. Am. J. Physiol. Heart Circ. Physiol. 298, H1079–H1087. doi: 10.1152/ajpheart.00306.2009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fallach, R., Shainberg, A., Avlas, O., Fainblut, M., Chepurko, Y., Porat, E., et al. (2010). Cardiomyocyte Toll-like receptor 4 is involved in heart dysfunction following septic shock or myocardial ischemia. J. Mol. Cell. Cardiol. 48, 1236–1244. doi: 10.1016/j.yjmcc.2010.02.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Flohe, L., Brigelius-Flohe, R., Saliou, C., Traber, M. G., and Packer, L. (1997). Redox regulation of NF-kappa B activation. Free Radic. Biol. Med. 22, 1115–1126. doi: 10.1016/S0891-5849(96)00501-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Frantz, S., Ertl, G., and Bauersachs, J. (2007). Mechanisms of disease: toll-like receptors in cardiovascular disease. Nat. Clin. Pract. Cardiovasc. Med. 4, 444–454. doi: 10.1038/ncpcardio0938
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Frantz, S., Kobzik, L., Kim, Y. D., Fukazawa, R., Medzhitov, R., Lee, R. T., et al. (1999). Toll4 (TLR4) expression in cardiac myocytes in normal and failing myocardium. J. Clin. Invest. 104, 271–280. doi: 10.1172/JCI6709
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ha, T., Liu, L., Kelley, J., Kao, R., Williams, D., and Li, C. (2011). Toll-like receptors: new players in myocardial ischemia/reperfusion injury. Antioxid. Redox Signal. 15, 1875–1893. doi: 10.1089/ars.2010.3723
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kiechl, S., Lorenz, E., Reindl, M., Wiedermann, C. J., Oberhollenzer, F., Bonora, E., et al. (2002). Toll-like receptor 4 polymorphisms and atherogenesis. N. Engl. J. Med. 347, 185–192. doi: 10.1056/NEJMoa012673
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kim, H. M., Park, B. S., Kim, J. I., Kim, S. E., Lee, J., Oh, S. C., et al. (2007a). Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist Eritoran. Cell 130, 906–917. doi: 10.1016/j.cell.2007.08.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kim, S. C., Ghanem, A., Stapel, H., Tiemann, K., Knuefermann, P., Hoeft, A., et al. (2007b). Toll-like receptor 4 deficiency: smaller infarcts, but no gain in function. BMC Physiol. 7:5. doi: 10.1186/1472-6793-7-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lemaitre, B., Nicolas, E., Michaut, L., Reichhart, J. M., and Hoffmann, J. A. (1996). The dorsoventral regulatory gene cassette spatzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell 86, 973–983. doi: 10.1016/S0092-8674(00)80172-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lin, E., Freedman, J. E., and Beaulieu, L. M. (2009). Innate immunity and toll-like receptor antagonists: a potential role in the treatment of cardiovascular diseases. Cardiovasc. Ther. 27, 117–123. doi: 10.1111/j.1755-5922.2009.00077.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lin, L., and Knowlton, A. A. (2014). Innate immunity and cardiomyocytes in ischemic heart disease. Life Sci. 100, 1–8. doi: 10.1016/j.lfs.2014.01.062
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mann, D. L. (2011). The emerging role of innate immunity in the heart and vascular system: for whom the cell tolls. Circ. Res. 108, 1133–1145. doi: 10.1161/CIRCRESAHA.110.226936
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mersmann, J., Berkels, R., Zacharowski, P., Tran, N., Koch, A., Iekushi, K., et al. (2010). Preconditioning by toll-like receptor 2 agonist Pam3CSK4 reduces CXCL1-dependent leukocyte recruitment in murine myocardial ischemia/reperfusion injury. Crit. Care Med. 38, 903–909. doi: 10.1097/CCM.0b013e3181ce50e6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Murry, C. E., Jennings, R. B., and Reimer, K. A. (1986). Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation 74, 1124–1136. doi: 10.1161/01.CIR.74.5.1124
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Oyama, J., Blais, C. Jr., Liu, X., Pu, M., Kobzik, L., Kelly, R. A., et al. (2004). Reduced myocardial ischemia-reperfusion injury in toll-like receptor 4-deficient mice. Circulation 109, 784–789. doi: 10.1161/01.CIR.0000112575.66565.84
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Scaffidi, P., Misteli, T., and Bianchi, M. E. (2002). Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 418, 191–195. doi: 10.1038/nature00858
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Scheibner, K. A., Lutz, M. A., Boodoo, S., Fenton, M. J., Powell, J. D., and Horton, M. R. (2006). Hyaluronan fragments act as an endogenous danger signal by engaging TLR2. J. Immunol. 177, 1272–1281. doi: 10.4049/jimmunol.177.2.1272
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shishido, T., Nozaki, N., Yamaguchi, S., Shibata, Y., Nitobe, J., Miyamoto, T., et al. (2003). Toll-like receptor-2 modulates ventricular remodeling after myocardial infarction. Circulation 108, 2905–2910. doi: 10.1161/01.CIR.0000101921.93016.1C
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vilahur, G., Hernandez-Vera, R., Molins, B., Casani, L., Duran, X., Padro, T., et al. (2009). Short-term myocardial ischemia induces cardiac modified C-reactive protein expression and proinflammatory gene (cyclo-oxygenase-2, monocyte chemoattractant protein-1, and tissue factor) upregulation in peripheral blood mononuclear cells. J. Thromb. Haemost. 7, 485–493. doi: 10.1111/j.1538-7836.2008.03244.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vilahur, G., Juan-Babot, O., Pena, E., Onate, B., Casani, L., and Badimon, L. (2011). Molecular and cellular mechanisms involved in cardiac remodeling after acute myocardial infarction. J. Mol. Cell. Cardiol. 50, 522–533. doi: 10.1016/j.yjmcc.2010.12.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, Y., Abarbanell, A. M., Herrmann, J. L., Weil, B. R., Poynter, J., Manukyan, M. C., et al. (2010). Toll-like receptor signaling pathways and the evidence linking toll-like receptor signaling to cardiac ischemia/reperfusion injury. Shock 34, 548–557. doi: 10.1097/SHK.0b013e3181e686f5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: myocardium, ischemia/reperfusion injury, innate immune response, toll-like receptor, cytokines, inflammation
Citation: Vilahur G and Badimon L (2014) Ischemia/reperfusion activates myocardial innate immune response: the key role of the toll-like receptor. Front. Physiol. 5:496. doi: 10.3389/fphys.2014.00496
Received: 05 November 2014; Accepted: 02 December 2014;
Published online: 18 December 2014.
Edited by:
Gerhild Euler, Justus-Liebig-University, GermanyReviewed by:
William A. Coetzee, New York University School of Medicine, USAThorsten Leucker, Johns Hopkins University, USA
Gerhild Euler, Justus-Liebig-University, Germany
Copyright © 2014 Vilahur and Badimon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gemma Vilahur, Cardiovascular Research Center, c/Sant Antoni MaClaret 167, 08025 Barcelona, Spain e-mail: gvilahur@csic-iccc.org