Retroperitoneal liposarcoma: current insights in diagnosis and treatment
 Lucas E. Matthyssens
Lucas E. Matthyssens David Creytens
David Creytens Wim P. Ceelen
Wim P. Ceelen- 1Department of Surgery, Ghent University Hospital, Ghent, Belgium
- 2Department of Pathology, Ghent University Hospital, Ghent, Belgium
Retroperitoneal liposarcoma (RLS) is a rare, biologically heterogeneous tumor that present considerable challenges due to its size and deep location. As a consequence, the majority of patients with high-grade RLS will develop locally recurrent disease following surgery, and this constitutes the cause of death in most patients. Here, we review current insights and controversies regarding histology, molecular biology, extent of surgery, (neo)adjuvant treatment, and systemic treatment including novel targeted agents in RLS.
Introduction
Anatomy of the Retroperitoneum
The retroperitoneum (RP) forms together with the pre-peritoneum the extraperitoneal space. The RP space is an almost virtual and expandable space, defined anteriorly by the peritoneal extensions anchoring the transverse colon, the small bowel, as well as the ascending and descending colon, part of the duodenum, part of the pancreas, and a part of the liver (1, 2). The RP can be divided into perirenal and (anterior and posterior) pararenal spaces and contains several vital structures: the retroperitoneal organs (the pancreas, kidneys, adrenals, and part of the duodenum, ascending and descending colon), the greater abdominal vessels, the abdominal lymphatics, six major nerves and the autonomic (sympathetic) lumbar chains, and the connective tissue of fasciae, with the White line of Toldt as the fusion between the mesocolon and posterior RP (1).
Masses encountered in the RP can be benign, primary malignant, or metastatic. Primary retroperitoneal tumors (PRT) are those originating in the RP space, but not from the RP organs. Probably, the first report of a PRT was by Giovanni Battista Morgagni (1682–1771), describing in 1761, a retroperitoneal lipomatous tumor found at the autopsy of a 60-year-old woman (3). But it was Jean Fréderic Lobstein (1777–1835) of Strasbourg who launched the actual term “PRT” in his Traité d’anatomie pathologique (1829) (4). About three out of four PRTs are malignant (2, 5). PRTs are classified by their similarity with a certain type of mesenchymal tissue, with up to 2/3 being of mesodermal origin. Over 80% of mesodermal PRTs are malignant. Soft tissue sarcomas (STS), defined by James Stephen Ewing (1866–1943) in his book Neoplastic Diseases (1919) as “unusual malignant tumors composed of cells of the mesodermal/connective tissue type” (6), represent an extremely diverse group of more than 50 different types and subtypes of neoplasms, derived from adipose tissue, muscle, connective, vascular, or deep skin tissue and also bone and cartilage (2, 7–9). Although neural tissue is of (neuro-)ectodermal origin, malignant peripheral nerve sheath tumors (MPNST) are very often classified under STS as well.
Natural History
Soft tissue sarcoma accounts for <1% of all malignant tumors in adults (2, 10), with an estimated incidence of 4–5/100,000/year in Europe (11). About 10–15% of adult STS are located in the RP (2, 12). Liposarcoma is the most common variant and accounts for 20% of all STS, and over 50% of RP sarcomas (13). Commonly classified on their histologic basis (14), STS are very heterogeneous and carry a varying prognosis. The natural behavior and outcome of STS are dependent of the age of the patient, anatomical site and depth, size, and resectability of the tumor, as well as of histology, grade, nodal disease, and distant metastasis (DM) (10). Most retroperitoneal soft tissue sarcomas (RPS), even of important size, rarely metastasize (2): only about 10% of RPS are found to have metastatic disease at presentation, which is mostly hematogenous and equally distributed to the lungs or the liver (2, 5). The presence of DM is an adverse prognostic factor for the outcome of all STS. In RPS, DM occurs in approximately 20–25% of patients and once DM is found, overall survival is poor, at a median of 13 months (10). DM in STS and RPS is largely dependent on the tumor’s malignancy grade (see below) (10). Because most RPS are low-grade, DM is rare, and the main problem is local control and recurrence. Their large size and deep location in an anatomically complex area containing a number of vital structures, makes the resectability of RPS difficult and sometimes impossible (5, 12). Patients who undergo complete (macroscopic) or even compartmental resection (R0 or R1) of the primary tumor have an improved prognosis with a 5-year overall survival of 54–70% (2, 15), yet 41–50% of these patients will demonstrate locally recurrent disease within 5 years after surgery (2, 5, 15). A review from the Memorial Sloan-Kettering Cancer Center (New York, NY, USA), the Royal Marsden Hospital (London, UK), and the French national multicenter study (10, 12, 13, 15) indicated that after more than 5 years, and even after complete macroscopic excision, local recurrence of RPS affects 60–70% of patients and is usually the cause of death (12).
Predisposing Factors
Most STS and especially RPS have no clearly identified cause (10). However, some predisposing factors have been identified: genetic alterations and exposure to radiation or chemical substances. The most important specific and non-specific genetic alterations predisposing to STS are listed in Table 1 (16). These genetically predisposed patients are even more at risk when exposed to ionizing radiation (17). In the general population, and especially in childhood (17–19), repeated computed tomography (CT)-scanning (18) and especially high dose ionizing radiation [as used in external beam radiation therapy (EBRT)] are associated with a higher risk of developing STS, with an estimated incidence of 5% after therapeutic radiation, e.g., for breast cancer, malignant lymphoma, and pediatric cancers (20, 21). The exact mechanism of STS development after EBRT remains, however, unknown (10). Most radiation-associated STS are high-grade/poorly differentiated and are found at the edge of the radiation field, with median latency periods of more than 8 years (range 6–20); mainly fibrosarcoma, osteogenic sarcoma, angiosarcoma, leiomyosarcoma, and undifferentiated pleomorphic sarcoma have been described after EBRT (10, 20). Toxic exposures to chemical agents have also lead to the development of STS, but this is at present mostly of historic interest and consisted mainly of exposure to phenoxyacetic acid/herbicides, thorium bromide/thorotrast, vinyl chloride, arsenic, asbestos, androgenic-anabolic steroids, dioxins, and chlorophenoles (20).
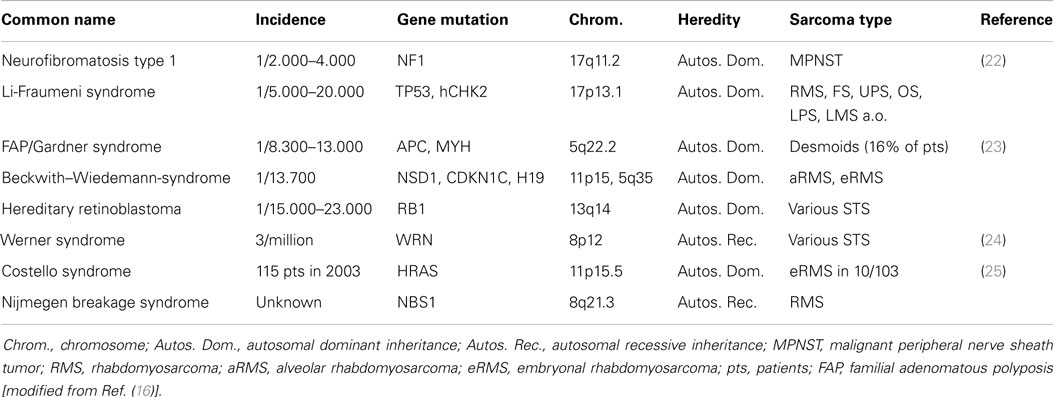
Table 1. Predisposing genetic alterations for soft tissue sarcoma.
Clinical Presentation
Patients presenting with RPS are usually in their mid-fifties (median age 56 years) (5, 12, 13, 15, 21, 26–36), but RPS have been described to occur at all ages (2–98 years) (2, 5, 13, 15, 21, 26, 28, 31–33, 35–40). The gender distribution is supposedly equal (10, 12, 21), although some large retrospective series suggest a small surplus of female patients (mean 1.26M:1F) (2, 5, 13, 15, 26, 31–35, 37, 39, 40). Because the RP is a deep, expandable space without many bony boundaries, slowly growing tumors generally do not quickly cause signs or symptoms and may therefore grow to an important size before being discovered by increased abdominal girth, a palpable lump, or because of compression (causing gastrointestinal, urologic, or neurological symptoms). The majority (>75%) of PRT and RPS present “late,” with an important size. RPS is probably the largest tumors found in the human body (2). In fact, RPS measuring <5 cm is considered rare (35). RPS generally measure >5 cm, and mostly >10 cm diameter at presentation (34, 36). In the largest series of prospectively followed RPS, Lewis et al. found 94% of these tumors exceeding 5 cm in diameter and 60% exceeding 10 cm (2, 5). About 20–50% of RPS even exceed 20 cm in diameter at the time of resection (2, 15, 26, 30–33, 40, 41). Although probably decreasing with time [because of more widespread use of CT and magnetic resonance imaging (MRI)] between 60% (2, 39) and 80% (5, 12) of patients are believed to present with a palpable abdominal mass, and half of the patients have “pain” at presentation (21).
Diagnostic Workup
The diagnosis and treatment of STS mandates a multidisciplinary approach ideally carried out in reference centers treating a high number of patients annually (11). Different imaging studies can be used in the evaluation of PRT/RPS: conventional radiographies of the abdomen usually indicate displacement of bowel and altered intestinal aeration, and may show signs of calcification in the tumoral mass (suggestive of teratoma). Ultrasound is useful as a quick first evaluator of abdominal complaints, but is of limited value for in-depth evaluation of RP masses, especially in adults with increased abdominal girth/obesity. Doppler/duplex ultrasound may offer additional information on the patency of the femoral and iliac vessels and of the inferior caval vein (ICV), especially in case of suspicion of partial or complete deep venous thrombosis due to vascular compression. The diagnostic investigation of choice to evaluate PRT/RPS is contrast-enhanced CT-scanning or MRI of the abdomen and pelvis (11, 12, 21, 42). They will determine the anatomical location of the tumor, its size, and probable origin, the relationship of the tumor to adjacent visceral and neurovascular structures, possible compression or invasion, and the presence or absence of transperitoneal spread or liver or lung metastases (12, 43). Liposarcomas demonstrate a characteristic appearance on CT and MRI with a predominantly fatty component (12). MRI does not cause added radiation exposure and may be specifically required under certain circumstances (12), e.g., in pediatric patients, in cases of myxoid or round cell liposarcoma (MRI of the spine, because of the higher risk for spinal metastasis compared to other STS), or in alveolar soft part sarcomas and angiosarcomas (MRI of the brain, for their propensity to metastasize to the brain especially in the presence of pulmonary metastasis) (44). MRI cannot, however, reliably distinguish between benign and malignant tissue, but diffusion-weighted MRI (DW-MRI) seems a very promising technique and is under study (12, 43, 45).
The “staging” investigation of choice for the detection of DM is contrast-enhanced CT-scan of the chest and abdomen (12). 18F-Fluorodeoxyglucose (FDG)-positron emission tomography (PET) – CT scan may provide additional functional/biological information about the retroperitoneal tumor and may possibly differentiate a high-grade from a low-grade STS (46). Apart from grading, FDG-PET may also aid in staging (detection of metastases), restaging and in the evaluation of treatment response and follow-up, by detecting residual masses or recurrences after attempted radical surgery for STS. In a recent study of 102 STS at UCLA, the tumor glycolytic phenotype correlated significantly with the histologic grade in 91% of tumors, which may offer prognostic significance – although FDG-PET could not reliably distinguish among French Fédération Nationale des Centers de Lutte contre le Cancer (FNCLCC)-grade 2 and grade 3 STS and the various subtypes. When regarding the liposarcomas studied, in 6 of 16 (38%), there was a SUVmax <2.5 g/mL, suggesting that for this STS-subtype, FDG-PET-based treatment monitoring might be difficult. Further prospective studies on the value of PET for STS are underway (44).
Classification, Staging, and Grading
The distribution of sarcoma subtypes in the RP differs from other localizations, with a predominant role (75–85%) for liposarcoma and leiomyosarcoma. RPS is classified using the World Health Organization (WHO) Classification of soft tissue tumors (Table 2) (14). Based on its histologic type and subtype, the tumor is classified into one of four categories: benign, intermediate (locally aggressive), intermediate (rarely metastasing), and malignant (14).

Table 2. WHO classification of soft tissue tumors of intermediate malignant potential and malignant soft tissue tumors.
Different staging systems have been in use for predicting the systemic outcomes of patients with STS, but a specific staging system for RPS is not (yet) available. The revised UICC/AJCC-7 cancer staging system for the prognostic classification of sarcomas is the most commonly used; since 1977 this includes the histologic grade (Table 3) (48). Other staging systems include the “surgical staging system” (SSS) by Enneking and the Musculoskeletal Tumor Society (49) and the postsurgical classification system by the Sarcoma Disease Management Team at Memorial Sloan-Kettering Cancer Center (MSK-system) (50).
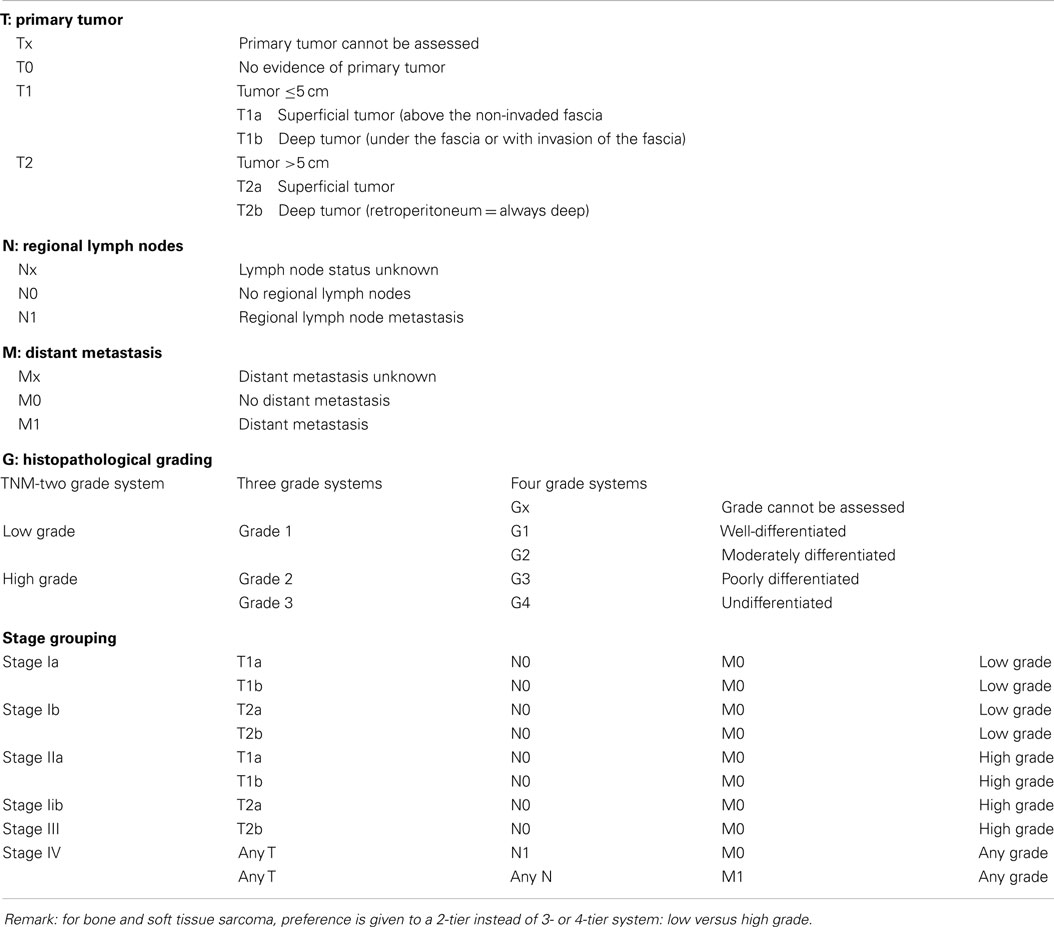
Table 3. Sarcoma staging system, seventh edition of the American Joint Committee on Cancer/International Union against Cancer (UICC/AJCC-7, 2010).
Histologic grade represents the most important indicator of metastatic risk and OS in adult STS. The main objective of grading is to select patients for adjuvant chemotherapy (51). The concept of histological grade in sarcoma was introduced by Broders in 1920 and since then, various 2-, 3-, or 4-tier grading systems have been in use. There is at present no single generally agreed upon grading system for STS. Since the 1980s, the FNCLCC (52, 53) and the US National Cancer Institute (NCI) (54) systems are the most commonly used grading systems for STS (Table 4). Both are 3-grade systems based on histologic tumor differentiation, mitotic rate/activity, and percentage of tumor necrosis. The NCI system also requires quantification of cellularity and pleomorphism for certain sarcoma subtypes, which is difficult to determine objectively. The UICC/AJCC-7 STS staging system is, however, not fully adapted for RPS. As the majority of RPS are large and deeply situated, the prognostic value of “T” (size and depth) is less applicable (all are T2b) and the same is true for “N,” as most RPS do not develop lymphatic metastasis. The system is not applicable for local recurrences (very common in RPS) and grading remains difficult with demonstrable interobserver discordances, even among experienced pathologists. Therefore, the surgical oncology team from MD Anderson Cancer Center (Houston, TX, USA) proposed in 2009 a novel practical “histology-based prognostic system” to predict overall survival in all RPS patients (34). This system stratifies RPS patients into three risk groups according to tumor histology, with “Atypical lipomatous tumor” (ALT, well-differentiated liposarcoma) having the best prognosis, “non-ALT liposarcoma” (non-ALT LPS) having the worst overall survival and “Other” histology (non-LPS) having an intermediate prognosis. This system is also applicable for recurrent disease, and further risk stratification can still be determined within each of the groups (34).

Table 4. The French Fédération Nationale des Centers de Lutte Contre le Cancer (FNCLCC) Grading System.
On the other hand, as for other neoplasms, molecular markers hold also great promise for refining our ability to establish early prognosis and to predict response to treatment in STS/RPS (“molecular grading”) (51). Molecular profiling analysis by microarray technology has been performed in STS and a 67-gene expression signature called CINSARC has recently been identified as a clinically applicable prognostic marker (51). However, the value of CINSARC for predicting the response to treatment is not yet known and will soon be validated in prospective independent series (51). Since almost a decade, cancer nomograms have been developed, mainly instigated by the Sarcoma Disease Management Team of MSKCC. Nomograms are being increasingly accepted to predict risk of recurrence and disease-specific death, and to aid the clinician in counseling patients and planning for surveillance and follow-up (13). Nomograms for RPS and liposarcoma of the RP have recently been published by different teams, based on the histologic subtype, margin of resection, contiguous organ resection, and age as prognostic markers for survival.
Histology and Molecular Biology
The histological classification of liposarcoma has evolved significantly over past several decades, in large part owing to the advances in our understanding of its molecular genetics. The recently updated World Health Organization (WHO) classification of soft tissue and bone tumors recognizes four major liposarcoma subtypes: atypical lipomatous tumor/well-differentiated liposarcoma [which includes the adipocytic (or lipoma-like), sclerosing, inflammatory and spindle cell variants], dedifferentiated liposarcoma, myxoid liposarcoma, and pleomorphic liposarcoma (Figures 1–4) (55, 56). These four main subgroups are characterized by distinctive morphologies, as well as unique genetic findings. A fifth subtype (the so-called “mixed or combined liposarcoma”), which was still a separate entity in the 2002 WHO classification, has been removed from the most recent 2013 WHO classification, based on the consensus view that those rare cases probably represent examples of (variants of) dedifferentiated liposarcoma. It is important to emphasize that atypical lipomatous tumor and well-differentiated liposarcoma are synonyms, describing lesions, which are identical both morphologically and karyotypically. Use of the term atypical lipomatous tumor is determined principally by tumor location and resectability. In sites such as the RP, it is usually impossible to obtain a wide tumor free surgical excision margin of more than 2 cm. In such cases, local recurrence is common and often leads to death, even in the absence of dedifferentiation or metastasis. At these sites, thus, the term well-differentiated liposarcoma is used rather than atypical lipomatous tumor (55, 56). Histopathology is the gold standard in the diagnostic traject of lipomatous tumors. In addition to tumor size and anatomic location, one of the most important determining factors for the prognosis of liposarcoma patients is the histological liposarcoma subtype, further underlining the importance of correct subclassification. However, establishing the correct lipomatous tumor subtype can be laborious and requires in some instances a histological assessment together with immunohistochemistry and molecular analyses using fluorescence in situ hybridization (FISH), polymerase chain reaction (PCR), multiplex ligation-dependent probe amplification (MLPA), and/or array comparative genomic hybridization (aCGH). Finally, with a growing number of molecularly targeted agents in oncology, molecular testing will become increasingly important in guiding treatment strategies of liposarcomas in the near future.
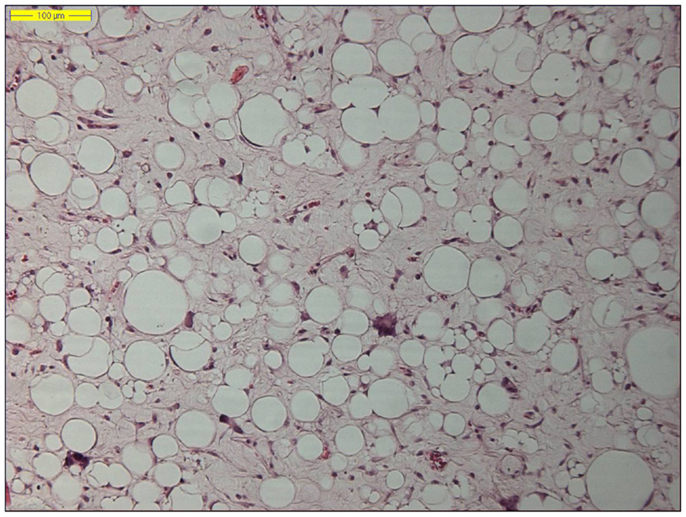
Figure 1. Histology of a lipoma-like well-differentiated liposarcoma (hematoxylin and eosin, original magnification 200×).
The two by far most common (lipo)sarcoma subtypes in the RP are the well-differentiated liposarcoma (Figure 1) and dedifferentiated liposarcoma, followed by the leiomyosarcoma. Primary myxoid liposarcomas, occurring predominantly in the lower limbs of young to middle-aged adults, are extremely rare and may be “non-existing” in the RP. Therefore, a diagnosis of primary retroperitoneal myxoid liposarcoma should be regarded with suspicion, as most such cases represent either metastatic myxoid liposarcoma or well-differentiated/dedifferentiated liposarcoma with myxoid stromal change (57, 58). Pleomorphic liposarcoma, defined as a high-grade pleomorphic sarcoma showing variable amounts of lipoblastic differentiation, arise most often in the limbs of elderly patients and are extremely rare in retroperitoneal location (59, 60).
Well-differentiated liposarcoma is a genetically distinct group of lesions. With the exception of the spindle cell variant, all well-differentiated liposarcoma subtypes share the same genetic aberration and are characterized by supernumerary ring and/or giant rod chromosomes containing amplified segments from the 12q13-15 region where several proto-oncogenes including murine double minute type 2 (MDM2), cyclin-dependent kinase 4 (CDK4), high-mobility AT-hook 2 (HMGA2), and tetraspanin 31 (TSPAN31 or SAS) are located (61–69). MDM2 is the most frequent amplified gene, close to 100%, and CDK4 is shown to be amplified in over 90% of cases (70, 71). Co-amplification of MDM2 and CDK4 is a common feature in well-differentiated liposarcoma and is thought to be the initiating “driving” factor in fat tumorigenesis, resulting in proliferation through combined effects upon p53 (by inactivating TP53) and the cell cycle (by RB1 phosphorylation), respectively. It has been suggested that CDK4 provides a selection advantage in well-differentiated liposarcoma and may contribute to transformation as CDK4 negative well-differentiated liposarcoma exhibit more favorable prognostic features (64, 70–72). Amplification of MDM2, CDK4, and HMGA2 can be detected by molecular techniques including FISH, PCR, MLPA, or aCGH techniques (68, 73–75). P53 mutations are rarely seen in well-differentiated and dedifferentiated liposarcomas, but are commonly in pleomorphic liposarcomas.
Barretina et al. showed 16.7% of pleomorphic liposarcoma cases had mutations in p53 (76). Similarly, high p53 mutations rates (approximately 60%) were observed in pleomorphic liposarcoma by Ghadimi et al. (77).
Moreover, identifying MDM2 amplification, as well as overexpression of the corresponding MDM2 protein by immunohistochemistry, has proved an adjunctive tool in the diagnosis of lipomatous neoplasms, especially in the diagnosis of a well-differentiated liposarcoma, because MDM2 amplification is absent in “ordinary” lipomas (Figures 2 and 3) (78–81). Molecular testing should be considered for “relapsing lipomas,” tumors with questionable cytologic atypia (pathologists tend to overestimate the degree of cytologic atypia), or for large lipomatous tumors (>15 cm) without diagnostic cytologic atypia (81).
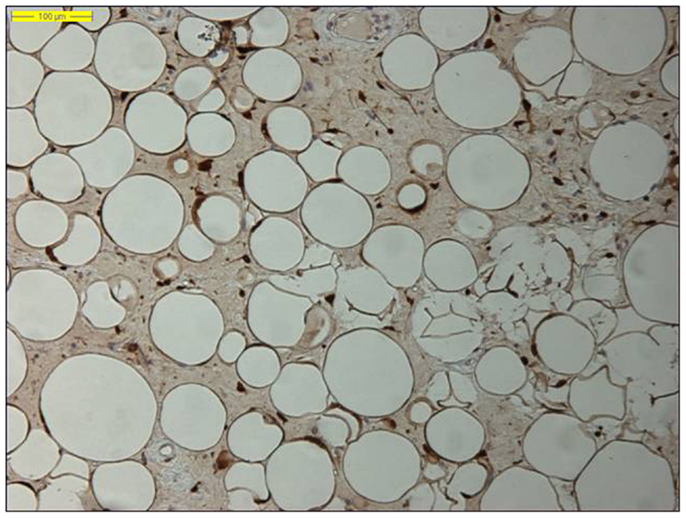
Figure 2. Nuclear MDM2 immunohistochemical overexpression in the atypical adipocytes in a lipoma-like well-differentiated liposarcoma (original magnification 200×).
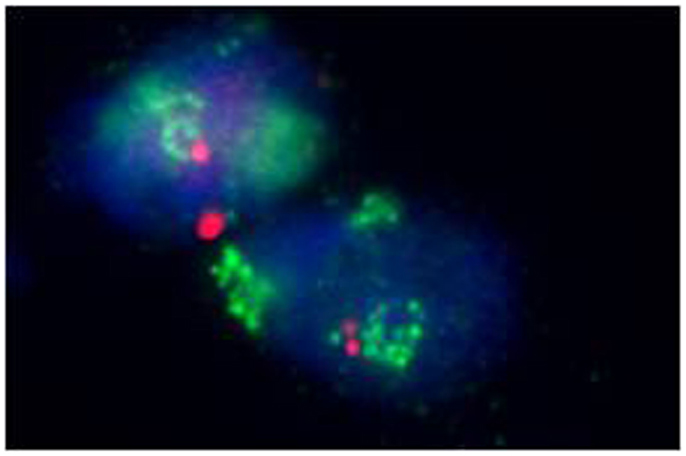
Figure 3. Amplification of the MDM2 gene in a well-differentiated liposarcoma (fluorescence in situ hybridization, FISH).
Dedifferentiated liposarcoma is a biologically fascinating lesion, in which morphology, genetics, and clinical behavior converge to define a distinctive clinicopathological entity. The term “tumor dedifferentiation” as established in 1971 by Dahlin and Beabout, characterizes “the morphological progression of a low-grade tumor to a less differentiated neoplasm with a more aggressive behavior” (82). Dedifferentiated liposarcoma is traditionally defined as “a non-lipogenic high-grade sarcoma arising from a well-differentiated liposarcoma that confers metastatic potential.” The term dedifferentiated liposarcoma was first introduced by Evans in 1979, describing a liposarcoma containing a well-differentiated liposarcoma component juxtaposed to areas of high-grade non-lipogenic sarcoma and was believed to occur from well-differentiated liposarcoma after several years (83). The RP is the most common location, outnumbering somatic soft tissue by at least 5/1 (55, 56, 84). More than 90% of dedifferentiated liposarcoma arises de novo (synchronous), while <10% occurs in recurrences (metachronous). Dedifferentiated areas in dedifferentiated liposarcoma exhibit a wide morphological spectrum (84). Histologically, most cases of dedifferentiated liposarcoma show areas of high-grade poorly differentiated sarcoma resembling high-grade myxofibrosarcoma, fibrosarcoma, malignant solitary fibrous tumor, or pleomorphic sarcoma NOS (Figure 4). In about 5–10% of cases, the dedifferentiated component shows divergent differentiation featuring myogenic, angiosarcomatous, or osteochondromatous components (85–88). Several recent studies have reported that most sarcomas diagnosed as poorly differentiated sarcomas and arising in the RP are, in fact, dedifferentiated liposarcomas and can now be diagnosed as such on the basis of MDM2 amplification even in challenging cases of a non-lipogenic undifferentiated sarcoma without an atypical adipocytic component (89–91). Like atypical lipomatous tumor/well-differentiated liposarcoma, dedifferentiated liposarcoma is characterized by presence of supernumary ring and/or giant rod chromosomes containing amplified segments from the 12q13-15 region (55, 56, 62, 63, 65, 66, 84). Intensive research has identified several oncogenes residing in this region, including MDM2, CDK4, HMGA2, TSPAN31 (SAS), YEATS4, miR-26a-2, CPM, OS1, OS9, CHOP (DDIT3), and GLI1 (63). The most evidence, to date, demonstrates an oncogenic role in dedifferentiated liposarcoma, like the atypical lipomatous tumor/well-differentiated liposarcoma, for MDM2, CDK4, HMGA2, and TSPAN31 (SAS) (55, 64, 70, 71, 84). Wang et al. described consistent amplification of the fibroblast growth factor receptor substrate 2 gene (FRS2) in dedifferentiated (and well-differentiated) liposarcoma (92). Recently, STAT6 (12q13) amplification and overexpression was described in a subset of dedifferentiated liposarcoma, further underlining the genomic complexity and heterogeneity of ring and giant marker chromosomes of this tumor type, particularly concerning amplicons originating from the chromosomal region 12q13-15 (93, 94). Despite its typically high-grade morphology, dedifferentiated liposarcoma is much less aggressive than other types of high-grade pleomorphic sarcoma (55, 56, 95, 96). Dedifferentiation is associated with a 15–20% metastatic rate; however, mortality is related more often to uncontrolled local recurrences than to metastatic spread. Therefore, it is of clinical importance to distinguish a dedifferentiated liposarcoma from a de novo high-grade pleomorphic sarcoma of some other type (97). A recent study by Thway et al. have suggested that the immunohistochemical trio of CDK4, MDM2, and the cell cycle regulator p16 is an useful ancillary diagnostic tool distinguishing dedifferentiated liposarcomas from pleomorphic and myxoid liposarcomas (Figures 5 and 6) (98).
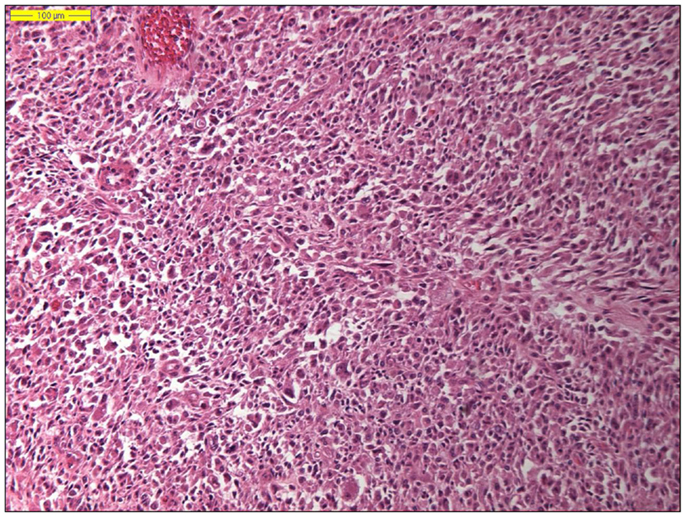
Figure 4. Histology of a dedifferentiated liposarcoma (hematoxylin and eosin, original magnification 100×).
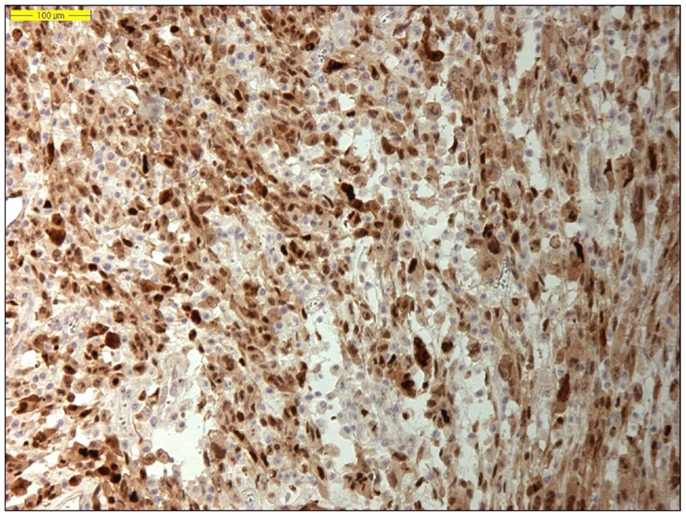
Figure 5. Nuclear MDM2 immunohistochemical overexpression in dedifferentiated liposarcoma (original magnification 200×).
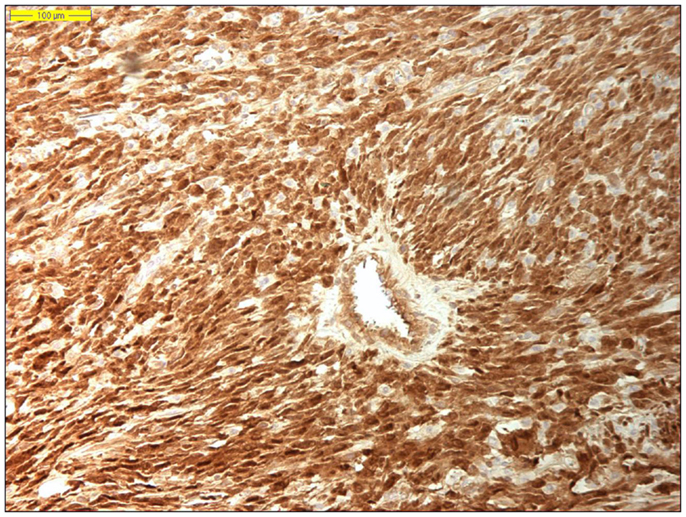
Figure 6. Nuclear CDK4 immunohistochemical overexpression in dedifferentiated liposarcoma (original magnification 200×).
The concept of dedifferentiation in liposarcoma has undergone an evolution in the last several years and the traditional views have been modified by the concept of low-grade dedifferentiation in dedifferentiated liposarcoma. Where it was once assumed that all dedifferentiated tumors manifested themselves as high-grade, undifferentiated sarcoma-like lesions, the concept of low-grade dedifferentiation has increasingly been recognized, with areas resembling low-grade myxofibrosarcoma, desmoid fibromatosis, well-differentiated fibrosarcoma, and even dermatofibrosarcoma protuberans (55, 56, 84). The significance of this lower grade of progression is, to date, not completely known and is still controversial. However, there is some suggestion that the lower grade progression carries a better prognosis than the high-grade undifferentiated type of dedifferentiated liposarcoma (55).
The mechanisms responsible for progression from well-differentiated liposarcoma to dedifferentiated liposarcoma are incompletely understood. Since MDM2 and CDK4 amplifications are present in both well-differentiated and dedifferentiated liposarcoma, the presence of these amplifications as such are not triggers for dedifferentiation in liposarcomas. As a group, dedifferentiated liposarcomas show more complex chromosomal aberrations than do well-differentiated liposarcomas. Chromosomal imbalances additionally to the 12q13-q15 amplicon, including amplifications in 1p32 (including JUN), 1q21-q24, and/or 6q23 (including the ASK1 or MAP3K5 gene), have been reported to be more frequent in dedifferentiated liposarcoma than in well-differentiated liposarcoma (69). Recent studies into the well-differentiated liposarcoma de-differentiation process have suggested a role for c-Jun N-terminal kinase (JNK) pathway (99). The proto-oncogene c-Jun encodes part of the activator protein transcription factor (AP-1) complex involved in cell proliferation, transformation, and apoptosis. ASK1 activates JNK ultimately leading to c-Jun activation and peroxisome proliferator-activated receptors (PPAR) gamma inactivation. PPAR gamma is involved in the adipocytic differentiation process and its inhibition may result in dedifferentiation. Co-amplification of 1p32 and 6q23 that contain c-Jun and apoptosis signaling kinase 1 (ASK1) are seen in dedifferentiated liposarcoma but not in well-differentiated liposarcoma (84, 99–103).
Treatment
Surgery
Surgery is the mainstay of treatment of non-metastatic retroperitoneal liposarcoma (RLS). Whenever possible, macroscopically complete resection should be aimed at, often requiring en-bloc removal of adjacent structures such as the abdominal wall, psoas, or paravertebral muscles. In an attempt to optimize the surgical approach to these patients and provide a standardized, reproducible technique, technical guidelines were recently provided by E-Surge, a master class in sarcoma surgery, and the EORTC soft tissue and bone sarcoma group (104). Areas of uncertainty include the necessity of pretreatment biopsy, and the impact of surgical radicality versus disease biology on local control and long-term survival.
Pretreatment Biopsy
In most patients with RLS, the iconographic appearance (location, density, displacement rather than invasion of adjacent organs) is nearly diagnostic and pretreatment biopsy therefore unnecessary. As a consequence, it has been argued that pretreatment biopsy does not offer any value in patients with a resectable retroperitoneal mass (105). In some patients, however, radiology may suggest a different pathology that may not require surgery as the first approach (lymphoma, Ewing sarcoma, GIST). Also, in patients at risk for incomplete resection and in whom neoadjuvant radiotherapy is planned, pretreatment histological confirmation is mandatory. In these patients, image guided core or fine needle aspiration biopsy are reliable and safe, and preferred over open or laparoscopic approaches, which may be associated with a higher risk of tumor spillage and may compromise future surgical strategy by altering tissue planes (106–109).
Extent of Surgery Versus Tumor Biology
In contrast to limb (lipo)sarcoma, removal of the entire tumor with a rim of normal tissue is usually precluded in (large) RLS due to adjacent large vessels, nerves, or bony structures. As a consequence, many patients develop locally recurrent disease in the abdomen, which constitutes the cause of death in approximately three out of four patients (Figure 7) (34). Several centers therefore advocate liberal compartmental, en-bloc resection of adjacent organs in order to reduce the risk of local relapse (30, 32). On the other hand, although all liposarcomas share the amplification of 12q13-15, resulting in overexpression of MDM2 and CDK4, the molecular biology of the disease is heterogeneous, and likely to be differing between limb and retroperitoneal disease locations, given the fact that recent molecular studies highlighted major ontogenetic differences between normal subcutaneous and visceral (including retroperitoneal) fat tissue (110). High grade, dedifferentiated tumors are at much higher risk to recur and spread systemically, and therefore, unlikely to benefit from extensive surgery (111).
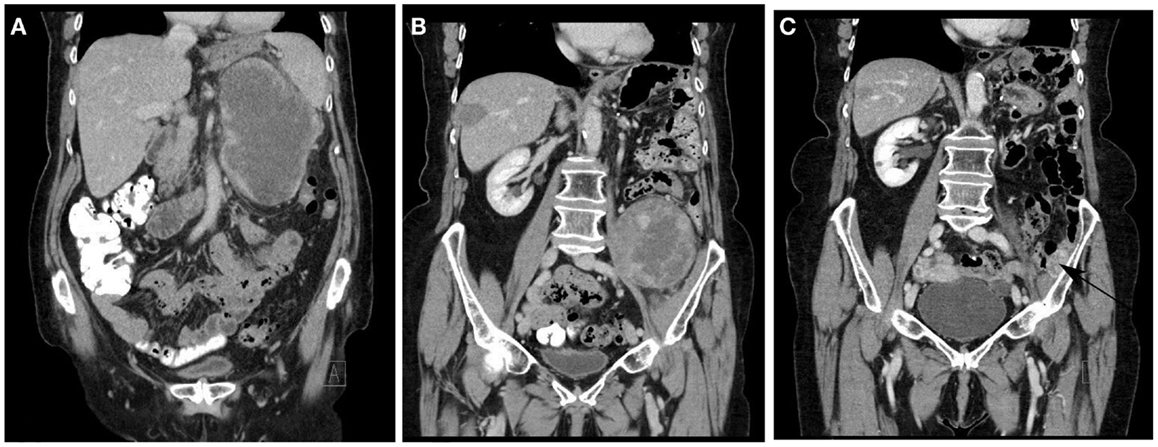
Figure 7. Typical disease course of dedifferentiated liposarcoma in an elderly patient, who presented with a recurrent RLS in February 2011 2 years after primary surgery (A). The patient was treated with neoadjuvant radiotherapy (45 Gy in fractions of 1.8 Gy) and macroscopically completes resection. In May 2012, a solitary metastasis in the right liver lobe (B) was treated with RF ablation. In September 2012, a solitary 1.6 cm metastasis was removed thoracoscopically from the right lower lobe. In May 2013, a second retroperitoneal recurrence developed (B) for which repeat macroscopically complete surgery was performed. In September 2014, a small recurrence was noted against the left iliac bone [(C) arrow] for which additional surgery is planned. No other local or metastatic locations were noted.
The published surgical experience is entirely based on retrospective analyses and difficult to interpret due to significant heterogeneity in terms of care setting (monocentric versus multicentric or multinational), sarcoma type, stage distribution, type of presentation (primary versus recurrent disease), staging system used, and pathology methods used (Table 5). Nevertheless, some observations are rather consistent. First, as is clear from the results of multivariate analyses and the associated estimates of the hazard or risk ratio, advanced tumor grade represents the most important adverse prognostic factor for overall survival as well as for local recurrence. Second, macroscopically incomplete, piecemeal resection, or tumor rupture are associated with a dismal outcome and should not be attempted unless for symptomatic reasons. The efficacy of extended, liberal resection is equivocal, while some authors have identified multiorgan resection and microscopic margin status (R0 versus R1) as independent prognosticators for local recurrence; others found that only tumor biology (grade) and macroscopically complete resection were associated with outcome. Obviously, interpretation of these data is hampered by the fact that in these large tumors, precise determination of R0 resection is not a sinecure. When considering the potential benefit of extensive surgery, it should be noted that a significant proportion of patients will develop multifocal recurrence, including at sites remote from the primary tumor location. Tseng and coworkers found that as many of 50% of patients with recurrent RLS presented with multifocal disease; importantly, type or extent of surgery did not predict recurrence outside of the resection field on univariate logistic regression analysis (37). Although tumor spill or incomplete resection may explain multifocal recurrence, it has been suggested that a “field change” of the entire intra-abdominal fat tissue may underly the observation of remote (out-of-field) recurrence (112). Genomic analyses of normal retroperitoneal fat as well as tumor samples may provide further insight into this phenomenon.
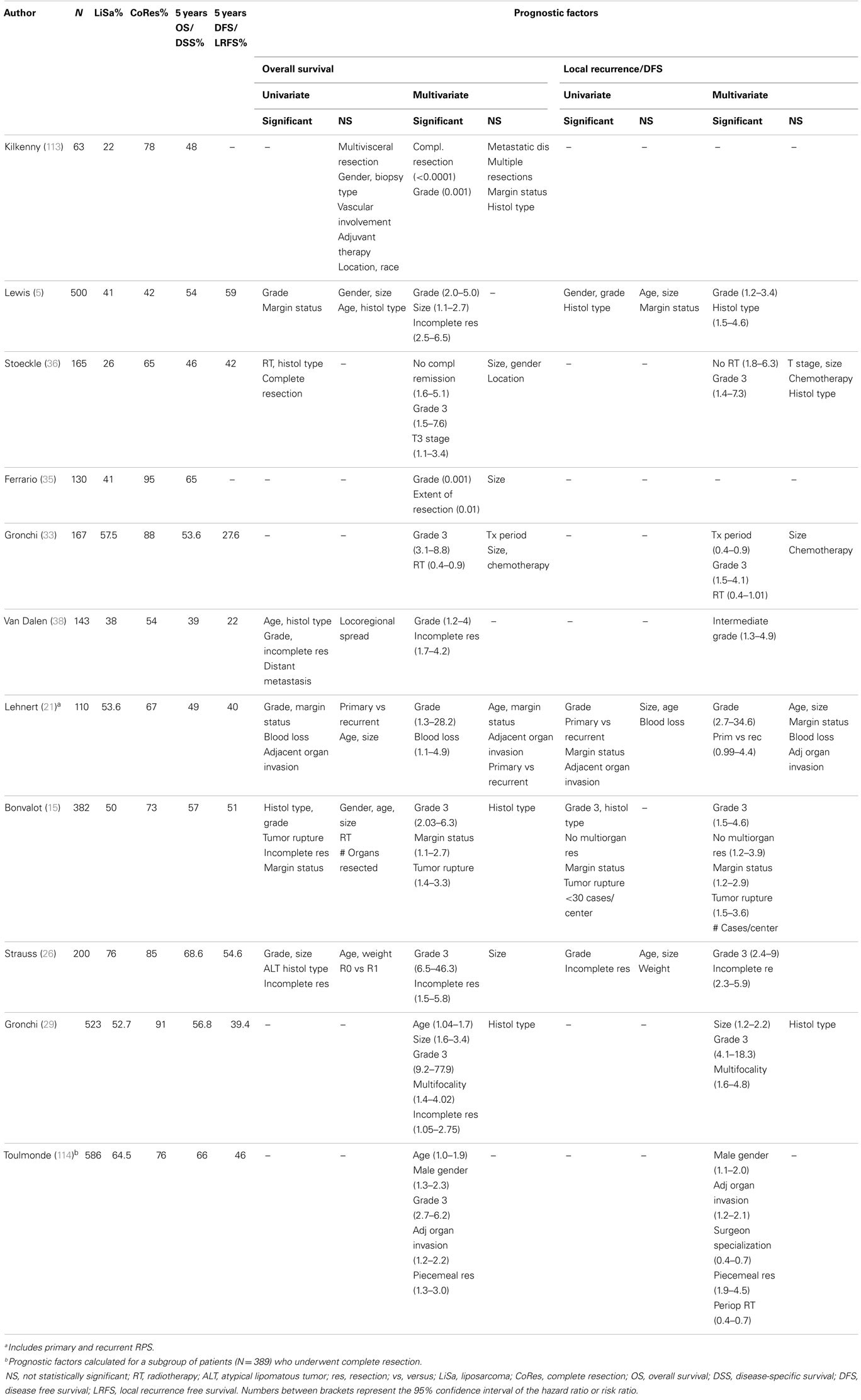
Table 5. Prognostic factors of local control and overall survival outcome in selected published series of surgically treated retroperitoneal (lipo)sarcoma.
Radiation Therapy
Even after optimal resection of RLS, local recurrence remains common and constitutes the most frequent cause of death. Therefore, adjuvant radiation therapy (RT) may constitute a valuable treatment option in order to improve local control, specifically with involved margins or high-grade tumors. In patients with soft tissue sarcoma of the extremity, two small randomized trials have shown that postoperatiive external beam radiotherapy or brachytherapy improve local control, but do not benefit overall survival (115–117). In retroperitoneal soft tissue sarcoma, a myriad of small trials has been published, which show marked variation in RT dose, fractionation, concurrent use of chemotherapy, delivery method (external beam or brachytherapy), timing (preoperative, intraoperative, or postoperative), and energy carrier (photons, electrons, protons, or carbon ions) (118).
Preoperative RT is usually regarded as the treatment sequence of choice. First, preoperative radiation helps to avoid damage to radiation sensitive structures and organs, which usually fill in the resection bed after removal of these large tumors. Second, treatment compliance is usually better and related toxicity less in the preoperative setting. Also, the biological effects of RT are enhanced in undisturbed, well perfused, and oxygenated tissue.
Table 6 illustrates published data on the use of preoperative RT in patients with retroperitoneal sarcoma. Most are small, retrospective series describing different histologies and treatment methods. Local control seems, on average, somewhat better compared to surgery alone series. Although the small numbers preclude any robust conclusion, most authors did not find any benefit of adding either IOERT or postoperative BT to the external beam RT. Only one small study compared surgery alone with preoperative external beam and intraoperative RT followed by surgery, and found that the combined modality resulted in improved local control without any difference in overall survival (119). Several authors have scrutinized data from the surveillance, epidemiology, and end results (SEER) database in an attempt to define the role of adjuvant RT in RLS (Table 7). The results are difficult to compare due to differences in inclusion period, inclusion criteria, case mix, and analytical method. As a general finding, adjuvant radiotherapy either did not benefit survival or did so in a subgroup of stage I patients only. Of note, the large majority of patients treated with radiotherapy were administered this treatment in the postoperative period.
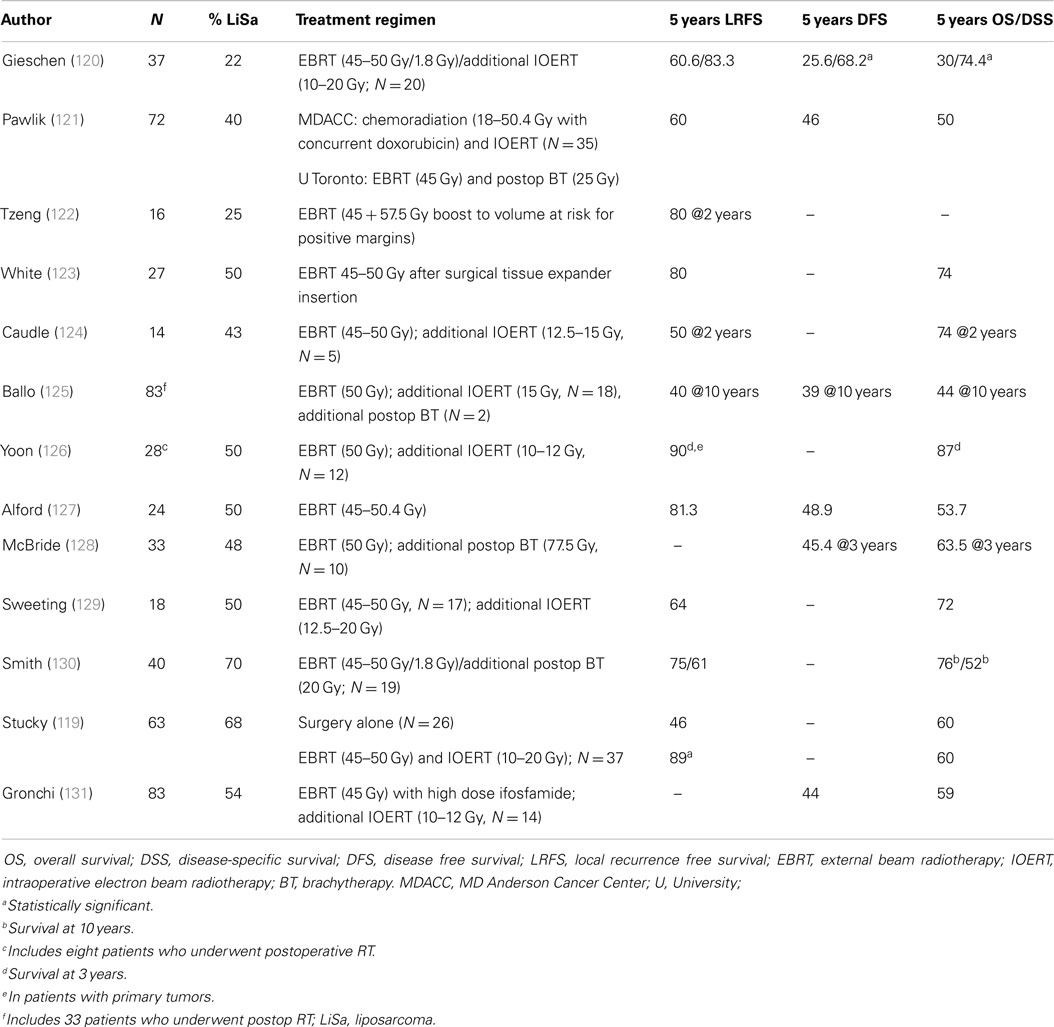
Table 6. Selected clinical studies of preoperative radiotherapy for retroperitoneal (lipo)sarcoma.
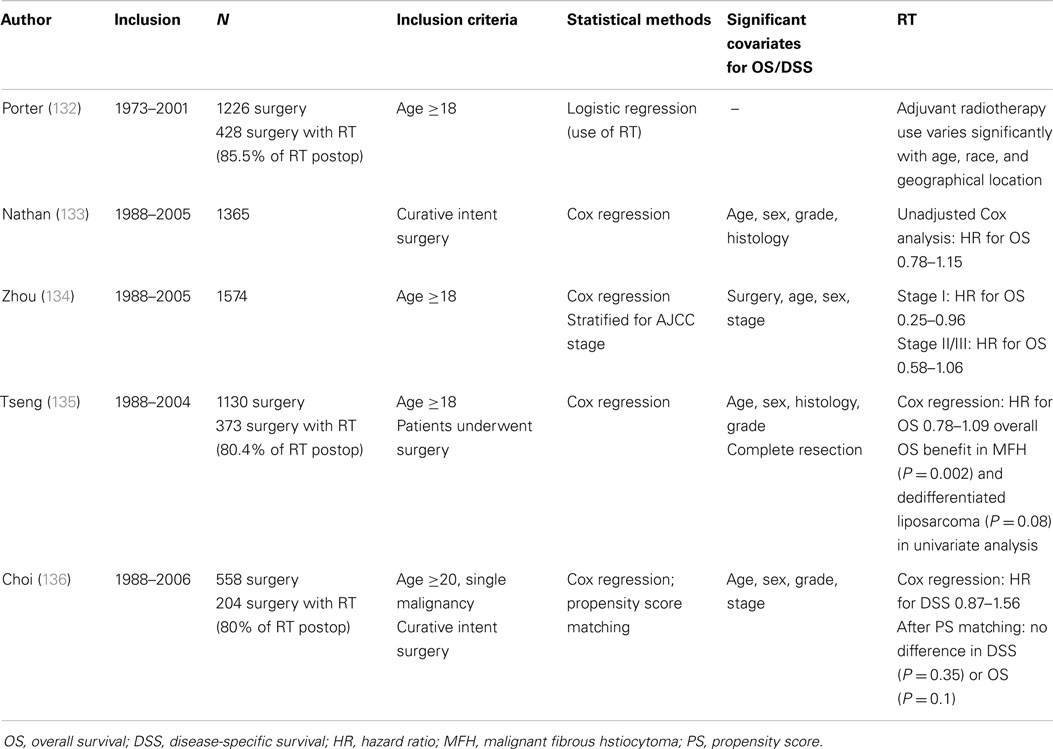
Table 7. Studies based on data from the surveillance, epidemiology, and end results (SEER) database in an attempt to define the role of adjuvant radiotherapy in retroperitoneal sarcoma.
To date, no randomized trials have been completed or published comparing surgery alone with combined surgery and RT. The American College of Surgeons Oncology Group (ACOSOG) initiated a trial (Z9031) in 2004 comparing surgery alone versus fractionated RT followed by surgery. The primary endpoint was progression free survival at 5 years. This trial was terminated due to poor accrual. The European Organization for Research and Treatment of Cancer (EORTC) protocol 62092, which started in 2012, randomizes patients to either en-bloc surgery alone versus fractionated RT (50.4 Gy in 28 fractions) followed by en-bloc surgery. The primary endpoint is abdominal recurrence free survival. With a planned sample size of N = 256, completion of the inclusion period will take at least until 2019.
Systemic Therapy
Chemotherapy has an established role in the palliative management of advanced or metastatic soft tissue sarcoma. Active agents include the anthracyclines (doxorubicin and epirubicin) and the alkylating agent ifosfamide (137). In patients with resistant disease, gemcitabine, docetaxel, trabectedin, and pazopanib were established as effective second or third line options over the last decade (138). STS are a very heterogeneous group, and chemosensitivity is determined by histological type and grade. Examples include the response of angiosarcomas to paclitaxel and pegylated liposomal doxorubicin, the response of leiomyosarcomas to gemcitabine and docetaxel, and the response of desmoid tumors to liposomal doxorubicin (138). Similarly, the response of liposarcoma to chemotherapy differs according to histological subtype and grade. Investigators from the Royal Marsden Hospital investigated response to chemotherapy in 88 patients with liposarcoma (43% located in the RP) (139). They found a significantly higher response rate in myxoid liposarcoma compared to all other liposarcomas (48 versus 18%, P = 0.012). The response rate was 25% in dedifferentiated liposarcoma, while none of the well-differentiated liposarcomas responded. Also, response was significantly better in patients with liposarcoma of the upper limb (75%) or lower limb (36%) compared to other locations (18%). Italiano and coworkers reported the role of chemotherapy in unresectable and/or metastatic well-differentiated and dedifferentiated liposarcoma (77.5% retroperitoneal) based on retrospective analysis of data from 10 centers (139). Seventy-three percent of the included 208 patients had at least one metastatic site. Using RECIST criteria, response was complete in 1% and partial in 11%, while stable disease and progression were seen in 48 and 39%, respectively. No difference was observed in response rate between WD and DD liposarcoma.
Because well-differentiated liposarcoma and dedifferentiated liposarcoma respond poorly to systemic chemotherapy, it is essential that novel molecular targets will be identified to provide new possibilities for therapies. The reported results of recent clinical trials for novel systemic therapies in advanced liposarcoma are overall encouraging. In the past decade, results from clinical trials have identified several novel systemic therapies in soft tissue sarcoma, many of which have potential efficacy in liposarcoma (5, 140). In contrast to conventional cytotoxic chemotherapies, which are non-specific, the majority of these novel therapies are based on the understanding of disease biology inherent to a given sarcoma histology, in many cases targeting a specific, aberrant genetic, or molecular pathway. For the majority of novel therapies, treatment efficacy is heavily dependent on subtype. Reported human studies and clinical trials for novel systemic therapies in liposarcoma are summarized in Table 8. Targeting MDM2 or CDK4 (MDM2 and CDK4 antagonists) in well-differentiated liposarcoma and dedifferentiated liposarcoma has been of interest for several years (5, 140–143). Based on the fact that well-differentiated liposarcoma and dedifferentiated liposarcoma are relatively resistant to systemic chemotherapy, MDM2 and CDK4 targeted therapy may be a very promising approach, especially for advanced or unresectable well-differentiated and dedifferentiated liposarcoma. A class of imidazoline compounds, termed nutlins, has been identified as potent and selective small-molecule MDM2 inhibitors. RG7112 (Hoffmann-La Roche) (Phase I, neoadjuvant) is a member of the nutlin family and is the first MDM2 antagonist to be assessed clinically. RG7112 is a potent inhibitor of p53-MDM2 binding that effectively stabilizes p53 protein, activates p53 signaling, and inhibits cancer cell growth (140, 141). Flavopiridol-CDK4 inhibitor and PD0332991-CDK4/CDK6 inhibitor (Pfizer) (Phase I) are potent CDK4 inhibitors, preventing downstream phosphorylation of the retinoblastoma (RB) protein. CDK4 inhibition would thus restore native cell cycle regulation and prevent uncontrolled tumor cell proliferation (140, 143). Several other interesting candidate targets for novel systemic therapies, including YEATS4, c-jun, JNK, and others, have been reported but have not yet been tested, to our knowledge, in the setting of a clinical trial (140, 144). PPAR are critical regulators of normal adipocyte differentiation. PPAR gamma is one of the three isoforms that forms a heterodimeric complex with the retinoid X receptor to regulate transcription of adipocyte-specific genes involved in the terminal adipocyte pathway. Activation of PPAR gamma by PPAR gamma agonists (troglitazone, rosiglitazone, efatutazone; phase I and II) represents an attractive target, particularly in dedifferentiated liposarcoma, myxoid/round cell liposarcoma and pleomorphic liposarcoma, as a mechanism to revert these subtypes to a well differentiated phenotype with potentially more indolent disease progression (5).
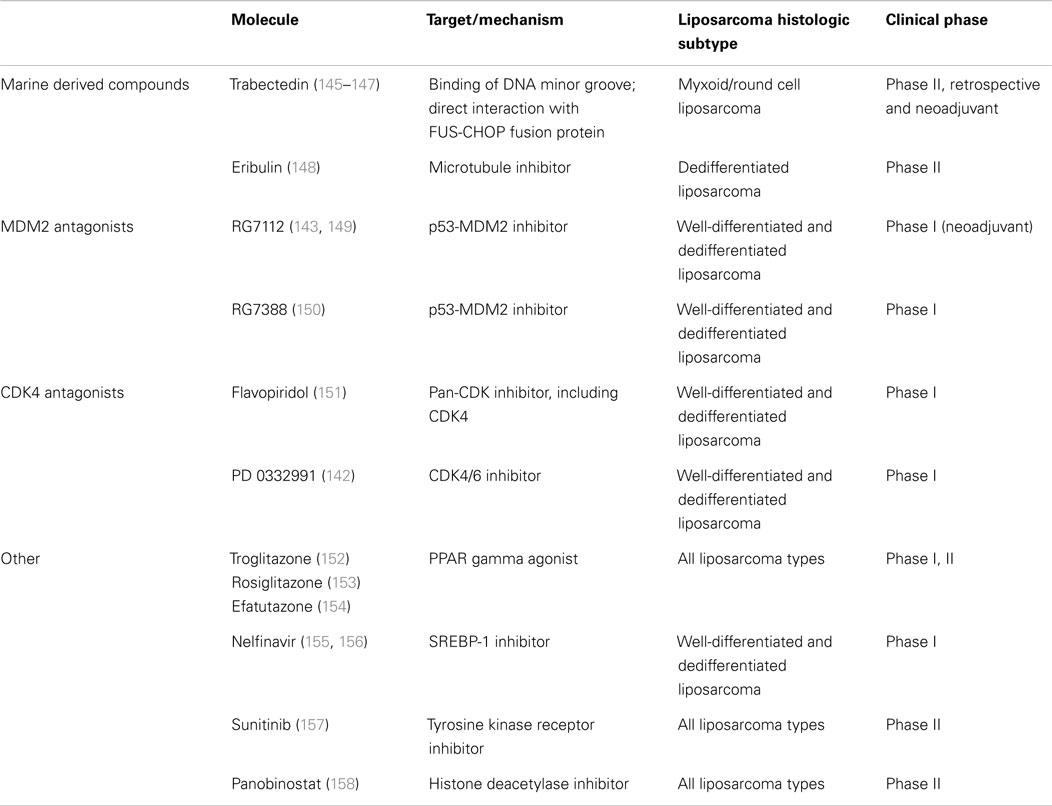
Table 8. Molecular therapeutic targets and agents in soft tissue sarcoma.
Tyrosine kinase receptors are a diverse family of surface molecules recognized for their critical role in regulating multiple aspects of carcinogenesis, tumor cell proliferation, and disease progression (e.g., angiogenesis, metastasis) across many solid tumor types. In the presence of a specific growth factor ligand, tyrosine kinase receptors and their associated downstream molecules are frequently over-expressed or mutated, leading to constitutive activation or aberrant signaling. In a single institution study of 48 patients at the Moffitt Cancer Center, Tariq Mahmood et al. reported impressive efficacy with sunitinib, especially for liposarcoma (157). It is unclear why sunitinib, but not pazopanib or sorafenib, has anti-tumor activity in liposarcoma despite having similar molecular targets (159, 160).
Conclusion
Retroperitoneal liposarcoma is a rare tumor, and exhibits considerable histological heterogeneity. Adequate staging and grading is essential in guiding the therapeutic approach. Macroscopically complete resection offers the best chance of prolonged recurrence free survival. The value of extensive, “compartmental” resection in these patients is at present not well defined, but influenced by disease biology. Preoperative radiotherapy may lower the risk of locally recurrent disease, and a prospective, randomized EORTC study is currently open for inclusion. In parallel with the unraveling of the molecular pathways underlying sarcoma genesis, several targeted agents are in active development that may contribute to the available systemic treatment options in the near future.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Mirilas P, Skandalakis JE. Surgical anatomy of the retroperitoneal spaces-part I: embryogenesis and anatomy. Am Surg (2009) 75(11):1091–7.
2. Liles JS, Tzeng CWD, Short JJ, Kulesza P, Heslin MJ. Retroperitoneal and intra-abdominal sarcoma. Curr Probl Surg (2009) 46(6):445–503. doi: 10.1067/j.cpsurg.2009.01.004
3. Morgagni J. De sedibus, et causis morborum per anatomen indagatis, Libri quinque. Venetia: Remondinius (1761).
5. Lewis JJ, Leung D, Woodruff JM, Brennan MF. Retroperitoneal soft-tissue sarcoma: analysis of 500 patients treated and followed at a single institution. Ann Surg (1998) 228(3):355–65. doi:10.1097/00000658-199809000-00008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Ewing J. Neoplastic Diseases. A Text-Book on Tumors. Philadelphia, PA: WB Saunders Company (1919).
7. Coindre JM. Grading of soft tissue sarcomas: review and update. Arch Pathol Lab Med (2006) 130(10):1448–53. doi:10.1043/1543-2165(2006)130[1448:gostsr]2.0.co;2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Coindre JM, Terrier P, Guillou L, Le Doussal V, Collin F, Ranchere D, et al. Predictive value of grade for metastasis development in the main histologic types of adult soft tissue sarcomas: a study of 1240 patients from the French Federation of Cancer Centers Sarcoma Group. Cancer (2001) 91(10):1914–26. doi:10.1002/1097-0142(20010515)91:10<1914::AID-CNCR1214>3.0.CO;2-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Weiss SWGJ. Enzinger and Weiss’s Soft Tissue Tumors. 5 ed. Philadelphia, PA: Mosby, Elsevier Inc (2008).
11. ESMO/European Sarcoma Network WG. Soft tissue and visceral sarcomas: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol (2012) 23(Suppl 7):vii92–9. doi:10.1093/annonc/mds253
13. Dalal KMM. Subtype specific prognostic nomogram for patients with primary liposarcoma of the retroperitoneum, extremity, or trunk. Ann Surg (2006) 244(3):381–91. doi:10.1097/01.sla.0000234795.98607.00
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Fletcher CDM, Unni KK, Mertens F, editors. World Health Organization Classification of Tumours. Pathology and Genetics of Tumours of Soft Tissue and Bone. Lyon: IARC Press (2002).
15. Bonvalot S, Rivoire M, Castaing M, Stoeckle E, Le Cesne A, Blay JY, et al. Primary retroperitoneal sarcomas: a multivariate analysis of surgical factors associated with local control. J Clin Oncol (2009) 27(1):31–7. doi:10.1200/JCO.2008.18.0802
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Garber JE, Offit K. Hereditary cancer predisposition syndromes. J Clin Oncol (2005) 23(2):276–92. doi:10.1200/JCO.2005.10.042
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Berrington de Gonzalez A, Kutsenko A, Rajaraman P. Sarcoma risk after radiation exposure. Clin Sarcoma Res (2012) 2(1):18. doi:10.1186/2045-3329-2-18
18. Krille L, Zeeb H, Jahnen A, Mildenberger P, Seidenbusch M, Schneider K, et al. Computed tomographies and cancer risk in children: a literature overview of CT practices, risk estimations and an epidemiologic cohort study proposal. Radiat Environ Biophys (2012) 51(2):103–11. doi:10.1007/s00411-012-0405-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Frush DP, Donnelly LF, Rosen NS. Computed tomography and radiation risks: what pediatric health care providers should know. Pediatrics (2003) 112(4):951–7. doi:10.1542/peds.112.4.951
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Lahat G, Lazar A, Lev D. Sarcoma epidemiology and etiology: potential environmental and genetic factors. Surg Clin North Am (2008) 88(3):451–81. doi:10.1016/j.suc.2008.03.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Lehnert T, Cardona S, Hinz U, Willeke F, Mechtersheimer G, Treiber M, et al. Primary and locally recurrent retroperitoneal soft-tissue sarcoma: local control and survival. Eur J Surg Oncol (2009) 35(9):986–93. doi:10.1016/j.ejso.2008.11.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Patil S, Chamberlain RS. Neoplasms associated with germline and somatic NF1 gene mutations. Oncologist (2012) 17(1):101–16. doi:10.1634/theoncologist.2010-0181
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. D’Orazio JA. Inherited cancer syndromes in children and young adults. J Pediatr Hematol Oncol (2010) 32(3):195–228. doi:10.1097/MPH.0b013e3181ced34c
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Lauper JM, Krause A, Vaughan TL, Monnat RJ Jr. Spectrum and risk of neoplasia in werner syndrome: a systematic review. PLoS One (2013) 8(4):e59709. doi:10.1371/journal.pone.0059709
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Hennekam RCM. Costello syndrome: an overview. Am J Med Genet C Semin Med Genet (2003) 117C(1):42–8. doi:10.1002/ajmg.c.10019
26. Strauss DC, Hayes AJ, Thway K, Moskovic EC, Fisher C, Thomas JM. Surgical management of primary retroperitoneal sarcoma. Br J Surg (2010) 97(5):698–706. doi:10.1002/bjs.6994
27. Karakousis CP, Velez AF, Emrich LJ. Management of retroperitoneal sarcomas and patient survival. Am J Surg (1985) 150(3):376–80. doi:10.1016/0002-9610(85)90083-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Bremjit PJ, Jones RL, Chai X, Kane G, Rodler ET, Loggers ET, et al. A contemporary large single-institution evaluation of resected retroperitoneal sarcoma. Ann Surg Oncol (2014) 21(7):2150–8. doi:10.1245/s10434-014-3616-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Gronchi A, Miceli R, Shurell E, Eilber FC, Eilber FR, Anaya DA, et al. Outcome prediction in primary resected retroperitoneal soft tissue sarcoma: histology-specific overall survival and disease-free survival nomograms built on major sarcoma center data sets. J Clin Oncol (2013) 31(13):1649–55. doi:10.1200/JCO.2012.44.3747
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Bonvalot S, Miceli R, Berselli M, Causeret S, Colombo C, Mariani L, et al. Aggressive surgery in retroperitoneal soft tissue sarcoma carried out at high-volume centers is safe and is associated with improved local control. Ann Surg Oncol (2010) 17(6):1507–14. doi:10.1245/s10434-010-1057-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Ardoino I, Miceli R, Berselli M, Mariani L, Biganzoli E, Fiore M, et al. Histology-specific nomogram for primary retroperitoneal soft tissue sarcoma. Cancer (2010) 116(10):2429–36. doi:10.1002/cncr.25057
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Gronchi A, Lo Vullo S, Fiore M, Mussi C, Stacchiotti S, Collini P, et al. Aggressive surgical policies in a retrospectively reviewed single-institution case series of retroperitoneal soft tissue sarcoma patients. J Clin Oncol (2009) 27(1):24–30. doi:10.1200/JCO.2008.17.8871
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Gronchi A, Casali PG, Fiore M, Mariani L, Lo Vullo S, Bertulli R, et al. Retroperitoneal soft tissue sarcomas: patterns of recurrence in 167 patients treated at a single institution. Cancer (2004) 100(11):2448–55. doi:10.1002/cncr.20269
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Anaya DA, Lahat G, Wang X, Xiao L, Tuvin D, Pisters PW, et al. Establishing prognosis in retroperitoneal sarcoma: a new histology-based paradigm. Ann Surg Oncol (2009) 16(3):667–75. doi:10.1245/s10434-008-0250-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Ferrario T, Karakousis CP. Retroperitoneal sarcomas: grade and survival. Arch Surg (2003) 138(3):248–51. doi:10.1001/archsurg.138.3.248
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Stoeckle E, Coindre JM, Bonvalot S, Kantor G, Terrier P, Bonichon F, et al. Prognostic factors in retroperitoneal sarcoma: a multivariate analysis of a series of 165 patients of the French Cancer Center Federation Sarcoma Group. Cancer (2001) 92(2):359–68. doi:10.1002/1097-0142(20010715)92:2<359::AID-CNCR1331>3.0.CO;2-Y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Tseng WW, Madewell JE, Wei W, Somaiah N, Lazar AJ, Ghadimi MP, et al. Locoregional disease patterns in well-differentiated and dedifferentiated retroperitoneal liposarcoma: implications for the extent of resection? Ann Surg Oncol (2014) 21(7):2136–43. doi:10.1245/s10434-014-3643-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. van Dalen T, Plooij JM, van Coevorden F, van Geel AN, Hoekstra HJ, Albus-Lutter C, et al. Long-term prognosis of primary retroperitoneal soft tissue sarcoma. Eur J Surg Oncol (2007) 33(2):234–8. doi:10.1016/j.ejso.2006.09.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Hassan I, Park SZ, Donohue JH, Nagorney DM, Kay PA, Nasciemento AG, et al. Operative management of primary retroperitoneal sarcomas: a reappraisal of an institutional experience. Ann Surg (2004) 239(2):244–50. doi:10.1097/01.sla.0000108670.31446.54
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Singer S, Antonescu CR, Riedel E, Brennan MF. Histologic subtype and margin of resection predict pattern of recurrence and survival for retroperitoneal liposarcoma. Ann Surg (2003) 238(3):358–70. doi:10.1097/01.sla.0000086542.11899.38
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Cormier JN, Pollock RE. Soft tissue sarcomas. CA Cancer J Clin (2004) 54(2):94–109. doi:10.3322/canjclin.54.2.94
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Zhu Z, Zhao XM, Zhao YF, Yang L, Zhao J, Dai JR, et al. Evaluation of CT findings for the differentiation of benign from malignant primary retroperitoneal tumors. Chin Med J (2014) 127(1):114–9. doi:10.3760/cma.j.issn.0366-6999.20132406
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Hughes TM, Spillane AJ. Imaging of soft tissue tumours. Br J Surg (2000) 87(3):259–60. doi:10.1046/j.1365-2168.2000.01412.x
44. Demetri GD, Antonia S, Benjamin RS, Bui MM, Casper ES, Conrad EU III, et al. Soft tissue sarcoma. J Natl Compr Canc Netw (2010) 8(6):630–74.
45. Landa J, Schwartz LH. Contemporary imaging in sarcoma. Oncologist (2009) 14(10):1021–38. doi:10.1634/theoncologist.2009-0194
46. Kitajima K, Kono A, Konishi J, Suenaga Y, Takahashi S, Sugimura K. F-18-FDG-PET/CT findings of retroperitoneal tumors: a pictorial essay. Jpn J Radiol (2013) 31(5):301–9. doi:10.1007/s11604-013-0192-x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Lyons LL, North PE, Mac-Moune Lai F, Stoler MH, Folpe AL, Weiss SW. Kaposiform hemangioendothelioma: a study of 33 cases emphasizing its pathologic, immunophenotypic, and biologic uniqueness from juvenile hemangioma. Am J Surg Pathol (2004) 28(5):559–68. doi:10.1097/00000478-200405000-00001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Sobin LHGM, Wittekind C, editors. UICC TNM Classification of Malignant Tumours, 7th Edition, 2010. 7 ed. Oxford: Wiley-Blackwell (2010). 310 p.
49. Enneking WF, Spanier SS, Goodman MA. A system for the surgical staging of musculoskeletal sarcoma. Clin Orthop Relat Res (1980) 153:106–20.
50. van Dalen T, Hennipman A, Van Coevorden F, Hoekstra HJ, van Geel BN, Slootweg P, et al. Evaluation of a clinically applicable post-surgical classification system for primary retroperitoneal soft-tissue sarcoma. Ann Surg Oncol (2004) 11(5):483–90. doi:10.1245/ASO.2004.09.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Neuville A, Chibon F, Coindre JM. Grading of soft tissue sarcomas: from histological to molecular assessment. Pathology (2014) 46(2):113–20. doi:10.1097/PAT.0000000000000048
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Trojani M, Contesso G, Coindre JM, Rouesse J, Bui NB, de Mascarel A, et al. Soft-tissue sarcomas of adults; study of pathological prognostic variables and definition of a histopathological grading system. Int J Cancer (1984) 33(1):37–42. doi:10.1002/ijc.2910330108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Guillou L, Coindre JM, Bonichon F, Bui NB, Terrier P, Collin F, et al. Comparative study of the National Cancer Institute and French Federation of Cancer Centers Sarcoma Group grading systems in a population of 410 adult patients with soft tissue sarcoma. J Clin Oncol (1997) 15(1):350–62.
54. Costa J, Wesley RA, Glatstein E, Rosenberg SA. The grading of soft tissue sarcomas. Results of a clinicohistopathologic correlation in a series of 163 cases. Cancer (1984) 53(3):530–41. doi:10.1002/1097-0142(19840201)53:3<530::AID-CNCR2820530327>3.0.CO;2-D
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Dei Tos AP. Liposarcomas: diagnostic pitfalls and new insights. Histopathology (2014) 64(1):38–52. doi:10.1111/his.12311
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Fletcher CD, Bridge JA, Hogendoorn P, Mertens F. World Health Organization Classification of Tumours; Pathology and Genetics. Tumours of Soft Tissue and Bone. Lyon: IARC Press (2013).
57. de Vreeze RS, de Jong D, Tielen IH, Ruijter HJ, Nederlof PM, Haas RL, et al. Primary retroperitoneal myxoid/round cell liposarcoma is a nonexisting disease: an immunohistochemical and molecular biological analysis. Mod Pathol (2009) 22(2):223–31. doi:10.1038/modpathol.2008.164
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Sioletic S, Dal Cin P, Fletcher CD, Hornick JL. Well-differentiated and dedifferentiated liposarcomas with prominent myxoid stroma: analysis of 56 cases. Histopathology (2013) 62(2):287–93. doi:10.1111/j.1365-2559.2012.04348.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Hornick JL, Bosenberg MW, Mentzel T, McMenamin ME, Oliveira AM, Fletcher CD. Pleomorphic liposarcoma: clinicopathologic analysis of 57 cases. Am J Surg Pathol (2004) 28(10):1257–67. doi:10.1097/01.pas.0000135524.73447.4a
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Wang L, Ren W, Zhou X, Sheng W, Wang J. Pleomorphic liposarcoma: a clinicopathological, immunohistochemical and molecular cytogenetic study of 32 additional cases. Pathol Int (2013) 63(11):523–31. doi:10.1111/pin.12104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Dal Cin P, Kools P, Sciot R, De Wever I, Van Damme B, Van de Ven W, et al. Cytogenetic and fluorescence in situ hybridization investigation of ring chromosomes characterizing a specific pathologic subgroup of adipose tissue tumors. Cancer Genet Cytogenet (1993) 68(2):85–90. doi:10.1016/0165-4608(93)90001-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Dei Tos AP, Doglioni C, Piccinin S, Sciot R, Furlanetto A, Boiocchi M, et al. Coordinated expression and amplification of the MDM2, CDK4, and HMGI-C genes in atypical lipomatous tumours. J Pathol (2000) 190(5):531–6. doi:10.1002/(SICI)1096-9896(200004)190:5<531::AID-PATH579>3.3.CO;2-N
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Gisselsson D, Hoglund M, Mertens F, Mitelman F, Mandahl N. Chromosomal organization of amplified chromosome 12 sequences in mesenchymal tumors detected by fluorescence in situ hybridization. Genes Chromosomes Cancer (1998) 23(3):203–12. doi:10.1002/(SICI)1098-2264(199811)23:3<203::AID-GCC1>3.0.CO;2-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Italiano A, Bianchini L, Keslair F, Bonnafous S, Cardot-Leccia N, Coindre JM, et al. HMGA2 is the partner of MDM2 in well-differentiated and dedifferentiated liposarcomas whereas CDK4 belongs to a distinct inconsistent amplicon. Int J Cancer (2008) 122(10):2233–41. doi:10.1002/ijc.23380
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Pedeutour F, Forus A, Coindre JM, Berner JM, Nicolo G, Michiels JF, et al. Structure of the supernumerary ring and giant rod chromosomes in adipose tissue tumors. Genes Chromosomes Cancer (1999) 24(1):30–41. doi:10.1002/(SICI)1098-2264(199901)24:1<30::AID-GCC5>3.3.CO;2-G
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Pedeutour F, Suijkerbuijk RF, Forus A, Van Gaal J, Van de Klundert W, Coindre JM, et al. Complex composition and co-amplification of SAS and MDM2 in ring and giant rod marker chromosomes in well-differentiated liposarcoma. Genes Chromosomes Cancer (1994) 10(2):85–94. doi:10.1002/gcc.2870100203
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Rosai J, Akerman M, Dal Cin P, DeWever I, Fletcher CD, Mandahl N, et al. Combined morphologic and karyotypic study of 59 atypical lipomatous tumors. Evaluation of their relationship and differential diagnosis with other adipose tissue tumors (a report of the CHAMP Study Group). Am J Surg Pathol (1996) 20(10):1182–9. doi:10.1097/00000478-199610000-00002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Sirvent N, Coindre JM, Maire G, Hostein I, Keslair F, Guillou L, et al. Detection of MDM2-CDK4 amplification by fluorescence in situ hybridization in 200 paraffin-embedded tumor samples: utility in diagnosing adipocytic lesions and comparison with immunohistochemistry and real-time PCR. Am J Surg Pathol (2007) 31(10):1476–89. doi:10.1097/PAS.0b013e3180581fff
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Tap WD, Eilber FC, Ginther C, Dry SM, Reese N, Barzan-Smith K, et al. Evaluation of well-differentiated/de-differentiated liposarcomas by high-resolution oligonucleotide array-based comparative genomic hybridization. Genes Chromosomes Cancer (2011) 50(2):95–112. doi:10.1002/gcc.20835
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Italiano A, Bianchini L, Gjernes E, Keslair F, Ranchere-Vince D, Dumollard JM, et al. Clinical and biological significance of CDK4 amplification in well-differentiated and dedifferentiated liposarcomas. Clin Cancer Res (2009) 15(18):5696–703. doi:10.1158/1078-0432.CCR-08-3185
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Louis-Brennetot C, Coindre JM, Ferreira C, Perot G, Terrier P, Aurias A. The CDKN2A/CDKN2B/CDK4/CCND1 pathway is pivotal in well-differentiated and dedifferentiated liposarcoma oncogenesis: an analysis of 104 tumors. Genes Chromosomes Cancer (2011) 50(11):896–907. doi:10.1002/gcc.20909
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Lee SE, Kim YJ, Kwon MJ, Choi DI, Lee J, Cho J, et al. High level of CDK4 amplification is a poor prognostic factor in well-differentiated and dedifferentiated liposarcoma. Histol Histopathol (2014) 29(1):127–38.
73. Creytens D, van Gorp J, Ferdinande L, Speel EJ, Libbrecht L. Detection of MDM2/CDK4 amplification in lipomatous soft tissue tumors from formalin-fixed, paraffin-embedded tissue: comparison of multiplex ligation-dependent probe amplification (MLPA) and fluorescence in situ hybridization (FISH). Diagn Mol Pathol (2014). doi:10.1097/PDM.0000000000000041
74. Shimada S, Ishizawa T, Ishizawa K, Matsumura T, Hasegawa T, Hirose T. The value of MDM2 and CDK4 amplification levels using real-time polymerase chain reaction for the differential diagnosis of liposarcomas and their histologic mimickers. Hum Pathol (2006) 37(9):1123–9. doi:10.1016/j.humpath.2006.04.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Weaver J, Downs-Kelly E, Goldblum JR, Turner S, Kulkarni S, Tubbs RR, et al. Fluorescence in situ hybridization for MDM2 gene amplification as a diagnostic tool in lipomatous neoplasms. Mod Pathol (2008) 21(8):943–9. doi:10.1038/modpathol.2008.84
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Barretina J, Taylor BS, Banerji S, Ramos AH, Lagos-Quintana M, Decarolis PL, et al. Subtype-specific genomic alterations define new targets for soft-tissue sarcoma therapy. Nat Genet (2010) 42(8):715–21. doi:10.1038/ng.619
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Ghadimi MP, Liu P, Peng T, Bolshakov S, Young ED, Torres KE, et al. Pleomorphic liposarcoma: clinical observations and molecular variables. Cancer (2011) 117(23):5359–69. doi:10.1002/cncr.26195
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Aleixo PB, Hartmann AA, Menezes IC, Meurer RT, Oliveira AM. Can MDM2 and CDK4 make the diagnosis of well differentiated/dedifferentiated liposarcoma? An immunohistochemical study on 129 soft tissue tumours. J Clin Pathol (2009) 62(12):1127–35. doi:10.1136/jcp.2009.070201
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Alshenawy H. Can HMGI-C be used as an aid with MDM2 and CDK4 to differentiate liposarcoma subtypes from their mimics? J Cancer Res Clin Oncol (2013) 139(6):1073–81. doi:10.1007/s00432-013-1420-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Binh MB, Sastre-Garau X, Guillou L, de Pinieux G, Terrier P, Lagace R, et al. MDM2 and CDK4 immunostainings are useful adjuncts in diagnosing well-differentiated and dedifferentiated liposarcoma subtypes: a comparative analysis of 559 soft tissue neoplasms with genetic data. Am J Surg Pathol (2005) 29(10):1340–7. doi:10.1097/01.pas.0000170343.09562.39
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Zhang H, Erickson-Johnson M, Wang X, Oliveira JL, Nascimento AG, Sim FH, et al. Molecular testing for lipomatous tumors: critical analysis and test recommendations based on the analysis of 405 extremity-based tumors. Am J Surg Pathol (2010) 34(9):1304–11. doi:10.1097/PAS.0b013e3181e92d0b
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Dahlin DC, Beabout JW. Dedifferentiation of low-grade chondrosarcomas. Cancer (1971) 28(2):461–6. doi:10.1002/1097-0142(197108)28:2<461::AID-CNCR2820280227>3.0.CO;2-U
83. Evans HL. Liposarcoma: a study of 55 cases with a reassessment of its classification. Am J Surg Pathol (1979) 3(6):507–23. doi:10.1097/00000478-197912000-00004
84. Coindre JM, Pedeutour F, Aurias A. Well-differentiated and dedifferentiated liposarcomas. Virchows Arch (2010) 456(2):167–79. doi:10.1007/s00428-009-0815-x
85. Aurello P, Virgilio E, Sirimarco D, Novi L, D’Angelo F, Ramacciato G. Dedifferentiated liposarcoma of the retroperitoneum with osteosarcomatous component. Int J Surg Pathol (2013) 21(3):314–5. doi:10.1177/1066896913476749
86. Binh MB, Guillou L, Hostein I, Chateau MC, Collin F, Aurias A, et al. Dedifferentiated liposarcomas with divergent myosarcomatous differentiation developed in the internal trunk: a study of 27 cases and comparison to conventional dedifferentiated liposarcomas and leiomyosarcomas. Am J Surg Pathol (2007) 31(10):1557–66. doi:10.1097/PAS.0b013e31804b4109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Evans HL, Khurana KK, Kemp BL, Ayala AG. Heterologous elements in the dedifferentiated component of dedifferentiated liposarcoma. Am J Surg Pathol (1994) 18(11):1150–7. doi:10.1097/00000478-199411000-00009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Yoshida A, Ushiku T, Motoi T, Shibata T, Fukayama M, Tsuda H. Well-differentiated liposarcoma with low-grade osteosarcomatous component: an underrecognized variant. Am J Surg Pathol (2010) 34(9):1361–6. doi:10.1097/PAS.0b013e3181ebcc45
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Chibon F, Mariani O, Derre J, Malinge S, Coindre JM, Guillou L, et al. A subgroup of malignant fibrous histiocytomas is associated with genetic changes similar to those of well-differentiated liposarcomas. Cancer Genet Cytogenet (2002) 139(1):24–9. doi:10.1016/S0165-4608(02)00614-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Coindre JM, Hostein I, Maire G, Derre J, Guillou L, Leroux A, et al. Inflammatory malignant fibrous histiocytomas and dedifferentiated liposarcomas: histological review, genomic profile, and MDM2 and CDK4 status favour a single entity. J Pathol (2004) 203(3):822–30. doi:10.1002/path.1579
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Coindre JM, Mariani O, Chibon F, Mairal A, De Saint Aubain Somerhausen N, Favre-Guillevin E, et al. Most malignant fibrous histiocytomas developed in the retroperitoneum are dedifferentiated liposarcomas: a review of 25 cases initially diagnosed as malignant fibrous histiocytoma. Mod Pathol (2003) 16(3):256–62. doi:10.1097/01.MP.0000056983.78547.77
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Wang X, Asmann YW, Erickson-Johnson MR, Oliveira JL, Zhang H, Moura RD, et al. High-resolution genomic mapping reveals consistent amplification of the fibroblast growth factor receptor substrate 2 gene in well-differentiated and dedifferentiated liposarcoma. Genes Chromosomes Cancer (2011) 50(11):849–58. doi:10.1002/gcc.20906
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Doyle LA, Tao D, Marino-Enriquez A. STAT6 is amplified in a subset of dedifferentiated liposarcoma. Mod Pathol (2014) 27(9):1231–7. doi:10.1038/modpathol.2013.247
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Doyle LA, Vivero M, Fletcher CD, Mertens F, Hornick JL. Nuclear expression of STAT6 distinguishes solitary fibrous tumor from histologic mimics. Mod Pathol (2014) 27(3):390–5. doi:10.1038/modpathol.2013.164
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Henricks WH, Chu YC, Goldblum JR, Weiss SW. Dedifferentiated liposarcoma: a clinicopathological analysis of 155 cases with a proposal for an expanded definition of dedifferentiation. Am J Surg Pathol (1997) 21(3):271–81. doi:10.1097/00000478-199703000-00002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. McCormick D, Mentzel T, Beham A, Fletcher CD. Dedifferentiated liposarcoma. Clinicopathologic analysis of 32 cases suggesting a better prognostic subgroup among pleomorphic sarcomas. Am J Surg Pathol (1994) 18(12):1213–23. doi:10.1097/00000478-199412000-00004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Goldblum JR. An approach to pleomorphic sarcomas: can we subclassify, and does it matter? Mod Pathol (2014) 27(Suppl 1):S39–46. doi:10.1038/modpathol.2013.174
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Thway K, Flora R, Shah C, Olmos D, Fisher C. Diagnostic utility of p16, CDK4, and MDM2 as an immunohistochemical panel in distinguishing well-differentiated and dedifferentiated liposarcomas from other adipocytic tumors. Am J Surg Pathol (2012) 36(3):462–9. doi:10.1097/PAS.0b013e3182417330
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. Mariani O, Brennetot C, Coindre JM, Gruel N, Ganem C, Delattre O, et al. JUN oncogene amplification and overexpression block adipocytic differentiation in highly aggressive sarcomas. Cancer Cell (2007) 11(4):361–74. doi:10.1016/j.ccr.2007.02.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Chibon F, Mariani O, Derre J, Mairal A, Coindre JM, Guillou L, et al. ASK1 (MAP3K5) as a potential therapeutic target in malignant fibrous histiocytomas with 12q14-q15 and 6q23 amplifications. Genes Chromosomes Cancer (2004) 40(1):32–7. doi:10.1002/gcc.20012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. Rieker RJ, Weitz J, Lehner B, Egerer G, Mueller A, Kasper B, et al. Genomic profiling reveals subsets of dedifferentiated liposarcoma to follow separate molecular pathways. Virchows Arch (2010) 456(3):277–85. doi:10.1007/s00428-009-0869-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
102. Singer S, Socci ND, Ambrosini G, Sambol E, Decarolis P, Wu Y, et al. Gene expression profiling of liposarcoma identifies distinct biological types/subtypes and potential therapeutic targets in well-differentiated and dedifferentiated liposarcoma. Cancer Res (2007) 67(14):6626–36. doi:10.1158/0008-5472.CAN-07-0584
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
103. Snyder EL, Sandstrom DJ, Law K, Fiore C, Sicinska E, Brito J, et al. c-Jun amplification and overexpression are oncogenic in liposarcoma but not always sufficient to inhibit the adipocytic differentiation programme. J Pathol (2009) 218(3):292–300. doi:10.1002/path.2564
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
104. Bonvalot S, Raut CP, Pollock RE, Rutkowski P, Strauss DC, Hayes AJ, et al. Technical considerations in surgery for retroperitoneal sarcomas: position paper from E-Surge, a master class in sarcoma surgery, and EORTC-STBSG. Ann Surg Oncol (2012) 19(9):2981–91. doi:10.1245/s10434-012-2342-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
105. Chew C, Reid R, O’Dwyer PJ. Value of biopsy in the assessment of a retroperitoneal mass. Surgeon (2006) 4(2):79–81. doi:10.1016/S1479-666X(06)80034-X
106. Miah AB, Hannay J, Benson C, Thway K, Messiou C, Hayes AJ, et al. Optimal management of primary retroperitoneal sarcoma: an update. Expert Rev Anticancer Ther (2014) 14(5):565–79. doi:10.1586/14737140.2014.883279
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
107. Chakrabarti I, Bhowmik S, Sinha MGM, Bera P. Ultrasound-guided aspiration cytology of retroperitoneal masses with histopathological corroboration: a study of 71 cases. J Cytol (2014) 31(1):15–9. doi:10.4103/0970-9371.130629
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. Hwang SY, Warrier S, Thompson S, Davidson T, Yang JL, Crowe P. Safety and accuracy of core biopsy in retroperitoneal sarcomas. Asia Pac J Clin Oncol (2013). doi:10.1111/ajco.12125
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
109. Guo Z, Kurtycz DF, De Las Casas LE, Hoerl HD. Radiologically guided percutaneous fine-needle aspiration biopsy of pelvic and retroperitoneal masses: a retrospective study of 68 cases. Diagn Cytopathol (2001) 25(1):43–9. doi:10.1002/dc.2000
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
110. Chau YY, Bandiera R, Serrels A, Martinez-Estrada OM, Qing W, Lee M, et al. Visceral and subcutaneous fat have different origins and evidence supports a mesothelial source. Nat Cell Biol (2014) 16(4):367–75. doi:10.1038/ncb2922
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
111. Gronchi A, Pollock RE. Quality of local treatment or biology of the tumor: which are the trump cards for loco-regional control of retroperitoneal sarcoma? Ann Surg Oncol (2013) 20(7):2111–3. doi:10.1245/s10434-013-2971-0
112. Strauss DC. Patterns of recurrence in retroperitoneal liposarcomas: reflecting surgical approach or tumor biology? Ann Surg Oncol (2014) 21(7):2113–6. doi:10.1245/s10434-014-3644-3
113. Kilkenny JW, Bland KI, Copeland EM. Retroperitoneal sarcoma: the University of Florida experience. J Am Coll Surg (1996) 182(4):329–39.
114. Toulmonde M, Bonvalot S, Ray-Coquard I, Stoeckle E, Riou O, Isambert N, et al. Retroperitoneal sarcomas: patterns of care in advanced stages, prognostic factors and focus on main histological subtypes: a multicenter analysis of the French Sarcoma Group. Ann Oncol (2014) 25(3):730–4. doi:10.1093/annonc/mdt576
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
115. Beane JD, Yang JC, White D, Steinberg SM, Rosenberg SA, Rudloff U. Efficacy of adjuvant radiation therapy in the treatment of soft tissue sarcoma of the extremity: 20-year follow-up of a randomized prospective trial. Ann Surg Oncol (2014) 21(8):2484–9. doi:10.1245/s10434-014-3732-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
116. Harrison LB, Franzese F, Gaynor JJ, Brennan MF. Long-term results of a prospective randomized trial of adjuvant brachytherapy in the management of completely resected soft tissue sarcomas of the extremity and superficial trunk. Int J Radiat Oncol Biol Phys (1993) 27(2):259–65. doi:10.1016/0360-3016(93)90236-O
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
117. Yang JC, Chang AE, Baker AR, Sindelar WF, Danforth DN, Topalian SL, et al. Randomized prospective study of the benefit of adjuvant radiation therapy in the treatment of soft tissue sarcomas of the extremity. J Clin Oncol (1998) 16(1):197–203.
118. Mohindra P, Neuman HB, Kozak KR. The role of radiation in retroperitoneal sarcomas. Curr Treat Options Oncol (2013) 14(3):425–41. doi:10.1007/s11864-013-0236-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
119. Stucky CCH, Wasif N, Ashman JB, Pockaj BA, Gunderson LL, Gray RJ. Excellent local control with preoperative radiation therapy, surgical resection, and intra-operative electron radiation therapy for retroperitoneal sarcoma. J Surg Oncol (2014) 109(8):798–803. doi:10.1002/jso.23576
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
120. Gieschen HL, Spiro IJ, Suit HD, Ott MJ, Rattner DW, Ancukiewicz M, et al. Long-term results of intraoperative electron beam radiotherapy for primary and recurrent retroperitoneal soft tissue sarcoma. Int J Radiat Oncol Biol Phys (2001) 50(1):127–31. doi:10.1016/S0360-3016(00)01589-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
121. Pawlik TM, Pisters PW, Mikula L, Feig BW, Hunt KK, Cormier JN, et al. Long-term results of two prospective trials of preoperative external beam radiotherapy for localized intermediate- or high-grade retroperitoneal soft tissue sarcoma. Ann Surg Oncol (2006) 13(4):508–17. doi:10.1245/ASO.2006.05.035
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
122. Tzeng CWD, Fiveash JB, Popple RA, Arnoletti JP, Russo SM, Urist MM, et al. Preoperative radiation therapy with selective dose escalation to the margin at risk for retroperitoneal sarcoma. Cancer (2006) 107(2):371–9. doi:10.1002/cncr.22005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
123. White JS, Biberdorf D, DiFrancesco LM, Kurien E, Temple W. Use of tissue expanders and pre-operative external beam radiotherapy in the treatment of retroperitoneal sarcoma. Ann Surg Oncol (2007) 14(2):583–90. doi:10.1245/s10434-006-9139-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
124. Caudle AS, Tepper JE, Calvo BF, Meyers MO, Goyal LK, Cance WG, et al. Complications associated with neoadjuvant radiotherapy in the multidisciplinary treatment of retroperitoneal sarcomas. Ann Surg Oncol (2007) 14(2):577–82. doi:10.1245/s10434-006-9248-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
125. Ballo MT, Zagars GK, Pollock RE, Benjamin RS, Feig BW, Cormier JN, et al. Retroperitoneal soft tissue sarcoma: an analysis of radiation and surgical treatment. Int J Radiat Oncol Biol Phys (2007) 67(1):158–63. doi:10.1016/j.ijrobp.2006.08.025
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
126. Yoon SS, Chen YL, Kirsch DG, Maduekwe UN, Rosenberg AE, Nielsen GP, et al. Proton-beam, intensity-modulated, and/or intraoperative electron radiation therapy combined with aggressive anterior surgical resection for retroperitoneal sarcomas. Ann Surg Oncol (2010) 17(6):1515–29. doi:10.1245/s10434-010-0935-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
127. Alford S, Choong P, Chander S, Henderson M, Powell G, Ngan S. Outcomes of preoperative radiotherapy and resection of retroperitoneal sarcoma. ANZ J Surg (2013) 83(5):336–41. doi:10.1111/j.1445-2197.2012.06211.x
128. McBride SM, Raut CP, Lapidus M, Devlin PM, Marcus KJ, Bertagnolli M, et al. Locoregional recurrence after preoperative radiation therapy for retroperitoneal sarcoma: adverse impact of multifocal disease and potential implications of dose escalation. Ann Surg Oncol (2013) 20(7):2140–7. doi:10.1245/s10434-013-2868-y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
129. Sweeting RS, Deal AM, Llaguna OH, Bednarski BK, Meyers MO, Yeh JJ, et al. Intraoperative electron radiation therapy as an important treatment modality in retroperitoneal sarcoma. J Surg Res (2013) 185(1):245–9. doi:10.1016/j.jss.2013.05.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
130. Smith MJ, Ridgway PF, Catton CN, Cannell AJ, O’Sullivan B, Mikula LA, et al. Combined management of retroperitoneal sarcoma with dose intensification radiotherapy and resection: long-term results of a prospective trial. Radiother Oncol (2014) 110(1):165–71. doi:10.1016/j.radonc.2013.10.041
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
131. Gronchi A, De Paoli A, Dani C, Merlo DF, Quagliuolo V, Grignani G, et al. Preoperative chemo-radiation therapy for localised retroperitoneal sarcoma: a phase I-II study from the Italian Sarcoma Group. Eur J Cancer (2014) 50(4):784–92. doi:10.1016/j.ejca.2013.11.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
132. Porter GA, Baxter NN, Pisters PWT. Retroperitoneal sarcoma: a population-based analysis of epidemiology, surgery, and radiotherapy. Cancer (2006) 106(7):1610–6. doi:10.1002/cncr.21761
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
133. Nathan H, Raut CP, Thornton K, Herman JM, Ahuja N, Schulick RD, et al. Predictors of survival after resection of retroperitoneal sarcoma: a population-based analysis and critical appraisal of the AJCC staging system. Ann Surg (2009) 250(6):970–6. doi:10.1097/SLA.0b013e3181b25183
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
134. Zhou Z, McDade TP, Simons JP, Ng SC, Lambert LA, Whalen GF, et al. Surgery and radiotherapy for retroperitoneal and abdominal sarcoma both necessary and sufficient. Arch Surg (2010) 145(5):426–31. doi:10.1001/archsurg.2010.70
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
135. Tseng WH, Martinez SR, Do L, Tamurian RM, Borys D, Canter RJ. Lack of survival benefit following adjuvant radiation in patients with retroperitoneal sarcoma: a SEER analysis. J Surg Res (2011) 168(2):E173–80. doi:10.1016/j.jss.2011.02.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
136. Choi AH, Barnholtz-Sloan JS, Kim JA. Effect of radiation therapy on survival in surgically resected retroperitoneal sarcoma: a propensity score-adjusted SEER analysis. Ann Oncol (2012) 23(9):2449–57. doi:10.1093/annonc/mdr616
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
137. Krikelis D, Judson I. Role of chemotherapy in the management of soft tissue sarcomas. Expert Rev Anticancer Ther (2010) 10(2):249–60. doi:10.1586/era.09.176
138. Constantinidou A, Pollack S, Loggers E, Rodler E, Jones RL. The evolution of systemic therapy in sarcoma. Expert Rev Anticancer Ther (2013) 13(2):211–23. doi:10.1586/era.12.161
139. Jones RL, Fisher C, Al-Muderis O, Judson IR. Differential sensitivity of liposarcoma subtypes to chemotherapy. Eur J Cancer (2005) 41(18):2853–60. doi:10.1016/j.ejca.2005.07.023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
140. Hoffman A, Lazar AJ, Pollock RE, Lev D. New frontiers in the treatment of liposarcoma, a therapeutically resistant malignant cohort. Drug Resist Updat (2011) 14(1):52–66. doi:10.1016/j.drup.2010.11.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
141. Constantinidou A, Pollack SM, Jones RL. MDM2 inhibition in liposarcoma: a step in the right direction. Lancet Oncol (2012) 13(11):1070–1. doi:10.1016/S1470-2045(12)70457-6
142. Dickson MA, Tap WD, Keohan ML, D’Angelo SP, Gounder MM, Antonescu CR, et al. Phase II trial of the CDK4 inhibitor PD0332991 in patients with advanced CDK4-amplified well-differentiated or dedifferentiated liposarcoma. J Clin Oncol (2013) 31(16):2024–8. doi:10.1200/JCO.2012.46.5476
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
143. Ray-Coquard I, Blay JY, Italiano A, Le Cesne A, Penel N, Zhi J, et al. Effect of the MDM2 antagonist RG7112 on the P53 pathway in patients with MDM2-amplified, well-differentiated or dedifferentiated liposarcoma: an exploratory proof-of-mechanism study. Lancet Oncol (2012) 13(11):1133–40. doi:10.1016/S1470-2045(12)70474-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
144. Conyers R, Young S, Thomas DM. Liposarcoma: molecular genetics and therapeutics. Sarcoma (2011) 2011:483154. doi:10.1155/2011/483154
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
145. Blay JY, Casali P, Nieto A, Tanovic A, Le Cesne A. Efficacy and safety of trabectedin as an early treatment for advanced or metastatic liposarcoma and leiomyosarcoma. Future Oncol (2014) 10(1):59–68. doi:10.2217/fon.13.163
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
146. Demetri GD, Chawla SP, von Mehren M, Ritch P, Baker LH, Blay JY, et al. Efficacy and safety of trabectedin in patients with advanced or metastatic liposarcoma or leiomyosarcoma after failure of prior anthracyclines and ifosfamide: results of a randomized phase II study of two different schedules. J Clin Oncol (2009) 27(25):4188–96. doi:10.1200/JCO.2008.21.0088
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
147. Gronchi A, Bui BN, Bonvalot S, Pilotti S, Ferrari S, Hohenberger P, et al. Phase II clinical trial of neoadjuvant trabectedin in patients with advanced localized myxoid liposarcoma. Ann Oncol (2012) 23(3):771–6. doi:10.1093/annonc/mdr265
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
148. Schoffski P, Ray-Coquard IL, Cioffi A, Bui NB, Bauer S, Hartmann JT, et al. Activity of eribulin mesylate in patients with soft-tissue sarcoma: a phase 2 study in four independent histological subtypes. Lancet Oncol (2011) 12(11):1045–52. doi:10.1016/S1470-2045(11)70230-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
149. Tovar C, Graves B, Packman K, Filipovic Z, Higgins B, Xia M, et al. MDM2 small-molecule antagonist RG7112 activates p53 signaling and regresses human tumors in preclinical cancer models. Cancer Res (2013) 73(8):2587–97. doi:10.1158/0008-5472.CAN-12-2807
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
150. Ding Q, Zhang Z, Liu JJ, Jiang N, Zhang J, Ross TM, et al. Discovery of RG7388, a potent and selective p53-MDM2 inhibitor in clinical development. J Med Chem (2013) 56(14):5979–83. doi:10.1021/jm400487c
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
151. Luke JJ, D’Adamo DR, Dickson MA, Keohan ML, Carvajal RD, Maki RG, et al. The cyclin-dependent kinase inhibitor flavopiridol potentiates doxorubicin efficacy in advanced sarcomas: preclinical investigations and results of a phase I dose-escalation clinical trial. Clin Cancer Res (2012) 18(9):2638–47. doi:10.1158/1078-0432.CCR-11-3203
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
152. Demetri GD, Fletcher CD, Mueller E, Sarraf P, Naujoks R, Campbell N, et al. Induction of solid tumor differentiation by the peroxisome proliferator-activated receptor-gamma ligand troglitazone in patients with liposarcoma. Proc Natl Acad Sci U S A (1999) 96(7):3951–6. doi:10.1073/pnas.96.7.3951
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
153. Debrock G, Vanhentenrijk V, Sciot R, Debiec-Rychter M, Oyen R, Van Oosterom A. A phase II trial with rosiglitazone in liposarcoma patients. Br J Cancer (2003) 89(8):1409–12. doi:10.1038/sj.bjc.6601306
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
154. Pishvaian MJ, Marshall JL, Wagner AJ, Hwang JJ, Malik S, Cotarla I, et al. A phase 1 study of efatutazone, an oral peroxisome proliferator-activated receptor gamma agonist, administered to patients with advanced malignancies. Cancer (2012) 118(21):5403–13. doi:10.1002/cncr.27526
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
155. Guan M, Fousek K, Jiang C, Guo S, Synold T, Xi B, et al. Nelfinavir induces liposarcoma apoptosis through inhibition of regulated intramembrane proteolysis of SREBP-1 and ATF6. Clin Cancer Res (2011) 17(7):1796–806. doi:10.1158/1078-0432.CCR-10-3216
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
156. Pan J, Mott M, Xi B, Hepner E, Guan M, Fousek K, et al. Phase I study of nelfinavir in liposarcoma. Cancer Chemother Pharmacol (2012) 70(6):791–9. doi:10.1007/s00280-012-1961-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
157. Mahmood ST, Agresta S, Vigil CE, Zhao X, Han G, D’Amato G, et al. Phase II study of sunitinib malate, a multitargeted tyrosine kinase inhibitor in patients with relapsed or refractory soft tissue sarcomas. Focus on three prevalent histologies: leiomyosarcoma, liposarcoma and malignant fibrous histiocytoma. Int J Cancer (2011) 129(8):1963–9. doi:10.1002/ijc.25843
158. Cassier PA, Lefranc A, Amela EY, Chevreau C, Bui BN, Lecesne A, et al. A phase II trial of panobinostat in patients with advanced pretreated soft tissue sarcoma. A study from the French Sarcoma Group. Br J Cancer (2013) 109(4):909–14. doi:10.1038/bjc.2013.442
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
159. Sleijfer S, Ray-Coquard I, Papai Z, Le Cesne A, Scurr M, Schoffski P, et al. Pazopanib, a multikinase angiogenesis inhibitor, in patients with relapsed or refractory advanced soft tissue sarcoma: a phase II study from the European organisation for research and treatment of cancer-soft tissue and bone sarcoma group (EORTC study 62043). J Clin Oncol (2009) 27(19):3126–32. doi:10.1200/JCO.2008.21.3223
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
160. von Mehren M, Rankin C, Goldblum JR, Demetri GD, Bramwell V, Ryan CW, et al. Phase 2 Southwest Oncology Group-directed intergroup trial (S0505) of sorafenib in advanced soft tissue sarcomas. Cancer (2012) 118(3):770–6. doi:10.1002/cncr.26334
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: liposarcoma, sarcoma, surgery, radiotherapy, MDM2
Citation: Matthyssens LE, Creytens D and Ceelen WP (2015) Retroperitoneal liposarcoma: current insights in diagnosis and treatment. Front. Surg. 2:4. doi: 10.3389/fsurg.2015.00004
Received: 30 October 2014; Accepted: 28 January 2015;
Published online: 10 February 2015.
Edited by:
Giulia Veronesi, European Institute of Oncology, ItalyReviewed by:
Tomoo Iwakuma, University of Kansas Medical Center, USAElisabetta Pennacchioli, European Institute of Oncology, Italy
Copyright: © 2015 Matthyssens, Creytens and Ceelen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wim P. Ceelen, Department of Gastrointestinal Surgery, UZ Gent, 2K12 IC, De Pintelaan 185, Ghent B-9000, Belgium e-mail: wim.ceelen@ugent.be