Endocrinology Unit, Centre for Cardiovascular Science, The Queen’s Medical Research Institute, University of Edinburgh, Edinburgh, Scotland
Numerous clinical studies associate an adverse prenatal environment with the development of cardio-metabolic disorders and neuroendocrine dysfunction, as well as an increased risk of psychiatric diseases in later life. Experimentally, prenatal exposure to stress or excess glucocorticoids in a variety of animal models can malprogram offspring physiology, resulting in a reduction in birth weight and subsequently increasing the likelihood of disorders of cardiovascular function, glucose homeostasis, hypothalamic–pituitary–adrenal (HPA) axis activity and anxiety-related behaviours in adulthood. During fetal development, placental 11β-hydroxysteroid dehydrogenase type 2 (11β-HSD2) provides a barrier to maternal glucocorticoids. Reduced placental 11β-HSD2 in human pregnancy correlates with lower birth weight and higher blood pressure in later life. Similarly, in animal models, inhibition or knockout of placental 11β-HSD2 lowers offspring birth weight, in part by reducing glucose delivery to the developing fetus in late gestation. Molecular mechanisms thought to underlie the programming effects of early life stress and glucocorticoids include epigenetic changes in target chromatin, notably affecting tissue-specific expression of the intracellular glucocorticoid receptor (GR). As such, excess glucocorticoids in early life can permanently alter tissue glucocorticoid signalling, effects which may have short-term adaptive benefits but increase the risk of later disease.
It is now widely recognized that exposure to an adverse environment during prenatal development can have lasting effects on an individual’s physiology and risk of disease. Maternal malnutrition, psychological stress or perturbed hormonal status (e.g. glucocorticoid overexposure or the hyperinsulinemia associated with gestational diabetes) transmit signals to the developing fetus and, depending on the timing of these exposures, permanently affect tissue structure and function. Although fetal responses to such exposures may impart an enhanced capacity to cope with the immediate stressor, such in utero ‘developmental programming’ can contribute a significant risk for a number of important adult diseases. For instance, adaptations to an intrauterine environment in which oxygen and nutrients may be limited has been suggested to promote the development of a ‘thrifty phenotype’, reducing fetal growth and favouring metabolic efficiency (Hales and Barker, 1992
).
In terms of developmental metabolic programming, numerous studies in both humans and animal models have shown that a poor intrauterine environment followed by postnatal overnutrition promotes the development of diseases including hypertension, insulin resistance, central obesity and type 2 diabetes (Gluckman et al., 2005
). Maternal stress during pregnancy is also associated with intrauterine growth restriction (IUGR) and an increased risk of premature birth (Rondo et al., 2003
), as well as with emotional and cognitive deficits in early life (Talge et al., 2007
). Thus, low birth weight as an indicator of a suboptimal intrauterine environment has been used extensively as a predictor of later disease risk (Barker et al., 1993
). Although low birth weight is a relatively crude marker of the prenatal environment, robust associations are found between reduced birth weight and increased risk of later disease in numerous populations worldwide (Whincup et al., 2008
). Furthermore, these associations are continuous across a wide range of normal birth weights, and act largely independently of other lifestyle factors such as socioeconomic status and current obesity (Barker, 2004
).
Two major hypotheses have been proposed to underlie these associations between adverse prenatal conditions and postnatal health – fetal malnutrition (Barker et al., 1993
) and overexposure to glucocorticoids (Edwards et al., 1993
; Seckl, 2004
). Importantly, these hypotheses are not mutually exclusive. Alterations in maternal nutrition or placental dysfunction, which result in reduced nutrient delivery to the fetus, also affect fetal glucocorticoid exposure. Conversely, antenatal stress or maternal administration of glucocorticoids affects feeding behaviour. Given their potent actions during development, excess glucocorticoids likely represent a common pathway by which poor environmental conditions are signalled from the mother to the fetus, triggering changes in offspring growth and permanently affecting tissue and organ function.
Glucocorticoids, steroid hormones produced predominantly by the adrenal gland, are key mediators of stress responses. Whilst the acute and chronic effects of pharmacological glucocorticoid excess are well-recognised (including induction of hyperglycemia, insulin resistance, hyperlipidemia, hypertension and dysphoria, with suppression of immune, inflammatory and cognitive processes), their role in the biology of the response to stress is more nuanced, with balanced homeostatic effects to facilitate short-term survival and recovery from challenge (Munck and Naray-Fejes-Toth, 1994
; McEwen, 2007
). In addition, glucocorticoids are crucial during fetal development for the maturation of tissues and organs, promoting cellular differentiation, and most notably acting during late gestation to stimulate surfactant production by the lung. This action is critical to prepare the fetus for extrauterine life, and it is for this reason that synthetic glucocorticoid treatment is so widely used in preterm pregnancies where lung immaturity threatens neonatal viability. Although these treatments greatly improve survival (Roberts and Dalziel, 2006
), they are not without adverse affects.
Numerous studies across a wide range of species have shown that prenatal treatment with glucocorticoids reduces birth weight and that these offspring are at increased risk of cardio-metabolic disease, hypothalamic–pituitary–adrenal (HPA) axis perturbations and affective disorders in later life (Seckl, 2004
). Moreover, in human pregnancies complicated by intrauterine growth restriction (IUGR), fetal cortisol levels are elevated at term (Goland et al., 1993
), associating reduced fetal growth rates with elevated glucocorticoids. The effects of exogenous glucocorticoids on birth weight are greatest when administered during periods of rapid growth, typically the later stages of pregnancy (Nyirenda et al., 1998
). It has been shown in rats that excess maternal glucocorticoids are key in mediating the effects of maternal stress or diet on offspring hypertension (Langley-Evans, 1997a
; Lesage et al., 2001
). Inhibition of maternal glucocorticoid synthesis using metyrapone in pregnant rats prevents the increased blood pressure seen in offspring of low protein diet fed rat dams. Replacement of corticosterone to the dam reinstated the programming effects, interestingly only in female offspring (Langley-Evans, 1997a
). Maternal adrenalectomy likewise prevented the reduction in fetal adrenal weights seen with maternal food restriction, however fetal body weights were still lower in adrenalectomised and food-restricted fetuses (Lesage et al., 2001
). These studies lend support to the idea that maternal glucocorticoids mediate at least a component of the programming effects on offspring HPA axis perturbations on fetal development.
A number of the adult diseases associated with the ‘low birth weight baby syndrome’ involve perturbations in key glucocorticoid-responsive tissues, notably liver, adipose tissue and brain. As described below, a re-setting of HPA axis sensitivity is thought to underlie many of the cardio-metabolic outcomes associated with low birth weight and glucocorticoid overexposure. Furthermore, many of the affective disorders that are linked to prenatal stress involve glucocorticoid-sensitive central nervous system (CNS) targets. Excess glucocorticoid exposure in late pregnancy can also induce long-lasting effects on peripheral tissue expression of glucocorticoid-sensitive genes. In rat models of prenatal glucocorticoid exposure or maternal malnutrition, the hepatic glucocorticoid receptor (GR) is upregulated, plausibly driving the observed increase in both the expression and activity of the gluconeogenic enzyme phosphoenolypyruvate carboyxkinase (PEPCK), and programming insulin resistance and impaired glucose tolerance in adult life (Hales et al., 1996
; Ozanne et al., 1996
; Nyirenda et al., 1998
).
It is clear that the effects of prenatal stress and glucocorticoid excess during pregnancy on subsequent physiological and psychological outcomes differ not only due to the timing of exposure, but also on the sex of the offspring. In human pregnancy, the placenta of female fetuses may impart a relative protection from glucocorticoid excess due to increased glucocorticoid inactivation (discussed in more detail below) compared with males (Clifton and Murphy, 2004
). Recent data in the rat has also shown that male, but not female, offspring exposed to stress in early gestation exhibited increased anxiety-related behaviours as adults, again changes associated with sex-differences in placental function (Mueller and Bale, 2008
). However, the precise mechanisms of sex-specific susceptibility to prenatal insults is not yet clear. Likewise, the issue of timing is clearly species specific, and presumably related to the expression of GR and mineralocorticoid receptors (MR) in relation to the exposure. These issues are reviewed in more detail elsewhere (Matthews et al., 2002
; Kapoor et al., 2008
, 2009
).
Placental 11β-Hydroxysteroid Dehydrogenase Type 2
In normal pregnancy, maternal glucocorticoid levels are markedly higher than those in the fetal circulation. The fetus is protected from the relatively high maternal glucocorticoid levels of pregnancy by the placental inactivation of active glucocorticoids (cortisol in humans, corticosterone in rodents) to its inactive 11-keto forms (cortisone and 11-dehydrocorticosterone, respectively) by the enzyme 11β-hydroxysteroid dehydrogenase type 2 (11β-HSD2). This enzyme acts as a ‘barrier’ to prevent premature or inappropriate action at glucocorticoid-responsive tissues during fetal development.
It has been suggested that a reduction in the expression or activity of placental 11β-HSD2, by leading to increased trans-placental passage of active glucocorticoids, reduces fetal growth. In support of this notion, in human and rodent pregnancy fetal weight is correlated with 11β-HSD2 activity (Stewart et al., 1995
; Lindsay et al., 1996
; Murphy et al., 2002
). In addition, mutations in the HSD11B2 gene in humans, although rare, markedly reduce birth weight (Dave-Sharma et al., 1998
). Numerous studies have shown that inhibition of 11β-HSD2 during pregnancy leads to a reduction in birth weight and the development of later hypertension and glucose intolerance (Edwards et al., 1993
; Lindsay et al., 1996
; Langley-Evans, 1997b
), as well as programming increased HPA axis activity and anxiety-related behaviours (Welberg et al., 2000
). More recently, genetic manipulations have shown that 11β-HSD2 knockout mice (11β-HSD2−/−) exhibit reduced birth weight and heightened anxiety in adulthood (Holmes et al., 2006
). Moreover, heterozygous matings of 11β-HSD2+/− animals have shown that allelic deficiency in fetal 11β-HSD2 and birth weight follow a dose–response pattern. Within the same litter from heterozygous crosses, homozygous knockout offspring (11β-HSD2−/−) are significantly lighter than wildtype littermates (11β-HSD2+/+), with heterozygotes intermediate, suggesting sensitive gene dosage effects (Holmes et al., 2006
; Wyrwoll et al., 2009
). Studies using this model have recently shown that placental transport function, clearly important in the determination of fetal growth and offspring birth weight, is impaired in late gestation in 11β-HSD2−/− fetuses (Wyrwoll et al., 2009
). It is not yet clear whether excess fetal glucocorticoids down-regulate placental function per se and thus reduce fetal growth, although one study in rats has suggested that reduced placental weight and glucose transport in response to maternal low protein diet precedes any significant reduction in fetal weight, suggesting that reduced nutrient transport is causal in IUGR (Jansson et al., 2006
). Alternatively, it may be that in response to fetal growth restriction (due to maternal malnutrition or elevated fetal glucocorticoids secondary to reduced placental 11β-HSD2), the placenta is able to up-regulate transport function and increase its efficiency as a compensatory mechanism to a point, but that this is insufficient to sustain normal fetal growth during the last stages of pregnancy when growth is most rapid.
Interestingly, it appears that there are gene-interaction effects on 11β-HSD2 function. Initial studies investigating the effects of a complete lack of 11β-HSD2 reported that although there was clearly an effect on blood pressure resulting from a lack of this gene, that fetal weight was unaltered (Kotelevtsev et al., 1999
). This has been suggested to be due perhaps to the crossed background used in this mouse model, compared with that of the more recent 11β-HSD2 transgenic models mentioned above which are maintained on the C57/Bl6 background (Seckl and Meaney, 2006
). More recently, a study using rats selectively bred to exhibit high or low levels of anxiety behaviour (HAB and LAB rats respectively) revealed that in response to repeated maternal stress throughout pregnancy, 11β-HSD2 activity was reduced only in the placentas of HAB offspring (Lucassen et al., 2009
). This segregation of behavioural traits in association with differences in molecular responses of a known glucocorticoid programming related gene are intriguing, however the identity of potential modifying genes are at present unknown.
Importantly, placental 11β-HSD2 may be a crucial node between the maternal environment and the fetus. Thus, maternal undernutrition or ingestion of a low protein diet (which also lower birth weight and leads to cardio-metabolic syndrome in the offspring) decreases the expression and/or activity of placental 11β-HSD2 (Langley-Evans et al., 1996
; Lesage et al., 2001
; Stocker et al., 2004
). Maternal stress similarly reduces placental 11β-HSD2 (Mairesse et al., 2007
). In vitro studies using placental cells lines have also indicated that a number of other factors, including hypoxia, catecholamines and proinflammatory cytokines, can down-regulate 11β-HSD2 activity (Hardy and Yang, 2002
; Chisaka et al., 2005
; Homan et al., 2006
). These findings are of particular relevance to human situations of placental insufficiency or maternal infection. A proposed model for fetal programming by altered placental function and/or glucocorticoid overexposure is indicated in Figure 1
.
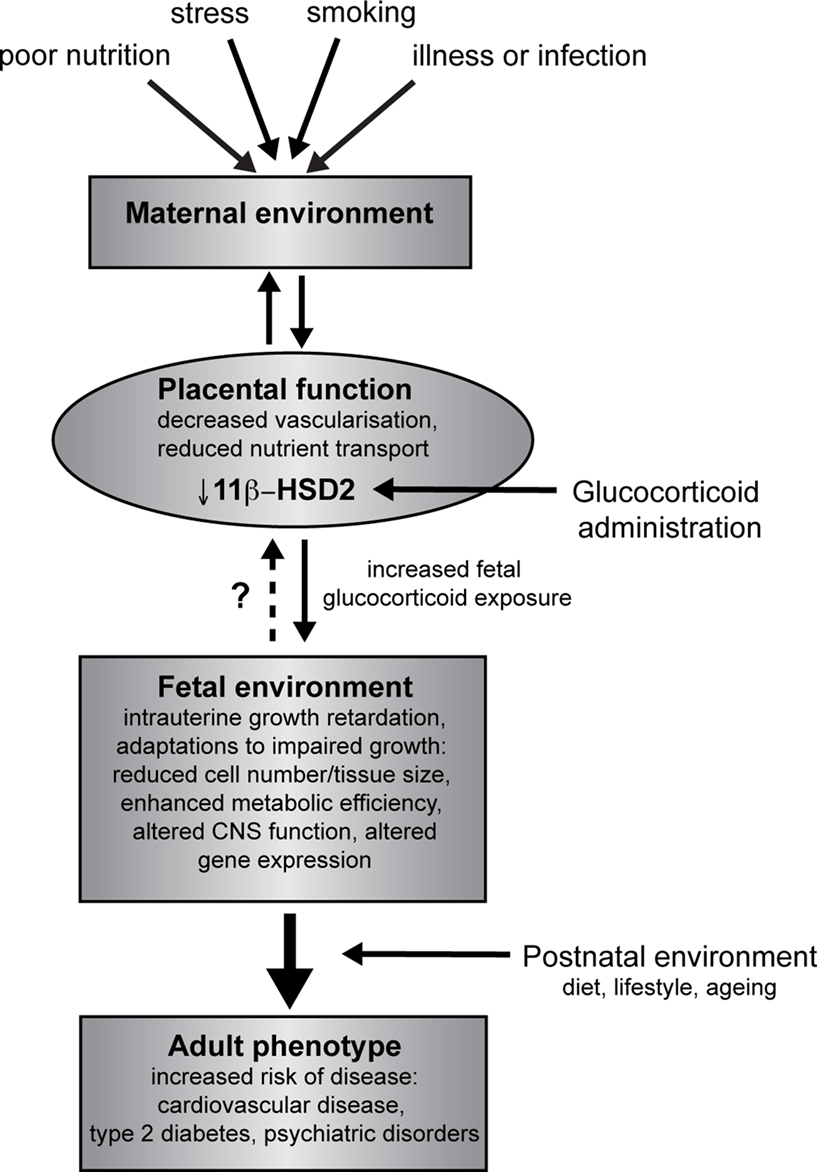
Figure 1. Overview of developmental programming. A perturbed maternal environment, due to poor diet, excessive stress or impaired health during pregnancy, impairs placental function and thus delivery of nutrients (critically glucose and oxygen) to the fetus. In addition, administration of exogenous glucocorticoids reduces the activity of placental 11β-HSD2, decreasing this barrier to excess glucocorticoids and reducing fetal growth. In response to these stressors, the developing fetus is able to adapt to the prevailing conditions in order to promote survival, altering tissue size, structure and function. These adaptations, although beneficial in the short term, when conflicted by postnatal conditions predispose to an increased risk of adult disease.
Glucocorticoid Receptor
During development, GRs are expressed from early embryonic life in most tissues. Glucocorticoid receptors are essential for offspring survival, as indicated by the lethal postnatal phenotype of GR null mice (Cole et al., 1995
). The use of transgenic mouse models has identified that variation in the level of GR alters stress responses and activity of the HPA axis. A 30–50% reduction in GR is associated with exaggerated HPA responses to stress (Pepin et al., 1992
; Michailidou et al., 2008
), whereas the generation of transgenic GR-overexpressing animals produces mice with a relatively stress-resistant phenotype (Reichardt et al., 2000
; Wei et al., 2004
).
Exposure of the fetus to stress or high levels of glucocorticoids – whether from exogenous or endogenous origins – can permanently affect GR expression. For instance, inhibition or deficiency of placental 11β-HSD2 has been shown to reduce hippocampal GR expression (Levitt et al., 1996
) but conversely increases amygdala GR mRNA levels (Welberg et al., 2000
). A reduction in hippocampal GR would be expected to reduce glucocorticoid negative feedback and lead to an overactive HPA axis. In contrast, increased GR expression in the amygdala, a key CNS structure involved in mediating fear and anxiety responses, associates with an anxiogenic phenotype in rodents (Welberg et al., 2000
).
Although present in almost all cells, there is considerable variation in GR expression between different tissues and, in the case of the CNS, even between different brain nuclei (Herman et al., 1989
). The receptor protein itself is encoded by exons 2–9, however the complexity of such tissue-specific regulation is conferred by the presence of multiple alternative untranslated first exons (McCormick et al., 2000
; Turner and Muller, 2005
). There is currently extensive evidence that at least a component of the programming effects induced by early life events involves alterations in expression of specific alternative first exons of GR to regulate tissue-specific changes in expression and function, at least in the brain.
In rats, exposure to high levels of postnatal care (high levels of licking and grooming by the dam) leads to increased GR mRNA expression in the hippocampus and reduced HPA axis responses to restraint stress as adults compared with offspring raised by low licking-grooming (low-LG) dams (Liu et al., 1997
). Postnatal handling, which removes the differences in hippocampal GR expression, eliminates these maternal programming effects (Meaney et al., 1989
). In addition, cross-fostering studies indicate that the effect on GR expression is directly related to the level of maternal care (Francis et al., 1999
). Whilst there is no difference at postnatal day 1 (P1) in gene expression patterns between low- and high-LG offspring, by P6 the differences are apparent (Weaver et al., 2004
). Thus, differences in exposure to nurturing behaviour in early life are sufficient to induce long-term programming of GR expression.
This increase in GR expression is mediated by activation of serotonergic (5-HT) pathways in response to tactile stimulation (ie, handling or maternal licking/grooming; Smythe et al., 1994
). Increased 5-HT signalling (acting via cyclic AMP-activated pathways) increases the expression of key transcription factors, notably nerve growth factor induced protein A (NGFI-A), which binds to and regulates activation of the GR promoter. In the hippocampus, NGFI-A binds promoter DNA, specifically within NGFI-A binding sites of exon 17, increasing expression of this mRNA transcript (Weaver et al., 2007
).
Mechanistically, differences in GR expression between the offspring of high- and low-LG females associate with differential methylation of the exon 17 GR promoter (Weaver et al., 2004
, 2007
). DNA hypermethylation, along with histone hypoacetylation, are epigenetic modifications associated with relatively inactive chromatin and reduced gene expression. In general, regions of the genome that are particularly susceptible to epigenetic modification include gene promoter regions, regulatory regions of imprinted genes and transposable elements (for review, see Jirtle and Skinner, 2007
). In low-LG offspring, the exon 17 promoter is hypermethylated, histone H3 hypoacetylated and there is reduced binding of the activating transcription factor NGFI-A. Conversely, in offspring of high-LG mothers, the reverse situation was found, with significantly lower levels of methylation, increased H3 acetylation and increased NGFI-A binding (Weaver et al., 2004
).
Although epigenetic DNA modifications are typically thought of as being stable once established, pharmacological manipulation of acetylation and methylation patterns have been shown in low- and high-LG offspring. Low GR expression in the low-LG offspring can be reversed by the central infusion of a histone deacetylase inhibitor, trichostatin A (TSA), which increases acetylation of histone H3, reduces methylation of the exon 17 region and increases NGFI-A binding in the adult rat brain (Weaver et al., 2004
). The higher levels of GR in high-LG offspring can be reduced by the administration of the methyl donor S-adenosyl-methionine (SAM), through increased DNA methylation of exon 17 (Weaver et al., 2005
). The combined results of these studies imply that there is a degree of plasticity that remains in the adult rodent brain for alterations in gene expression by epigenetic modification. Crucial appears to be transcription factor binding to critical DNA response elements at key sensitive developmental ages to maintain open chromatin for the lifespan.
Recent data from human studies have indicated that these same hippocampal GR targets might also be subject to programming by early life events. Analysis of brain tissue from suicide victims who were abused as children indicated a higher level of NR3C1 exon 1F methylation (the human homolog of the rat exon 17 region) compared with non-abused suicide victims or control subjects (McGowan et al., 2009
). These findings lend further support to the idea that long-term stress and behavioural outcomes might be influenced by events in early life, and are consistent with previously reported HPA axis disturbances, induced by aversive conditions in childhood, on subsequent adult psychiatric health (Heim and Nemeroff, 2001
). This mechanism, whilst intriguing, remains as yet to be shown in systems outside the brain. However, it is a plausible ‘cause’ of aspects of developmental programming phenomena.
Corticotropin Releasing Hormone
A critical component of the central HPA axis (detailed below), corticotrophin releasing hormone (CRH) has also been proposed as a mediator of the effects of early life stress on later cognitive and behavioural outcomes. In humans CRH is produced by the placenta and released into the maternal and fetal circulation (Goland et al., 1993
). High concentrations of CRH are found in growth-retarded fetuses, and elevated maternal CRH levels are associated with decreased gestational length and an increased risk of preterm delivery (Wadhwa et al., 1998
). In contrast to the negative feedback actions of glucocorticoids on the expression and release of central CRH, placental production is enhanced by glucocorticoids (King et al., 2001
). Hence, maternal stress, accompanied by elevated CRH and glucocorticoid concentrations, affect both the length of pregnancy and the hormonal environment of the developing fetus.
Administration of CRH to pregnant rat dams reduces both maternal body weight during pregnancy and offspring weight during the first two postnatal weeks, as well as enhancing offspring behavioural responses to a stressor in the early neonatal period (Williams et al., 1995
), effects reminiscent of the actions of maternal stress (Williams et al., 1998
). Therefore, it was suggested that at least a component of stress/glucocorticoid programming effects might be mediated through CRH actions on the developing fetal brain.
It is perhaps noteworthy that in terms of hippocampal development, the last trimester of human pregnancy is equated with the first postnatal week in the rat (Avishai-Eliner et al., 2002
). Although detailed analysis of the ontogeny of GR expression in the human fetal brain has not been reported, in the rat there is a relatively high expression of GR from midgestation onwards (Diaz et al., 1998
) and a rapid rise in GR and MR expression after birth (Matthews et al., 2002
). Hence, the last week of gestation and early postnatal life are considered particularly sensitive periods in rodent development in terms of glucocorticoid-mediated programming.
In support of this, handling of neonatal rat pups (an intervention which stimulates maternal care) reduced hypothalamic CRH expression and enhanced hippocampal GR expression in adult animals, and reduced the stress hormone responses to an acute stressor (Plotsky and Meaney, 1993
; Avishai-Eliner et al., 2001
). Interestingly, handling induces a distinct temporal pattern of gene expression, with reduction in paraventricular nuclei (PVN) CRH expression as early as postnatal day 9 (P9) preceding the increase in GR (which emerged between P23–45). The same pattern of adult gene expression was shown to be induced by postnatal administration of a CRH-receptor antagonist (Fenoglio et al., 2005
) implicating this as a key molecular mechanism in the emergence of early life programming of the HPA axis function.
In animal models, maternal dietary restriction, antenatal glucocorticoid administration and prenatal stress are associated with elevated blood pressure and exaggerated stress responses in offspring, indicative of reduced negative feedback of HPA axis activity (Langley-Evans, 1997a
; Doyle et al., 2000
; de Vries et al., 2007
). Under conditions of stress, activation of the HPA axis results in increased production and release of corticotrophin-releasing hormone (CRH) and arginine vasopressin (AVP) from the PVN. These hypophysiotropic hormones are released into the portal blood system and act on cells of the anterior pituitary to stimulate adrenocorticotropic hormone (ACTH) expression and release, which in turn increases glucocorticoid production at the adrenals. Subsequently, increased levels of circulating glucocorticoids feed back to inhibit HPA axis activity and prevent excessive production of stress hormones. Negative feedback occurs at the level of the brain and at the pituitary, and in particular inhibitory inputs derive from hippocampal GR-expressing neurons to inhibit PVN neurohormone release (refer Figure 2
A).
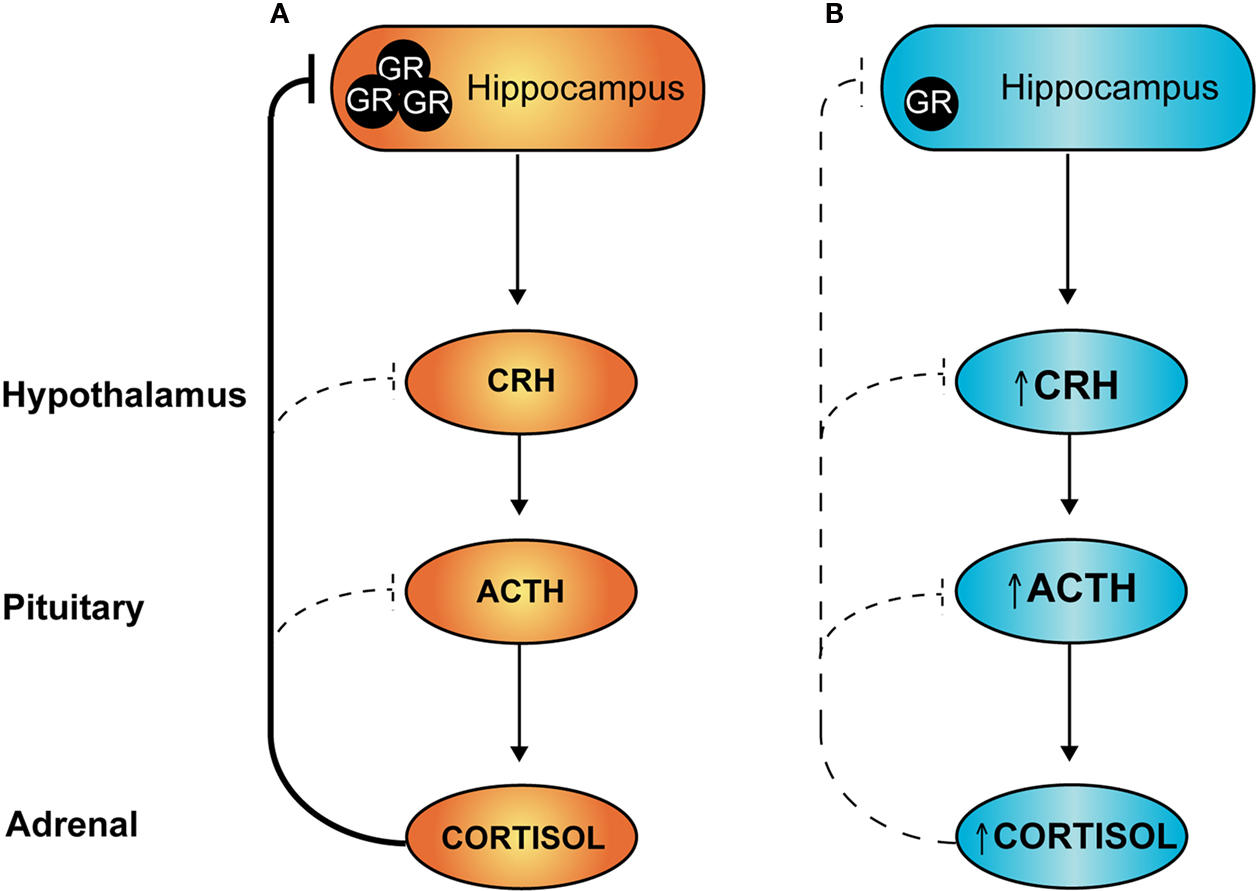
Figure 2. Hypothalamic–pituitary–adrenal (HPA) axis: programming by early life stress. Under conditions of stress, sustained activation of the HPA axis is attenuated via negative feedback actions of circulating glucocorticoids. At least a component of this attenuation occurs through activation of hippocampal glucocorticoid receptors (GR), as shown in (A) above. Individuals exposed to stress during early development and/or excess glucocorticoids have reduced expression of GR in the hippocampus (B) leading to a loss of this feedback inhibition and an overactive HPA axis, both in the basal state and under conditions of stress.
In programming models, reduced negative feedback of the HPA axis is thought to be due, at least in part, to a reduction in hippocampal glucocorticoid receptor (GR) signalling. Administration of dexamethasone to pregnant rats programmes offspring hypertension and increases basal corticosterone levels, and is associated with a reduction in hippocampal GR expression (Levitt et al., 1996
). The relative lack of hippocampal GR would be predicted to reduce HPA axis negative feedback, leading to over-activity and elevation of circulating glucocorticoids (as depicted in Figure 2
B). Such chronic mild glucocorticoid excess is anticipated, over time, to produce adverse metabolic, cardiovascular and neuropsychiatric effects, as seen in individuals with Cushing’s syndrome.
As well as central effects, fetal overexposure to glucocorticoids is also associated with alterations in the structure and function of numerous tissues. In terms of blood pressure regulation, prenatal glucocorticoid administration or maternal low protein diet leads to a permanent reduction in nephron number, hypersensitivity to the effects of vasoconstrictors and increased local GR expression in kidney tissue (Bertram et al., 2001
). The effects of maternal low protein diet on kidney development have been proposed to act through increased fetal glucocorticoid exposure, secondary to an increase in transplacental passage of maternal glucocorticoids (Langley-Evans et al., 1996
). As discussed previously, altered placental function is thought to be key in the programming of offspring phenotype.
So, exposure of the feto-placental unit to excess exogenous or endogenous glucocorticoids can have permanent effects on the offspring, but does maternal stress per se exert sufficient effect to impact the fetus in a similar way, especially as physiological glucocorticoids are substrates for inactivation by placental 11β-HSD2? In a number of human studies, prenatal stress has been associated with low birth weight, increased risk of premature delivery and impaired neurodevelopment in early life (Talge et al., 2007
). Self-reported anxiety during pregnancy in a large cohort of women in the UK is associated with increased salivary cortisol levels in offspring at 10 years of age (O’Connor et al., 2005
). A common behavioural outcome of pregnancies affected by prenatal stress is an increased incidence of attention deficit hyperactivity disorder (ADHD; O’Connor et al., 2002
; van den Bergh et al., 2006
). These findings in humans are also supported by non-human primate studies, where maternal prenatal stress is associated with attention deficits in offspring (van Os and Selten, 1998
; Schneider et al., 2002
).
Exposure to traumatic events during pregnancy have also been associated with increased rates of IUGR, HPA axis dysregulation as well as increased risk of certain psychiatric disorders. Offspring of pregnant women who were exposed to but survived the September 11th 2001 attacks on the World Trade Center showed greater incidence of IUGR compared with control populations (Berkowitz et al., 2003
). In addition, maternal cortisol levels were reduced in those mothers who developed post-traumatic stress disorder (PTSD), and intriguingly this trait was observed in 1-year-old offspring who were in utero during the event (Yehuda et al., 2005
). Crucially, these associations were confined to those exposed in the third trimester, whereas the offspring of women developing similar PTSD but exposed earlier in gestation did not show HPA alterations. These findings are partly echoed by studies of offspring of Holocaust survivors with PTSD who show altered cortisol levels and metabolism though the offspring themselves have no medical or neuropsychiatric illness (Yehuda, 2002
). These studies lend support to the idea that there is transmission of perturbed HPA axis function from mother to fetus during gestation, although they do not rule out the possibility that genetic differences are mediating these transgenerational associations.
A growing number of studies indicate that the effects of intrauterine exposures (including nutritional imbalance, hormone exposure, heavy metal or other toxins or endocrine disruptors) can be passed on to subsequent generations, without further exposure of the F1 generation. In order for these effects to be inherited, such exposure must induce either stable chromosomal alterations or involve epigenetic modification that is maintained through germ cell maturation (Jirtle and Skinner, 2007
).
As mentioned above, offspring of females administered dexamethasone during the last week of pregnancy exhibit lower birth weight and dysregulated glucose homeostasis in adulthood, in association with alterations in hepatic gluconeogenic enzymes (Nyirenda et al., 1998
). When these F1 animals are mated, their offspring (F2) also have lower birth weight, disrupted glucose homeostasis and elevated liver PEPCK in the absence of any gestational manipulation (Drake et al., 2005
). However, the F3 generation is unaffected. This most likely reflects a transgenerational effect as a result of alterations in primordial germ cell formation in the F1 generation. This effect seems to persist through fertilisation and development. It follows either the female or the male line, the latter suggesting it is stable through meiosis since the exposed fetal testis of the F1 male will not begin germ cell development until puberty. Presumably, the glucocorticoid exposure that these animals experienced prenatally affected both somatic and germ cell differentiation, leading to these exposed offspring developing the observed phenotype but also programming gametes and affecting the next generation of offspring. Again, the question of timing is important. In the rat, gonadal sex determination occurs around E12–E15, and exposures that coincide with this period are likely to affect gamete formation. This has been shown repeatedly to be the case in studies looking at the effects of administering endocrine disruptors during early fetal life. Administration of the antiandrogenic agent vinclozolin to pregnant rats from E8–E15 resulted in increased spermatogenic apoptosis, reduced male fertility and increased rates of adult disease in the F1 offspring (Anway et al., 2005
). These defects are inherited in subsequent generations, from F2–F4 and further can be paternally transmitted. This is particularly of interest as it moves the field of developmental programming to reconsider paternal effects, as until relatively recently it has been the influence of the maternal environment and uterine events that have received most attention.
Developmental plasticity allows the adaptation of an organism to environmental changes in early life, doubtless to facilitate immediate survival and perhaps also to ‘predict’ the likely challenges in later life. As highlighted in the present review, disordered HPA axis function and regulation is a common outcome in most, if not all, models of developmental programming. It is clear that alterations in glucocorticoid signalling in early life can induce persistent effects on later phenotype and disease risk.
That glucocorticoid exposure is implicated in these programming effects is perhaps intuitive – alterations in neonatal levels of stress hormones signal to the developing HPA axis a prediction of what might be encountered in adult life. It may be that the set-point for long-term regulation of the HPA axis, by means of altered GR expression, is determined by early life levels of the corresponding ligand. From an evolutionary perspective the phenotypic outcomes of these exposures appear to ‘make sense’. Glucocorticoid or stress-exposed offspring are adapted to a relatively challenging life – metabolic efficiency, earlier age of puberty, elevated blood pressure and heightened responses to stress will enable survival to reproductive age under conditions of food scarcity and predation. In contemporary humans these postnatal factors in most developed societies rarely pertain, and consequently the actual adult environment often conflicts with these adaptations. This may plausibly promote later disease, especially after the reproductive years. Whether prenatal stress in humans can induce transmissible alterations in cardiovascular, endocrine or behavioural traits is yet to be determined, but follow-up analysis of those exposed to in utero stressors will address these questions. Also key will be to determine whether there may be windows during development in which preventative or therapeutic interventions can reverse such adverse programming effects.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Avishai-Eliner, S., Eghbal-Ahmadi, M., Tabachnik, E., Brunson, K. L., and Baram, T. Z. (2001). Down-regulation of hypothalamic corticotropin-releasing hormone messenger ribonucleic acid (mRNA) precedes early-life experience-induced changes in hippocampal glucocorticoid receptor mRNA. Endocrinology 142, 89–97.
Bertram, C., Trowern, A. R., Copin, N., Jackson, A. A., and Whorwood, C. B. (2001). The maternal diet during pregnancy programs altered expression of the glucocorticoid receptor and type 2 11beta-hydroxysteroid dehydrogenase: potential molecular mechanisms underlying the programming of hypertension in utero. Endocrinology 142, 2841–2853.
Chisaka, H., Johnstone, J. F., Premyslova, M., Manduch, Z., and Challis, J. R. (2005). Effect of pro-inflammatory cytokines on expression and activity of 11beta-hydroxysteroid dehydrogenase type 2 in cultured human term placental trophoblast and human choriocarcinoma JEG-3 cells. J. Soc. Gynecol. Investig. 12, 303–309.
Cole, T. J., Blendy, J. A., Monaghan, A. P., Krieglstein, K., Schmid, W., Aguzzi, A., Fantuzzi, G., Hummler, E., Unsicker, K., and Schutz, G. (1995). Targeted disruption of the glucocorticoid receptor gene blocks adrenergic chromaffin cell development and severely retards lung maturation. Genes Dev. 9, 1608–1621.
Dave-Sharma, S., Wilson, R. C., Harbison, M. D., Newfield, R., Azar, M. R., Krozowski, Z. S., Funder, J. W., Shackleton, C. H., Bradlow, H. L., Wei, J. Q., Hertecant, J., Moran, A., Neiberger, R. E., Balfe, J. W., Fattah, A., Daneman, D., Akkurt, H. I., De Santis, C., and New, M. I. (1998). Examination of genotype and phenotype relationships in 14 patients with apparent mineralocorticoid excess. J. Clin. Endocrinol. Metab. 83, 2244–2254.
de Vries, A., Holmes, M. C., Heijnis, A., Seier, J. V., Heerden, J., Louw, J., Wolfe-Coote, S., Meaney, M. J., Levitt, N. S., and Seckl, J. R. (2007). Prenatal dexamethasone exposure induces changes in nonhuman primate offspring cardiometabolic and hypothalamic-pituitary-adrenal axis function. J. Clin. Invest. 117, 1058–1067.
Fenoglio, K. A., Brunson, K. L., Avishai-Eliner, S., Stone, B. A., Kapadia, B. J., and Baram, T. Z. (2005). Enduring, handling-evoked enhancement of hippocampal memory function and glucocorticoid receptor expression involves activation of the corticotropin-releasing factor type 1 receptor. Endocrinology 146, 4090–4096.
Holmes, M. C., Abrahamsen, C. T., French, K. L., Paterson, J. M., Mullins, J. J., and Seckl, J. R. (2006). The mother or the fetus? 11beta-hydroxysteroid dehydrogenase type 2 null mice provide evidence for direct fetal programming of behavior by endogenous glucocorticoids. J. Neurosci. 26, 3840–3844.
Jansson, N., Pettersson, J., Haafiz, A., Ericsson, A., Palmberg, I., Tranberg, M., Ganapathy, V., Powell, T. L., and Jansson, T. (2006). Down-regulation of placental transport of amino acids precedes the development of intrauterine growth restriction in rats fed a low protein diet. J. Physiol. 576, 935–946.
Lesage, J., Blondeau, B., Grino, M., Breant, B., and Dupouy, J. P. (2001). Maternal undernutrition during late gestation induces fetal overexposure to glucocorticoids and intrauterine growth retardation, and disturbs the hypothalamo–pituitary adrenal axis in the newborn rat. Endocrinology 142, 1692–1702.
Lucassen, P. J., Bosch, O. J., Jousma, E., Kromer, S. A., Andrew, R., Seckl, J. R., and Neumann, I. D. (2009). Prenatal stress reduces postnatal neurogenesis in rats selectively bred for high, but not low, anxiety: possible key role of placental 11beta-hydroxysteroid dehydrogenase type 2. Eur. J. Neurosci. 29, 97–103.
McCormick, J. A., Lyons, V., Jacobson, M. D., Noble, J., Diorio, J., Nyirenda, M., Weaver, S., Ester, W., Yau, J. L., Meaney, M. J., Seckl, J. R., and Chapman, K. E. (2000). 5’-Heterogeneity of glucocorticoid receptor messenger RNA is tissue specific: differential regulation of variant transcripts by early-life events. Mol. Endocrinol. 14, 506–517.
Michailidou, Z., Carter, R. N., Marshall, E., Sutherland, H. G., Brownstein, D. G., Owen, E., Cockett, K., Kelly, V., Ramage, L., Al-Dujaili, E. A., Ross, M., Maraki, I., Newton, K., Holmes, M. C., Seckl, J. R., Morton, N. M., Kenyon, C. J., and Chapman, K. E. (2008). Glucocorticoid receptor haploinsufficiency causes hypertension and attenuates hypothalamic–pituitary–adrenal axis and blood pressure adaptions to high-fat diet. FASEB J. 22, 3896–3907.
Nyirenda, M. J., Lindsay, R. S., Kenyon, C. J., Burchell, A., and Seckl, J. R. (1998). Glucocorticoid exposure in late gestation permanently programs rat hepatic phosphoenolpyruvate carboxykinase and glucocorticoid receptor expression and causes glucose intolerance in adult offspring. J. Clin. Invest. 101, 2174–2181.
Stewart, P. M., Rogerson, F. M., and Mason, J. I. (1995). Type 2 11 beta-hydroxysteroid dehydrogenase messenger ribonucleic acid and activity in human placenta and fetal membranes: its relationship to birth weight and putative role in fetal adrenal steroidogenesis. J. Clin. Endocrinol. Metab. 80, 885–890.
Stocker, C., O’Dowd, J., Morton, N. M., Wargent, E., Sennitt, M. V., Hislop, D., Glund, S., Seckl, J. R., Arch, J. R., and Cawthorne, M. A. (2004). Modulation of susceptibility to weight gain and insulin resistance in low birthweight rats by treatment of their mothers with leptin during pregnancy and lactation. Int. J. Obes. Relat. Metab. Disord. 28, 129–136.
Weaver, I. C., D’Alessio, A. C., Brown, S. E., Hellstrom, I. C., Dymov, S., Sharma, S., Szyf, M., and Meaney, M. J. (2007). The transcription factor nerve growth factor-inducible protein a mediates epigenetic programming: altering epigenetic marks by immediate-early genes. J. Neurosci. 27, 1756–1768.
Whincup, P. H., Kaye, S. J., Owen, C. G., Huxley, R., Cook, D. G., Anazawa, S., Barrett-Connor, E., Bhargava, S. K., Birgisdottir, B. E., Carlsson, S., de Rooij, S. R., Dyck, R. F., Eriksson, J. G., Falkner, B., Fall, C., Forsen, T., Grill, V., Gudnason, V., Hulman, S., Hypponen, E., Jeffreys, M., Lawlor, D. A., Leon, D. A., Minami, J., Mishra, G., Osmond, C., Power, C., Rich-Edwards, J. W., Roseboom, T. J., Sachdev, H. S., Syddall, H., Thorsdottir, I., Vanhala, M., Wadsworth, M., and Yarbrough, D. E. (2008). Birth weight and risk of type 2 diabetes: a systematic review. JAMA 300, 2886–2897.