
Mitochondrial Determinants of Anti-Cancer Drug-Induced Cardiotoxicity

 , ,
, ,  ,
,
Abstract
:1. Introduction
2. Mitochondria and Heart Physio-Pathology
2.1. Mitochondrial Quality Control
2.1.1. Ubiquitin Proteasome System (UPS)
2.1.2. Mitophagy
2.2. Mitochondrial Dynamics
3. Cardiac Mitochondrial Dysfunction Secondary to Anti-Cancer Drug Treatments
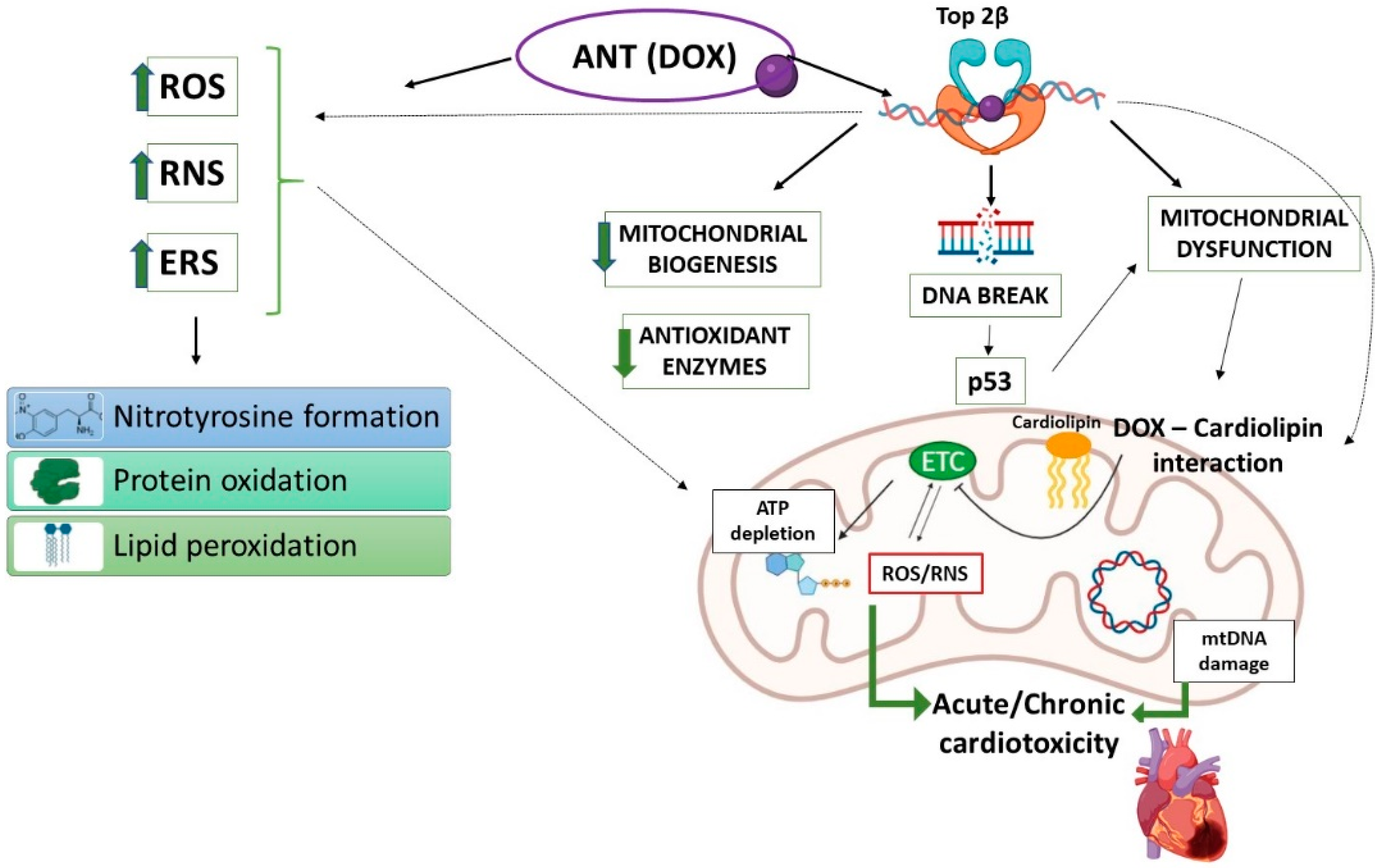
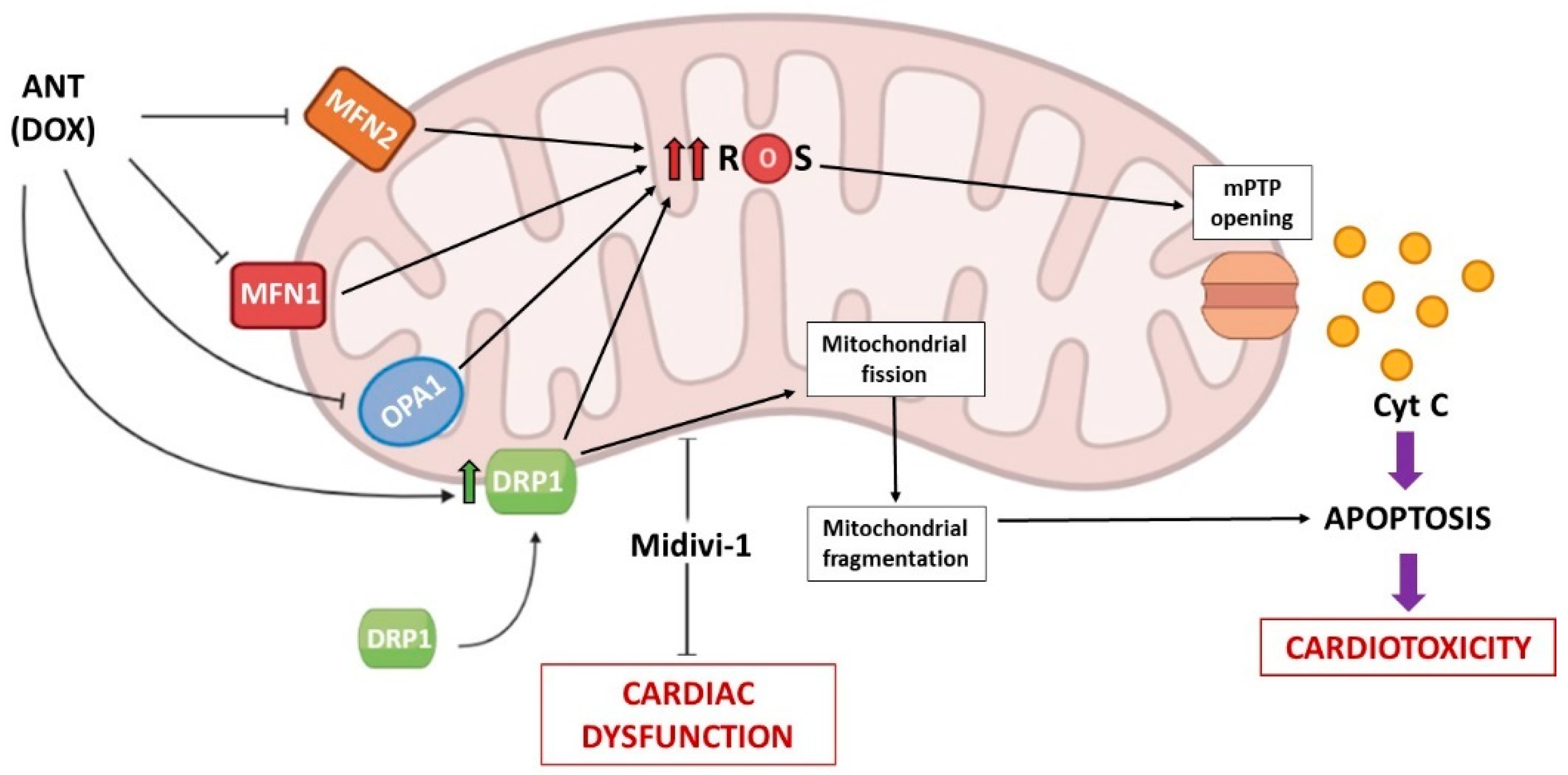
3.1. Anthracyclines (ANTs)
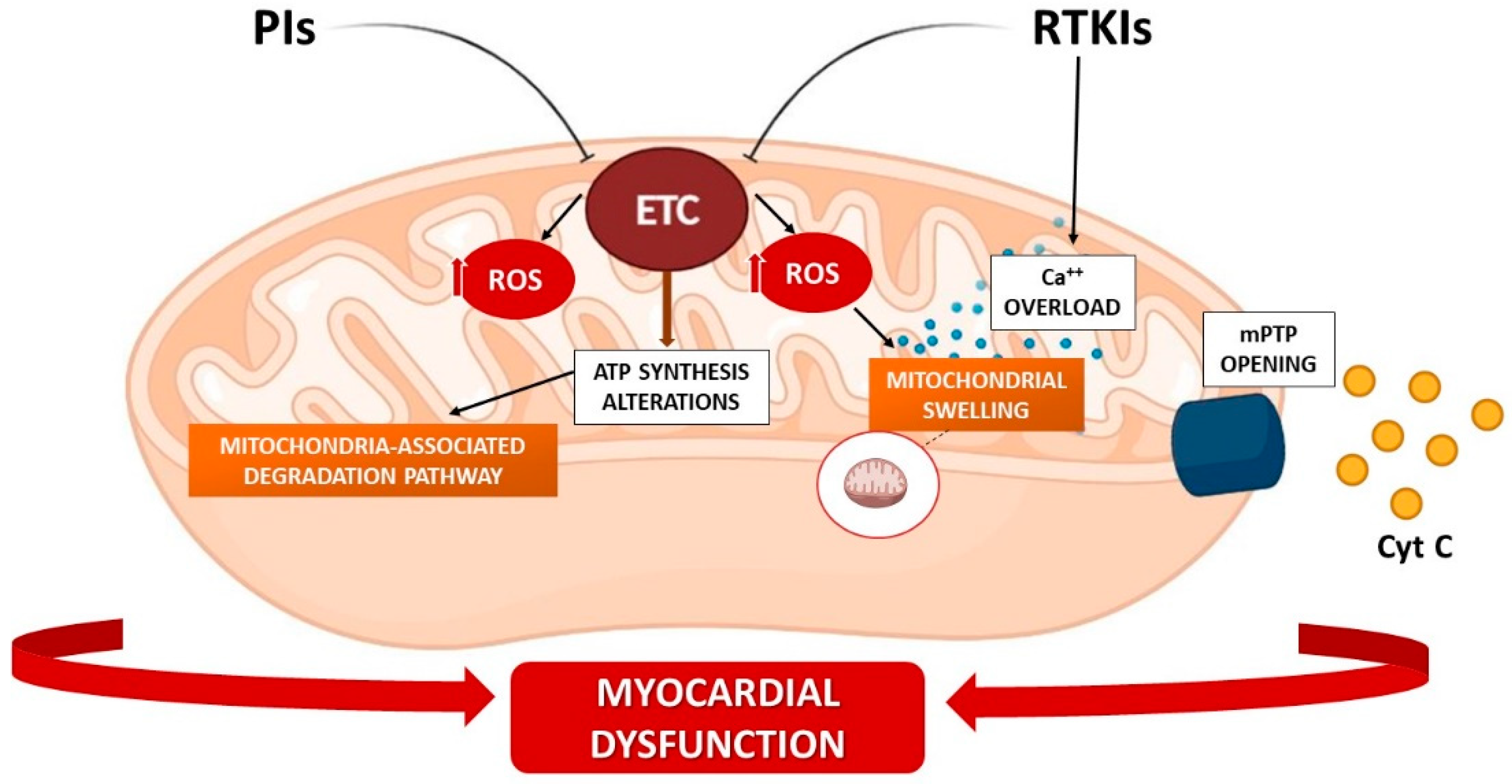
3.2. RTK Inhibitors (RTKIs)
3.3. Proteasome Inhibitors (PIs)
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AMPK | AMP-activated protein kinase |
| ANTs | anthracyclines |
| ARE | antioxidant response element |
| BCL-2 | B cell lymphoma-2 |
| CHF | chronic heart failure |
| CMDL-1 | cardiomyocyte mitochondrial dynamic-related lncRNA 1 |
| CML | chronic myelogenous leukemia |
| CVAEs | cardiovascular adverse events |
| CVDs | cardiovascular diseases |
| DOX | doxorubicin |
| Drp1 | dynamin-related protein 1 |
| ER | endoplasmic reticulum |
| Fis1 | fission protein 1 |
| FoxO | Forkhead box subgroup O |
| GPER | G-protein coupled receptor 30 |
| GSH | glutathione |
| hiPSC-CM | human-induced pluripotent stem cell-derived cardiomyocytes |
| I/R | ischemia/reperfusion |
| IMM | inner mitochondrial membrane |
| Keap1 | kelch-like ECH-associated protein 1 |
| LIR | LC-3-interacting region |
| MAD | mitochondria-associated degradation |
| MFF | mitochondrial fission factor |
| MFN1 | mitofusin-1 |
| MFN2 | mitofusin 2 |
| Midivi-1 | mitochondrial division inhibitor-1 |
| MM | multiple myeloma |
| mtDNA | mitochondrial DNA |
| NFAT | nuclear factor of activated T-cells |
| Nfe2l2 | nuclear factor erythroid derived 2 like 2 |
| Nrf2 | nuclear factor erythroid 2–related factor 2 |
| OMM | outer mitochondrial membrane |
| OPA1 | optic atrophy 1 |
| PDK | pyruvate dehydrogenase kinase |
| PGC-1α | peroxisome proliferator-activated receptor gamma coactivator 1-α |
| PGC-1β | peroxisome proliferator-activated receptor gamma coactivator 1-β |
| Ph+ B-ALL | Philadelphia chromosome-positive B-acute lymphoblastic leukemia |
| PINK1 | PTEN-induced putative kinase 1 |
| PIs | proteasome inhibitors |
| RNS | reactive nitrogen species |
| ROS | reactive oxygen species |
| RTKIs | receptor tyrosine kinase inhibitors |
| RTKs | Receptor tyrosine kinases |
| SOD | superoxide dismutase |
| Top | topoisomerase |
| TXNRD | thioredoxin reductase |
| UBD | ubiquitin binding domain |
| UCP-2 | uncoupling protein 2 |
| UCP-3 | uncoupling protein 3 |
| UPS | ubiquitin–proteasome system |
| URPmt | mitochondrial unfolded protein response |
| VEGF | vascular endothelial growth factor |
References
- Pasqua, T.; Rocca, C.; Giglio, A.; Angelone, T. Cardiometabolism as an Interlocking Puzzle between the Healthy and Diseased Heart: New Frontiers in Therapeutic Applications. J. Clin. Med. 2021, 10, 721. [Google Scholar] [CrossRef]
- Nguyen, B.Y.; Ruiz-Velasco, A.; Bui, T.; Collins, L.; Wang, X.; Liu, W. Mitochondrial function in the heart: The insight into mechanisms and therapeutic potentials. J. Cereb. Blood Flow Metab. 2019, 176, 4302–4318. [Google Scholar] [CrossRef]
- Cadete, V.J.; Vasam, G.; Menzies, K.J.; Burelle, Y. Mitochondrial quality control in the cardiac system: An integrative view. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2019, 1865, 782–796. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Shkurat, T.P.; Melnichenko, A.A.; Grechko, A.V.; Orekhov, A.N. The role of mitochondrial dysfunction in cardiovascular disease: A brief review. Ann. Med. 2018, 50, 121–127. [Google Scholar] [CrossRef]
- Marín-García, J.; Akhmedov, A.T. Mitochondrial dynamics and cell death in heart failure. Heart Fail. Rev. 2016, 21, 123–136. [Google Scholar] [CrossRef]
- Liesa, M.; Palacín, M.; Zorzano, A. Mitochondrial Dynamics in Mammalian Health and Disease. Physiol. Rev. 2009, 89, 799–845. [Google Scholar] [CrossRef] [Green Version]
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 2010, 11, 872–884. [Google Scholar] [CrossRef]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [Green Version]
- Friedman, J.R.; Nunnari, J. Mitochondrial form and function. Nature 2014, 505, 335–343. [Google Scholar] [CrossRef] [Green Version]
- Rosca, M.G.; Hoppel, C.L. Mitochondrial dysfunction in heart failure. Heart Fail. Rev. 2012, 18, 607–622. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.; Tian, R. Mitochondrial dysfunction in pathophysiology of heart failure. J. Clin. Investig. 2018, 128, 3716–3726. [Google Scholar] [CrossRef] [Green Version]
- Cardinale, D.; Colombo, A.; Lamantia, G.; Colombo, N.; Civelli, M.; De Giacomi, G.; Rubino, M.; Veglia, F.; Fiorentini, C.; Cipolla, C.M. Anthracycline-Induced Cardiomyopathy: Clinical Relevance and Response to Pharmacologic Therapy. J. Am. Coll. Cardiol. 2010, 55, 213–220. [Google Scholar] [CrossRef] [Green Version]
- Randle, P.; Garland, P.; Hales, C.; Newsholme, E. The glucose fatty-acid cycle its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1963, 281, 785–789. [Google Scholar] [CrossRef]
- Varga, Z.; Ferdinandy, P.; Liaudet, L.; Pacher, P. Drug-induced mitochondrial dysfunction and cardiotoxicity. Am. J. Physiol. Circ. Physiol. 2015, 309, H1453–H1467. [Google Scholar] [CrossRef] [Green Version]
- Zamorano, J.L.; Lancellotti, P.; Rodriguez Munoz, D.; Aboyans, V.; Asteggiano, R.; Galderisi, M.; Habib, G.; Lenihan, D.J.; Lip, G.Y.H.; Lyon, A.R.; et al. ESC position paper on cancer treatments and cardiovascular toxicity developed under the auspices of the ESC committee for practice guidelines: The task force for cancer treatments and cardiovascular toxicity of the European Society of Cardiology (ESC). Eur. Heart J. 2016, 37, 2768–2801. [Google Scholar] [CrossRef]
- Natarajan, V.; Chawla, R.; Mah, T.; Vivekanandan, R.; Tan, S.Y.; Sato, P.Y.; Mallilankaraman, K. Mitochondrial Dysfunction in Age-Related Metabolic Disorders. Proteomics 2020, 20, e1800404. [Google Scholar] [CrossRef]
- Koklesova, L.; Liskova, A.; Samec, M.; Zhai, K.; Al-Ishaq, R.K.; Bugos, O.; Šudomová, M.; Biringer, K.; Pec, M.; Adamkov, M.; et al. Protective Effects of Flavonoids against Mitochondriopathies and Associated Pathologies: Focus on the Predictive Approach and Personalized Prevention. Int. J. Mol. Sci. 2021, 22, 8649. [Google Scholar] [CrossRef]
- Randle, P.J.; Priestman, D.A.; Mistry, S.C.; Halsall, A. Glucose fatty acid interactions and the regulation of glucose disposal. J. Cell. Biochem. 1994, 55, 1–11. [Google Scholar] [CrossRef]
- Holmgren, D.; Wahlander, H.; Eriksson, B.; Oldfors, A.; Holme, E.; Tulinius, M. Cardiomyopathy in children with mitochondrial disease Clinical course and cardiological findings. Eur. Heart J. 2003, 24, 280–288. [Google Scholar] [CrossRef]
- Murphy, E.; Ardehali, H.; Balaban, R.S.; DiLisa, F.; Dorn, G.W., 2nd; Kitsis, R.N.; Otsu, K.; Ping, P.; Rizzuto, R.; Sack, M.N.; et al. Mitochondrial Function, Biology, and Role in Disease: A Scientific Statement from the American Heart Association. Circ. Res. 2016, 118, 1960–1991. [Google Scholar] [CrossRef]
- Rocca, C.; Pasqua, T.; Boukhzar, L.; Anouar, Y.; Angelone, T. Progress in the emerging role of selenoproteins in cardiovascular disease: Focus on endoplasmic reticulum-resident selenoproteins. Cell Mol. Life Sci. 2019, 76, 3969–3985. [Google Scholar] [CrossRef]
- Rocca, C.; Boukhzar, L.; Granieri, M.C.; Alsharif, I.; Mazza, R.; Lefranc, B.; Tota, B.; Leprince, J.; Cerra, M.C.; Anouar, Y.; et al. A selenoprotein T-derived peptide protects the heart against ischaemia/reperfusion injury through inhibition of apoptosis and oxidative stress. Acta Physiol. 2018, 223, e13067. [Google Scholar] [CrossRef]
- Peoples, J.N.; Saraf, A.; Ghazal, N.; Pham, T.T.; Kwong, J.Q. Mitochondrial dysfunction and oxidative stress in heart disease. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef]
- Radi, R.; Turrens, J.; Chang, L.; Bush, K.; Crapo, J.; Freeman, B. Detection of catalase in rat heart mitochondria. J. Biol. Chem. 1991, 266, 22028–22034. [Google Scholar] [CrossRef]
- Andreyev, A.Y.; Kushnareva, Y.E.; Murphy, A.N.; Starkov, A. Mitochondrial ROS metabolism: 10 Years later. Biochemistry 2015, 80, 517–531. [Google Scholar] [CrossRef] [Green Version]
- Marí, M.; Morales, A.; Colell, A.; García-Ruiz, C.; Fernández-Checa, J.C. Mitochondrial Glutathione, a Key Survival Antioxidant. Antioxid. Redox Signal. 2009, 11, 2685–2700. [Google Scholar] [CrossRef] [Green Version]
- Bhandari, S. Impact of intravenous iron on cardiac and skeletal oxidative stress and cardiac mitochondrial function in experimental uraemia chronic kidney disease. Front. Biosci. 2021, 26, 442. [Google Scholar] [CrossRef]
- Fenton, H.J.H. Oxidation of tartaric acid in presence of iron. J. Chem. Soc. Trans. 1894, 65, 899–910. [Google Scholar] [CrossRef] [Green Version]
- Masaki, H.; Okano, Y.; Sakurai, H. Differential role of catalase and glutathione peroxidase in cultured human fibroblasts under exposure of H2O2 or ultraviolet B light. Arch. Dermatol. Res. 1998, 290, 113–118. [Google Scholar] [CrossRef]
- Logan, A.; Pell, V.R.; Shaffer, K.J.; Evans, C.; Stanley, N.; Robb, E.L.; Prime, T.A.; Chouchani, E.T.; Cochemé, H.M.; Fearnley, I.M.; et al. Assessing the Mitochondrial Membrane Potential in Cells and In Vivo using Targeted Click Chemistry and Mass Spectrometry. Cell Metab. 2016, 23, 379–385. [Google Scholar] [CrossRef] [Green Version]
- Jonckheere, A.I.; Smeitink, J.A.M.; Rodenburg, R.J.T. Mitochondrial ATP synthase: Architecture, function and pathology. J. Inherit. Metab. Dis. 2011, 35, 211–225. [Google Scholar] [CrossRef] [Green Version]
- Perry, S.W.; Norman, J.P.; Barbieri, J.; Brown, E.B.; Gelbard, H.A. Mitochondrial membrane potential probes and the proton gradient: A practical usage guide. BioTechniques 2011, 50, 98–115. [Google Scholar] [CrossRef]
- Arruda, A.P.; Hotamisligil, G.S. Calcium homeostasis and organelle function in the pathogenesis of obesity and diabetes. Cell Metab. 2015, 22, 381–397. [Google Scholar] [CrossRef] [Green Version]
- Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-Timme, C.A.; Sancak, Y.; Bao, X.R.; Strittmatter, L.; Goldberger, O.; Bogorad, R.L.; et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 2011, 476, 341–345. [Google Scholar] [CrossRef] [Green Version]
- De Stefani, D.; Raffaello, A.; Teardo, E.; Szabò, I.; Rizzuto, R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011, 476, 336–340. [Google Scholar] [CrossRef]
- Luongo, T.S.; Lambert, J.P.; Gross, P.; Nwokedi, M.; Lombardi, A.A.; Shanmughapriya, S. The mitochondrial Na+/Ca2+ exchanger is essential for Ca2+ homeostasis and viability. Nature 2017, 545, 93–97. [Google Scholar] [CrossRef] [Green Version]
- Eisner, D.A.; Caldwell, J.L.; Kistamás, K.; Trafford, A.W. Calcium and Excitation-Contraction Coupling in the Heart. Circ. Res. 2017, 121, 181–195. [Google Scholar] [CrossRef]
- Sheu, S.S.; Jou, M.J. Mitochondrial free Ca2+ concentration in living cells. J. Bioenerg. Biomembr. 1994, 26, 487–493. [Google Scholar] [CrossRef]
- Olson, M.L.; Chalmers, S.; McCarron, J.G. Mitochondrial organization and Ca2+ uptake. Biochem. Soc. Trans. 2012, 40, 158–167. [Google Scholar] [CrossRef] [Green Version]
- Patergnani, S.; Suski, J.M.; Agnoletto, C.; Bononi, A.; Bonora, M.; De Marchi, E.; Giorgi, C.; Marchi, S.; Missiroli, S.; Poletti, F.; et al. Calcium signaling around Mitochondria Associated Membranes (MAMs). Cell Commun. Signal. 2011, 9, 19. [Google Scholar] [CrossRef] [Green Version]
- Del Re, D.P.; Amgalan, D.; Linkermann, A.; Liu, Q.; Kitsis, R.N. Fundamental Mechanisms of Regulated Cell Death and Implications for Heart Disease. Physiol. Rev. 2019, 99, 1765–1817. [Google Scholar] [CrossRef]
- Chipuk, J.E.; Moldoveanu, T.; Llambi, F.; Parsons, M.J.; Green, D.R. The BCL-2 Family Reunion. Mol. Cell 2010, 37, 299–310. [Google Scholar] [CrossRef]
- Gavathiotis, E.; Reyna, D.E.; Davis, M.L.; Bird, G.H.; Walensky, L.D. BH3-Triggered Structural Reorganization Drives the Activation of Proapoptotic BAX. Mol. Cell 2010, 40, 481–492. [Google Scholar] [CrossRef] [Green Version]
- Du, C.; Fang, M.; Li, Y.; Li, L.; Wang, X. Smac, a Mitochondrial Protein that Promotes Cytochrome c–Dependent Caspase Activation by Eliminating IAP Inhibition. Cell 2000, 102, 33–42. [Google Scholar] [CrossRef] [Green Version]
- Verhagen, A.M.; Ekert, P.G.; Pakusch, M.; Silke, J.; Connolly, L.M.; Reid, G.E.; Moritz, R.L.; Simpson, R.J.; Vaux, D.L. Identification of DIABLO, a Mammalian Protein that Promotes Apoptosis by Binding to and Antagonizing IAP Proteins. Cell 2000, 102, 43–53. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, Y.; Imai, Y.; Nakayama, H.; Takahashi, K.; Takio, K.; Takahashi, R. A Serine Protease, HtrA2, Is Released from the Mitochondria and Interacts with XIAP, Inducing Cell Death. Mol. Cell 2001, 8, 613–621. [Google Scholar] [CrossRef]
- Chipuk, J.E.; McStay, G.P.; Bharti, A.; Kuwana, T.; Clarke, C.J.; Siskind, L.J.; Obeid, L.M.; Green, D.R. Sphingolipid Metabolism Cooperates with BAK and BAX to Promote the Mitochondrial Pathway of Apoptosis. Cell 2012, 148, 988–1000. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Nijhawan, D.; Budihardjo, I.; Srinivasula, S.M.; Ahmad, M.; Alnemri, E.S.; Wang, X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 1997, 91, 479–489. [Google Scholar] [CrossRef] [Green Version]
- Zou, H.; Henzel, W.; Liu, X.; Lutschg, A.; Wang, X. Apaf-1, a Human Protein Homologous to C. elegans CED-4, Participates in Cytochrome c–Dependent Activation of Caspase-3. Cell 1997, 90, 405–413. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Kim, C.N.; Yang, J.; Jemmerson, R.; Wang, X. Induction of Apoptotic Program in Cell-Free Extracts: Requirement for dATP and Cytochrome c. Cell 1996, 86, 147–157. [Google Scholar] [CrossRef] [Green Version]
- Edlich, F.; Banerjee, S.; Suzuki, M.; Cleland, M.M.; Arnoult, D.; Wang, C.; Neutzner, A.; Tjandra, N.; Youle, R.J. Bcl-xL Retrotranslocates Bax from the Mitochondria into the Cytosol. Cell 2011, 145, 104–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minoia, M.; Boncoraglio, A.; Vinet, J.; Morelli, F.F.; Brunsting, J.F.; Poletti, A.; Krom, S.; Reits, E.; Kampinga, H.H.; Carra, S. BAG3 induces the sequestration of proteasomal clients into cytoplasmic puncta: Implications for a proteasome-toautophagy switch. Autophagy 2014, 10, 1603–1621. [Google Scholar] [CrossRef] [Green Version]
- Hammerling, B.C.; Gustafsson, Å.B. Mitochondrial quality control in the myocardium: Cooperation between protein degradation and mitophagy. J. Mol. Cell. Cardiol. 2014, 75, 122–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bragoszewski, P.; Turek, M.; Chacinska, A. Control of mitochondrial biogenesis and function by the ubiquitin–proteasome system. Open Biol. 2017, 7, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciechanover, A.; Stanhill, A. The complexity of recognition of ubiquitinated substrates by the 26S proteasome. Biochim. Biophys. Acta 2014, 1843, 86–96. [Google Scholar] [CrossRef] [Green Version]
- Kwon, Y.T.; Ciechanover, A. The Ubiquitin Code in the Ubiquitin-Proteasome System and Autophagy. Trends Biochem. Sci. 2017, 42, 873–886. [Google Scholar] [CrossRef]
- Kodroń, A.; Mussulini, B.H.; Pilecka, I.; Chacińska, A. The ubiquitin-proteasome system and its crosstalk with mitochondria as therapeutic targets in medicine. Pharmacol. Res. 2021, 163, 105248. [Google Scholar] [CrossRef]
- Karbowski, M.; Youle, R.J. Regulating mitochondrial outer membrane proteins by ubiquitination and proteasomal degradation. Curr. Opin. Cell Biol. 2011, 23, 476–482. [Google Scholar] [CrossRef] [Green Version]
- Quiles, J.M.; Gustafsson, Å.B. Mitochondrial Quality Control and Cellular Proteostasis: Two Sides of the Same Coin. Front. Physiol. 2020, 11, 515. [Google Scholar] [CrossRef]
- Szczepanowska, K.; Maiti, P.; Kukat, A.; Hofsetz, E.; Nolte, H.; Senft, K.; Becker, C.; Ruzzenente, B.; Hornig-Do, H.-T.; Wibom, R.; et al. CLPP coordinates mitoribosomal assembly through the regulation of ERAL 1 levels. EMBO J. 2016, 35, 2566–2583. [Google Scholar] [CrossRef] [Green Version]
- Weinhäupl, K.; Lindau, C.; Hessel, A.; Wang, Y.; Schütze, C.; Jores, T.; Melchionda, L.; Schönfisch, B.; Kalbacher, H.; Bersch, B.; et al. Structural Basis of Membrane Protein Chaperoning through the Mitochondrial Intermembrane Space. Cell 2018, 175, 1365–1379.e25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavie, J.; De Belvalet, H.; Sonon, S.; Ion, A.M.; Dumon, E.; Melser, S.; Lacombe, D.; Dupuy, J.-W.; Lalou, C.; Bénard, G. Ubiquitin-Dependent Degradation of Mitochondrial Proteins Regulates Energy Metabolism. Cell Rep. 2018, 23, 2852–2863. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, C.; Katus, H.A.; Doroudgar, S. Protein Misfolding in Cardiac Disease. Circulation 2019, 139, 2085–2088. [Google Scholar] [CrossRef] [PubMed]
- Ranek, M.J.; Zheng, H.; Huang, W.; Kumarapeli, A.R.; Li, J.; Liu, J.; Wang, X. Genetically induced moderate inhibition of 20S proteasomes in cardiomyocytes facilitates heart failure in mice during systolic overload. J. Mol. Cell. Cardiol. 2015, 85, 273–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Predmore, J.M.; Wang, P.; Davis, F.; Bartolone, S.; Westfall, M.; Dyke, D.B.; Pagani, F.; Powell, S.R.; Day, S.M. Ubiquitin Proteasome Dysfunction in Human Hypertrophic and Dilated Cardiomyopathies. Circulation 2010, 121, 997–1004. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Q.; Gao, W.; Du, F.; Wang, X. Mule/ARF-BP1, a BH3-Only E3 Ubiquitin Ligase, Catalyzes the Polyubiquitination of Mcl-1 and Regulates Apoptosis. Cell 2005, 121, 1085–1095. [Google Scholar] [CrossRef] [Green Version]
- Münch, C.; Harper, J.W. Mitochondrial unfolded protein response controls matrix pre-RNA processing and translation. Nature 2016, 534, 710–713. [Google Scholar] [CrossRef] [Green Version]
- Tahrir, F.G.; Langford, D.; Amini, S.; Mohseni Ahooyi, T.; Khalili, K. Mitochondrial quality control in cardiac cells: Mechanisms and role in cardiac cell injury and disease. J. Cell. Physiol. 2019, 234, 8122–8133. [Google Scholar] [CrossRef]
- Gustafsson, Å.B.; Dorn, G.W., 2nd. Evolving and Expanding the Roles of Mitophagy as a Homeostatic and Pathogenic Process. Physiol. Rev. 2019, 99, 853–892. [Google Scholar] [CrossRef]
- Tong, M.; Saito, T.; Zhai, P.; Oka, S.-I.; Mizushima, W.; Nakamura, M.; Ikeda, S.; Shirakabe, A.; Sadoshima, J. Mitophagy Is Essential for Maintaining Cardiac Function During High Fat Diet-Induced Diabetic Cardiomyopathy. Circ. Res. 2019, 124, 1360–1371. [Google Scholar] [CrossRef]
- Billia, F.; Hauck, L.; Konecny, F.; Rao, V.; Shen, J.; Mak, T.W. PTENinducible kinase 1 (PINK1)/Park6 is indispensable for normal heart function. Proc. Natl. Acad. Sci. USA 2011, 108, 9572–9577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirakabe, A.; Zhai, P.; Ikeda, Y.; Saito, T.; Maejima, Y.; Hsu, C.-P.; Nomura, M.; Egashira, K.; Levine, B.; Sadoshima, J. Drp1-Dependent Mitochondrial Autophagy Plays a Protective Role Against Pressure Overload–Induced Mitochondrial Dysfunction and Heart Failure. Circulation 2016, 133, 1249–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddall, H.K.; Yellon, D.M.; Ong, S.-B.; Mukherjee, U.A.; Burke, N.; Hall, A.R.; Angelova, P.R.; Ludtmann, M.H.; Deas, E.; Davidson, S.M.; et al. Loss of PINK1 Increases the Heart’s Vulnerability to Ischemia-Reperfusion Injury. PLoS ONE 2013, 8, e62400. [Google Scholar] [CrossRef]
- Kubli, D.A.; Zhang, X.; Lee, Y.; Hanna, R.A.; Quinsay, M.N.; Nguyen, C.K.; Jimenez, R.; Petrosyan, S.; Murphy, A.N.; Gustafsson, A.B. Parkin Protein Deficiency Exacerbates Cardiac Injury and Reduces Survival following Myocardial Infarction. J. Biol. Chem. 2013, 288, 915–926. [Google Scholar] [CrossRef] [Green Version]
- Truban, D.; Hou, X.; Caulfield, T.R.; Fiesel, F.C.; Springer, W. PINK1, Parkin, and Mitochondrial Quality Control: What can we Learn about Parkinson’s Disease Pathobiology? J. Park. Dis. 2017, 7, 13–29. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Dorn, G.W. PINK1-Phosphorylated Mitofusin 2 Is a Parkin Receptor for Culling Damaged Mitochondria. Science 2013, 340, 471–475. [Google Scholar] [CrossRef] [Green Version]
- Gong, G.; Song, M.; Csordas, G.; Kelly, D.P.; Matkovich, S.J.; Dorn, G.W., 2nd. Parkin-mediated mitophagy directs perinatal cardiac metabolic maturation in mice. Science 2015, 350, aad2459. [Google Scholar] [CrossRef] [Green Version]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef]
- Kawajiri, S.; Saiki, S.; Sato, S.; Sato, F.; Hatano, T.; Eguchi, H.; Hattori, N. PINK1 is recruited to mitochondria with parkin and associates with LC3 in mitophagy. FEBS Lett. 2010, 584, 1073–1079. [Google Scholar] [CrossRef] [Green Version]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.A.; Outzen, H.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [Green Version]
- Narendra, D.; Tanaka, A.; Suen, D.-F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, M.; Chacko, L.A.; Joseph, J.P.; Ananthanarayanan, V. Mitochondrial dynamics, positioning and function mediated by cytoskeletal interactions. Cell Mol. Life Sci. 2021, 78, 3969–3986. [Google Scholar] [CrossRef] [PubMed]
- El-Hattab, A.W.; Suleiman, J.; Almannai, M.; Scaglia, F. Mitochondrial dynamics: Biological roles, molecular machinery, and related diseases. Mol. Genet. Metab. 2018, 125, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Gerald, W.D., II. Evolving Concepts of Mitochondrial Dynamics. Ann. Rev. Physiol. 2019, 81, 1–17. [Google Scholar]
- Cipolat, S.; De Brito, O.M.; Dal Zilio, B.; Scorrano, L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc. Natl. Acad. Sci. USA 2004, 101, 15927–15932. [Google Scholar] [CrossRef] [Green Version]
- Smirnova, E.; Griparic, L.; Shurland, D.-L.; van der Bliek, A.M. Dynamin-related Protein Drp1 Is Required for Mitochondrial Division in Mammalian Cells. Mol. Biol. Cell 2001, 12, 2245–2256. [Google Scholar] [CrossRef] [Green Version]
- Li, A.; Gao, M.; Jiang, W.; Qin, Y.; Gong, G. Mitochondrial Dynamics in Adult Cardiomyocytes and Heart Diseases. Front. Cell Dev. Biol. 2020, 8. [Google Scholar] [CrossRef]
- Yin, Y.; Shen, H. Advances in Cardiotoxicity Induced by Altered Mitochondrial Dynamics and Mitophagy. Front. Cardiovasc. Med. 2021, 8. [Google Scholar] [CrossRef]
- Gorini, S.; De Angelis, A.; Berrino, L.; Malara, N.; Rosano, G.; Ferraro, E. Chemotherapeutic Drugs and Mitochondrial Dysfunction: Focus on Doxorubicin, Trastuzumab, and Sunitinib. Oxid. Med. Cell. Longev. 2018, 2018, 7582730, Correction in Oxid. Med. Cell. Longev. 2019, 2019, 9601435. [Google Scholar] [CrossRef]
- Larsen, S.; Nielsen, J.; Hansen, C.N.; Nielsen, L.B.; Wibrand, F.; Stride, N.; Schrøder, H.D.; Boushel, R.; Helge, J.W.; Dela, F.; et al. Biomarkers of mitochondrial content in skeletal muscle of healthy young human subjects. J. Physiol. 2012, 590, 3349–3360. [Google Scholar] [CrossRef]
- Gilliam, L.A.; St Clair, D.K. Chemotherapy-Induced Weakness and Fatigue in Skeletal Muscle: The Role of Oxidative Stress. Antioxid. Redox Signal. 2011, 15, 2543–2563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorensen, J.C.; Cheregi, B.D.; Timpani, C.; Nurgali, K.; Hayes, A.; Rybalka, E. Mitochondria: Inadvertent targets in chemotherapy-induced skeletal muscle toxicity and wasting? Cancer Chemother. Pharmacol. 2016, 78, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Rocca, C.; Pasqua, T.; Cerra, M.C.; Angelone, T. Cardiac Damage in Anthracyclines Therapy: Focus on Oxidative Stress and Inflammation. Antioxid. Redox Signal. 2020, 32, 1081–1097. [Google Scholar] [CrossRef] [PubMed]
- Minotti, G.; Menna, P.; Salvatorelli, E.; Cairo, G.; Gianni, L. Anthracyclines: Molecular Advances and Pharmacologic Developments in Antitumor Activity and Cardiotoxicity. Pharmacol. Rev. 2004, 56, 185–229. [Google Scholar] [CrossRef] [Green Version]
- Fischer, F.; Hamann, A.; Osiewacz, H.D. Mitochondrial quality control: An integrated network of pathways. Trends Biochem. Sci. 2012, 37, 284–292. [Google Scholar] [CrossRef]
- Kim, C.-W.; Choi, K.-C. Effects of anticancer drugs on the cardiac mitochondrial toxicity and their underlying mechanisms for novel cardiac protective strategies. Life Sci. 2021, 277, 119607. [Google Scholar] [CrossRef]
- Egea, J.; Fabregat, I.; Frapart, Y.M.; Ghezzi, P.; Görlach, A.; Kietzmann, T.; Kubaichuk, K.; Knaus, U.G.; Lopez, M.G.; Olaso-Gonzalez, G.; et al. European contribution to the study of ROS: A summary of the findings and prospects for the future from the COST action BM1203 (EU-ROS). Redox Biol. 2017, 13, 94–162. [Google Scholar] [CrossRef] [Green Version]
- Tewey, K.M.; Rowe, T.C.; Yang, L.; Halligan, B.D.; Liu, L.F. Adriamycin-Induced DNA Damage Mediated by Mammalian DNA Topoisomerase II. Science 1984, 226, 466–468. [Google Scholar] [CrossRef]
- Henriksen, P.A. Anthracycline cardiotoxicity: An update on mechanisms, monitoring and prevention. Heart 2018, 104, 971–977. [Google Scholar] [CrossRef]
- Gianni, L.; Herman, E.H.; Lipshultz, S.E.; Minotti, G.; Sarvazyan, N.; Sawyer, D.B. Anthracycline Cardiotoxicity: From Bench to Bedside. J. Clin. Oncol. 2008, 26, 3777–3784. [Google Scholar] [CrossRef] [Green Version]
- Eschenhagen, T.; Force, T.; Ewer, M.S.; De Keulenaer, G.; Suter, T.M.; Anker, S.D.; Avkiran, M.; de Azambuja, E.; Balligand, J.-L.; Brutsaert, D.L.; et al. Cardiovascular side effects of cancer therapies: A position statement from the Heart Failure Association of the European Society of Cardiology. Eur. J. Heart Fail. 2011, 13, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Zhou, Y.; Liu, W. Precision cardio-oncology: Understanding the cardiotoxicity of cancer therapy. NPJ Precis. Oncol. 2017, 1, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ewer, M.S.; Lippman, S.M. Type II Chemotherapy-Related Cardiac Dysfunction: Time to Recognize a New Entity. J. Clin. Oncol. 2005, 23, 2900–2902. [Google Scholar] [CrossRef] [PubMed]
- Tocchetti, C.G.; Cadeddu, C.; Di Lisi, D.; Femminò, S.; Madonna, R.; Mele, D.; Monte, I.; Novo, G.; Penna, C.; Pepe, A.; et al. From Molecular Mechanisms to Clinical Management of Antineoplastic Drug-Induced Cardiovascular Toxicity: A Translational Overview. Antioxid. Redox Signal. 2019, 30, 2110–2153. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Liu, X.; Bawa-Khalfe, T.; Lu, L.-S.; Lyu, Y.L.; Liu, L.F.; Yeh, E.T.H. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat. Med. 2012, 18, 1639–1642. [Google Scholar] [CrossRef]
- Vejpongsa, P.; Yeh, E.T. Prevention of anthracycline-induced cardiotoxicity: Challenges and opportunities. J. Am. Coll. Cardiol. 2014, 64, 938–945. [Google Scholar] [CrossRef] [Green Version]
- Borowitz, J.; Rathinavelu, A.; Kanthasamy, A.; Wilsbacher, J.; Isom, G. Accumulation of Labeled Cyanide in Neuronal Tissue. Toxicol. Appl. Pharmacol. 1994, 129, 80–85. [Google Scholar] [CrossRef]
- Montaigne, D.; Marechal, X.; Preau, S.; Baccouch, R.; Modine, T.; Fayad, G.; Lancel, S.; Neviere, R. Doxorubicin induces mitochondrial permeability transition and contractile dysfunction in the human myocardium. Mitochondrion 2011, 11, 22–26. [Google Scholar] [CrossRef]
- Ferrero, M.; Ferrero, E.; Gaja, G.; Bernelli-Zazzera, A. Adriamycin: Energy metabolism and mitochondrial oxidations in the heart of treated rabbits. Biochem. Pharmacol. 1976, 25, 125–130. [Google Scholar] [CrossRef]
- Zhou, S.; Starkov, A.; Froberg, M.K.; Leino, R.L.; Wallace, K.B. Cumulative and irreversible cardiac mitochondrial dysfunction induced by doxorubicin. Cancer Res. 2001, 61, 771–777. [Google Scholar]
- Davies, K.J.; Doroshow, J.H. Redox cycling of anthracyclines by cardiac mitochondria. I. Anthracycline radical formation by NADH dehydrogenase. J. Biol. Chem. 1986, 261, 3060–3067. [Google Scholar] [CrossRef]
- Pereira, G.C.; Pereira, S.P.; Tavares, L.C.; Carvalho, F.S.; Magalhães-Novais, S.; Barbosa, I.A.; Santos, M.S.; Bjork, J.; Moreno, A.J.; Wallace, K.B.; et al. Cardiac cytochrome c and cardiolipin depletion during anthracycline-induced chronic depression of mitochondrial function. Mitochondrion 2016, 30, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Wallace, K.B. Adriamycin-induced interference with cardiac mitochondrial calcium homeostasis. Cardiovasc. Toxicol. 2007, 7, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Rocca, C.; Scavello, F.; Colombo, B.; Gasparri, A.M.; Dallatomasina, A.; Granieri, M.C.; Amelio, D.; Pasqua, T.; Cerra, M.C.; Tota, B.; et al. Physiological levels of chromogranin A prevent doxorubicin-induced cardiotoxicity without impairing its anticancer activity. FASEB J. 2019, 33, 7734–7747. [Google Scholar] [CrossRef]
- Cova, D.; De Angelis, L.; Monti, E.; Piccinini, F. Subcellular Distribution of Two Spin Trapping Agents in Rat Heart: Possible Explanation for Their Different Protective Effects against Doxorubicin-Induced Cardiotoxicity. Free Radic. Res. Commun. 1992, 15, 353–360. [Google Scholar] [CrossRef]
- Goormaghtigh, E.; Huart, P.; Praet, M.; Brasseur, R.; Ruysschaert, J.-M. Structure of the adriamycin-cardiolipin complex: Role in mitochondrial toxicity. Biophys. Chem. 1990, 35, 247–257. [Google Scholar] [CrossRef]
- El-Hafidi, M.; Correa, F.; Zazueta, C. Mitochondrial dysfunction in metabolic and cardiovascular diseases associated with cardiolipin remodeling. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2020, 1866, 165744. [Google Scholar] [CrossRef]
- Goormaghtigh, E.; Huart, P.; Brasseur, R.; Ruysschaert, J.-M. Mechanism of inhibition of mitochondrial enzymatic complex I–III by adriamycin derivatives. Biochim. Biophys. Acta (BBA) Biomembr. 1986, 861, 83–94. [Google Scholar] [CrossRef]
- Wallace, K.B.; Sardão, V.A.; Oliveira, P.J. Mitochondrial Determinants of Doxorubicin-Induced Cardiomyopathy. Circ. Res. 2020, 126, 926–941. [Google Scholar] [CrossRef]
- Nordgren, K.K.; Wallace, K.B. Keap1 redox-dependent regulation of doxorubicin-induced oxidative stress response in cardiac myoblasts. Toxicol. Appl. Pharmacol. 2014, 274, 107–116. [Google Scholar] [CrossRef]
- Sampaio, S.F.; Branco, A.F.; Wojtala, A.; Vega-Naredo, I.; Wieckowski, M.R.; Oliveira, P.J. p66Shc signaling is involved in stress responses elicited by anthracycline treatment of rat cardiomyoblasts. Arch. Toxicol. 2016, 90, 1669–1684. [Google Scholar] [CrossRef] [PubMed]
- Nabati, M.; Janbabai, G.; Baghyari, S.; Esmaili, K.; Yazdani-Charati, J. Cardioprotective Effects of Carvedilol in Inhibiting Doxorubicin-induced Cardiotoxicity. J. Cardiovasc. Pharmacol. 2017, 69, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Waldner, R.; Laschan, C.; Lohninger, A.; Gessner, M.; Tüchler, H.; Huemer, M.; Spiegel, W.; Karlic, H. Effects of doxorubicin-containing chemotherapy and a combination with l-carnitine on oxidative metabolism in patients with non-Hodgkin lymphoma. J. Cancer Res. Clin. Oncol. 2005, 132, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, R.; Ramirez, M.C.; Nes, K.; Schuster, A.; Aguayo, R.; Morales, M.; Ramos, C.; Hasson, D.; Sotomayor, C.G.; Henriquez, P.; et al. Prevention of doxorubicin-induced Cardiotoxicity by pharmacological non-hypoxic myocardial preconditioning based on Docosahexaenoic Acid (DHA) and carvedilol direct antioxidant effects: Study protocol for a pilot, randomized, double-blind, controlled trial (CarDHA trial). Trials 2020, 21, 137. [Google Scholar] [CrossRef]
- Macedo, A.V.; Hajjar, L.A.; Lyon, A.R.; Nascimento, B.R.; Putzu, A.; Rossi, L.; Costa, R.B.; Landoni, G.; Nogueira-Rodrigues, A.; Ribeiro, A.L. Efficacy of Dexrazoxane in Preventing Anthracycline Cardiotoxicity in Breast Cancer. JACC CardioOncol. 2019, 1, 68–79. [Google Scholar] [CrossRef]
- Kheiri, B.; Abdalla, A.; Osman, M.; Haykal, T.; Chahine, A.; Ahmed, S.; Osman, K.; Hassan, M.; Bachuwa, G.; Bhatt, D.L. Meta-Analysis of Carvedilol for the Prevention of Anthracycline-Induced Cardiotoxicity. Am. J. Cardiol. 2018, 122, 1959–1964. [Google Scholar] [CrossRef]
- Bosch, X.; Rovira, M.; Sitges, M.; Domènech, A.; Ortiz-Pérez, J.T.; de Caralt, T.M.; Morales-Ruiz, M.; Perea, R.J.; Monzó, M.; Esteve, J. Enalapril and carvedilol for preventing chemotherapy-induced left ventricular systolic dysfunction in patients with malignant hemopathies: The OVERCOME trial (preventiOn of left Ventricular dysfunction with Enalapril and caRvedilol in patients submitted to intensive ChemOtherapy for the treatment of Malignant hEmopathies). J. Am. Coll. Cardiol. 2013, 61, 2355–2362. [Google Scholar]
- Vincent, D.T.; Ibrahim, Y.F.; Espey, M.G.; Suzuki, Y.J. The role of antioxidants in the era of cardio-oncology. Cancer Chemother. Pharmacol. 2013, 72, 1157–1168. [Google Scholar] [CrossRef] [Green Version]
- Fabiani, I.; Aimo, A.; Grigoratos, C.; Castiglione, V.; Gentile, F.; Saccaro, L.F.; Arzilli, C.; Cardinale, D.; Passino, C.; Emdin, M. Oxidative stress and inflammation: Determinants of anthracycline cardiotoxicity and possible therapeutic targets. Heart Fail. Rev. 2021, 26, 881–890. [Google Scholar] [CrossRef]
- Bugger, H.; Guzman, C.; Zechner, C.; Palmeri, M.; Russell, K.S.; Russell, R.R. Uncoupling protein downregulation in doxorubicin-induced heart failure improves mitochondrial coupling but increases reactive oxygen species generation. Cancer Chemother. Pharmacol. 2010, 67, 1381–1388. [Google Scholar] [CrossRef] [Green Version]
- Walder, K.; Norman, R.A.; Hanson, R.; Schrauwen, P.; Neverova, M.; Jenkinson, C.P.; Easlick, J.; Warden, C.H.; Pecqueur, C.; Raimbault, S.; et al. Association between uncoupling protein polymorphisms (UCP2-UCP3) and energy metabolism/obesity in Pima indians. Hum. Mol. Genet. 1998, 7, 1431–1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvalho, R.A.; Sousa, R.P.; Cadete, V.J.; Lopaschuk, G.D.; Palmeira, C.M.; Bjork, J.A.; Wallace, K.B. Metabolic remodeling associated with subchronic doxorubicin cardiomyopathy. Toxicology 2010, 270, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Berthiaume, J.M.; Wallace, K.B. Persistent Alterations to the Gene Expression Profile of the Heart Subsequent to Chronic Doxorubicin Treatment. Cardiovasc. Toxicol. 2007, 7, 178–191. [Google Scholar] [CrossRef] [PubMed]
- Bansal, N.; Amdani, S.M.; Hutchins, K.K.; Lipshultz, S.E. Cardiovascular disease in survivors of childhood cancer. Curr. Opin. Pediatr. 2018, 30, 628–638. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Tao, A.; Song, J.; Liu, Q.; Wang, H.; Rui, T. Doxorubicin-induced cardiomyocyte apoptosis: Role of mitofusin 2. Int. J. Biochem. Cell Biol. 2017, 88, 55–59. [Google Scholar] [CrossRef]
- Samant, S.A.; Zhang, H.J.; Hong, Z.; Pillai, V.B.; Sundaresan, N.R.; Wolfgeher, D.; Archer, S.L.; Chan, D.C.; Gupta, M.P. SIRT3 Deacetylates and Activates OPA1 To Regulate Mitochondrial Dynamics during Stress. Mol. Cell. Biol. 2013, 34, 807–819. [Google Scholar] [CrossRef] [Green Version]
- Aung, L.H.H.; Li, R.; Prabhakar, B.S.; Li, P. Knockdown of Mtfp1 can minimize doxorubicin cardiotoxicity by inhibiting Dnm1l-mediated mitochondrial fission. J. Cell. Mol. Med. 2017, 21, 3394–3404. [Google Scholar] [CrossRef] [Green Version]
- Marques-Aleixo, I.; Alves, E.; Torrella, J.R.; Oliveira, P.; Magalhães, J.; Ascensao, A. Exercise and Doxorubicin Treatment Modulate Cardiac Mitochondrial Quality Control Signaling. Cardiovasc. Toxicol. 2018, 18, 43–55. [Google Scholar] [CrossRef]
- Xia, Y.; Chen, Z.; Chen, A.; Fu, M.; Dong, Z.; Hu, K.; Yang, X.; Zou, Y.; Sun, A.; Qian, J.; et al. LCZ696 improves cardiac function via alleviating Drp1-mediated mitochondrial dysfunction in mice with doxorubicin-induced dilated cardiomyopathy. J. Mol. Cell. Cardiol. 2017, 108, 138–148. [Google Scholar] [CrossRef]
- Zhuang, X.; Sun, X.; Zhou, H.; Zhang, S.; Zhong, X.; Xu, X.; Guo, Y.; Xiong, Z.; Liu, M.; Lin, Y.; et al. Klotho attenuated Doxorubicin-induced cardiomyopathy by alleviating Dynamin-related protein 1-mediated mitochondrial dysfunction. Mech. Ageing Dev. 2021, 195, 111442. [Google Scholar] [CrossRef]
- Lim, K.; Lu, T.-S.; Molostvov, G.; Lee, C.; Lam, F.; Zehnder, D.; Hsiao, L.-L. Vascular Klotho Deficiency Potentiates the Development of Human Artery Calcification and Mediates Resistance to Fibroblast Growth Factor 23. Circulation 2012, 125, 2243–2255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, J.; Cha, S.-K.; An, S.-W.; Kuro-O, M.; Birnbaumer, L.; Huang, C.-L. Cardioprotection by Klotho through downregulation of TRPC6 channels in the mouse heart. Nat. Commun. 2012, 3, 1238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catanzaro, M.P.; Weiner, A.; Kaminaris, A.; Li, C.; Cai, F.; Zhao, F.; Kobayashi, S.; Kobayashi, T.; Huang, Y.; Sesaki, H.; et al. Doxorubicin-induced cardiomyocyte death is mediated by unchecked mitochondrial fission and mitophagy. FASEB J. 2019, 33, 11096–11108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taguchi, N.; Ishihara, N.; Jofuku, A.; Oka, T.; Mihara, K. Mitotic Phosphorylation of Dynamin-related GTPase Drp1 Participates in Mitochondrial Fission. J. Biol. Chem. 2007, 282, 11521–11529. [Google Scholar] [CrossRef] [Green Version]
- Cereghetti, G.M.; Stangherlin, A.; de Brito, O.M.; Chang, C.-R.; Blackstone, C.; Bernardi, P.; Scorrano, L. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc. Natl. Acad. Sci. USA 2008, 105, 15803–15808. [Google Scholar] [CrossRef] [Green Version]
- Aung, L.H.H.; Chen, X.; Jumbo, J.C.C.; Li, Z.; Wang, S.-Y.; Zhao, C.; Liu, Z.; Wang, Y.; Li, P. Cardiomyocyte mitochondrial dynamic-related lncRNA 1 (CMDL-1) may serve as a potential therapeutic target in doxorubicin cardiotoxicity. Mol. Ther. Nucleic Acids 2021, 25, 638–651. [Google Scholar] [CrossRef]
- Zhou, L.; Li, R.; Liu, C.; Sun, T.; Aung, L.H.H.; Chen, C.; Gao, J.; Zhao, Y.; Wang, K. Foxo3a inhibits mitochondrial fission and protects against doxorubicin-induced cardiotoxicity by suppressing MIEF2. Free Radic. Biol. Med. 2017, 104, 360–370. [Google Scholar] [CrossRef]
- Chen, L.; Gong, Q.; Stice, J.P.; Knowlton, A.A. Mitochondrial OPA1, apoptosis, and heart failure. Cardiovasc. Res. 2009, 84, 91–99. [Google Scholar] [CrossRef] [Green Version]
- Lemmon, M.A.; Schlessinger, J. Cell Signaling by Receptor Tyrosine Kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [Green Version]
- Saraon, P.; Pathmanathan, S.; Snider, J.; Lyakisheva, A.; Wong, V.; Stagljar, I. Receptor tyrosine kinases and cancer: Oncogenic mechanisms and therapeutic approaches. Oncogene 2021, 40, 4079–4093. [Google Scholar] [CrossRef]
- Hubbard, S.R. Structural analysis of receptor tyrosine kinases. Prog. Biophys. Mol. Biol. 1999, 71, 343–358. [Google Scholar] [CrossRef]
- Robinson, D.R.; Wu, Y.-M.; Lin, S.-F. The protein tyrosine kinase family of the human genome. Oncogene 2000, 19, 5548–5557. [Google Scholar] [CrossRef] [Green Version]
- Roberti, M.; Bottegoni, G. Non-ATP Competitive Protein Kinase Inhibitors. Curr. Med. Chem. 2010, 17, 2804–2821. [Google Scholar] [CrossRef]
- Force, T.; Krause, D.S.; Van Etten, R.A. Molecular mechanisms of cardiotoxicity of tyrosine kinase inhibition. Nat. Cancer 2007, 7, 332–344. [Google Scholar] [CrossRef] [PubMed]
- Varricchi, G.; Ameri, P.; Cadeddu, C.; Ghigo, A.; Madonna, R.; Marone, G.; Mercurio, V.; Monte, I.; Novo, G.; Parrella, P.; et al. Antineoplastic Drug-Induced Cardiotoxicity: A Redox Perspective. Front. Physiol. 2018, 9, 167. [Google Scholar] [CrossRef] [PubMed]
- Di Lisi, D.; Madonna, R.; Zito, C.; Bronte, E.; Badalamenti, G.; Parrella, P.; Monte, I.; Tocchetti, C.G.; Russo, A.; Novo, G. Anticancer therapy-induced vascular toxicity: VEGF inhibition and beyond. Int. J. Cardiol. 2017, 227, 11–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, T.F.; Rupnick, M.A.; Kerkela, R.; Dallabrida, S.M.; Zurakowski, D.; Nguyen, L.; Woulfe, K.; Pravda, E.; Cassiola, F.; Desai, J.; et al. Cardiotoxicity associated with tyrosine kinase inhibitor sunitinib. Lancet 2007, 370, 2011–2019. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z. ErbB Receptors and Cancer. Methods Mol. Biol. 2017, 1652, 3–35. [Google Scholar]
- Kenigsberg, B.; Jain, V.; Barac, A. Cardio-oncology Related to Heart Failure: Epidermal Growth Factor Receptor Target-Based Therapy. Heart Fail Clin. 2017, 13, 297–309. [Google Scholar] [CrossRef]
- Grazette, L.P.; Boecker, W.; Matsui, T.; Semigran, M.; Force, T.; Hajjar, R.J.; Rosenzweig, A. Inhibition of ErbB2 causes mitochondrial dysfunction in cardiomyocytes: Implications for herceptin-induced cardiomyopathy. J. Am. Coll. Cardiol. 2004, 44, 2231–2238. [Google Scholar] [CrossRef] [Green Version]
- Crone, S.A.; Zhao, Y.Y.; Fan, L.; Gu, Y.; Minamisawa, S.; Liu, Y.; Peterson, K.L.; Chen, J.; Kahn, R.; Condorelli, G.; et al. ErbB2 is essential in the prevention of dilated cardiomyopathy. Nat. Med. 2002, 8, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Kerkelä, R.; Grazette, L.; Yacobi, R.; Iliescu, C.; Patten, R.; Beahm, C.; Walters, B.; Shevtsov, S.; Pesant, S.; Clubb, F.J.; et al. Cardiotoxicity of the cancer therapeutic agent imatinib mesylate. Nat. Med. 2006, 12, 908–916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerkela, R.; Woulfe, K.C.; Durand, J.B.; Vagnozzi, R.; Kramer, D.; Chu, T.F.; Beahm, C.; Chen, M.H.; Force, T. Sunitinib-induced cardiotoxicity is mediated by off-target inhibition of AMP-activated protein kinase. Clin. Transl. Sci. 2009, 2, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Will, Y.; Dykens, J.A.; Nadanaciva, S.; Hirakawa, B.; Jamieson, J.; Marroquin, L.D.; Hynes, J.; Patyna, S.; Jessen, B.A. Effect of the Multitargeted Tyrosine Kinase Inhibitors Imatinib, Dasatinib, Sunitinib, and Sorafenib on Mitochondrial Function in Isolated Rat Heart Mitochondria and H9c2 Cells. Toxicol. Sci. 2008, 106, 153–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellor, H.R.; Bell, A.R.; Valentin, J.-P.; Roberts, R.R.A. Cardiotoxicity Associated with Targeting Kinase Pathways in Cancer. Toxicol. Sci. 2010, 120, 14–32. [Google Scholar] [CrossRef]
- Huang, W.S.; Metcalf, C.A.; Sundaramoorthi, R.; Wang, Y.; Zou, D.; Thomas, R.M.; Zhu, X.; Cai, L.; Wen, D.; Liu, S.; et al. Discovery of 3-[2-(imidazo [1,2-b]pyridazin-3-yl)ethynyl]-4-methyl-N-{4-[(4-methylpiperazin-1-yl)methyl]-3-(trifluoromethyl)phenyl}benzamide (AP24534), a potent, orally active pan-inhibitor of breakpoint cluster region-abelson (BCR-ABL) kinase including the T315I gatekeeper mutant. J. Med. Chem. 2010, 53, 4701–4719. [Google Scholar]
- Talbert, D.R.; Doherty, K.R.; Trusk, P.B.; Moran, D.M.; Shell, S.A.; Bacus, S. A Multi-parameter In Vitro Screen in Human Stem Cell-Derived Cardiomyocytes Identifies Ponatinib-Induced Structural and Functional Cardiac Toxicity. Toxicol. Sci. 2015, 143, 147–155. [Google Scholar] [CrossRef] [Green Version]
- Weng, Z.; Luo, Y.; Yang, X.; Greenhaw, J.J.; Li, H.; Xie, L.; Mattes, W.; Shi, Q. Regorafenib impairs mitochondrial functions, activates AMP-activated protein kinase, induces autophagy, and causes rat hepatocyte necrosis. Toxicology 2015, 327, 10–21. [Google Scholar] [CrossRef] [Green Version]
- Thomson, M. Evidence of undiscovered cell regulatory mechanisms: Phosphoproteins and protein kinases in mitochondria. Cell. Mol. Life Sci. 2002, 59, 213–219. [Google Scholar] [CrossRef]
- Wang, X.; Shen, X.; Yan, Y.; Li, H. Pyruvate dehydrogenase kinases (PDKs): An overview toward clinical applications. Biosci. Rep. 2021, 41, 20204402. [Google Scholar] [CrossRef]
- Chaar, M.; Kamta, J.; Ait-Oudhia, S. Mechanisms, monitoring, and management of tyrosine kinase inhibitors-associated cardiovascular toxicities. OncoTargets Ther. 2018, 11, 6227–6237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Sheehan, R.P.; Palmer, A.C.; Everley, R.A.; Boswell, S.A.; Ron-Harel, N.; Ringel, A.E.; Holton, K.M.; Jacobson, C.A.; Erickson, A.R.; et al. Adaptation of Human iPSC-Derived Cardiomyocytes to Tyrosine Kinase Inhibitors Reduces Acute Cardiotoxicity via Metabolic Reprogramming. Cell Syst. 2019, 8, 412–426.e7. [Google Scholar] [CrossRef] [PubMed]
- Doenst, T.; Nguyen, T.D.; Abel, E.D. Cardiac metabolism in heart failure: Implications beyond ATP production. Circ. Res. 2013, 113, 709–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alhoshani, A.; Alanazi, F.E.; Alotaibi, M.R.; Attwa, M.W.; Kadi, A.A.; Aldhfyan, A.; Akhtar, S.; Hourani, S.; Agouni, A.; Zeidan, A.; et al. EGFR Inhibitor Gefitinib Induces Cardiotoxicity through the Modulation of Cardiac PTEN/Akt/FoxO3a Pathway and Reactive Metabolites Formation: In Vivo and in Vitro Rat Studies. Chem. Res. Toxicol. 2020, 33, 1719–1728. [Google Scholar] [CrossRef]
- Barton, M.; Filardo, E.; Lolait, S.J.; Thomas, P.; Maggiolini, M.; Prossnitz, E.R. Twenty years of the G protein-coupled estrogen receptor GPER: Historical and personal perspectives. J. Steroid Biochem. Mol. Biol. 2018, 176, 4–15. [Google Scholar] [CrossRef] [Green Version]
- Lappano, R.; De Marco, P.; De Francesco, E.M.; Chimento, A.; Pezzi, V.; Maggiolini, M. Cross-talk between GPER and growth factor signaling. J. Steroid Biochem. Mol. Biol. 2013, 137, 50–56. [Google Scholar] [CrossRef]
- Rocca, C.; Femminò, S.; Aquila, G.; Granieri, M.C.; De Francesco, E.M.; Pasqua, T.; Rigiracciolo, D.C.; Fortini, F.; Cerra, M.C.; Maggiolini, M.; et al. Notch1 Mediates Preconditioning Protection Induced by GPER in Normotensive and Hypertensive Female Rat Hearts. Front. Physiol. 2018, 9, 521. [Google Scholar] [CrossRef]
- Recchia, A.G.; De Francesco, E.M.; Vivacqua, A.; Sisci, D.; Panno, M.L.; Andò, S.; Maggiolini, M. The G Protein-coupled Receptor 30 Is Up-regulated by Hypoxia-inducible Factor-1α (HIF-1α) in Breast Cancer Cells and Cardiomyocytes. J. Biol. Chem. 2011, 286, 10773–10782. [Google Scholar] [CrossRef] [Green Version]
- De Francesco, E.M.; Rocca, C.; Scavello, F.; Amelio, D.; Pasqua, T.; Rigiracciolo, D.C.; Scarpelli, A.; Avino, S.; Cirillo, F.; Amodio, N.; et al. Protective Role of GPER Agonist G-1 on Cardiotoxicity Induced by Doxorubicin. J. Cell. Physiol. 2017, 232, 1640–1649. [Google Scholar] [CrossRef]
- Narayan, V.; Keefe, S.; Haas, N.; Wang, L.; Puzanov, I.; Putt, M.; Catino, A.; Fang, J.; Agarwal, N.; Hyman, D.; et al. Prospective Evaluation of Sunitinib-Induced Cardiotoxicity in Patients with Metastatic Renal Cell Carcinoma. Clin. Cancer Res. 2017, 23, 3601–3609. [Google Scholar] [CrossRef] [Green Version]
- Heath, E.I.; Infante, J.; Lewis, L.D.; Luu, T.; Stephenson, J.; Tan, A.R.; Kasubhai, S.; Lorusso, P.; Ma, B.; Suttle, A.B.; et al. A randomized, double-blind, placebo-controlled study to evaluate the effect of repeated oral doses of pazopanib on cardiac conduction in patients with solid tumors. Cancer Chemother. Pharmacol. 2013, 71, 565–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdel-Rahman, O.; Fouad, M. Risk of cardiovascular toxicities in patients with solid tumors treated with sorafenib: An updated systematic review and meta-analysis. Future Oncol. 2014, 10, 1981–1992. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.; Ding, M.; Li, X.; Zhou, X.; Zhu, Q.; Varela-Ramirez, A.; Yi, C. Comparative evaluation of cardiovascular risks among nine FDA-approved VEGFR-TKIs in patients with solid tumors: A Bayesian network analysis of randomized controlled trials. J. Cancer Res. Clin. Oncol. 2021, 147, 2407–2420. [Google Scholar] [CrossRef]
- Casavecchia, G.; Galderisi, M.; Novo, G.; Gravina, M.; Santoro, C.; Agricola, E.; Capalbo, S.; Zicchino, S.; Cameli, M.; De Gennaro, L.; et al. Early diagnosis, clinical management, and follow-up of cardiovascular events with ponatinib. Heart Fail. Rev. 2020, 25, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.E.; Kim, D.-W.; Pinilla-Ibarz, J.; Le Coutre, P.; Paquette, R.; Chuah, C.; Nicolini, F.E.; Apperley, J.F.; Khoury, H.J.; Talpaz, M.; et al. A Phase 2 Trial of Ponatinib in Philadelphia Chromosome–Positive Leukemias. N. Engl. J. Med. 2013, 369, 1783–1796. [Google Scholar] [CrossRef] [Green Version]
- Iacovelli, R.; Ciccarese, C.; Fornarini, G.; Massari, F.; Bimbatti, D.; Mosillo, C.; Rebuzzi, S.E.; DI Nunno, V.; Grassi, M.; Fantinel, E.; et al. Cabozantinib-related cardiotoxicity: A prospective analysis in a real-world cohort of metastatic renal cell carcinoma patients. Br. J. Clin. Pharmacol. 2019, 85, 1283–1289. [Google Scholar] [CrossRef]
- Milling, R.V.; Grimm, D.; Krüger, M.; Grosse, J.; Kopp, S.; Bauer, J.; Infanger, M.; Wehland, M. Pazopanib, Cabozantinib, and Vandetanib in the Treatment of Progressive Medullary Thyroid Cancer with a Special Focus on the Adverse Effects on Hypertension. Int. J. Mol. Sci. 2018, 19, 3258. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.D.; le Coutre, P.; Schwarz, M.; Grille, P.; Levitin, M.; Fateh-Moghadam, S.; Giles, F.J.; Dörken, B.; Haverkamp, W.; Köhncke, C. Clinical cardiac safety profile of nilotinib. Haematologica 2012, 97, 883–889. [Google Scholar] [CrossRef] [Green Version]
- Motzer, R.J.; Escudier, B.; Tomczak, P.; Hutson, T.E.; Michaelson, D.; Negrier, S.; Oudard, S.; Gore, M.E.; Tarazi, J.; Hariharan, S.; et al. Axitinib versus sorafenib as second-line treatment for advanced renal cell carcinoma: Overall survival analysis and updated results from a randomised phase 3 trial. Lancet Oncol. 2013, 14, 552–562. [Google Scholar] [CrossRef]
- Rousseau, A.; Bertolotti, A. Regulation of proteasome assembly and activity in health and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 697–712. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, L.; Amodio, N.; Gulla, A.; Anderson, K.C. Harnessing the Immune System against Multiple Myeloma: Challenges and Opportunities. Front. Oncol. 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Taiana, E.; Cantafio, M.G.; Favasuli, V.; Bandini, C.; Viglietto, G.; Piva, R.; Neri, A.; Amodio, N. Genomic Instability in Multiple Myeloma: A “Non-Coding RNA” Perspective. Cancers 2021, 13, 2127. [Google Scholar] [CrossRef]
- Annunziata, C.M.; Davis, R.E.; Demchenko, Y.; Bellamy, W.; Gabrea, A.; Zhan, F.; Lenz, G.; Hanamura, I.; Wright, G.; Xiao, W.; et al. Frequent engagement of the classical and alternative NF-kappaB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell 2007, 12, 115–130. [Google Scholar] [CrossRef] [Green Version]
- Motegi, A.; Murakawa, Y.; Takeda, S. The vital link between the ubiquitin–proteasome pathway and DNA repair: Impact on cancer therapy. Cancer Lett. 2009, 283, 1–9. [Google Scholar] [CrossRef]
- Amodio, N.; Bellizzi, D.; Leotta, M.; Raimondi, L.; Biamonte, L.; D’Aquila, P.; Di Martino, M.T.; Calimeri, T.; Rossi, M.; Lionetti, M.; et al. miR-29b induces SOCS-1 expression by promoter demethylation and negatively regulates migration of multiple myeloma and endothelial cells. Cell Cycle 2013, 12, 3650–3662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amodio, N.; D’Aquila, P.; Passarino, G.; Tassone, P.; Bellizzi, D. Epigenetic modifications in multiple myeloma: Recent advances on the role of DNA and histone methylation. Expert Opin. Ther. Targets 2016, 21, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Gandolfi, S.; Laubach, J.P.; Hideshima, T.; Chauhan, D.; Anderson, K.C.; Richardson, P.G. The proteasome and proteasome inhibitors in multiple myeloma. Cancer Metastasis Rev. 2017, 36, 561–584. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, S.V.; Kumar, S. Multiple myeloma current treatment algorithms. Blood Cancer J. 2020, 10, 94. [Google Scholar] [CrossRef] [PubMed]
- Ito, S. Proteasome Inhibitors for the Treatment of Multiple Myeloma. Cancers 2020, 12, 265. [Google Scholar] [CrossRef] [Green Version]
- Cole, D.C.; Frishman, W.H. Cardiovascular Complications of Proteasome Inhibitors Used in Multiple Myeloma. Cardiol. Rev. 2018, 26, 122–129. [Google Scholar] [CrossRef]
- Nowis, D.; Mączewski, M.; Mackiewicz, U.; Kujawa, M.; Ratajska, A.; Wieckowski, M.; Wilczynski, G.; Malinowska, M.; Bil, J.; Salwa, P.; et al. Cardiotoxicity of the Anticancer Therapeutic Agent Bortezomib. Am. J. Pathol. 2010, 176, 2658–2668. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Li, J.; Huang, W.; Su, H.; Liang, Q.; Tian, Z.; Horak, K.M.; Molkentin, J.D.; Wang, X. Proteasome functional insufficiency activates the calcineurin–NFAT pathway in cardiomyocytes and promotes maladaptive remodelling of stressed mouse hearts. Cardiovasc. Res. 2010, 88, 424–433. [Google Scholar] [CrossRef] [Green Version]
- Hasinoff, B.B.; Patel, D.; Wu, X. Molecular Mechanisms of the Cardiotoxicity of the Proteasomal-Targeted Drugs Bortezomib and Carfilzomib. Cardiovasc. Toxicol. 2017, 17, 237–250. [Google Scholar] [CrossRef]
- Kisselev, A.F.; Goldberg, A.L. Proteasome inhibitors: From research tools to drug candidates. Chem. Biol. 2001, 8, 739–758. [Google Scholar] [CrossRef] [Green Version]
- Mårtensson, C.U.; Priesnitz, C.; Song, J.; Ellenrieder, L.; Doan, K.N.; Boos, F.; Floerchinger, A.; Zufall, N.; Oeljeklaus, S.; Warscheid, B.; et al. Mitochondrial protein translocation-associated degradation. Nature 2019, 569, 679–683. [Google Scholar] [CrossRef]
- Pokorna, Z.; Jirkovsky, E.; Hlavackova, M.; Jansova, H.; Jirkovska, A.; Lencova-Popelova, O.; Brazdova, P.; Kubes, J.; Sotakova-Kasparova, D.; Mazurova, Y.; et al. In vitro and in vivo investigation of cardiotoxicity associated with anticancer proteasome inhibitors and their combination with anthracycline. Clin. Sci. 2019, 133, 1827–1844. [Google Scholar] [CrossRef] [PubMed]
- Pancheri, E.; Guglielmi, V.; Wilczynski, G.M.; Malatesta, M.; Tonin, P.; Tomelleri, G.; Nowis, D.; Vattemi, G. Non-Hematologic Toxicity of Bortezomib in Multiple Myeloma: The Neuromuscular and Cardiovascular Adverse Effects. Cancers 2020, 12, 2540. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Yin, J.; Wei, J.; Shang, Z. Incidence and Risk of Cardiotoxicity Associated with Bortezomib in the Treatment of Cancer: A Systematic Review and Meta-Analysis. PLoS ONE 2014, 9, e87671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laubach, J.P.; Moslehi, J.J.; Francis, S.A.; San Miguel, J.F.; Sonneveld, P.; Orlowski, R.Z.; Moreau, P.; Rosiñol, L.; Faber, E.A., Jr.; Voorhees, P.; et al. A retrospective analysis of 3954 patients in phase 2/3 trials of bortezomib for the treatment of multiple myeloma: Towards providing a benchmark for the cardiac safety profile of proteasome inhibition in multiple myeloma. Br. J. Haematol. 2017, 178, 547–560. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, D.J.; Chen, Q.; Voorhees, P.M.; Strader, J.S.; Shenk, K.D.; Sun, C.M.; Demo, S.D.; Bennett, M.K.; van Leeuwen, F.; Chanan-Khan, A.A.; et al. Potent activity of carfilzomib, a novel, irreversible inhibitor of the ubiquitin-proteasome pathway, against preclinical models of multiple myeloma. Blood 2007, 110, 3281–3290. [Google Scholar] [CrossRef] [Green Version]
- Demo, S.D.; Kirk, C.J.; Aujay, M.A.; Buchholz, T.J.; Dajee, M.; Ho, M.N.; Jiang, J.; Laidig, G.J.; Lewis, E.R.; Parlati, F.; et al. Antitumor Activity of PR-171, a Novel Irreversible Inhibitor of the Proteasome. Cancer Res. 2007, 67, 6383–6391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, D.; Martin, T.; Nooka, A.; Harvey, R.D.; Vij, R.; Niesvizky, R.; Badros, A.Z.; Jagannath, S.; McCulloch, L.; Rajangam, K.; et al. Integrated safety profile of single-agent carfilzomib: Experience from 526 patients enrolled in 4 phase II clinical studies. Haematology 2013, 98, 1753–1761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, A.K.; Rajkumar, S.V.; Dimopoulos, M.A.; Masszi, T.; Špička, I.; Oriol, A.; Hájek, R.; Rosiñol, L.; Siegel, D.S.; Mihaylov, G.G.; et al. Carfilzomib, lenalidomide, and dexamethasone for relapsed multiple myeloma. N. Engl. J. Med. 2015, 372, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Siegel, D.S.; Dimopoulos, M.A.; Ludwig, H.; Facon, T.; Goldschmidt, H.; Jakubowiak, A.; Miguel, J.S.; Obreja, M.; Blaedel, J.; Stewart, A.K. Improvement in Overall Survival with Carfilzomib, Lenalidomide, and Dexamethasone in Patients with Relapsed or Refractory Multiple Myeloma. J. Clin. Oncol. 2018, 36, 728–734. [Google Scholar] [CrossRef] [PubMed]
- Atrash, S.; Tullos, A.; Panozzo, S.; Bhutani, M.S.; Van Rhee, F.; Barlogie, B.; Usmani, S.Z. Cardiac complications in relapsed and refractory multiple myeloma patients treated with carfilzomib. Blood Cancer J. 2015, 5, e272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waxman, A.J.; Clasen, S.C.; Garfall, A.L.; Carver, J.R.; Vogl, D.T.; O’Quinn, R.; Cohen, A.D.; Stadtmauer, E.A.; Ky, B.; Weiss, B.M. Carfilzomib-associated cardiovascular adverse events: A systematic review and meta-analysis. J. Clin. Oncol. 2017, 35, 8018. [Google Scholar] [CrossRef]
- Cornell, R.F.; Ky, B.; Weiss, B.M.; Dahm, C.N.; Gupta, D.K.; Du, L.; Carver, J.R.; Cohen, A.D.; Engelhardt, B.G.; Garfall, A.; et al. Prospective Study of Cardiac Events During Proteasome Inhibitor Therapy for Relapsed Multiple Myeloma. J. Clin. Oncol. 2019, 37, 1946–1955. [Google Scholar] [CrossRef]
- Chauhan, D.; Tian, Z.; Zhou, B.; Kuhn, D.; Orlowski, R.; Raje, N.; Richardson, P.; Anderson, K.C. In Vitro and In Vivo Selective Antitumor Activity of a Novel Orally Bioavailable Proteasome Inhibitor MLN9708 against Multiple Myeloma Cells. Clin. Cancer Res. 2011, 17, 5311–5321. [Google Scholar] [CrossRef] [Green Version]
- Kupperman, E.; Lee, E.C.; Cao, Y.; Bannerman, B.; Fitzgerald, M.; Berger, A.; Yu, J.; Yang, Y.; Hales, P.; Bruzzese, F.; et al. Evaluation of the Proteasome Inhibitor MLN9708 in Preclinical Models of Human Cancer. Cancer Res. 2010, 70, 1970–1980. [Google Scholar] [CrossRef] [Green Version]
- Moreau, P.; Masszi, T.; Grzasko, N.; Bahlis, N.J.; Hansson, M.; Pour, L.; Sandhu, I.; Ganly, P.; Baker, B.W.; Jackson, S.R.; et al. Oral Ixazomib, Lenalidomide, and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2016, 374, 1621–1634. [Google Scholar] [CrossRef]
- Wu, P.; Oren, O.; Gertz, M.A.; Yang, E.H. Proteasome Inhibitor-Related Cardiotoxicity: Mechanisms, Diagnosis, and Management. Curr. Oncol. Rep. 2020, 22, 66. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Wu, C.; Lei, M.; Yan, Q.; Zhang, H.; Zhang, L.; Wang, X.; Yang, Y.; Li, J.; Zhu, Y.; et al. Anti-tumor activity of a novel proteasome inhibitor D395 against multiple myeloma and its lower cardiotoxicity compared with carfilzomib. Cell Death Dis. 2021, 12, 429. [Google Scholar] [CrossRef]
- Paradzik, T.; Bandini, C.; Mereu, E.; Labrador, M.; Taiana, E.; Amodio, N.; Neri, A.; Piva, R. The Landscape of Signaling Pathways and Proteasome Inhibitors Combinations in Multiple Myeloma. Cancers 2021, 13, 1235. [Google Scholar] [CrossRef] [PubMed]
- Diwadkar, S.; Patel, A.A.; Fradley, M.G. Bortezomib-Induced Complete Heart Block and Myocardial Scar: The Potential Role of Cardiac Biomarkers in Monitoring Cardiotoxicity. Case Rep. Cardiol. 2016, 2016, 3456287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meseeha, M.G.; Kolade, V.O.; Attia, M.N. Partially reversible bortezomib-induced cardiotoxicity: An unusual cause of acute cardiomyopathy. J. Community Hosp. Intern. Med. Persp. 2015, 5, 28982. [Google Scholar] [CrossRef] [Green Version]
- Voortman, J.; Giaccone, G. Severe reversible cardiac failure after bortezomib treatment combined with chemotherapy in a non-small cell lung cancer patient: A case report. BMC Cancer 2006, 6, 129. [Google Scholar] [CrossRef] [Green Version]
- Danhof, S.; Schreder, M.; Rasche, L.; Strifler, S.; Einsele, H.; Knop, S. ‘Real-life’ experience of reapproval carfilzomib-based therapy in myeloma—Analysis of cardiac toxicity and predisposing factors. Eur. J. Haematol. 2016, 97, 25–32. [Google Scholar] [CrossRef]
- Hahn, V.S.; Zhang, K.W.; Sun, L.; Narayan, V.; Lenihan, D.J.; Ky, B. Heart Failure with Targeted Cancer Therapies: Mechanisms and Cardioprotection. Circ. Res. 2021, 128, 1576–1593. [Google Scholar] [CrossRef]
- Jouni, H.; Aubry, M.C.; Lacy, M.Q.; Rajkumar, S.V.; Kumar, S.K.; Frye, R.L.; Herrmann, J. Ixazomib cardiotoxicity: A possible class effect of proteasome inhibitors. Am. J. Hematol. 2017, 92, 220–221. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Tyrosine Kinase Inhibitor | Molecular Target | Type of Study | Type of Cancer | Cardiotoxic Effect | Ref. |
|---|---|---|---|---|---|
| Sunitinib | Multi-tyrosine kinases (VEGFR, PDGFR, c-KIT) | Phase I/II clinical trial Multicenter prospective study | Imatinib-resistant, metastatic, gastrointestinal stromal tumors metastatic renal cell carcinoma | Left ventricular dysfunction congestive heart failure hypertension | [157,180] |
| Pazopanib | Multi-tyrosine kinases (VEGFR, PDGFR, c-KIT) | Randomized, double-blind, placebo-controlled study | Advanced solid tumors | Hypertension reduction in heart rate small prolongation of the QTc interval | [181] |
| Sorafenib | Multi-tyrosine kinases (VEGFR, PDGFR, FLT3) | Systematic review and meta-analysis | Renal cell carcinoma melanoma | Hypertension myocardial infarction ischemia acute coronary syndrome rarely heart failure | [182] |
| Regorafenib | Multi-tyrosine kinases (VEGFR1-3, PDGFR-β, FGFR) | Meta-analysis of 45 RTCs | Solid tumors | Hypertension generally few cardiovascular side effects | [183] |
| Ponatinib | Multi-tyrosine kinases FGFR, PDGFR, and VEGFR | Phase II clinical trial Review | Chronic myeloid leukemia; Philadelphia chromosome-positive leukemias | Arterial thrombotic events | [184,185] |
| Cabozantinib | Flt-3, RET, MET | Multicenter prospective study Review | Metastatic renal cell carcinoma medullary thyroid cancer | Modest risk of developing left ventricular systolic dysfunction hypertension | [186,187] |
| Nilotinib | PDGFR, CSF-1R, | Retrospective study | Chronic myeloid leukemia | Accelerated atherosclerosis peripheral arterial occlusive disease (PAOD) QTc prolongation. | [188] |
| Axitinib | VEGFR | Clinical trial | Metastatic renal cell carcinoma | Hypertension myocardial infarction | [189] |
| Proteasome Inhibitors | Mechanism of Action | Type of Study | Type of Cancer | Cardiotoxic Effects | Potential Preventive/Cardioprotective Strategies | Ref. |
|---|---|---|---|---|---|---|
| Bortezomib | Slowly-reversible inhibitor of β5 and β5i subunits | Systematic review and meta-analysis of 25 prospective phase II/III trials | Untreated multiple myeloma | Heart failure, conduction disorders, arrhythmias, ischemic heart disease, pericardial effusion and orthostatic hypotension | Assessment of cardiac function, evaluation of serum biomarkers of heart failure; Evaluation of atrial fibrillation history; Identification of cardiovascular risk factors; Use of β-blockers, ACE inhibitors, angiotensin II receptor blockers, apremilast (PDE4 inhibitor), metformin, PKG activator | [201,203,204,205,207,221,224,225,226,227,228] |
| Carfilzomib | Irreversible inhibitor of β5 and β5i subunits | Phase III trial (ASPIRE trial) Prospective, observational study (PROTECT trial) | Relapsed and refractory multiple myeloma | Arrhythmias, heart failure, cardiomyopathy, ischemic heart disease | [212,217,221,224,225,226,227,228] | |
| Ixazomib | Reversible inhibitor of β5 and β5i subunits, inhibition of β1 and β2 subunits at high concentration | Randomized phase III trial (TOURMALINE-MM1 trial) | Relapsed and refractory multiple myeloma | Heart failure | [220,221,224,225,226,227,228,229] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rocca, C.; De Francesco, E.M.; Pasqua, T.; Granieri, M.C.; De Bartolo, A.; Gallo Cantafio, M.E.; Muoio, M.G.; Gentile, M.; Neri, A.; Angelone, T.; et al. Mitochondrial Determinants of Anti-Cancer Drug-Induced Cardiotoxicity. Biomedicines 2022, 10, 520. https://doi.org/10.3390/biomedicines10030520
Rocca C, De Francesco EM, Pasqua T, Granieri MC, De Bartolo A, Gallo Cantafio ME, Muoio MG, Gentile M, Neri A, Angelone T, et al. Mitochondrial Determinants of Anti-Cancer Drug-Induced Cardiotoxicity. Biomedicines. 2022; 10(3):520. https://doi.org/10.3390/biomedicines10030520
Chicago/Turabian StyleRocca, Carmine, Ernestina Marianna De Francesco, Teresa Pasqua, Maria Concetta Granieri, Anna De Bartolo, Maria Eugenia Gallo Cantafio, Maria Grazia Muoio, Massimo Gentile, Antonino Neri, Tommaso Angelone, and et al. 2022. "Mitochondrial Determinants of Anti-Cancer Drug-Induced Cardiotoxicity" Biomedicines 10, no. 3: 520. https://doi.org/10.3390/biomedicines10030520