
Update on Molecular Genetics of Gastrointestinal Stromal Tumors


Diagnostic and Research Institute of Pathology, Medical University of Graz, 8010 Graz, Austria
*
Author to whom correspondence should be addressed.
Diagnostics 2021, 11(2), 194; https://doi.org/10.3390/diagnostics11020194
Submission received: 17 December 2020
/
Revised: 21 January 2021
/
Accepted: 23 January 2021
/
Published: 28 January 2021
(This article belongs to the Special Issue Molecular Classification of Soft Tissue and Bone Tumors)
Abstract
:Gastrointestinal stromal tumors (GISTs) are the most common mesenchymal tumors of the gastrointestinal tract. The majority are sporadic, solitary tumors that harbor mutually exclusive KIT or PDGFRA gain-of-function mutations. The type of mutation in addition to risk stratification corresponds to the biological behavior of GIST and response to treatment. Up to 85% of pediatric GISTs and 10–15% of adult GISTs are devoid of these (KIT/PDGFRA) mutations and are referred to as wild-type GISTs (wt-GIST). It has been shown that these wt-GISTs are a heterogeneous tumor group with regard to their clinical behavior and molecular profile. Recent advances in molecular pathology helped to further sub-classify the so-called “wt-GISTs”. Based on their significant clinical and molecular heterogeneity, wt-GISTs are divided into a syndromic and a non-syndromic (sporadic) subgroup. Recently, the use of succinate dehydrogenase B (SDHB) by immunohistochemistry has been used to stratify GIST into an SDHB-retained and an SDHB-deficient group. In this review, we focus on GIST sub-classification based on clinicopathologic, and molecular findings and discuss the known and yet emerging prognostic and predictive genetic alterations. We also give insights into the limitations of targeted therapy and highlight the mechanisms of secondary resistance.
1. Introduction
Gastrointestinal stromal tumors (GISTs) are the most common mesenchymal neoplasms of the gastrointestinal tract with an annual incidence of approximately 10–15 cases per million [1,2]. They recapitulate the interstitial cells of Cajal (ICC) lineage/differentiation [3,4]. GISTs most frequently occur in the stomach followed by the small intestine (including duodenum) and rarely affect the colon/rectum and the esophagus. Metastases mainly occur in the liver and peritoneum [5]. They usually present sporadically in older adults (median age 60–65 years) with slight male predominance [6]. Up to 2% of GISTs occur in children with girls being more frequently affected. In addition, the association of GIST with various syndromes has been described, including neurofibromatosis-1 (NF1), familial GISTs, Carney triad (CT) and Carney Stratakis Syndrome (CSS) [1,7,8,9,10].
In 1998, Hirota et al. published a landmark paper demonstrating that activating mutations in the KIT gene, a transmembrane receptor with tyrosine kinase activity, is an oncogenic driver event in GISTs development [11] and in 2003, Heinrich et al. identified alternative mutations in the platelet-derived growth factor receptor alpha (PDGFRA) gene, as the second most common driver mutation in GISTs, showing that KIT and PDGFRA mutations are mutually exclusive [12]. Knowledge about the underlying genetic alterations revealed possible targeted treatment with tyrosine kinase inhibitors (TKIs) such as imatinib and sunitinib [13,14,15]. Continuous research efforts helped to further elucidate molecular insights of this disease and allowed the development of new treatment options based on the underlying molecular signature [14,16,17,18,19].
Nevertheless, the exact pathologic classification of GIST is the backbone of GIST treatment and is based on H&E (hematoxylin & eosin) morphology, immunohistochemistry (KIT, DOG1), risk stratification according to Miettinen and molecular testing at least in cases where treatment with TKI is planned [5,20].
In general, based on cytomorphology, three different morphologic patterns can be seen [2,21,22]. GISTs with spindle cell morphology (~70% of cases) are composed of cells with palely eosinophilic fibrillary cytoplasm, ovoid nuclei and ill-defined cell borders commonly with a syncytial appearance (Figure 1A). The cells are arranged in short fascicles or whorls. Epithelioid GISTs (up to 20% of cases) are composed of round cells with eosinophilic to clear cytoplasm arranged in sheets and nests (Figure 1B). GISTs with mixed morphology (~10%) are composed of both spindle and epithelioid cells. The cellularity is highly variable and collagenous, sclerotic or myxoid stromal changes can be seen in each subtype. The main differential diagnoses for all patterns are summarized in Table 1. Spindle cell GISTs can show nuclear palisading (Figure 1C), a storiform growth pattern (Figure 1D), and prominent paranuclear vacuolation (Figure 1E), a morphologic feature by far more commonly found in GISTs than in smooth muscle tumors. Epithelioid tumors can demonstrate a clear cytoplasm and a prominent plasmacytoid morphology (Figure 1F). The vast majority of GISTs are characterized by uniform and monotonous tumor cells. Nevertheless, pleomorphic GISTs and dedifferentiated GISTs are rarely seen (Figure 1G,H) [21,23,24,25,26,27].
In total, 95% of “classic” GIST express KIT by immunohistochemistry [28]. In addition, expression of CD34 in about 60–70%, smooth muscle actin (SMA) in 30–40%, S-100 protein in 5%, and desmin or keratin in 1–2% have been reported [22]. Moreover, discovered on GIST-1 (DOG1), a calcium-activated chloride channel protein has been shown useful to detect KIT negative tumors [29,30,31]. Specifically, the clone K9 showed the highest sensitivity and specificity for both KIT-positive and -negative tumors [32].
It is well known that approximately 80% of GISTs harbor activating mutations in the KIT or PDGFRA genes that are responsible for the up-regulation of crucial signaling pathways including MAPK and PI3K-AKT [2,12,33,34]. On the other hand, GISTs lacking KIT and PDGFRA mutations are referred to as “wild-type” (wt)-GISTs [35,36]. These tumors differ from KIT and PDGFRA-mutant GISTs with regard to their clinical behavior and heterogenetic molecular profile. Over the past few years, advances in molecular pathology helped to elucidate alternative molecular drivers in the non KIT- non PDGFRA- mutated so-called “wt”-GIST group. Alternative mutations, structural chromosomal and epigenetic changes have been demonstrated in this group making the molecular classification more complex. Recent insights about the crucial role of the SDH-complex, especially in the pathobiology of pediatric GISTs, helped to divide GISTs by immunohistochemistry in a succinate dehydrogenase B (SDHB)-retained and SDHB-deficient subgroup. This widely available screening approach can facilitate decisions on further molecular testing strategies.
This review will focus on the molecular genetics of classic KIT/PDGFRA mutated GIST and on the sub-classification of the wt-GIST group based on recent molecular findings. In addition, emerging prognostic and predictive genetic alterations will be discussed. Furthermore, insights into the limitations of targeted therapy and the mechanisms of secondary resistance will be highlighted.
2. Molecular Classification
Recent advances in molecular pathology led to a better sub-classification of GISTs into an SDH-competent and an SDH-deficient group (by using an SDHB immunohistochemistry (IHC)), regardless of responsible mutation being acquired or inherited (Figure 2).
The SDH-competent tumor group includes: (i) KIT- and PDGFRA-mutated GISTs, (ii) GISTs with mutations in BRAF, NF1, HRAS, NRAS, and (iii) GISTs with exceedingly rare reported mutations in ARID1A, ARID1B, CBL, FGFR1, ATR, LTK, SUFU, PARK2, ZNF217, KRAS, MEN1 and PIK3CA. Additionally, (iiii) GISTs harboring structural chromosomal changes such as FGFR1-HOOK3, FGFR1-TACC1, ETV6–NTRK3, KIT-PDGFRA and PRKAR1B-BRAF are placed within the group of the SDH-competent GIST [9,37,38,39,40,41].
The SDH-deficient tumor group includes wt-GISTs in association with CT, CSS or sporadic pediatric and so-called “young adult” GISTs [5,9,10,42].
The frequency of the most common genetic alterations found in GIST is presented in Table 2.
2.1. KIT/PDGFRA-Mutated GIST
2.1.1. KIT-Mutated GISTs
KIT is a proto-oncogene and encodes the 145-kDa receptor tyrosine kinase (RTK) KIT, a transmembrane receptor with tyrosine kinase (TK) activity located on chromosome 4q11-q12 [5]. It is a member of the type III RTK family (together with PDGFRA, PDGFRB, the macrophage colony-stimulating-factor 1 receptor (CSF1R) and the receptor-type tyrosine-protein kinase FLT3 [43]. KIT consists of two main regions, the receptor regulatory domains (dimerization domain in the extracellular (EC) region, the transmembrane region and the juxta-membrane domain (JM) as well as the enzymatic domains the intracellular tyrosine kinase domains (TK[I] and TK[II]) (Figure 3) [44].
The RTK-KIT plays an important role in cell proliferation and differentiation (including Cajal cells) [3,45,46] and therefore plays a crucial role in the development of tumors (especially GIST, acute myeloid leukemia and melanoma) when it is mutated or upregulated [44]. These changes result in constant activation of the TK with consequent phosphorylation of substrate proteins without the presence of the corresponding ligand, called stem cell factor, influencing the intracellular signal transduction cascades, such as Ras/Raf/MAPK and PI3K/AKT pathways and leading to a constant autonomous activation causing an uncontrolled proliferation and inhibition of apoptosis [44,47,48].
Most of the KIT mutations in GISTs are somatic and are found in up to 80% of all tumors (Figure 2), rarely families with germline mutations have been described [8,34]. The mutations are found in different gene regions, including exons 8, 9, 11, 13, 14, 15, and 17. Mutations in exon 11 are the most frequent and are affecting the juxta-membrane domain (Figure 3). They are mostly caused by in-frame deletions within codon Gln550 and Glu560 (known as hot spot regions), missense-point-mutations mainly affecting codon Trp557, Val559, Val560 or Leu576 and duplications (especially in the 3′end) [21,49]. Deletions affecting codons 557–558 of exon 11 of the KIT gene have been reported in up to 28% of all GISTs and have been associated with high-risk tumors, having higher mitotic index (>5/50 HPF) and larger (>5 cm) tumor size [34]. Tumors showing this molecular profile occur equally in gastric and non- gastric location and arise in patients usually younger than 60 years. The local recurrence rate is lower compared to KIT exon 9, PDGFRA exon 18 and other KIT exon 11-mutated tumors. However, the prognostic power seems to be confirmed to the gastric location [34,50,51,52].
According to the Polish Registry, GIST with intron10/exon11 junction deletions (resulting in pK550_K558 deletion) and homo/hemizygous KIT exon 11 mutant GIST are rare tumors, accounting for 1.4% and 4% of all GISTs, respectively. Both are high-risk tumors with presumed aggressive/metastatic behavior and early metastatic disease/disseminated disease at presentation, respectively [34]. In contrast, single nucleotide substitutions and duplications (exclusively gastric location) are associated with benign clinical outcome [34,51]. Similar findings were very recently published by Shen et al. [53].
The mutations in exon 9 are found in approximately 10% of cases and are interfering with the extracellular parts (duplications of Ala502- Tyr503) (Figure 3). These tumors are commonly located in the small bowel and are often associated with a more aggressive phenotype [49]. Primary KIT mutations can also occur in exon 13 (TK[I]: ATP binding pocket) and exon 17 (TK[II]: kinase activation loop), but these mutations are rare (~2%) and data are quite limited [55]. KIT exon 13 and exon 17 mutant GISTs are more frequently found in the small bowel, usually have spindle cell morphology and most of them have the same behavior when compared to other GISTs. Gastric KIT exon 13 mutant is an exception as they tend to be slightly larger and more aggressive than gastric GISTs on average [55,56].
2.1.2. PDGFRA-Mutated GISTs
PDGFRA is a typical RTK and is, as well as KIT, located on chromosome 4q11-q12. Together with its ligand platelet-derived growth factor (PDGF), it is responsible for many physiological processes of growing and development in the human body. The receptor is similarly constructed as KIT (Figure 3). The PDGFs are divided into five isoforms: PDGF-AA, PDGF-AB, PDGF-BB, PDG-FCC and PDGF-DD, and bind to the receptors PDGFRA and PDGFRB [57]. Particularly, the PDGFRA is important for lung, skin, intestine, skeleton, gonads and is an essential factor in embryonic development. Normally, after binding to the receptor, phosphorylation activates signal cascades (Ras/Raf/MAPK and PI3K) [57]. Due to genetic aberrations, the PDGF signal is unrestrained active in neoplastic cells, which leads to ligand-independent phosphorylation and therefore uninhibited proliferation. Additionally, it plays a role in the epithelial-mesenchymal-transformation. PDGFRA-mutated GIST account for approximately 8–10% of GISTs (Figure 2); however, their lower representation in clinical trials can be explained by a comparatively benign clinical behavior of these tumors.
Most frequently, mutations are localized in the exon 18 (Figure 3) that codes for the activation loop in the TK domain and represents about 80% of the PDGFRA-mutated GISTs [58]. Mostly, these are missense mutations that result in the substitution of Asp to Val in codon 842 (D842V), a mutation known to be imatinib-resistant [16,51]. Further mutations are described in exon 14 and rarely in exon 12. Exon 14 correlates to the TK domain and exon 12 to the juxta-membrane. Typically, PDGFRA-mutated GISTs show an epithelioid pattern and are located in the stomach [12,29,31]. PDGFRA exon 18 mutation status correlates with an extremely favorable disease outcome compared to KIT exon 9 mutations and KIT deletions involving codons 557 and/or 558 of exon 11 [50]. Nevertheless, cases in the stomach that progressed (11 of 14 cases) carried an exon 18 PDGFRA D842V substitution [50].
2.1.3. GIST Genomic Progression Model
In most GISTs (including micro GISTs), KIT, PDGFRA, NF1 or SDH mutations are the initiating oncogenic drivers. However, additional stepwise accumulation of chromosomal aberrations is necessary/essential for further tumor progression. The earliest aberration found in up to 70% of cases is the loss of 14q [59]. Recently, somatic homozygous inactivating mutations of the chromosome 14q of the MYC-associated factor X (MAX) gene has been identified as a common early step in the progression of GISTs (microGISTs and low-risk GISTs) [60,61]. The inactivation of the MAX tumor suppressor leads to a p16 inactivation and an increase of proliferation in early tumors. In intermediate and high-risk GISTs further alterations have been described, namely, losses of 22q, 1p, 15q, 13q and 9p (spanning CDKN2A or p16INK4A) and/or gains at 5q, 8q, 16q and 20q [62,63,64,65,66]. These cell cycle dysregulating events result in inactivating mutations in other tumor suppressors such as p16, RB1, TP53 and cause the transition to high-grade GISTs [67,68,69]. Furthermore, it has been demonstrated that inactivation of dystrophin, encoded by the DMD gene on Xp21.1, contributes to permissiveness for metastatic behavior in GIST and was found in approximately 90% of metastatic GISTs [70].
2.1.4. Resistance Mechanisms in GIST
The vast majority of patients with unresectable or metastatic GIST respond to imatinib treatment. Treatment response can be demonstrated on CT scan as a reduction of the tumor mass or as decreased FDG uptake on a PET scan.
Resistance to imatinib, primary and secondary, can be partially explained by a conformational shift in the kinase domain of KIT and PDGFRA that favor the activated state [71]. Imatinib can only bind to the inactive conformation of both the KIT and PDGFRA receptors. For example, the PDGFRA D842V mutation, a known imatinib resistance mutation, results in a distortion of the kinase activation loop, thus strongly tilting the protein conformation in favor of the activated structure.
Tumor progression within the first 6 month of treatment is known as primary resistance. In this group patients with GISTs harboring a PDGFRA D842V mutation are therefore over-represented in the primary resistant GIST group as well as wt-GIST and KIT exon 9-mutated GISTs initially treated with only 400 mg of imatinib [16,51].
Although the majority of patients show a good response or stable disease under imatinib treatment, tumor progression in one or more lesions usually occur after 12–36 months. This finding is called secondary resistance and is most frequently caused by secondary acquired mutations in the KIT kinase domain. Rarely, other resistance mechanisms including KIT/PDGFRA genomic amplification and activation of alternative oncogenes have been reported [51,72]. Secondary KIT kinase mutations are non-randomly distributed single nucleotide substitutions affecting codons in the ATP binding pocket (exons 13 and 14) and the kinase activation loop (exon 17 and 18) (see Figure 4) [72]. Acquired secondary resistance mutations are described in up to two-thirds of GISTs progressing after treatment with tyrosine kinase inhibitors TKI [73,74,75,76]. A very common secondary resistance mutation is the V654A in exon 13 of the KIT gene [72,77]. Secondary mutations are found to be significantly more common in GISTs with primary KIT exon 11 mutations than in those with exon 9 mutations. In tumors with primary PDGFRA mutations, secondary mutations in exon 18 with a primary mutation in exon 12 have been rarely described [77]. While sunitinib has been shown to be effective against secondary mutations located in the ATP binding pocket (exon 13 and 14), this drug is not effective against mutations in the kinase activation loop (exon 17 and 18) based on in vitro and in vivo studies [14,77]. Recently, Zhang et al. have demonstrated that cabozantinib, in a mouse model with a V654A second site KIT mutation, might be a more effective drug in overcoming secondary resistance than sunitinib. In addition, they concluded that second-side mutations are not only responsible for drug resistance, but also for changing the oncogenic potential and activation of different signaling pathways, in this case, KIT-dependent STAT activation [78]. Unfortunately, knowledge about the substantial inter- and intralesional heterogeneity of TKI resistance mutations in metastases of patients treated with imatinib alone or imatinib and sunitinib challenges the potential treatment options and tissue selection for mutational analysis [72]. Therefore, the use of liquid biopsy has been shown to be feasible to search for resistance mutations [79].
Recently, a new treatment option with a potent KIT/PDGFRA inhibitor avapritinib, with substantial clinical activity in patients with the PDGFRA D842V mutation, became available [17]. However, secondary resistance against avapritinib can occur and is caused by secondary PDGFRA mutations in exons 13, 14 and 15 (PDGFRA kinase domain) that interfere with the avapritinib binding site [81].
2.1.5. Prognosis and Mutational Status in Treatment-Naïve GIST
The prognostic value of mutational status was nicely demonstrated in the study analyzing a series of 451 untreated primary localized GIST for KIT, PDGFRA and BRAF mutations finding that the mutational status is a significant prognostic indicator of overall survival (OS) [6]. Based on multivariable Cox regression models, the authors identified three distinct molecular risk groups. Group I, consisting of PDGFRA exon 12, BRAF and KIT exon 13-mutated cases, exhibited the best clinical outcome. Group II, the intermediate-risk group, included KIT exon 17, PDGFRA exon 18 D842V and PDGFRA exon 14-mutated GISTs. Group III, displayed the worst clinical outcome and was comprised of KIT exon 9 and exon 11 and PDGFRA non-D842V exon 18 mutant GISTs [6]. This study highlights the prognostic impact of the mutational status in the natural history of GIST. Therefore, the inclusion of molecular data together with risk stratification criteria can clearly help to enhance the decision-making process, especially in the adjuvant setting.
2.1.6. Genetic Subtypes of GIST—Impact on Treatment Response
In the last two decades, a growing body of evidence showed that the mutational status in GIST is a strong predictive indicator of response to treatment. Imatinib mesylate (STI571, Gleevec™, Novartis Pharmaceuticals, Basel, Switzerland) is an oral selective inhibitor of a number of TK including KIT, PDGFR, ABL and BCR-ABL. Since 2000, it has been used in GIST therapy in patients with metastatic/advanced disease and has become a paradigm in the treatment of solid tumors with targeted therapy, tremendously changing the survival of these patients [82]. Subsequently, other TKI, namely, sunitinib, regorafenib, and very recently ripretinib and avapritinib, have been approved and are mainly used in an advanced (recurrent and metastatic) disease after ineffective imatinib treatment or in the context of selective mutations like PDGFRA D842V [17,18,20,83].
Studies demonstrated the prognostic significance and prediction to treatment response in certain types of mutations detected in GIST (see Table 1). Tumors with common KIT exon 11 mutations, at codon 557/558, especially if located in the stomach, are associated with more aggressive behavior, higher risk of disease recurrence and increased risk of developing metastases [50,51,84]. Therefore, including this molecular information especially in the decision process if a neoadjuvant therapy showed be given seems to be of relevance. Tumors with a mutation in KIT exon 9 were shown to be imatinib sensitive; however, these GIST require a double dose (800 mg/daily) [14]. In PDGFRA-mutated tumors, substitutions involving codon D842 in exon 18 (including D842V, RD841-842KI, DI842-843IM) are primarily resistant to both, imatinib and sunitinib [12,14,77]. In contrast, further mutations in exon 18, including D842Y, D846Y, N848K, Y849K, HSN845-848P and mutations in exon 14 of the PDGFRA gene are sensitive to imatinib [12]. However, very recently avapritinib has been approved for the treatment of advanced PDGFRA D842V-mutant GIST [17].
In the pediatric GIST group, disease progression has been shown to occur later if they are on sunitinib therapy than on imatinib, leading to the conclusion that these patients benefit more from sunitinib therapy in the first line of treatment [15,85].
Taking all this into account, the European Society for Medical Oncology (ESMO) recommend routine analysis of GIST associated mutations for better planning of adjuvant therapy, with the possible exclusion of <2 cm non-rectal GISTs [20].
2.1.7. Morphological Changes after TKI Therapy
Treatment with TKI can influence GIST morphology [23,27]. A dramatic decrease in tumor cellularity as well as prominent stromal alterations including marked sclerosis and myxoid change can occur. However, in the vast majority of cases, the cytomorphology remains comparable with the primary tumor. Nevertheless, marked changes in the tumor morphology have been described. Very rare but well described is the so-called dedifferentiated GIST, frequently showing an abrupt transition from a “classic” GIST morphology in a dedifferentiated component.
Dedifferentiation in GIST is commonly associated with long term TKI treatment but can also occur de novo [23,24,25,27]. The dedifferentiated component shows an anaplastic/pleomorphic appearance, high nuclear atypia, high mitotic activity, and necrosis. Various histologic patterns have been reported in this context including rhabdomyosarcoma [25,86], angiosarcoma [23], or undifferentiated pleomorphic and spindle sarcoma. Usually, dedifferentiation is not associated with additional mutations in the original driver oncogene. Instead, these tumors show genetic instability, indicated by LOH or low-level KIT amplification [23]. Therefore, the dedifferentiation process might be caused by the activation of alternative pathways driven by KIT-independent oncogenic mechanisms. Nevertheless, the possibility of dedifferentiation in GISTs should always be considered when an undifferentiated sarcoma component is seen in the gastrointestinal tract. In this context, extensive sampling of tumor tissue, patients’ clinical history and molecular analysis are diagnostically helpful.
2.1.8. Familial GIST
Familial GIST syndrome is defined by germline mutation of KIT or PDGFRA, the occurrence of multiple GISTs, hyperpigmentation, mast cell tumors and dysphagia due to ICC hyperplasia [87,88]. To date, approximately 50 cases have been described. The most common KIT and PDGFRA mutations observed in individuals with familial GIST are summarized in Table 3 [8,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101].
The vast majority of the patients develop multiple GISTs by middle age. The morphology of tumors in this setting is similar to sporadic GISTs and they are caused by a monoclonal proliferation of tumor cells. Additionally, diffuse proliferation of the Cajal cell population is found causing ICC hyperplasia, representing, however, a non-neoplastic polyclonal nature [88,102].
2.2. Wild-Type GIST
Approximately 10–12% of all GISTs lack mutations in KIT and PDGFRA and are called wt-GISTs. Over the last few years, it became apparent that this group is heterogeneous with regards to clinical phenotype and molecular characteristics [103]. Based on recent advances in molecular pathology, wt-GISTs can be sub-classified in an SDH-competent and an SDH-deficient group, irrespective of whether they are sporadic or familial/genetic [9]. Immunohistochemical screening for SDH deficiency became a powerful and convenient screening tool to stratify GIST patients (especially pediatric and young adult patients with GIST located in the stomach) into these two groups (see Figure 5).
At this point, genetic testing using DNA- and RNA-NGS can be used to exclude the known genetic changes (mutations and fusions) in GIST. The workflow applicable in routine practice is shown in Figure 6. GISTs lacking these known molecular changes should be collected for research in specialized centers.
2.2.1. SDH-Competent wt-GISTs
NF1-Mutant GIST
NF1 is an inherited, autosomal dominant disease phenotypically characterized by multiple café-au-lait spots, Lisch nodules, freckling neurofibromas, and occasional development of malignant peripheral nerve sheath tumors. Approximately 7% of patients with NF1 develop GIST during their lifespan [7]. NF1-associated GISTs are commonly multicentric (with or without multinodular growth pattern), predominantly located in the small intestine and lack KIT and PDGFRA mutations [104,105]. These tumors express KIT, DOG1 and SDHB by IHC [106]. NF1-associated GISTs frequently demonstrate loss of heterozygosity at 14q and 22q similar to sporadic KIT- and PDGFRA-mutated GIST [107]. In addition, it has been suggested that somatic inactivating NF1 mutations outside the context of NF1 may be the oncogenic mechanism for a subset of sporadic adult wt-GIST [60,104,105]. Moreover, an NF1- mutant GISTs may harbor the additional cancer-related mutations, like inactivating Notch pathway mutations including NOTCH2, MAML2 and CDC73, most frequently found in tumors at the duodenal-jejunal flexure (ligament of Treitz) [105].
BRAF, KRAS and PIK3CA-Mutant GISTs
BRAF mutations (V600E) have been found in approximately 8–13% of wt-GISTs, and to date, BRAF and KIT/ PDGFRA mutations seem to be mutually exclusive [35,41,108,109]. According to available data, pediatric and young adult wt-GISTs are practically devoid of BRAF mutations, with only a single pediatric wt-GIST reported as BRAF-mutated so far [110]. BRAF-mutant GISTs equally affect men and women, are commonly associated with small bowel manifestation amd show spindle cell morphology and variable clinical behavior. Currently, no clinical or prognostic correlations have been linked to BRAF mutation status [111].
KRAS-mutated GISTs are exceedingly rare and the clinicopathologic features are not fully elucidated yet [112].
PIK3CA mutant GISTs are also exceedingly rare. In the largest study to date 10 (8 primary and 2 metastatic GISTs) out of 529 imatinib-naïve GISTs demonstrated PIK3CA mutations. PIK3CA mutations were associated with large tumor size and aggressive clinical behavior [113]. As the number of reported patients is small, further studies with good follow up is required.
GISTs with ETV6-NTRK3-Fusion
NTRK gene fusions appear to be primary oncogenic drivers in NTRK-rearranged tumors. Although GISTs with ETV6-NTRK3 fusions are exceedingly rare, approved highly potent NTRK-inhibitors are now available. The excellent clinical response and outcome in patients harboring NTRK-rearranged tumors treated with these agents have been demonstrated in several studies [114,115,116]. Even though pan-TRK IHC is wildly used as a reliable and affordable screening method for the detection of NTRK-fusions in most of the pathology departments [117,118], it has its limitations. The positive expression has been reported in a subset of neoplasms with neuronal and smooth muscle differentiation, as well as GIST, where diffuse, moderate to strong cytoplasmic pan-TRK expression has been described (Figure 7) with lack of an NTRK1-3-fusions by RNA sequencing [119]. Therefore, fusion analysis should be performed in SDHB-retained and mutation-negative cases, to enable optimal patients management especially in the metastatic setting.
2.2.2. SDH-Deficient wt-GISTs
A unique group within the wt-GISTs are the SDH-deficient GISTs. The key oncogenetic mechanism of these tumors is an energy metabolism defect in the SDH complex (mitochondrial complex or succinate reductase) which is composed of 4 subunits SDHA, SDHB, SDHC and SDHD, mapping to 5p15.33, 1p36.13, 1q23.3, and 11q23.1, respectively. The SDH enzyme is a key enzyme in the Krebs cycle and electron transport chain. It is a highly conserved heterotetrameric protein that consists of SDHA and SDHB being part of the catalytic unit and SDHC and SDHD are membrane-anchoring subunits. Genetic or epigenetic alterations in any of the subunits lead to accumulation of succinate which is a competitive inhibitor of α-ketoglutarate- dependent dioxygenases, including the TET family of 5-methylcytosine hydroxylases [120]. Members of the TET family are active DNA demethylases that convert 5-methylcytosine to 5-hydroxymethylcytosine, and inhibition of TET activities can lead to aberrant DNA methylation in GISTs. A genome-wide DNA methylation analysis of SDH-deficient GISTs revealed greater DNA hypermethylation than in GISTs with KIT mutation [121]. Activated cellular pathways are leading to increased angiogenesis and cellular proliferation are activated [122]. Accumulation of succinate causes stabilization of HIF1-α, which controls oncogene transcription [123].
Insulin-like growth factor 1 receptor (IGF1R) is overexpressed in KIT/PDGFR wt-GISTs and is particularly elevated in SDH-deficient GISTs [124,125,126]. The IGF family is composed of two ligands (IGF1 and IGF2), two receptors (IGFR1 and IGFR1) and 6 IGF binding proteins (IGFBPs). Activation of IGFR results in activation of downstream signals, including the MAPK and PI3K/AKT pathways [127]. Inhibition of IGF1R induces apoptosis and represses AKT and MAPK signaling in GIST cells, which implicates the IGF signal in the development of SDH-deficient GISTs [128]. Very recently, it has been shown that two RTK genes (KIT and FGF receptor 1 (FGFR1)), as well as FGF3 and FGF4 oncogenes are most highly expressed in SDH-deficient tumors [129]. These findings may explain the poor response of TKIs in SDH-deficient GISTs.
Alterations of a single SDH subunit can be reliably detectable by loss of SDHB expression using IHC. Therefore, SDHB IHC can be used as a convenient tissue-based screening method for genetic and epigenetic alterations in the SDH complex [103].
SDH-deficient GISTs usually occur in patients younger than 40 years of age, have a female predilection, occur in the stomach (most commonly antrum) and have a spectrum of behavior from indolent to progressive. The tumors show characteristic morphologic features including a multinodular growth pattern (Figure 8A), the occurrence of multiple tumors, lymphovascular involvement and lymph node metastasis. Morphologically, these tumors are epithelioid or mixed epithelioid/spindled (Figure 8B). Mitotic activity may reach more than 5 per 5mm 2; however, risk stratification, according to Miettinen, is not working in this context [130]. Tumors consistently express KIT and DOG1 by IHC (Figure 8C) and show loss of SDHB staining (Figure 8D). However, the mechanism of KIT activation in SDH-deficient GISTs remains unclear. They lack the canonical chromosomal alterations observed in KIT/PDGFRA/NF1-mutant GISTs (i.e., loss of 14q, 22q, 1p, and 15q) and, instead, may show 1q deletion, presumably involving the SDHC locus [131,132].
2.2.3. Syndromic SDH-Deficient wt-GIST
Carney Triad (CT)
CT is a very rare disease with the synchronous or metachronous occurrence of at least three different tumor entities; GIST, paraganglioma (PGL), and pulmonary chondroma. CT is never inherited and affects mostly females. Most cases of CT show down-regulation of SDH through site-specific hypermethylation of the SDHC gene [133,134]. In most cases, the SDHx epigenetic downregulation leads to downstream activation of the HIF signaling pathway. Overexpression of insulin-like growth factor receptor type 1 (IGFR1) at the protein level has been found in the majority of SDH deficient GIST; however, the exact molecular mechanism for this overexpression is currently not known.
Killian et al. studied SDH deactivation through genome-wide DNA methylation and expression study. This study included 59 SDH-deficient GIST and showed that 94% of tumors lacking SDH mutations showed SDHC promoter-specific CpG island hypermethylation and subsequent gene silencing. This fact led to the hypothesis that SDHC epimutation could be the main molecular mechanism that leads to succinate dehydrogenase enzyme dysfunction in SDH-deficient GIST that lack SDH mutations [121]. In 2016, Boikos et al. reported that 84/95 wt-GISTs lacked SDHB expression by IHC. Molecular analysis of the SDHB deficient GISTs revealed that 2/3 of the cases demonstrated mutations in SDH subunits whereas 1/3 showed SDHC promoter methylation. Mutations in SDH subunits were associated with CSS whereas SDHC promoter methylation was the main molecular characteristic of GIST in CT [9]. Only a few cases of CT are reported to have SDH subunit mutations, suggesting a partial overlap between the two conditions. Nevertheless, both lead to increased methylation of the entire genome in these tumors [9].
Carney-Stratakis Syndrome (CSS)
CSS is characterized by gastric multifocal GISTs and PGL. CSS shows an autosomal-dominant, with incomplete penetrance, inheritance pattern affecting both genders during childhood and adolescence. SDH deficiency is caused by inactivating germline mutations or large deletions in the SDHB, SDHC or SDHD (rarely SDHA) genes encoding the subunits B, C or D of the SDH enzyme [10,135,136].
In CSS, in contrast to CT, DNA methylation patterns were identified only at a few of the CpGs located close to the SDHB gene [133]. In these patients, the SDHC gene promoter was completely unmethylated in all screened CpG sites supporting the hypothesis that the CSS is a truly different entity from CT.
SDHA-Deficient wt-GIST
Approximately 30% of SDH-deficient GISTs demonstrate loss of expression for SDHB and SDHA by IHC. Loss of SDHA expression is a strong indicator of mutations in the SDHA gene. Most of the cases demonstrate germline mutations. The most common SDHA mutation detected in these patients is the c.91C4T; p.R31X. Simultaneous allelic loss at the SDHA locus at 5p15 has been detected with comparative genomic hybridization. Mutations in this tumor suppressor follow a classic 2-hit hypothesis. Loss of SDHA protein expression is associated with both truncating and missense germline mutations. SDHA mutation associated GISTs occur at an older age than other SDH-deficient GISTs, with the median age of presentation in the largest series being 34 years [42,135].
2.2.4. Treatment Options in GISTs without Currently Druggable Target
Surgical management is considered the main treatment option for non-metastatic wt-GIST and it should also be considered as a treatment option in the metastatic setting, in the so-called wt-GIST group, if a druggable target (see molecular screening approach) cannot be detected [137]. Systemic treatment in metastatic wt-GIST showed no objective tumor response to imatinib, but a superior response to sunitinib, especially in the pediatric GIST group [138].
The SDH-deficient GIST group is mainly composed of pediatric GIST patients including patients with CSS and CT, whereas only a small subset of sporadic adult (especially young adult) patients fall into this group. The underlining genetics for loss of the SDHB expression by IHC is heterogeneous including somatic mutations and germline mutations in SDHA/B/C/D as well as promotor hypermethylation or deletions [9,42,133].
3. Conclusions
In the last two decades, molecular pathology has massively improved our understanding of GIST development. Identification of targetable genetic alterations has subsequently changed treatment approaches and brought survival benefits for the vast majority of GIST patients. Mutational analyses have been shown to have a prognostic and therapeutic impact. Understanding of drug resistance mechanisms contributed to the development of novel therapeutic strategies/targets. Avapritinib is one example of a new generation TKI that greatly improved treatment, especially for patients with a PDGFRA D842V mutation. Nowadays, comprehensive NGS based molecular testing strategies can facilitate to detect clinically relevant targets, including NTRK fusions. Therefore, a comprehensive molecular workup in a specialized center is needed if the common KIT/PDGFRA mutations in a GIST cannot be detected.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Soreide, K.; Sandvik, O.M.; Soreide, J.A.; Giljaca, V.; Jureckova, A.; Bulusu, V.R. Global epidemiology of gastrointestinal stromal tumours (gist): A systematic review of population-based cohort studies. Cancer Epidemiol. 2016, 40, 39–46. [Google Scholar] [CrossRef] [Green Version]
- Liegl-Atzwanger, B.; Fletcher, J.A.; Fletcher, C.D. Gastrointestinal stromal tumors. Virchows Arch. Int. J. Pathol. 2010, 456, 111–127. [Google Scholar] [CrossRef]
- Kindblom, L.G.; Remotti, H.E.; Aldenborg, F.; Meis-Kindblom, J.M. Gastrointestinal pacemaker cell tumor (gipact): Gastrointestinal stromal tumors show phenotypic characteristics of the interstitial cells of cajal. Am. J. Pathol. 1998, 152, 1259–1269. [Google Scholar] [PubMed]
- Barajas-López, C.; Berezin, I.; Daniel, E.E.; Huizinga, J.D. Pacemaker activity recorded in interstitial cells of cajal of the gastrointestinal tract. Am. J. Physiol. 1989, 257, C830–C835. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, M.; Lasota, J. Gastrointestinal stromal tumors: Pathology and prognosis at different sites. Semin. Diagn. Pathol. 2006, 23, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Rossi, S.; Miceli, R.; Messerini, L.; Bearzi, I.; Mazzoleni, G.; Capella, C.; Arrigoni, G.; Sonzogni, A.; Sidoni, A.; Toffolatti, L.; et al. Natural history of imatinib-naive gists: A retrospective analysis of 929 cases with long-term follow-up and development of a survival nomogram based on mitotic index and size as continuous variables. Am. J. Surg. Pathol. 2011, 35, 1646–1656. [Google Scholar] [CrossRef]
- Miettinen, M.; Fetsch, J.F.; Sobin, L.H.; Lasota, J. Gastrointestinal stromal tumors in patients with neurofibromatosis 1: A clinicopathologic and molecular genetic study of 45 cases. Am. J. Surg. Pathol. 2006, 30, 90–96. [Google Scholar] [CrossRef]
- Nishida, T.; Hirota, S.; Taniguchi, M.; Hashimoto, K.; Isozaki, K.; Nakamura, H.; Kanakura, Y.; Tanaka, T.; Takabayashi, A.; Matsuda, H.; et al. Familial gastrointestinal stromal tumours with germline mutation of the kit gene. Nat. Genet. 1998, 19, 323–324. [Google Scholar] [CrossRef]
- Boikos, S.A.; Pappo, A.S.; Killian, J.K.; LaQuaglia, M.P.; Weldon, C.B.; George, S.; Trent, J.C.; von Mehren, M.; Wright, J.A.; Schiffman, J.D.; et al. Molecular subtypes of kit/pdgfra wild-type gastrointestinal stromal tumors: A report from the national institutes of health gastrointestinal stromal tumor clinic. JAMA Oncol. 2016, 2, 922–928. [Google Scholar] [CrossRef] [Green Version]
- Belinsky, M.G.; Rink, L.; von Mehren, M. Succinate dehydrogenase deficiency in pediatric and adult gastrointestinal stromal tumors. Front. Oncol. 2013, 3, 117. [Google Scholar] [CrossRef] [Green Version]
- Hirota, S.; Isozaki, K.; Moriyama, Y.; Hashimoto, K.; Nishida, T.; Ishiguro, S.; Kawano, K.; Hanada, M.; Kurata, A.; Takeda, M.; et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 1998, 279, 577–580. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, M.C.; Corless, C.L.; Duensing, A.; McGreevey, L.; Chen, C.J.; Joseph, N.; Singer, S.; Griffith, D.J.; Haley, A.; Town, A.; et al. Pdgfra activating mutations in gastrointestinal stromal tumors. Science 2003, 299, 708–710. [Google Scholar] [CrossRef] [PubMed]
- Druker, B.J.; Tamura, S.; Buchdunger, E.; Ohno, S.; Segal, G.M.; Fanning, S.; Zimmermann, J.; Lydon, N.B. Effects of a selective inhibitor of the abl tyrosine kinase on the growth of bcr-abl positive cells. Nat. Med. 1996, 2, 561–566. [Google Scholar] [CrossRef]
- Heinrich, M.C.; Owzar, K.; Corless, C.L.; Hollis, D.; Borden, E.C.; Fletcher, C.D.; Ryan, C.W.; von Mehren, M.; Blanke, C.D.; Rankin, C.; et al. Correlation of kinase genotype and clinical outcome in the north american intergroup phase iii trial of imatinib mesylate for treatment of advanced gastrointestinal stromal tumor: Calgb 150105 study by cancer and leukemia group b and southwest oncology group. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2008, 26, 5360–5367. [Google Scholar]
- Janeway, K.A.; Albritton, K.H.; Van Den Abbeele, A.D.; D’Amato, G.Z.; Pedrazzoli, P.; Siena, S.; Picus, J.; Butrynski, J.E.; Schlemmer, M.; Heinrich, M.C.; et al. Sunitinib treatment in pediatric patients with advanced gist following failure of imatinib. Pediatric Blood Cancer 2009, 52, 767–771. [Google Scholar] [CrossRef]
- Heinrich, M.C.; Corless, C.L.; Demetri, G.D.; Blanke, C.D.; von Mehren, M.; Joensuu, H.; McGreevey, L.S.; Chen, C.J.; Van den Abbeele, A.D.; Druker, B.J.; et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2003, 21, 4342–4349. [Google Scholar] [CrossRef]
- Heinrich, M.C.; Jones, R.L.; von Mehren, M.; Schöffski, P.; Serrano, C.; Kang, Y.K.; Cassier, P.A.; Mir, O.; Eskens, F.; Tap, W.D.; et al. Avapritinib in advanced pdgfra d842v-mutant gastrointestinal stromal tumour (navigator): A multicentre, open-label, phase 1 trial. Lancet Oncol. 2020, 21, 935–946. [Google Scholar] [CrossRef]
- Demetri, G.D.; van Oosterom, A.T.; Garrett, C.R.; Blackstein, M.E.; Shah, M.H.; Verweij, J.; McArthur, G.; Judson, I.R.; Heinrich, M.C.; Morgan, J.A.; et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: A randomised controlled trial. Lancet (Lond. Engl.) 2006, 368, 1329–1338. [Google Scholar] [CrossRef]
- Demetri, G.D.; Reichardt, P.; Kang, Y.K.; Blay, J.Y.; Rutkowski, P.; Gelderblom, H.; Hohenberger, P.; Leahy, M.; von Mehren, M.; Joensuu, H.; et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (grid): An international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet (Lond. Engl.) 2013, 381, 295–302. [Google Scholar] [CrossRef] [Green Version]
- Casali, P.G.; Abecassis, N.; Aro, H.T.; Bauer, S.; Biagini, R.; Bielack, S.; Bonvalot, S.; Boukovinas, I.; Bovee, J.; Brodowicz, T.; et al. Gastrointestinal stromal tumours: Esmo-euracan clinical practice guidelines for diagnosis, treatment and follow-up. Ann. Oncol. Off. J. Eur. Soc. Med Oncol. 2018, 29, iv68–iv78. [Google Scholar] [CrossRef]
- Miettinen, M.; Lasota, J. Gastrointestinal stromal tumors: Review on morphology, molecular pathology, prognosis, and differential diagnosis. Arch. Pathol. Lab. Med. 2006, 130, 1466–1478. [Google Scholar] [PubMed]
- Fletcher, C.D.; Berman, J.J.; Corless, C.; Gorstein, F.; Lasota, J.; Longley, B.J.; Miettinen, M.; O’Leary, T.J.; Remotti, H.; Rubin, B.P.; et al. Diagnosis of gastrointestinal stromal tumors: A consensus approach. Int. J. Surg. Pathol. 2002, 10, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Antonescu, C.R.; Romeo, S.; Zhang, L.; Nafa, K.; Hornick, J.L.; Nielsen, G.P.; Mino-Kenudson, M.; Huang, H.Y.; Mosquera, J.M.; Dei Tos, P.A.; et al. Dedifferentiation in gastrointestinal stromal tumor to an anaplastic kit-negative phenotype: A diagnostic pitfall: Morphologic and molecular characterization of 8 cases occurring either de novo or after imatinib therapy. Am. J. Surg. Pathol. 2013, 37, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Díaz Delgado, M.; Hernández Amate, A.; Pereira Gallardo, S.; Jaramillo, S.; Virizuela Echaburu, J.A.; González-Cámpora, R.J. Gastrointestinal stromal tumors: Morphological, immunohistochemical and molecular changes associated with kinase inhibitor therapy. Pathol. Oncol. Res. POR 2011, 17, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Liegl, B.; Hornick, J.L.; Antonescu, C.R.; Corless, C.L.; Fletcher, C.D. Rhabdomyosarcomatous differentiation in gastrointestinal stromal tumors after tyrosine kinase inhibitor therapy: A novel form of tumor progression. Am. J. Surg. Pathol. 2009, 33, 218–226. [Google Scholar] [CrossRef]
- Choi, J.J.; Sinada-Bottros, L.; Maker, A.V.; Weisenberg, E. Dedifferentiated gastrointestinal stromal tumor arising de novo from the small intestine. Pathol. Res. Pract. 2014, 210, 264–266. [Google Scholar] [CrossRef] [PubMed]
- Pauwels, P.; Debiec-Rychter, M.; Stul, M.; De Wever, I.; Van Oosterom, A.T.; Sciot, R. Changing phenotype of gastrointestinal stromal tumours under imatinib mesylate treatment: A potential diagnostic pitfall. Histopathology 2005, 47, 41–47. [Google Scholar] [CrossRef]
- Sarlomo-Rikala, M.; Kovatich, A.J.; Barusevicius, A.; Miettinen, M. Cd117: A sensitive marker for gastrointestinal stromal tumors that is more specific than cd34. Mod. Pathol. Off. J. United States Can. Acad. Pathol. Inc 1998, 11, 728–734. [Google Scholar]
- Liegl, B.; Hornick, J.L.; Corless, C.L.; Fletcher, C.D. Monoclonal antibody dog1.1 shows higher sensitivity than kit in the diagnosis of gastrointestinal stromal tumors, including unusual subtypes. Am. J. Surg. Pathol. 2009, 33, 437–446. [Google Scholar] [CrossRef]
- Miettinen, M.; Wang, Z.F.; Lasota, J. Dog1 antibody in the differential diagnosis of gastrointestinal stromal tumors: A study of 1840 cases. Am. J. Surg. Pathol. 2009, 33, 1401–1408. [Google Scholar] [CrossRef]
- Espinosa, I.; Lee, C.H.; Kim, M.K.; Rouse, B.T.; Subramanian, S.; Montgomery, K.; Varma, S.; Corless, C.L.; Heinrich, M.C.; Smith, K.S.; et al. A novel monoclonal antibody against dog1 is a sensitive and specific marker for gastrointestinal stromal tumors. Am. J. Surg. Pathol. 2008, 32, 210–218. [Google Scholar] [CrossRef]
- Lopes, L.F.; West, R.B.; Bacchi, L.M.; van de Rijn, M.; Bacchi, C.E. Dog1 for the diagnosis of gastrointestinal stromal tumor (gist): Comparison between 2 different antibodies. Appl. Immunohistochem. Mol. Morphol. AIMM 2010, 18, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Rubin, B.P.; Singer, S.; Tsao, C.; Duensing, A.; Lux, M.L.; Ruiz, R.; Hibbard, M.K.; Chen, C.J.; Xiao, S.; Tuveson, D.A.; et al. Kit activation is a ubiquitous feature of gastrointestinal stromal tumors. Cancer Res. 2001, 61, 8118–8121. [Google Scholar] [PubMed]
- Wozniak, A.; Rutkowski, P.; Piskorz, A.; Ciwoniuk, M.; Osuch, C.; Bylina, E.; Sygut, J.; Chosia, M.; Rys, J.; Urbanczyk, K.; et al. Prognostic value of kit/pdgfra mutations in gastrointestinal stromal tumours (gist): Polish clinical gist registry experience. Ann. Oncol. Off. J. Eur. Soc. Med Oncol. 2012, 23, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Agaram, N.P.; Wong, G.C.; Guo, T.; Maki, R.G.; Singer, S.; Dematteo, R.P.; Besmer, P.; Antonescu, C.R. Novel v600e braf mutations in imatinib-naive and imatinib-resistant gastrointestinal stromal tumors. Genes Chromosomes Cancer 2008, 47, 853–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janeway, K.A.; Liegl, B.; Harlow, A.; Le, C.; Perez-Atayde, A.; Kozakewich, H.; Corless, C.L.; Heinrich, M.C.; Fletcher, J.A. Pediatric kit wild-type and platelet-derived growth factor receptor alpha-wild-type gastrointestinal stromal tumors share kit activation but not mechanisms of genetic progression with adult gastrointestinal stromal tumors. Cancer Res. 2007, 67, 9084–9088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, E.; Chmielecki, J.; Tang, C.M.; Wang, K.; Heinrich, M.C.; Kang, G.; Corless, C.L.; Hong, D.; Fero, K.E.; Murphy, J.D.; et al. Fgfr1 and ntrk3 actionable alterations in “wild-type” gastrointestinal stromal tumors. J. Transl. Med. 2016, 14, 339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pantaleo, M.A.; Astolfi, A.; Di Battista, M.; Heinrich, M.C.; Paterini, P.; Scotlandi, K.; Santini, D.; Catena, F.; Manara, M.C.; Nannini, M.; et al. Insulin-like growth factor 1 receptor expression in wild-type gists: A potential novel therapeutic target. Int. J. Cancer 2009, 125, 2991–2994. [Google Scholar] [CrossRef]
- Charo, L.M.; Burgoyne, A.M.; Fanta, P.T.; Patel, H.; Chmielecki, J.; Sicklick, J.K.; McHale, M.T. A novel prkar1b-braf fusion in gastrointestinal stromal tumor guides adjuvant treatment decision-making during pregnancy. J. Natl. Compr. Cancer Netw. JNCCN 2018, 16, 238–242. [Google Scholar] [CrossRef] [Green Version]
- Brenca, M.; Rossi, S.; Polano, M.; Gasparotto, D.; Zanatta, L.; Racanelli, D.; Valori, L.; Lamon, S.; Dei Tos, A.P.; Maestro, R. Transcriptome sequencing identifies etv6-ntrk3 as a gene fusion involved in gist. J. Pathol. 2016, 238, 543–549. [Google Scholar] [CrossRef]
- Hostein, I.; Faur, N.; Primois, C.; Boury, F.; Denard, J.; Emile, J.F.; Bringuier, P.P.; Scoazec, J.Y.; Coindre, J.M. Braf mutation status in gastrointestinal stromal tumors. Am. J. Clin. Pathol. 2010, 133, 141–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, A.J.; Remillard, S.P.; Zhang, Y.X.; Doyle, L.A.; George, S.; Hornick, J.L. Loss of expression of sdha predicts sdha mutations in gastrointestinal stromal tumors. Mod. Pathol. Off. J. United States Can. Acad. Pathol. Inc 2013, 26, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Hanks, S.K.; Quinn, A.M.; Hunter, T. The protein kinase family: Conserved features and deduced phylogeny of the catalytic domains. Science 1988, 241, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Abbaspour Babaei, M.; Kamalidehghan, B.; Saleem, M.; Huri, H.Z.; Ahmadipour, F. Receptor tyrosine kinase (c-kit) inhibitors: A potential therapeutic target in cancer cells. Drug Des. Dev. Ther. 2016, 10, 2443–2459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeda, H.; Yamagata, A.; Nishikawa, S.; Yoshinaga, K.; Kobayashi, S.; Nishi, K.; Nishikawa, S. Requirement of c-kit for development of intestinal pacemaker system. Development (Camb. Engl.) 1992, 116, 369–375. [Google Scholar]
- Orr-Urtreger, A.; Avivi, A.; Zimmer, Y.; Givol, D.; Yarden, Y.; Lonai, P. Developmental expression of c-kit, a proto-oncogene encoded by the w locus. Development (Camb. Engl.) 1990, 109, 911–923. [Google Scholar]
- Heinrich, M.C.; Rubin, B.P.; Longley, B.J.; Fletcher, J.A. Biology and genetic aspects of gastrointestinal stromal tumors: Kit activation and cytogenetic alterations. Hum. Pathol. 2002, 33, 484–495. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.J.; Li, H.T.; Yu, J.P.; Li, Y.M.; Han, X.P.; Chen, P.; Yu, W.W.; Chen, W.K.; Jiao, Z.Y.; Liu, H.B. Identification of key genes and associated pathways in kit/pdgfra wild-type gastrointestinal stromal tumors through bioinformatics analysis. Mol. Med. Rep. 2018, 18, 4499–4515. [Google Scholar] [CrossRef] [Green Version]
- Antonescu, C.R.; Sommer, G.; Sarran, L.; Tschernyavsky, S.J.; Riedel, E.; Woodruff, J.M.; Robson, M.; Maki, R.; Brennan, M.F.; Ladanyi, M.; et al. Association of kit exon 9 mutations with nongastric primary site and aggressive behavior: Kit mutation analysis and clinical correlates of 120 gastrointestinal stromal tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2003, 9, 3329–3337. [Google Scholar]
- Wozniak, A.; Rutkowski, P.; Schöffski, P.; Ray-Coquard, I.; Hostein, I.; Schildhaus, H.U.; Le Cesne, A.; Bylina, E.; Limon, J.; Blay, J.Y.; et al. Tumor genotype is an independent prognostic factor in primary gastrointestinal stromal tumors of gastric origin: A european multicenter analysis based on conticagist. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 6105–6116. [Google Scholar] [CrossRef] [Green Version]
- Debiec-Rychter, M.; Sciot, R.; Le Cesne, A.; Schlemmer, M.; Hohenberger, P.; van Oosterom, A.T.; Blay, J.Y.; Leyvraz, S.; Stul, M.; Casali, P.G.; et al. Kit mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur. J. Cancer (Oxf. Engl. 1990) 2006, 42, 1093–1103. [Google Scholar] [CrossRef] [PubMed]
- Steigen, S.E.; Eide, T.J.; Wasag, B.; Lasota, J.; Miettinen, M. Mutations in gastrointestinal stromal tumors--a population-based study from northern norway. APMIS Acta Pathol. Microbiol. Immunol. Scand. 2007, 115, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.Y.; Ma, X.L.; Wang, M.; Zhuang, C.; Ni, B. Exon 11 homozygous mutations and intron 10/exon 11 junction deletions in the kit gene are associated with poor prognosis of patients with gastrointestinal stromal tumors. Cancer Med. 2020, 9, 6485–6496. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, I.M.; Mariño-Enríquez, A.; Fletcher, J.A. What is new in gastrointestinal stromal tumor? Adv. Anat. Pathol. 2017, 24, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Lasota, J.; Corless, C.L.; Heinrich, M.C.; Debiec-Rychter, M.; Sciot, R.; Wardelmann, E.; Merkelbach-Bruse, S.; Schildhaus, H.U.; Steigen, S.E.; Stachura, J.; et al. Clinicopathologic profile of gastrointestinal stromal tumors (gists) with primary kit exon 13 or exon 17 mutations: A multicenter study on 54 cases. Mod. Pathol. Off. J. United States Can. Acad. Pathol. Inc. 2008, 21, 476–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szucs, Z.; Thway, K.; Fisher, C.; Bulusu, R.; Constantinidou, A.; Benson, C.; van der Graaf, W.T.; Jones, R.L. Molecular subtypes of gastrointestinal stromal tumors and their prognostic and therapeutic implications. Future Oncol. (Lond. Engl.) 2017, 13, 93–107. [Google Scholar] [CrossRef] [Green Version]
- Fredriksson, L.; Li, H.; Eriksson, U. The pdgf family: Four gene products form five dimeric isoforms. Cytokine Growth Factor Rev. 2004, 15, 197–204. [Google Scholar] [CrossRef]
- Corless, C.L.; Schroeder, A.; Griffith, D.; Town, A.; McGreevey, L.; Harrell, P.; Shiraga, S.; Bainbridge, T.; Morich, J.; Heinrich, M.C. Pdgfra mutations in gastrointestinal stromal tumors: Frequency, spectrum and in vitro sensitivity to imatinib. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2005, 23, 5357–5364. [Google Scholar] [CrossRef]
- Breiner, J.A.; Meis-Kindblom, J.; Kindblom, L.G.; McComb, E.; Liu, J.; Nelson, M.; Bridge, J.A. Loss of 14q and 22q in gastrointestinal stromal tumors (pacemaker cell tumors). Cancer Genet. Cytogenet. 2000, 120, 111–116. [Google Scholar] [CrossRef]
- Belinsky, M.G.; Rink, L.; Cai, K.Q.; Capuzzi, S.J.; Hoang, Y.; Chien, J.; Godwin, A.K.; von Mehren, M. Somatic loss of function mutations in neurofibromin 1 and myc associated factor x genes identified by exome-wide sequencing in a wild-type gist case. BMC Cancer 2015, 15, 887. [Google Scholar] [CrossRef] [Green Version]
- Schaefer, I.M.; Wang, Y.; Liang, C.W.; Bahri, N.; Quattrone, A.; Doyle, L.; Mariño-Enríquez, A.; Lauria, A.; Zhu, M.; Debiec-Rychter, M.; et al. Max inactivation is an early event in gist development that regulates p16 and cell proliferation. Nat. Commun. 2017, 8, 14674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wozniak, A.; Sciot, R.; Guillou, L.; Pauwels, P.; Wasag, B.; Stul, M.; Vermeesch, J.R.; Vandenberghe, P.; Limon, J.; Debiec-Rychter, M. Array cgh analysis in primary gastrointestinal stromal tumors: Cytogenetic profile correlates with anatomic site and tumor aggressiveness, irrespective of mutational status. Genes Chromosomes Cancer 2007, 46, 261–276. [Google Scholar] [CrossRef] [PubMed]
- El-Rifai, W.; Sarlomo-Rikala, M.; Andersson, L.C.; Knuutila, S.; Miettinen, M. DNA sequence copy number changes in gastrointestinal stromal tumors: Tumor progression and prognostic significance. Cancer Res. 2000, 60, 3899–3903. [Google Scholar] [PubMed]
- Gunawan, B.; Bergmann, F.; Höer, J.; Langer, C.; Schumpelick, V.; Becker, H.; Füzesi, L. Biological and clinical significance of cytogenetic abnormalities in low-risk and high-risk gastrointestinal stromal tumors. Hum. Pathol. 2002, 33, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Gunawan, B.; von Heydebreck, A.; Sander, B.; Schulten, H.J.; Haller, F.; Langer, C.; Armbrust, T.; Bollmann, M.; Gasparov, S.; Kovac, D.; et al. An oncogenetic tree model in gastrointestinal stromal tumours (gists) identifies different pathways of cytogenetic evolution with prognostic implications. J. Pathol. 2007, 211, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, I.M.; Delfs, C.; Cameron, S.; Gunawan, B.; Agaimy, A.; Ghadimi, B.M.; Haller, F. Chromosomal aberrations in primary pdgfra-mutated gastrointestinal stromal tumors. Hum. Pathol. 2014, 45, 85–97. [Google Scholar] [CrossRef]
- Schneider-Stock, R.; Boltze, C.; Lasota, J.; Miettinen, M.; Peters, B.; Pross, M.; Roessner, A.; Günther, T. High prognostic value of p16ink4 alterations in gastrointestinal stromal tumors. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2003, 21, 1688–1697. [Google Scholar] [CrossRef]
- Merten, L.; Agaimy, A.; Moskalev, E.A.; Giedl, J.; Kayser, C.; Geddert, H.; Schaefer, I.M.; Cameron, S.; Werner, M.; Ströbel, P.; et al. Inactivating mutations of rb1 and tp53 correlate with sarcomatous histomorphology and metastasis/recurrence in gastrointestinal stromal tumors. Am. J. Clin. Pathol. 2016, 146, 718–726. [Google Scholar] [CrossRef] [Green Version]
- Romeo, S.; Debiec-Rychter, M.; Van Glabbeke, M.; Van Paassen, H.; Comite, P.; Van Eijk, R.; Oosting, J.; Verweij, J.; Terrier, P.; Schneider, U.; et al. Cell cycle/apoptosis molecule expression correlates with imatinib response in patients with advanced gastrointestinal stromal tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 4191–4198. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Marino-Enriquez, A.; Bennett, R.R.; Zhu, M.; Shen, Y.; Eilers, G.; Lee, J.C.; Henze, J.; Fletcher, B.S.; Gu, Z.; et al. Dystrophin is a tumor suppressor in human cancers with myogenic programs. Nat. Genet. 2014, 46, 601–606. [Google Scholar] [CrossRef] [Green Version]
- Roskoski, R., Jr. Structure and regulation of kit protein-tyrosine kinase--the stem cell factor receptor. Biochem. Biophys. Res. Commun. 2005, 338, 1307–1315. [Google Scholar] [CrossRef] [PubMed]
- Liegl, B.; Kepten, I.; Le, C.; Zhu, M.; Demetri, G.D.; Heinrich, M.C.; Fletcher, C.D.; Corless, C.L.; Fletcher, J.A. Heterogeneity of kinase inhibitor resistance mechanisms in gist. J. Pathol. 2008, 216, 64–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinrich, M.C.; Corless, C.L.; Blanke, C.D.; Demetri, G.D.; Joensuu, H.; Roberts, P.J.; Eisenberg, B.L.; von Mehren, M.; Fletcher, C.D.; Sandau, K.; et al. Molecular correlates of imatinib resistance in gastrointestinal stromal tumors. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2006, 24, 4764–4774. [Google Scholar] [CrossRef]
- Agaram, N.P.; Besmer, P.; Wong, G.C.; Guo, T.; Socci, N.D.; Maki, R.G.; DeSantis, D.; Brennan, M.F.; Singer, S.; DeMatteo, R.P.; et al. Pathologic and molecular heterogeneity in imatinib-stable or imatinib-responsive gastrointestinal stromal tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2007, 13, 170–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prenen, H.; Cools, J.; Mentens, N.; Folens, C.; Sciot, R.; Schöffski, P.; Van Oosterom, A.; Marynen, P.; Debiec-Rychter, M. Efficacy of the kinase inhibitor su11248 against gastrointestinal stromal tumor mutants refractory to imatinib mesylate. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2006, 12, 2622–2627. [Google Scholar] [CrossRef] [Green Version]
- Wardelmann, E.; Merkelbach-Bruse, S.; Pauls, K.; Thomas, N.; Schildhaus, H.U.; Heinicke, T.; Speidel, N.; Pietsch, T.; Buettner, R.; Pink, D.; et al. Polyclonal evolution of multiple secondary kit mutations in gastrointestinal stromal tumors under treatment with imatinib mesylate. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2006, 12, 1743–1749. [Google Scholar] [CrossRef] [Green Version]
- Heinrich, M.C.; Maki, R.G.; Corless, C.L.; Antonescu, C.R.; Harlow, A.; Griffith, D.; Town, A.; McKinley, A.; Ou, W.B.; Fletcher, J.A.; et al. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2008, 26, 5352–5359. [Google Scholar] [CrossRef]
- Zhang, J.Q.; Bosbach, B.; Loo, J.K.; Vitiello, G.A. The v654a second-site kit mutation increases tumor oncogenesis and stat activation in a mouse model of gastrointestinal stromal tumor. Oncogene 2020, 39, 7153–7165. [Google Scholar] [CrossRef]
- Dalle Fratte, C.; Guardascione, M.; De Mattia, E.; Borsatti, E.; Boschetto, R.; Farruggio, A.; Canzonieri, V.; Romanato, L.; Borsatti, R.; Gagno, S.; et al. Clonal selection of a novel deleterious tp53 somatic mutation discovered in ctdna of a kit/pdgfra wild-type gastrointestinal stromal tumor resistant to imatinib. Front. Pharmacol. 2020, 11, 36. [Google Scholar] [CrossRef] [Green Version]
- Serrano, C.; Fletcher, J.A. Overcoming heterogenity in imatinib-resistant gastrointestinal stromal tumor. Oncotarget 2019, 10, 6286–6287. [Google Scholar] [CrossRef]
- Grunewald, S.; Klug, L.R.; Muhlenberg, T. Resistance to avapritinib in pdgfra-driven gist is caused by secondary mutations in the pdgfra kinase domain. Cancer Discov. 2021, 11, 108–125. [Google Scholar] [CrossRef] [PubMed]
- Joensuu, H.; Roberts, P.J.; Sarlomo-Rikala, M.; Andersson, L.C.; Tervahartiala, P.; Tuveson, D.; Silberman, S.; Capdeville, R.; Dimitrijevic, S.; Druker, B.; et al. Effect of the tyrosine kinase inhibitor sti571 in a patient with a metastatic gastrointestinal stromal tumor. N. Engl. J. Med. 2001, 344, 1052–1056. [Google Scholar] [CrossRef] [PubMed]
- Blay, J.Y.; Serrano, C.; Heinrich, M.C.; Zalcberg, J.; Bauer, S.; Gelderblom, H.; Schöffski, P.; Jones, R.L.; Attia, S.; D’Amato, G.; et al. Ripretinib in patients with advanced gastrointestinal stromal tumours (invictus): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2020, 21, 923–934. [Google Scholar] [CrossRef]
- Wardelmann, E.; Losen, I.; Hans, V.; Neidt, I.; Speidel, N.; Bierhoff, E.; Heinicke, T.; Pietsch, T.; Büttner, R.; Merkelbach-Bruse, S. Deletion of trp-557 and lys-558 in the juxtamembrane domain of the c-kit protooncogene is associated with metastatic behavior of gastrointestinal stromal tumors. Int. J. Cancer 2003, 106, 887–895. [Google Scholar] [CrossRef]
- Rutkowski, P.; Magnan, H.; Chou, A.J.; Benson, C. Treatment of gastrointestinal stromal tumours in paediatric and young adult patients with sunitinib: A multicentre case series. BMC Cancer 2017, 17, 717. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Khalili, M. Case report of rhabdomyosarcomatous transformation of a primary gastrointestinal stromal tumor (gist). BMC Cancer 2019, 19, 913. [Google Scholar] [CrossRef] [Green Version]
- Chompret, A.; Kannengiesser, C.; Barrois, M.; Terrier, P.; Dahan, P.; Tursz, T.; Lenoir, G.M.; Bressac-De Paillerets, B. Pdgfra germline mutation in a family with multiple cases of gastrointestinal stromal tumor. Gastroenterology 2004, 126, 318–321. [Google Scholar] [CrossRef]
- Hirota, S.; Nishida, T.; Isozaki, K.; Taniguchi, M.; Nishikawa, K.; Ohashi, A.; Takabayashi, A.; Obayashi, T.; Okuno, T.; Kinoshita, K.; et al. Familial gastrointestinal stromal tumors associated with dysphagia and novel type germline mutation of kit gene. Gastroenterology 2002, 122, 1493–1499. [Google Scholar] [CrossRef]
- Beghini, A.; Tibiletti, M.G.; Roversi, G.; Chiaravalli, A.M.; Serio, G.; Capella, C.; Larizza, L. Germline mutation in the juxtamembrane domain of the kit gene in a family with gastrointestinal stromal tumors and urticaria pigmentosa. Cancer 2001, 92, 657–662. [Google Scholar] [CrossRef]
- Lasota, J.; Miettinen, M. A new familial gist identified. Am. J. Surg. Pathol. 2006, 30, 1342. [Google Scholar] [CrossRef]
- Robson, M.E.; Glogowski, E.; Sommer, G.; Antonescu, C.R.; Nafa, K.; Maki, R.G.; Ellis, N.; Besmer, P.; Brennan, M.; Offit, K. Pleomorphic characteristics of a germ-line kit mutation in a large kindred with gastrointestinal stromal tumors, hyperpigmentation, and dysphagia. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2004, 10, 1250–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartmann, K.; Wardelmann, E.; Ma, Y.; Merkelbach-Bruse, S.; Preussner, L.M.; Woolery, C.; Baldus, S.E.; Heinicke, T.; Thiele, J.; Buettner, R.; et al. Novel germline mutation of kit associated with familial gastrointestinal stromal tumors and mastocytosis. Gastroenterology 2005, 129, 1042–1046. [Google Scholar] [CrossRef] [PubMed]
- Arima, J.; Hiramatsu, M.; Taniguchi, K.; Kobayashi, T.; Tsunematsu, I.; Kagota, S.; Sakane, J.; Suzuki, Y.; Hirota, S. Multiple gastrointestinal stromal tumors caused by a novel germline kit gene mutation (asp820gly): A case report and literature review. Gastric Cancer Off. J. Int. Gastric Cancer Assoc. Jpn. Gastric Cancer Assoc. 2020, 23, 760–764. [Google Scholar] [CrossRef] [PubMed]
- Fornasarig, M.; Gasparotto, D.; Foltran, L.; Campigotto, M. A novel kindred with familial gastrointestinal stromal tumors caused by a rare kit germline mutation (n655k): Clinico-pathological presentation and tki sensitivity. J. Pers. Med. 2020, 10, 234. [Google Scholar] [CrossRef]
- Kang, D.Y.; Park, C.K.; Choi, J.S.; Jin, S.Y.; Kim, H.J.; Joo, M.; Kang, M.S.; Moon, W.S.; Yun, K.J.; Yu, E.S.; et al. Multiple gastrointestinal stromal tumors: Clinicopathologic and genetic analysis of 12 patients. Am. J. Surg. Pathol. 2007, 31, 224–232. [Google Scholar] [CrossRef]
- Neuhann, T.M.; Mansmann, V.; Merkelbach-Bruse, S.; Klink, B.; Hellinger, A.; Höffkes, H.G.; Wardelmann, E.; Schildhaus, H.U.; Tinschert, S. A novel germline kit mutation (p.L576p) in a family presenting with juvenile onset of multiple gastrointestinal stromal tumors, skin hyperpigmentations, and esophageal stenosis. Am. J. Surg. Pathol. 2013, 37, 898–905. [Google Scholar] [CrossRef]
- Bachet, J.B.; Landi, B.; Laurent-Puig, P.; Italiano, A.; Le Cesne, A.; Lévy, P.; Safar, V.; Duffaud, F.; Blay, J.Y.; Emile, J.F. Diagnosis, prognosis and treatment of patients with gastrointestinal stromal tumour (gist) and germline mutation of kit exon 13. Eur. J. Cancer (Oxf. Engl. 1990) 2013, 49, 2531–2541. [Google Scholar] [CrossRef]
- Graham, J.; Debiec-Rychter, M.; Corless, C.L.; Reid, R.; Davidson, R.; White, J.D. Imatinib in the management of multiple gastrointestinal stromal tumors associated with a germline kit k642e mutation. Arch. Pathol. Lab. Med. 2007, 131, 1393–1396. [Google Scholar]
- Thalheimer, A.; Schlemmer, M.; Bueter, M.; Merkelbach-Bruse, S.; Schildhaus, H.U.; Buettner, R.; Hartung, E.; Thiede, A.; Meyer, D.; Fein, M.; et al. Familial gastrointestinal stromal tumors caused by the novel kit exon 17 germline mutation n822y. Am. J. Surg. Pathol. 2008, 32, 1560–1565. [Google Scholar] [CrossRef]
- Pasini, B.; Matyakhina, L.; Bei, T.; Muchow, M.; Boikos, S.; Ferrando, B.; Carney, J.A.; Stratakis, C.A. Multiple gastrointestinal stromal and other tumors caused by platelet-derived growth factor receptor alpha gene mutations: A case associated with a germline v561d defect. J. Clin. Endocrinol. Metab. 2007, 92, 3728–3732. [Google Scholar] [CrossRef] [Green Version]
- Manley, P.N.; Abu-Abed, S.; Kirsch, R.; Hawrysh, A.; Perrier, N.; Feilotter, H.; Pollett, A.; Riddell, R.H.; Hookey, L.; Walia, J.S. Familial pdgfra-mutation syndrome: Somatic and gastrointestinal phenotype. Hum. Pathol. 2018, 76, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Hirota, S.; Isozaki, K.; Sun, H.; Ohashi, A.; Kinoshita, K.; O’Brien, P.; Kapusta, L.; Dardick, I.; Obayashi, T.; et al. Polyclonal nature of diffuse proliferation of interstitial cells of cajal in patients with familial and multiple gastrointestinal stromal tumours. Gut 2002, 51, 793–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miettinen, M.; Wang, Z.F.; Sarlomo-Rikala, M.; Osuch, C.; Rutkowski, P.; Lasota, J. Succinate dehydrogenase-deficient gists: A clinicopathologic, immunohistochemical, and molecular genetic study of 66 gastric gists with predilection to young age. Am. J. Surg. Pathol. 2011, 35, 1712–1721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasparotto, D.; Rossi, S.; Polano, M.; Tamborini, E.; Lorenzetto, E.; Sbaraglia, M.; Mondello, A.; Massani, M.; Lamon, S.; Bracci, R.; et al. Quadruple-negative gist is a sentinel for unrecognized neurofibromatosis type 1 syndrome. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 273–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgoyne, A.M.; De Siena, M.; Alkhuziem, M.; Tang, C.M.; Medina, B.; Fanta, P.T.; Belinsky, M.G.; von Mehren, M.; Thorson, J.A.; Madlensky, L.; et al. Duodenal-jejunal flexure gi stromal tumor frequently heralds somatic nf1 and notch pathway mutations. JCO Precis. Oncol. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Gaal, J.; Stratakis, C.A.; Carney, J.A.; Ball, E.R.; Korpershoek, E.; Lodish, M.B.; Levy, I.; Xekouki, P.; van Nederveen, F.H.; den Bakker, M.A.; et al. Sdhb immunohistochemistry: A useful tool in the diagnosis of carney-stratakis and carney triad gastrointestinal stromal tumors. Mod. Pathol. Off. J. United States Can. Acad. Pathol. Inc 2011, 24, 147–151. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Tobo, T.; Nakamori, M.; Imamura, M.; Kojima, A.; Oda, Y.; Nakamura, N.; Takahira, T.; Yao, T.; Tsuneyoshi, M. Neurofibromatosis type 1-related gastrointestinal stromal tumors: A special reference to loss of heterozygosity at 14q and 22q. J. Cancer Res. Clin. Oncol. 2009, 135, 791–798. [Google Scholar] [CrossRef]
- Agaimy, A.; Terracciano, L.M.; Dirnhofer, S.; Tornillo, L.; Foerster, A.; Hartmann, A.; Bihl, M.P. V600e braf mutations are alternative early molecular events in a subset of kit/pdgfra wild-type gastrointestinal stromal tumours. J. Clin. Pathol. 2009, 62, 613–616. [Google Scholar] [CrossRef]
- Daniels, M.; Lurkin, I.; Pauli, R.; Erbstösser, E.; Hildebrandt, U.; Hellwig, K.; Zschille, U.; Lüders, P.; Krüger, G.; Knolle, J.; et al. Spectrum of kit/pdgfra/braf mutations and phosphatidylinositol-3-kinase pathway gene alterations in gastrointestinal stromal tumors (gist). Cancer Lett. 2011, 312, 43–54. [Google Scholar] [CrossRef]
- Nannini, M.; Biasco, G.; Astolfi, A.; Pantaleo, M.A. An overview on molecular biology of kit/pdgfra wild type (wt) gastrointestinal stromal tumours (gist). J. Med Genet. 2013, 50, 653–661. [Google Scholar] [CrossRef]
- Falchook, G.S.; Trent, J.C.; Heinrich, M.C.; Beadling, C.; Patterson, J.; Bastida, C.C.; Blackman, S.C.; Kurzrock, R. Braf mutant gastrointestinal stromal tumor: First report of regression with braf inhibitor dabrafenib (gsk2118436) and whole exomic sequencing for analysis of acquired resistance. Oncotarget 2013, 4, 310–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miranda, C.; Nucifora, M.; Molinari, F.; Conca, E.; Anania, M.C.; Bordoni, A.; Saletti, P.; Mazzucchelli, L.; Pilotti, S.; Pierotti, M.A.; et al. Kras and braf mutations predict primary resistance to imatinib in gastrointestinal stromal tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 1769–1776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lasota, J.; Felisiak-Golabek, A.; Wasag, B.; Kowalik, A.; Zięba, S.; Chłopek, M.; Wang, Z.F.; Coates, T.; Kopczynski, J.; Gozdz, S.; et al. Frequency and clinicopathologic profile of pik3ca mutant gists: Molecular genetic study of 529 cases. Mod. Pathol. Off. J. United States Can. Acad. Pathol. Inc 2016, 29, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.; Laetsch, T.W.; Kummar, S.; DuBois, S.G.; Lassen, U.N.; Demetri, G.D.; Nathenson, M.; Doebele, R.C.; Farago, A.F.; Pappo, A.S.; et al. Efficacy of larotrectinib in trk fusion-positive cancers in adults and children. N. Engl. J. Med. 2018, 378, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Doebele, R.C.; Drilon, A.; Paz-Ares, L.; Siena, S.; Shaw, A.T.; Farago, A.F.; Blakely, C.M.; Seto, T.; Cho, B.C.; Tosi, D.; et al. Entrectinib in patients with advanced or metastatic ntrk fusion-positive solid tumours: Integrated analysis of three phase 1-2 trials. Lancet Oncol. 2020, 21, 271–282. [Google Scholar] [CrossRef]
- Hong, D.S.; DuBois, S.G.; Kummar, S.; Farago, A.F.; Albert, C.M.; Rohrberg, K.S.; van Tilburg, C.M.; Nagasubramanian, R.; Berlin, J.D.; Federman, N.; et al. Larotrectinib in patients with trk fusion-positive solid tumours: A pooled analysis of three phase 1/2 clinical trials. N. Engl. J. Med. 2020, 21, 531–540. [Google Scholar] [CrossRef]
- Marchiò, C.; Scaltriti, M.; Ladanyi, M.; Iafrate, A.J.; Bibeau, F.; Dietel, M.; Hechtman, J.F.; Troiani, T.; López-Rios, F.; Douillard, J.Y.; et al. Esmo recommendations on the standard methods to detect ntrk fusions in daily practice and clinical research. Ann. Oncol. Off. J. Eur. Soc. Med Oncol. 2019, 30, 1417–1427. [Google Scholar] [CrossRef] [Green Version]
- Demetri, G.D.; Antonescu, C.R.; Bjerkehagen, B.; Bovée, J.; Boye, K.; Chacón, M.; Dei Tos, A.P.; Desai, J.; Fletcher, J.A.; Gelderblom, H.; et al. Diagnosis and management of tropomyosin receptor kinase (trk) fusion sarcomas: Expert recommendations from the world sarcoma network. Ann. Oncol. Off. J. Eur. Soc. Med Oncol. 2020, 31, 1506–1517. [Google Scholar] [CrossRef]
- Brčić, I.; Godschachner, T.M.; Bergovec, M.; Igrec, J.; Till, H.; Lackner, H.; Scheipl, S.; Kashofer, K.; Brodowicz, T.; Leithner, A.; et al. Broadening the spectrum of ntrk rearranged mesenchymal tumors and usefulness of pan-trk immunohistochemistry for identification of ntrk fusions. Mod. Pathol. Off. J. United States Can. Acad. Pathol. Inc 2021, 34, 396–407. [Google Scholar] [CrossRef]
- Mason, E.F.; Hornick, J.L. Succinate dehydrogenase deficiency is associated with decreased 5-hydroxymethylcytosine production in gastrointestinal stromal tumors: Implications for mechanisms of tumorigenesis. Mod. Pathol. Off. J. United States Can. Acad. Pathol. Inc 2013, 26, 1492–1497. [Google Scholar] [CrossRef] [Green Version]
- Killian, J.K.; Kim, S.Y.; Miettinen, M.; Smith, C.; Merino, M.; Tsokos, M.; Quezado, M.; Smith, W.I., Jr.; Jahromi, M.S.; Xekouki, P.; et al. Succinate dehydrogenase mutation underlies global epigenomic divergence in gastrointestinal stromal tumor. Cancer Discov. 2013, 3, 648–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gill, A.J. Succinate dehydrogenase (sdh)-deficient neoplasia. Histopathology 2018, 72, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Gill, A.J. Succinate dehydrogenase (sdh) and mitochondrial driven neoplasia. Pathology 2012, 44, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Belinsky, M.G.; Rink, L.; Cai, K.Q.; Ochs, M.F.; Eisenberg, B.; Huang, M.; von Mehren, M.; Godwin, A.K. The insulin-like growth factor system as a potential therapeutic target in gastrointestinal stromal tumors. Cell Cycle (Georget. Tex.) 2008, 7, 2949–2955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, A.; Chen, J.; Clarkson, A.; Samra, J.S.; Clifton-Bligh, R.J.; Hugh, T.J.; Gill, A.J. Succinate dehydrogenase-deficient gists are characterized by igf1r overexpression. Mod. Pathol. Off. J. United States Can. Acad. Pathol. Inc 2012, 25, 1307–1313. [Google Scholar] [CrossRef] [Green Version]
- Lasota, J.; Wang, Z.; Kim, S.Y.; Helman, L.; Miettinen, M. Expression of the receptor for type i insulin-like growth factor (igf1r) in gastrointestinal stromal tumors: An immunohistochemical study of 1078 cases with diagnostic and therapeutic implications. Am. J. Surg. Pathol. 2013, 37, 114–119. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Rohan, T. Role of the insulin-like growth factor family in cancer development and progression. J. Natl. Cancer Inst. 2000, 92, 1472–1489. [Google Scholar] [CrossRef]
- Tarn, C.; Rink, L.; Merkel, E.; Flieder, D.; Pathak, H.; Koumbi, D.; Testa, J.R.; Eisenberg, B.; von Mehren, M.; Godwin, A.K. Insulin-like growth factor 1 receptor is a potential therapeutic target for gastrointestinal stromal tumors. Proc. Natl. Acad. Sci. USA 2008, 105, 8387–8392. [Google Scholar] [CrossRef] [Green Version]
- Flavahan, W.A.; Drier, Y.; Johnstone, S.E.; Hemming, M.L.; Tarjan, D.R.; Hegazi, E.; Shareef, S.J.; Javed, N.M.; Raut, C.P.; Eschle, B.K.; et al. Altered chromosomal topology drives oncogenic programs in sdh-deficient gists. Nature 2019, 575, 229–233. [Google Scholar] [CrossRef]
- Mason, E.F.; Hornick, J.L. Conventional risk stratification fails to predict progression of succinate dehydrogenase-deficient gastrointestinal stromal tumors: A clinicopathologic study of 76 cases. Am. J. Surg. Pathol. 2016, 40, 1616–1621. [Google Scholar] [CrossRef]
- Stratakis, C.A.; Carney, J.A. The triad of paragangliomas, gastric stromal tumours and pulmonary chondromas (carney triad), and the dyad of paragangliomas and gastric stromal sarcomas (carney-stratakis syndrome): Molecular genetics and clinical implications. J. Intern. Med. 2009, 266, 43–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matyakhina, L.; Bei, T.A.; McWhinney, S.R.; Pasini, B.; Cameron, S.; Gunawan, B.; Stergiopoulos, S.G.; Boikos, S.; Muchow, M.; Dutra, A.; et al. Genetics of carney triad: Recurrent losses at chromosome 1 but lack of germline mutations in genes associated with paragangliomas and gastrointestinal stromal tumors. J. Clin. Endocrinol. Metab. 2007, 92, 2938–2943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haller, F.; Moskalev, E.A.; Faucz, F.R.; Barthelmeß, S.; Wiemann, S.; Bieg, M.; Assie, G.; Bertherat, J.; Schaefer, I.M.; Otto, C.; et al. Aberrant DNA hypermethylation of sdhc: A novel mechanism of tumor development in carney triad. Endocr. Relat. Cancer 2014, 21, 567–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carney, J.A. Gastric stromal sarcoma, pulmonary chondroma, and extra-adrenal paraganglioma (carney triad): Natural history, adrenocortical component, and possible familial occurrence. Mayo Clin. Proc. 1999, 74, 543–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miettinen, M.; Killian, J.K.; Wang, Z.F.; Lasota, J.; Lau, C.; Jones, L.; Walker, R.; Pineda, M.; Zhu, Y.J.; Kim, S.Y.; et al. Immunohistochemical loss of succinate dehydrogenase subunit a (sdha) in gastrointestinal stromal tumors (gists) signals sdha germline mutation. Am. J. Surg. Pathol. 2013, 37, 234–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Settas, N.; Faucz, F.R.; Stratakis, C.A. Succinate dehydrogenase (sdh) deficiency, carney triad and the epigenome. Mol. Cell. Endocrinol. 2018, 469, 107–111. [Google Scholar] [CrossRef]
- Demetri, G.D.; von Mehren, M.; Antonescu, C.R.; DeMatteo, R.P.; Ganjoo, K.N.; Maki, R.G.; Pisters, P.W.; Raut, C.P.; Riedel, R.F.; Schuetze, S.; et al. Nccn task force report: Update on the management of patients with gastrointestinal stromal tumors. J. Natl. Compr. Cancer Netw. JNCCN 2010, 8 (Suppl. 2), S1–S41. [Google Scholar] [CrossRef] [Green Version]
- Murray, M.; Hatcher, H.; Jessop, F.; Williams, D.; Carroll, N.; Bulusu, R.; Judson, I. Treatment of wild-type gastrointestinal stromal tumor (wt-gist) with imatinib and sunitinib. Pediatric Blood Cancer 2008, 50, 386–388. [Google Scholar] [CrossRef]
Figure 1.
Morphology and immunohistochemical findings in GIST (Gastrointestinal Stroma Tumor). (A) Spindle cell GIST (inset: IHC DOG1+). (B) Epithelioid GIST (inset: IHC DOG1+). (C) Spindle cell GISTs with nuclear palisading, (D) GIST with storiform growth pattern, and (E) GIST with prominent paranuclear vacuolation. (F) Epithelioid GIST with a prominent plasmacytoid morphology. (G) Pleomorphic GIST (inset: IHC KIT+). (H) Dedifferentiated GIST (inset: IHC DOG1− and KIT−). IHC (immunohistochemistry); −(negative); +(positive).
Figure 1.
Morphology and immunohistochemical findings in GIST (Gastrointestinal Stroma Tumor). (A) Spindle cell GIST (inset: IHC DOG1+). (B) Epithelioid GIST (inset: IHC DOG1+). (C) Spindle cell GISTs with nuclear palisading, (D) GIST with storiform growth pattern, and (E) GIST with prominent paranuclear vacuolation. (F) Epithelioid GIST with a prominent plasmacytoid morphology. (G) Pleomorphic GIST (inset: IHC KIT+). (H) Dedifferentiated GIST (inset: IHC DOG1− and KIT−). IHC (immunohistochemistry); −(negative); +(positive).

Figure 2.
Sub-classification of GISTs into a succinate dehydrogenase (SDH)-competent and an SDH-deficient group by using an SDHB IHC #. Legend: CSS: Carney–Stratakis Syndrome; CT: Carney triad; CT *: in some cases, mutations described [9]; wt: wild type. Grey rectangle: DNA- and RNA sequencing in a specialized center.
Figure 2.
Sub-classification of GISTs into a succinate dehydrogenase (SDH)-competent and an SDH-deficient group by using an SDHB IHC #. Legend: CSS: Carney–Stratakis Syndrome; CT: Carney triad; CT *: in some cases, mutations described [9]; wt: wild type. Grey rectangle: DNA- and RNA sequencing in a specialized center.

Figure 3.
Graphical representation of KIT and PDGFRA transmembrane tyrosine kinase receptors with frequency and localization/distribution of primary mutations found in sporadic GIST (adapted from [53,54]).

Figure 4.
Secondary KIT mutations and predictive response to most frequently used TKIs (IM:imatinib; SU: sunitinib; RE: regorafenib). Adapted from [80].
Figure 4.
Secondary KIT mutations and predictive response to most frequently used TKIs (IM:imatinib; SU: sunitinib; RE: regorafenib). Adapted from [80].

Figure 5.
Diagnostic algorithm in wild-type gastrointestinal stromal tumors (wt-GISTs). Legend: Assoc.: associated; CSS: Carney–Stratakis Syndrome; CT: Carney triad; IHC: immunohistochemistry; PC: pulmonary chondroma; PGL: paraganglioma; SDHB: Succinate dehydrogenase B; wt: wild type; *: rarely reported.
Figure 5.
Diagnostic algorithm in wild-type gastrointestinal stromal tumors (wt-GISTs). Legend: Assoc.: associated; CSS: Carney–Stratakis Syndrome; CT: Carney triad; IHC: immunohistochemistry; PC: pulmonary chondroma; PGL: paraganglioma; SDHB: Succinate dehydrogenase B; wt: wild type; *: rarely reported.

Figure 6.
Diagnostic and molecular testing algorithm for GIST. * Our GIST mutation-panel includes following genes: PDGFA, KRAS, NRAS, HRAS, BRAF, KIT (Exon 8,9,10,11,12,13,17,18), PDGFRB (Exon 12,13,14,17,18), TP53 (Exons 4–10), SDHA, SDHB, SDHC, SDHD, NF1, CDKN2A and RB1. ** If available, mutation-panel is performed. TKI: Tyrosin Kinase Inhibitor.
Figure 6.
Diagnostic and molecular testing algorithm for GIST. * Our GIST mutation-panel includes following genes: PDGFA, KRAS, NRAS, HRAS, BRAF, KIT (Exon 8,9,10,11,12,13,17,18), PDGFRB (Exon 12,13,14,17,18), TP53 (Exons 4–10), SDHA, SDHB, SDHC, SDHD, NF1, CDKN2A and RB1. ** If available, mutation-panel is performed. TKI: Tyrosin Kinase Inhibitor.

Figure 7.
Immunohistochemical staining with pan-TRK antibody in GIST shows diffuse cytoplasmic and membranous expression. (Scale bar shows 0.1 mm).
Figure 7.
Immunohistochemical staining with pan-TRK antibody in GIST shows diffuse cytoplasmic and membranous expression. (Scale bar shows 0.1 mm).

Figure 8.
Morphology and immunohistochemical findings in SDHB-deficient GIST. (A) The gastric tumor with multilobulated/plexiform growth pattern. (B) The tumor is composed of epithelioid cells with a syncytial appearance. (C,D) Immunohistochemically, the tumor shows positivity for KIT and DOG1 (C), while the expression of SDHB is lost (D); the cytoplasmic stain of the endothelial cells shows the positive internal control).
Figure 8.
Morphology and immunohistochemical findings in SDHB-deficient GIST. (A) The gastric tumor with multilobulated/plexiform growth pattern. (B) The tumor is composed of epithelioid cells with a syncytial appearance. (C,D) Immunohistochemically, the tumor shows positivity for KIT and DOG1 (C), while the expression of SDHB is lost (D); the cytoplasmic stain of the endothelial cells shows the positive internal control).


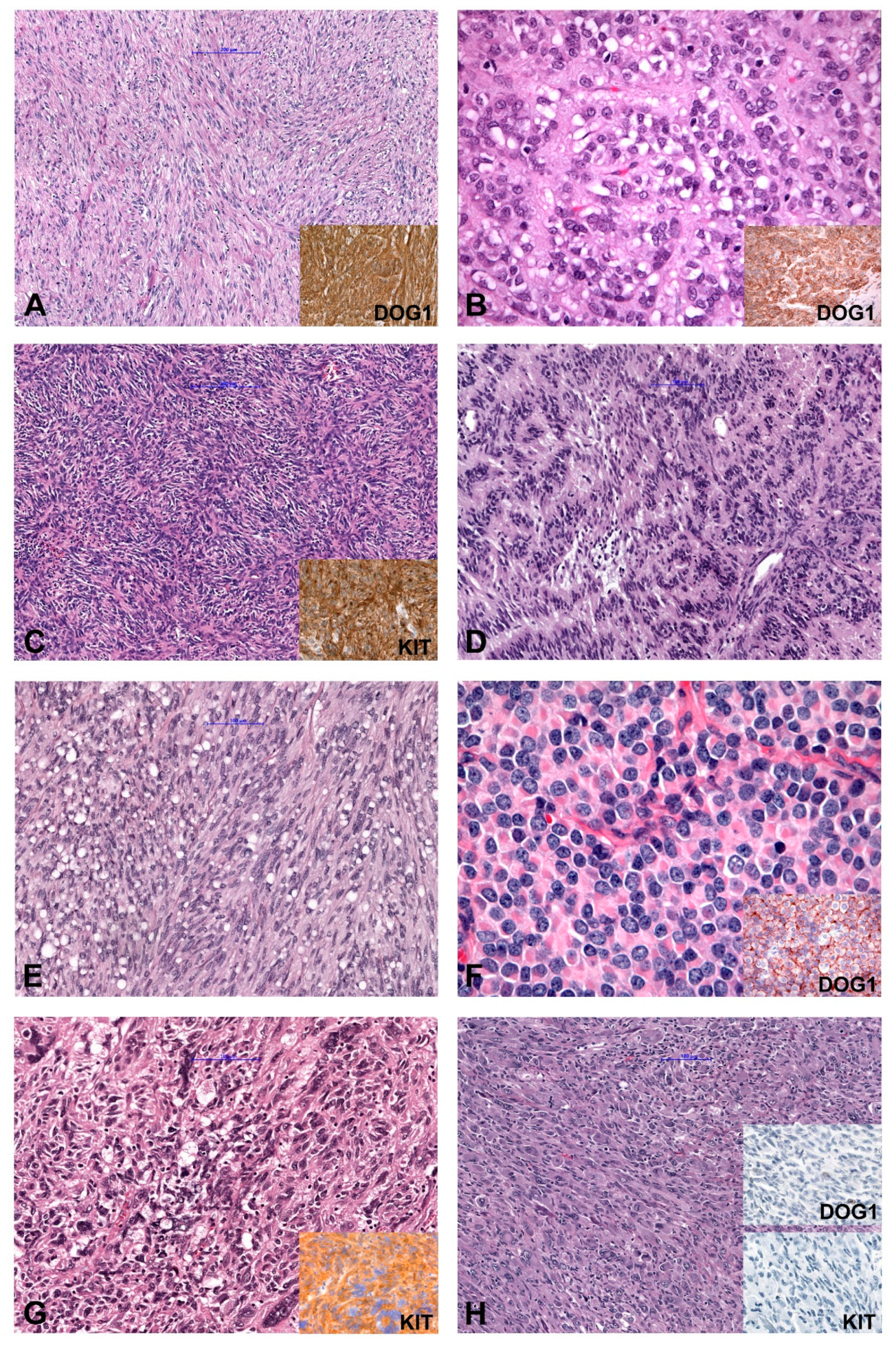{kind=link}
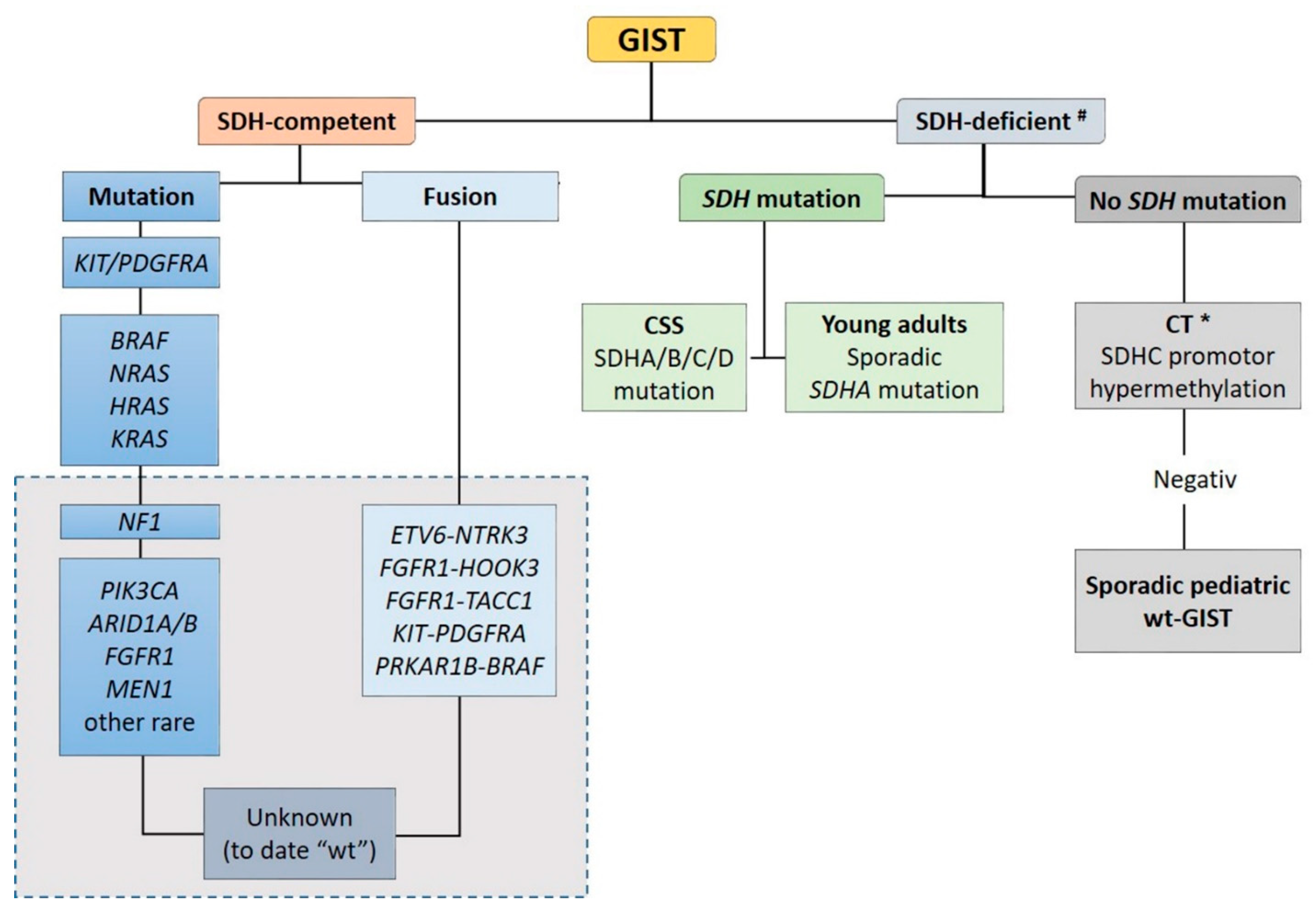{kind=link}
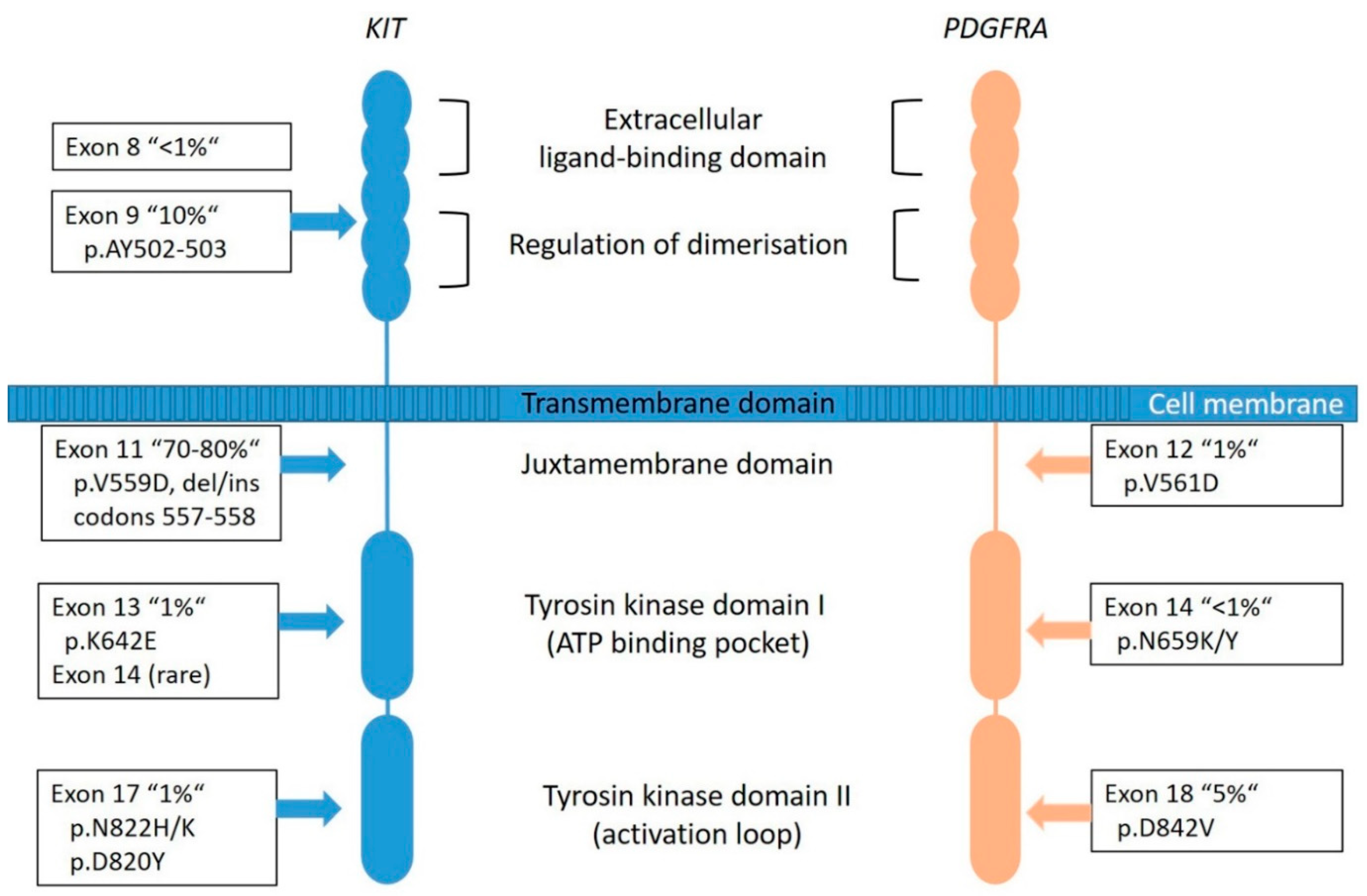{kind=link}
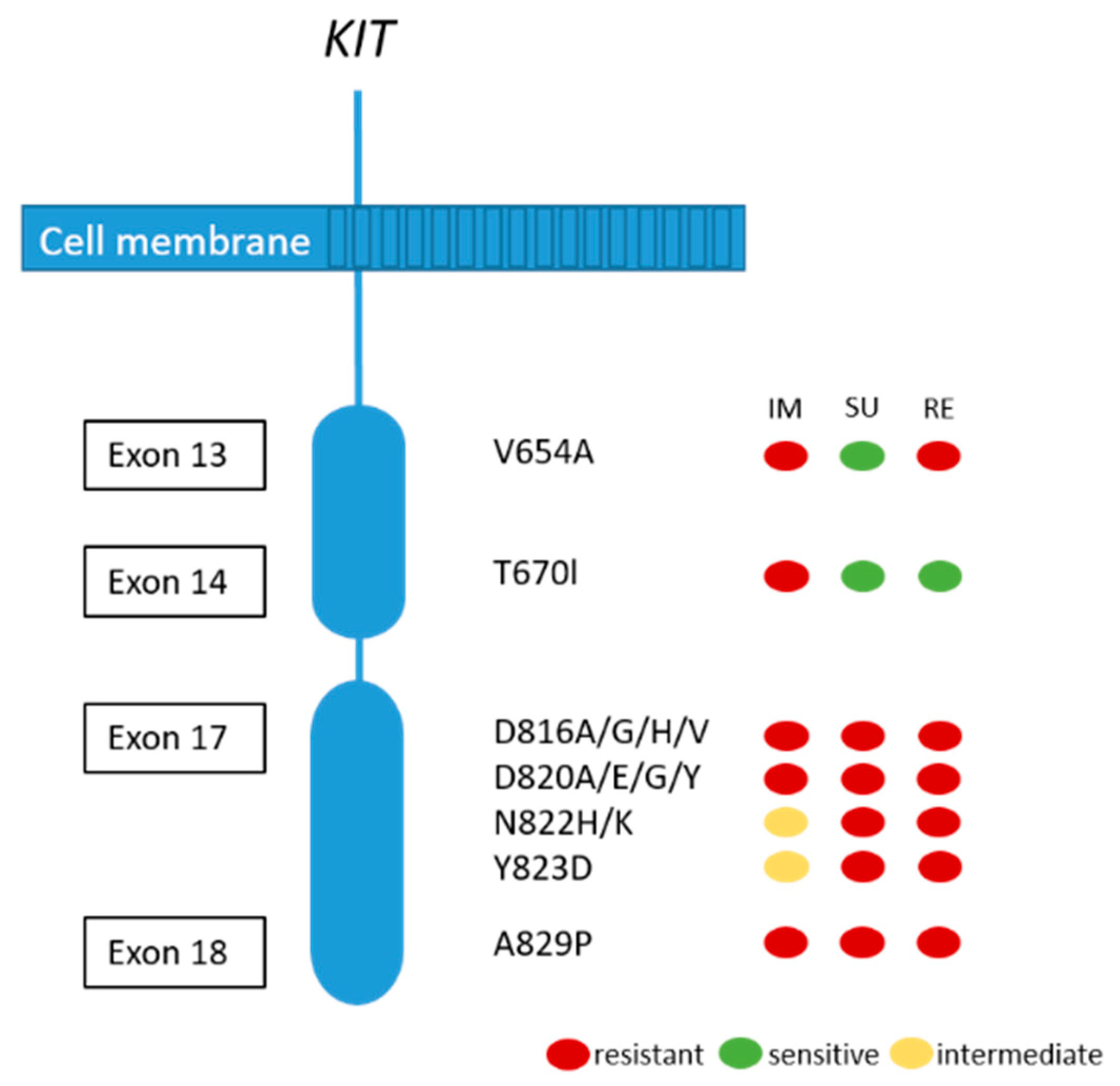{kind=link}
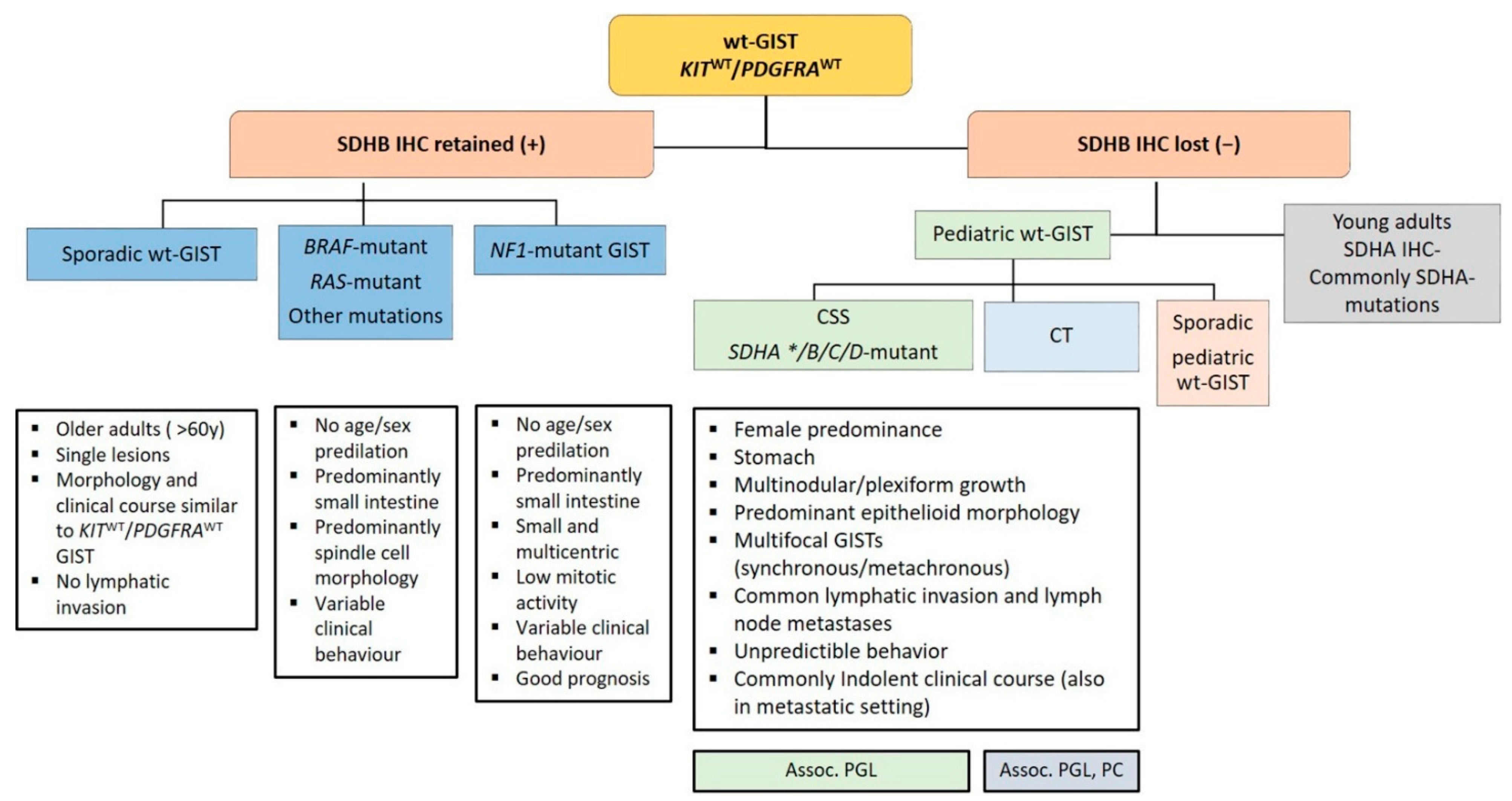{kind=link}
{kind=link}
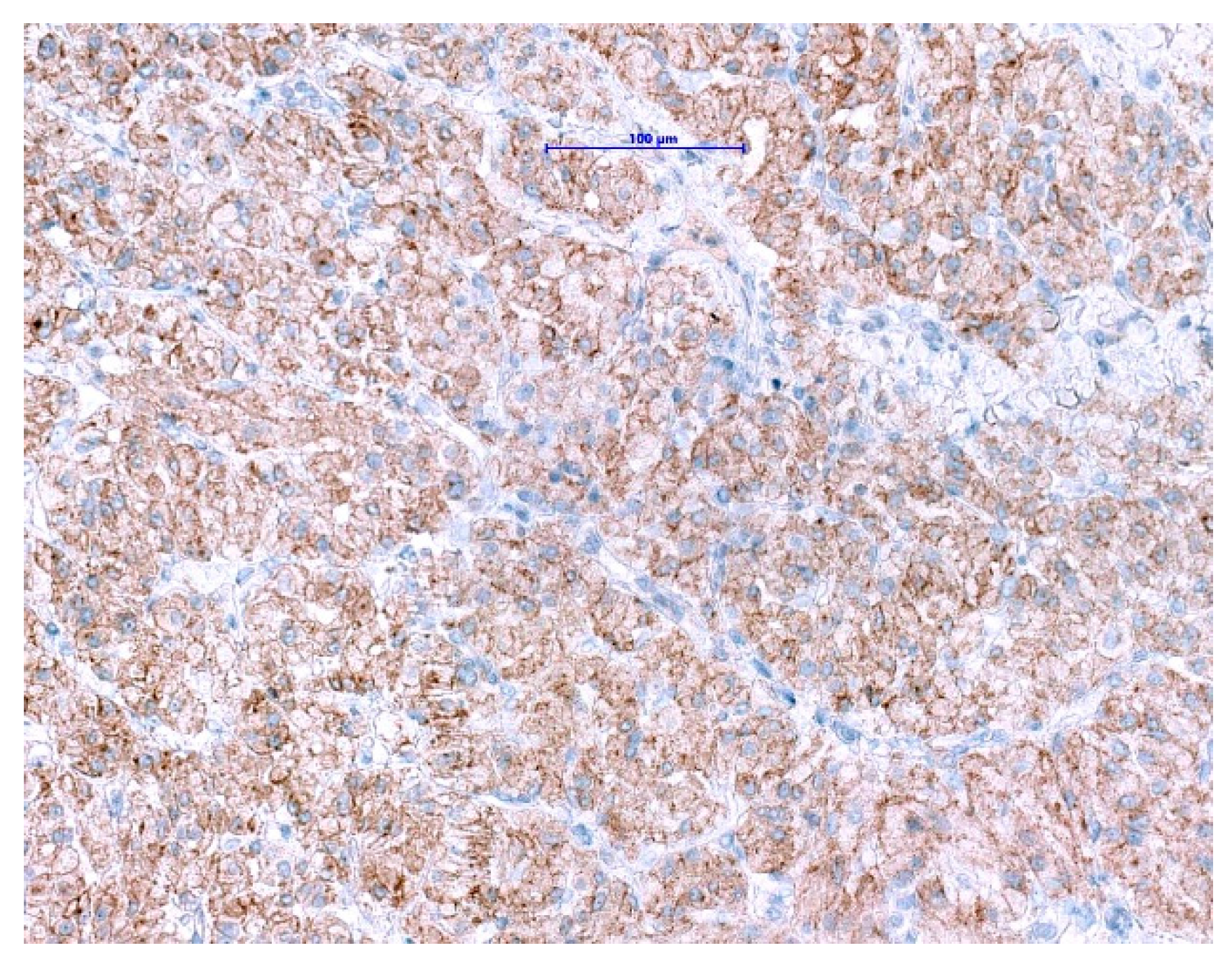{kind=link}
{kind=link}
Table 1.
Summary of the most frequent differential diagnosis for spindle cell, epithelioid and mixed GIST, including immunohistochemistry and molecular findings.
Table 1.
Summary of the most frequent differential diagnosis for spindle cell, epithelioid and mixed GIST, including immunohistochemistry and molecular findings.
| Morphology | KIT/DOG1 IHC | Diagnosis | Additional Positive IHC | Helpful Genetic Alteration(s) |
|---|---|---|---|---|
| Spindle cell | KIT+, DOG1+ | GIST | CD34 (70%), SMA (30%) | KIT, PDGFRA and others |
| KIT− (or very weak), DOG1- | Leiomyoma /Leiomyosarcoma | Desmin, SMA, caldesmon | ||
| Schwanomma | S100, SOX10 (nuclear) | |||
| Solitary fibrous tumor | CD34, STAT6 (nuclear) | NAB2-STAT6 | ||
| Fibromatosis | beta-catenin (nuclear) | CTNNB1 or APC mutation | ||
| IMT | ALK | ALK and ROS1 (rare) rearrangements | ||
| DDLPS | MDM2, CDK4 | MDM2, CDK4 amplification by FISH | ||
| Inflammatory fibroid polyp | CD34 | PDGFRA mutations | ||
| Epithelioid/mixed | KIT+, DOG1+ | GIST | SDHB retained/deficient (stomach) | KIT, PDGFRA, SDHA-D, SDHC promotor hypermethylation |
| KIT+/−, DOG1+/− | GIST | PDGFRA, KIT mutations | ||
| KIT−, DOG1− | PEComa | SMA, HMB45, MelanA, Desmin, TFE3 (subset of cases) | TSC2 mutation, TFE3-fusions | |
| Melanoma metastasis (can be KIT+) | SOX10, S100, HMB45, Melan A | cave: the common BRAFV600E mutation can be also found in GIST | ||
| Glomus tumor | SMA, caldesmon | NOTCH rearrangements and/or BRAF mutations (p.Val600Glu) | ||
| Neuroendocrine neoplasms | cytokeratin, synaptophysin, chromogranin A | DAXX, ATRX, p53, RB1 mutations |
Legend: ALK: anaplastic lymphoma kinase; DDLPS: dedifferentiated liposarcoma; FISH: fluorescence in situ hybridization; GIST: gastrointestinal stromal tumor; IMT: inflammatory myofibroblastic tumor; PEComa: perivascular epithelioid tumor; SDHB: succinate dehydrogenase B; SMA: smooth muscle aktin.
Table 2.
Anatomic distribution, frequency and treatment response of the most common GISTs groups.
| Genetic Type | Frequency | Anatomic Location | Treatment | |
|---|---|---|---|---|
| KIT mutations | ||||
| Exon 8 | <0.1% | |||
| Exon 9 | 6% | small & large bowel | Imatinib sensitiv (800 mg/d) | |
| Exon 11 | 66% | all locations | Imatinib sensitive | |
| Exon 13 | 1% | all locations | usually secondary mutation resistant to imatinib, responds to sunitinib | |
| Exon 17 | <1% | all locations | secondary mutation resistant to imatinib and sunitinib; have been shown to respond to other TKI like regorafenib | |
| PDGFRAmutations | ||||
| Exon 12 | 1% | all locations | ||
| Exon 14 | <1% | stomach | Imatinib sensitiv | |
| Exon 18 D842V | 6% | stomach | Imatinib/sunitinib resistant; good respons to avapritinib | |
| Exon 18 others | 1% | all locations | response to imatinib reported | |
| KIT/PDGFRA “wild-type” | Limited responses to imatinib Possible response to other TKIs (limited data) | |||
| SDHB IHC+/SDH-competent | NF1 mutation (assoc. with RD) | <1% | small bowel | |
| NRAS/HRAS/KRAS mutations | <1% | all locations (limited data) | ||
| BRAF mutation | 1% | most commonly stomach | ||
| Other rare mutations/fusions | all locations (limited data) | |||
| SDHB IHC−/SDH-deficient | SDHA/B/C/D mutations (CSS) | 2% | stomach | |
| Part of the CT * | 1% | stomach | ||
| SDHA mutation (young adults) | stomach | |||
| Sporadic pediatric wt- GIST | 1% | stomach |
Legend: CSS: Carney–Stratakis Syndrome; CT: Carney triad; *: most cases show promotor hypermethylation.
Table 3.
Most common KIT and PDGFRA mutations found in familial GIST.
| KIT Mutations | |
|---|---|
| Exon 8 | p.D419del |
| Exon 9 | p.K509I |
| Exon 11 | p.W557R p.V559A p.D579del p.V560G p.V560A p.L576P |
| Exon 13 | p.K642E p.N655K |
| Exon 17 | p.D820Y p.D820G p.N822Y |
| PDGFRA mutations | |
| Exon 12 | p.V561D p.Y555C |
| Exon 14 | p.P653L |
| Exon 18 | p.D842Y p.D846V |
Adapted from [94].
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Brčić, I.; Argyropoulos, A.; Liegl-Atzwanger, B. Update on Molecular Genetics of Gastrointestinal Stromal Tumors. Diagnostics 2021, 11, 194. https://doi.org/10.3390/diagnostics11020194
AMA Style
Brčić I, Argyropoulos A, Liegl-Atzwanger B. Update on Molecular Genetics of Gastrointestinal Stromal Tumors. Diagnostics. 2021; 11(2):194. https://doi.org/10.3390/diagnostics11020194
Chicago/Turabian StyleBrčić, Iva, Alexandra Argyropoulos, and Bernadette Liegl-Atzwanger. 2021. "Update on Molecular Genetics of Gastrointestinal Stromal Tumors" Diagnostics 11, no. 2: 194. https://doi.org/10.3390/diagnostics11020194
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.