Tumor Heterogeneity: Mechanisms and Bases for a Reliable Application of Molecular Marker Design

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
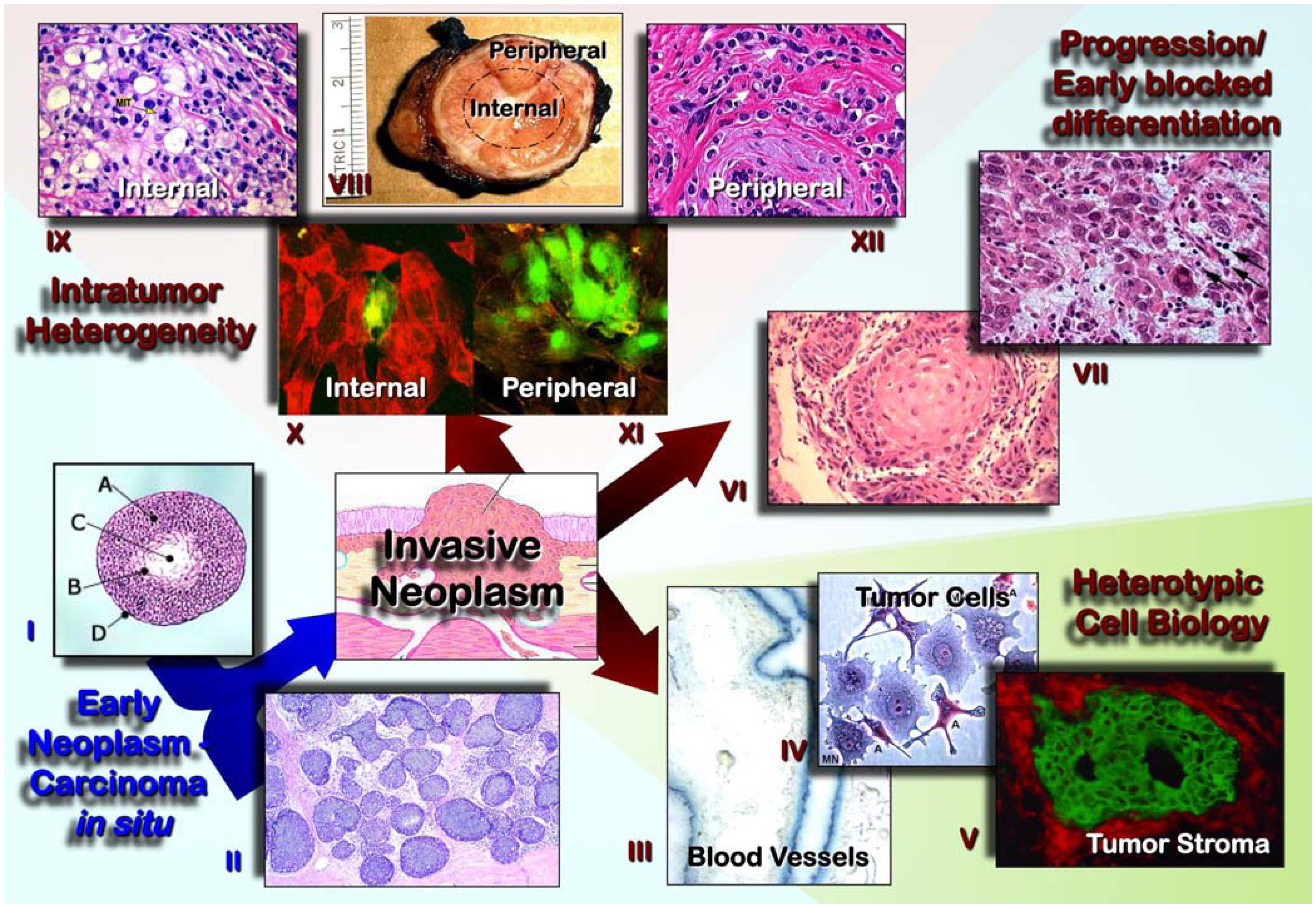
2. Tumor Cell Segregation
2.1. Clonal Origin and Expansions. Role in the Natural History of Neoplasms, Tumor Progression, and Intra-Tumor Clonal Diversity
2.1.1. Early Neoplasms, Precancerous Lesions and Progression
2.2. Cancer Stem Cells and Plasticity
2.2.1. Phenotypic Plasticity
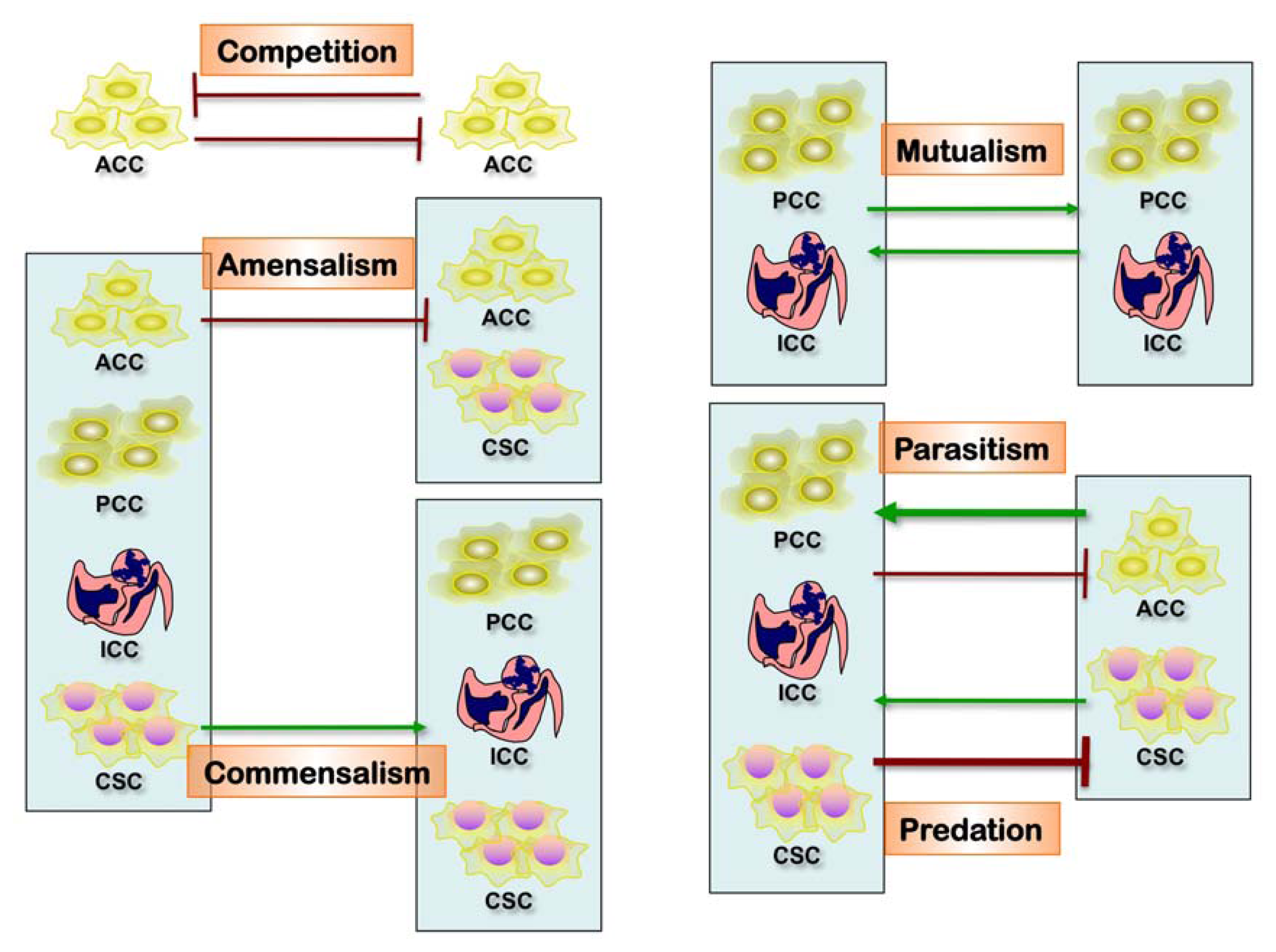
2.3. Interactions of Distinct Tumor Clones. Clonal Evolution and Progression
2.3.1. Tumor Evolution as Byproduct of Clonal Heterogeneity
2.3.2. Biological Interactions among Distinct Tumor Clones
3. Tumor Components
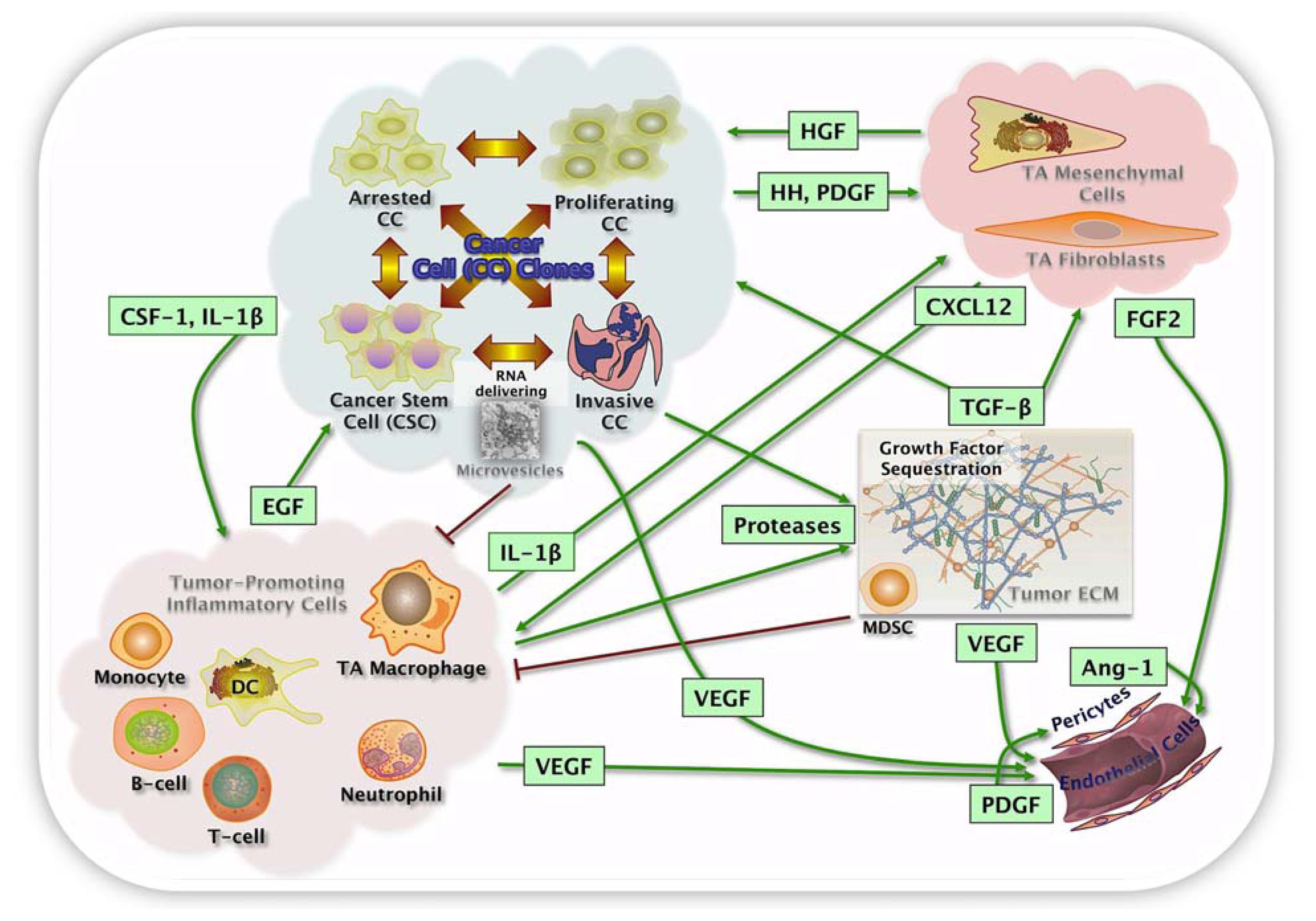
3.1. Cellular Interactions and Microenvironment
- The altered/initiated cell is already endowed with some degree of inherent growth autonomy and starts to replicate unchecked, forming a focal lesion and then, after a number of further steps, a full blown cancer [84]. The analysis of several multistage models of cancer induction has led to the conclusion that initiation per se does not result in any significant growth of pre-neoplastic and/or neoplastic lesions, and the appearance of the latter is heavily dependent on the presence of a promoting/selective environment [85]. Furthermore, transplantation experiments have convincingly demonstrated that different types of altered cells do not display any evidence of growth autonomy when transferred in a normal tissue environment of young animals in vivo [67]. By analogy, altered, putative initiated cells can be found in the skin of several healthy human subjects, suggesting that their presence per se is not necessarily associated with selective clonal growth, and additional (promoting/selective) influences must be enforced when the latter does occur [86]. Also epidemiologic evidence on smoking and lung cancer suggests that clonal expansion of cells is much more relevant than early mutations [87].
- (II) The other possibility is that the single altered cell does not express any significant degree of growth autonomy and is still under the control of normal homeostatic mechanisms; if this is the case, its selective clonal growth must be linked to the dynamics of cell turnover typical of the tissue where it resides. Thus, specific alterations of these dynamics could translate into a promoting effect for any putative altered cells present in that tissue. It is self-apparent that, within this perspective, the tissue microenvironment surrounding rare initiated/altered cells is given a central role in their selective emergence as focal proliferative lesions. In this context the neoplastic development is a biologic process that is not directly caused by the inciting agent acting on a passive target, but results from the interaction of living structures (cells, tissues and organs) with those agents [88,89].
3.1.1. Promoting Potential of a Growth-Constrained Tissue Microenvironment
3.1.2. Molecular Analysis of Selectogenic Microenvironments
3.1.3. Stress and the Mutator Phenotype
3.1.4. Chronic Inflammation, Myeloid-Derived Cells in Tissue and Tumor Microenvironment
3.1.5. From Tissue Microenvironment to Tumor Microenvironment
3.2. Heterogeneity, Microenvironment and Metastasis
4. Mechanisms Involved in Intra-Tumor Heterogeneity and Progression
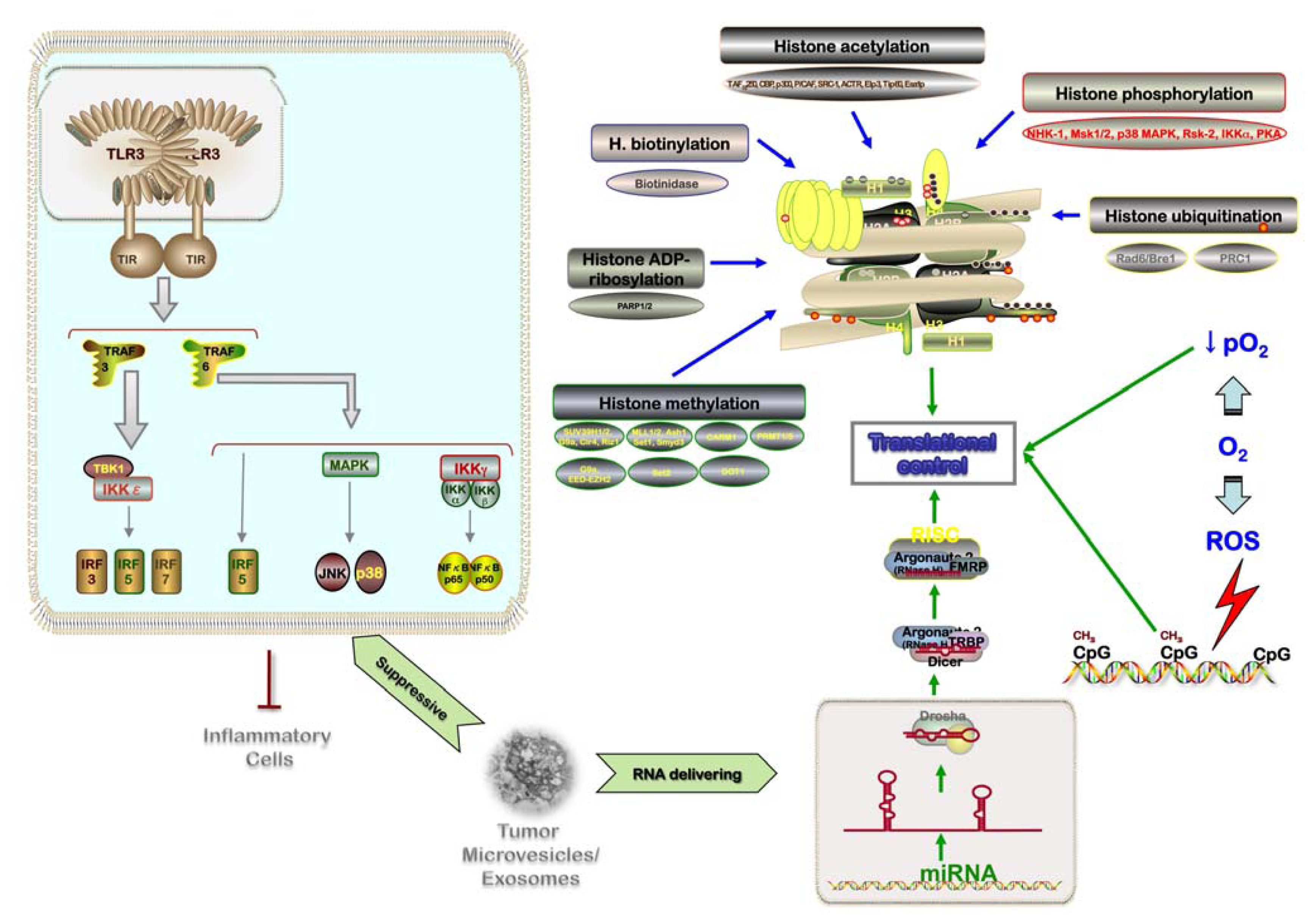
4.1. Hypoxia: Intratumor Variability and Influence on Metastasis
4.2. Regulation of Gene Expression: Exosomes and Epigenetics
5. Clinical Implications
5.1. Diagnosis: Sampling and Genetic Targets
5.1.1. Sampling Issues
5.1.2. Focused Approaches
5.1.3. Genome-Wide Approaches
5.1.4. Issues of Quantity and Quality
5.2. Therapeutic Response
6. Conclusions and Future Directions
6.1. Future Directions
- Conflict of InterestThe author declares no conflict of interest. I apologize to all authors whose publication I may have omitted.
References
- Fidler, I.J.; Hart, I.R. Biological diversity in metastatic neoplasms: Origins and implications. Science 1982, 217, 998–1003. [Google Scholar]
- Heppner, G.H.; Miller, F.R. The cellular basis of tumor progression. Int. Rev. Cytol 1998, 177, 1–56. [Google Scholar]
- Talmadge, J.E.; Wolman, S.R.; Fidler, I.J. Evidence for the clonal origin of spontaneous metastases. Science 1982, 217, 361–363. [Google Scholar]
- Diaz-Cano, S.J. General morphological and biological features of neoplasms: Integration of molecular findings. Histopathology 2008, 53, 1–19. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar]
- Kinzler, K.W.; Vogelstein, B. Lessons from hereditary colorectal cancer. Cell 1996, 87, 159–170. [Google Scholar]
- Blanes, A.; Diaz-Cano, S.J. Complementary analysis of microsatellite tumor profile and mismatch repair defects in colorectal carcinomas. World J. Gastroenterol 2006, 12, 5932–5940. [Google Scholar]
- Blanes, A.; Diaz-Cano, S.J. DNA and kinetic heterogeneity during the clonal evolution of adrenocortical proliferative lesions. Hum. Pathol 2006, 37, 1295–1303. [Google Scholar]
- Blanes, A.; Sanchez-Carrillo, J.J.; Diaz-Cano, S.J. Topographic molecular profile of pheochromocytomas: Role of somatic down-regulation of mismatch repair. J. Clin. Endocrinol. Metab 2006, 91, 1150–1158. [Google Scholar]
- Pozo, L.; Sanchez-Carrillo, J.J.; Martinez, A.; Blanes, A.; Diaz-Cano, S.J. Differential kinetic features by tumour topography in cutaneous small-cell neuroendocrine (Merkel cell) carcinomas. J. Eur. Acad. Dermatol. Venereol 2007, 21, 1220–1228. [Google Scholar]
- Kinzler, K.W.; Vogelstein, B. Landscaping the cancer terrain. Science 1998, 280, 1036–1037. [Google Scholar]
- Maley, C.C.; Galipeau, P.C.; Finley, J.C.; Wongsurawat, V.J.; Li, X.; Sanchez, C.A.; Paulson, T.G.; Blount, P.L.; Risques, R.A.; Rabinovitch, P.S.; et al. Genetic clonal diversity predicts progression to esophageal adenocarcinoma. Nat. Genet 2006, 38, 468–473. [Google Scholar]
- Iwasa, Y.; Michor, F. Evolutionary dynamics of intratumor heterogeneity. PLoS One 2011, 6. [Google Scholar] [CrossRef]
- Parmigiani, G.; Boca, S.; Lin, J.; Kinzler, K.W.; Velculescu, V.; Vogelstein, B. Design and analysis issues in genome-wide somatic mutation studies of cancer. Genomics 2009, 93, 17–21. [Google Scholar]
- Diaz-Cano, S.J. Clonality studies in the analysis of adrenal medullary proliferations: Application principles and limitations. Endocr. Pathol 1998, 9, 301–316. [Google Scholar]
- Diaz-Cano, S.J. Designing a molecular analysis of clonality in tumours. J. Pathol 2000, 191, 343–344. [Google Scholar]
- Diaz-Cano, S.J.; Blanes, A.; Wolfe, H.J. PCR techniques for clonality assays. Diagn. Mol. Pathol 2001, 10, 24–33. [Google Scholar]
- Pozo-Garcia, L.; Diaz-Cano, S.J. Clonal origin and expansions in neoplasms: Biologic and technical aspects must be considered together. Am. J. Pathol 2003, 162, 353–354. [Google Scholar]
- Leedham, S.J.; Wright, N.A. Human tumour clonality assessment—Flawed but necessary. J. Pathol 2008, 215, 351–354. [Google Scholar]
- Arif, S.; Blanes, A.; Diaz-Cano, S.J. Hashimoto’s thyroiditis shares features with early papillary thyroid carcinoma. Histopathology 2002, 41, 357–362. [Google Scholar]
- Blanes, A.; Rubio, J.; Martinez, A.; Wolfe, H.J.; Diaz-Cano, S.J. Kinetic profiles by topographic compartments in muscle-invasive transitional cell carcinomas of the bladder: Role of TP53 and NF1 genes. Am. J. Clin. Pathol 2002, 118, 93–100. [Google Scholar]
- Diaz-Cano, S.J.; Blanes, A.; Rubio, J.; Matilla, A.; Wolfe, H.J. Molecular evolution and intratumor heterogeneity by topographic compartments in muscle-invasive transitional cell carcinoma of the urinary bladder. Lab. Invest 2000, 80, 279–289. [Google Scholar]
- Diaz-Cano, S.J.; de Miguel, M.; Blanes, A.; Tashjian, R.; Galera, H.; Wolfe, H.J. Clonality as expression of distinctive cell kinetics patterns in nodular hyperplasias and adenomas of the adrenal cortex. Am. J. Pathol 2000, 156, 311–319. [Google Scholar]
- Diaz-Cano, S.J.; de Miguel, M.; Blanes, A.; Tashjian, R.; Galera, H.; Wolfe, H.J. Clonal patterns in phaeochromocytomas and MEN-2A adrenal medullary hyperplasias: Histological and kinetic correlates. J. Pathol 2000, 192, 221–228. [Google Scholar]
- Jones, P.A. DNA methylation and cancer. Cancer Res 1986, 46, 461–466. [Google Scholar]
- Jones, P.A.; Baylin, S.B. The epigenomics of cancer. Cell 2007, 128, 683–692. [Google Scholar]
- Diaz-Cano, S.J. Are PCR artifacts in microdissected samples preventable? Hum. Pathol 2001, 32, 1415–1416. [Google Scholar]
- Diaz-Cano, S.J.; de Miguel, M.; Blanes, A.; Galera, H.; Wolfe, H.J. Contribution of the microvessel network to the clonal and kinetic profiles of adrenal cortical proliferative lesions. Hum. Pathol 2001, 32, 1232–1239. [Google Scholar]
- Blanes, A.; Rubio, J.; Sanchez-Carrillo, J.J.; Diaz-Cano, S.J. Coexistent intraurothelial carcinoma and muscle-invasive urothelial carcinoma of the bladder: Clonality and somatic down-regulation of DNA mismatch repair. Hum. Pathol 2009, 40, 988–997. [Google Scholar]
- Diaz-Cano, S.J.; de Miguel, M.; Blanes, A.; Tashjian, R.; Wolfe, H.J. Germline RET 634 mutation positive MEN 2A-related C-cell hyperplasias have genetic features consistent with intraepithelial neoplasia. J. Clin. Endocrinol. Metab 2001, 86, 3948–3957. [Google Scholar]
- Rubio, J.; Blanes, A.; Sanchez-Carrillo, J.J.; Diaz-Cano, S.J. Microsatellite abnormalities and somatic down-regulation of mismatch repair characterize nodular-trabecular muscle-invasive urothelial carcinoma of the bladder. Histopathology 2007, 51, 458–467. [Google Scholar]
- Polyak, K.; Haviv, I.; Campbell, I.G. Co-evolution of tumor cells and their microenvironment. Trends Genet 2009, 25, 30–38. [Google Scholar]
- Husain, E.A.; Mein, C.; Pozo, L.; Blanes, A.; Diaz-Cano, S.J. Heterogeneous topographic profiles of kinetic and cell cycle regulator microsatellites in atypical (dysplastic) melanocytic nevi. Mod. Pathol 2011, 24, 471–486. [Google Scholar]
- Visvader, J.E.; Lindeman, G.J. Cancer stem cells in solid tumours: Accumulating evidence and unresolved questions. Nat. Rev. Cancer 2008, 8, 755–768. [Google Scholar]
- Rossi, D.J.; Jamieson, C.H.; Weissman, I.L. Stems cells and the pathways to aging and cancer. Cell 2008, 132, 681–696. [Google Scholar]
- Tan, B.T.; Park, C.Y.; Ailles, L.E.; Weissman, I.L. The cancer stem cell hypothesis: A work in progress. Lab. Invest 2006, 86, 1203–1207. [Google Scholar]
- Ailles, L.E.; Weissman, I.L. Cancer stem cells in solid tumors. Curr. Opin. Biotechnol 2007, 18, 460–466. [Google Scholar]
- Cho, R.W.; Clarke, M.F. Recent advances in cancer stem cells. Curr. Opin. Genet. Dev 2008, 18, 48–53. [Google Scholar]
- Kelly, P.N.; Dakic, A.; Adams, J.M.; Nutt, S.L.; Strasser, A. Tumor growth need not be driven by rare cancer stem cells. Science 2007, 317, 337. [Google Scholar]
- Kern, S.E.; Shibata, D. The fuzzy math of solid tumor stem cells: A perspective. Cancer Res 2007, 67, 8985–8988. [Google Scholar]
- Park, S.Y.; Gonen, M.; Kim, H.J.; Michor, F.; Polyak, K. Cellular and genetic diversity in the progression of in situ human breast carcinomas to an invasive phenotype. J. Clin. Invest 2010, 120, 636–644. [Google Scholar]
- Marotta, L.L.; Almendro, V.; Marusyk, A.; Shipitsin, M.; Schemme, J.; Walker, S.R.; Bloushtain-Qimron, N.; Kim, J.J.; Choudhury, S.A.; Maruyama, R.; et al. The JAK2/STAT3 signaling pathway is required for growth of CD44CD24 stem cell-like breast cancer cells in human tumors. J. Clin. Invest 2011, 121, 2723–2735. [Google Scholar]
- Hill, R.P. Identifying cancer stem cells in solid tumors: Case not proven. Cancer Res 2006, 66, 1891–1895. [Google Scholar]
- Burkert, J.; Wright, N.A.; Alison, M.R. Stem cells and cancer: An intimate relationship. J. Pathol 2006, 209, 287–297. [Google Scholar]
- Garcia, S.B.; Park, H.S.; Novelli, M.; Wright, N.A. Field cancerization, clonality, and epithelial stem cells: The spread of mutated clones in epithelial sheets. J. Pathol 1999, 187, 61–81. [Google Scholar]
- Poulsom, R.; Alison, M.R.; Forbes, S.J.; Wright, N.A. Adult stem cell plasticity. J. Pathol 2002, 197, 441–456. [Google Scholar]
- Durrett, R.; Foo, J.; Leder, K.; Mayberry, J.; Michor, F. Intratumor heterogeneity in evolutionary models of tumor progression. Genetics 2011, 188, 461–477. [Google Scholar]
- Spencer, S.L.; Gaudet, S.; Albeck, J.G.; Burke, J.M.; Sorger, P.K. Non-genetic origins of cell-to-cell variability in TRAIL-induced apoptosis. Nature 2009, 459, 428–432. [Google Scholar]
- Sanz-Moreno, V.; Gadea, G.; Ahn, J.; Paterson, H.; Marra, P.; Pinner, S.; Sahai, E.; Marshall, C.J. Rac activation and inactivation control plasticity of tumor cell movement. Cell 2008, 135, 510–523. [Google Scholar]
- Nowell, P.C. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28. [Google Scholar]
- Shah, N.P.; Nicoll, J.M.; Nagar, B.; Gorre, M.E.; Paquette, R.L.; Kuriyan, J.; Sawyers, C.L. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell 2002, 2, 117–125. [Google Scholar]
- Corless, C.L.; Heinrich, M.C. Molecular pathobiology of gastrointestinal stromal sarcomas. Annu. Rev. Pathol 2008, 3, 557–586. [Google Scholar]
- Edwards, S.L.; Brough, R.; Lord, C.J.; Natrajan, R.; Vatcheva, R.; Levine, D.A.; Boyd, J.; Reis-Filho, J.S.; Ashworth, A. Resistance to therapy caused by intragenic deletion in BRCA2. Nature 2008, 451, 1111–1115. [Google Scholar]
- Blagosklonny, M.V. Oncogenic resistance to growth-limiting conditions. Nat. Rev. Cancer 2002, 2, 221–225. [Google Scholar]
- Laconi, E.; Doratiotto, S.; Vineis, P. The microenvironments of multistage carcinogenesis. Semin. Cancer Biol 2008, 18, 322–329. [Google Scholar]
- Laconi, E.; Sonnenschein, C. Cancer development at tissue level. Semin. Cancer Biol 2008, 18, 303–304. [Google Scholar]
- Bissell, M.J.; Labarge, M.A. Context, tissue plasticity, and cancer: Are tumor stem cells also regulated by the microenvironment? Cancer Cell 2005, 7, 17–23. [Google Scholar]
- Polyak, K. Heterogeneity in breast cancer. J. Clin. Invest 2011, 121, 3786–3788. [Google Scholar]
- Merlo, L.M.; Pepper, J.W.; Reid, B.J.; Maley, C.C. Cancer as an evolutionary and ecological process. Nat. Rev. Cancer 2006, 6, 924–935. [Google Scholar]
- Marusyk, A.; DeGregori, J. Declining cellular fitness with age promotes cancer initiation by selecting for adaptive oncogenic mutations. Biochim. Biophys. Acta 2008, 1785, 1–11. [Google Scholar]
- Lowe, S.W.; Cepero, E.; Evan, G. Intrinsic tumour suppression. Nature 2004, 432, 307–315. [Google Scholar]
- Zhang, J.; Grindley, J.C.; Yin, T.; Jayasinghe, S.; He, X.C.; Ross, J.T.; Haug, J.S.; Rupp, D.; Porter-Westpfahl, K.S.; Wiedemann, L.M.; et al. PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature 2006, 441, 518–522. [Google Scholar]
- Michor, F.; Iwasa, Y.; Nowak, M.A. Dynamics of cancer progression. Nat. Rev. Cancer 2004, 4, 197–205. [Google Scholar]
- Michor, F.; Polyak, K. The origins and implications of intratumor heterogeneity. Cancer Prev. Res. (Phila.) 2010, 3, 1361–1364. [Google Scholar]
- Marusyk, A.; Casas-Selves, M.; Henry, C.J.; Zaberezhnyy, V.; Klawitter, J.; Christians, U.; DeGregori, J. Irradiation alters selection for oncogenic mutations in hematopoietic progenitors. Cancer Res 2009, 69, 7262–7269. [Google Scholar]
- Laconi, S.; Pani, P.; Pillai, S.; Pasciu, D.; Sarma, D.S.; Laconi, E. A growth-constrained environment drives tumor progression invivo. Proc. Natl. Acad. Sci. USA 2001, 98, 7806–7811. [Google Scholar]
- Nagy, J.D. Competition and natural selection in a mathematical model of cancer. Bull. Math. Biol 2004, 66, 663–687. [Google Scholar]
- Devireddy, L.R.; Gazin, C.; Zhu, X.; Green, M.R. A cell-surface receptor for lipocalin 24p3 selectively mediates apoptosis and iron uptake. Cell 2005, 123, 1293–1305. [Google Scholar]
- Guba, M.; Cernaianu, G.; Koehl, G.; Geissler, E.K.; Jauch, K.W.; Anthuber, M.; Falk, W.; Steinbauer, M. A primary tumor promotes dormancy of solitary tumor cells before inhibiting angiogenesis. Cancer Res 2001, 61, 5575–5579. [Google Scholar]
- Axelrod, R.; Axelrod, D.E.; Pienta, K.J. Evolution of cooperation among tumor cells. Proc. Natl. Acad. Sci. USA 2006, 103, 13474–13479. [Google Scholar]
- Mallepell, S.; Krust, A.; Chambon, P.; Brisken, C. Paracrine signaling through the epithelial estrogen receptor alpha is required for proliferation and morphogenesis in the mammary gland. Proc. Natl. Acad. Sci. USA 2006, 103, 2196–2201. [Google Scholar]
- Godar, S.; Ince, T.A.; Bell, G.W.; Feldser, D.; Donaher, J.L.; Bergh, J.; Liu, A.; Miu, K.; Watnick, R.S.; Reinhardt, F.; et al. Growth-inhibitory and tumor- suppressive functions of p53 depend on its repression of CD44 expression. Cell 2008, 134, 62–73. [Google Scholar]
- McAllister, S.S.; Gifford, A.M.; Greiner, A.L.; Kelleher, S.P.; Saelzler, M.P.; Ince, T.A.; Reinhardt, F.; Harris, L.N.; Hylander, B.L.; Repasky, E.A.; et al. Systemic endocrine instigation of indolent tumor growth requires osteopontin. Cell 2008, 133, 994–1005. [Google Scholar]
- Yamamoto, K.; Hanada, R.; Kikuchi, A.; Ichikawa, M.; Aihara, T.; Oguma, E.; Moritani, T.; Shimanuki, Y.; Tanimura, M.; Hayashi, Y. Spontaneous regression of localized neuroblastoma detected by mass screening. J. Clin. Oncol 1998, 16, 1265–1269. [Google Scholar]
- Axelrod, R.; Hamilton, W.D. The evolution of cooperation. Science 1981, 211, 1390–1396. [Google Scholar]
- Lyons, J.G.; Lobo, E.; Martorana, A.M.; Myerscough, M.R. Clonal diversity in carcinomas: Its implications for tumour progression and the contribution made to it by epithelial-mesenchymal transitions. Clin. Exp. Metastasis 2008, 25, 665–677. [Google Scholar]
- Morrison, S.J.; Spradling, A.C. Stem cells and niches: Mechanisms that promote stem cell maintenance throughout life. Cell 2008, 132, 598–611. [Google Scholar]
- Reddig, P.J.; Juliano, R.L. Clinging to life: Cell to matrix adhesion and cell survival. Cancer Metastasis Rev 2005, 24, 425–439. [Google Scholar]
- Tlsty, T.D.; Coussens, L.M. Tumor stroma and regulation of cancer development. Annu. Rev. Pathol 2006, 1, 119–150. [Google Scholar]
- Baithun, S.I.; Naase, M.; Blanes, A.; Diaz-Cano, S.J. Molecular and kinetic features of transitional cell carcinomas of the bladder: Biological and clinical implications. Virchows Arch 2001, 438, 289–297. [Google Scholar]
- Laconi, E. The evolving concept of tumor microenvironments. Bioessays 2007, 29, 738–744. [Google Scholar]
- Huang, L.E.; Bindra, R.S.; Glazer, P.M.; Harris, A.L. Hypoxia-induced genetic instability— Calculated mechanism underlying tumor progression. J. Mol. Med. (Berl.) 2007, 85, 139–148. [Google Scholar]
- Hahn, W.C.; Counter, C.M.; Lundberg, A.S.; Beijersbergen, R.L.; Brooks, M.W.; Weinberg, R.A. Creation of human tumour cells with defined genetic elements. Nature 1999, 400, 464–468. [Google Scholar]
- Rubin, H. Microenvironmental regulation of the initiated cell. Adv. Cancer Res 2003, 90, 1–62. [Google Scholar]
- Mudgil, A.V.; Segal, N.; Andriani, F.; Wang, Y.; Fusenig, N.E.; Garlick, J.A. Ultraviolet B irradiation induces expansion of intraepithelial tumor cells in a tissue model of early cancer progression. J. Invest. Dermatol 2003, 121, 191–197. [Google Scholar]
- Schollnberger, H.; Manuguerra, M.; Bijwaard, H.; Boshuizen, H.; Altenburg, H.P.; Rispens, S.M.; Brugmans, M.J.; Vineis, P. Analysis of epidemiological cohort data on smoking effects and lung cancer with a multi-stage cancer model. Carcinogenesis 2006, 27, 1432–1444. [Google Scholar]
- Farber, E.; Rubin, H. Cellular adaptation in the origin and development of cancer. Cancer Res 1991, 51, 2751–2761. [Google Scholar]
- Cristini, V.; Frieboes, H.B.; Gatenby, R.; Caserta, S.; Ferrari, M.; Sinek, J. Morphologic instability and cancer invasion. Clin. Cancer Res 2005, 11, 6772–6779. [Google Scholar]
- Cayama, E.; Tsuda, H.; Sarma, D.S.; Farber, E. Initiation of chemical carcinogenesis requires cell proliferation. Nature 1978, 275, 60–62. [Google Scholar]
- Breivik, J. The evolutionary origin of genetic instability in cancer development. Semin. Cancer Biol 2005, 15, 51–60. [Google Scholar]
- West, R.B.; Rubin, B.P.; Miller, M.A.; Subramanian, S.; Kaygusuz, G.; Montgomery, K.; Zhu, S.; Marinelli, R.J.; De Luca, A.; Downs-Kelly, E.; et al. A landscape effect in tenosynovial giant-cell tumor from activation of CSF1 expression by a translocation in a minority of tumor cells. Proc. Natl. Acad. Sci. USA 2006, 103, 690–695. [Google Scholar]
- van Vugt, M.A.; Bras, A.; Medema, R.H. Restarting the cell cycle when the checkpoint comes to a halt. Cancer Res 2005, 65, 7037–7040. [Google Scholar]
- Jonason, A.S.; Kunala, S.; Price, G.J.; Restifo, R.J.; Spinelli, H.M.; Persing, J.A.; Leffell, D.J.; Tarone, R.E.; Brash, D.E. Frequent clones of p53-mutated keratinocytes in normal human skin. Proc. Natl. Acad. Sci. USA 1996, 93, 14025–14029. [Google Scholar]
- Laconi, E.; Pani, P.; Farber, E. The resistance phenotype in the development and treatment of cancer. Lancet Oncol 2000, 1, 235–241. [Google Scholar]
- Marongiu, F.; Doratiotto, S.; Montisci, S.; Pani, P.; Laconi, E. Liver repopulation and carcinogenesis: Two sides of the same coin? Am. J. Pathol 2008, 172, 857–864. [Google Scholar]
- Campisi, J. Aging and cancer cell biology, 2007. Aging Cell 2007, 6, 261–263. [Google Scholar]
- Sharpless, N.E.; DePinho, R.A. How stem cells age and why this makes us grow old. Nat. Rev. Mol. Cell Biol 2007, 8, 703–713. [Google Scholar]
- McCullough, K.D.; Coleman, W.B.; Smith, G.J.; Grishan, J.W. Age-dependent regulation of the tumorigenic potential of neoplastically transformed rat liver epithelial cells by the liver microenvironment. Cancer Res 1994, 54, 3668–3671. [Google Scholar]
- Pasciu, D.; Montisci, S.; Greco, M.; Doratiotto, S.; Pitzalis, S.; Pani, P.; Laconi, S; Laconi, E. Aging is associated with increased clonogenic potential in rat liver in vivo. Aging Cell 2006, 5, 373–377. [Google Scholar]
- Krtolica, A.; Parrinello, S.; Lockett, S.; Desprez, P.Y.; Campisi, J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: A link between cancer and aging. Proc. Natl. Acad. Sci. USA 2001, 98, 12072–12077. [Google Scholar]
- Liu, D.; Hornsby, P.J. Senescent human fibroblasts increase the early growth of xenograft tumors via matrix metalloproteinase secretion. Cancer Res 2007, 67, 3117–3126. [Google Scholar]
- Trost, T.M.; Lausch, E.U.; Fees, S.A.; Schmitt, S.; Enklaar, T.; Reutzel, D.; Brixel, L.R.; Schmidtke, P.; Maringer, M.; Schiffer, I.B.; et al. Premature senescence is a primary fail-safe mechanism of ERBB2-driven tumorigenesis in breast carcinoma cells. Cancer Res 2005, 65, 840–849. [Google Scholar]
- Philip, M.; Rowley, D.A.; Schreiber, H. Inflammation as a tumor promoter in cancer induction. Semin. Cancer Biol 2004, 14, 433–439. [Google Scholar]
- Hussain, S.P.; Harris, C.C. Inflammation and cancer: An ancient link with novel potentials. Int. J. Cancer 2007, 121, 2373–2380. [Google Scholar]
- Prehn, R.T. Regeneration versus neoplastic growth. Carcinogenesis 1997, 18, 1439–1444. [Google Scholar]
- Correa, P.; Houghton, J. Carcinogenesis of Helicobacter pylori. Gastroenterology 2007, 133, 659–672. [Google Scholar]
- De Marzo, A.M.; Marchi, V.L.; Epstein, J.I.; Nelson, W.G. Proliferative inflammatory atrophy of the prostate: Implications for prostatic carcinogenesis. Am. J. Pathol 1999, 155, 1985–1992. [Google Scholar]
- De Marzo, A.M.; Nakai, Y.; Nelson, W.G. Inflammation, atrophy, and prostate carcinogenesis. Urol. Oncol 2007, 25, 398–400. [Google Scholar]
- Fox, J.G.; Wang, T.C. Inflammation, atrophy, and gastric cancer. J. Clin. Invest 2007, 117, 60–69. [Google Scholar]
- Harpaz, N. Neoplastic precursor lesions related to the development of cancer in inflammatory bowel disease. Gastroenterol. Clin. North Am 2007, 36. [Google Scholar]
- Rass, U.; Ahel, I.; West, S.C. Defective DNA repair and neurodegenerative disease. Cell 2007, 130, 991–1004. [Google Scholar]
- de Boer, J.; Andressoo, J.O.; de Wit, J.; Huijmans, J.; Beems, R.B.; van Steeg, H.; Weeda, G.; van der Horst, G.T.; van Leeuwen, W.; Themmen, A.P.; et al. Premature aging in mice deficient in DNA repair and transcription. Science 2002, 296, 1276–1279. [Google Scholar]
- Bielas, J.H.; Loeb, L.A. Mutator phenotype in cancer: Timing and perspectives. Environ. Mol. Mutagen 2005, 45, 206–213. [Google Scholar]
- Bardelli, A.; Cahill, D.P.; Lederer, G.; Speicher, M.R.; Kinzler, K.W.; Vogelstein, B.; Lengauer, C. Carcinogen-specific induction of genetic instability. Proc. Natl. Acad. Sci. USA 2001, 98, 5770–5775. [Google Scholar]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar]
- Ellis, L.M.; Hicklin, D.J. VEGF-targeted therapy: Mechanisms of anti-tumour activity. Nat. Rev. Cancer 2008, 8, 579–591. [Google Scholar]
- Yang, L.; Huang, J.; Ren, X.; Gorska, A.E.; Chytil, A.; Aakre, M.; Carbone, D.P.; Matrisian, L.M.; Richmond, A.; Lin, P.C.; et al. Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell 2008, 13, 23–35. [Google Scholar]
- Gabrilovich, D. Mechanisms and functional significance of tumour-induced dendritic-cell defects. Nat Rev. Immunol 2004, 4, 941–952. [Google Scholar]
- Marx, J. Cancer biology. All in the stroma: Cancer’s Cosa Nostra. Science 2008, 320, 38–41. [Google Scholar]
- Marx, J. Cancer immunology. Cancer’s bulwark against immune attack: MDS cells. Science 2008, 319, 154–156. [Google Scholar]
- Nonaka, K.; Saio, M.; Suwa, T.; Frey, A.B.; Umemura, N.; Imai, H.; Ouyang, G.F.; Osada, S.; Balazs, M.; Adany, R.; et al. Skewing the Th cell phenotype toward Th1 alters the maturation of tumor-infiltrating mononuclear phagocytes. J. Leukoc. Biol 2008, 84, 679–688. [Google Scholar]
- Umemura, N.; Saio, M.; Suwa, T.; Kitoh, Y.; Bai, J.; Nonaka, K.; Ouyang, G.-F.; Okada, M.; Balazs, M.; Adany, R.; et al. Tumor-infiltrating myeloid-derived suppressor cells are pleiotropic-inflamed monocytes/macrophages that bear M1- and M2-type characteristics. J. Leukoc. Biol 2008, 83, 1136–1144. [Google Scholar]
- Mantovani, A.; Sica, A.; Allavena, P.; Garlanda, C.; Locati, M. Tumor-associated macrophages and the related myeloid-derived suppressor cells as a paradigm of the diversity of macrophage activation. Hum. Immunol 2009, 70, 325–330. [Google Scholar]
- Sica, A.; Larghi, P.; Mancino, A.; Rubino, L.; Porta, C.; Totaro, M.G.; Rimoldi, M.; Biswas, S.K.; Allavena, P.; Mantovani, A. Macrophage polarization in tumour progression. Semin. Cancer Biol 2008, 18, 349–355. [Google Scholar]
- Mantovani, A.; Sica, A. Macrophages, innate immunity and cancer: Balance, tolerance, and diversity. Curr. Opin. Immunol 2010, 22, 231–237. [Google Scholar]
- Sinha, P.; Clements, V.K.; Miller, S.; Ostrand-Rosenberg, S. Tumor immunity: A balancing act between T cell activation, macrophage activation and tumor-induced immune suppression. Cancer Immunol. Immunother 2005, 54, 1137–1142. [Google Scholar]
- Sinha, P.; Clements, V.K.; Ostrand-Rosenberg, S. Reduction of myeloid-derived suppressor cells and induction of M1 macrophages facilitate the rejection of established metastatic disease. J. Immunol 2005, 174, 636–645. [Google Scholar]
- Sinha, P.; Clements, V.K.; Ostrand-Rosenberg, S. Interleukin-13-regulated M2 macrophages in combination with myeloid suppressor cells block immune surveillance against metastasis. Cancer Res 2005, 65, 11743–11751. [Google Scholar]
- Lewis, C.E.; Pollard, J.W. Distinct role of macrophages in different tumor microenvironments. Cancer Res 2006, 66, 605–612. [Google Scholar]
- Yang, L.; DeBusk, L.M.; Fukuda, K.; Fingleton, B.; Green-Jarvis, B.; Shyr, Y.; Matrisian, L.M.; Carbone, D.P.; Lin, P.C. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell 2004, 6, 409–421. [Google Scholar]
- Sinha, P.; Clements, V.K.; Bunt, S.K.; Albelda, S.M.; Ostrand-Rosenberg, S. Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J. Immunol 2007, 179, 977–983. [Google Scholar]
- Condeelis, J.; Pollard, J.W. Macrophages: Obligate partners for tumor cell migration, invasion, and metastasis. Cell 2006, 124, 263–266. [Google Scholar]
- Larsen, M.; Tazzyman, S.; Lund, E.L.; Junker, N.; Lewis, C.E.; Kristjansen, P.E.; Murdoch, C. Hypoxia-induced secretion of macrophage migration-inhibitory factor from MCF-7 breast cancer cells is regulated in a hypoxia-inducible factor-independent manner. Cancer Lett 2008, 265, 239–249. [Google Scholar]
- Bosco, M.C.; Puppo, M.; Blengio, F.; Fraone, T.; Cappello, P.; Giovarelli, M.; Varesio, L. Monocytes and dendritic cells in a hypoxic environment: Spotlights on chemotaxis and migration. Immunobiology 2008, 213, 733–749. [Google Scholar]
- Youn, J.I.; Nagaraj, S.; Collazo, M.; Gabrilovich, D.I. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J. Immunol 2008, 181, 5791–5802. [Google Scholar]
- Movahedi, K.; Guilliams, M.; van den Bossche, J.; van den Bergh, R.; Gysemans, C.; Beschin, A.; De Baetselier, P.; Van Ginderachter, J.A. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood 2008, 111, 4233–4244. [Google Scholar]
- Kusmartsev, S.; Gabrilovich, D.I. Inhibition of myeloid cell differentiation in cancer: The role of reactive oxygen species. J. Leukoc. Biol 2003, 74, 186–196. [Google Scholar]
- Sinha, P.; Clements, V.K.; Fulton, A.M.; Ostrand-Rosenberg, S. Prostaglandin E2 promotes tumor progression by inducing myeloid-derived suppressor cells. Cancer Res 2007, 67, 4507–4513. [Google Scholar]
- Bronte, V.; Serafini, P.; Mazzoni, A.; Segal, D.M.; Zanovello, P. L-arginine metabolism in myeloid cells controls T-lymphocyte functions. Trends Immunol 2003, 24, 302–306. [Google Scholar]
- Almand, B.; Clark, J.I.; Nikitina, E.; van Beynen, J.; English, N.R.; Knight, S.C.; Carbone, D.P.; Gabrilovich, D.I. Increased production of immature myeloid cells in cancer patients: A mechanism of immunosuppression in cancer. J. Immunol 2001, 166, 678–689. [Google Scholar]
- Baniyash, M. TCR zeta-chain downregulation: Curtailing an excessive inflammatory immune response. Nat. Rev. Immunol 2004, 4, 675–687. [Google Scholar]
- Bunt, S.K.; Sinha, P.; Clements, V.K.; Leips, J.; Ostrand-Rosenberg, S. Inflammation induces myeloid-derived suppressor cells that facilitate tumor progression. J. Immunol 2006, 176, 284–290. [Google Scholar]
- Bunt, S.K.; Yang, L.; Sinha, P.; Clements, V.K.; Leips, J.; Ostrand-Rosenberg, S. Reduced inflammation in the tumor microenvironment delays the accumulation of myeloid-derived suppressor cells and limits tumor progression. Cancer Res 2007, 67, 10019–10026. [Google Scholar]
- Simpson, A.J. The natural somatic mutation frequency and human carcinogenesis. Adv. Cancer Res 1997, 71, 209–240. [Google Scholar]
- Farber, E.; Cameron, R. The sequential analysis of cancer development. Adv. Cancer Res 1980, 31, 125–226. [Google Scholar]
- Bindra, R.S.; Glazer, P.M. Genetic instability and the tumor microenvironment: Towards the concept of microenvironment-induced mutagenesis. Mutat. Res 2005, 569, 75–85. [Google Scholar]
- Sneddon, J.B.; Werb, Z. Location, location, location: The cancer stem cell niche. Cell Stem Cell 2007, 1, 607–611. [Google Scholar]
- Calabrese, C.; Poppleton, H.; Kocak, M.; Hogg, T.L.; Fuller, C.; Hamner, B.; Oh, E.Y.; Gaber, M.W.; Finklestein, D.; Allen, M.; et al. A perivascular niche for brain tumor stem cells. Cancer Cell 2007, 11, 69–82. [Google Scholar]
- Veeravagu, A.; Bababeygy, S.R.; Kalani, M.Y.; Hou, L.C.; Tse, V. The cancer stem cell-vascular niche complex in brain tumor formation. Stem Cells Dev 2008, 17, 859–867. [Google Scholar]
- Gilbertson, R.J.; Gutmann, D.H. Tumorigenesis in the brain: Location, location, location. Cancer Res 2007, 67, 5579–5582. [Google Scholar]
- Gilbertson, R.J.; Rich, J.N. Making a tumour’s bed: Glioblastoma stem cells and the vascular niche. Nat. Rev. Cancer 2007, 7, 733–736. [Google Scholar]
- Croker, A.K.; Goodale, D.; Chu, J.; Postenka, C.; Hedley, B.D.; Hess, D.A.; Allan, A.L. High aldehyde dehydrogenase and expression of cancer stem cell markers selects for breast cancer cells with enhanced malignant and metastatic ability. J. Cell. Mol. Med 2009, 13, 2236–2252. [Google Scholar]
- Croker, A.K.; Allan, A.L. Cancer stem cells: Implications for the progression and treatment of metastatic disease. J. Cell. Mol. Med 2008, 12, 374–390. [Google Scholar]
- Campbell, I.; Qiu, W.; Haviv, I. Genetic changes in tumour microenvironments. J. Pathol 2011, 223, 450–458. [Google Scholar]
- Korkaya, H.; Liu, S.; Wicha, M.S. Breast cancer stem cells, cytokine networks, and the tumor microenvironment. J. Clin. Invest 2011, 121, 3804–3809. [Google Scholar]
- Weinberg, R.A. Coevolution in the tumor microenvironment. Nat. Genet 2008, 40, 494–495. [Google Scholar]
- Whiteside, T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar]
- Ben-Baruch, A. Host microenvironment in breast cancer development: Inflammatory cells, cytokines and chemokines in breast cancer progression: Reciprocal tumor-microenvironment interactions. Breast Cancer Res 2003, 5, 31–36. [Google Scholar]
- Zipin-Roitman, A.; Meshel, T.; Sagi-Assif, O.; Shalmon, B.; Avivi, C.; Pfeffer, R.M.; Witz, I.P.; Ben-Baruch, A. CXCL10 promotes invasion-related properties in human colorectal carcinoma cells. Cancer Res 2007, 67, 3396–3405. [Google Scholar]
- Malumbres, M.; Barbacid, M. To cycle or not to cycle: A critical decision in cancer. Nat. Rev. Cancer 2001, 1, 222–231. [Google Scholar]
- Hersey, P.; Zhang, X.D. How melanoma cells evade trail-induced apoptosis. Nat. Rev. Cancer 2001, 1, 142–150. [Google Scholar]
- Anderson, A.R.; Hassanein, M.; Branch, K.M.; Lu, J.; Lobdell, N.A.; Maier, J.; Basanta, D.; Weidow, B.; Narasanna, A.; Arteaga, C.L.; et al. Microenvironmental independence associated with tumor progression. Cancer Res 2009, 69, 8797–8806. [Google Scholar]
- Anderson, A.R.; Rejniak, K.A.; Gerlee, P.; Quaranta, V. Microenvironment driven invasion: A multiscale multimodel investigation. J. Math. Biol 2009, 58, 579–624. [Google Scholar]
- Anderson, A.R.; Weaver, A.M.; Cummings, P.T.; Quaranta, V. Tumor morphology and phenotypic evolution driven by selective pressure from the microenvironment. Cell 2006, 127, 905–915. [Google Scholar]
- Jimenez, J.J.; Blanes, A.; Diaz-Cano, S.J. Microsatellite instability in colon cancer. N. Engl. J. Med 2003, 349, 1774–1776. [Google Scholar]
- Diaz-Cano, S.J. Molecular mechanisms in melanoma. N. Engl. J. Med 2006, 355, 1395–1396. [Google Scholar]
- Pozo, L.; Husein, E.; Blanes, A.; Diaz-Cano, S.J. The correlation of regression with the grade of dysplasia (atypia) in melanocytic naevi. Histopathology 2008, 52, 387–389. [Google Scholar]
- Husemann, Y.; Geigl, J.B.; Schubert, F.; Musiani, P.; Meyer, M.; Burghart, E.; Forni, G.; Eils, R.; Fehm, T.; Riethmüller, G.; et al. Systemic spread is an early step in breast cancer. Cancer Cell 2008, 13, 58–68. [Google Scholar]
- Arif, S.; Patel, J.; Blanes, A.; Diaz-Cano, S.J. Cytoarchitectural and kinetic features in the histological evaluation of follicular thyroid neoplasms. Histopathology 2007, 50, 750–763. [Google Scholar]
- Boone, C.W.; Kelloff, G.J.; Freedman, L.S. Intraepithelial and postinvasive neoplasia as a stochastic continuum of clonal evolution, and its relationship to mechanisms of chemopreventive drug action. J. Cell. Biochem. Suppl 1993, 17G, 14–25. [Google Scholar]
- Namba, R.; Maglione, J.E.; Davis, R.R.; Baron, C.A.; Liu, S.; Carmack, C.E.; Young, L.J.T.; Borowsky, A.D.; Cardiff, R.D.; Gregg, J.P. Heterogeneity of mammary lesions represent molecular differences. BMC Cancer 2006, 6. [Google Scholar] [CrossRef]
- Steeg, P.S. Tumor metastasis: Mechanistic insights and clinical challenges. Nat. Med 2006, 12, 895–904. [Google Scholar]
- Steeg, P.S. Cancer: Micromanagement of metastasis. Nature 2007, 449, 671–673. [Google Scholar]
- Talmadge, J.E. Clonal selection of metastasis within the life history of a tumor. Cancer Res 2007, 67, 11471–11475. [Google Scholar]
- Christofori, G. New signals from the invasive front. Nature 2006, 441, 444–450. [Google Scholar]
- Gupta, G.P.; Massague, J. Cancer metastasis: Building a framework. Cell 2006, 127, 679–695. [Google Scholar]
- Nguyen, D.X. Tracing the origins of metastasis. J Pathol 2011, 223, 195–204. [Google Scholar]
- Nguyen, D.X.; Massague, J. Genetic determinants of cancer metastasis. Nat. Rev. Genet 2007, 8, 341–352. [Google Scholar]
- Li, F.; Tiede, B.; Massague, J.; Kang, Y. Beyond tumorigenesis: Cancer stem cells in metastasis. Cell Res 2007, 17, 3–14. [Google Scholar]
- Wicha, M.S. Cancer stem cells and metastasis: Lethal seeds. Clin. Cancer Res 2006, 12, 5606–5607. [Google Scholar]
- Kaplan, R.N.; Psaila, B.; Lyden, D. Bone marrow cells in the ‘pre-metastatic niche’: Within bone and beyond. Cancer Metastasis Rev 2006, 25, 521–529. [Google Scholar]
- Kaplan, R.N.; Rafii, S.; Lyden, D. Preparing the “soil”: The premetastatic niche. Cancer Res 2006, 66, 11089–11093. [Google Scholar]
- Kaplan, R.N.; Riba, R.D.; Zacharoulis, S.; Bramley, A.H.; Vincent, L.; Costa, C.; MacDonald, D.D.; Jin, D.K.; Shido, K.; Kerns, S.A.; et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 2005, 438, 820–827. [Google Scholar]
- Bissig, H.; Richter, J.; Desper, R.; Meier, V.; Schraml, P.; Schäffer, A.A.; Sauter, G.; Mihatsch, M.J.; Moch, H. Evaluation of the clonal relationship between primary and metastatic renal cell carcinoma by comparative genomic hybridization. Am. J. Pathol 1999, 155, 267–274. [Google Scholar]
- Cheng, L.; Bostwick, D.G.; Li, G.; Wang, Q.; Hu, N.; Vortmeyer, A.O.; Zhuang, Z. Allelic imbalance in the clonal evolution of prostate carcinoma. Cancer 1999, 85, 2017–2022. [Google Scholar]
- Kuukasjarvi, T.; Karhu, R.; Tanner, M.; Kähkönen, M.; Schäffer, A.; Nupponen, N.; Pennanen, S.; Kallioniemi, A.; Kallioniemi, O.P.; Isola, J. Genetic heterogeneity and clonal evolution underlying development of asynchronous metastasis in human breast cancer. Cancer Res 1997, 57, 1597–1604. [Google Scholar]
- Ramaswamy, S.; Ross, K.N.; Lander, E.S.; Golub, T.R. A molecular signature of metastasis in primary solid tumors. Nat. Genet 2003, 33, 49–54. [Google Scholar]
- Weigelt, B.; Glas, A.M.; Wessels, L.F.; Witteveen, A.T.; Peterse, J.L.; van’t Veer, L.J. Gene expression profiles of primary breast tumors maintained in distant metastases. Proc. Natl. Acad. Sci. USA 2003, 100, 15901–15905. [Google Scholar]
- Weigelt, B.; Hu, Z.; He, X.; Livasy, C.; Carey, L.A.; Ewend, M.G.; Glas, A.M.; Perou, C.M.; van’t Veer, L.J. Molecular portraits and 70-gene prognosis signature are preserved throughout the metastatic process of breast cancer. Cancer Res 2005, 65, 9155–9158. [Google Scholar]
- Liu, W.; Laitinen, S.; Khan, S.; Vihinen, M.; Kowalski, J.; Yu, G.; Chen, L.; Ewing, C.M.; Eisenberger, M.A. Copy number analysis indicates monoclonal origin of lethal metastatic prostate cancer. Nat. Med 2009, 15, 559–565. [Google Scholar]
- Ruijter, E.T.; van de Kaa, C.A.; Schalken, J.A.; Debruyne, F.M.; Ruiter, D.J. Histological grade heterogeneity in multifocal prostate cancer. Biological and clinical implications. J. Pathol 1996, 180, 295–299. [Google Scholar]
- Alvarado, C.; Beitel, L.K.; Sircar, K.; Aprikian, A.; Trifiro, M.; Gottlieb, B. Somatic mosaicism and cancer: A micro-genetic examination into the role of the androgen receptor gene in prostate cancer. Cancer Res 2005, 65, 8514–8518. [Google Scholar]
- Shah, S.P.; Morin, R.D.; Khattra, J.; Prentice, L.; Pugh, T.; Burleigh, A.; Delaney, A.; Gelmon, K.; Guliany, R.; Senz, J.; et al. Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution. Nature 2009, 461, 809–813. [Google Scholar]
- Holmgren, L.; O’Reilly, M.S.; Folkman, J. Dormancy of micrometastases: Balanced proliferation and apoptosis in the presence of angiogenesis suppression. Nat. Med 1995, 1, 149–153. [Google Scholar]
- Braun, S.; Pantel, K.; Muller, P.; Janni, W.; Hepp, F.; Kentenich, C.R.; Gastroph, S.; Wischnik, A.; Dimpfl, T.; Kindermann, G.; et al. Cytokeratin-positive cells in the bone marrow and survival of patients with stage I, II, or III breast cancer. N. Engl. J. Med 2000, 342, 525–533. [Google Scholar]
- Diaz-Cano, S.J. Bone marrow metastases in breast cancer. N. Engl. J. Med 2000, 343, 577. [Google Scholar]
- Passlick, B.; Izbicki, J.R.; Kubuschok, B.; Nathrath, W.; Thetter, O.; Pichlmeier, U.; Schweiberer, L.; Riethmüller, G.; Pantel, K. Immunohistochemical assessment of individual tumor cells in lymph nodes of patients with non-small-cell lung cancer. J. Clin. Oncol 1994, 12, 1827–1832. [Google Scholar]
- Schlimok, G.; Funke, I.; Holzmann, B.; Göttlinger, G.; Schmidt, G.; Häuser, H.; Swierkot, S.; Warnecke, H.H.; Schneider, B.; Koprowski, H. Micrometastatic cancer cells in bone marrow: In vitro detection with anti-cytokeratin and in vivo labeling with anti-17-1A monoclonal antibodies. Proc. Natl. Acad. Sci. USA 1987, 84, 8672–8676. [Google Scholar]
- Smith, B.L. Approaches to breast-cancer staging. N. Engl. J. Med 2000, 342, 580–581. [Google Scholar]
- Klein, C.A.; Schmidt-Kittler, O.; Schardt, J.A.; Pantel, K.; Speicher, M.R.; Riethmuller, G. Comparative genomic hybridization, loss of heterozygosity, and DNA sequence analysis of single cells. Proc. Natl. Acad. Sci. USA 1999, 96, 4494–4499. [Google Scholar]
- Klein, C.A.; Blankenstein, T.J.; Schmidt-Kittler, O.; Petronio, M.; Polzer, B.; Stoecklein, N.H.; Riethmüller, G. Genetic heterogeneity of single disseminated tumour cells in minimal residual cancer. Lancet 2002, 360, 683–689. [Google Scholar]
- Schmidt-Kittler, O.; Ragg, T.; Daskalakis, A.; Granzow, M.; Ahr, A.; Blankenstein, T.J.F.; Kaufmann, M.; Diebold, J.; Arnholdt, H.; Müller, P.; et al. From latent disseminated cells to overt metastasis: Genetic analysis of systemic breast cancer progression. Proc. Natl. Acad. Sci. USA 2003, 100, 7737–7742. [Google Scholar]
- Bedenne, L.; Michel, P.; Bouche, O.; Milan, C.; Mariette, C.; Conroy, T.; Pezet, D.; Roullet, B.; Seitz, J.-F.; Herr, J.-P.; et al. Chemoradiation followed by surgery compared with chemoradiation alone in squamous cancer of the esophagus: FFCD 9102. J. Clin. Oncol 2007, 25, 1160–1168. [Google Scholar]
- Stoecklein, N.H.; Hosch, S.B.; Bezler, M.; Stern, F.; Hartmann, C.H.; Vay, C.; Siegmund, A.; Scheunemann, P.; Schurr, P.; Knoefel, W.T.; et al. Direct genetic analysis of single disseminated cancer cells for prediction of outcome and therapy selection in esophageal cancer. Cancer Cell 2008, 13, 441–453. [Google Scholar]
- Podsypanina, K.; Du, Y.C.; Jechlinger, M.; Beverly, L.J.; Hambardzumyan, D.; Varmus, H. Seeding and propagation of untransformed mouse mammary cells in the lung. Science 2008, 321, 1841–1844. [Google Scholar]
- Gadi, V.K.; Nelson, J.L. Fetal microchimerism in women with breast cancer. Cancer Res 2007, 67, 9035–9038. [Google Scholar]
- Solakoglu, O.; Maierhofer, C.; Lahr, G.; Breit, E.; Scheunemann, P.; Heumos, I.; Pichlmeier, U.; Schlimok, G.; Oberneder, R.; Köllermann, M.W.; et al. Heterogeneous proliferative potential of occult metastatic cells in bone marrow of patients with solid epithelial tumors. Proc. Natl. Acad. Sci. USA 2002, 99, 2246–2251. [Google Scholar]
- Gangnus, R.; Langer, S.; Breit, E.; Pantel, K.; Speicher, M.R. Genomic profiling of viable and proliferative micrometastatic cells from early-stage breast cancer patients. Clin Cancer Res 2004, 10, 3457–3464. [Google Scholar]
- Abbruzzese, J.L.; Abbruzzese, M.C.; Hess, K.R.; Raber, M.N.; Lenzi, R.; Frost, P. Unknown primary carcinoma: Natural history and prognostic factors in 657 consecutive patients. J. Clin. Oncol 1994, 12, 1272–1280. [Google Scholar]
- Klein, C.A.; Holzel, D. Systemic cancer progression and tumor dormancy: Mathematical models meet single cell genomics. Cell Cycle 2006, 5, 1788–1798. [Google Scholar]
- Sieber, O.M.; Tomlinson, S.R.; Tomlinson, I.P. Tissue, cell and stage specificity of (epi)mutations in cancers. Nat. Rev. Cancer 2005, 5, 649–655. [Google Scholar]
- Fialkow, P.J. Clonal origin of human tumors. Annu. Rev. Med 1979, 30, 135–143. [Google Scholar]
- Weinberg, R.A. The Biology of Cancer, 1st ed; Garland Science: New York, NY, USA, 2007; p. 796. [Google Scholar]
- Novelli, M.R.; Williamson, J.A.; Tomlinson, I.P.; Elia, G.; Hodgson, S.V.; Talbot, I.C.; Bodmer, W.F.; Wright, N.A. Polyclonal origin of colonic adenomas in an XO/XY patient with FAP. Science 1996, 272, 1187–1190. [Google Scholar]
- Leroi, A.M.; Koufopanou, V.; Burt, A. Cancer selection. Nat. Rev. Cancer 2003, 3, 226–231. [Google Scholar]
- Diaz-Cano, S.J. Kinetic topographical heterogeneity in follicular thyroid neoplasms and growth patterns. Histopathology 2007, 51, 416–418. [Google Scholar]
- Gonzalez-Garcia, I.; Sole, R.V.; Costa, J. Metapopulation dynamics and spatial heterogeneity in cancer. Proc. Natl. Acad. Sci. USA 2002, 99, 13085–13089. [Google Scholar]
- Gatenby, R.A.; Silva, A.S.; Gillies, R.J.; Frieden, B.R. Adaptive therapy. Cancer Res 2009, 69, 4894–4903. [Google Scholar]
- Gatenby, R.A.; Vincent, T.L. An evolutionary model of carcinogenesis. Cancer Res 2003, 63, 6212–6220. [Google Scholar]
- Chan, D.A.; Giaccia, A.J. Hypoxia, gene expression, and metastasis. Cancer Metastasis Rev 2007, 26, 333–339. [Google Scholar]
- Evans, S.M.; Hahn, S.M.; Magarelli, D.P.; Koch, C.J. Hypoxic heterogeneity in human tumors: EF5 binding, vasculature, necrosis, and proliferation. Am. J. Clin. Oncol 2001, 24, 467–472. [Google Scholar]
- Martinive, P.; Defresne, F.; Quaghebeur, E.; Daneau, G.; Crokart, N.; Grégoire, V.; Gallez, B.; Dessy, C.; Feron, O. Impact of cyclic hypoxia on HIF-1alpha regulation in endothelial cells--new insights for anti-tumor treatments. FEBS J 2009, 276, 509–518. [Google Scholar]
- Kim, Y.; Lin, Q.; Glazer, P.M.; Yun, Z. Hypoxic tumor microenvironment and cancer cell differentiation. Curr. Mol. Med 2009, 9, 425–434. [Google Scholar]
- Harris, A.L. Hypoxia--a key regulatory factor in tumour growth. Nat. Rev. Cancer 2002, 2, 38–47. [Google Scholar]
- Subarsky, P.; Hill, R.P. The hypoxic tumour microenvironment and metastatic progression. Clin. Exp. Metastasis 2003, 20, 237–250. [Google Scholar]
- Pouyssegur, J.; Dayan, F.; Mazure, N.M. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature 2006, 441, 437–443. [Google Scholar]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar]
- Struckmann, K.; Mertz, K.; Steu, S.; Storz, M.; Staller, P.; Krek, W.; Schraml, P.; Moch, H. pVHL co-ordinately regulates CXCR4/CXCL12 and MMP2/MMP9 expression in human clear-cell renal cell carcinoma. J Pathol 2008, 214, 464–471. [Google Scholar]
- Keith, B.; Simon, M.C. Hypoxia-inducible factors, stem cells, and cancer. Cell 2007, 129, 465–472. [Google Scholar]
- Bos, R.; Zhong, H.; Hanrahan, C.F.; Mommers, E.C.; Semenza, G.L.; Pinedo, H.M.; Abeloff, M.D.; Simons, J.W.; van Diest, P.J.; van der Wall, E. Levels of hypoxia-inducible factor-1 alpha during breast carcinogenesis. J. Natl. Cancer Inst 2001, 93, 309–314. [Google Scholar]
- Semenza, G.L. Angiogenesis in ischemic and neoplastic disorders. Annu. Rev. Med 2003, 54, 17–28. [Google Scholar]
- Serrati, S.; Margheri, F.; Fibbi, G.; Di Cara, G.; Minafra, L.; Pucci-Minafra, I.; Liotta, F.; Annunziato, F.; Pucci, M.; Del Rosso, M. Endothelial cells and normal breast epithelial cells enhance invasion of breast carcinoma cells by CXCR-4-dependent up-regulation of urokinase-type plasminogen activator receptor (uPAR, CD87) expression. J. Pathol 2008, 214, 545–554. [Google Scholar]
- Jankowski, K.; Kucia, M.; Wysoczynski, M.; Reca, R.; Zhao, D.; Trzyna, E.; Trent, J.; Peiper, S.; Zembala, M.; Ratajczak, J.; et al. Both hepatocyte growth factor (HGF) and stromal-derived factor-1 regulate the metastatic behavior of human rhabdomyosarcoma cells, but only HGF enhances their resistance to radiochemotherapy. Cancer Res 2003, 63, 7926–7935. [Google Scholar]
- Libura, J.; Drukala, J.; Majka, M.; Tomescu, O.; Navenot, J.M.; Kucia, M.; Marquez, L.; Peiper, S.C.; Barr, F.G.; Janowska-Wieczorek, A.; et al. CXCR4-SDF-1 signaling is active in rhabdomyosarcoma cells and regulates locomotion, chemotaxis, and adhesion. Blood 2002, 100, 2597–2606. [Google Scholar]
- Maeda, S.; Shinchi, H.; Kurahara, H.; Mataki, Y.; Maemura, K.; Sato, M.; Natsugoe, S.; Aikou, T.; Takao, S. CD133 expression is correlated with lymph node metastasis and vascular endothelial growth factor-C expression in pancreatic cancer. Br. J. Cancer 2008, 98, 1389–1397. [Google Scholar]
- Brabletz, T.; Jung, A.; Spaderna, S.; Hlubek, F.; Kirchner, T. Opinion: Migrating cancer stem cells - an integrated concept of malignant tumour progression. Nat. Rev. Cancer 2005, 5, 744–749. [Google Scholar]
- Kang, Y.; Massague, J. Epithelial-mesenchymal transitions: Twist in development and metastasis. Cell 2004, 118, 277–279. [Google Scholar]
- Diaz-Cano, S.J. Re: Pomerance et al. High-level expression, activation, and subcellular localization of p38-MAP kinase in thyroid neoplasms. J. Pathol. 2006, 209, 298–306. J. Pathol 2006, 210, 133–134. [Google Scholar]
- Murphy, P.M. Chemokines and the molecular basis of cancer metastasis. N. Engl. J. Med 2001, 345, 833–835. [Google Scholar]
- Stainier, D. No stem cell is an islet (yet). N. Engl. J. Med 2006, 354, 521–523. [Google Scholar]
- Zlotnik, A. New insights on the role of CXCR4 in cancer metastasis. J. Pathol 2008, 215, 211–213. [Google Scholar]
- DeNardo, D.G.; Johansson, M.; Coussens, L.M. Immune cells as mediators of solid tumor metastasis. Cancer Metastasis Rev 2008, 27, 11–18. [Google Scholar]
- Hiratsuka, S.; Watanabe, A.; Sakurai, Y.; Akashi-Takamura, S.; Ishibashi, S.; Miyake, K.; Shibuya, M.; Akira, S.; Aburatani, H.; Maru, Y. The S100A8-serum amyloid A3-TLR4 paracrine cascade establishes a pre-metastatic phase. Nat. Cell Biol 2008, 10, 1349–1355. [Google Scholar]
- Bornstein, S.R.; Hornsby, P.J. What can we learn from gene expression profiling for adrenal tumor management? J. Clin. Endocrinol. Metab 2005, 90, 1900–1902. [Google Scholar]
- Garber, M.E.; Troyanskaya, O.G.; Schluens, K.; Petersen, S.; Thaesler, Z.; Pacyna-Gengelbach, M.; van de Rijn, M.; Rosen, G.D.; Perou, C.M.; Whyte, R.I.; et al. Diversity of gene expression in adenocarcinoma of the lung. Proc. Natl. Acad. Sci. USA 2001, 98, 13784–13789. [Google Scholar]
- Steeg, P.S. New insights into the tumor metastatic process revealed by gene expression profiling. Am. J. Pathol 2005, 166, 1291–1294. [Google Scholar]
- van de Rijn, M.; Rubin, B.P. Gene expression studies on soft tissue tumors. Am. J. Pathol 2002, 161, 1531–1534. [Google Scholar]
- Van’T Veer, L.J.; Paik, S.; Hayes, D.F. Gene expression profiling of breast cancer: A new tumor marker. J. Clin. Oncol 2005, 23, 1631–1635. [Google Scholar]
- Vaupel, P.; Harrison, L. Tumor hypoxia: Causative factors, compensatory mechanisms, and cellular response. Oncologist 2004, 9, 4–9. [Google Scholar]
- Pennacchietti, S.; Michieli, P.; Galluzzo, M.; Mazzone, M.; Giordano, S.; Comoglio, P.M. Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell 2003, 3, 347–361. [Google Scholar]
- Kulshreshtha, R.; Davuluri, R.V.; Calin, G.A.; Ivan, M. A microRNA component of the hypoxic response. Cell Death Differ 2008, 15, 667–671. [Google Scholar]
- Ouatas, T.; Salerno, M.; Palmieri, D.; Steeg, P.S. Basic and translational advances in cancer metastasis: Nm23. J. Bioenerg. Biomembr 2003, 35, 73–79. [Google Scholar]
- Salerno, M.; Ouatas, T.; Palmieri, D.; Steeg, P.S. Inhibition of signal transduction by the nm23 metastasis suppressor: Possible mechanisms. Clin. Exp. Metastasis 2003, 20, 3–10. [Google Scholar]
- Steeg, P.S. Metastasis suppressors alter the signal transduction of cancer cells. Nat. Rev. Cancer 2003, 3, 55–63. [Google Scholar]
- Steeg, P.S.; Ouatas, T.; Halverson, D.; Palmieri, D.; Salerno, M. Metastasis suppressor genes: Basic biology and potential clinical use. Clin. Breast Cancer 2003, 4, 51–62. [Google Scholar]
- Wulfkuhle, J.D.; Paweletz, C.P.; Steeg, P.S.; Petricoin, E.F., 3rd; Liotta, L. Proteomic approaches to the diagnosis, treatment, and monitoring of cancer. Adv. Exp. Med. Biol. 2003, 532, 59–68. [Google Scholar]
- Diaz-Cano, S.J. Paratumoral gene expression profiles: Promising markers of malignancy in melanocytic lesions. Br. J. Dermatol 2011, 165, 702–703. [Google Scholar]
- Wachsman, W.; Morhenn, V.; Palmer, T.; Walls, L.; Hata, T.; Zalla, J.; Scheinberg, R.; Sofen, H.; Mraz, S.; Gross, K.; et al. Noninvasive genomic detection of melanoma. Br. J. Dermatol 2011, 164, 797–806. [Google Scholar]
- Peinado, H.; Lavotshkin, S.; Lyden, D. The secreted factors responsible for pre-metastatic niche formation: Old sayings and new thoughts. Semin. Cancer Biol 2011, 21, 139–146. [Google Scholar]
- György, B.; Szabó, T.G.; Pásztói, M.; Pál, Z.; Misják, P.; Aradi, B.; László, V.; Pállinger, E.; Pap, E.; Kittel, A.; et al. Membrane vesicles, current state-of-the-art: Emerging role of extracellular vesicles. Cell. Mol. Life Sci 2011, 68, 2667–2688. [Google Scholar]
- Valadi, H.; Ekstrom, K.; Bossios, A.; Sjostrand, M.; Lee, J.J.; Lotvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol 2007, 9, 654–659. [Google Scholar]
- Mathivanan, S.; Ji, H.; Simpson, R.J. Exosomes: Extracellular organelles important in intercellular communication. J. Proteomics 2010, 73, 1907–1920. [Google Scholar]
- Hendrix, A.; Westbroek, W.; Bracke, M.; De Wever, O. An ex(o)citing machinery for invasive tumor growth. Cancer Res 2010, 70, 9533–9537. [Google Scholar]
- Whiteside, T.L.; Mandapathil, M.; Szczepanski, M.; Szajnik, M. Mechanisms of tumor escape from the immune system: Adenosine-producing Treg, exosomes and tumor-associated TLRs. Bull. Cancer 2011, 98, E25–E31. [Google Scholar]
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev Genet 2011, 12, 861–874. [Google Scholar]
- Corcoran, C.; Friel, A.M.; Duffy, M.J.; Crown, J.; O’Driscoll, L. Intracellular and extracellular microRNAs in breast cancer. Clin. Chem 2011, 57, 18–32. [Google Scholar]
- Krutovskikh, V.A.; Herceg, Z. Oncogenic microRNAs (OncomiRs) as a new class of cancer biomarkers. Bioessays 2010, 32, 894–904. [Google Scholar]
- Curtin, J.A.; Fridlyand, J.; Kageshita, T.; Patel, H.N.; Busam, K.J.; Kutzner, H.; Cho, K.H.; Aiba, S.; Bröcker, E.B.; LeBoit, P.E.; et al. Distinct sets of genetic alterations in melanoma. N. Engl. J. Med 2005, 353, 2135–2147. [Google Scholar]
- Lee, J.H.; Choi, J.W.; Kim, Y.S. Frequencies of BRAF and NRAS mutations are different in histological types and sites of origin of cutaneous melanoma: A meta-analysis. Br. J. Dermatol 2011, 164, 776–784. [Google Scholar]
- Guo, Y.; Feng, Y.; Trivedi, N.S.; Huang, S. Medusa structure of the gene regulatory network: Dominance of transcription factors in cancer subtype classification. Exp. Biol. Med. (Maywood) 2011, 236, 628–636. [Google Scholar]
- Guo, Z.; Wu, F.; Asplund, A.; Hu, X.; Mazurenko, N.; Kisseljov, F.; Pontén, J.; Wilander, E. Analysis of intratumoral heterogeneity of chromosome 3p deletions and genetic evidence of polyclonal origin of cervical squamous carcinoma. Mod. Pathol 2001, 14, 54–61. [Google Scholar]
- Esteller, M. Epigenetics in cancer. N. Engl. J. Med 2008, 358, 1148–1159. [Google Scholar]
- Feinberg, A.P. The epigenetics of cancer etiology. Semin. Cancer Biol 2004, 14, 427–432. [Google Scholar]
- Eden, A.; Gaudet, F.; Waghmare, A.; Jaenisch, R. Chromosomal instability and tumors promoted by DNA hypomethylation. Science 2003, 300, 455. [Google Scholar]
- Gaudet, F.; Hodgson, J.G.; Eden, A.; Jackson-Grusby, L.; Dausman, J.; Gray, J.W.; Leonhardt, H.; Jaenisch, R. Induction of tumors in mice by genomic hypomethylation. Science 2003, 300, 489–492. [Google Scholar]
- Karpf, A.R.; Matsui, S. Genetic disruption of cytosine DNA methyltransferase enzymes induces chromosomal instability in human cancer cells. Cancer Res 2005, 65, 8635–8639. [Google Scholar]
- Bestor, T.H. Transposons reanimated in mice. Cell 2005, 122, 322–325. [Google Scholar]
- Esteller, M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat. Rev. Genet 2007, 8, 286–298. [Google Scholar]
- Esteller, M.; Herman, J.G. Generating mutations but providing chemosensitivity: The role of O6-methylguanine DNA methyltransferase in human cancer. Oncogene 2004, 23, 1–8. [Google Scholar]
- Nieto, M.; Samper, E.; Fraga, M.F.; Gonzalez de Buitrago, G.; Esteller, M.; Serrano, M. The absence of p53 is critical for the induction of apoptosis by 5-aza-2′-deoxycytidine. Oncogene 2004, 23, 735–743. [Google Scholar]
- Agrelo, R.; Cheng, W.H.; Setien, F.; Ropero, S.; Espada, J.; Fraga, M.F.; Herranz, M.; M.F.; Sanchez-Cespedes, M.; Artiga, M.J.; et al. Epigenetic inactivation of the premature aging Werner syndrome gene in human cancer. Proc. Natl. Acad. Sci. USA 2006, 103, 8822–8827. [Google Scholar]
- Ballestar, E.; Paz, M.F.; Valle, L.; Wei, S.; Fraga, M.F.; Espada, J.; Cigudosa, J.C.; Huang, T.H.; Esteller, M. Methyl-CpG binding proteins identify novel sites of epigenetic inactivation in human cancer. EMBO J 2003, 22, 6335–6345. [Google Scholar]
- Fraga, M.F.; Ballestar, E.; Villar-Garea, A.; Boix-Chornet, M.; Espada, J.; Schotta, G.; Bonaldi, T.; Haydon, C.; Ropero, S.; Petrie, K.; et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet 2005, 37, 391–400. [Google Scholar]
- He, L.; Hannon, G.J. MicroRNAs: Small RNAs with a big role in gene regulation. Nat. Rev. Genet 2004, 5, 522–531. [Google Scholar]
- Lujambio, A.; Ropero, S.; Ballestar, E.; Lujambio, A.; Ropero, S.; Ballestar, E.; Fraga, M.F.; Cerrato, C.; Setién, F.; Casado, S.; et al. Genetic unmasking of an epigenetically silenced microRNA in human cancer cells. Cancer Res 2007, 67, 1424–1429. [Google Scholar]
- Nakamura, T.; Kuwai, T.; Kitadai, Y.; Sasaki, T.; Fan, D.; Coombes, K.R.; Kim, S.J.; Fidler, I.J. Zonal heterogeneity for gene expression in human pancreatic carcinoma. Cancer Res 2007, 67, 7597–7604. [Google Scholar]
- Shipitsin, M.; Campbell, L.L.; Argani, P.; Weremowicz, S.; Bloushtain-Qimron, N.; Yao, J.; Nikolskaya, T.; Serebryiskaya, T.; Beroukhim, R.; Hu, M.; et al. Molecular definition of breast tumor heterogeneity. Cancer Cell 2007, 11, 259–273. [Google Scholar]
- Tomlinson, I.P.; Lambros, M.B.; Roylance, R.R. Loss of heterozygosity analysis: Practically and conceptually flawed? Genes Chromosomes Cancer 2002, 34, 349–353. [Google Scholar]
- Califano, J.; van der Riet, P.; Westra, W.; Nawroz, H.; Clayman, G.; Piantadosi, S.; Corio, R.; Lee, D.; Greenberg, B.; Koch, W.; et al. Genetic progression model for head and neck cancer: Implications for field cancerization. Cancer Res 1996, 56, 2488–2492. [Google Scholar]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.-M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar]
- Wood, L.D.; Parsons, D.W.; Jones, S.; Lin, J.; Sjöblom, T.; Leary, R.J.; Shen, D.; Boca, S.M.; Barber, T.; Ptak, J.; et al. The genomic landscapes of human breast and colorectal cancers. Science 2007, 318, 1108–1113. [Google Scholar]
- Siegmund, K.D.; Marjoram, P.; Woo, Y.J.; Tavare, S.; Shibata, D. Inferring clonal expansion and cancer stem cell dynamics from DNA methylation patterns in colorectal cancers. Proc. Natl. Acad. Sci. USA 2009, 106, 4828–4833. [Google Scholar]
- Diaz-Cano, S. PCR-based alternative for diagnosis of immunoglobulin heavy chain gene rearrangement: Principles, practice, and polemics. Diagn. Mol. Pathol 1996, 5, 3–9. [Google Scholar]
- Brady, S.P.; Magro, C.M.; Diaz-Cano, S.J.; Wolfe, H.J. Analysis of clonality of atypical cutaneous lymphoid infiltrates associated with drug therapy by PCR/DGGE. Hum. Pathol 1999, 30, 130–136. [Google Scholar]
- Campbell, P.J.; Pleasance, E.D.; Stephens, P.J.; Dicks, E.; Rance, R.; Goodhead, I.; Follows, G.A.; Green, A.R.; Futreal, P.A.; Stratton, M.R. Subclonal phylogenetic structures in cancer revealed by ultra-deep sequencing. Proc. Natl. Acad. Sci. USA 2008, 105, 13081–13086. [Google Scholar]
- Mullighan, C.G.; Phillips, L.A.; Su, X.; Ma, J.; Miller, C.B.; Shurtleff, S.A.; Downing, J.R. Genomic analysis of the clonal origins of relapsed acute lymphoblastic leukemia. Science 2008, 322, 1377–1380. [Google Scholar]
- Panzer-Grumayer, E.R.; Cazzaniga, G.; van der Velden, V.H.; del Giudice, L.; Peham, M.; Mann, G.; Eckert, C.; Schrauder, A.; Germano, G.; Harbott, J.; et al. Immunogenotype changes prevail in relapses of young children with TEL-AML1-positive acute lymphoblastic leukemia and derive mainly from clonal selection. Clin. Cancer Res 2005, 11, 7720–7727. [Google Scholar]
- Teixeira, M.R.; Pandis, N.; Bardi, G.; Andersen, J.A.; Heim, S. Karyotypic comparisons of multiple tumorous and macroscopically normal surrounding tissue samples from patients with breast cancer. Cancer Res 1996, 56, 855–859. [Google Scholar]
- Teixeira, M.R.; Pandis, N.; Bardi, G.; Andersen, J.A.; Mitelman, F.; Heim, S. Clonal heterogeneity in breast cancer: Karyotypic comparisons of multiple intra- and extra-tumorous samples from 3 patients. Int. J. Cancer 1995, 63, 63–68. [Google Scholar]
- Giaretti, W.; Monaco, R.; Pujic, N.; Rapallo, A.; Nigro, S.; Geido, E. Intratumor heterogeneity of K-ras2 mutations in colorectal adenocarcinomas: Association with degree of DNA aneuploidy. Am. J. Pathol 1996, 149, 237–245. [Google Scholar]
- Coons, S.W.; Johnson, P.C.; Shapiro, J.R. Cytogenetic and flow cytometry DNA analysis of regional heterogeneity in a low grade human glioma. Cancer Res 1995, 55, 1569–1577. [Google Scholar]
- Kallioniemi, O.P. Comparison of fresh and paraffin-embedded tissue as starting material for DNA flow cytometry and evaluation of intratumor heterogeneity. Cytometry 1988, 9, 164–169. [Google Scholar]
- Lage, J.M.; Leamon, J.H.; Pejovic, T.; Hamann, S.; Lacey, M.; Dillon, D.; Segraves, R.; Vossbrinck, B.; González, A.; Pinkel, D.; et al. Whole genome analysis of genetic alterations in small DNA samples using hyperbranched strand displacement amplification and array-CGH. Genome Res 2003, 13, 294–307. [Google Scholar]
- Leith, J.T.; Michelson, S.; Faulkner, L.E.; Bliven, S.F. Growth properties of artificial heterogeneous human colon tumors. Cancer Res 1987, 47, 1045–1051. [Google Scholar]
- Farber, L.; Efrati, E.; Elkin, H.; Peerless, Y.; Sabo, E.; Ben-Izhak, O.; Hershkovitz, D. Molecular morphometric analysis shows relative intra-tumoural homogeneity for KRAS mutations in colorectal cancer. Virchows Arch 2011, 459, 487–493. [Google Scholar]
- Kuwai, T.; Nakamura, T.; Kim, S.J.; Sasaki, T.; Kitadai, Y.; Langley, R.R.; Fan, D.; Hamilton, S.R.; Fidler, I.J. Intratumoral heterogeneity for expression of tyrosine kinase growth factor receptors in human colon cancer surgical specimens and orthotopic tumors. Am. J. Pathol 2008, 172, 358–366. [Google Scholar]
- Inda, M.M.; Bonavia, R.; Mukasa, A.; Narita, Y.; Sah, D.W.Y.; Vandenberg, S.; Brennan, C.; Johns, T.G.; Bachoo, R.; Hadwiger, P.; et al. Tumor heterogeneity is an active process maintained by a mutant EGFR-induced cytokine circuit in glioblastoma. Genes Dev 2010, 24, 1731–1745. [Google Scholar]
- Konishi, N.; Hiasa, Y.; Matsuda, H.; Tao, M.; Tsuzuki, T.; Hayashi, I.; Kitahori, Y.; Shiraishi, T.; Yatani, R.; Shimazaki, J.; et al. Intratumor cellular heterogeneity and alterations in ras oncogene and p53 tumor suppressor gene in human prostate carcinoma. Am. J. Pathol 1995, 147, 1112–1122. [Google Scholar]
- Maley, C.C.; Reid, B.J.; Forrest, S. Cancer prevention strategies that address the evolutionary dynamics of neoplastic cells: Simulating benign cell boosters and selection for chemosensitivity. Cancer Epidemiol. Biomarkers Prev 2004, 13, 1375–1384. [Google Scholar]
- Cottu, P.H.; Asselah, J.; Lae, M.; Pierga, J.Y.; Diéras, V.; Mignot, L.; Sigal-Zafrani, B.; Vincent-Salomon, A. Intratumoral heterogeneity of HER2/neu expression and its consequences for the management of advanced breast cancer. Ann. Oncol 2008, 19, 595–597. [Google Scholar]




© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Diaz-Cano, S.J. Tumor Heterogeneity: Mechanisms and Bases for a Reliable Application of Molecular Marker Design. Int. J. Mol. Sci. 2012, 13, 1951-2011. https://doi.org/10.3390/ijms13021951
Diaz-Cano SJ. Tumor Heterogeneity: Mechanisms and Bases for a Reliable Application of Molecular Marker Design. International Journal of Molecular Sciences. 2012; 13(2):1951-2011. https://doi.org/10.3390/ijms13021951
Chicago/Turabian StyleDiaz-Cano, Salvador J. 2012. "Tumor Heterogeneity: Mechanisms and Bases for a Reliable Application of Molecular Marker Design" International Journal of Molecular Sciences 13, no. 2: 1951-2011. https://doi.org/10.3390/ijms13021951