AKIP1, a Cardiac Hypertrophy Induced Protein that Stimulates Cardiomyocyte Growth via the Akt Pathway

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
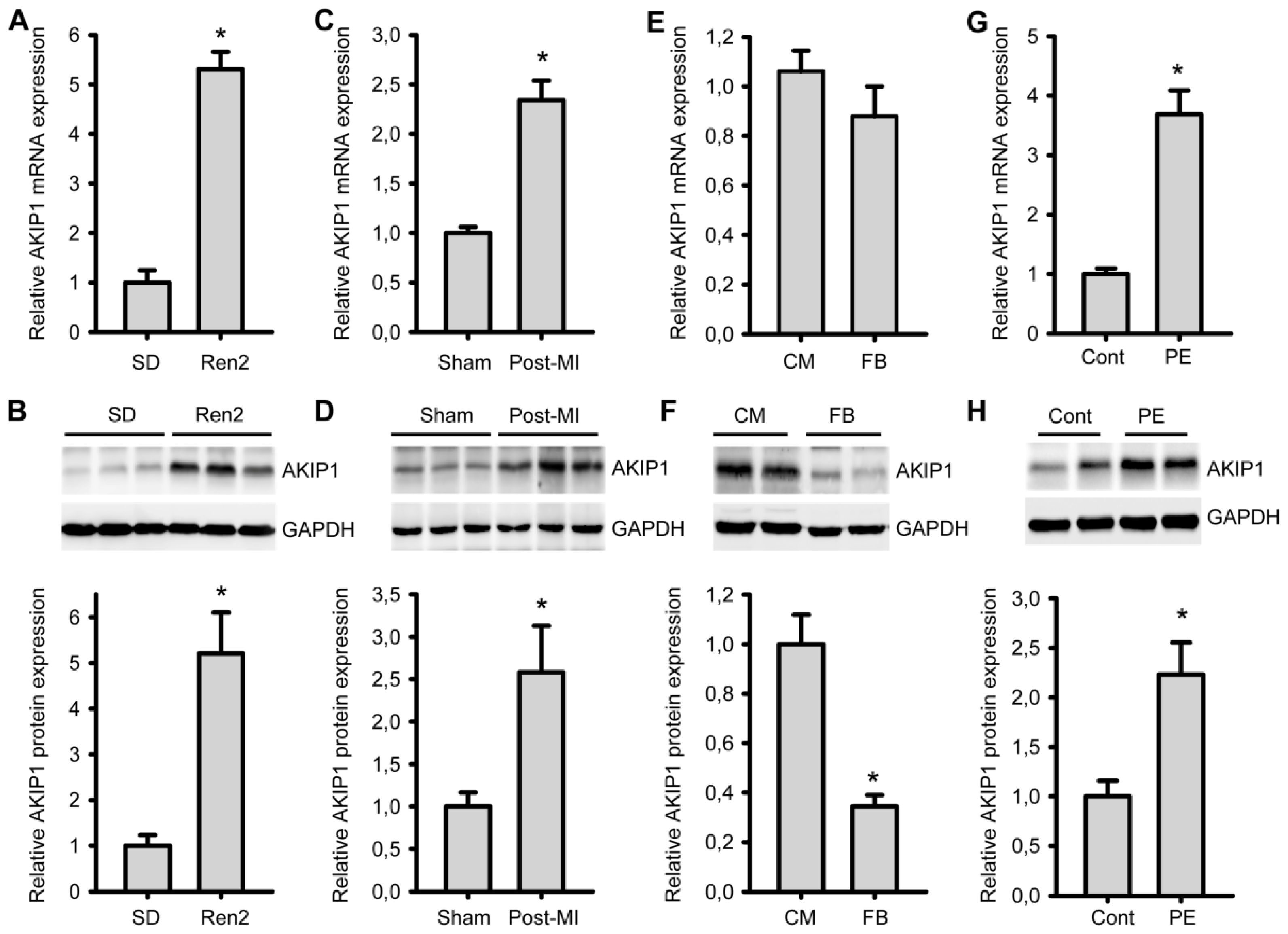
2.1. AKIP1 Expression Is Upregulated in Cardiac Hypertrophy/HF Models
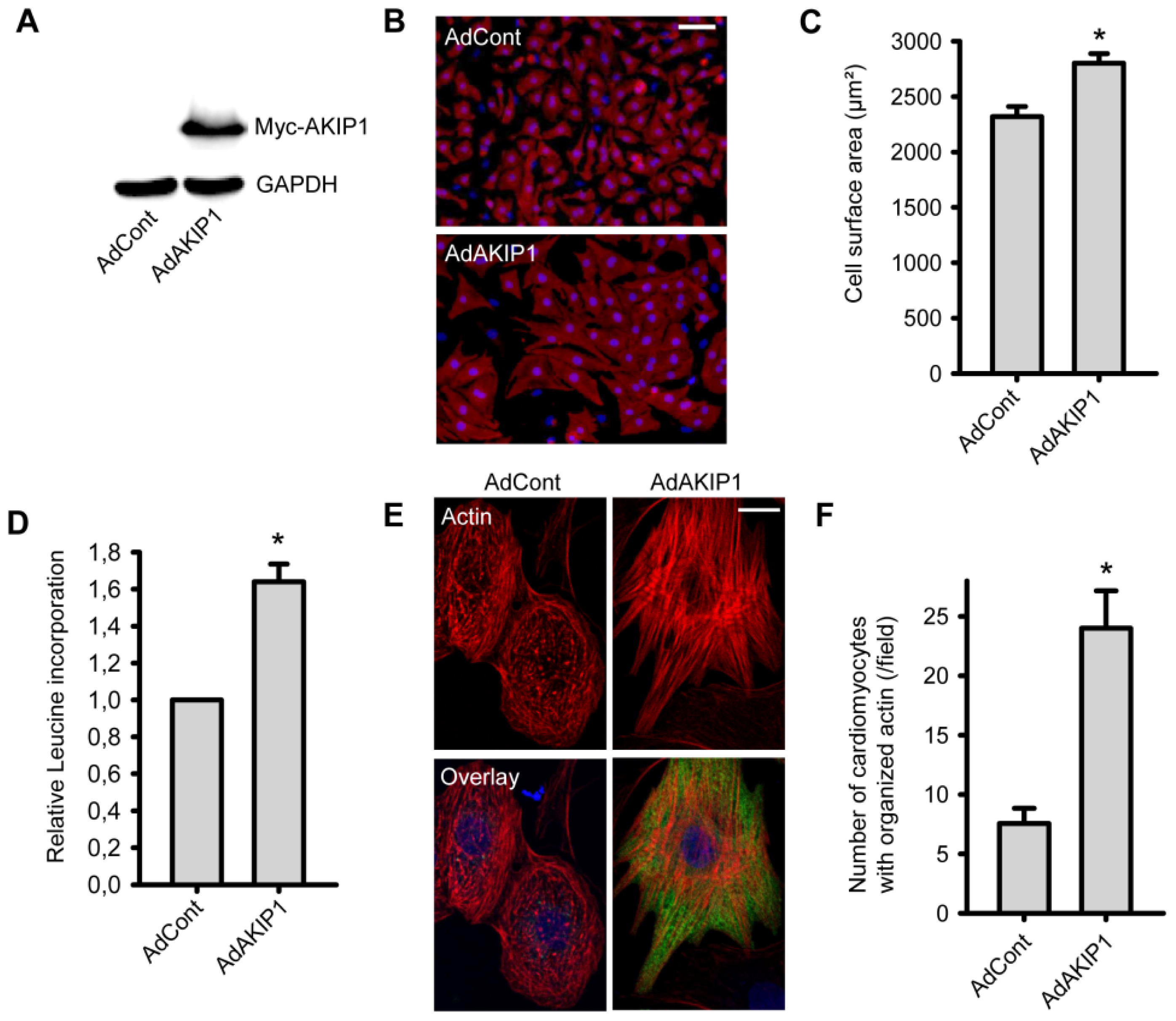
2.2. Overexpression of AKIP1 Stimulates Cardiomyocyte Hypertrophy
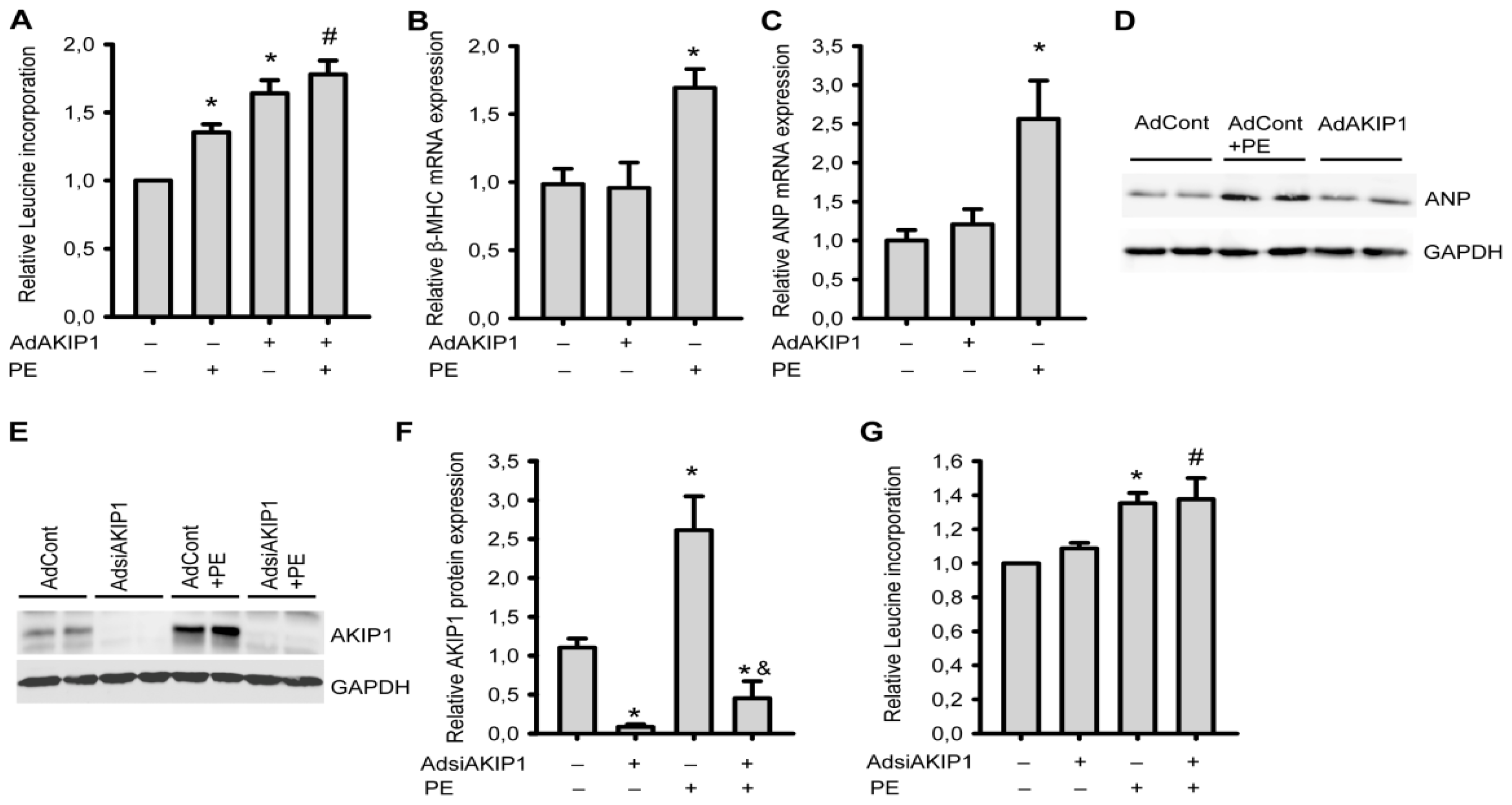
2.3. AKIP1 Is Not Required for Hypertrophy Induced by Neurohormonal Signals
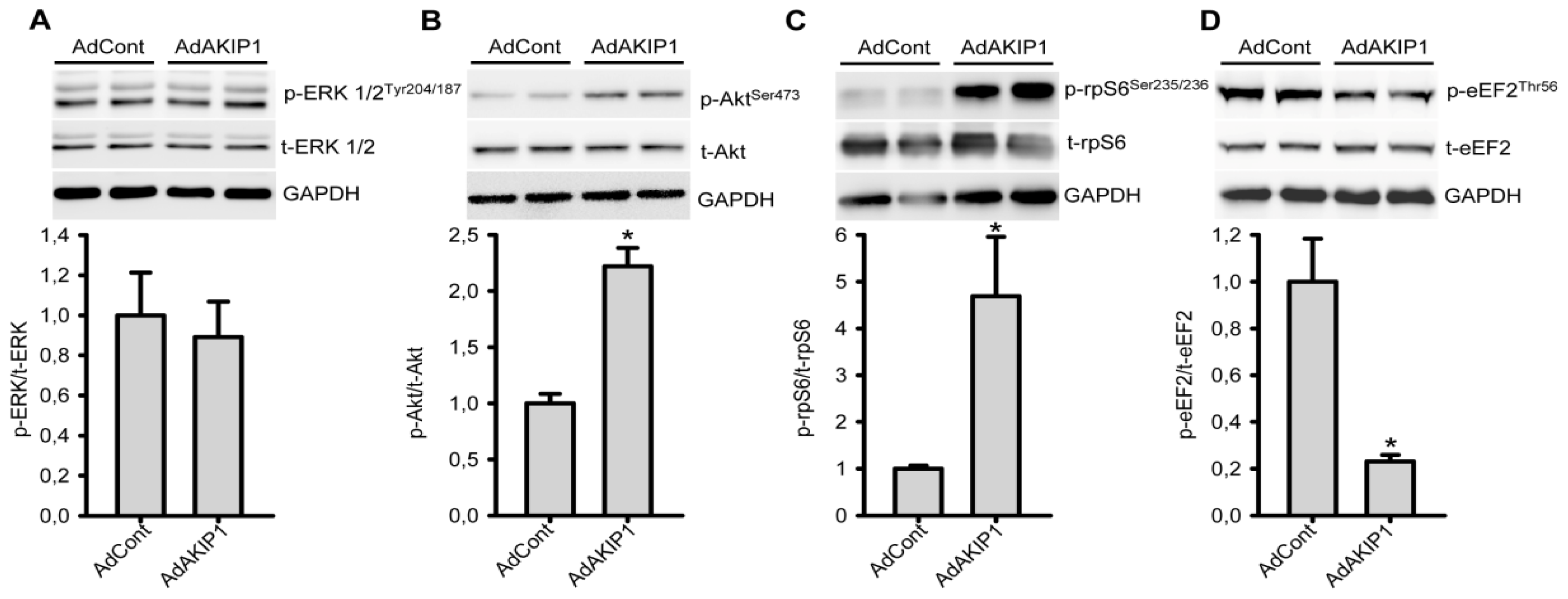
2.4. AKIP1 Specifically Stimulates Akt Kinase and Downstream Factors Controlling Protein Synthesis
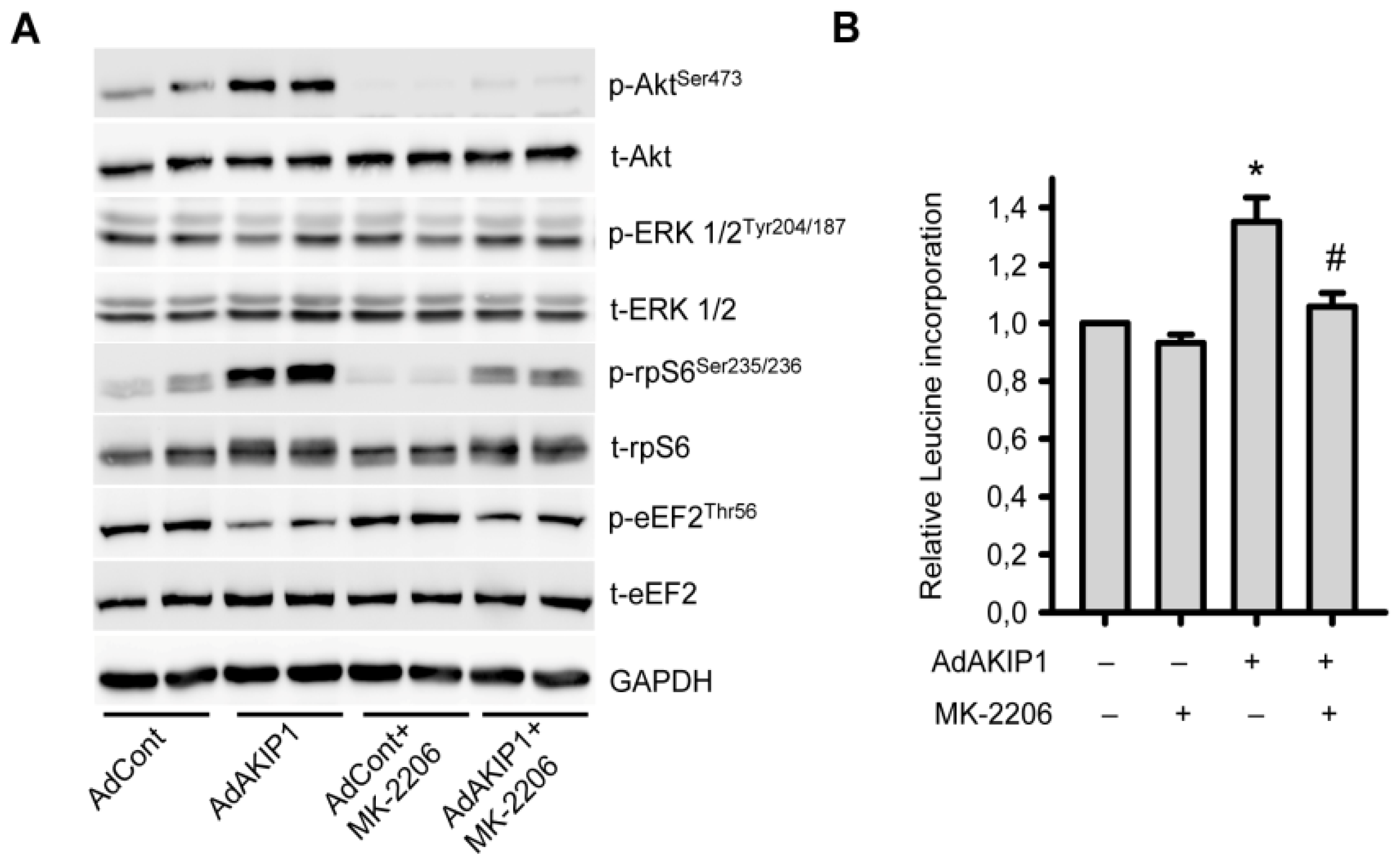
2.5. Akt Activity Is Essential for AKIP1 Induced Protein Translation and Hypertrophy Development
3. Discussion
4. Materials and Methods
4.1. Isolation and Culturing of Primary Cardiomyocytes
4.2. Animal Studies
4.3. Generation and Purification of Antibody
4.4. Generation of Recombinant Adenovirus and Transient Infection
4.5. Real Time PCR
4.6. Protein Synthesis Assay
4.7. Immunofluorescence (IF) and Cell Size Measurement
4.8. Western Blot
4.9. Statistical Analysis
5. Conclusions
Supplementary Information
ijms-14-21378-s001.pdf




Acknowledgments
Conflicts of Interest
References
- Hill, J.; Olson, E. Cardiac plasticity. N. Engl. J. Med 2008, 358, 1370–1380. [Google Scholar]
- Maillet, M.; van Berlo, J.H.; Molkentin, J.D. Molecular basis of physiological heart growth: Fundamental concepts and new players. Nat. Rev. Mol. Cell Biol 2012, 14, 38–48. [Google Scholar]
- Frey, N.; Olson, E.N. Cardiac hypertrophy: The good, the bad, and the ugly. Annu. Rev. Physiol 2003, 65, 45–79. [Google Scholar]
- Bernardo, B.C.; Weeks, K.L.; Pretorius, L.; McMullen, J.R. Molecular distinction between physiological and pathological cardiac hypertrophy: Experimental findings and therapeutic strategies. Pharmacol. Ther 2010, 128, 191–227. [Google Scholar]
- Lorell, B.H.; Carabello, B.A. Left ventricular hypertrophy: Pathogenesis, detection, and prognosis. Circulation 2000, 102, 470–479. [Google Scholar]
- Bueno, O.F.; de Windt, L.J.; Tymitz, K.M.; Witt, S.A.; Kimball, T.R.; Klevitsky, R.; Hewett, T.E.; Jones, S.P.; Lefer, D.J.; Peng, C.F.; et al. The MEK1-ERK1/2 signaling pathway promotes compensated cardiac hypertrophy in transgenic mice. EMBO J 2000, 19, 6341–6350. [Google Scholar]
- Chung, E.; Yeung, F.; Leinwand, L.A. Akt and MAPK signaling mediate pregnancy-induced cardiac adaptation. J. Appl. Physiol 2012, 112, 1564–1575. [Google Scholar]
- DeBosch, B.; Treskov, I.; Lupu, T.S.; Weinheimer, C.; Kovacs, A.; Courtois, M.; Muslin, A.J. Akt1 is required for physiological cardiac growth. Circulation 2006, 113, 2097–2104. [Google Scholar]
- Taniyama, Y.; Ito, M.; Sato, K.; Kuester, C.; Veit, K.; Tremp, G.; Liao, R.; Colucci, W.S.; Ivashchenko, Y.; Walsh, K.; et al. Akt3 overexpression in the heart results in progression from adaptive to maladaptive hypertrophy. J. Mol. Cell. Cardiol 2005, 38, 375–385. [Google Scholar]
- Sastri, M.; Haushalter, K.J.; Panneerselvam, M.; Chang, P.; Fridolfsson, H.; Finley, J.C.; Ng, D.; Schilling, J.M.; Miyanohara, A.; Day, M.E.; et al. A kinase interacting protein (AKIP1) is a key regulator of cardiac stress. Proc. Natl. Acad. Sci. USA 2013, 110, e387–e396. [Google Scholar]
- Sastri, M.; Barraclough, D.; Carmichael, P.; Taylor, S. A-kinase-interacting protein localizes protein kinase A in the nucleus. Proc. Natl. Acad. Sci. USA 2005, 102, 349–354. [Google Scholar]
- Gao, F.; Cheng, J.; Shi, T.; Yeh, E.T. Neddylation of a breast cancer-associated protein recruits a class III histone deacetylase that represses NFkappaB-dependent transcription. Nat. Cell Biol 2006, 8, 1171–1177. [Google Scholar]
- Gao, N.; Asamitsu, K.; Hibi, Y.; Ueno, T.; Okamoto, T. AKIP1 enhances NF-kappaB-dependent gene expression by promoting the nuclear retention and phosphorylation of p65. J. Biol. Chem 2008, 283, 7834–7843. [Google Scholar]
- Yu, K.P.; Itokawa, T.; Zhu, M.L.; Syam, S.; Seth, A.; Insogna, K. Breast cancer-associated gene 3 (BCA3) is a novel rac1-interacting protein. J. Bone Miner. Res 2007, 22, 628–637. [Google Scholar]
- Leung, T.; Ngan, H. Interaction of TAp73 and breast cancer-associated gene 3 enhances the sensitivity of cervical cancer cells in response to irradiation-induced apoptosis. Cancer Res 2010, 70, 6486–6496. [Google Scholar]
- Gao, N.; Hibi, Y.; Cueno, M.; Asamitsu, K.; Okamoto, T. A-kinase-interacting protein 1 (AKIP1) acts as a molecular determinant of PKA in NF-kappaB signaling. J. Biol. Chem 2010, 285, 28097–28104. [Google Scholar]
- Zimmerman, R.; Peng, D.J.; Lanz, H.; Zhang, Y.H.; Danen-Van Oorschot, A.; Qu, S.; Backendorf, C.; Noteborn, M. PP2A inactivation is a crucial step in triggering apoptin-induced tumor-selective cell killing. Cell Death Dis 2012, 3. [Google Scholar] [CrossRef]
- Kitching, R.; Li, H.; Wong, M.; Kanaganayakam, S.; Kahn, H.; Seth, A. Characterization of a novel human breast cancer associated gene (BCA3) encoding an alternatively spliced proline-rich protein. Biochim. Biophys. Acta 2003, 1625, 116–121. [Google Scholar]
- Lu, B.; Yu, H.; Zwartbol, M.; Ruifrok, W.P.; van Gilst, W.H.; de Boer, R.A.; Sillje, H.H. Identification of hypertrophy and heart failure associated genes by combining in vitro and in vivo models. Physiol. Genomics 2012, 44, 443–454. [Google Scholar]
- Leon, D.A.; Canaves, J.M. In silico study of breast cancer associated gene 3 using LION target engine and other tools. BioTechniques 2003, 35. [Google Scholar]
- Ruifrok, W.P.; Qian, C.; Sillje, H.H.; van Goor, H.; van Veldhuisen, D.J.; van Gilst, W.H.; de Boer, R.A. Heart failure-associated anemia: Bone marrow dysfunction and response to erythropoietin. J. Mol. Med. (Berl.) 2011, 89, 377–387. [Google Scholar]
- Westenbrink, B.D.; Lipsic, E.; van der Meer, P.; van der Harst, P.; Oeseburg, H.; Du Marchie Sarvaas, G.J.; Koster, J.; Voors, A.A.; van Veldhuisen, D.J.; van Gilst, W.H.; et al. Erythropoietin improves cardiac function through endothelial progenitor cell and vascular endothelial growth factor mediated neovascularization. Eur. Heart J 2007, 28, 2018–2027. [Google Scholar]
- De Boer, R.A.; Pokharel, S.; Flesch, M.; van Kampen, D.A.; Suurmeijer, A.J.; Boomsma, F.; van Gilst, W.H.; van Veldhuisen, D.J.; Pinto, Y.M. Extracellular signal regulated kinase and SMAD signaling both mediate the angiotensin II driven progression towards overt heart failure in homozygous TGR(mRen2)27. J. Mol. Med. (Berl.) 2004, 82, 678–687. [Google Scholar]
- Shubeita, H.E.; McDonough, P.M.; Harris, A.N.; Knowlton, K.U.; Glembotski, C.C.; Brown, J.H.; Chien, K.R. Endothelin induction of inositol phospholipid hydrolysis, sarcomere assembly, and cardiac gene expression in ventricular myocytes. A paracrine mechanism for myocardial cell hypertrophy. J. Biol. Chem 1990, 265, 20555–20562. [Google Scholar]
- Ruvinsky, I.; Meyuhas, O. Ribosomal protein S6 phosphorylation: From protein synthesis to cell size. Trends Biochem. Sci 2006, 31, 342–348. [Google Scholar]
- Kaul, G.; Pattan, G.; Rafeequi, T. Eukaryotic elongation factor-2 (eEF2): Its regulation and peptide chain elongation. Cell Biochem. Funct 2011, 29, 227–234. [Google Scholar]
- Giusti, B.; Marini, M.; Rossi, L.; Lapini, I.; Magi, A.; Capalbo, A.; Lapalombella, R.; di Tullio, S.; Samaja, M.; Esposito, F.; et al. Gene expression profile of rat left ventricles reveals persisting changes following chronic mild exercise protocol: Implications for cardioprotection. BMC Genomics 2009, 10. [Google Scholar] [CrossRef]
- Lu, B.; Tigchelaar, W.; Ruifrok, W.P.; van Gilst, W.H.; de Boer, R.A.; Sillje, H.H. DHRS7c, a novel cardiomyocyte-expressed gene that is down-regulated by adrenergic stimulation and in heart failure. Eur. J. Heart Fail 2012, 14, 5–13. [Google Scholar]
- Datta, S.R.; Brunet, A.; Greenberg, M.E. Cellular survival: A play in three Akts. Genes Dev 1999, 13, 2905–2927. [Google Scholar]
- Song, G.; Ouyang, G.; Bao, S. The activation of Akt/PKB signaling pathway and cell survival. J. Cell. Mol. Med 2005, 9, 59–71. [Google Scholar]
- DeBosch, B.J.; Muslin, A.J. Insulin signaling pathways and cardiac growth. J. Mol. Cell. Cardiol 2008, 44, 855–864. [Google Scholar]
- Fischer, P.; Hilfiker-Kleiner, D. Survival pathways in hypertrophy and heart failure: The gp130-STAT axis. Basic Res. Cardiol 2007, 102, 393–411. [Google Scholar]
- Shao, Z.; Bhattacharya, K.; Hsich, E.; Park, L.; Walters, B.; Germann, U.; Wang, Y.M.; Kyriakis, J.; Mohanlal, R.; Kuida, K.; et al. c-Jun N-terminal kinases mediate reactivation of Akt and cardiomyocyte survival after hypoxic injury in vitro and in vivo. Circ. Res. 2006, 98, 111–118. [Google Scholar]
- Barron, A.J.; Finn, S.G.; Fuller, S.J. Chronic activation of extracellular-signal-regulated protein kinases by phenylephrine is required to elicit a hypertrophic response in cardiac myocytes. Biochem. J 2003, 371, 71–79. [Google Scholar]
- Prasad, A.M.; Ma, H.; Sumbilla, C.; Lee, D.I.; Klein, M.G.; Inesi, G. Phenylephrine hypertrophy, Ca2+-ATPase (SERCA2), and Ca2+ signaling in neonatal rat cardiac myocytes. Am. J. Physiol. Cell Physiol 2007, 292, C2269–C2275. [Google Scholar]
- Chaanine, A.H.; Jeong, D.; Liang, L.; Chemaly, E.R.; Fish, K.; Gordon, R.E.; Hajjar, R.J. JNK modulates FOXO3a for the expression of the mitochondrial death and mitophagy marker BNIP3 in pathological hypertrophy and in heart failure. Cell Death Dis 2012, 3. [Google Scholar] [CrossRef]
- Haq, S.; Choukroun, G.; Lim, H.; Tymitz, K.M.; del Monte, F.; Gwathmey, J.; Grazette, L.; Michael, A.; Hajjar, R.; Force, T.; et al. Differential activation of signal transduction pathways in human hearts with hypertrophy versus advanced heart failure. Circulation 2001, 103, 670–677. [Google Scholar]
- Lu, B.; Mahmud, H.; Maass, A.H.; Yu, B.; van Gilst, W.H.; de Boer, R.A.; Sillje, H.H. The Plk1 inhibitor BI 2536 temporarily arrests primary cardiac fibroblasts in mitosis and generates aneuploidy in vitro. PLoS One 2010, 5, e12963. [Google Scholar]
- Westenbrink, B.D.; Ruifrok, W.P.; Voors, A.A.; Tilton, R.G.; van Veldhuisen, D.J.; Schoemaker, R.G.; van Gilst, W.H.; de Boer, R.A. Vascular endothelial growth factor is crucial for erythropoietin-induced improvement of cardiac function in heart failure. Cardiovasc. Res 2010, 87, 30–39. [Google Scholar]
- Lee, M.A.; Bohm, M.; Paul, M.; Bader, M.; Ganten, U.; Ganten, D. Physiological characterization of the hypertensive transgenic rat TGR(mREN2)27. Am. J. Physiol 1996, 270, E919–E929. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Yu, H.; Tigchelaar, W.; Lu, B.; Van Gilst, W.H.; De Boer, R.A.; Westenbrink, B.D.; Silljé, H.H.W. AKIP1, a Cardiac Hypertrophy Induced Protein that Stimulates Cardiomyocyte Growth via the Akt Pathway. Int. J. Mol. Sci. 2013, 14, 21378-21393. https://doi.org/10.3390/ijms141121378
Yu H, Tigchelaar W, Lu B, Van Gilst WH, De Boer RA, Westenbrink BD, Silljé HHW. AKIP1, a Cardiac Hypertrophy Induced Protein that Stimulates Cardiomyocyte Growth via the Akt Pathway. International Journal of Molecular Sciences. 2013; 14(11):21378-21393. https://doi.org/10.3390/ijms141121378
Chicago/Turabian StyleYu, Hongjuan, Wardit Tigchelaar, Bo Lu, Wiek H. Van Gilst, Rudolf A. De Boer, B. Daan Westenbrink, and Herman H. W. Silljé. 2013. "AKIP1, a Cardiac Hypertrophy Induced Protein that Stimulates Cardiomyocyte Growth via the Akt Pathway" International Journal of Molecular Sciences 14, no. 11: 21378-21393. https://doi.org/10.3390/ijms141121378