
Vasa Vasorum in Atherosclerosis and Clinical Significance

Abstract
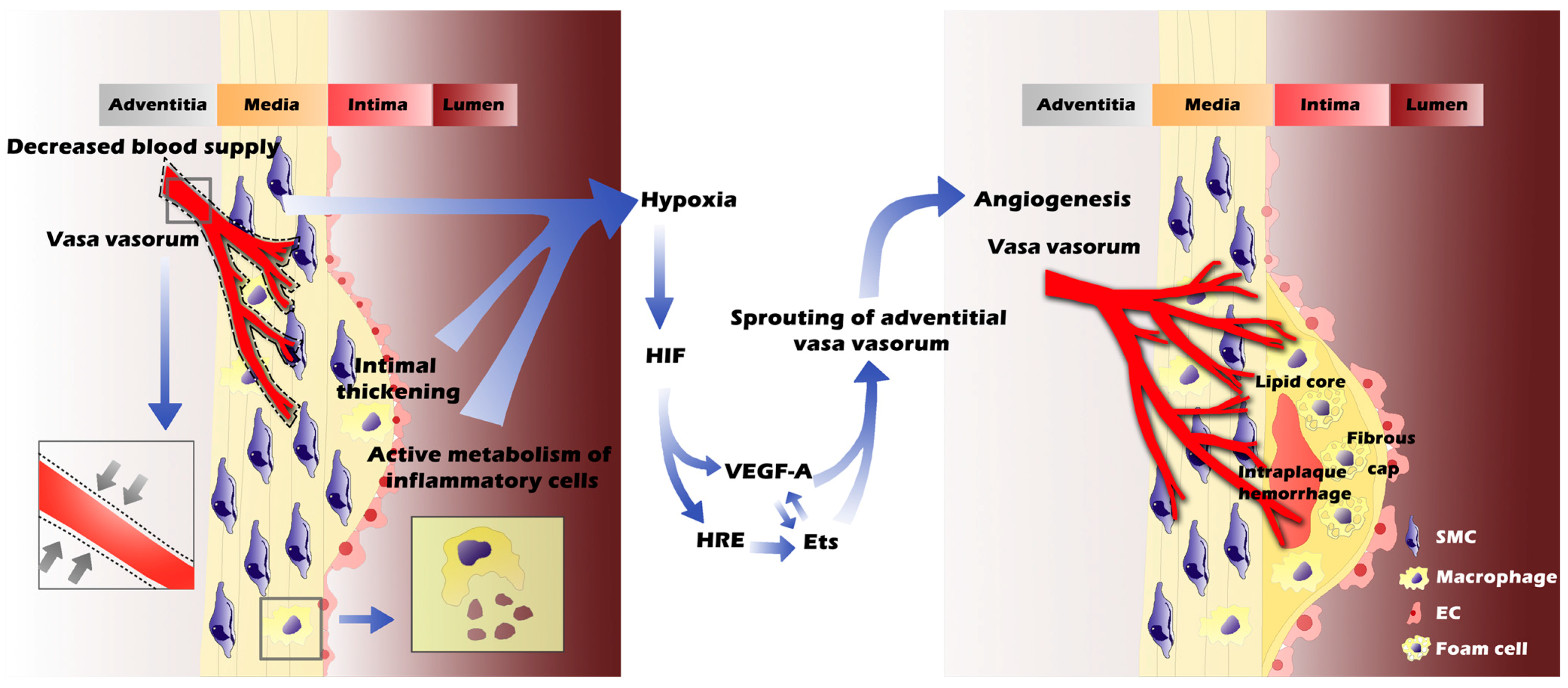
:1. Introduction
2. Structure and Function of Vasa Vasorum
2.1. Structure

2.2. Function
3. Stimuli of Vasa Vasorum Neovascularization
3.1. Hypoxia

3.2. Inflammation
3.3. Lipids
4. Factors Leading to Immature and Fragile Vasa Vasorum
4.1. Imbalance of Angiogenic Factors in Proteolytic Environment
4.2. Further Exacerbation of Neovessel Damage: Iron, Cholesterol Crystal and Proteases
5. Vasa Vasorum Imaging in Atherosclerosis Plaques
5.1. Anatomic Imaging of Vasa Vasorum in Atherosclerosis
5.2. Molecular Imaging of Vasa Vasorum in Atherosclerosis

{kind=link}
{kind=link}
| Imaging Modality | Spatial Resolution | Temporal Resolution | Targets (Reference) | Species | In Vivo Imaging | Histological Validation | Results |
|---|---|---|---|---|---|---|---|
| Planar gamma camera | cm3 | Hours | 123I-labelled MMP inhibitors [147] | Apoe−/− mice | + | + | Signal of 123I-labelled inhibitors into plaques in high cholesterol animals was 2.72-folds of the control |
| Autoradiograph | μm3 | Milliseconds | ED-B [148] | Apoe−/− mice | − | − | 125I-labeled monoclonal antibodies against ED-B identified the angiogenesis in atherosclerotic plaques ex vivo |
| 99mTc-MT1-MMP mAb [149] | WHHLMI rabbits | + | + | The highest accumulation of 99mTc-MT1-MMP mAb was found in atheromatous lesions in comparison with stable lesions | |||
| MRI | mm3 | Seconds | Integrin αvβ3 | Male New Zealand White (NZW) rabbits [151] | + | + | Paramagnetic gadolinium-based nanoparticles showed strong enhancement in atheroscletotic lesions that was twice of the non-targeted nanoparticles |
| JCR:LA-cp rats [152] | + | + | The enhancement of ανβ3-targeted nanoparticles was preserved in benfluorex treating group | ||||
| Humans [153,154,155] | + | + | Targeted gadolinium compounds detected VV, total area and Ktrans of the enhancement could be quantitative parameters | ||||
| CEUS | μm3 | Milliseconds | ET-1/VEGFsp receptor [158] | Tg25 (hCETP) Dahl-S rats | + | + | Expanded VV in transgenic rats with carotid artery disease were detected by targeted microbubbles |
| VEGFR-2 [159] | Female nude mice | + | − | VEGFR-2 targeted microbubbles were able to evaluate anti-angiogenic effect of sorafenib |
6. Anti- and Pro-Angiogenic Therapies on Vasa Vasorum
6.1. Anti-Angiogenic Therapies
| Compounds | Year | Functions | Species | Possible Mechanisms | Results |
|---|---|---|---|---|---|
| ET-A | 1993 [173], 2002 [174] | Inhibiting ET-1 receptor | Female domestic pigs | Inhibiting mitogenic activity of SMCs | Elevated VV density in hypercholesterolemia pigs were greatly preserved by ~32% after ET-A treatment |
| Fumagillin nanoparticle | 2006 [175] | An anti-angiogenic agent that targets αvβ3 integrin | Male NZW rabbits | Not investigate | MRI enhancement and the numbers of microvessels are decreased in fumagillin-treated cholesterol-fed rabbits |
| rPAI-123 | 2009 [176], 2011 [177] | A truncated isoform of plasminogen activator inhibitor-1 (PAI-1) | Female ApoB-48−/−/Ldlr−/− mice | Reducing FGF-2 expression. Increasing plasmin and MMP activities on degrading compounds (i.e., perlecan, nidogen, fibrin) that produce supportive scaffold in the extracellular milieu, leading to apoptosis of ECs | A 37% reduction in total vessel area and a 43% reduction in vessel length of the second order VV are observed as a result of apoptosis of ECs |
| c3Ado | 2009 [178] | An anti-proliferative and anti-inflammatory drug | Apoe−/−/Ldl−/− double knockout mice | Preventing the proliferation and migration of ECs | VV neovascularization is inhibited dose dependently |
6.2. Pro-Angiogenic Therapies
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mozaffarian, D.; Benjamin, E.J.; Go, A.S.; Arnett, D.K.; Blaha, M.J.; Cushman, M.; de Ferranti, S.; Despres, J.P.; Fullerton, H.J.; Howard, V.J.; et al. Heart disease and stroke statistics—2015 update: A report from the American Heart Association. Circulation 2015, 131, e29–e322. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.J.; Richardson, P.D.; Woolf, N.; Katz, D.R.; Mann, J. Risk of thrombosis in human atherosclerotic plaques: Role of extracellular lipid, macrophage, and smooth muscle cell content. Br. Heart J. 1993, 69, 377–381. [Google Scholar] [CrossRef] [PubMed]
- De Boer, O.J.; van der Wal, A.C.; Teeling, P.; Becker, A.E. Leucocyte recruitment in rupture prone regions of lipid-rich plaques: A prominent role for neovascularization? Cardiovasc. Res. 1999, 41, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Staub, D.; Patel, M.B.; Tibrewala, A.; Ludden, D.; Johnson, M.; Espinosa, P.; Coll, B.; Jaeger, K.A.; Feinstein, S.B. Vasa vasorum and plaque neovascularization on contrast-enhanced carotid ultrasound imaging correlates with cardiovascular disease and past cardiovascular events. Stroke J. Cereb. Circ. 2010, 41, 41–47. [Google Scholar] [CrossRef]
- Takano, M.; Mizuno, K.; Okamatsu, K.; Yokoyama, S.; Ohba, T.; Sakai, S. Mechanical and structural characteristics of vulnerable plaques: Analysis by coronary angioscopy and intravascular ultrasound. J. Am. Coll. Cardiol. 2001, 38, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Ritman, E.L.; Lerman, A. The dynamic vasa vasorum. Cardiovasc. Res. 2007, 75, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Koester, W. Endareritis and arteritis. Berl. Klin. Wochenschr. 1876, 13, 454–455. [Google Scholar]
- Paterson, J.C. Vascularization and hemorrhage of the intima of arteriosclerotic coronary arteries. Arch. Pathol. 1936, 22, 313–324. [Google Scholar]
- Patterson, J.C. Capillary rupture with intimal hemorrhage as a causative factor in coronary thrombosis. Arch. Pathol. 1938, 25, 474–487. [Google Scholar]
- Barger, A.C.; Beeuwkes, R., 3rd; Lainey, L.L.; Silverman, K.J. Hypothesis: Vasa vasorum and neovascularization of human coronary arteries. A possible role in the pathophysiology of atherosclerosis. N. Engl. J. Med. 1984, 310, 175–177. [Google Scholar] [CrossRef] [PubMed]
- Gossl, M.; Versari, D.; Hildebrandt, H.A.; Bajanowski, T.; Sangiorgi, G.; Erbel, R.; Ritman, E.L.; Lerman, L.O.; Lerman, A. Segmental heterogeneity of vasa vasorum neovascularization in human coronary atherosclerosis. JACC Cardiovasc. Imaging 2010, 3, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.M.; Sangiorgi, G.; Ritman, E.L.; McKenna, C.; Holmes, D.R., Jr.; Schwartz, R.S.; Lerman, A. Enhanced coronary vasa vasorum neovascularization in experimental hypercholesterolemia. J. Clin. Investig. 1998, 101, 1551–1556. [Google Scholar] [CrossRef] [PubMed]
- Dunmore, B.J.; McCarthy, M.J.; Naylor, A.R.; Brindle, N.P. Carotid plaque instability and ischemic symptoms are linked to immaturity of microvessels within plaques. J. Vasc. Surg. 2007, 45, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Sluimer, J.C.; Kolodgie, F.D.; Bijnens, A.P.; Maxfield, K.; Pacheco, E.; Kutys, B.; Duimel, H.; Frederik, P.M.; van Hinsbergh, V.W.; Virmani, R.; et al. Thin-walled microvessels in human coronary atherosclerotic plaques show incomplete endothelial junctions relevance of compromised structural integrity for intraplaque microvascular leakage. J. Am. Coll. Cardiol. 2009, 53, 1517–1527. [Google Scholar] [CrossRef] [PubMed]
- Khurana, R.; Zhuang, Z.; Bhardwaj, S.; Murakami, M.; de Muinck, E.; Yla-Herttuala, S.; Ferrara, N.; Martin, J.F.; Zachary, I.; Simons, M. Angiogenesis-dependent and independent phases of intimal hyperplasia. Circulation 2004, 110, 2436–2443. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, J.; Lerman, L.O.; Rodriguez-Porcel, M.; Holmes, D.R., Jr.; Richardson, D.M.; Ritman, E.L.; Lerman, A. Coronary vasa vasorum neovascularization precedes epicardial endothelial dysfunction in experimental hypercholesterolemia. Cardiovasc. Res. 2001, 51, 762–766. [Google Scholar] [CrossRef] [PubMed]
- Maiellaro, K.; Taylor, W.R. The role of the adventitia in vascular inflammation. Cardiovasc. Res. 2007, 75, 640–648. [Google Scholar] [CrossRef] [PubMed]
- Clarke, J.A. An X-ray microscopic study of the postnatal development of the vasa vasorum of normal human coronary arteries. Acta Anat. 1966, 64, 506–516. [Google Scholar] [CrossRef] [PubMed]
- Wolinsky, H.; Glagov, S. Nature of species differences in the medial distribution of aortic vasa vasorum in mammals. Circ. Res. 1967, 20, 409–421. [Google Scholar] [CrossRef] [PubMed]
- Schoenenberger, F.; Mueller, A. On the vascularization of the bovine aortic wall. Helv. Physiol. Pharmacol. Acta 1960, 18, 136–150. [Google Scholar] [PubMed]
- Gossl, M.; Rosol, M.; Malyar, N.M.; Fitzpatrick, L.A.; Beighley, P.E.; Zamir, M.; Ritman, E.L. Functional anatomy and hemodynamic characteristics of vasa vasorum in the walls of porcine coronary arteries. Anat. Rec. Part A Discov. Mol. Cell. Evol. Biol. 2003, 272, 526–537. [Google Scholar] [CrossRef]
- Fleiner, M.; Kummer, M.; Mirlacher, M.; Sauter, G.; Cathomas, G.; Krapf, R.; Biedermann, B.C. Arterial neovascularization and inflammation in vulnerable patients: Early and late signs of symptomatic atherosclerosis. Circulation 2004, 110, 2843–2850. [Google Scholar] [CrossRef] [PubMed]
- Bitar, R.; Moody, A.R.; Leung, G.; Symons, S.; Crisp, S.; Butany, J.; Rowsell, C.; Kiss, A.; Nelson, A.; Maggisano, R. In vivo 3D high-spatial-resolution MR imaging of intraplaque hemorrhage. Radiology 2008, 249, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Acoltzin Vidal, C.; Maldonado Villasenor, I.; Rodriguez Cisneros, L.; Muniz Murguia, J.J. Diminished vascular density in the aortic wall. Morphological and functional characteristics of atherosclerosis. Arch. Cardiol. Mexico 2004, 74, 176–180. [Google Scholar]
- Rademakers, T.; Douma, K.; Hackeng, T.M.; Post, M.J.; Sluimer, J.C.; Daemen, M.J.; Biessen, E.A.; Heeneman, S.; van Zandvoort, M.A. Plaque-associated vasa vasorum in aged apolipoprotein E-deficient mice exhibit proatherogenic functional features in vivo. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Gossl, M.; Malyar, N.M.; Rosol, M.; Beighley, P.E.; Ritman, E.L. Impact of coronary vasa vasorum functional structure on coronary vessel wall perfusion distribution. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H2019–H2026. [Google Scholar] [CrossRef] [PubMed]
- Han, D.G. The innateness of coronary artery: Vasa vasorum. Med. Hypotheses 2010, 74, 443–444. [Google Scholar] [CrossRef] [PubMed]
- Moss, A.J.; Samuelson, P.; Angell, C.; Minken, S.L. Polarographic evaluation of transmural oxygen availabitlity in intact muscular arteries. J. Atheroscler. Res. 1968, 8, 803–810. [Google Scholar] [CrossRef] [PubMed]
- Crawford, D.W.; Back, L.H.; Cole, M.A. In vivo oxygen transport in the normal rabbit femoral arterial wall. J. Clin. Investig. 1980, 65, 1498–1508. [Google Scholar] [CrossRef] [PubMed]
- Bratzler, R.L.; Chisolm, G.M.; Colton, C.K.; Smith, K.A.; Lees, R.S. The distribution of labeled low-density lipoproteins across the rabbit thoracic aorta in vivo. Atherosclerosis 1977, 28, 289–307. [Google Scholar] [CrossRef] [PubMed]
- Scotland, R.S.; Vallance, P.J.; Ahluwalia, A. Endogenous factors involved in regulation of tone of arterial vasa vasorum: Implications for conduit vessel physiology. Cardiovasc. Res. 2000, 46, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Scotland, R.; Vallance, P.; Ahluwalia, A. Endothelin alters the reactivity of vasa vasorum: Mechanisms and implications for conduit vessel physiology and pathophysiology. Br. J. Pharmacol. 1999, 128, 1229–1234. [Google Scholar] [CrossRef] [PubMed]
- Ohhira, A.; Ohhashi, T. Effects of aortic pressure and vasoactive agents on the vascular resistance of the vasa vasorum in canine isolated thoracic aorta. J. Physiol. 1992, 453, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Vio, A.; Gozzetti, G.; Reggiani, A. Importance of the vasa vasorum in the healing processes of arterial sutures. (Experimental study on the dog). Boll. Soc. Ital. Biol. Sper. 1967, 43, 88–90. [Google Scholar] [PubMed]
- Winternitz, M.C.; Thomas, R.M.; LeCompte, P.M. The Biology of Arteriosclerosis; Springfield: Prince George’s County, MA, USA, 1938. [Google Scholar]
- Ribatti, D.; Levi-Schaffer, F.; Kovanen, P.T. Inflammatory angiogenesis in atherogenesis—A double-edged sword. Ann. Med. 2008, 40, 606–621. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.K.; Armstrong, M.L.; Heistad, D.D. Vasa vasorum in atherosclerotic coronary arteries: Responses to vasoactive stimuli and regression of atherosclerosis. Circ. Res. 1988, 62, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Sano, M.; Sasaki, T.; Hirakawa, S.; Sakabe, J.; Ogawa, M.; Baba, S.; Zaima, N.; Tanaka, H.; Inuzuka, K.; Yamamoto, N.; et al. Lymphangiogenesis and angiogenesis in abdominal aortic aneurysm. PLoS ONE 2014, 9, e89830. [Google Scholar] [CrossRef] [PubMed]
- Davie, N.J.; Gerasimovskaya, E.V.; Hofmeister, S.E.; Richman, A.P.; Jones, P.L.; Reeves, J.T.; Stenmark, K.R. Pulmonary artery adventitial fibroblasts cooperate with vasa vasorum endothelial cells to regulate vasa vasorum neovascularization: A process mediated by hypoxia and endothelin-1. Am. J. Pathol. 2006, 168, 1793–1807. [Google Scholar] [CrossRef] [PubMed]
- Bjornheden, T.; Levin, M.; Evaldsson, M.; Wiklund, O. Evidence of hypoxic areas within the arterial wall in vivo. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 870–876. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, Y.; Chen, Y.X.; Kinukawa, N.; Sueishi, K. Distributions of diffuse intimal thickening in human arteries: Preferential expression in atherosclerosis-prone arteries from an early age. Virchows Arch. Int. J. Pathol. 2002, 441, 279–288. [Google Scholar] [CrossRef]
- Zemplenyi, T.; Crawford, D.W.; Cole, M.A. Adaptation to arterial wall hypoxia demonstrated in vivo with oxygen microcathodes. Atherosclerosis 1989, 76, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Jarvilehto, M.; Tuohimaa, P. Vasa vasorum hypoxia: Initiation of atherosclerosis. Med. Hypotheses 2009, 73, 40–41. [Google Scholar] [CrossRef] [PubMed]
- Den Hartog, J.P. Strength of Materials; Dover Publications, Inc.: New York, NY, USA, 1949; p. 323. [Google Scholar]
- Bjornheden, T.; Bondjers, G. Oxygen consumption in aortic tissue from rabbits with diet-induced atherosclerosis. Arteriosclerosis 1987, 7, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Sluimer, J.C.; Gasc, J.M.; van Wanroij, J.L.; Kisters, N.; Groeneweg, M.; Sollewijn Gelpke, M.D.; Cleutjens, J.P.; van den Akker, L.H.; Corvol, P.; Wouters, B.G.; et al. Hypoxia, hypoxia-inducible transcription factor, and macrophages in human atherosclerotic plaques are correlated with intraplaque angiogenesis. J. Am. Coll. Cardiol. 2008, 51, 1258–1265. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.M.; Sangiorgi, G.; Ritman, E.L.; Lerman, A.; McKenna, C.; Virmani, R.; Edwards, W.D.; Holmes, D.R.; Schwartz, R.S. Adventitial vasa vasorum in balloon-injured coronary arteries: Visualization and quantitation by a microscopic three-dimensional computed tomography technique. J. Am. Coll. Cardiol. 1998, 32, 2072–2079. [Google Scholar] [CrossRef] [PubMed]
- Gossl, M.; Versari, D.; Mannheim, D.; Ritman, E.L.; Lerman, L.O.; Lerman, A. Increased spatial vasa vasorum density in the proximal LAD in hypercholesterolemia—Implications for vulnerable plaque-development. Atherosclerosis 2007, 192, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z. Atherosclerosis and atheroma plaque rupture: Imaging modalities in the visualization of vasa vasorum and atherosclerotic plaques. Sci. World J. 2014, 2014, 312764. [Google Scholar]
- Gossl, M.; Versari, D.; Lerman, L.O.; Chade, A.R.; Beighley, P.E.; Erbel, R.; Ritman, E.L. Low vasa vasorum densities correlate with inflammation and subintimal thickening: Potential role in location--determination of atherogenesis. Atherosclerosis 2009, 206, 362–368. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.S.; Kiriakidis, S.; Sandison, A.; Paleolog, E.M.; Davies, A.H. Hypoxia-inducible factor pathway and diseases of the vascular wall. J. Vasc. Surg. 2013, 58, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L.; Agani, F.; Booth, G.; Forsythe, J.; Iyer, N.; Jiang, B.H.; Leung, S.; Roe, R.; Wiener, C.; Yu, A. Structural and functional analysis of hypoxia-inducible factor 1. Kidney Int. 1997, 51, 553–555. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, M.; Abe, M.; Kurosawa, H.; Hida, W.; Shirato, K.; Sato, Y. Hypoxia induces transcription factor ETS-1 via the activity of hypoxia-inducible factor-1. Biochem. Biophys. Res. Commun. 2001, 289, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Leung, D.W.; Cachianes, G.; Kuang, W.J.; Goeddel, D.V.; Ferrara, N. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science 1989, 246, 1306–1309. [Google Scholar] [CrossRef] [PubMed]
- Ushio-Fukai, M. VEGF signaling through NADPH oxidase-derived ROS. Antioxid. Redox Signal. 2007, 9, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Shweiki, D.; Itin, A.; Soffer, D.; Keshet, E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature 1992, 359, 843–845. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, F.; Kai, H.; Tokuda, K.; Shibata, R.; Kusaba, K.; Tahara, N.; Niiyama, H.; Nagata, T.; Imaizumi, T. Hypoxia-inducible factor-1α/vascular endothelial growth factor pathway for adventitial vasa vasorum formation in hypertensive rat aorta. Hypertension 2002, 39, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Joukov, V.; Pajusola, K.; Kaipainen, A.; Chilov, D.; Lahtinen, I.; Kukk, E.; Saksela, O.; Kalkkinen, N.; Alitalo, K. A novel vascular endothelial growth factor, VEGF-C, is a ligand for the Flt4 (VEGFR-3) and KDR (VEGFR-2) receptor tyrosine kinases. EMBO J. 1996, 15, 290–298. [Google Scholar] [PubMed]
- Achen, M.G.; Jeltsch, M.; Kukk, E.; Makinen, T.; Vitali, A.; Wilks, A.F.; Alitalo, K.; Stacker, S.A. Vascular endothelial growth factor D (VEGF-D) is a ligand for the tyrosine kinases VEGF receptor 2 (Flk1) and VEGF receptor 3 (Flt4). Proc. Natl. Acad. Sci. USA 1998, 95, 548–553. [Google Scholar] [CrossRef] [PubMed]
- Kaipainen, A.; Korhonen, J.; Mustonen, T.; van Hinsbergh, V.W.; Fang, G.H.; Dumont, D.; Breitman, M.; Alitalo, K. Expression of the fms-like tyrosine kinase 4 gene becomes restricted to lymphatic endothelium during development. Proc. Natl. Acad. Sci. USA 1995, 92, 3566–3570. [Google Scholar] [CrossRef] [PubMed]
- Kukk, E.; Lymboussaki, A.; Taira, S.; Kaipainen, A.; Jeltsch, M.; Joukov, V.; Alitalo, K. VEGF-C receptor binding and pattern of expression with VEGFR-3 suggests a role in lymphatic vascular development. Development 1996, 122, 3829–3837. [Google Scholar] [PubMed]
- Partanen, T.A.; Arola, J.; Saaristo, A.; Jussila, L.; Ora, A.; Miettinen, M.; Stacker, S.A.; Achen, M.G.; Alitalo, K. VEGF-C and VEGF-D expression in neuroendocrine cells and their receptor, VEGFR-3, in fenestrated blood vessels in human tissues. FASEB J. 2000, 14, 2087–2096. [Google Scholar] [CrossRef] [PubMed]
- Rutanen, J.; Leppanen, P.; Tuomisto, T.T.; Rissanen, T.T.; Hiltunen, M.O.; Vajanto, I.; Niemi, M.; Hakkinen, T.; Karkola, K.; Stacker, S.A.; et al. Vascular endothelial growth factor-D expression in human atherosclerotic lesions. Cardiovasc. Res. 2003, 59, 971–979. [Google Scholar] [CrossRef] [PubMed]
- Belgore, F.; Blann, A.; Neil, D.; Ahmed, A.S.; Lip, G.Y. Localisation of members of the vascular endothelial growth factor (VEGF) family and their receptors in human atherosclerotic arteries. J. Clin. Pathol. 2004, 57, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Vuorio, T.; Nurmi, H.; Moulton, K.; Kurkipuro, J.; Robciuc, M.R.; Ohman, M.; Heinonen, S.E.; Samaranayake, H.; Heikura, T.; Alitalo, K.; et al. Lymphatic vessel insufficiency in hypercholesterolemic mice alters lipoprotein levels and promotes atherogenesis. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1162–1170. [Google Scholar] [CrossRef] [PubMed]
- Iwasaka, C.; Tanaka, K.; Abe, M.; Sato, Y. Ets-1 regulates angiogenesis by inducing the expression of urokinase-type plasminogen activator and matrix metalloproteinase-1 and the migration of vascular endothelial cells. J. Cell. Physiol. 1996, 169, 522–531. [Google Scholar] [CrossRef] [PubMed]
- Hashiya, N.; Jo, N.; Aoki, M.; Matsumoto, K.; Nakamura, T.; Sato, Y.; Ogata, N.; Ogihara, T.; Kaneda, Y.; Morishita, R. In vivo evidence of angiogenesis induced by transcription factor Ets-1: Ets-1 is located upstream of angiogenesis cascade. Circulation 2004, 109, 3035–3041. [Google Scholar] [CrossRef] [PubMed]
- Higashida, T.; Kanno, H.; Nakano, M.; Funakoshi, K.; Yamamoto, I. Expression of hypoxia-inducible angiogenic proteins (hypoxia-inducible factor-1α, vascular endothelial growth factor, and E26 transformation-specific-1) and plaque hemorrhage in human carotid atherosclerosis. J. Neurosurg. 2008, 109, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Kitange, G.; Shibata, S.; Tokunaga, Y.; Yagi, N.; Yasunaga, A.; Kishikawa, M.; Naito, S. Ets-1 transcription factor-mediated urokinase-type plasminogen activator expression and invasion in glioma cells stimulated by serum and basic fibroblast growth factors. Lab. Investig. J. Tech. Methods Pathol. 1999, 79, 407–416. [Google Scholar]
- Paumelle, R.; Tulasne, D.; Kherrouche, Z.; Plaza, S.; Leroy, C.; Reveneau, S.; Vandenbunder, B.; Fafeur, V. Hepatocyte growth factor/scatter factor activates the ETS1 transcription factor by a RAS-RAF-MEK-ERK signaling pathway. Oncogene 2002, 21, 2309–2319. [Google Scholar] [CrossRef] [PubMed]
- Langheinrich, A.C.; Kampschulte, M.; Scheiter, F.; Dierkes, C.; Stieger, P.; Bohle, R.M.; Weidner, W. Atherosclerosis, inflammation and lipoprotein glomerulopathy in kidneys of apoE−/−/LDL−/− double knockout mice. BMC Nephrol. 2010, 11. [Google Scholar] [CrossRef]
- Kumamoto, M.; Nakashima, Y.; Sueishi, K. Intimal neovascularization in human coronary atherosclerosis: Its origin and pathophysiological significance. Hum. Pathol. 1995, 26, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, A.; Shoji, K.; Tsuruda, T.; Furukoji, E.; Takahashi, M.; Nishihira, K.; Tamura, S.; Asada, Y. Medial and adventitial macrophages are associated with expansive atherosclerotic remodeling in rabbit femoral artery. Histol. Histopathol. 2008, 23, 127–136. [Google Scholar] [PubMed]
- Sluimer, J.C.; Daemen, M.J. Novel concepts in atherogenesis: Angiogenesis and hypoxia in atherosclerosis. J. Pathol. 2009, 218, 7–29. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.J.; Dean, R.T.; Jessup, W. Free and esterified oxysterol: Formation during copper-oxidation of low density lipoprotein and uptake by macrophages. J. Lipid Res. 1996, 37, 320–335. [Google Scholar] [PubMed]
- Ho-Tin-Noe, B.; le Dall, J.; Gomez, D.; Louedec, L.; Vranckx, R.; El-Bouchtaoui, M.; Legres, L.; Meilhac, O.; Michel, J.B. Early atheroma-derived agonists of peroxisome proliferator-activated receptor-gamma trigger intramedial angiogenesis in a smooth muscle cell-dependent manner. Circ. Res. 2011, 109, 1003–1014. [Google Scholar] [CrossRef] [PubMed]
- Sahebkar, A.; Watts, G.F. New LDL-cholesterol lowering therapies: Pharmacology, clinical trials, and relevance to acute coronary syndromes. Clin. Ther. 2013, 35, 1082–1098. [Google Scholar] [CrossRef] [PubMed]
- Lusis, A.J. Atherosclerosis. Nature 2000, 407, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Kolodgie, F.D.; Gold, H.K.; Burke, A.P.; Fowler, D.R.; Kruth, H.S.; Weber, D.K.; Farb, A.; Guerrero, L.J.; Hayase, M.; Kutys, R.; et al. Intraplaque hemorrhage and progression of coronary atheroma. N. Engl. J. Med. 2003, 349, 2316–2325. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Nagata, D.; Hirata, Y.; Tabata, Y.; Nagai, R.; Sata, M. Augmented angiogenesis in adventitia promotes growth of atherosclerotic plaque in apolipoprotein E-deficient mice. Atherosclerosis 2011, 215, 366–373. [Google Scholar] [CrossRef] [PubMed]
- Moulton, K.S. Angiogenesis in atherosclerosis: Gathering evidence beyond speculation. Curr. Opin. Lipidol. 2006, 17, 548–555. [Google Scholar] [CrossRef] [PubMed]
- Ricote, M.; Li, A.C.; Willson, T.M.; Kelly, C.J.; Glass, C.K. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature 1998, 391, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Liu, C.; Miller, Y.I. Zebrafish models of dyslipidemia: Relevance to atherosclerosis and angiogenesis. Transl. Res. J. Lab. Clin. Med. 2014, 163, 99–108. [Google Scholar] [CrossRef]
- Salomon, R.G.; Hong, L.; Hollyfield, J.G. Discovery of carboxyethylpyrroles (CEPs): Critical insights into AMD, autism, cancer, and wound healing from basic research on the chemistry of oxidized phospholipids. Chem. Res. Toxicol. 2011, 24, 1803–1816. [Google Scholar] [CrossRef] [PubMed]
- West, X.Z.; Malinin, N.L.; Merkulova, A.A.; Tischenko, M.; Kerr, B.A.; Borden, E.C.; Podrez, E.A.; Salomon, R.G.; Byzova, T.V. Oxidative stress induces angiogenesis by activating TLR2 with novel endogenous ligands. Nature 2010, 467, 972–976. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, T.K.; Stoll, L.L.; Denning, G.M.; Harrelson, A.; Blomkalns, A.L.; Idelman, G.; Rothenberg, F.G.; Neltner, B.; Romig-Martin, S.A.; Dickson, E.W.; et al. Proinflammatory phenotype of perivascular adipocytes: Influence of high-fat feeding. Circ. Res. 2009, 104, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Barandier, C.; Montani, J.P.; Yang, Z. Mature adipocytes and perivascular adipose tissue stimulate vascular smooth muscle cell proliferation: Effects of aging and obesity. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H1807–H1813. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Xu, T.Y.; Guan, Y.F.; Su, D.F.; Fan, G.R.; Miao, C.Y. Perivascular adipose tissue-derived visfatin is a vascular smooth muscle cell growth factor: Role of nicotinamide mononucleotide. Cardiovasc. Res. 2009, 81, 370–380. [Google Scholar] [CrossRef] [PubMed]
- Manka, D.; Chatterjee, T.K.; Stoll, L.L.; Basford, J.E.; Konaniah, E.S.; Srinivasan, R.; Bogdanov, V.Y.; Tang, Y.; Blomkalns, A.L.; Hui, D.Y.; Weintraub, N.L. Transplanted perivascular adipose tissue accelerates injury-induced neointimal hyperplasia: Role of monocyte chemoattractant protein-1. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1723–1730. [Google Scholar] [CrossRef] [PubMed]
- Rajsheker, S.; Manka, D.; Blomkalns, A.L.; Chatterjee, T.K.; Stoll, L.L.; Weintraub, N.L. Crosstalk between perivascular adipose tissue and blood vessels. Curr. Opin. Pharmacol. 2010, 10, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Touchard, A.; Henry, T.D.; Sangiorgi, G.; Spagnoli, L.G.; Mauriello, A.; Conover, C.; Schwartz, R.S. Extracellular proteases in atherosclerosis and restenosis. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1119–1127. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.Q.; Mao, Y.; Wang, B.; Lu, X.T.; Bai, W.W.; Sun, Y.Y.; Liu, Y.; Liu, H.M.; Zhang, L.; Zhao, Y.X.; et al. Specific matrix metalloproteinases play different roles in intraplaque angiogenesis and plaque instability in rabbits. PLoS ONE 2014, 9, e107851. [Google Scholar] [CrossRef] [PubMed]
- Svensson, P.A.; Olson, F.J.; Hagg, D.A.; Ryndel, M.; Wiklund, O.; Karlstrom, L.; Hulthe, J.; Carlsson, L.M.; Fagerberg, B. Urokinase-type plasminogen activator receptor is associated with macrophages and plaque rupture in symptomatic carotid atherosclerosis. Int. J. Mol. Med. 2008, 22, 459–464. [Google Scholar] [PubMed]
- Mattock, K.L.; Gough, P.J.; Humphries, J.; Burnand, K.; Patel, L.; Suckling, K.E.; Cuello, F.; Watts, C.; Gautel, M.; Avkiran, M.; et al. Legumain and cathepsin-L expression in human unstable carotid plaque. Atherosclerosis 2010, 208, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Kornmark, L.; Jonasson, L.; Forssell, C.; Yuan, X.M. Cathepsin L is significantly associated with apoptosis and plaque destabilization in human atherosclerosis. Atherosclerosis 2009, 202, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Le Dall, J.; Ho-Tin-Noe, B.; Louedec, L.; Meilhac, O.; Roncal, C.; Carmeliet, P.; Germain, S.; Michel, J.B.; Houard, X. Immaturity of microvessels in haemorrhagic plaques is associated with proteolytic degradation of angiogenic factors. Cardiovasc. Res. 2010, 85, 184–193. [Google Scholar]
- Leclercq, A.; Houard, X.; Philippe, M.; Ollivier, V.; Sebbag, U.; Meilhac, O.; Michel, J.B. Involvement of intraplaque hemorrhage in atherothrombosis evolution via neutrophil protease enrichment. J. Leukoc. Biol. 2007, 82, 1420–1429. [Google Scholar] [CrossRef] [PubMed]
- Maisonpierre, P.C.; Suri, C.; Jones, P.F.; Bartunkova, S.; Wiegand, S.J.; Radziejewski, C.; Compton, D.; McClain, J.; Aldrich, T.H.; Papadopoulos, N.; et al. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science 1997, 277, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Lam, T.; Boudreau, N.J.; Bollen, A.W.; Lawton, M.T.; Young, W.L. Abnormal balance in the angiopoietin-tie2 system in human brain arteriovenous malformations. Circ. Res. 2001, 89, 111–113. [Google Scholar] [CrossRef] [PubMed]
- Anagnostopoulos, A.; Eleftherakis-Papaiakovou, V.; Kastritis, E.; Tsionos, K.; Bamias, A.; Meletis, J.; Dimopoulos, M.A.; Terpos, E. Serum concentrations of angiogenic cytokines in Waldenstrom macroglobulinaemia: The ration of angiopoietin-1 to angiopoietin-2 and angiogenin correlate with disease severity. Br. J. Haematol. 2007, 137, 560–568. [Google Scholar] [CrossRef] [PubMed]
- Post, S.; Peeters, W.; Busser, E.; Lamers, D.; Sluijter, J.P.; Goumans, M.J.; de Weger, R.A.; Moll, F.L.; Doevendans, P.A.; Pasterkamp, G.; et al. Balance between angiopoietin-1 and angiopoietin-2 is in favor of angiopoietin-2 in atherosclerotic plaques with high microvessel density. J. Vasc. Res. 2008, 45, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. Contribution of neovascularization and intraplaque haemorrhage to atherosclerotic plaque progression and instability. Acta Physiol. 2015, 213, 539–553. [Google Scholar] [CrossRef]
- Nagy, E.; Eaton, J.W.; Jeney, V.; Soares, M.P.; Varga, Z.; Galajda, Z.; Szentmiklosi, J.; Mehes, G.; Csonka, T.; Smith, A.; et al. Red cells, hemoglobin, heme, iron, and atherogenesis. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1347–1353. [Google Scholar] [CrossRef] [PubMed]
- Balla, J.; Jacob, H.S.; Balla, G.; Nath, K.; Eaton, J.W.; Vercellotti, G.M. Endothelial-cell heme uptake from heme proteins: Induction of sensitization and desensitization to oxidant damage. Proc. Natl. Acad. Sci. USA 1993, 90, 9285–9289. [Google Scholar] [CrossRef] [PubMed]
- Potor, L.; Banyai, E.; Becs, G.; Soares, M.P.; Balla, G.; Balla, J.; Jeney, V. Atherogenesis may involve the prooxidant and proinflammatory effects of ferryl hemoglobin. Oxid. Med. Cell. Longev. 2013, 2013. [Google Scholar] [CrossRef]
- Juckett, M.B.; Balla, J.; Balla, G.; Jessurun, J.; Jacob, H.S.; Vercellotti, G.M. Ferritin protects endothelial cells from oxidized low density lipoprotein in vitro. Am. J. Pathol. 1995, 147, 782–789. [Google Scholar] [PubMed]
- Lee, F.Y.; Lee, T.S.; Pan, C.C.; Huang, A.L.; Chau, L.Y. Colocalization of iron and ceroid in human atherosclerotic lesions. Atherosclerosis 1998, 138, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, A.L. Modified hemoglobins produce venular interendothelial gaps and albumin leakage in the rat mesentery. Am. J. Physiol. 1999, 277, H650–H659. [Google Scholar] [PubMed]
- Abela, G.S.; Aziz, K.; Vedre, A.; Pathak, D.R.; Talbott, J.D.; Dejong, J. Effect of cholesterol crystals on plaques and intima in arteries of patients with acute coronary and cerebrovascular syndromes. Am. J. Cardiol. 2009, 103, 959–968. [Google Scholar] [CrossRef] [PubMed]
- Kaartinen, M.; Penttila, A.; Kovanen, P.T. Mast cells accompany microvessels in human coronary atheromas: Implications for intimal neovascularization and hemorrhage. Atherosclerosis 1996, 123, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Ambrose, J.A.; Tannenbaum, M.A.; Alexopoulos, D.; Hjemdahl-Monsen, C.E.; Leavy, J.; Weiss, M.; Borrico, S.; Gorlin, R.; Fuster, V. Angiographic progression of coronary artery disease and the development of myocardial infarction. J. Am. Coll. Cardiol. 1988, 12, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Hyafil, F.; Cornily, J.C.; Feig, J.E.; Gordon, R.; Vucic, E.; Amirbekian, V.; Fisher, E.A.; Fuster, V.; Feldman, L.J.; Fayad, Z.A. Noninvasive detection of macrophages using a nanoparticulate contrast agent for computed tomography. Nat. Med. 2007, 13, 636–641. [Google Scholar] [CrossRef] [PubMed]
- Romero, J.M.; Pizzolato, R.; Atkinson, W.; Meader, A.; Jaimes, C.; Lamuraglia, G.; Jaff, M.R.; Buonanno, F.; Delgado Almandoz, J.; Gonzalez, R.G. Vasa vasorum enhancement on computerized tomographic angiography correlates with symptomatic patients with 50% to 70% carotid artery stenosis. Stroke J. Cereb. Circ. 2013, 44, 3344–3349. [Google Scholar] [CrossRef]
- Sadeghi, M.M.; Glover, D.K.; Lanza, G.M.; Fayad, Z.A.; Johnson, L.L. Imaging atherosclerosis and vulnerable plaque. J. Nucl. Med. 2010, 51, 51S–65S. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; van der Steen, A.F.; Lancee, C.T.; Cespedes, I.; Bom, N. Blood flow imaging and volume flow quantitation with intravascular ultrasound. Ultrasound Med. Biol. 1998, 24, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Moritz, R.; Eaker, D.R.; Anderson, J.L.; Kline, T.L.; Jorgensen, S.M.; Lerman, A.; Ritman, E.L. IVUS detection of vasa vasorum blood flow distribution in coronary artery vessel wall. JACC Cardiovasc. Imaging 2012, 5, 935–940. [Google Scholar] [CrossRef] [PubMed]
- Papaioannou, T.G.; Vavuranakis, M.; Androulakis, A.; Lazaros, G.; Kakadiaris, I.; Vlaseros, I.; Naghavi, M.; Kallikazaros, I.; Stefanadis, C. In-vivo imaging of carotid plaque neoangiogenesis with contrast-enhanced harmonic ultrasound. Int. J. Cardiol. 2009, 134, e110–e112. [Google Scholar] [CrossRef] [PubMed]
- O’Malley, S.M.; Vavuranakis, M.; Naghavi, M.; Kakadiaris, I.A. Intravascular ultrasound-based imaging of vasa vasorum for the detection of vulnerable atherosclerotic plaque. Med. Image Comput. Comput. Assist. Interv. MICCAI 2005, 8 Pt 1, 343–351. [Google Scholar]
- Vavuranakis, M.; Kakadiaris, I.A.; O’Malley, S.M.; Papaioannou, T.G.; Sanidas, E.A.; Naghavi, M.; Carlier, S.; Tousoulis, D.; Stefanadis, C. A new method for assessment of plaque vulnerability based on vasa vasorum imaging, by using contrast-enhanced intravascular ultrasound and differential image analysis. Int. J. Cardiol. 2008, 130, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Goertz, D.E.; Frijlink, M.E.; Tempel, D.; van Damme, L.C.; Krams, R.; Schaar, J.A.; Ten Cate, F.J.; Serruys, P.W.; de Jong, N.; van der Steen, A.F. Contrast harmonic intravascular ultrasound: A feasibility study for vasa vasorum imaging. Investig. Radiol. 2006, 41, 631–638. [Google Scholar] [CrossRef]
- Goertz, D.E.; Frijlink, M.E.; Tempel, D.; Bhagwandas, V.; Gisolf, A.; Krams, R.; de Jong, N.; van der Steen, A.F. Subharmonic contrast intravascular ultrasound for vasa vasorum imaging. Ultrasound Med. Biol. 2007, 33, 1859–1872. [Google Scholar] [CrossRef] [PubMed]
- Goertz, D.E.; Frijlink, M.E.; de Jong, N.; van der Steen, A.F. Nonlinear intravascular ultrasound contrast imaging. Ultrasound Med. Biol. 2006, 32, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Magnoni, M.; Coli, S.; Marrocco-Trischitta, M.M.; Melisurgo, G.; de Dominicis, D.; Cianflone, D.; Chiesa, R.; Feinstein, S.B.; Maseri, A. Contrast-enhanced ultrasound imaging of periadventitial vasa vasorum in human carotid arteries. Eur. J. Echocardiogr. 2009, 10, 260–264. [Google Scholar] [CrossRef] [PubMed]
- Shah, F.; Balan, P.; Weinberg, M.; Reddy, V.; Neems, R.; Feinstein, M.; Dainauskas, J.; Meyer, P.; Goldin, M.; Feinstein, S.B. Contrast-enhanced ultrasound imaging of atherosclerotic carotid plaque neovascularization: A new surrogate marker of atherosclerosis? Vasc. Med. 2007, 12, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Schinkel, A.F.; Krueger, C.G.; Tellez, A.; Granada, J.F.; Reed, J.D.; Hall, A.; Zang, W.; Owens, C.; Kaluza, G.L.; Staub, D.; et al. Contrast-enhanced ultrasound for imaging vasa vasorum: Comparison with histopathology in a swine model of atherosclerosis. Eur. J. Echocardiogr. 2010, 11, 659–664. [Google Scholar] [CrossRef] [PubMed]
- Coli, S.; Magnoni, M.; Sangiorgi, G.; Marrocco-Trischitta, M.M.; Melisurgo, G.; Mauriello, A.; Spagnoli, L.; Chiesa, R.; Cianflone, D.; Maseri, A. Contrast-enhanced ultrasound imaging of intraplaque neovascularization in carotid arteries: Correlation with histology and plaque echogenicity. J. Am. Coll. Cardiol. 2008, 52, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.C.; Carr, C.L.; Davidson, B.P.; Ellegala, D.; Xie, A.; Ammi, A.; Belcik, T.; Lindner, J.R. Temporal characterization of the functional density of the vasa vasorum by contrast-enhanced ultrasonography maximum intensity projection imaging. JACC Cardiovasc. Imaging 2010, 3, 1265–1272. [Google Scholar] [CrossRef] [PubMed]
- Moguillansky, D.; Leng, X.; Carson, A.; Lavery, L.; Schwartz, A.; Chen, X.; Villanueva, F.S. Quantification of plaque neovascularization using contrast ultrasound: A histologic validation. Eur. Heart J. 2011, 32, 646–653. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Hu, S.; Sun, Y.; Yu, H.; Han, X.; Cheng, W.; Ban, X.; Zhang, S.; Yu, B.; Jang, I.K. Vasa vasorum and plaque progression, and responses to atorvastatin in a rabbit model of atherosclerosis: Contrast-enhanced ultrasound imaging and intravascular ultrasound study. Heart 2013, 99, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Deyama, J.; Nakamura, T.; Takishima, I.; Fujioka, D.; Kawabata, K.; Obata, J.E.; Watanabe, K.; Watanabe, Y.; Saito, Y.; Mishina, H.; et al. Contrast-enhanced ultrasound imaging of carotid plaque neovascularization is useful for identifying high-risk patients with coronary artery disease. Circ. J. 2013, 77, 1499–1507. [Google Scholar] [CrossRef] [PubMed]
- Vavuranakis, M.; Sigala, F.; Vrachatis, D.A.; Papaioannou, T.G.; Filis, K.; Kavantzas, N.; Kalogeras, K.I.; Massoura, C.; Toufektzian, L.; Kariori, M.G.; et al. Quantitative analysis of carotid plaque vasa vasorum by CEUS and correlation with histology after endarterectomy. VASA Z. Gefasskrankh. 2013, 42, 184–195. [Google Scholar] [CrossRef]
- Kubo, T.; Tanaka, A.; Ino, Y.; Kitabata, H.; Shiono, Y.; Akasaka, T. Assessment of coronary atherosclerosis using optical coherence tomography. J. Atheroscler. Thromb. 2014, 21, 895–903. [Google Scholar] [CrossRef] [PubMed]
- Kume, T.; Akasaka, T.; Kawamoto, T.; Watanabe, N.; Toyota, E.; Neishi, Y.; Sukmawan, R.; Sadahira, Y.; Yoshida, K. Assessment of coronary intima—Media thickness by optical coherence tomography: Comparison with intravascular ultrasound. Circ. J. 2005, 69, 903–907. [Google Scholar] [CrossRef] [PubMed]
- Kitabata, H.; Tanaka, A.; Kubo, T.; Takarada, S.; Kashiwagi, M.; Tsujioka, H.; Ikejima, H.; Kuroi, A.; Kataiwa, H.; Ishibashi, K.; et al. Relation of microchannel structure identified by optical coherence tomography to plaque vulnerability in patients with coronary artery disease. Am. J. Cardiol. 2010, 105, 1673–1678. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.H.; Sun, C.; Vuong, B.; Lee, K.K.; Mariampillai, A.; Marotta, T.R.; Spears, J.; Montanera, W.J.; Herman, P.R.; Kiehl, T.R.; et al. Endovascular optical coherence tomography intensity kurtosis: Visualization of vasa vasorum in porcine carotid artery. Biomed. Opt. Express 2012, 3, 388–399. [Google Scholar] [CrossRef] [PubMed]
- Tearney, G.J.; Regar, E.; Akasaka, T.; Adriaenssens, T.; Barlis, P.; Bezerra, H.G.; Bouma, B.; Bruining, N.; Cho, J.M.; Chowdhary, S.; et al. Consensus standards for acquisition, measurement, and reporting of intravascular optical coherence tomography studies: A report from the International Working Group for Intravascular Optical Coherence Tomography Standardization and Validation. J. Am. Coll. Cardiol. 2012, 59, 1058–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimiya, K.; Matsumoto, Y.; Takahashi, J.; Uzuka, H.; Odaka, Y.; Nihei, T.; Hao, K.; Tsuburaya, R.; Ito, K.; Shimokawa, H. In vivo visualization of adventitial vasa vasorum of the human coronary artery on optical frequency domain imaging. Validation study. Circ. J. 2014, 78, 2516–2518. [Google Scholar] [CrossRef] [PubMed]
- Aoki, T.; Rodriguez-Porcel, M.; Matsuo, Y.; Cassar, A.; Kwon, T.G.; Franchi, F.; Gulati, R.; Kushwaha, S.S.; Lennon, R.J.; Lerman, L.O.; et al. Evaluation of coronary adventitial vasa vasorum using 3D optical coherence tomography—Animal and human studies. Atherosclerosis 2015, 239, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Dobrucki, L.W.; Sinusas, A.J. PET and SPECT in cardiovascular molecular imaging. Nat. Rev. Cardiol. 2010, 7, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Tawakol, A.; Migrino, R.Q.; Bashian, G.G.; Bedri, S.; Vermylen, D.; Cury, R.C.; Yates, D.; LaMuraglia, G.M.; Furie, K.; Houser, S.; et al. In vivo 18F-fluorodeoxyglucose positron emission tomography imaging provides a noninvasive measure of carotid plaque inflammation in patients. J. Am. Coll. Cardiol. 2006, 48, 1818–1824. [Google Scholar] [CrossRef] [PubMed]
- Folco, E.J.; Sheikine, Y.; Rocha, V.Z.; Christen, T.; Shvartz, E.; Sukhova, G.K.; di Carli, M.F.; Libby, P. Hypoxia but not inflammation augments glucose uptake in human macrophages: Implications for imaging atherosclerosis with 18Fluorine-labeled 2-deoxy-d-glucose positron emission tomography. J. Am. Coll. Cardiol. 2011, 58, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Porcel, M.; Cai, W.; Gheysens, O.; Willmann, J.K.; Chen, K.; Wang, H.; Chen, I.Y.; He, L.; Wu, J.C.; Li, Z.B.; et al. Imaging of VEGF receptor in a rat myocardial infarction model using PET. J. Nucl. Med. 2008, 49, 667–673. [Google Scholar] [CrossRef] [PubMed]
- Meoli, D.F.; Sadeghi, M.M.; Krassilnikova, S.; Bourke, B.N.; Giordano, F.J.; Dione, D.P.; Su, H.; Edwards, D.S.; Liu, S.; Harris, T.D.; et al. Noninvasive imaging of myocardial angiogenesis following experimental myocardial infarction. J. Clin. Investig. 2004, 113, 1684–1691. [Google Scholar] [CrossRef] [PubMed]
- Janssen, M.L.; Oyen, W.J.; Dijkgraaf, I.; Massuger, L.F.; Frielink, C.; Edwards, D.S.; Rajopadhye, M.; Boonstra, H.; Corstens, F.H.; Boerman, O.C. Tumor targeting with radiolabeled αvβ3 integrin binding peptides in a nude mouse model. Cancer Res. 2002, 62, 6146–6151. [Google Scholar] [PubMed]
- Kopka, K.; Breyholz, H.J.; Wagner, S.; Law, M.P.; Riemann, B.; Schroer, S.; Trub, M.; Guilbert, B.; Levkau, B.; Schober, O.; et al. Synthesis and preliminary biological evaluation of new radioiodinated MMP inhibitors for imaging MMP activity in vivo. Nucl. Med. Biol. 2004, 31, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Furumoto, S.; Takashima, K.; Kubota, K.; Ido, T.; Iwata, R.; Fukuda, H. Tumor detection using 18F-labeled matrix metalloproteinase-2 inhibitor. Nucl.Med. Biol. 2003, 30, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Schafers, M.; Riemann, B.; Kopka, K.; Breyholz, H.J.; Wagner, S.; Schafers, K.P.; Law, M.P.; Schober, O.; Levkau, B. Scintigraphic imaging of matrix metalloproteinase activity in the arterial wall in vivo. Circulation 2004, 109, 2554–2559. [Google Scholar] [CrossRef] [PubMed]
- Matter, C.M.; Schuler, P.K.; Alessi, P.; Meier, P.; Ricci, R.; Zhang, D.; Halin, C.; Castellani, P.; Zardi, L.; Hofer, C.K.; et al. Molecular imaging of atherosclerotic plaques using a human antibody against the extra-domain B of fibronectin. Circ. Res. 2004, 95, 1225–1233. [Google Scholar] [CrossRef] [PubMed]
- Kuge, Y.; Takai, N.; Ogawa, Y.; Temma, T.; Zhao, Y.; Nishigori, K.; Ishino, S.; Kamihashi, J.; Kiyono, Y.; Shiomi, M.; et al. Imaging with radiolabelled anti-membrane type 1 matrix metalloproteinase (MT1-MMP) antibody: Potentials for characterizing atherosclerotic plaques. Eur. J. Nucl. Med. Mol. Imaging 2010, 37, 2093–2104. [Google Scholar] [CrossRef] [PubMed]
- Aoki, S.; Aoki, K.; Ohsawa, S.; Nakajima, H.; Kumagai, H.; Araki, T. Dynamic MR imaging of the carotid wall. J. Magn. Reson. Imaging JMRI 1999, 9, 420–427. [Google Scholar] [CrossRef]
- Winter, P.M.; Morawski, A.M.; Caruthers, S.D.; Fuhrhop, R.W.; Zhang, H.; Williams, T.A.; Allen, J.S.; Lacy, E.K.; Robertson, J.D.; Lanza, G.M.; et al. Molecular imaging of angiogenesis in early-stage atherosclerosis with αvβ3-integrin-targeted nanoparticles. Circulation 2003, 108, 2270–2274. [Google Scholar] [CrossRef] [PubMed]
- Cai, K.; Caruthers, S.D.; Huang, W.; Williams, T.A.; Zhang, H.; Wickline, S.A.; Lanza, G.M.; Winter, P.M. MR molecular imaging of aortic angiogenesis. JACC Cardiovasc. Imaging 2010, 3, 824–832. [Google Scholar] [CrossRef] [PubMed]
- Kerwin, W.; Hooker, A.; Spilker, M.; Vicini, P.; Ferguson, M.; Hatsukami, T.; Yuan, C. Quantitative magnetic resonance imaging analysis of neovasculature volume in carotid atherosclerotic plaque. Circulation 2003, 107, 851–856. [Google Scholar] [CrossRef] [PubMed]
- Kerwin, W.S.; Oikawa, M.; Yuan, C.; Jarvik, G.P.; Hatsukami, T.S. MR imaging of adventitial vasa vasorum in carotid atherosclerosis. Magn. Reson. Med. 2008, 59, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Song, Y.; Chen, H.; Kerwin, W.S.; Hippe, D.S.; Dong, L.; Chen, M.; Zhou, C.; Hatsukami, T.S.; Yuan, C. Adventitial perfusion and intraplaque hemorrhage: A dynamic contrast-enhanced MRI study in the carotid artery. Stroke J. Cereb. Circ. 2013, 44, 1031–1036. [Google Scholar] [CrossRef]
- Grobner, T. Gadolinium—A specific trigger for the development of nephrogenic fibrosing dermopathy and nephrogenic systemic fibrosis? Nephrol. Dial. Transpl. 2006, 21, 1104–1108. [Google Scholar] [CrossRef]
- Feinstein, S.B. Contrast ultrasound imaging of the carotid artery vasa vasorum and atherosclerotic plaque neovascularization. J. Am. Coll. Cardiol. 2006, 48, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Decano, J.L.; Moran, A.M.; Ruiz-Opazo, N.; Herrera, V.L. Molecular imaging of vasa vasorum neovascularization via DEspR-targeted contrast-enhanced ultrasound micro-imaging in transgenic atherosclerosis rat model. Mol. Imaging Biol. MIB 2011, 13, 1096–1106. [Google Scholar] [CrossRef]
- Baron Toaldo, M.; Salvatore, V.; Marinelli, S.; Palama, C.; Milazzo, M.; Croci, L.; Venerandi, L.; Cipone, M.; Bolondi, L.; Piscaglia, F. Use of VEGFR-2 targeted ultrasound contrast agent for the early evaluation of response to sorafenib in a mouse model of hepatocellular carcinoma. Mol. Imaging Biol. MIB 2015, 17, 29–37. [Google Scholar] [CrossRef]
- Araldi, E.; Chamorro-Jorganes, A.; van Solingen, C.; Fernandez-Hernando, C.; Suarez, Y. Therapeutic potential of modulating microRNAs in atherosclerotic vascular disease. Curr. Vasc. Pharmacol. 2013, in press. [Google Scholar]
- O’Reilly, M.S.; Holmgren, L.; Shing, Y.; Chen, C.; Rosenthal, R.A.; Moses, M.; Lane, W.S.; Cao, Y.; Sage, E.H.; Folkman, J. Angiostatin: A novel angiogenesis inhibitor that mediates the suppression of metastases by a Lewis lung carcinoma. Cell 1994, 79, 315–328. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, M.S.; Boehm, T.; Shing, Y.; Fukai, N.; Vasios, G.; Lane, W.S.; Flynn, E.; Birkhead, J.R.; Olsen, B.R.; Folkman, J. Endostatin: An endogenous inhibitor of angiogenesis and tumor growth. Cell 1997, 88, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Moulton, K.S.; Heller, E.; Konerding, M.A.; Flynn, E.; Palinski, W.; Folkman, J. Angiogenesis inhibitors endostatin or TNP-470 reduce intimal neovascularization and plaque growth in apolipoprotein E-deficient mice. Circulation 1999, 99, 1726–1732. [Google Scholar] [CrossRef] [PubMed]
- Moulton, K.S.; Vakili, K.; Zurakowski, D.; Soliman, M.; Butterfield, C.; Sylvin, E.; Lo, K.M.; Gillies, S.; Javaherian, K.; Folkman, J. Inhibition of plaque neovascularization reduces macrophage accumulation and progression of advanced atherosclerosis. Proc. Natl. Acad. Sci. USA 2003, 100, 4736–4741. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, K.; Magnusson, P.; Dixelius, J.; Claesson-Welsh, L.; Cross, M.J. Angiostatin and endostatin inhibit endothelial cell migration in response to FGF and VEGF without interfering with specific intracellular signal transduction pathways. FEBS Lett. 2003, 536, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Stefanadis, C.; Toutouzas, K.; Stefanadi, E.; Kolodgie, F.; Virmani, R.; Kipshidze, N. First experimental application of bevacizumab-eluting PC coated stent for inhibition of vasa vasorum of atherosclerotic plaque: Angiographic results in a rabbit atheromatic model. Hell. J. Cardiol. HJC 2006, 47, 7–10. [Google Scholar]
- Luttun, A.; Tjwa, M.; Moons, L.; Wu, Y.; Angelillo-Scherrer, A.; Liao, F.; Nagy, J.A.; Hooper, A.; Priller, J.; de Klerck, B.; et al. Revascularization of ischemic tissues by PlGF treatment, and inhibition of tumor angiogenesis, arthritis and atherosclerosis by anti-Flt1. Nat. Med. 2002, 8, 831–840. [Google Scholar] [PubMed]
- Khurana, R.; Moons, L.; Shafi, S.; Luttun, A.; Collen, D.; Martin, J.F.; Carmeliet, P.; Zachary, I.C. Placental growth factor promotes atherosclerotic intimal thickening and macrophage accumulation. Circulation 2005, 111, 2828–2836. [Google Scholar] [CrossRef] [PubMed]
- Fischer, C.; Jonckx, B.; Mazzone, M.; Zacchigna, S.; Loges, S.; Pattarini, L.; Chorianopoulos, E.; Liesenborghs, L.; Koch, M.; de Mol, M.; et al. Anti-PlGF inhibits growth of VEGF(R)-inhibitor-resistant tumors without affecting healthy vessels. Cell 2007, 131, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.D.; Kim, H.R.; Chesler, L.; Tsao-Wu, G.; Bouck, N.; Polverini, P.J. Inhibition of angiogenesis by tissue inhibitor of metalloproteinase. J. Cell. Physiol. 1994, 160, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Gossl, M.; Herrmann, J.; Tang, H.; Versari, D.; Galili, O.; Mannheim, D.; Rajkumar, S.V.; Lerman, L.O.; Lerman, A. Prevention of vasa vasorum neovascularization attenuates early neointima formation in experimental hypercholesterolemia. Basic Res. Cardiol. 2009, 104, 695–706. [Google Scholar] [CrossRef] [PubMed]
- Kampschulte, M.; Gunkel, I.; Stieger, P.; Sedding, D.G.; Brinkmann, A.; Ritman, E.L.; Krombach, G.A.; Langheinrich, A.C. Thalidomide influences atherogenesis in aortas of ApoE−/−/LDLR−/− double knockout mice: A nano-CT study. Int. J. Cardiovasc. Imaging 2014, 30, 795–802. [Google Scholar] [CrossRef] [PubMed]
- Dashwood, M.R.; Barker, S.G.; Muddle, J.R.; Yacoub, M.H.; Martin, J.F. [125I]-endothelin-1 binding to vasa vasorum and regions of neovascularization in human and porcine blood vessels: A possible role for endothelin in intimal hyperplasia and atherosclerosis. J. Cardiovasc. Pharmacol. 1993, 22, S343–S347. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, J.; Best, P.J.; Ritman, E.L.; Holmes, D.R.; Lerman, L.O.; Lerman, A. Chronic endothelin receptor antagonism prevents coronary vasa vasorum neovascularization in experimental hypercholesterolemia. J. Am. Coll. Cardiol. 2002, 39, 1555–1561. [Google Scholar] [CrossRef] [PubMed]
- Winter, P.M.; Neubauer, A.M.; Caruthers, S.D.; Harris, T.D.; Robertson, J.D.; Williams, T.A.; Schmieder, A.H.; Hu, G.; Allen, J.S.; Lacy, E.K.; et al. Endothelial αvβ3 integrin-targeted fumagillin nanoparticles inhibit angiogenesis in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2103–2109. [Google Scholar] [CrossRef] [PubMed]
- Drinane, M.; Mollmark, J.; Zagorchev, L.; Moodie, K.; Sun, B.; Hall, A.; Shipman, S.; Morganelli, P.; Simons, M.; Mulligan-Kehoe, M.J. The antiangiogenic activity of rPAI-123 inhibits vasa vasorum and growth of atherosclerotic plaque. Circ. Res. 2009, 104, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Mollmark, J.; Ravi, S.; Sun, B.; Shipman, S.; Buitendijk, M.; Simons, M.; Mulligan-Kehoe, M.J. Antiangiogenic activity of rPAI-123 promotes vasa vasorum regression in hypercholesterolemic mice through a plasmin-dependent mechanism. Circ. Res. 2011, 108, 1419–1428. [Google Scholar] [CrossRef] [PubMed]
- Langheinrich, A.C.; Sedding, D.G.; Kampschulte, M.; Moritz, R.; Wilhelm, J.; Haberbosch, W.G.; Ritman, E.L.; Bohle, R.M. 3-Deazaadenosine inhibits vasa vasorum neovascularization in aortas of ApoE−/−/LDLR−/− double knockout mice. Atherosclerosis 2009, 202, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.T.; Marshall, J.L.; Weiss, S.R.; Huang, C.Y.; Warren, J.L.; Freedman, A.N.; Fu, A.Z.; Sansbury, L.B.; Potosky, A.L. Bevacizumab use and risk of cardiovascular adverse events among elderly patients with colorectal cancer receiving chemotherapy: A population-based study. Ann. Oncol. 2013, 24, 1574–1579. [Google Scholar] [CrossRef] [PubMed]
- Colon, G.P.; Deveikis, J.P.; Dickinson, L.D. Revascularization of occluded internal carotid arteries by hypertrophied vasa vasorum: Report of four cases. Neurosurgery 1999, 45, 634–637. [Google Scholar] [CrossRef] [PubMed]
- Shanahan, C.M.; Weissberg, P.L. Smooth muscle cell phenotypes in atherosclerotic lesions. Curr. Opin. Lipidol. 1999, 10, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.H.; Herrmann, J.; Lerman, L.O.; Holmes, D.R., Jr.; Napoli, C.; Ritman, E.L.; Lerman, A. Simvastatin preserves the structure of coronary adventitial vasa vasorum in experimental hypercholesterolemia independent of lipid lowering. Circulation 2002, 105, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Urbich, C.; Dernbach, E.; Zeiher, A.M.; Dimmeler, S. Double-edged role of statins in angiogenesis signaling. Circ. Res. 2002, 90, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Chaldakov, G.N.; Stankulov, I.S.; Fiore, M.; Ghenev, P.I.; Aloe, L. Nerve growth factor levels and mast cell distribution in human coronary atherosclerosis. Atherosclerosis 2001, 159, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Asanome, A.; Kawabe, J.; Matsuki, M.; Kabara, M.; Hira, Y.; Bochimoto, H.; Yamauchi, A.; Aonuma, T.; Takehara, N.; Watanabe, T.; et al. Nerve growth factor stimulates regeneration of perivascular nerve, and induces the maturation of microvessels around the injured artery. Biochem. Biophys. Res. Commun. 2014, 443, 150–155. [Google Scholar] [CrossRef] [PubMed]
- Mollmark, J.I.; Park, A.J.; Kim, J.; Wang, T.Z.; Katzenell, S.; Shipman, S.L.; Zagorchev, L.G.; Simons, M.; Mulligan-Kehoe, M.J. Fibroblast growth factor-2 is required for vasa vasorum plexus stability in hypercholesterolemic mice. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2644–2651. [Google Scholar] [CrossRef] [PubMed]
- Lindner, V.; Lappi, D.A.; Baird, A.; Majack, R.A.; Reidy, M.A. Role of basic fibroblast growth factor in vascular lesion formation. Circ. Res. 1991, 68, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Six, I.; Mouquet, F.; Corseaux, D.; Bordet, R.; Letourneau, T.; Vallet, B.; Dosquet, C.C.; Dupuis, B.; Jude, B.; Bertrand, M.E.; et al. Protective effects of basic fibroblast growth factor in early atherosclerosis. Growth Factors 2004, 22, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Che, J.; Okigaki, M.; Takahashi, T.; Katsume, A.; Adachi, Y.; Yamaguchi, S.; Matsunaga, S.; Takeda, M.; Matsui, A.; Kishita, E.; et al. Endothelial FGF receptor signaling accelerates atherosclerosis. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H154–H161. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Shi, H.M.; Fan, W.H.; Luo, X.P.; Jin, B.; Li, Y. The effects of simvastatin on angiogenesis: Studied by an original model of atherosclerosis and acute myocardial infarction in rabbit. Mol. Biol. Rep. 2011, 38, 3821–3828. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, J.; Lu, X.; Shi, G.-P. Vasa Vasorum in Atherosclerosis and Clinical Significance. Int. J. Mol. Sci. 2015, 16, 11574-11608. https://doi.org/10.3390/ijms160511574
Xu J, Lu X, Shi G-P. Vasa Vasorum in Atherosclerosis and Clinical Significance. International Journal of Molecular Sciences. 2015; 16(5):11574-11608. https://doi.org/10.3390/ijms160511574
Chicago/Turabian StyleXu, Junyan, Xiaotong Lu, and Guo-Ping Shi. 2015. "Vasa Vasorum in Atherosclerosis and Clinical Significance" International Journal of Molecular Sciences 16, no. 5: 11574-11608. https://doi.org/10.3390/ijms160511574