
A Novel Nanoprobe for Multimodal Imaging Is Effectively Incorporated into Human Melanoma Metastatic Cell Lines

Abstract
:1. Introduction
2. Results and Discussion
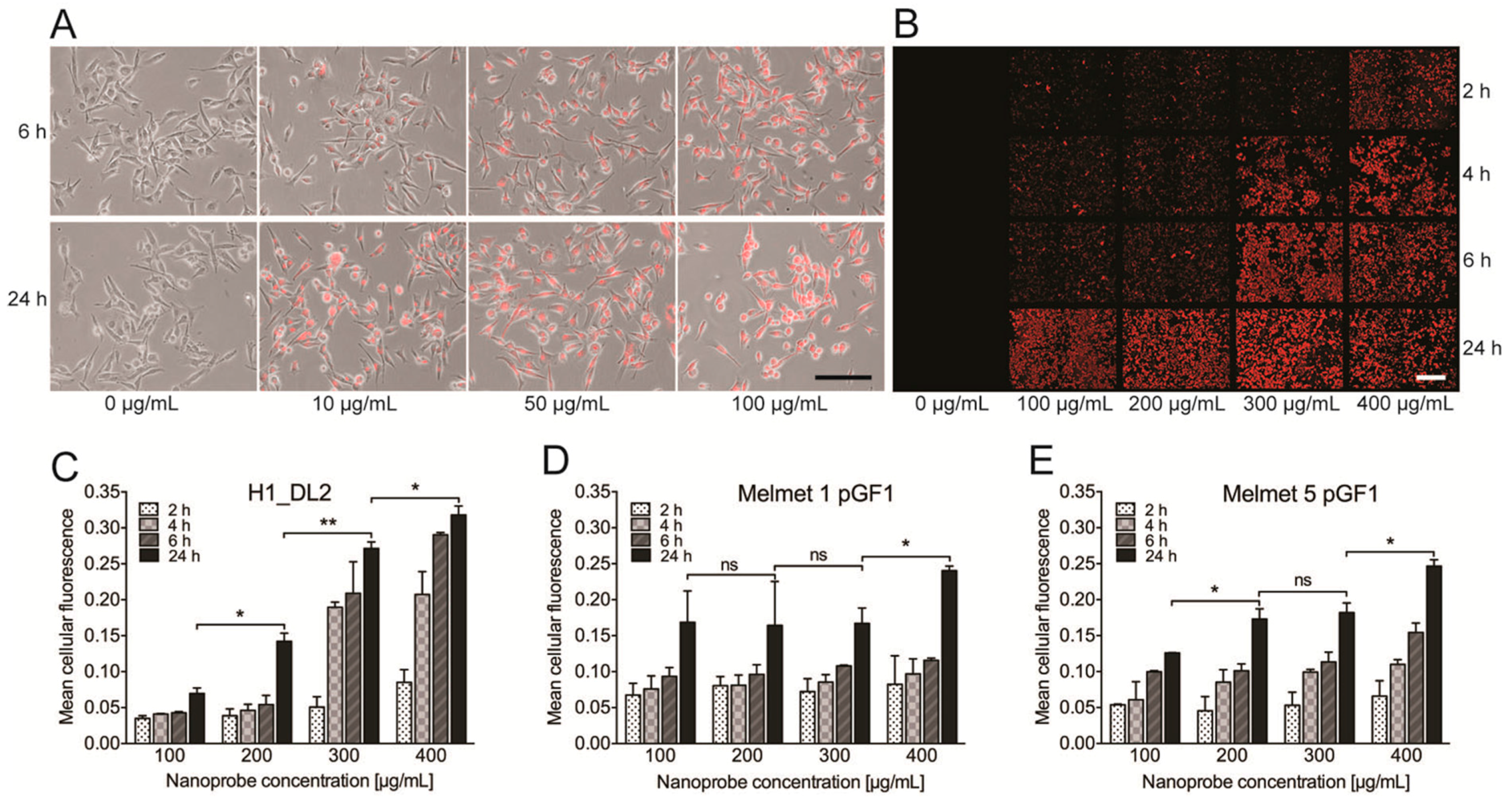
2.1. The Glycogen Nanoprobe Is Efficiently Internalized into the Metastatic Melanoma Cell Lines

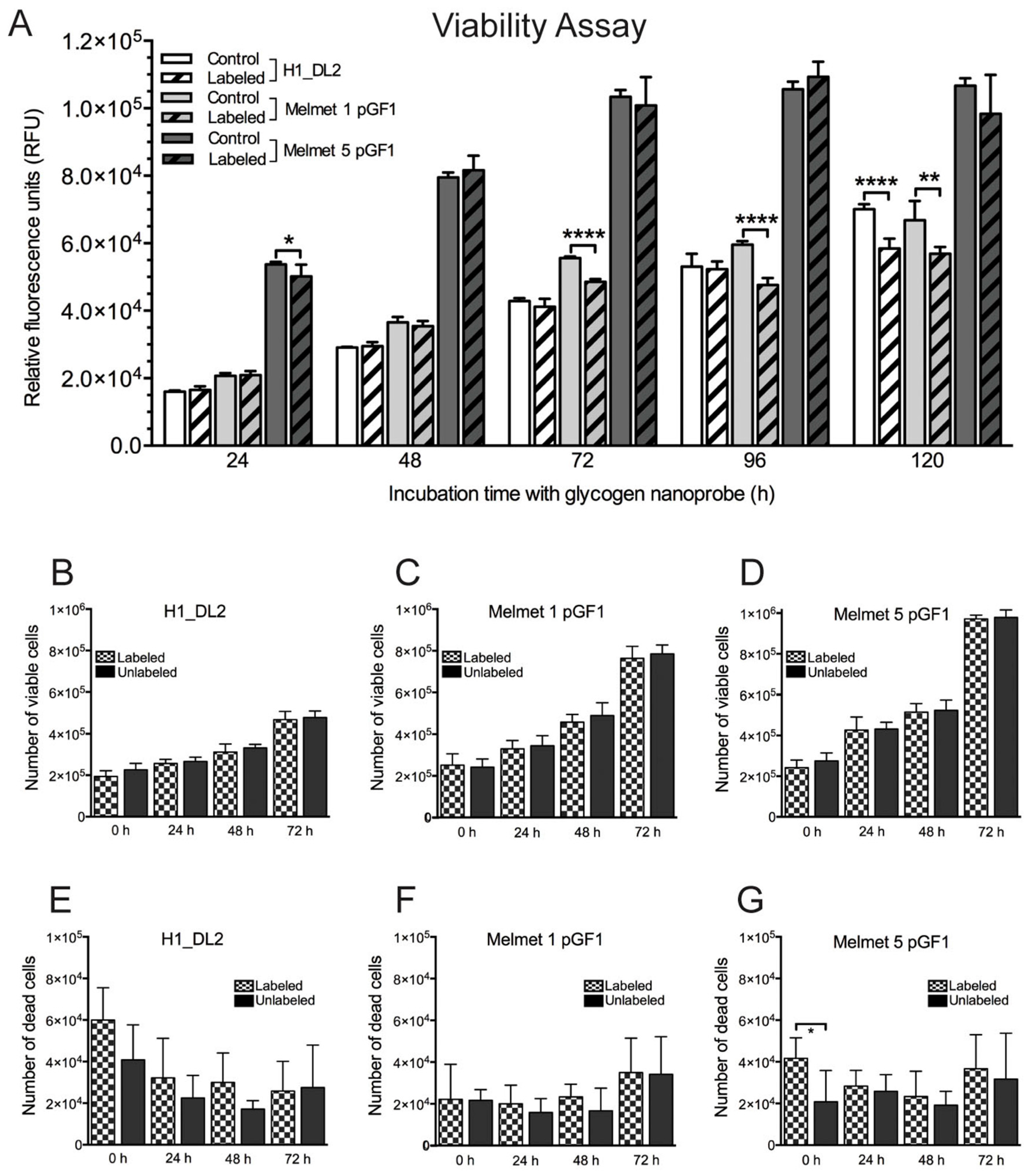
2.2. The Glycogen Nanoprobe Does Not Affect Short-Term Cell Viability in Vitro

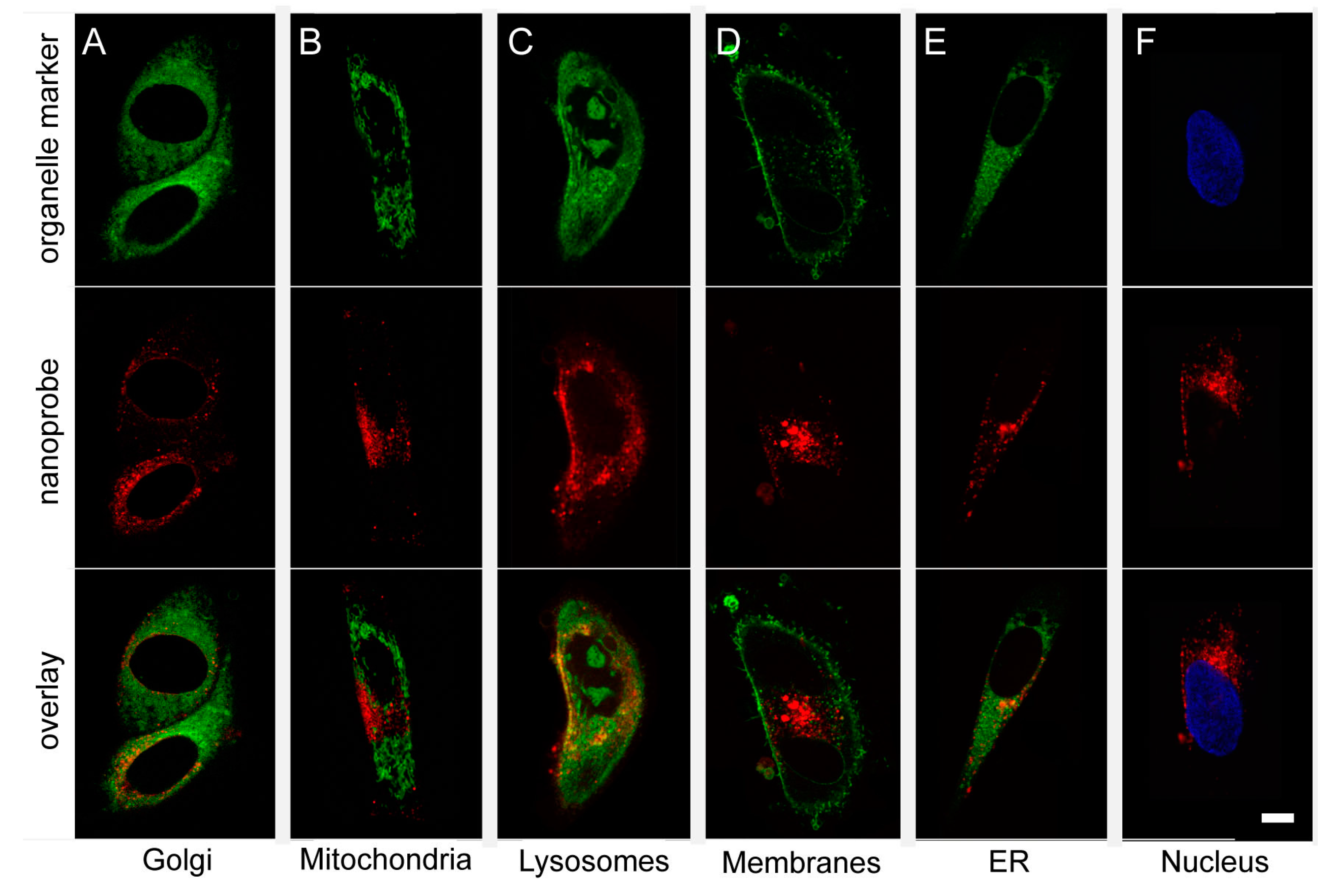
2.3. The Glycogen Nanoprobe Is Localized in Cytoplasm and Lysosomes


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organelle | Mean Pearson’s Correlation Coefficient | n | SD |
|---|---|---|---|
| Golgi apparatus | 0.0283 | 2 | 0.0063 |
| Mitochondria | 0.0574 | 1 | 0 |
| Lysosome | 0.5614 | 5 | 0.0849 |
| Plasma membrane | 0.0166 | 5 | 0.1003 |
| Endoplasmatic reticulum | 0.2839 | 3 | 0.0535 |
| Nucleus | 0.1117 | 11 | 0.2049 |
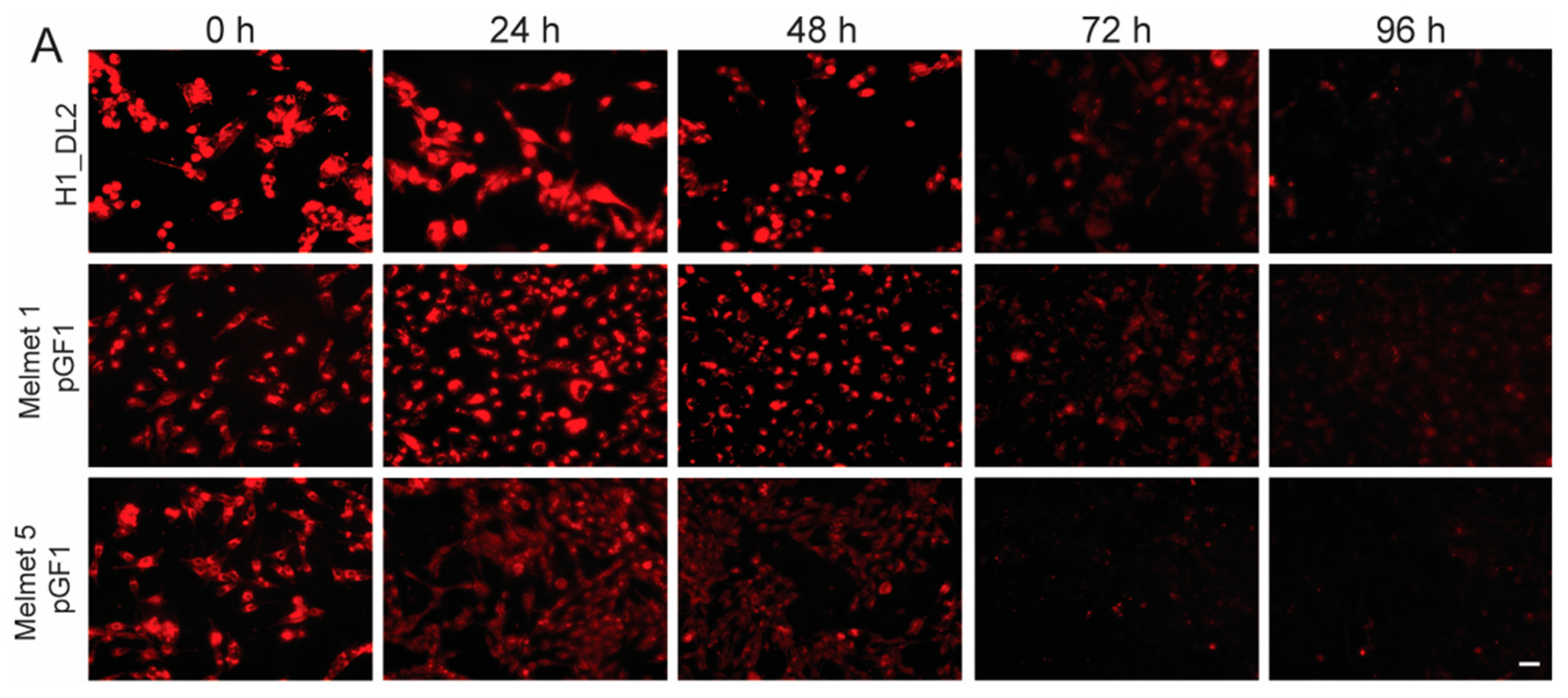
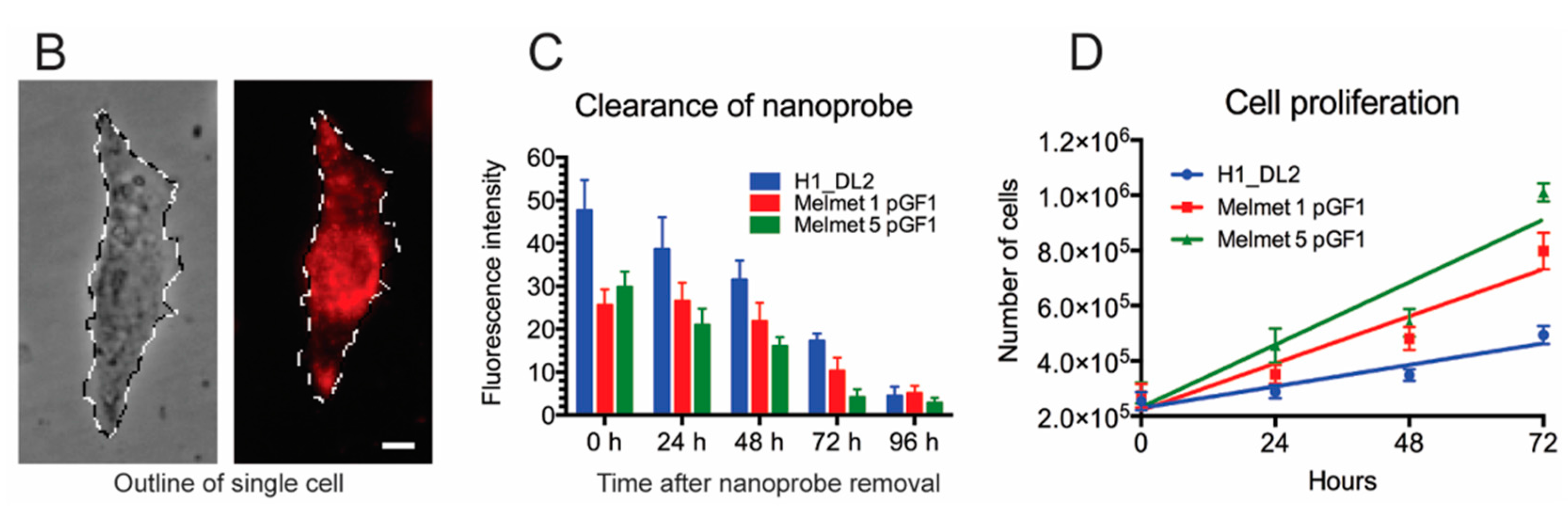
2.4. Intracellular Glycogen Nanoprobe Can Be Detected up to 96 h after Labeling
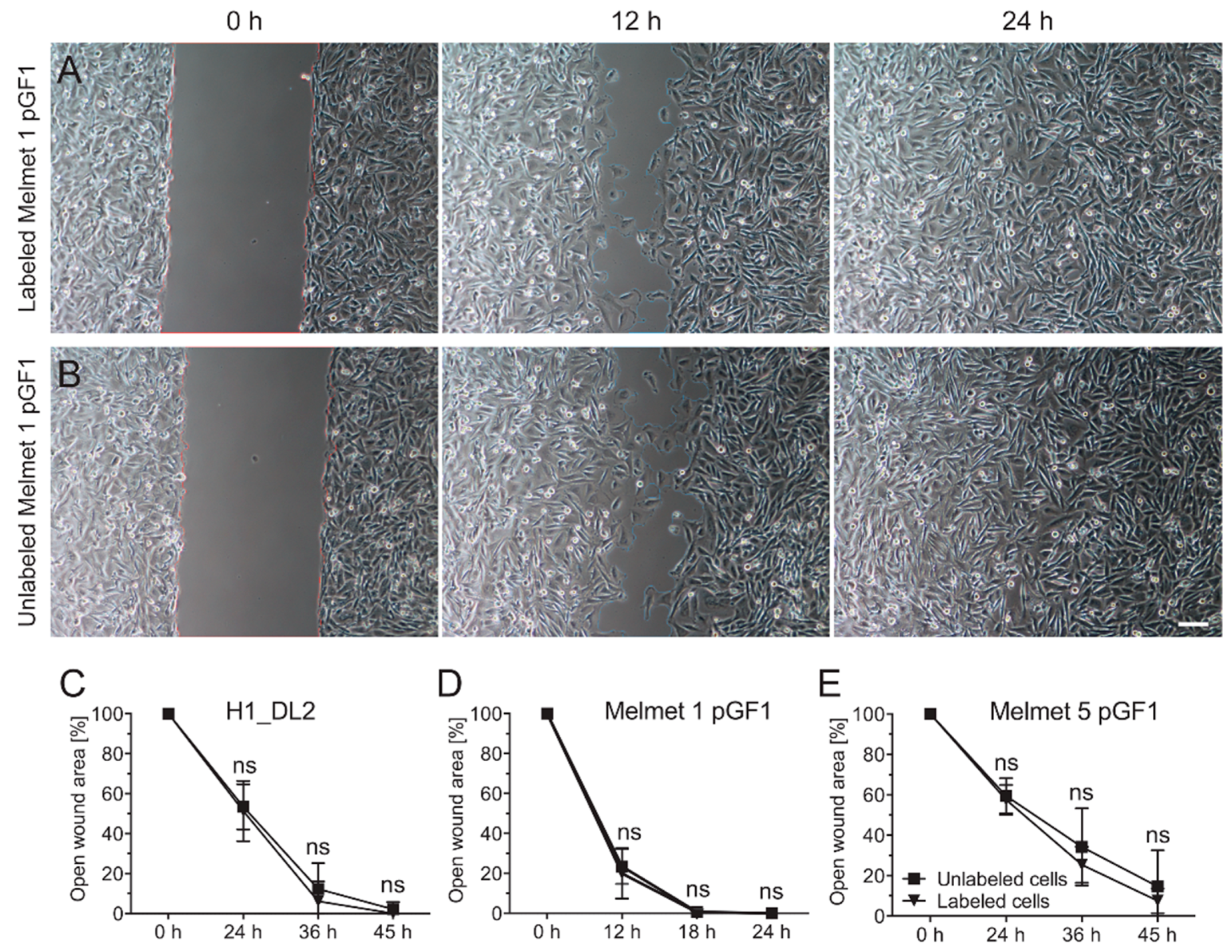
2.5. Labeling with the Glycogen Nanoprobe Does Not Influence in Vitro Cell Migration



2.6. The Surface Charge of the Nanoprobe Is Minimally Affected by pH
2.7. The Nanoprobe Is Stable in Blood Plasma for at Least 60 min
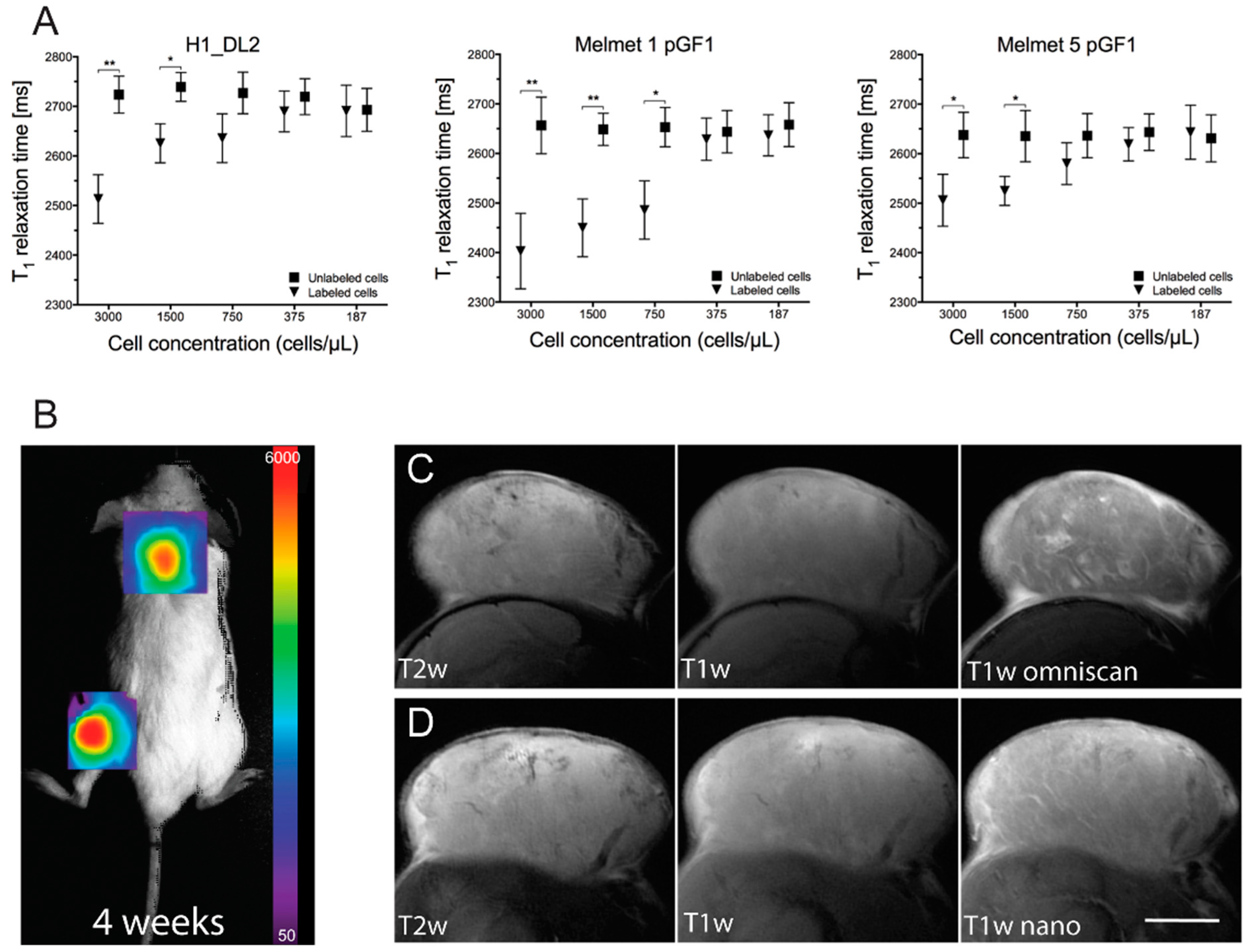
2.8. The Glycogen Nanoprobe Is an Effective Contrast Agent in Vitro

2.9. The Glycogen Nanoprobe Effectively Enhances MRI T1 Contrast in Subcutaneous Tumors
2.10. Discussion
3. Experimental Section
3.1. Glycogen Nanoprobe
3.2. Cell Lines and Cell Culture
3.3. Cellular Internalization of the Nanoprobe
3.4. Cell Viability
3.5. Subcellular Internalization of the Nanoprobe
3.6. Nanoprobe Clearance
3.7. Study of in Vitro Cell Migration
3.8. Measurement of Zeta-Potential
3.9. Measurement of Stability of the Glycogen Nanoprobe in Blood Plasma by Fluorescence Lifetime Correlation Spectroscopy (FLCS)
3.10. In Vitro MR Relaxometry
3.11. Animal Model and Tumor Cell Injections
3.12. Bioluminescence Imaging (BLI) of Tumor Development
3.13. Pilot in Vivo MRI Experiment
3.14. Statistical Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Spano, D.; Zollo, M. Tumor microenvironment: A main actor in the metastasis process. Clin. Exp. Metastasis 2012, 29, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Fink, K.R.; Fink, J.R. Imaging of brain metastases. Surg. Neurol. Int. 2013, 4 (Suppl. 4), S209–S219. [Google Scholar] [PubMed]
- Giovannini, E.; Lazzeri, P.; Milano, A.; Gaeta, M.C.; Ciarmiello, A. Clinical applications of choline PET/CT in brain tumors. Curr. Pharm. Des. 2015, 21, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Thorsen, F.; Fite, B.; Mahakian, L.M.; Seo, J.W.; Qin, S.; Harrison, V.; Johnson, S.; Ingham, E.; Caskey, C.; Sundstrøm, T.; et al. Multimodal imaging enables early detection and characterization of changes in tumor permeability of brain metastases. J. Control. Release 2013, 172, 812–822. [Google Scholar] [CrossRef] [PubMed]
- Jang, E.S.; Lee, S.Y.; Cha, E.J.; Sun, I.C.; Kwon, I.C.; Kim, D.; Kim, Y.I.; Kim, K.; Ahn, C.H. Fluorescent dye labeled iron oxide/silica core/shell nanoparticle as a multimodal imaging probe. Pharm. Res. 2014, 12, 3371–3378. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Zhao, J.; Conti, P.S.; Chen, K. Radiolabeled nanoparticles for multimodality tumor imaging. Theranostics 2014, 4, 290–306. [Google Scholar] [CrossRef] [PubMed]
- Khemtong, C.; Kessinger, C.W.; Gao, J. Polymeric nanomedicine for cancer MR imaging and drug delivery. Chem. Commun. 2009, 24, 3497–3510. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.E.; Koo, H.; Sun, I.C.; Ryu, J.H.; Kim, K.; Kwon, I.C. Multifunctional nanoparticles for multimodal imaging and theragnosis. Chem. Soc. Rev. 2012, 41, 2656–2672. [Google Scholar] [CrossRef] [PubMed]
- Sumer, B.; Gao, J. Theranostic nanomedicine for cancer. Nanomedicine 2008, 3, 137–140. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Yang, H.; Pan, W.; Diao, W.; Tang, B. A tumour mRNA-triggered nanocarrier for multimodal cancer cell imaging and therapy. Chem. Commun. 2014, 50, 7473–7476. [Google Scholar] [CrossRef] [PubMed]
- Laurent, S.; Saei, A.A.; Behzadi, S.; Panahifar, A.; Mahmoudi, M. Superparamagnetic iron oxide nanoparticles for delivery of therapeutic agents: Opportunities and challenges. Exp. Opin. Drug Deliv. 2014, 9, 1449–1470. [Google Scholar] [CrossRef] [PubMed]
- Jain, K.K. Nanobiotechnology-based strategies for crossing the blood-brain barrier. Nanomedicine 2012, 7, 1225–1233. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, P.M.; Chiocca, E.A. Nanotechnology-based strategies for the diagnosis and treatment of intracranial neoplasms. World Neurosurg. 2013, 80, 53–55. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Kievit, F.M.; Veiseh, O.; Arami, H.; Stephen, Z.R.; Fang, C.; Liu, Y.; Ellenbogen, R.G.; Zhang, M. Targeted cell uptake of a noninternalizing antibody through conjugation to iron oxide nanoparticles in primary central nervous system lymphoma. World Neurosurg. 2013, 80, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Yuk, S.H.; Oh, K.S.; Cho, S.H.; Lee, B.S.; Kim, S.Y.; Kwak, B.K.; Kim, K.; Kwon, I.C. Glycol chitosan/heparin immobilized iron oxide nanoparticles with a tumor-targeting characteristic for magnetic resonance imaging. Biomacromolecules 2011, 12, 2335–2343. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.I.; Kim, J.C.; Kim, Y.H.; Tae, G.; Lee, S.Y.; Kim, K.; Kwon, I.C. The effect of surface functionalization of PLGA nanoparticles by heparin- or chitosan-conjugated pluronic on tumor targeting. J. Control. Release 2010, 143, 374–382. [Google Scholar] [CrossRef] [PubMed]
- Mansa, R.; Detellier, C. Preparation and characterization of guar-montmorillonite nanocomposites. Materials 2013, 6, 5199–5216. [Google Scholar] [CrossRef]
- Wanga, C.; Huang, Y. Facile preparation of fluorescent Ag-clusters–chitosan-hybrid nanocomposites for bio-applications. New J. Chem. 2014, 38, 657–662. [Google Scholar] [CrossRef]
- Win, K.Y.; Feng, S.S. Effects of particle size and surface coating on cellular uptake of polymeric nanoparticles for oral delivery of anticancer drugs. Biomaterials 2005, 26, 2713–2722. [Google Scholar] [CrossRef] [PubMed]
- Sakhtianchi, R.; Minchin, R.F.; Lee, K.B.; Alkilany, A.M.; Serpooshan, V.; Mahmoudi, M. Exocytosis of nanoparticles from cells: Role in cellular retention and toxicity. Adv. Colloid Interface Sci. 2013, 201–202, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Ridley, A.J.; Schwartz, M.A.; Burridge, K.; Firtel, R.A.; Ginsberg, M.H.; Borisy, G.; Parsons, J.T.; Horwitz, A.R. Cell migration: Integrating signals from front to back. Science 2003, 302, 1704–1709. [Google Scholar] [CrossRef] [PubMed]
- Filippov, S.K.; Sedlacek, O.; Bogomolova, A.; Vetrik, M.; Jirak, D.; Kovar, J.; Kucka, J.; Bals, S.; Turner, S.; Stepanek, P.; et al. Glycogen as a biodegradable construction nanomaterial for in vivo use. Macromol. Biosci. 2012, 12, 1731–1738. [Google Scholar] [CrossRef] [PubMed]
- Naahidi, S.; Jafari, M.; Edalat, F.; Raymond, K.; Khademhosseini, A.; Chen, P. Biocompatibility of engineered nanoparticles for drug delivery. J. Control. Release 2013, 166, 182–194. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Li, J.; Lykotrafitis, G.; Bao, G.; Suresh, S. Size-dependent endocytosis of nanoparticles. Adv. Mater. 2009, 21, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Barua, S.; Rege, K. Cancer-cell-phenotype-dependent differential intracellular trafficking of unconjugated quantum dots. Small 2009, 5, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Kelf, T.A.; Sreenivasan, V.K.; Sun, J.; Kim, E.J.; Goldys, E.M.; Zvyagin, A.V. Non-specific cellular uptake of surface-functionalized quantum dots. Nanotechnology 2010, 21, 285105. [Google Scholar] [CrossRef] [PubMed]
- Chithrani Devika, B.; Chan, W.C.W. Elucidating the mechanism of cellular uptake and removal of protein-coated gold nanoparticles of different sizes and shapes. Nano Lett. 2007, 7, 1542–1550. [Google Scholar] [CrossRef] [PubMed]
- Rima, W.; Sancey, L.; Aloy, M.T.; Armandy, E.; Alcantara, G.B.; Epicier, T.; Malchere, A.; Joly-Pottuz, L.; Mowat, P.; Lux, F.; et al. Internalization pathways into cancer cells of gadolinium-based radiosensitizing nanoparticles. Biomaterials 2013, 34, 181–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Bauer, H.; Callahan, J.; Kopečková, P.; Pan, H.; Kopeček, J. Endocytic uptake of a large array of HPMA copolymers: Elucidation into the dependence on the physicochemical characteristics. J. Control. Release 2010, 143, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, J.C.; Favre, C.; Gomis, R.R.; Fernández-Novell, J.M.; Garcı́a-Rocha, M.; de la Iglesia, N.; Cid, E.; Guinovart, J.J. Control of glycogen deposition. FEBS Lett. 2003, 546, 127–132. [Google Scholar] [CrossRef]
- Glaumann, H.; Fredzell, J.; Jubner, A.; Ericsson, J.L.E. Uptake and degradation of glycogen by Kupffer cells. Exp. Mol. Pathol. 1979, 31, 70–80. [Google Scholar] [CrossRef]
- Rejman, J.; Oberle, V.; Zuhorn, I.S.; Hoekstra, D. Size-dependent internalization of particles via the pathways of clathrin- and caveolae-mediated endocytosis. Biochem. J. 2004, 377 Pt 1, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Fenouille, N.; Tichet, M.; Dufies, M.; Pottier, A.; Mogha, A.; Soo, J.K.; Rocchi, S.; Mallavialle, A.; Galibert, M.D.; Khammari, A.; et al. The epithelial-mesenchymal transition (EMT) regulatory factor SLUG (SNAI2) is a downstream target of SPARC and AKT in promoting melanoma cell invasion. PLoS ONE 2012, 7, e40378. [Google Scholar] [CrossRef] [PubMed]
- Eikenes, L.; Bruland, O.S.; Brekken, C.; Davies Cde, L. Collagenase increases the transcapillary pressure gradient and improves the uptake and distribution of monoclonal antibodies in human osteosarcoma xenografts. Cancer Res. 2004, 64, 4768–4773. [Google Scholar] [CrossRef] [PubMed]
- Sundstrøm, T.; Daphu, I.; Wendelbo, I.; Hodneland, E.; Lundervold, A.; Immervoll, H.; Skaftnesmo, K.O.; Babic, M.; Jendelova, P.; Syková, E.; et al. Automated tracking of nanoparticle-labeled melanoma cells improves the predictive power of a brain metastasis model. Cancer Res. 2013, 73, 2445–2456. [Google Scholar] [CrossRef] [PubMed]
- Terreno, E.; Geninatti Crich, S.; Belfiore, S.; Biancone, L.; Cabella, C.; Esposito, G.; Manazza, A.D.; Aime, S. Effect of the intracellular localization of a Gd-based imaging probe on the relaxation enhancement of water protons. Magn. Res. Med. 2006, 55, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Chacko, A.-M.; Li, C.; Pryma, D.A.; Brem, S.; Coukos, G.; Muzykantov, V. Targeted delivery of antibody-based therapeutic and imaging agents to CNS tumors: Crossing the blood-brain barrier divide. Exp. Opin. Drug Deliv. 2013, 10, 907–926. [Google Scholar] [CrossRef] [PubMed]
- Morris, D.L. Some effects of the intravenous injection of corn glycogen into rabbits. J. Biol. Chem. 1943, 148, 699–706. [Google Scholar]
- Czerney, P. Dyomics—Colours for life. In Fluorescent Dyes for Bioanalytical and Hightech Applications, 7th ed.; Dyomics GmbH: Jena, Germany, 2011; Volume 7, p. 84. [Google Scholar]
- Wang, J.; Daphu, I.; Pedersen, P.H.; Miletic, H.; Hovland, R.; Mørk, S.; Bjerkvig, R.; Tiron, C.; McCormack, E.; Micklem, D.; et al. A novel brain metastases model developed in immunodeficient rats closely mimics the growth of metastatic brain tumours in patients. Neuropathol. Appl. Neurobiol. 2011, 37, 189–205. [Google Scholar] [CrossRef] [PubMed]
- Prasmickaite, L.; Skrbo, N.; Høifødt, H.K.; Suo, Z.; Engebraten, O.; Gullestad, H.P.; Aamdal, S.; Fodstad, Ø.; Maelandsmo, G.M. Human malignant melanoma harbours a large fraction of highly clonogenic cells that do not express markers associated with cancer stem cells. Pigment Cell Melanoma Res. 2010, 23, 449–451. [Google Scholar] [CrossRef] [PubMed]
- Pawley, J.B. Handbook of Biological Confocal Microscopy, 3rd ed.; Springer Science + Business Media: New York, NY, USA, 2006. [Google Scholar]
- Kapusta, P.; Macháň, R.; Benda, A.; Hof, M. Fluorescence lifetime correlation spectroscopy (FLCS): Concepts, applications and outlook. Int. J. Mol. Sci. 2012, 13, 12890–12910. [Google Scholar] [CrossRef] [PubMed]
- Daphu, I.; Sundstrøm, T.; Horn, S.; Huszthy, P.C.; Niclou, S.P.; Sakariassen, P.Ø.; Immervoll, H.; Miletic, H.; Bjerkvig, R.; Thorsen, F. In vivo animal models for studying brain metastasis: Value and limitations. Clin. Exp. Metastasis 2013, 30, 695–710. [Google Scholar] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aasen, S.N.; Pospisilova, A.; Eichler, T.W.; Panek, J.; Hruby, M.; Stepanek, P.; Spriet, E.; Jirak, D.; Skaftnesmo, K.O.; Thorsen, F. A Novel Nanoprobe for Multimodal Imaging Is Effectively Incorporated into Human Melanoma Metastatic Cell Lines. Int. J. Mol. Sci. 2015, 16, 21658-21680. https://doi.org/10.3390/ijms160921658
Aasen SN, Pospisilova A, Eichler TW, Panek J, Hruby M, Stepanek P, Spriet E, Jirak D, Skaftnesmo KO, Thorsen F. A Novel Nanoprobe for Multimodal Imaging Is Effectively Incorporated into Human Melanoma Metastatic Cell Lines. International Journal of Molecular Sciences. 2015; 16(9):21658-21680. https://doi.org/10.3390/ijms160921658
Chicago/Turabian StyleAasen, Synnøve Nymark, Aneta Pospisilova, Tilo Wolf Eichler, Jiri Panek, Martin Hruby, Petr Stepanek, Endy Spriet, Daniel Jirak, Kai Ove Skaftnesmo, and Frits Thorsen. 2015. "A Novel Nanoprobe for Multimodal Imaging Is Effectively Incorporated into Human Melanoma Metastatic Cell Lines" International Journal of Molecular Sciences 16, no. 9: 21658-21680. https://doi.org/10.3390/ijms160921658