
Ghrelin Attenuates Liver Fibrosis through Regulation of TGF-β1 Expression and Autophagy

Abstract
:1. Introduction
2. Results
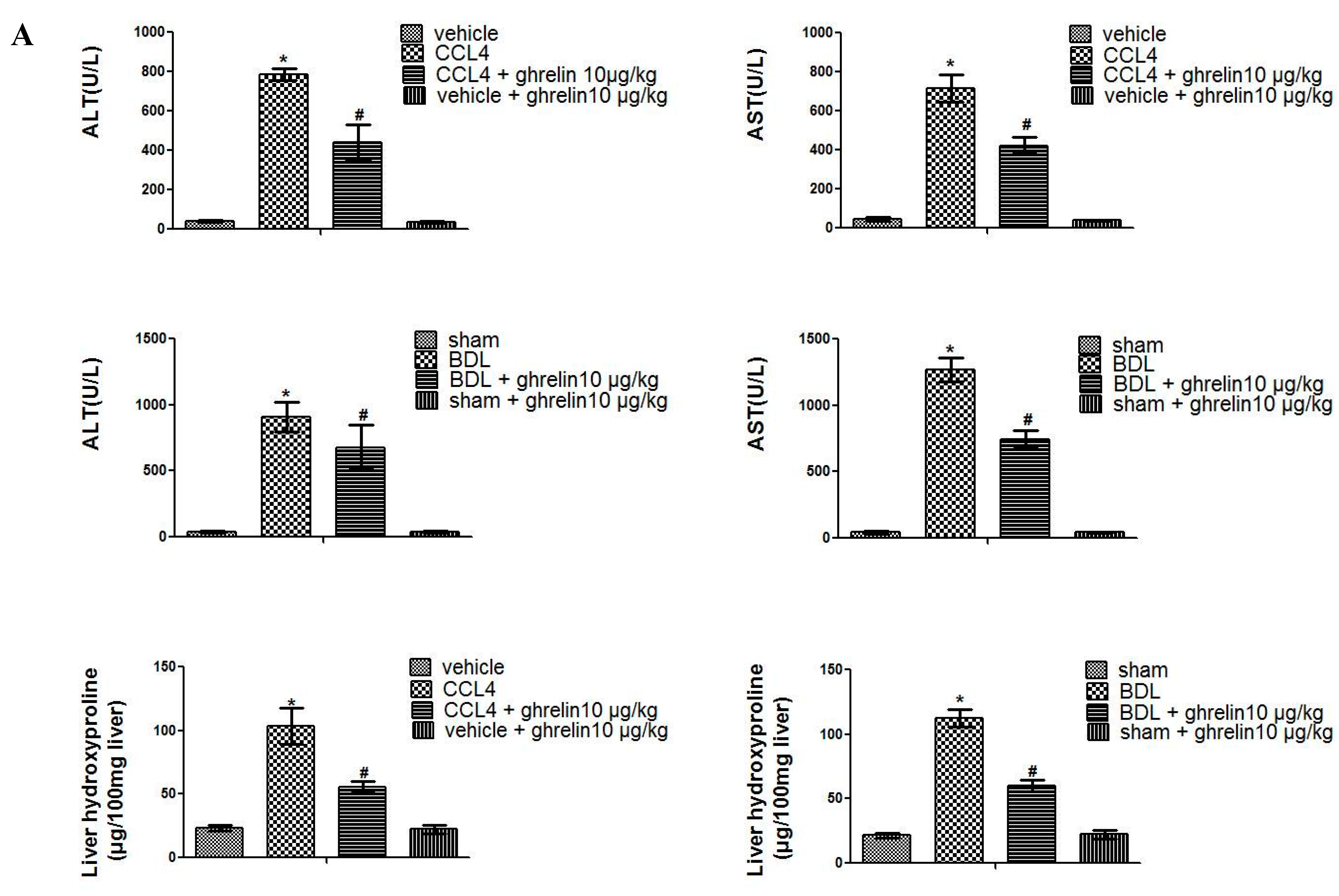
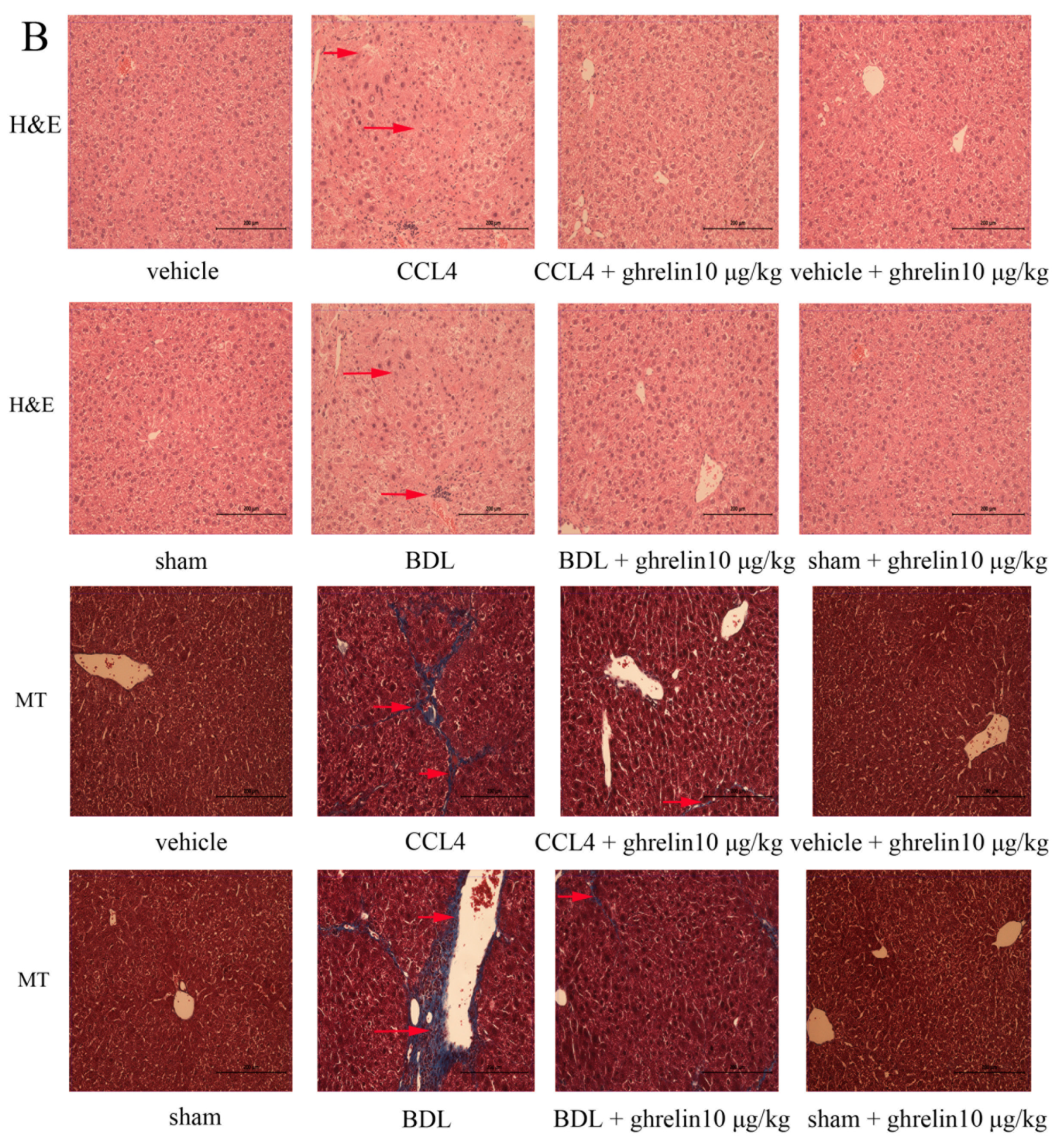
2.1. Liver Fibrosis Was Reduced by Ghrelin Administration in Both CCl4- and BDL-Induced Liver Fibrosis Mouse Models


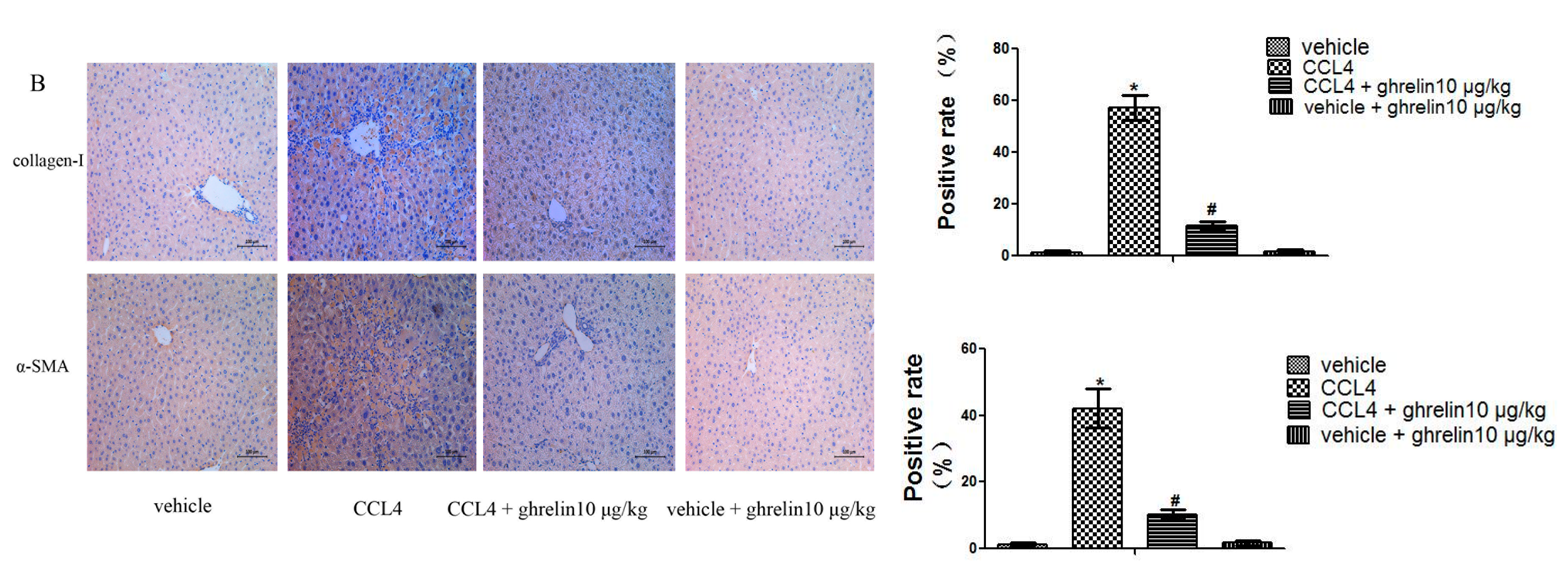
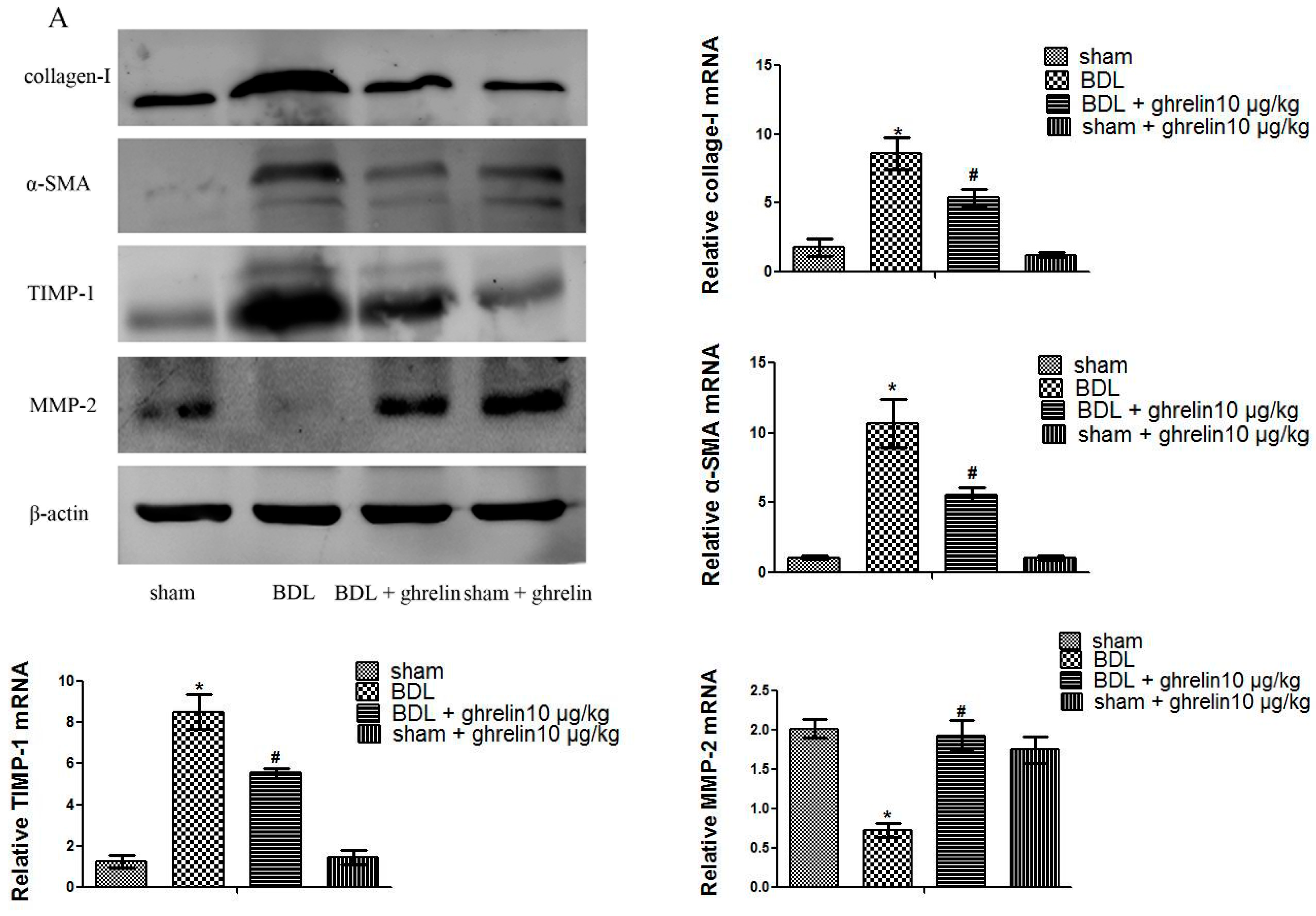
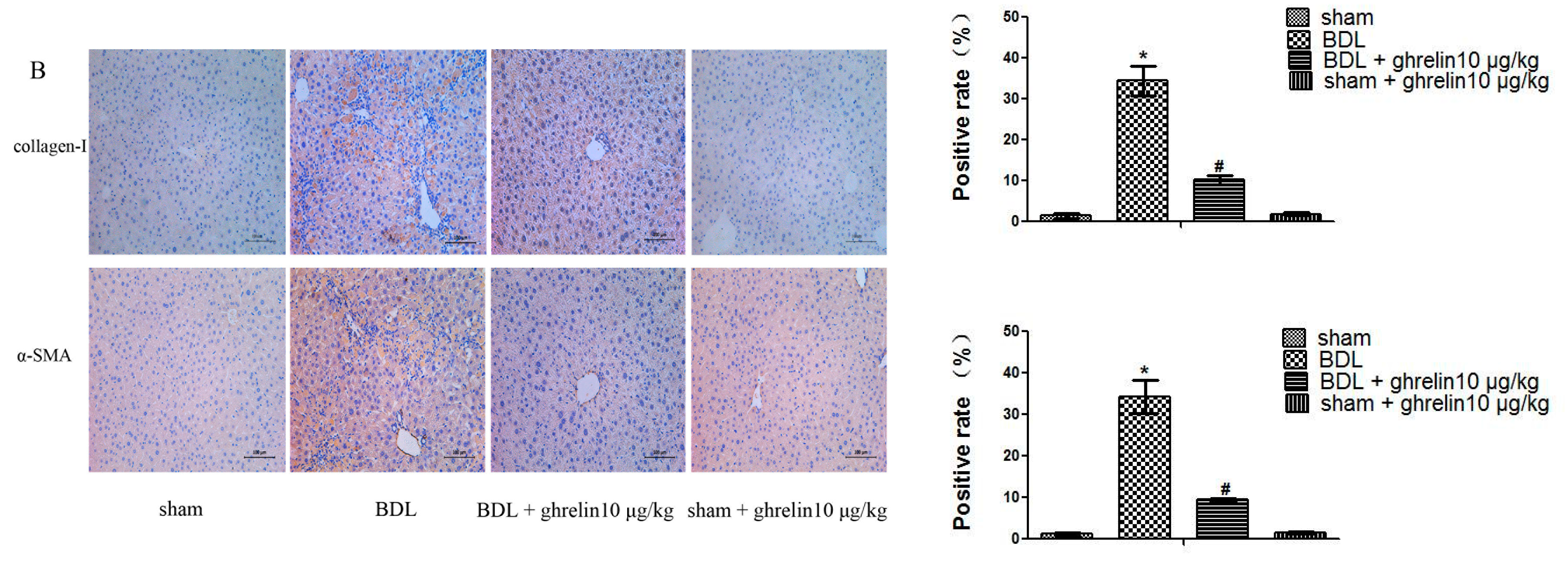
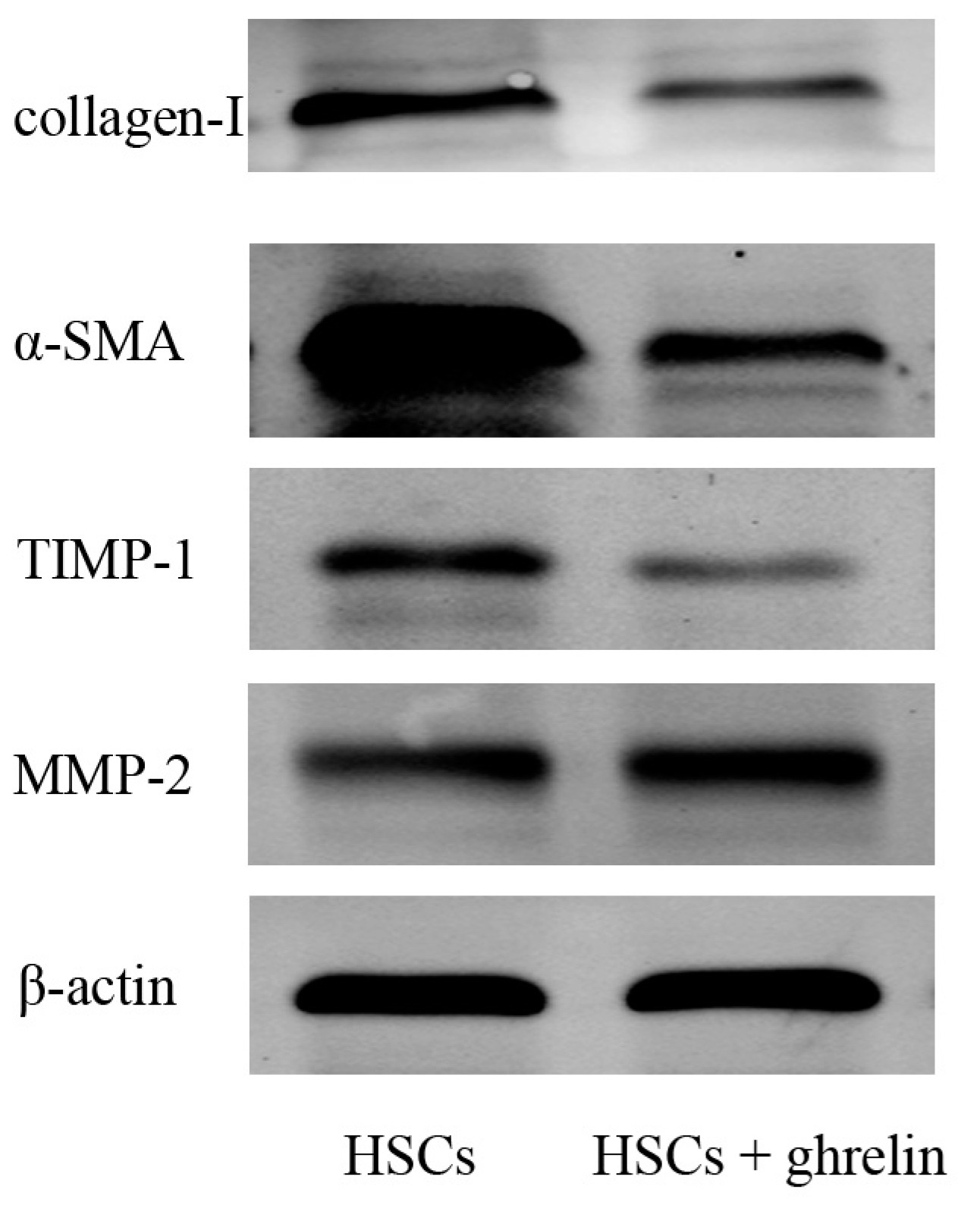
2.2. Ghrelin Inhibits Activation of HSCs in Liver Fibrosis and Regulates the Balance of MMP2 and TIPM1





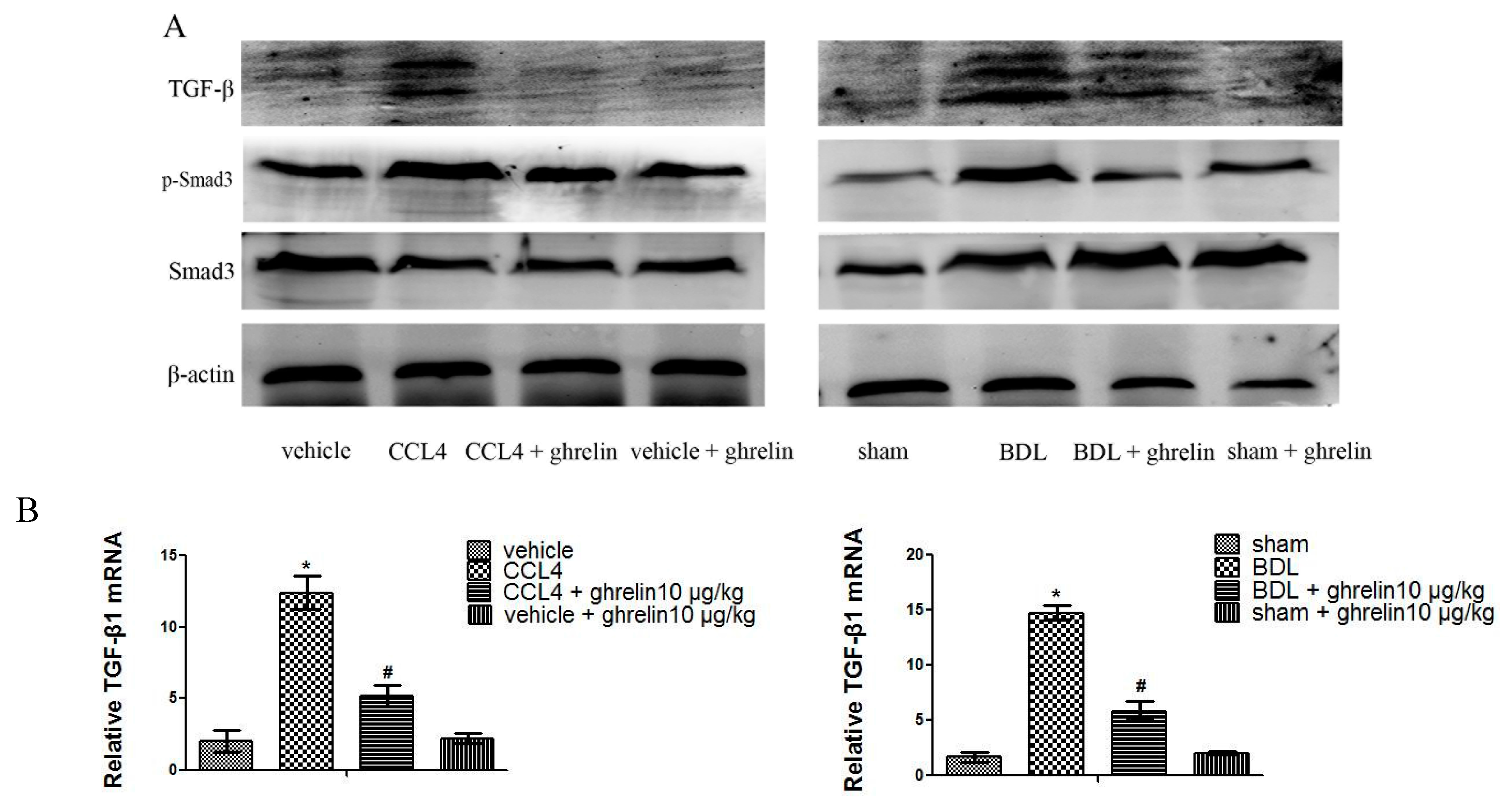
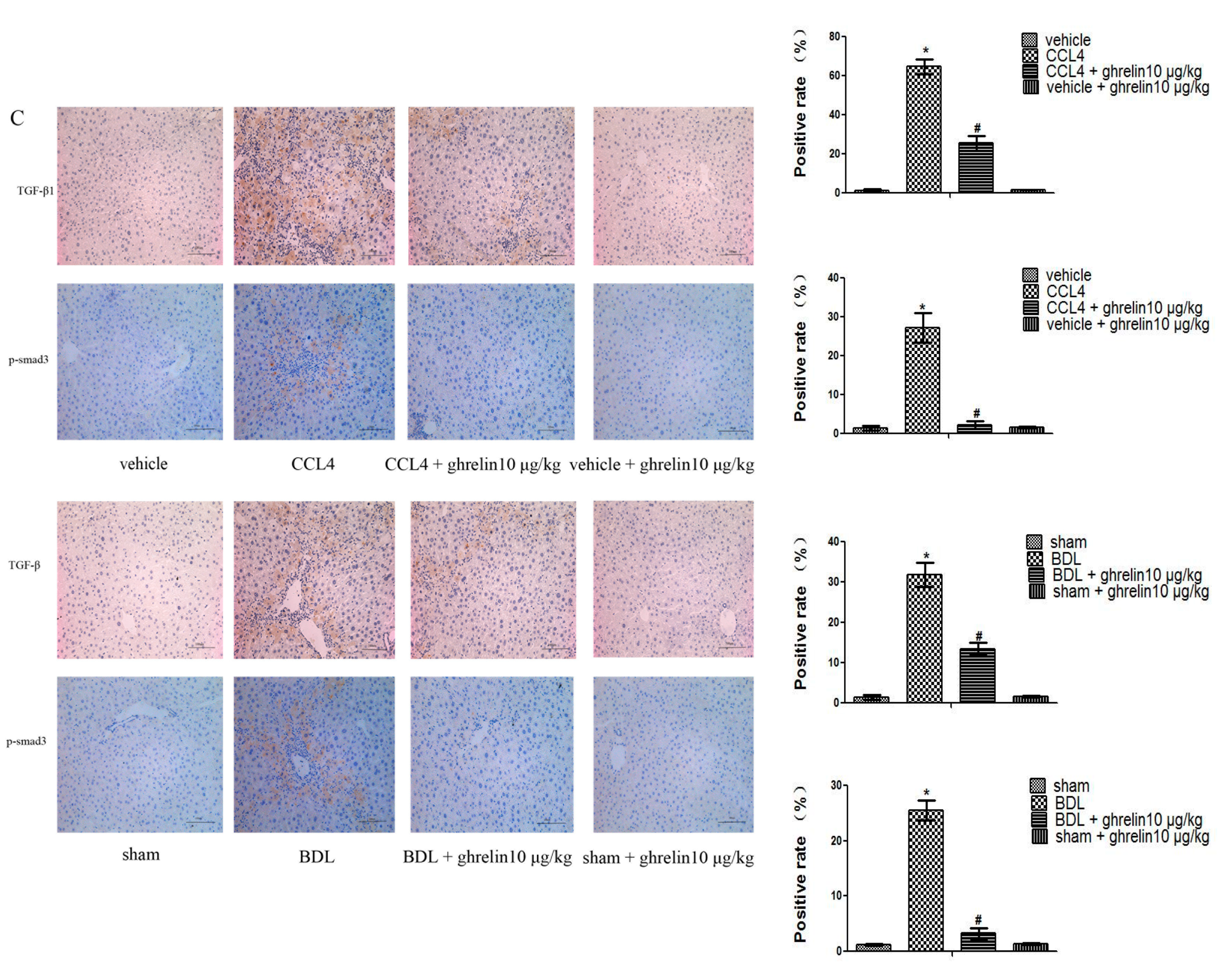
2.3. Ghrelin Regulates the TGF-β1/Smad3 Signaling Pathway in Liver Fibrosis
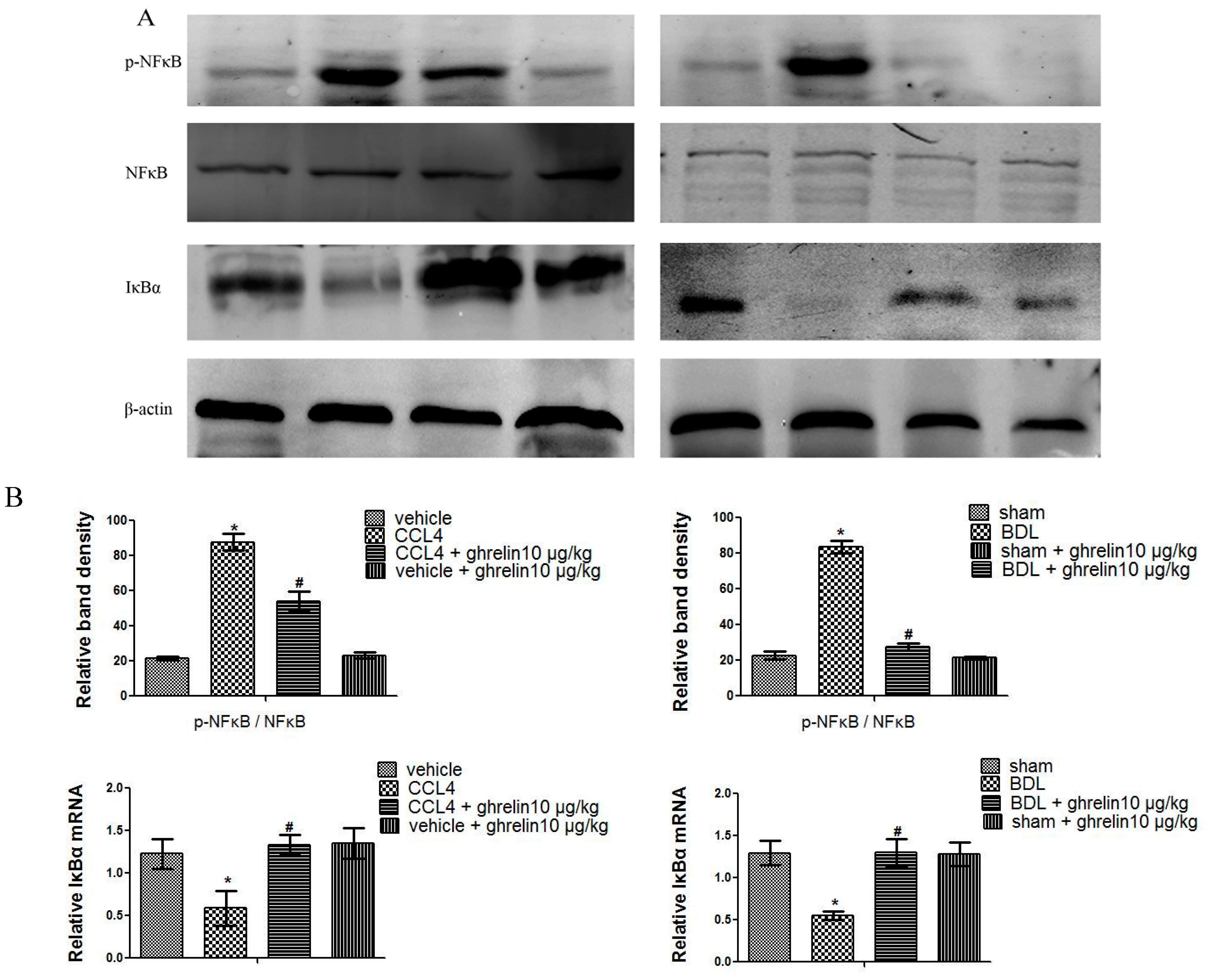
2.4. Ghrelin Attenuates Liver Injury via Inhibition of the NF-κB Signaling Pathway in Liver Fibrosis
2.5. Ghrelin Inhibits the Autophagy Process in Liver Fibrosis




3. Discussion
4. Experimental Section
4.1. Study Approval
4.2. Reagents
4.3. Animals and Treatment
4.4. Cell Culture and Treatment
4.5. Biochemical Analysis
4.6. Histopathological Analysis
4.7. Immunohistochemical Analysis
4.8. Western Blot Analysis
4.9. RNA Isolation and Real-Time Quantitative Reverse Transcriptase Polymerase Chain Reaction (qRT-PCR)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Sequence (5′-3′) | |
|---|---|---|
| Collagen I | Forward | CAATGGCACGGCTGTGTGCG |
| Reverse | AGCACTCGCCCTCCCGTCTT | |
| α-SMA | Forward | CCCAGACATCAGGGAGTAATGG |
| Reverse | TCTATCGGATACTTCAGCGTCA | |
| TIMP1 | Forward | CGAGACCACCTTATACCAGCG |
| Reverse | ATGACTGGGGTGTAGGCGTA | |
| MMP2 | Forward | GGACAAGTGGTCCGCGTAAA |
| Reverse | CCGACCGTTGAACAGGAAGG | |
| TGF-β1 | Forward | CCACCTGCAAGACCATCGAC |
| Reverse | CTGGCGAGCCTTAGTTTGGAC | |
| IκBα | Forward | GCCCCGCACAGCCATGTTTC |
| Reverse | AGCGGACAGGCGAGGAGAGC | |
| LC3 | Forward | GACCGCTGTAAGGAGGTGC |
| Reverse | AGAAGCCGAAGGTTTCTTGGG | |
| β-actin | Forward | GGCTGTATTCCCCTCCATCG |
| Reverse | CCAGTTGGTAACAATGCCATGT | |
4.10. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dranoff, J.A.; Wells, R.G. Portal fibroblasts: Underappreciated mediators of biliary fibrosis. Hepatology 2010, 5, 1438–4144. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. The cellular basis of hepatic fibrosis—Mechanisms and treatment strategies. N. Engl. J. Med. 1993, 328, 1828–1835. [Google Scholar] [PubMed]
- Sherman, M.; Klein, A. AASLD single-topic research conference on hepatocellular carcinoma: Conference proceedings. Hepatology 2004, 40, 1465–1473. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.S.; Kim, W.R. The global impact of hepatic fibrosis and end-stage liver disease. Clin. Liver Dis. 2008, 12, 733–746. [Google Scholar] [CrossRef] [PubMed]
- De Minicis, S.; Seki, E.; Uchinami, H.; Kluwe, J.; Zhang, Y.; Brenner, D.A.; Schwabe, R.F. Gene expression profiles during hepatic stellate cell activation in culture and in vivo. Gastroenterology 2007, 132, 1937–1946. [Google Scholar] [CrossRef] [PubMed]
- Cassiman, D.; Libbrecht, L.; Desmet, V.; Denef, C.; Roskams, T. Hepatic stellate cell/myofibroblast subpopulations in fibrotic human and rat livers. J. Hepatol. 2002, 36, 200–209. [Google Scholar] [CrossRef]
- Friedman, S.L. Mechanisms of hepatic fibrogenesis. Gastroenterology 2008, 134, 1655–1669. [Google Scholar] [CrossRef] [PubMed]
- Knittel, T.; Mehde, M.; Kobold, D.; Saile, B.; Dinter, C.; Ramadori, G. Expression patterns of matrix metalloproteinases and their inhibitors in parenchymal and non-parenchymal cells of rat liver: Regulation by TNF-α and TGF-β. J. Hepatol. 1999, 30, 48–60. [Google Scholar] [CrossRef]
- Massagué, J.; Blain, S.W.; Lo, R.S. TGFβ signaling in growth control, cancer, and heritable disorders. Cell 2000, 103, 295–309. [Google Scholar] [CrossRef]
- Friedman, S.L. Liver fibrosis—From bench to bedside. J. Hepatol. 2003, 38, 38–53. [Google Scholar] [CrossRef]
- Gressner, A.M.; Weiskirchen, R.; Breitkopf, K.; Dooley, S. Roles of TGF-β in hepatic fibrosis. Front. Biosci. 2002, 7, 793–807. [Google Scholar] [CrossRef]
- Sentíes-Gómez, M.D.; Gálvez-Gastélum, F.J.; Meza-García, E.; Armendáriz-Borunda, J. Hepatic fibrosis: Role of matrix metalloproteases and TGFβ. Gac. Med. Mex. 2005, 141, 315–322. [Google Scholar] [PubMed]
- Jaeschke, H. Mechanisms of liver injury. II. Mechanisms of neutrophil-induced liver cell injury during hepatic ischemia-reperfusion and other acute inflammatory conditions. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G1083–G1088. [Google Scholar] [CrossRef] [PubMed]
- Elsharkawy, A.M.; Mann, D.A. Nuclear factor-κB and the hepatic inflammation-fibrosis-cancer axis. Hepatology 2007, 46, 590–597. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Rautou, P.E.; Mansouri, A.; Lebrec, D.; Durand, F.; Valla, D.; Moreau, R. Autophagy in liver diseases. J. Hepatol. 2010, 53, 1123–1134. [Google Scholar] [CrossRef] [PubMed]
- Murrow, L.; Debnath, J. Autophagy as a stress-response and quality-control mechanism: Implications for cell injury and human disease. Annu. Rev. Pathol. 2013, 8, 105–137. [Google Scholar] [CrossRef] [PubMed]
- Thoen, L.F.; Guimaraes, E.L.; Grunsven, L.A. Autophagy: A new player in hepatic stellate cell activation. Autophagy 2012, 8, 126–128. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Gea, V.; Ghiassi-Nejad, Z.; Rozenfeld, R.; Gordon, R.; Fiel, M.I.; Yue, Z.; Czaja, M.J.; Friedman, S.L. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology 2012, 142, 938–946. [Google Scholar] [CrossRef] [PubMed]
- Kojima, M.; Hosoda, H.; Date, Y.; Nakazato, M.; Matsuo, H.; Kangawa, K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature 1999, 402, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Gnanapavan, S.; Kola, B.; Bustin, S.A.; Morris, D.G.; McGee, P.; Fairclough, P.; Bhattacharya, S.; Carpenter, R.; Grossman, A.B.; Korbonits, M. The tissue distribution of the mRNA of ghrelin and subtypes of its receptor, GHS-R, in humans. J. Clin. Endocrinol. Metab. 2002, 87, 2988–2991. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Y.; Asakawa, A.; Fujimiya, M.; Lee, S.D.; Inui, A. Ghrelin gene products and the regulation of food intake and gut motility. Pharmacol. Rev. 2009, 61, 430–481. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Xue, C. Ghrelin inhibits the development of acute pancreatitis and nuclear factor κB activation in pancreas and liver. Pancreas 2009, 38, 752–757. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.X.; Wu, D.; Wang, X.; Chen, H.L.; Chen, J.X.; Wang, X.X.; Wang, X.L.; Gan, L.; Guo, Z.Y.; Shi, G.X.; et al. Ghrelin protects against cobalt chloride-induced hypoxic injury in cardiac H9c2 cells by inhibiting oxidative stress and inducing autophagy. Peptides 2012, 38, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Cheyuo, C.; Jacob, A.; Wang, P. Ghrelin-mediated sympathoinhibition and suppression of inflammation in sepsis. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E265–E272. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Yoon, C.Y.; Jang, P.G.; Park, Y.J.; Shin, C.S.; Park, H.S.; Ryu, J.W.; Pak, Y.K.; Park, J.Y.; Lee, K.U.; et al. The mitogenic and antiapoptotic actions of ghrelin in 3T3-L1 adipocytes. Mol. Endocrinol. 200 2004, 18, 2291–2301. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Li, Z.; Wang, Z.; Li, Y.; Zhao, J.; Mulholland, M.; Zhang, W. Ghrelin contributes to protection of hepatocellular injury induced by ischaemia/reperfusion. Liver Int. 2014, 34, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Moreno, M.; Chaves, J.F.; Sancho-Bru, P.; Ramalho, F.; Ramalho, L.N.; Mansego, M.L.; Ivorra, C.; Dominguez, M.; Conde, L.; Millán, C.; et al. Ghrelin attenuates hepatocellular injury and liver fibrogenesis in rodents and influences fibrosis progression in humans. Hepatology 2010, 51, 974–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei, X.M.; Yung, B.Y.; Yip, S.P.; Ying, M.; Benzie, I.F.; Siu, P.M. Desacyl ghrelin prevents doxorubicin-induced myocardial fibrosis and apoptosis via the GHSR-independent pathway. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E311–E323. [Google Scholar] [CrossRef] [PubMed]
- Shek, F.W.; Benyon, R.C. How can transforming growth factor β be targeted usefully to combat liver fibrosis? Eur. J. Gastroenterol. Hepatol. 2004, 16, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Gaça, M.D.; Swenson, E.S.; Vellucci, V.F.; Reiss, M.; Wells, R.G. Smads 2 and 3 are differentially activated by transforming growth factor-β (TGF-β) in quiescent and activated hepatic stellate cells. Constitutive nuclear localization of Smads in activated cells is TGF-β-independent. J. Biol. Chem. 2003, 278, 11721–11728. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Hu, H.; Yin, J.Q. Therapeutic strategies against TGF-β signaling pathway in hepatic fibrosis. Liver Int. 2006, 26, 8–22. [Google Scholar] [CrossRef] [PubMed]
- Benyon, R.C.; Arthur, M.J. Extracellular matrix degradation and the role of hepatic stellate cells. Semin. Liver. Dis. 2001, 21, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Schuppan, D.; Afdhal, N.H. Liver cirrhosis. Lancet 2008, 371, 838–851. [Google Scholar] [CrossRef]
- Shintani, T.; Klionsky, D.J. Autophagy in health and disease: A double-edged sword. Science 2004, 306, 990–995. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.P.; Pei, X.M.; Sin, T.K.; Yip, S.P.; Yung, B.Y.; Chan, L.W.; Wong, C.S.; Siu, P.M. Acylated and unacylated ghrelin inhibit doxorubicin-induced apoptosis in skeletal muscle. Acta Physiol. 2014, 211, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Pei, X.M.; Yung, B.Y.; Yip, S.P.; Chan, L.W.; Wong, C.S.; Ying, M.; Siu, P.M. Protective effects of desacyl ghrelin on diabetic cardiomyopathy. Acta Diabetol. 2015, 52, 293–306. [Google Scholar] [CrossRef] [PubMed]
- Tam, B.T.; Pei, X.M.; Yung, B.Y.; Yip, S.P.; Chan, L.W.; Wong, C.S.; Siu, P.M. Unacylated ghrelin restores insulin and autophagic signaling in skeletal muscle of diabetic mice. Pflugers. Arch. 2015, 1, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.E.; Ding, Y.; Kim, S.I. TGF-β signaling via TAK1 pathway: Role in kidney fibrosis. Semin. Nephrol. 2012, 32, 244–252. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mao, Y.; Zhang, S.; Yu, F.; Li, H.; Guo, C.; Fan, X. Ghrelin Attenuates Liver Fibrosis through Regulation of TGF-β1 Expression and Autophagy. Int. J. Mol. Sci. 2015, 16, 21911-21930. https://doi.org/10.3390/ijms160921911
Mao Y, Zhang S, Yu F, Li H, Guo C, Fan X. Ghrelin Attenuates Liver Fibrosis through Regulation of TGF-β1 Expression and Autophagy. International Journal of Molecular Sciences. 2015; 16(9):21911-21930. https://doi.org/10.3390/ijms160921911
Chicago/Turabian StyleMao, Yuqing, Shaoren Zhang, Fujun Yu, Huanqing Li, Chuanyong Guo, and Xiaoming Fan. 2015. "Ghrelin Attenuates Liver Fibrosis through Regulation of TGF-β1 Expression and Autophagy" International Journal of Molecular Sciences 16, no. 9: 21911-21930. https://doi.org/10.3390/ijms160921911