
Hepatitis B Virus X Protein and Hepatocarcinogenesis

Abstract
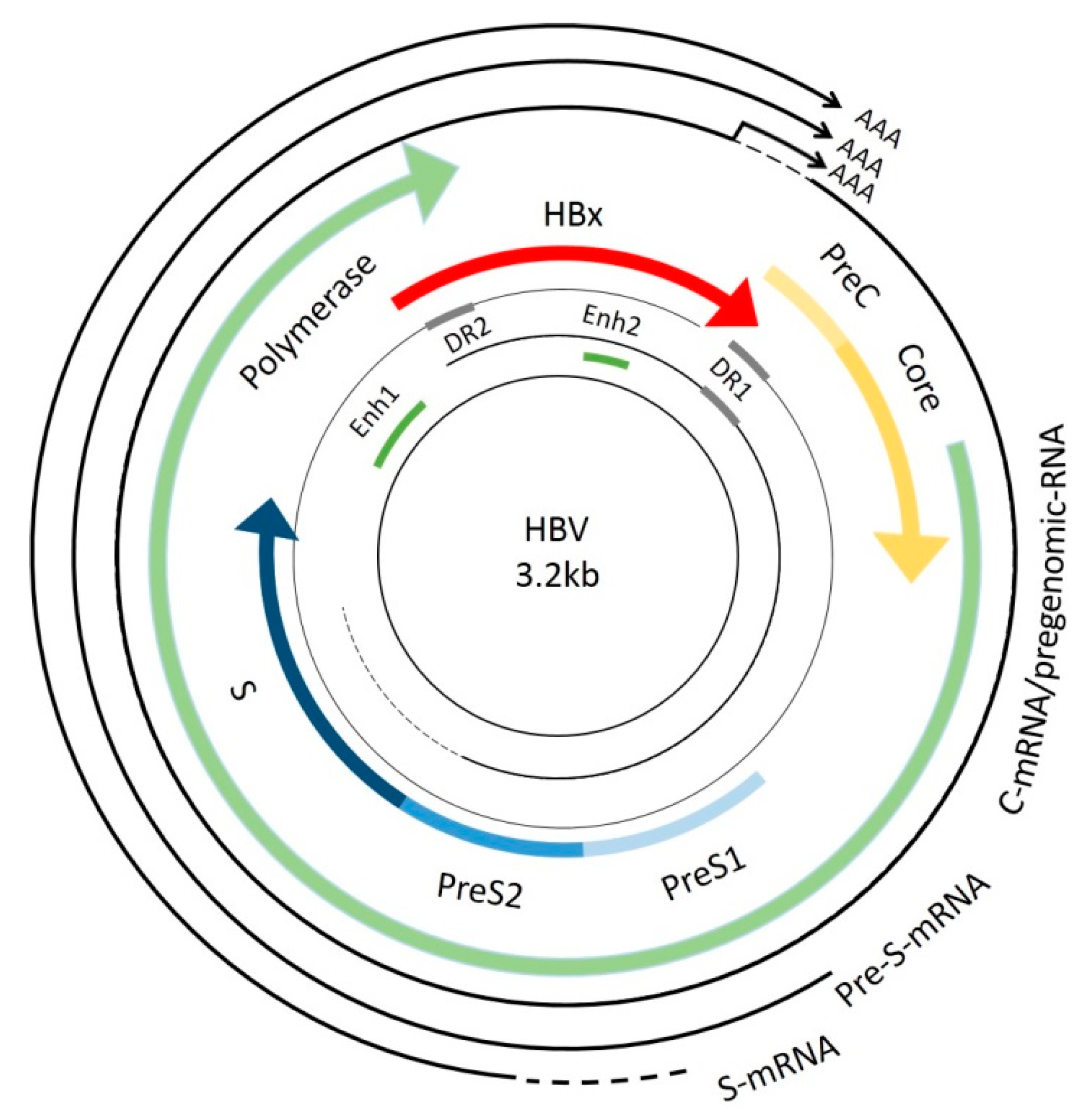
:1. Introduction
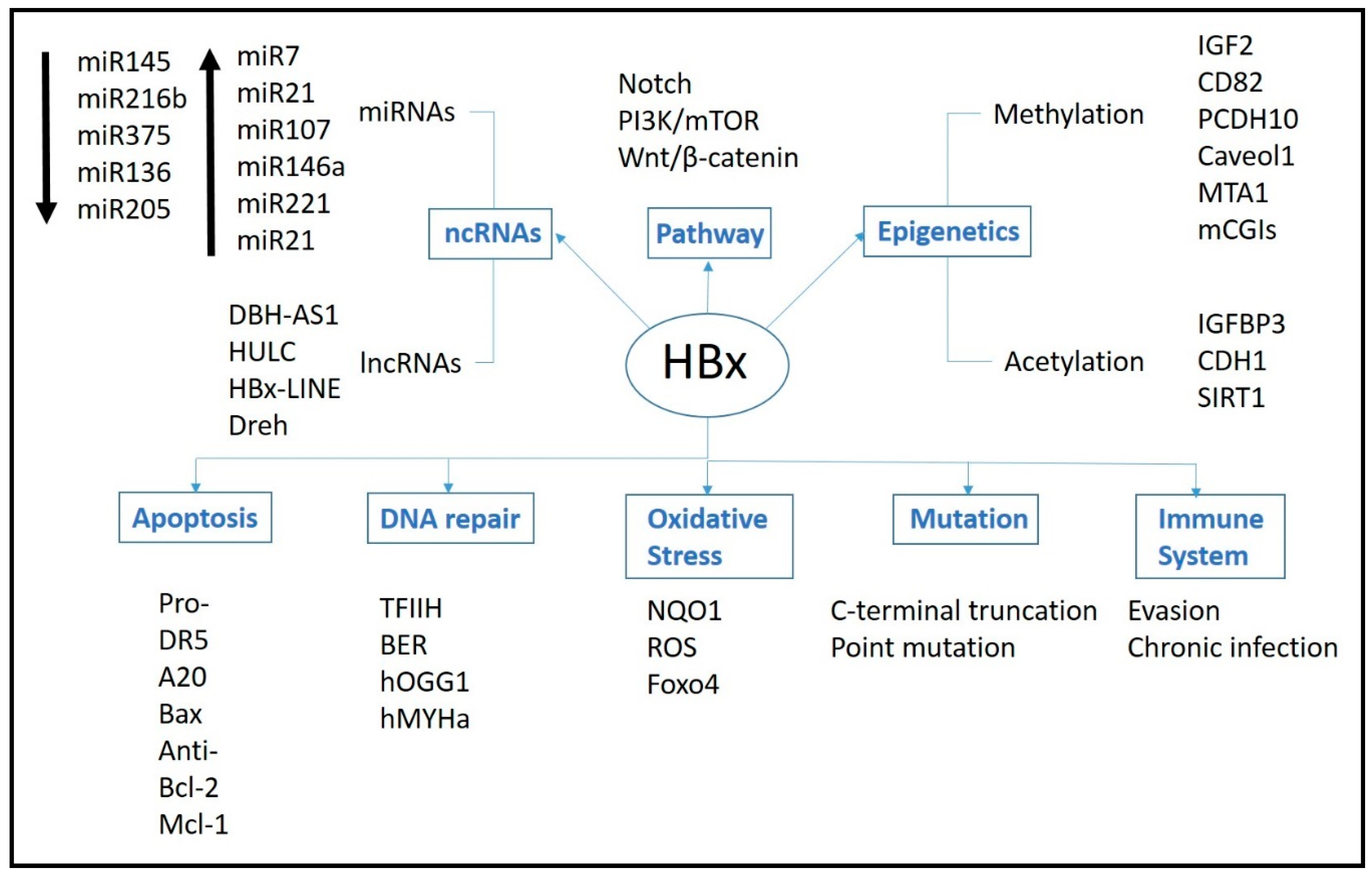
2. Mechanism of HBx Protein
3. Pathways
3.1. Signal Pathways
3.2. DNA Repair
3.3. Oxidative Stress
3.4. Immune System
3.5. Apoptosis
3.5.1. Pro-Apoptosis
3.5.2. Anti-Apoptosis
4. Epigenetics
4.1. Methylation
4.2. Acetylation
4.3. Non-Coding RNAs
4.3.1. miRNAs
4.3.2. LncRNAs
5. Genetic
5.1. Integration
5.2. HBx Gene Mutation
6. Conclusions and Prospects
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Torre, L.A.; Siegel, R.L.; Ward, E.M.; Jemal, A. Global cancer incidence and mortality rates and trends—An update. Cancer Epidemiol. Biomark. Prev. 2015. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Kudo, M.; Izumi, N.; Ichida, T.; Ku, Y.; Kokudo, N.; Sakamoto, M.; Takayama, T.; Nakashima, O.; Matsui, O.; Matsuyama, Y. Report of the 19th follow-up survey of primary liver cancer in Japan. Hepatol. Res. 2016, 46, 372–390. [Google Scholar] [CrossRef] [PubMed]
- Parkin, D.M. The global health burden of infection-associated cancers in the year 2002. Int. J. Cancer 2006, 118, 3030–3044. [Google Scholar] [CrossRef] [PubMed]
- Seeger, C.; Mason, W.S. Hepatitis B virus biology. Microbiol. Mol. Biol. Rev. 2000, 64, 51–68. [Google Scholar] [CrossRef] [PubMed]
- Minor, M.M.; Slagle, B.L. Hepatitis B virus HBx protein interactions with the ubiquitin proteasome system. Viruses 2014, 6, 4683–4702. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Abdel-Hafiz, H.; Suhail, M.; Al-Mars, A.; Zakaria, M.K.; Fatima, K.; Ahmad, S.; Azhar, E.; Chaudhary, A.; Qadri, I. Hepatitis B virus, HBx mutants and their role in hepatocellular carcinoma. World J. Gastroenterol. 2014, 20, 10238–10248. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.D.; Wang, Y.; Ye, L.H. Hepatitis B virus X protein accelerates the development of hepatoma. Cancer Biol. Med. 2014, 11, 182–190. [Google Scholar] [PubMed]
- Peng, Z.; Zhang, Y.; Gu, W.; Wang, Z.; Li, D.; Zhang, F.; Qiu, G.; Xie, K. Integration of the hepatitis B virus X fragment in hepatocellular carcinoma and its effects on the expression of multiple molecules: A key to the cell cycle and apoptosis. Int. J. Oncol. 2005, 26, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Hwang, G.Y.; Lin, C.Y.; Huang, L.M.; Wang, Y.H.; Wang, J.C.; Hsu, C.T.; Yang, S.S.; Wu, C.C. Detection of the hepatitis B virus X protein (HBx) antigen and anti-HBx antibodies in cases of human hepatocellular carcinoma. J. Clin. Microbiol. 2003, 41, 5598–5603. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Xiong, Y.; Wang, Y.; Wang, Y.; Zheng, G.; Xu, H. Hepatitis B virus X protein activates notch signaling by its effects on Notch1 and Notch4 in human hepatocellular carcinoma. Int. J. Oncol. 2016, 48, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Guo, J.; Li, W.; Lu, Y.; Fu, S.; Xie, X.; Xia, H.; Dong, X.; Chen, Y.; Quan, M.; et al. Hepatitis B virus X protein induces expression of α-fetoprotein and activates PI3K/mTOR signaling pathway in liver cells. Oncotarget 2015, 6, 12196–12208. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.; Chen, L.; Shan, X.; Shan, X.; Tang, J.; Zhou, F.; Chen, Q.; Quan, H.; Nie, D.; Zhang, W.; et al. Epigenetic silencing of SFRP1 and SFRP5 by hepatitis B virus X protein enhances hepatoma cell tumorigenicity through Wnt signaling pathway. Int. J. Cancer 2014, 135, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Qadri, I.; Fatima, K.; Abde, L.H.H. Hepatitis B virus X protein impedes the DNA repair via its association with transcription factor, TFIIH. BMC Microbiol. 2011, 11, 48. [Google Scholar] [CrossRef] [PubMed]
- Van Hemert, F.J.; van de Klundert, M.A.; Lukashov, V.V.; Kootstra, N.A.; Berkhout, B.; Zaaijer, H.L. Protein x of hepatitis B virus: Origin and structure similarity with the central domain of DNA glycosylase. PLoS ONE 2011, 6, e23392. [Google Scholar] [CrossRef] [PubMed]
- Cheng, B.; Zheng, Y.; Guo, X.; Wang, Y.; Liu, C. Hepatitis B viral x protein alters the biological features and expressions of DNA repair enzymes in LO2 cells. Liver Int. Off. J. Int. Assoc. Study Liver 2010, 30, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.L.; Wang, D.; Peng, X.E.; Chen, Y.L.; Zheng, D.L.; Chen, W.N.; Lin, X. Epigenetic silencing of NAD(P)H: Quinone oxidoreductase 1 by hepatitis B virus X protein increases mitochondrial injury and cellular susceptibility to oxidative stress in hepatoma cells. Free Radic. Biol. Med. 2013, 65, 632–644. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.Y.; Kim, Y.J. C-terminal region of hbx is crucial for mitochondrial DNA damage. Cancer Lett. 2013, 331, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Srisuttee, R.; Koh, S.S.; Park, E.H.; Cho, I.R.; Min, H.J.; Jhun, B.H.; Yu, D.Y.; Park, S.; Park do, Y.; Lee, M.O.; et al. Up-regulation of Foxo4 mediated by hepatitis B virus X protein confers resistance to oxidative stress-induced cell death. Int. J. Mol. Med. 2011, 28, 255–260. [Google Scholar] [PubMed]
- Hong, Y.; Zhou, L.; Xie, H.; Zheng, S. Innate immune evasion by hepatitis B virus-mediated downregulation of TRIF. Biochem. Biophys. Res. Commun. 2015, 463, 719–725. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Jung, S.Y.; Hodgson, A.J.; Madden, C.R.; Qin, J.; Slagle, B.L. Hepatitis B virus regulatory HBx protein binds to adaptor protein IPS-1 and inhibits the activation of β interferon. J. Virol. 2011, 85, 987–995. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.K.; Kim, S.Y.; Seong, J.K.; Cheong, J. Hepatitis B virus X increases immune cell recruitment by induction of chemokine SDF-1. FEBS Lett. 2014, 588, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Kong, F.; You, H.; Zhao, J.; Liu, W.; Hu, L.; Luo, W.; Hu, W.; Tang, R.; Zheng, K. The enhanced expression of death receptor 5 (DR5) mediated by HBV x protein through NF-κB pathway is associated with cell apoptosis induced by (TNF-α related apoptosis inducing ligand) trail in hepatoma cells. Virol. J. 2015, 12, 192. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Huang, C.; Wang, Y.; Lu, Z.; Zhuang, N.; Zhao, D.; He, J.; Shi, L. Hepatitis B virus X protein sensitizes trail-induced hepatocyte apoptosis by inhibiting the E3 ubiquitin ligase A20. PLoS ONE 2015, 10, e0127329. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Zhang, X.; Hu, D.; Feng, T.; Li, H.; Lu, Y.; Huang, J. Hepatitis B virus X (HBx) play an anti-apoptosis role in hepatic progenitor cells by activating Wnt/β-catenin pathway. Mol. Cell. Biochem. 2013, 383, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.Y.; Tang, S.H.; Wu, S.L.; Luo, Y.H.; Cao, M.R.; Zhou, H.K.; Jiang, X.W.; Shu, J.C.; Bie, C.Q.; Huang, S.M.; et al. Epigenetic modulation of insulin-like growth factor-II overexpression by hepatitis B virus X protein in hepatocellular carcinoma. Am. J. Cancer Res. 2015, 5, 956–978. [Google Scholar] [PubMed]
- Yu, G.; Bing, Y.; Li, W.; Xia, L.; Liu, Z. Hepatitis B virus inhibits the expression of CD82 through hypermethylation of its promoter in hepatoma cells. Mol. Med. Rep. 2014, 10, 2580–2586. [Google Scholar] [PubMed]
- Fang, S.; Huang, S.F.; Cao, J.; Wen, Y.A.; Zhang, L.P.; Ren, G.S. Silencing of PCDH10 in hepatocellular carcinoma via de novo DNA methylation independent of HBV infection or HBx expression. Clin. Exp. Med. 2013, 13, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Lu, Q.; Dong, J.; Li, X.; Ma, K.; Cai, L. Hepatitis B virus X protein suppresses caveolin-1 expression in hepatocellular carcinoma by regulating DNA methylation. BMC Cancer 2012, 12, 353. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.H.; Na, H.; Na, T.Y.; Shin, Y.K.; Seong, J.K.; Lee, M.O. Epigenetic control of metastasis-associated protein 1 gene expression by hepatitis B virus X protein during hepatocarcinogenesis. Oncogenesis 2012, 1, e25. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.M.; Lee, Y.G.; Bae, J.B.; Choi, J.K.; Tayama, C.; Hata, K.; Yun, Y.; Seong, J.K.; Kim, Y.J. HBx induces hypomethylation of distal intragenic CPG islands required for active expression of developmental regulators. Proc. Natl. Acad. Sci. USA 2014, 111, 9555–9560. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Song, X.; Li, Y.; Tan, D.; Liu, G. Hepatitis B virus X protein upregulates DNA methyltransferase 3a/3b and enhances SOCS-1CPG island methylation. Mol. Med. Rep. 2016, 13, 301–308. [Google Scholar] [PubMed]
- Qiu, X.; Zhang, L.; Lu, S.; Song, Y.; Lao, Y.; Hu, J.; Fan, H. Upregulation of DNMT1 mediated by HBx suppresses RASSF1A expression independent of DNA methylation. Oncol. Rep. 2014, 31, 202–208. [Google Scholar] [PubMed]
- Shon, J.K.; Shon, B.H.; Park, I.Y.; Lee, S.U.; Fa, L.; Chang, K.Y.; Shin, J.H.; Lee, Y.I. Hepatitis B virus-x protein recruits histone deacetylase 1 to repress insulin-like growth factor binding protein 3 transcription. Virus Res. 2009, 139, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Arzumanyan, A.; Friedman, T.; Kotei, E.; Ng, I.O.; Lian, Z.; Feitelson, M.A. Epigenetic repression of E-cadherin expression by hepatitis B virus X antigen in liver cancer. Oncogene 2012, 31, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Srisuttee, R.; Koh, S.S.; Kim, S.J.; Malilas, W.; Boonying, W.; Cho, I.R.; Jhun, B.H.; Ito, M.; Horio, Y.; Seto, E.; et al. Hepatitis B virus X (HBx) protein upregulates β-catenin in a human hepatic cell line by sequestering sirt1 deacetylase. Oncol. Rep. 2012, 28, 276–282. [Google Scholar] [PubMed]
- Gao, F.; Sun, X.; Wang, L.; Tang, S.; Yan, C. Downregulation of microRNA-145 caused by hepatitis B virus X protein promotes expression of CUL5 and contributes to pathogenesis of hepatitis B virus-associated hepatocellular carcinoma. Cell. Physiol. Biochem. Int. J. Exp. Cell Physiol. Biochem. Pharmacol. 2015, 37, 1547–1559. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.S.; Yen, C.J.; Chen, Y.J.; Chen, J.Y.; Wang, L.Y.; Chiu, S.J.; Shih, W.L.; Ho, C.Y.; Wei, T.T.; Pan, H.L.; et al. MiRNA-7/21/107 contribute to HBx-induced hepatocellular carcinoma progression through suppression of maspin. Oncotarget 2015, 6, 25962–25974. [Google Scholar] [CrossRef] [PubMed]
- Li, J.F.; Dai, X.P.; Zhang, W.; Sun, S.H.; Zeng, Y.; Zhao, G.Y.; Kou, Z.H.; Guo, Y.; Yu, H.; Du, L.Y.; et al. Upregulation of microRNA-146a by hepatitis B virus X protein contributes to hepatitis development by downregulating complement factor H. mBio 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.Y.; Zhou, S.J.; Deng, Y.L.; Zhang, Z.Y.; Zhang, E.L.; Wu, Z.B.; Huang, Z.Y.; Chen, X.P. miR-216b is involved in pathogenesis and progression of hepatocellular carcinoma through HBx-miR-216B-IGF2BP2 signaling pathway. Cell Death Dis. 2015, 6, E1670. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.J.; Tang, Y.S.; Huang, S.F.; Ai, J.G.; Wang, H.X.; Zhang, L.P. Hbx protein-induced upregulation of microRNA-221 promotes aberrant proliferation in HBv related hepatocellular carcinoma by targeting estrogen receptor-α. Oncol. Rep. 2015, 33, 792–798. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Wang, W.; Huang, Y.; Wu, J.; Chen, M.; Cui, P.; Zhang, W.; Zhang, Y. HBx elevates oncoprotein AEG-1 expression to promote cell migration by downregulating miR-375 and miR-136 in malignant hepatocytes. DNA Cell Biol. 2014, 33, 715–722. [Google Scholar] [CrossRef] [PubMed]
- Damania, P.; Sen, B.; Dar, S.B.; Kumar, S.; Kumari, A.; Gupta, E.; Sarin, S.K.; Venugopal, S.K. Hepatitis B virus induces cell proliferation via HBx-induced microRNA-21 in hepatocellular carcinoma by targeting programmed cell death protein4 (PDCD4) and phosphatase and tensin homologue (PTEN). PLoS ONE 2014, 9, e91745. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.; Wang, Y.; Sun, B.; Xiao, Z.; Ye, L.; Zhang, X. MiR-205 modulates abnormal lipid metabolism of hepatoma cells via targeting acyl-CoA synthetase long-chain family member 1 (ACSL1) mRNA. Biochem. Biophys. Res. Commun. 2014, 444, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.; Xiao, Z.; Sun, B.; Wang, Y.; Zheng, M.; Ye, L.; Zhang, X. Involvement of cholesterol in hepatitis B virus X protein-induced abnormal lipid metabolism of hepatoma cells via up-regulating miR-205-targeted ACSL4. Biochem. Biophys. Res. Commun. 2014, 445, 651–655. [Google Scholar] [CrossRef] [PubMed]
- Shan, X.; Ren, M.; Chen, K.; Huang, A.; Tang, H. Regulation of the microRNA processor DGCR8 by hepatitis B virus proteins via the transcription factor YY1. Arch. Virol. 2015, 160, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.L.; Ren, T.Y.; Cao, S.W.; Zheng, S.H.; Hu, X.M.; Hu, Y.W.; Lin, L.; Chen, J.; Zheng, L.; Wang, Q. HBx-related long non-coding RNA DBH-AS1 promotes cell proliferation and survival by activating MAPK signaling in hepatocellular carcinoma. Oncotarget 2015, 6, 33791–33804. [Google Scholar] [PubMed]
- Du, Y.; Kong, G.; You, X.; Zhang, S.; Zhang, T.; Gao, Y.; Ye, L.; Zhang, X. Elevation of highly up-regulated in liver cancer (HULC) by hepatitis B virus X protein promotes hepatoma cell proliferation via down-regulating p18. J. Biol. Chem. 2012, 287, 26302–26311. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.F.; Guo, Y.J.; Zhao, C.X.; Yuan, S.X.; Wang, Y.; Tang, G.N.; Zhou, W.P.; Sun, S.H. Hepatitis B virus X protein (HBx)-related long noncoding RNA (lncRNA) down-regulated expression by HBx (DREH) inhibits hepatocellular carcinoma metastasis by targeting the intermediate filament protein vimentin. Hepatol. 2013, 57, 1882–1892. [Google Scholar] [CrossRef] [PubMed]
- Lau, C.C.; Sun, T.; Ching, A.K.; He, M.; Li, J.W.; Wong, A.M.; Co, N.N.; Chan, A.W.; Li, P.S.; Lung, R.W.; et al. Viral-human chimeric transcript predisposes risk to liver cancer development and progression. Cancer Cell 2014, 25, 335–349. [Google Scholar] [CrossRef] [PubMed]
- Sze, K.M.; Chu, G.K.; Lee, J.M.; Ng, I.O. C-terminal truncated hepatitis B virus X protein is associated with metastasis and enhances invasiveness by c-Jun/matrix metalloproteinase protein 10 activation in hepatocellular carcinoma. Hepatology 2013, 57, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Ma, N.F.; Lau, S.H.; Hu, L.; Xie, D.; Wu, J.; Yang, J.; Wang, Y.; Wu, M.C.; Fung, J.; Bai, X.; et al. Cooh-terminal truncated HBV X protein plays key role in hepatocarcinogenesis. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2008, 14, 5061–5068. [Google Scholar] [CrossRef] [PubMed]
- Lizzano, R.A.; Yang, B.; Clippinger, A.J.; Bouchard, M.J. The C-terminal region of the hepatitis B virus X protein is essential for its stability and function. Virus Res. 2011, 155, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Li, S.K.; Ho, S.F.; Tsui, K.W.; Fung, K.P.; Waye, M.Y. Identification of functionally important amino acid residues in the mitochondria targeting sequence of hepatitis B virus X protein. Virology 2008, 381, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.Y.; Chai, S.; Tong, M.; Guan, X.Y.; Lin, C.H.; Ching, Y.P.; Xie, D.; Cheng, A.S.; Ma, S. C-terminal truncated hepatitis B virus X protein promotes hepatocellular carcinogenesis through induction of cancer and stem cell-like properties. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.P.; Hu, B.G.; Ye, C.; Ho, R.L.; Chen, G.G.; Lai, P.B. Hbx mutants differentially affect the activation of hypoxia-inducible factor-1α in hepatocellular carcinoma. Br. J. Cancer 2014, 110, 1066–1073. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Liu, S.; Zhao, Y.; Guo, Z.; Xu, J. X protein mutations in hepatitis b virus DNA predict postoperative survival in hepatocellular carcinoma. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2014, 35, 10325–10331. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Wei, F.; Li, C.; Lv, G.; Wang, G.; Liu, T.; Bellail, A.C.; Hao, C. Combination of mTOR and EGFR kinase inhibitors blocks mTORC1 and mTORC2 kinase activity and suppresses the progression of colorectal carcinoma. PLoS ONE 2013, 8, e73175. [Google Scholar] [CrossRef] [PubMed]
- Nagahashi, M.; Matsuda, Y.; Moro, K.; Tsuchida, J.; Soma, D.; Hirose, Y.; Kobayashi, T.; Kosugi, S.I.; Takabe, K.; Komatsu, M.; et al. DNA damage response and sphingolipid signaling in liver diseases. Surg. Today 2015. [Google Scholar] [CrossRef] [PubMed]
- Van de Klundert, M.A.; van Hemert, F.J.; Zaaijer, H.L.; Kootstra, N.A. The hepatitis B virus X protein inhibits thymine DNA glycosylase initiated base excision repair. PLoS ONE 2012, 7, e48940. [Google Scholar] [CrossRef] [PubMed]
- Shih, P.H.; Yeh, C.T.; Yen, G.C. Anthocyanins induce the activation of phase ii enzymes through the antioxidant response element pathway against oxidative stress-induced apoptosis. J. Agric. Food Chem. 2007, 55, 9427–9435. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Meng, X.M.; Huang, C.; Wu, B.M.; Zhang, L.; Lv, X.W.; Li, J. Long noncoding RNAs: Novel insights into hepatocelluar carcinoma. Cancer Lett. 2014, 344, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Carey, M.; Workman, J.L. The role of chromatin during transcription. Cell 2007, 128, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Yang, W.; Song, J.; Wu, Y.; Ni, B. Hepatitis B virus X protein-induced aberrant epigenetic modifications contributing to human hepatocellular carcinoma pathogenesis. Mol. Cell. Biol. 2013, 33, 2810–2816. [Google Scholar] [CrossRef] [PubMed]
- Kew, M.C. Hepatitis B virus X protein in the pathogenesis of hepatitis B virus-induced hepatocellular carcinoma. J. Gastroenterol. Hepatol. 2011, 26, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Hu, W.; Hu, J.; Wu, S.; Li, J.; Luo, Y.; Cao, M.; Zhou, H.; Jiang, X. Hepatitis B virus X protein promotes p3 transcript expression of the insulin-like growth factor 2 gene via inducing hypomethylation of p3 promoter in hepatocellular carcinoma. Liver Int. 2015, 35, 608–619. [Google Scholar] [CrossRef] [PubMed]
- Liz, J.; Esteller, M. LncRNAs and microRNAs with a role in cancer development. Biochim. Biophys. Acta 2016, 1859, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Bechmann, L.P.; Hannivoort, R.A.; Gerken, G.; Hotamisligil, G.S.; Trauner, M.; Canbay, A. The interaction of hepatic lipid and glucose metabolism in liver diseases. J. Hepatol. 2012, 56, 952–964. [Google Scholar] [CrossRef] [PubMed]
- Toh, S.T.; Jin, Y.; Liu, L.; Wang, J.; Babrzadeh, F.; Gharizadeh, B.; Ronaghi, M.; Toh, H.C.; Chow, P.K.; Chung, A.Y.; et al. Deep sequencing of the hepatitis B virus in hepatocellular carcinoma patients reveals enriched integration events, structural alterations and sequence variations. Carcinogenesis 2013, 34, 787–798. [Google Scholar] [CrossRef] [PubMed]
- Iavarone, M.; Trabut, J.B.; Delpuech, O.; Carnot, F.; Colombo, M.; Kremsdorf, D.; Brechot, C.; Thiers, V. Characterisation of hepatitis B virus X protein mutants in tumour and non-tumour liver cells using laser capture microdissection. J. Hepatol. 2003, 39, 253–261. [Google Scholar] [CrossRef]
- Wang, Y.; Lau, S.H.; Sham, J.S.; Wu, M.C.; Wang, T.; Guan, X.Y. Characterization of hbv integrants in 14 hepatocellular carcinomas: Association of truncated X gene and hepatocellular carcinogenesis. Oncogene 2004, 23, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Tu, H.; Bonura, C.; Giannini, C.; Mouly, H.; Soussan, P.; Kew, M.; Paterlini-Brechot, P.; Brechot, C.; Kremsdorf, D. Biological impact of natural cooh-terminal deletions of hepatitis B virus X protein in hepatocellular carcinoma tissues. Cancer Res. 2001, 61, 7803–7810. [Google Scholar] [PubMed]
- Kumar, V.; Jayasuryan, N.; Kumar, R. A truncated mutant (residues 58–140) of the hepatitis B virus X protein retains transactivation function. Proc. Natl. Acad. Sci. USA 1996, 93, 5647–5652. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Wang, J.; Wang, Y.; Wang, A.; Guo, H.; Wei, F.; Mehta, S.R.; Espitia, S.; Smith, D.M.; Liu, L.; et al. A novel mutant 10ALA/ARG together with mutant 144SER/ARG of hepatitis B virus X protein involved in hepatitis B virus-related hepatocarcinogenesis in HEPG2 cell lines. Cancer Lett. 2016, 371, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Tai, J.; Wang, G.; Liu, T.; Wang, L.; Lin, C.; Li, F. Effects of siRNA targeting c-Myc and VEGF on human colorectal cancer volo cells. J. Biochem. Mol. Toxicol. 2012, 26, 499–505. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Group | Sub-Group | Target | Mechanism | Reference |
|---|---|---|---|---|
| Pathways | Signaling pathway | Notch1,Notch4 | 1. Activate Notch pathway by receptor Notch1 and Notch4, inducing cell growth, cell cycle prograssion and anti-apoptosis | Gao et al. [11] |
| AFP | 2. Induce AFP expression to activate PI3K/mTOR pathway, resulting in the promotion of progression, invasion and metastasis of cancer cells | Zhu et al. [12] | ||
| SFRP1, SFRP5 | 3. Inhibit Wnt/β-catenin pathway by reducing its two ligands, SFRP1 and SFRP5, resulting in EMT | Xie et al. [13] | ||
| DNA repair | TFIIH | 1. Reduce DNA repaire capacity by interfering TFIIH | Qadri et al. [14] | |
| DNA glycosylases | 2. Has a similar structure of DNA glycosylases but doesn't have the capapility in DNA repair | Hement et al. [15] | ||
| hOGG1, hMYHa | 3. Inhibit DNA repair by hindering DNA repair enzyme hOGG1 and hMYHa | Cheng et al. [16] | ||
| Oxidative stress | NQO1 | 1. Induce mitochondria injury and oxidative stress by downregulating NQO1 | Wu et al. [17] | |
| mitochondria DNA | 2. Induce ROS and damage mitochondria DNA | Jung et al. [18] | ||
| Foxo4 | 3. Enhance resistances to oxidative stress-induced cell death by upregulating Foxo4 | Srisuttee et al. [19] | ||
| Immune | TRIF | 1. Enable HBV replication and evasion from innate immunity by reducing TRIF expression | Hong et al. [20] | |
| IPS-1 | 2. Bind to IPS-1 and diminish IFN-β signaling | Kumar et al. [21] | ||
| SDF-1 | 3. Recruit immune cells into liver by inducing SDF-1 via endoplasmic reticulum stress | Cho et al. [22] | ||
| Apoptosis | DR5 | 1. Promote TRAIL induced apoptosis by increasing DR5 | Kong et al. [23] | |
| A20 | 2. Sensitize TRAIL induced apoptosis by inhibiting caspase-8 inhibitor A20 | Zhang et al. [24] | ||
| Bcl-2, Mcl-1 | 3. Inhibit apoptosis by increasing apoptosis inhibition gene Bcl-2 and Mcl-1 | Shen et al. [25] | ||
| Epegnetics | Methylation | IGF-2 | 1. Hypomethylation the promoter of IGF-2 promoter | Liu et al. [26] |
| CD82 | 2. Hypermethylation the promoter of metastasis-inhibit gene CD82 | Yu et al. [27] | ||
| PCDH10 | 3. Hypermethylation the promoter of tumor suppressor gene PCDH10 | Fang et al. [28] | ||
| Caveolin-1 | 4. Hypermethylation the promoter of tumor suppressor gene Caveolin-1 | Yan et al. [29] | ||
| MTA1 | 5. Hypermethylation the promoter of tumor suppressor gene MTA1 | Lee et al. [30] | ||
| mCGIs | 6. Hypomethylation of mCGIs then influence cell differenciation | Lee et al. [31] | ||
| SOCS-1 | 7. Hypermethylation the promoter of tumor supprssor gene SOCS-1 | Fu et al. [32] | ||
| RASSF1A | 8. Hypermethylation the promoter of tumor suppressor gene RASSF1A | Qiu et al. [33] | ||
| Acetylation | SP1 | 1. Deacetylation of SP1 then romotes cell survival, transformation, and progression to cancer | Shon et al. [34] | |
| CDH1 | 2. Deacetylation of CDH1 promoter then inhibit metastasis | Arzumanyan et al. [35] | ||
| SIRT1 | 3. Attenuate the interaction between SIRT1 and β-catenin then protecting β-catenin from degradation | Srisuttee et al. [36] | ||
| miRNAs | miR-145 | 1. Induce CUL5 by down regulation of miR-145 then promote cell growth | Gao et al. [37] | |
| miR-7,21,107 | 2. Induce maspin by down regulation of miR-7,21,107 then promote migration, ivasion and chemoresistance | Chen et al. [38] | ||
| miR-146a | 3. Inhibit CFH by up regulation of miR-146a then enhance the alternative pathway of complement activation | Li et al. [39] | ||
| miR-216b | 4. Induce IGFBP2 by down regulation of miR-216b then increase cell proliferation | Liu et al. [40] | ||
| miR-221 | 5. Inhibit ERα, which is a protective factor against HCC, by up regulation of miR-221 | Chen et al. [41] | ||
| miR-136,375 | 6. Induce AEG-1 by down regulation of miR-136 and 375 then promote cell migration | Zhao et al. [42] | ||
| miR-21 | 7. Inhibit PDCD4 and PTEN by up regulation of miR-21 then increase cell proliferation | Damania et al. [43] | ||
| miR-205 | 8. Upregulate ACSL1/4 by inhibit miR-205 then affect lipid metabolism | Cui et al. [44,45] | ||
| DGCR8 | 9. Inhibit miRNA processor DGCR8 and interfer miRNA production | Shan et al. [46] | ||
| LncRNAs | DBH-AS1 | 1. Induce DBH-AS1 expression which activates ERK/p38/JNK MAPK signalling pathway | Huang et al. [47] | |
| HULC | 2. Up regulate HULC which promotes cell proliferation | Du et al. [48] | ||
| Dreh | 3. Inhibit HCC metastasis by downregulation of Dreh | Huang et al. [49] | ||
| LINE1 | 4. HBx-LINE1 fusion exerts lncRNA function and indeces EMT | Lau et al. [50] | ||
| Mutation | C-terminal truncation | MMP10 | 1. Increse invasion by activating MMP10 | Sze et al. [51] |
| HBx | 2. Promote proliferation and inhibit apoptotic frequency | Ma et al. [52] | ||
| HBx | 3. Decrease HBx steady level and slow HBV replication | Lizzano et al. [53] | ||
| mitochondrial | 4. Target mitichondrial then aggregate it at perinuclear space | Li et al. [54] | ||
| CSC | 5. Enhance stemness of CSC and drug resistancy | Ng et al. [55] | ||
| Point mutation | p53 | 1. A10R-S144R arrests cell cycle and attenuate p53 binding | Liu et al. [56] | |
| HIF-1 | 2. K130M/V131I strengthens the transcriptional activity of HIF-1 | Xie et al. [57] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, S.; Koh, S.S.Y.; Lee, C.G.L. Hepatitis B Virus X Protein and Hepatocarcinogenesis. Int. J. Mol. Sci. 2016, 17, 940. https://doi.org/10.3390/ijms17060940
Liu S, Koh SSY, Lee CGL. Hepatitis B Virus X Protein and Hepatocarcinogenesis. International Journal of Molecular Sciences. 2016; 17(6):940. https://doi.org/10.3390/ijms17060940
Chicago/Turabian StyleLiu, Shuaichen, Samantha S. Y. Koh, and Caroline G. L. Lee. 2016. "Hepatitis B Virus X Protein and Hepatocarcinogenesis" International Journal of Molecular Sciences 17, no. 6: 940. https://doi.org/10.3390/ijms17060940