
Role of Toll-Like Receptor Signaling in the Pathogenesis of Graft-versus-Host Diseases

Abstract
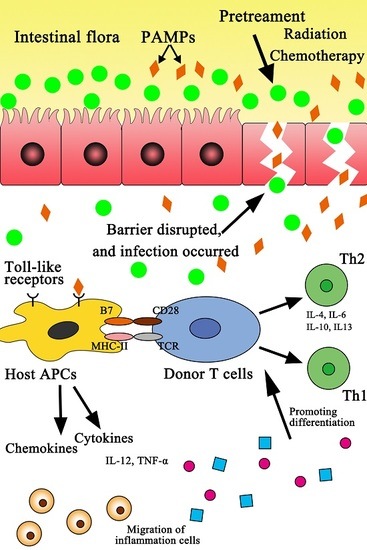
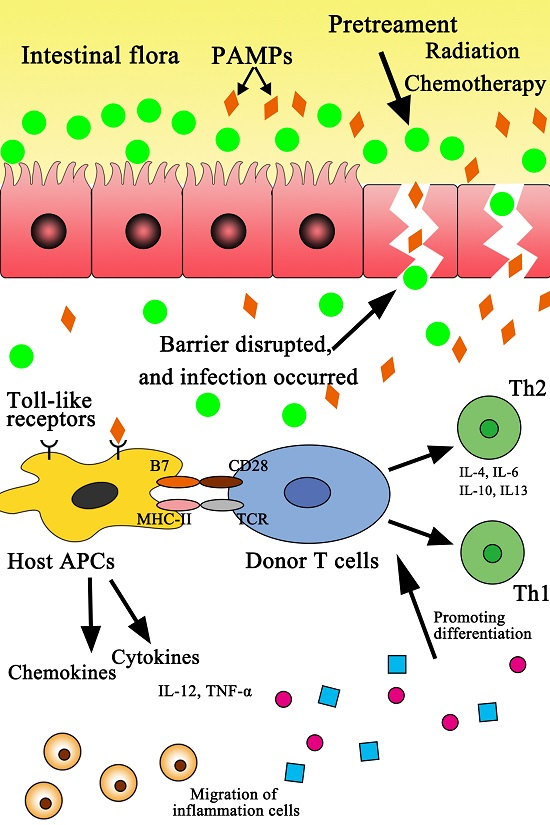
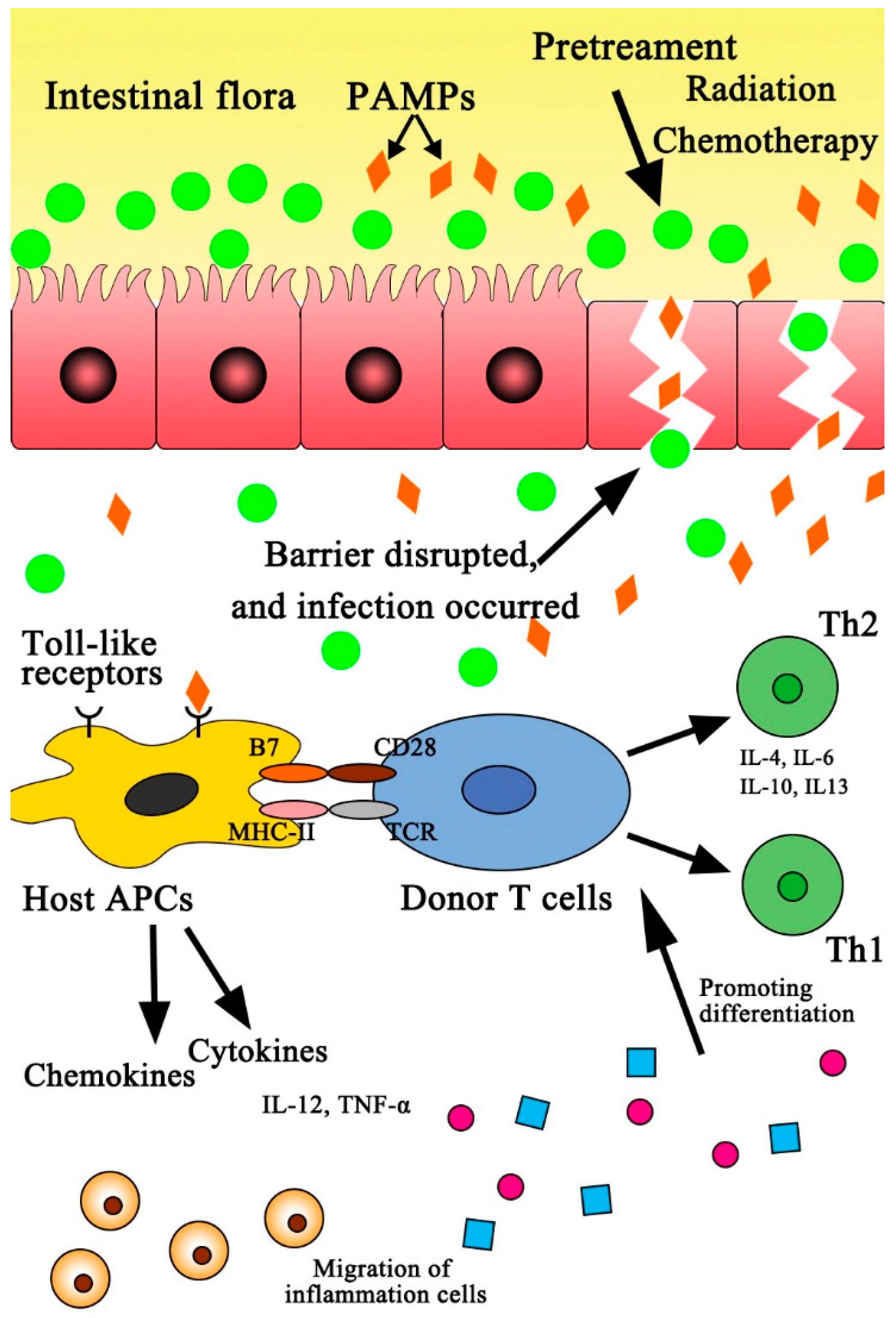
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction to Toll-Like Receptors (TLRs)
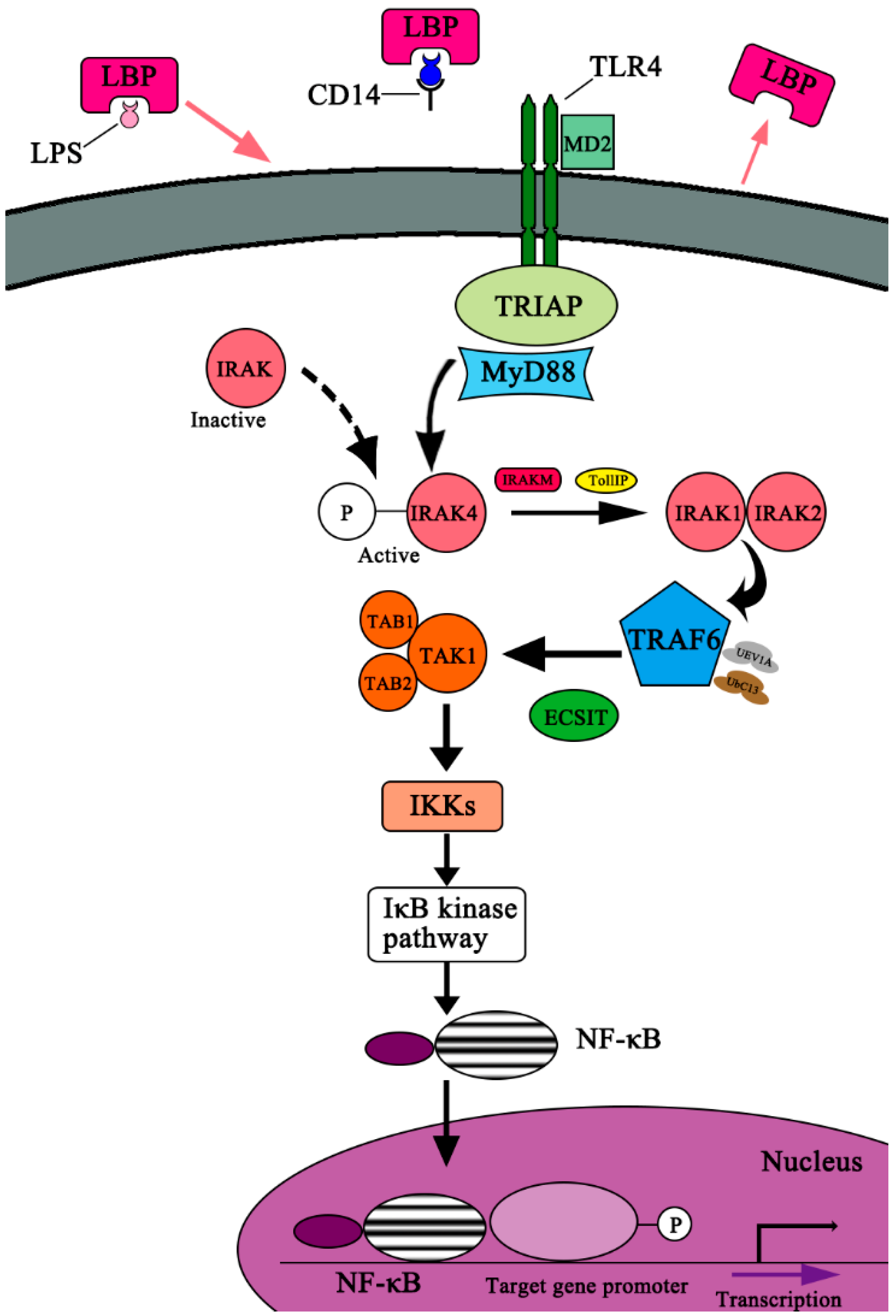
2. TLR4 Signaling Pathway Activated by LPS
3. The TLR9 Signaling Pathway
4. Other TLR Signaling Pathways
5. Inhibition of TLR Signaling Pathways
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mo, X.D.; Huang, X.J. Life quality related to health related after allogenic transplantation. Chin. J. Hematol. 2012, 33, 968–971. [Google Scholar]
- Van Bekkum, D.W.; Knaan, S. Role of bacterial microflora in development of intestinal lesions from graft-versus-host reaction. J. Natl. Cancer Inst. 1977, 58, 787–790. [Google Scholar] [PubMed]
- Poutsiaka, D.D.; Munson, D.; Price, L.L.; Chan, G.W.; Snydman, D.R. Blood stream infection (BSI) and acute GVHD after hematopoietic stem cell transplantation (HSCT) are associated. Bone Marrow Transplant. 2011, 46, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Reikine, S.; Nguyen, J.B.; Modis, Y. Pattern recognition and signaling mechanisms of RIG-I and MDA5. Front. Immunol. 2014, 5, 342. [Google Scholar] [CrossRef] [PubMed]
- Saxena, M.; Yeretssian, G. NOD-like receptors: Master regulators of inflammation and cancer. Front. Immunol. 2014, 5, 327. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.; Golenbock, D.; Bowie, A.G. The history of Toll-like receptors—Redefining innate immunity. Nat. Rev. Immunol. 2013, 13, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Ramadan, A.; Paczesny, S. Various forms of tissue damage and danger signals following hematopoietic stem-cell transplantation. Front. Immunol. 2015, 6, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duran-Struuck, R.; Reddy, P. Biological advances in acute graft-versus-host disease after allogeneic hematopoietic stem cell transplantation. Transplantation 2008, 85, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Johansson, J.E.; Ekman, T. Gut toxicity during hemopoietic stem cell transplantation may predict acute graft-versus-host disease severity in patients. Dig. Dis. Sci. 2007, 52, 2340–2345. [Google Scholar] [CrossRef] [PubMed]
- Wells, J.M.; Loonen, L.M.P.; Karczewski, J.M. The role of innate signaling in the homeostasis of tolerance and immunity in the intestine. Int. J. Med. Microbiol. 2010, 300, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Penack, O.; Holler, E.; van den Brink, M.R. Graft-versus-host disease: Regulation by microbe-associated molecules and innate immune receptors. Blood 2010, 115, 1865–1872. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Takeuchi, O.; Akira, S. Recognition of lipopeptides by Toll-like receptors. J. Endotoxin Res. 2002, 8, 459–463. [Google Scholar] [CrossRef] [PubMed]
- Lemaitre, B.; Nicolas, E.; Michaut, L.; Reichhart, J.M.; Hoffmann, J.A. The dorsoventral regulatory gene cassette spatzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell 1996, 86, 973–983. [Google Scholar] [CrossRef]
- Akira, S.; Takeda, K.; Kaisho, T. Toll-like receptors: Critical proteins linking innate and acquired immunity. Nat. Immunol. 2001, 2, 675–680. [Google Scholar] [CrossRef] [PubMed]
- Kumar, H.; Kawai, T.; Akira, S. Toll-like receptors and innate immunity. Biochem. Biophys. Res. Commun. 2009, 388, 621–625. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.W.; Luo, Y.; Lai, X.Y.; Shi, J.M.; Tan, Y.M.; He, J.S.; Xie, W.Z.; Zheng, W.Y.; Ye, X.J.; Yu, X.H.; et al. Donor TLR9 gene tagSNPs influence susceptibility to aGVHD and CMV reactivation in the allo-HSCT setting without polymorphisms in the TLR4 and NOD2 genes. Bone Marrow Transplant. 2014, 49, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Fukata, M.; Michelsen, K.S.; Eri, R.; Thomas, L.S.; Hu, B.; Lukasek, K.; Nast, C.C.; Lechago, J.; Xu, R.; Najki, Y.; et al. Toll-like receptor-4 is required for intestinal response to epithelial injury and limiting bacterial translocation in a murine model of acute colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 288, 1055–1065. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.Y.; Lee, Y.K.; Lee, S.E.; Ju, J.M.; Eom, K.S.; Kim, Y.J.; Chung, N.G.; Jeong, D.C.; Park, G.; Choi, E.Y.; et al. MyD88 in donor bone marrow cells is critical for protection from acute intestinal graft-versus-host disease. Mucosal Immunol. 2016, 9, 730–743. [Google Scholar] [CrossRef] [PubMed]
- Abreu, M.T. Toll-like receptor signaling in the intestinal epithelium: How bacterial recognition shapes intestinal function. Nat. Rev. Immunol. 2010, 10, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Gong, F.L.; Xiong, S.D.; Li, Z.Y. Medical Immunology, 3rd ed.; Science Press: Wuhan, China, 2008; pp. 134–135. [Google Scholar]
- Frazao, J.B.; Errante, P.R.; Condino-Neto, A. Toll-like receptors’ pathway disturbances are associated with increased susceptibility to infections in humans. Arch. Immunol. Ther. Exp. 2013, 61, 427–443. [Google Scholar] [CrossRef] [PubMed]
- Kenny, E.F.; O’Neill, L.A. Signalling adaptors used by Toll-like receptors: An update. Cytokine 2008, 43, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, D.R. Toll-like receptors and other links between innate and acquired alloimmunity. Curr. Opin. Immunol. 2004, 16, 538–544. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Ildstad, S.T. A novel role of innate immune responses (toll-like receptor-4) in triggering graft-versus-host disease. Transplantation 2010, 90, 1052–1053. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Cai, Z. TLR4 and organ transplant rejection. J. Zhejiang Univ. (Med. Sci.) 2011, 40, 495–500. [Google Scholar]
- Lorenz, E.; Schwartz, D.A.; Martin, P.J.; Gooley, T.; Lin, M.T.; Chien, J.W.; Hansen, J.A.; Clark, J.G. Association of TLR4 mutations and the risk for acute GVHD after HLA-matched-sibling hematopoietic stem cell transplantation. Biol. Blood Marrow Transplant. 2001, 7, 384–387. [Google Scholar] [CrossRef] [PubMed]
- Elmaagacli, A.H.; Koldehoff, M.; Hindahl, H.; Steckel, N.K.; Trenschel, R.; Peceny, R.; Ottinger, H.; Rath, P.M.; Ross, R.S.; Roggendorf, M.; et al. Mutations in innate immune system NOD2/CARD15 and TLR-4 (Thr399Ile) genes influence the risk for severe acute graft-versus-host disease in patients who underwent an allogeneic transplantation. Transplantation 2006, 81, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, I.; Hamblin, M.T.; McBride, C.; Beutler, B.; Di Rienzo, A. Excess of rare amino acid polymorphisms in the Toll-like receptor 4 in humans. Genetics 2001, 158, 1657–1664. [Google Scholar] [PubMed]
- Weng, J.; Lai, P.; Geng, S.; Luo, C.; Wu, S.; Ling, W.; Deng, C.; Huang, X.; Lu, Z.; Du, X. Role of Toll-like receptor 4 signaling in cutaneous chronic graft-versus-host disease. Clin. Transplant. 2015, 29, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Beelen, D.W.; Elmaagacli, A.; Müller, K.D.; Hirche, H.; Schaefer, U.W. Influence of intestinal bacterial decontamination using metronidazole and ciprofloxacin or ciprofloxacin alone on the development of acute graft-versus-host disease after marrow transplantation in patients with hematologic malignancies: Final results and long-term follow-up of an open-label prospective randomized trial. Blood 1999, 93, 3267–3275. [Google Scholar] [PubMed]
- Imado, T.; Iwasaki, T.; Kitano, S.; Satake, A.; Kuroiwa, T.; Tsunemi, S.; Sano, H. The protective role of host Toll-like receptor-4 in acute graft-versus-host disease. Transplantation 2010, 90, 1063–1070. [Google Scholar] [CrossRef] [PubMed]
- Fukata, M.; Chen, A.; Klepper, A.; Krishnareddy, S.; Vamadevan, A.S.; Thomas, L.S.; Xu, R.; Inoue, H.; Arditi, M.; Dannenberg, A.J.; et al. Cox-2 is regulated by Toll-like receptor-4 (TLR4) signaling: Role in proliferation and apoptosis in the intestine. Gastroenterology 2006, 131, 862–877. [Google Scholar] [CrossRef] [PubMed]
- Kuroiwa, T.; Kakishita, E.; Hamano, T.; Kataoka, Y.; Seto, Y.; Iwata, N.; Kaneda, Y.; Matsumoto, K.; Nakamura, T.; Ueki, T.; et al. Hepatocyte growth factor ameliorates acute graft-versus-host-disease and promotes hematopoietic function. J. Clin. Investig. 2001, 107, 1365–1373. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, K.; Wagner, H.; Takakura, Y. Role of immunostimulatory DNA and TLR9 ingene therapy. Crit. Rev. Ther. Drug Carr. Syst. 2006, 23, 89–110. [Google Scholar] [CrossRef]
- Elmaagacli, A.H.; Steckel, N.; Ditschkowski, M.; Hegerfeldt, Y.; Ottinger, H.; Trenschel, R.; Koldehoff, M.; Beelen, D.W. Toll-like receptor 9, NOD2 and IL23R gene polymorphisms influenced outcome in AML patients transplanted from HLA-identical sibling donors. Bone Marrow Transplant. 2011, 46, 702–708. [Google Scholar] [CrossRef] [PubMed]
- Duramad, O.; Fearon, K.L.; Chang, B.; Chan, J.H.; Gregorio, J.; Coffman, R.L.; Barrat, F.J. Inhibitors of TLR-9 act on multiple cell subsets in mouse and man in vitro and prevent death in vivo from systemic inflammation. J. Immunol. 2005, 174, 5193–5200. [Google Scholar] [CrossRef] [PubMed]
- Elmaagacli, A.H.; Koldehoff, M.; Beelen, D.W. Improved outcome of hematopoietic SCT in patients with homozygous gene variant of Toll-like receptor 9. Bone Marrow Transplant. 2009, 44, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.A.; Ehrhardt, M.J.; Lees, C.J.; Panoskaltsis-Mortari, A.; Krieg, A.M.; Sharpe, A.H.; Murphy, W.J.; Serody, J.S.; Hemmi, H.; Akira, S.; et al. TLR agonists regulate alloresponses and uncover a critical role for donor APCs in allogeneic bone marrow rejection. Blood 2008, 112, 3508–3516. [Google Scholar] [CrossRef] [PubMed]
- Heimesaat, M.M.; Nogai, A.; Bereswill, S.; Plickert, R.; Fischer, A.; Loddenkemper, C.; Steinhoff, U.; Tchaptchet, S.; Thiel, E.; Freudenberg, M.A.; et al. MyD88/TLR9 mediated immunopathology and gut microbiota dynamics in a novel murine model of intestinal graft-versus-host disease. Gut 2010, 59, 1079–1087. [Google Scholar] [CrossRef] [PubMed]
- Calcaterra, C.; Sfondrini, L.; Rossini, A.; Sommariva, M.; Rumio, C.; Ménard, S.; Balsari, A. Critical role of TLR9 in acute graft-versus-host disease. J. Immunol. 2008, 181, 6132–6139. [Google Scholar] [CrossRef] [PubMed]
- Ewaschuk, J.B.; Backer, J.L.; Churchill, T.A.; Obermeier, F.; Krause, D.O.; Madsen, K.L. Surface expression of Toll-like receptor 9 is upregulated on intestinal epithelial cells in response to pathogenicbacterial DNA. Infect Immun. 2007, 75, 2572–2579. [Google Scholar] [CrossRef] [PubMed]
- She, K.; Gilman, A.L.; Aslanian, S.; Shimizu, H.; Krailo, M.; Chen, Z.; Reid, G.S.; Wall, D.; Goldman, F.; Schultz, K.R. Altered Toll-like receptor 9 responses in circulating B cells at the onset of extensive chronic graft-versus-host disease. Biol. Blood Marrow Transplant. 2007, 13, 386–397. [Google Scholar] [CrossRef] [PubMed]
- Sivula, J.; Cordova, Z.M.; Tuimala, J.; Jaatinen, T.; Partanen, J.; Volin, L.; Turpeinen, H. Toll-Like receptor gene polymorphisms confer susceptibility to graft-versus-host disease in allogenic hematopoietic stem cell transplantation. Scand. J. Immunol. 2012, 76, 336–341. [Google Scholar] [CrossRef] [PubMed]
- Ehlers, M.; Ravetch, J.V. Opposing effects of Toll-like receptor stimulation induce autoimmunity or tolerance. Trends Immunol. 2007, 28, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Jasperson, L.K.; Bucher, C.; Panoskaltsis-Mortari, A.; Mellor, A.L.; Munn, D.H.; Blazar, B.R. Inducing the tryptophan catabolic pathway, indoleamine 2,3-dioxygenase (IDO), for suppression of graft-versus-host disease (GVHD) lethality. Blood 2009, 114, 5062–5070. [Google Scholar] [CrossRef] [PubMed]
- Cooke, K.R.; Hill, G.R.; Crawford, J.M.; Bungard, D.; Brinson, Y.S.; Delmonte, J., Jr.; Ferrara, J.L. Tumor necrosis factor-α production to lipopolysaccharide stimulation by donor cells predicts the severity of experimental acute graft-versus-host disease. J. Clin. Investig. 1998, 102, 1882–1891. [Google Scholar] [CrossRef] [PubMed]
- Sawitzki, B.; Brunstein, C.; Meisel, C.; Schumann, J.; Vogt, K.; Appelt, C.; Curtsinger, J.M.; Verneris, M.R.; Miller, J.S.; Wagner, J.E.; et al. Prevention of graft-versus-host disease by adoptive T regulatory therapy is associated with active repression of peripheral blood Toll-like receptor 5 mRNA expression. Biol. Blood Marrow Transplant. 2014, 20, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.Y.; Ryu, D.B.; Lee, S.E.; Park, G.; Choi, E.Y.; Min, C.K. Differential effect of MyD88 signal in donor T cells on graft-versus-leukemia effect and graft-versus-host disease after experimental allogeneic Stem cell transplantation. Mol. Cells 2015, 38, 966–974. [Google Scholar] [PubMed]
- Zeiser, R. Activation of innate immunity in graft-versus-host disease: Implications for Novel Targets? Oncol. Res. Treat. 2015, 38, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Nestel, F.P.; Price, K.S.; Seemayer, T.A.; Lapp, W.S. Macrophage priming and lipopolysaccharide-triggered release of tumor necrosis factor α during graft-versus-host disease. J. Exp. Med. 1992, 175, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Cooke, K.R.; Gerbitz, A.; Crawford, J.M.; Teshima, T.; Hill, G.R.; Tesolin, A.; Rossignol, D.P.; Ferrara, J.L. LPS antagonism reduces graft-versus-host disease and preserves graft-versus-leukemia activity after experimental bone marrow transplantation. J. Clin. Investig. 2001, 107, 1581–1589. [Google Scholar] [CrossRef] [PubMed]
- Garantziotis, S.; Palmer, S.M.; Snyder, L.D.; Ganous, T.; Chen, B.J.; Wang, T.; Cook, D.N.; Schwartz, D.A. Alloimmune lung injury induced by local innate immune activation through inhaled lipopolysaccharide. Transplantation 2007, 84, 1012–1019. [Google Scholar] [CrossRef] [PubMed]
- Brennan, T.V.; Lin, L.; Huang, X.; Cardona, D.M.; Li, Z.; Dredge, K.; Chao, N.J.; Yang, Y. Heparan sulfate, an endogenous TLR4 agonist, promotes acute GVHD after allogeneic stem cell transplantation. Blood 2012, 120, 2899–2908. [Google Scholar] [CrossRef] [PubMed]
- Loiarro, M.; Capolunghi, F.; Fantò, N.; Gallo, G.; Campo, S.; Arseni, B.; Carsetti, R.; Carminati, P.; de Santis, R.; Ruggiero, V.; et al. Pivotal Advance: Inhibition of MyD88 dimerization and recruitment of IRAK1 and IRAK4 by a novel peptidomimetic compound. J. Leukoc. Biol. 2007, 82, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Schmaltz, C.; Alpdogan, O.; Horndasch, K.J.; Muriglan, S.J.; Kappel, B.J.; Teshima, T.; Ferrara, J.L.; Burakoff, S.J.; van den Brink, M.R. Differential use of Fas ligand and perforin cytotoxic pathways by donor T cells in graft-versus-host disease and graft-versus-leukemia effect. Blood 2001, 97, 2886–2895. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.G.; Sergio, J.J.; Pearson, D.A.; Szot, G.L.; Shimizu, A.; Sykes, M. Interleukin-12 preserves the graft-versus-leukemia effect of allogeneic CD8 T cells while inhibiting CD4-dependent graft-versus-host disease in mice. Blood 1997, 90, 4651–4660. [Google Scholar] [PubMed]
- Dubberke, E.R.; Hollands, J.M.; Georgantopoulos, P.; Augustin, K.; DiPersio, J.F.; Mundy, L.M.; Khoury, H.J. Vancomycin-resistant enterococcal bloodstream infections on a hematopoietic stem cell transplant unit: Are the sick getting sicker? Bone Marrow Transplant. 2006, 38, 813–819. [Google Scholar] [CrossRef] [PubMed]


© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tu, S.; Zhong, D.; Xie, W.; Huang, W.; Jiang, Y.; Li, Y. Role of Toll-Like Receptor Signaling in the Pathogenesis of Graft-versus-Host Diseases. Int. J. Mol. Sci. 2016, 17, 1288. https://doi.org/10.3390/ijms17081288
Tu S, Zhong D, Xie W, Huang W, Jiang Y, Li Y. Role of Toll-Like Receptor Signaling in the Pathogenesis of Graft-versus-Host Diseases. International Journal of Molecular Sciences. 2016; 17(8):1288. https://doi.org/10.3390/ijms17081288
Chicago/Turabian StyleTu, Sanfang, Danli Zhong, Weixin Xie, Wenfa Huang, Yangyang Jiang, and Yuhua Li. 2016. "Role of Toll-Like Receptor Signaling in the Pathogenesis of Graft-versus-Host Diseases" International Journal of Molecular Sciences 17, no. 8: 1288. https://doi.org/10.3390/ijms17081288