
Cancer Cell Colonisation in the Bone Microenvironment

Abstract
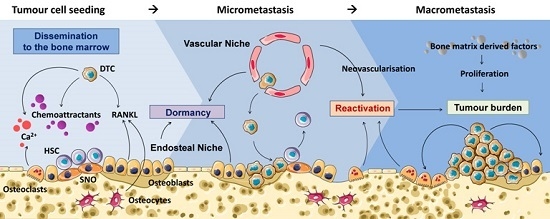
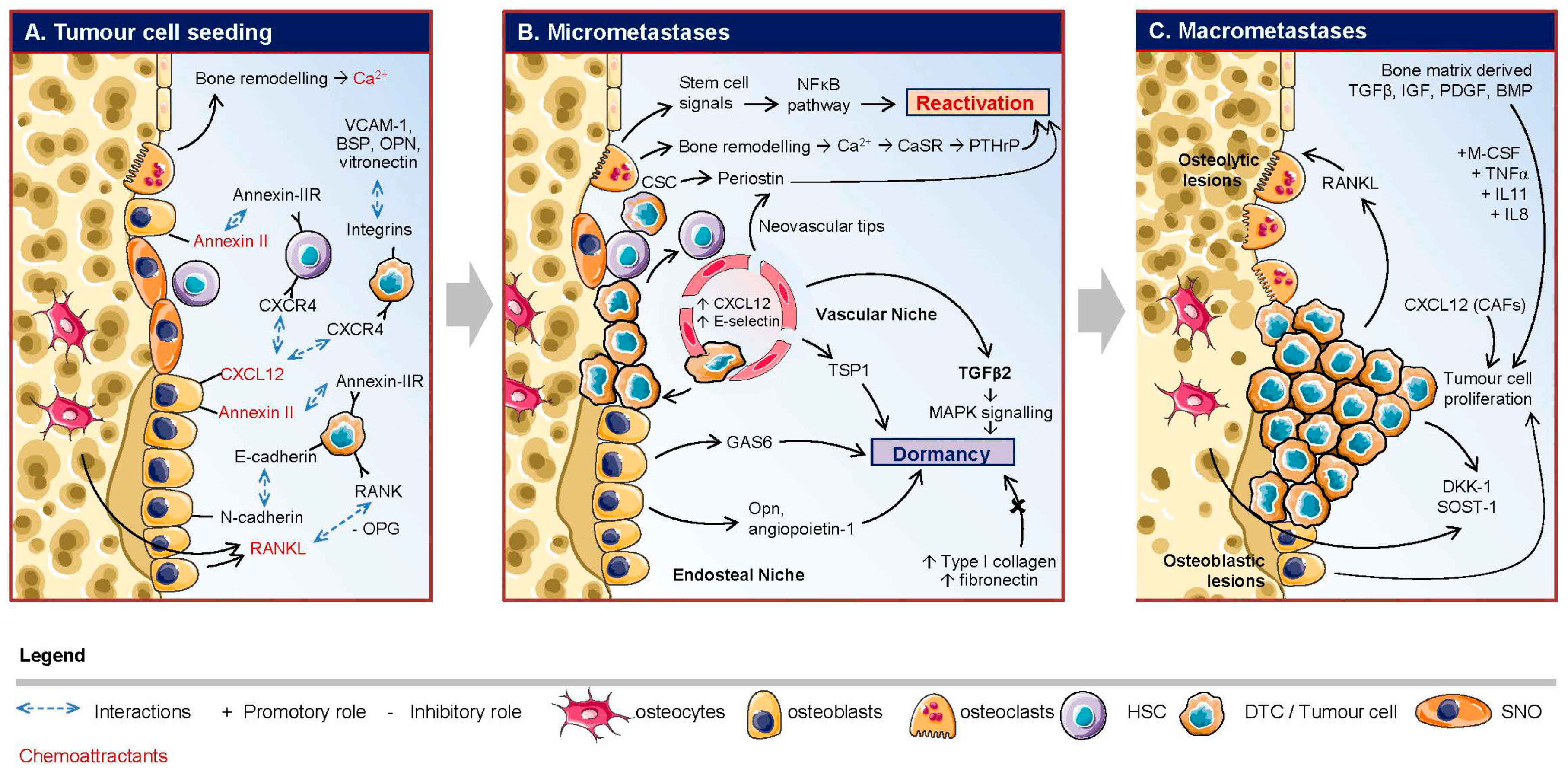
:
1. Introduction
2. Osteotropism
2.1. Cancer Cell Migration to the Bone
2.2. Cell Adhesion
3. Disseminated Tumour Cells (DTCs) in the Bone Marrow
Bone Marrow Niches
4. The Fate of DTCs in the Bone Marrow
4.1. Tumour Dormancy
4.2. Survival
4.3. Reactivation
5. Tumour Outgrowth and Secretion of Factors
6. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| BMP-7 | Bone morphogenic protein 7 |
| CAF | Cancer associated fibroblasts |
| CaSR | Calcium sensing receptor |
| CSC | Cancer Stem Cell |
| CXCL12 | C–X–C motif chemokine ligand 12 |
| CXCR4 | Couple chemokine (C–X–C) receptor type 4 |
| DTC | Disseminated tumour cell |
| EMT | Epithelial-to-mesenchymal transition |
| GAS6 | Growth arrest-specific 6 |
| HSC | Hematopoietic stem cell |
| ID1 or 3 | Inhibitor of differentiation 1 or 3 |
| IGF1 | Insulin-like growth factor 1 |
| NFκB | Nuclear factor κB |
| OPN | Osteopontin |
| PDGF | Platelet-derived growth factor |
| PTHrP | Parathyroid hormone-related protein |
| RANK | Receptor Activator of NFκB |
| RANKL | Receptor activator of NFκB ligand |
| SNO | Spindle-shaped N-cadherin+ osteoblast |
| SOST-1 | Sclerostin 1 |
| TGFβ | Transforming growth factor β |
| TSP-1 | Thrombospondin-1 |
| VCAM1 | Vascular cell adhesion molecule-1 |
References
- Coleman, R.E. Management of bone metastases. Oncologist 2000, 5, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.E. Metastatic bone disease: Clinical features, pathophysiology and treatment strategies. Cancer Treat. Rev. 2001, 27, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.E. Skeletal complications of malignancy. Cancer 1997, 80, 1588–1594. [Google Scholar] [CrossRef]
- Erler, J.T.; Bennewith, K.L.; Cox, T.R.; Lang, G.; Bird, D.; Koong, A.; Le, Q.T.; Giaccia, A.J. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell 2009, 15, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Croset, M.; Goehrig, D.; Frackowiak, A.; Bonnelye, E.; Ansieau, S.; Puisieux, A.; Clézardin, P. TWIST1 expression in breast cancer cells facilitates bone metastasis formation. J. Bone Miner. Res. 2014, 29, 1886–1899. [Google Scholar] [CrossRef] [PubMed]
- Sahay, D.; Leblanc, R.; Grunewald, T.G.P.; Ambatipudi, S.; Ribeiro, J.; Clézardin, P.; Peyruchaud, O. The LPA1/ZEB1/miR-21-activation pathway regulates metastasis in basal breast cancer. Oncotarget 2015, 6, 20604–20620. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Dave, B.; Mittal, V.; Tan, N.M.; Chang, J.C. Epithelial-mesenchymal transition, cancer stem cells and treatment resistance. Breast Cancer Res. 2012, 14. [Google Scholar] [CrossRef] [PubMed]
- Gunasinghe, N.P.; Wells, A.; Thompson, E.W.; Hugo, H.J. Mesenchymal-epithelial transition (MET) as a mechanism for metastatic colonisation in breast cancer. Cancer Metastasis Rev. 2012, 31, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Bendinelli, P.; Maroni, P.; Matteucci, E.; Desiderio, M.A. HGF and TGFβ1 differently influenced Wwox regulatory function on Twist program for mesenchymal-epithelial transition in bone metastatic versus parental breast carcinoma cells. Mol. Cancer 2015, 14. [Google Scholar] [CrossRef] [PubMed]
- Sanders, J.L.; Chattopadhyay, N.; Kifor, O.; Yamaguchi, T.; Butters, R.R.; Brown, E.M. Extracellular calcium-sensing receptor expression and its potential role in regulating parathyroid hormone-related peptide secretion in human breast cancer cell lines. Endocrinology 2000, 141, 4357–4364. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Takyar, F.M.; Swan, K.; Jeong, J.; VanHouten, J.; Sullivan, C.A.; Dann, P.; Yu, H.; Fiaschi-Taesch, N.; Chang, W.; et al. Calcium-sensing receptor (CaSR) promotes breast cancer by stimulating intracrine actions of parathyroid hormone-related protein. Cancer Res. 2016, 76, 5348–5360. [Google Scholar] [CrossRef] [PubMed]
- Fidler, I.J. The pathogenesis of cancer metastasis: the “seed and soil” hypothesis revisited. Nat. Rev. Cancer 2003, 3, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Siegel, P.M.; Shu, W.; Drobnjak, M.; Kakonen, S.M.; Cordón-Cardo, C.; Guise, T.A.; Massagué, J. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 2003, 3, 537–549. [Google Scholar] [CrossRef]
- Müller, A.; Homey, B.; Soto, H.; Ge, N.; Catron, D.; Buchanan, M.E.; McClanahan, T.; Murphy, E.; Yuan, W.; Wagner, S.N.; et al. Involvement of chemokine receptors in breast cancer metastasis. Nature 2001, 410, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Ha, H.K.; Lee, W.; Park, H.J.; Lee, S.D.; Lee, J.Z.; Chung, M.K. Clinical significance of CXCL16/CXCR6 expression in patients with prostate cancer. Mol. Med. Rep. 2011, 4, 419–424. [Google Scholar] [PubMed]
- Golan, K.; Kollet, O.; Lapidot, T. Dynamic cross talk between S1P and CXCL12 regulates hematopoietic stem cells migration, development and bone remodeling. Pharm. Basel. Switz. 2013, 6, 1145–1169. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.C.P.; Luker, K.E.; Garbow, J.R.; Prior, J.L.; Jackson, E.; Piwnica-Worms, D.; Luker, G.D. CXCR4 regulates growth of both primary and metastatic breast cancer. Cancer Res. 2004, 64, 8604–8612. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Cheng, G.; Hao, M.; Zheng, J.; Zhou, X.; Zhang, J.; Taichman, R.S.; Pienta, K.J.; Wang, J. CXCL12/CXCR4/CXCR7 chemokine axis and cancer progression. Cancer Metastasis Rev. 2010, 29, 709–722. [Google Scholar] [CrossRef] [PubMed]
- Richert, M.M.; Vaidya, K.S.; Mills, C.N.; Wong, D.; Korz, W.; Hurst, D.R.; Welch, D.R. Inhibition of CXCR4 by CTCE-9908 inhibits breast cancer metastasis to lung and bone. Oncol. Rep. 2009, 21, 761–767. [Google Scholar] [PubMed]
- Hu, W.; Zhen, X.; Xiong, B.; Wang, B.; Zhang, W.; Zhou, W. CXCR6 is expressed in human prostate cancer in vivo and is involved in the in vitro invasion of PC3 and LNCap cells. Cancer Sci. 2008, 99, 1362–1369. [Google Scholar] [CrossRef] [PubMed]
- Saidak, Z.; Boudot, C.; Abdoune, R.; Petit, L.; Brazier, M.; Mentaverri, R.; Kamel, S. Extracellular calcium promotes the migration of breast cancer cells through the activation of the calcium sensing receptor. Exp. Cell Res. 2009, 315, 2072–2080. [Google Scholar] [CrossRef] [PubMed]
- Adams, G.B.; Chabner, K.T.; Alley, I.R.; Olson, D.P.; Szczepiorkowski, Z.M.; Poznansky, M.C.; Kos, C.H.; Pollak, M.R.; Brown, E.M.; Scadden, D.T. Stem cell engraftment at the endosteal niche is specified by the calcium-sensing receptor. Nature 2006, 439, 599–603. [Google Scholar] [CrossRef] [PubMed]
- Mundy, G.R. Metastasis to bone: causes, consequences and therapeutic opportunities. Nat. Rev. Cancer 2002, 2, 584–593. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.R.; Mundy, G.R. Advances in osteoclast biology: Old findings and new insights from mouse models. Nat. Rev. Rheumatol. 2011, 7, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Santini, D.; Schiavon, G.; Vincenzi, B.; Gaeta, L.; Pantano, F.; Russo, A.; Ortega, C.; Porta, C.; Galluzzo, S.; Armento, G.; et al. Receptor activator of NF-κB (RANK) expression in primary tumors associates with bone metastasis occurrence in breast cancer patients. PLoS ONE 2011, 6, e19234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, D.H.; Nakashima, T.; Sanchez, O.H.; Kozieradzki, I.; Komarova, S.V.; Sarosi, I.; Morony, S.; Rubin, E.; Sarao, R.; Hojilla, C.V.; et al. Regulation of cancer cell migration and bone metastasis by RANKL. Nature 2006, 440, 692–696. [Google Scholar] [CrossRef] [PubMed]
- Boyce, B.F.; Xing, L. Functions of RANKL/RANK/OPG in bone modeling and remodeling. Arch Biochem. Biophys. 2008, 473, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Arrigoni, C.; de Luca, P.; Gilardi, M.; Previdi, S.; Broggini, M.; Moretti, M. Direct but not indirect co-culture with osteogenically differentiated human bone marrow stromal cells increases RANKL/OPG ratio in human breast cancer cells generating bone metastases. Mol. Cancer. 2014, 21. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, T.; Utsuyama, M.; Hirokawa, K. Expression profiles of receptor activator of nuclear factor κB ligand, receptor activator of nuclear factor κB, and osteoprotegerin messenger RNA in aged and ovariectomized rat bones. J. Bone Miner Res. 2001, 16, 1416–1425. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, T.; Hayashi, M.; Fukunaga, T.; Kurata, K.; Oh-hora, M.; Feng, J.Q.; Bonewald, L.F.; Kodama, T.; Wutz, A.; Wagner, E.F.; et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat. Med. 2011, 17, 1231–1234. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Onal, M.; Jilka, R.L.; Weinstein, R.S.; Manolagas, S.C.; O’Brien, C.A. Matrix-embedded cells control osteoclast formation. Nat. Med. 2011, 17, 1235–1241. [Google Scholar] [CrossRef] [PubMed]
- Shiozawa, Y.; Havens, A.M.; Jung, Y.; Ziegler, A.M.; Pedersen, E.A.; Wang, J.; Wang, J.; Lu, G.; Roodman, G.D.; Loberg, R.D.; et al. Annexin II/annexin II receptor axis regulates adhesion, migration, homing, and growth of prostate cancer. J. Cell Biochem. 2008, 105, 370–380. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.A.; Jiang, W.G. Loss of tight junction barrier function and its role in cancer metastasis. Biochim. Biophys. Acta. 2009, 1788, 872–891. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yu, C.; Gao, X.; Welte, T.; Muscarella, A.M.; Tian, L.; Zhao, H.; Zhao, Z.; Du, S.; Tao, J.; et al. The osteogenic niche promotes early-stage bone colonization of disseminated breast cancer cells. Cancer Cell. 2015, 27, 193–210. [Google Scholar] [CrossRef] [PubMed]
- Clëzardin, P. Integrins in bone metastasis formation and potential therapeutic implications. Curr. Cancer Drug Targets 2009, 9, 801–806. [Google Scholar] [CrossRef] [PubMed]
- Sung, V.; Stubbs, J.T.; Fisher, L.; Aaron, A.D.; Thompson, E.W. Bone sialoprotein supports breast cancer cell adhesion proliferation and migration through differential usage of the αvβ3 and αvβ5 integrins. J. Cell Physiol. 1998, 176, 482–494. [Google Scholar] [CrossRef]
- Zhao, Y.; Bachelier, R.; Treilleux, I.; Pujuguet, P.; Peyruchaud, O.; Baron, R.; Clément-Lacroix, P.; Clézardin, P. Tumor αvβ3 integrin is a therapeutic target for breast cancer bone metastases. Cancer Res. 2007, 67, 5821–5830. [Google Scholar] [CrossRef] [PubMed]
- Sloan, E.K.; Pouliot, N.; Stanley, K.L.; Chia, J.; Moseley, J.M.; Hards, D.K.; Anderson, R.L. Tumor-specific expression of αvβ3 integrin promotes spontaneous metastasis of breast cancer to bone. Breast Cancer Res. 2006, 8. [Google Scholar] [CrossRef] [PubMed]
- Michigami, T.; Shimizu, N.; Williams, P.J.; Niewolna, M.; Dallas, S.L.; Mundy, G.R.; Yoneda, T. Cell-cell contact between marrow stromal cells and myeloma cells via VCAM-1 and α4β1-integrin enhances production of osteoclast-stimulating activity. Blood 2000, 96, 1953–1960. [Google Scholar] [PubMed]
- Malanchi, I.; Santamaria-Martínez, A.; Susanto, E.; Peng, H.; Lehr, H.A.; Delaloye, J.F.; Huelsken, J. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature 2012, 481, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.H.F.; Wang, Q.; Gerald, W.; Hudis, C.A.; Norton, L.; Smid, M.; Foekens, J.A.; Massagué, J. Latent bone metastasis in breast cancer tied to Src-dependent survival signals. Cancer Cell 2009, 16, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.H.; Jin, X.; Malladi, S.; Zou, Y.; Wen, Y.H.; Brogi, E.; Smid, M.; Foekens, J.A.; Massagué, J. Selection of bone metastasis seeds by mesenchymal signals in the primary tumor stroma. Cell 2013, 15, 1060–1073. [Google Scholar] [CrossRef] [PubMed]
- Winkler, I.G.; Barbier, V.; Nowlan, B.; Jacobsen, R.N.; Forristal, C.E.; Patton, J.T.; Magnani, J.L.; Lévesque, J.P. Vascular niche E-selectin regulates hematopoietic stem cell dormancy, self renewal and chemoresistance. Nat. Med. 2012, 18, 1651–1657. [Google Scholar] [CrossRef] [PubMed]
- Shiozawa, Y.; Pedersen, E.A.; Patel, L.R.; Ziegler, A.M.; Havens, A.M.; Jung, Y.; Wang, J.; Zalucha, S.; Loberg, R.D.; Pienta, K.J.; et al. GAS6/AXL axis regulates prostate cancer invasion, proliferation, and survival in the bone marrow niche. Neoplasia 2010, 12, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Bhatia, R. Molecular pathways: Stem cell quiescence. Clin. Cancer Res. 2011, 17, 4936–4941. [Google Scholar] [CrossRef] [PubMed]
- Lamorte, S.; Remédio, L.; Dias, S. Communication between bone marrow niches in normal bone marrow function and during hemopathies progression. Hematol. Rev. 2009, 1. [Google Scholar] [CrossRef]
- Ghajar, C.M.; Peinado, H.; Mori, H.; Matei, I.R.; Evason, K.J.; Brazier, H.; Almeida, D.; Koller, A.; Hajjar, K.A.; Stainier, D.Y.; et al. The perivascular niche regulates breast tumor dormancy. Nat. Cell. Biol. 2013, 15, 807–817. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Okuda, H.; Xing, F.; Pandey, P.R.; Watabe, M.; Hirota, S.; Pai, S.K.; Liu, W.; Fukuda, K.; Chambers, C.; et al. Bone morphogenetic protein 7 in dormancy and metastasis of prostate cancer stem-like cells in bone. J. Exp. Med. 2011, 208, 2641–2655. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Xing, F.; Liu, Y.; Wu, K.; Said, N.; Pochampally, R.; Shiozawa, Y.; Lin, H.K.; Balaji, K.C.; Watabe, K. Secreted protein acidic and rich in cysteine (SPARC) mediates metastatic dormancy of prostate cancer in the bone. J. Biol. Chem. 2016, 291, 19351–19363. [Google Scholar] [CrossRef] [PubMed]
- Bragado, P.; Estrada, Y.; Parikh, F.; Krause, S.; Capobianco, C.; Farina, H.G.; Schewe, D.M.; Aguirre-Ghiso, J.A. TGF-β2 dictates disseminated tumour cell fate in target organs through TGF-β-RIII and p38α/β signalling. Nat. Cell Biol. 2013, 15, 1351–1361. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Mu, E.; Wei, Y.; Riethdorf, S.; Yang, Q.; Yuan, M.; Yan, J.; Hua, Y.; Tiede, B.J.; Lu, X.; et al. VCAM-1 promotes osteolytic expansion of indolent bone micrometastasis of breast cancer by engaging α4β1-positive osteoclast progenitors. Cancer Cell 2011, 20, 701–714. [Google Scholar] [CrossRef] [PubMed]
- Braun, S.; Vogl, F.D.; Naume, B.; Janni, W.; Osborne, M.P.; Coombes, R.C.; Schlimok, G.; Diel, I.J.; Gerber, B.; Gebauer, G.; et al. A pooled analysis of bone marrow micrometastasis in breast cancer. N. Engl. J. Med. 2005, 353, 793–802. [Google Scholar] [CrossRef] [PubMed]
- Loeb, S.; Feng, Z.; Ross, A.; Trock, B.J.; Humphreys, E.B.; Walsh, P.C. Can we stop prostate specific antigen testing 10 years after radical prostatectomy? J. Urol. 2011, 186, 500–505. [Google Scholar] [CrossRef] [PubMed]
- Morrissey, C.; Roudier, M.P.; Dowell, A.; True, L.D.; Ketchanji, M.; Welty, C.; Corey, E.; Lange, P.H.; Higano, C.S.; Vessella, R.L. Effects of androgen deprivation therapy and bisphosphonate treatment on bone in patients with metastatic castration-resistant prostate cancer: results from the University Of Washington Rapid Autopsy Series. J. Bone Miner Res. 2013, 28, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Lam, H.M.; Vessella, R.L.; Morrissey, C. The role of the microenvironment-dormant prostate disseminated tumor cells in the bone marrow. Drug. Discov. Today 2014, 11, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Mansi, J.L.; Gogas, H.; Bliss, J.M.; Gazet, J.C.; Berger, U.; Coombes, R.C. Outcome of primary-breast-cancer patients with micrometastases: A long-term follow-up study. Lancet 1999, 354, 197–202. [Google Scholar] [CrossRef]
- Lilleby, W.; Stensvold, A.; Mills, I.G.; Nesland, J.M. Disseminated tumor cells and their prognostic significance in nonmetastatic prostate cancer patients. Int. J. Cancer 2013, 133, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Morgan, T.M.; Lange, P.H.; Porter, M.P.; Lin, D.W.; Ellis, W.J.; Gallaher, I.S.; Vessella, R.L. Disseminated tumor cells in prostate cancer patients after radical prostatectomy and without evidence of disease predicts biochemical recurrence. Clin. Cancer Res. 2009, 15, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.E.; Gregory, W.; Marshall, H.; Wilson, C.; Holen, I. The metastatic microenvironment of breast cancer: Clinical implications. Breast 2013, 22, S50–S56. [Google Scholar] [CrossRef] [PubMed]
- Ehninger, A.; Trumpp, A. The bone marrow stem cell niche grows up: Mesenchymal stem cells and macrophages move in. J. Exp. Med. 2011, 208, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Kusumbe, A.P.; Ramasamy, S.K.; Itkin, T.; Mäe, M.A.; Langen, U.H.; Betsholtz, C.; Lapidot, T.; Adams, R.H. Age-dependent modulation of vascular niches for haematopoietic stem cells. Nature 2016, 532, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Broxmeyer, H.E.; Orschell, C.M.; Clapp, D.W.; Hangoc, G.; Cooper, S.; Plett, P.A.; Liles, W.C.; Li, X.; Graham-Evans, B.; Campbell, T.B.; et al. Rapid mobilization of murine and human hematopoietic stem and progenitor cells with AMD3100, a CXCR4 antagonist. J. Exp. Med. 2005, 201, 1307–1318. [Google Scholar] [CrossRef] [PubMed]
- Haider, M.T.; Holen, I.; Dear, T.N.; Hunter, K.; Brown, H.K. Modifying the osteoblastic niche with zoledronic acid in vivo—Potential implications for breast cancer bone metastasis. Bone 2014, 66, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Shiozawa, Y.; Pedersen, E.A.; Havens, A.M.; Jung, Y.; Mishra, A.; Joseph, J.; Kim, J.K.; Patel, L.R.; Ying, C.; Ziegler, A.M.; et al. Human prostate cancer metastases target the hematopoietic stem cell niche to establish footholds in mouse bone marrow. J. Clin. Investig. 2011, 121, 1298–1312. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Reeves, K.J.; Brown, H.K.; Fowles, A.C.; Docherty, F.E.; Ottewell, P.D.; Croucher, P.I.; Holen, I.; Eaton, C.L. The frequency of osteolytic bone metastasis is determined by conditions of the soil, not the number of seeds; evidence from in vivo models of breast and prostate cancer. J. Exp. Clin. Cancer Res. 2015, 34. [Google Scholar] [CrossRef] [PubMed]
- Price, T.T.; Burness, M.L.; Sivan, A.; Warner, M.J.; Cheng, R.; Lee, C.H.; Olivere, L.; Comatas, K.; Magnani, J.; Lyerly, H.K.; et al. Dormant breast cancer micrometastases reside in specific bone marrow niches that regulate their transit to and from bone. Sci. Transl. Med. 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Docherty, F.E.; Brown, H.K.; Reeves, K.J.; Fowles, A.C.; Ottewell, P.D.; Dear, T.N.; Holen, I.; Croucher, P.I.; Eaton, C.L. Prostate cancer cells preferentially home to osteoblast-rich areas in the early stages of bone metastasis: evidence from in vivo models. J. Bone Miner Res. 2014, 29, 2688–2696. [Google Scholar] [CrossRef] [PubMed]
- Ghajar, C.M. Metastasis prevention by targeting the dormant niche. Nat. Rev. Cancer 2015, 15, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Croucher, P.I.; McDonald, M.M.; Martin, T.J. Bone metastasis: The importance of the neighbourhood. Nat. Rev. Cancer 2016, 16, 373–386. [Google Scholar] [CrossRef] [PubMed]
- Sosa, M.S.; Bragado, P.; Aguirre-Ghiso, J.A. Mechanisms of disseminated cancer cell dormancy: An awakening field. Nat. Rev. Cancer 2014, 14, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Anthony, B.A.; Link, D.C. Regulation of hematopoietic stem cells by bone marrow stromal cells. Trends Immunol. 2014, 35, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Wang, J.; Shiozawa, Y.; McGee, S.; Kim, J.; Jung, Y.; Joseph, J.; Berry, J.E.; Havens, A.; Pienta, K.J.; et al. Hypoxia stabilizes GAS6/Axl signaling in metastatic prostate cancer. Mol. Cancer Res. 2012, 10, 703–712. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Deng, C.; Li, Y.P. TGF-β and BMP signaling in osteoblast differentiation and bone formation. Int. J. Biol. Sci. 2012, 8, 272–288. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.X.; Bos, P.D.; Massagué, J. Metastasis: From dissemination to organ-specific colonization. Nat. Rev. Cancer 2009, 9, 274–284. [Google Scholar] [CrossRef] [PubMed]
- Becker, S.; Becker-Pergola, G.; Wallwiener, D.; Solomayer, E.F.; Fehm, T. Detection of cytokeratin-positive cells in the bone marrow of breast cancer patients undergoing adjuvant therapy. Breast Cancer Res. Treat. 2006, 97, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.W.; Lee, A.; Shientag, L.; Yu, J.; Dong, Y.; Kao, G.; Al-Mehdi, A.B.; Bernhard, E.J.; Muschel, R.J. Apoptosis: An early event in metastatic inefficiency. Cancer Res. 2001, 61, 333–338. [Google Scholar] [PubMed]
- Minn, A.J.; Kang, Y.; Serganova, I.; Gupta, G.P.; Giri, D.D.; Doubrovin, M.; Ponomarev, V.; Gerald, W.L.; Blasberg, R.; Massagué, J. Distinct organ-specific metastatic potential of individual breast cancer cells and primary tumors. J. Clin. Investig. 2005, 115, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J.; Obenauf, A.C. Metastatic colonization by circulating tumour cells. Nature 2016, 529, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Eck, S.M.; Côté, A.L.; Winkelman, W.D.; Brinckerhoff, C.E. CXCR4 and matrix metalloproteinase-1 are elevated in breast carcinoma-associated fibroblasts and in normal mammary fibroblasts exposed to factors secreted by breast cancer cells. Mol. Cancer Res. 2009, 7, 1033–1044. [Google Scholar] [CrossRef] [PubMed]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 2005, 121, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Prajapati, P.; Lambert, D.W. Cancer-associated fibroblasts—Not-so-innocent bystanders in metastasis to bone? J. Bone Oncol. 2016. [Google Scholar] [CrossRef]
- Ottewell, P.D.; Wang, N.; Meek, J.; Fowles, C.A.; Croucher, P.I.; Eaton, C.L.; Holen, I. Castration-induced bone loss triggers growth of disseminated prostate cancer cells in bone. Endocr. Relat. Cancer 2014, 21, 769–781. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Zhou, H.; Fong-Yee, C.; Modzelewski, J.R.K.; Seibel, M.J.; Dunstan, C.R. Bone resorption increases tumour growth in a mouse model of osteosclerotic breast cancer metastasis. Clin. Exp. Metastasis 2008, 25, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Ottewell, P.D.; Wang, N.; Brown, H.K.; Reeves, K.J.; Fowles, C.A.; Croucher, P.I.; Eaton, C.L.; Holen, I. Zoledronic acid has differential antitumor activity in the pre- and postmenopausal bone microenvironment in vivo. Clin. Cancer Res. 2014, 20, 2922–2932. [Google Scholar] [CrossRef] [PubMed]
- Ottewell, P.D.; O’Donnell, L.; Holen, I. Molecular alterations that drive breast cancer metastasis to bone. BoneKEy Rep. 2015, 18. [Google Scholar] [CrossRef] [PubMed]
- Croucher, P.I.; Parker, B.S.; Corcoran, N.; Rogers, M.J. Bone turnover markers and prostate cancer: Not just a measure of bone disease? Eur. Urol. 2015, 68, 51–52. [Google Scholar] [CrossRef] [PubMed]
- Lipton, A.; Chapman, J.A.W.; Demers, L.; Shepherd, L.E.; Han, L.; Wilson, C.F.; Pritchard, K.I.; Leitzel, K.E.; Ali, S.M.; Pollak, M. Elevated bone turnover predicts for bone metastasis in postmenopausal breast cancer: Results of NCIC CTG MA. 14. J. Clin. Oncol. 2011, 29, 3605–3610. [Google Scholar] [CrossRef] [PubMed]
- Roodman, G.D. High bone turnover markers predict poor outcome in patients with bone metastasis. J. Clin. Oncol. 2005, 23, 4821–4822. [Google Scholar] [CrossRef] [PubMed]
- Anders, C.K.; Johnson, R.; Litton, J.; Phillips, M.; Bleyer, A. Breast cancer before age 40 years. Semin. Oncol. 2009, 36, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.W.; Porter, M.; Montgomery, B. Treatment and survival outcomes in young men diagnosed with prostate cancer: A population-based cohort study. Cancer 2009, 115, 2863–2871. [Google Scholar] [CrossRef] [PubMed]
- Rakhra, K.; Bachireddy, P.; Zabuawala, T.; Zeiser, R.; Xu, L.; Kopelman, A.; Fan, A.C.; Yang, Q.; Braunstein, L.; Crosby, E.; et al. CD4(+) T cells contribute to the remodeling of the microenvironment required for sustained tumor regression upon oncogene inactivation. Cancer Cell 2010, 18, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Baschuk, N.; Rautela, J.; Parker, B.S. Bone specific immunity and its impact on metastasis. BONEKey Rep. 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Le Gall, C.; Bellahcène, A.; Bonnelye, E.; Gasser, J.A.; Castronovo, V.; Green, J.; Zimmermann, J.; Clézardin, P. A cathepsin K inhibitor reduces breast cancer induced osteolysis and skeletal tumor burden. Cancer Res. 2007, 67, 9894–9902. [Google Scholar] [CrossRef] [PubMed]
- Bellahcène, A.; Bachelier, R.; Detry, C.; Lidereau, R.; Clézardin, P.; Castronovo, V. Transcriptome analysis reveals an osteoblast-like phenotype for human osteotropic breast cancer cells. Breast Cancer Res. Treat. 2007, 101, 135–148. [Google Scholar] [CrossRef] [PubMed]
- Pawelek, J.M.; Chakraborty, A.K. Fusion of tumour cells with bone marrow-derived cells: A unifying explanation for metastasis. Nat. Rev. Cancer 2008, 8, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Lawson, M.A.; McDonald, M.M.; Kovacic, N.; Khoo, W.H.; Terry, R.L.; Down, J.; Kaplan, W.; Paton-Hough, J.; Fellows, C.; Pettitt, J.A.; et al. Osteoclasts control reactivation of dormant myeloma cells by remodelling the endosteal niche. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Barkan, D.; El Touny, L.H.; Michalowski, A.M.; Smith, J.A.; Chu, I.; Davis, A.S.; Webster, J.D.; Hoover, S.; Simpson, R.M.; Gauldie, J.; et al. Metastatic growth from dormant cells induced by a col-I-enriched fibrotic environment. Cancer Res. 2010, 70, 5706–5716. [Google Scholar] [CrossRef] [PubMed]
- Aguirre-Ghiso, J.A.; Liu, D.; Mignatti, A.; Kovalski, K.; Ossowski, L. Urokinase receptor and fibronectin regulate the ERKMAPK to p38MAPK activity ratios that determine carcinoma cell proliferation or dormancy in vivo. Mol. Biol. Cell 2001, 12, 863–879. [Google Scholar] [CrossRef] [PubMed]
- Christensen, J.; Shastri, V.P. Matrix-metalloproteinase-9 is cleaved and activated by Cathepsin K. BMC Res. Notes 2015, 8. [Google Scholar] [CrossRef] [PubMed]
- Kollet, O.; Dar, A.; Shivtiel, S.; Kalinkovich, A.; Lapid, K.; Sztainberg, Y.; Tesio, M.; Samstein, R.M.; Goichberg, P.; Spiegel, A.; et al. Osteoclasts degrade endosteal components and promote mobilization of hematopoietic progenitor cells. Nat. Med. 2006, 12, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Swarbrick, A.; Roy, E.; Allen, T.; Bishop, J.M. Id1 cooperates with oncogenic Ras to induce metastatic mammary carcinoma by subversion of the cellular senescence response. Proc. Natl. Acad. Sci. USA 2008, 105, 5402–5407. [Google Scholar] [CrossRef] [PubMed]
- Stankic, M.; Pavlovic, S.; Chin, Y.; Brogi, E.; Padua, D.; Norton, L.; Massagué, J.; Benezra, R. TGF-β-Id1 signaling opposes Twist1 and promotes metastatic colonization via a mesenchymal-to-epithelial transition. Cell Rep. 2013, 5, 1228–1242. [Google Scholar] [CrossRef] [PubMed]
- Brown, H.K.; Ottewell, P.D.; Evans, C.A.; Holen, I. Location matters: Osteoblast and osteoclast distribution is modified by the presence and proximity to breast cancer cells in vivo. Clin. Exp. Metastasis 2012, 29, 927–938. [Google Scholar] [CrossRef] [PubMed]
- Buijs, J.T.; Stayrook, K.R.; Guise, T.A. The role of TGF-β in bone metastasis: novel therapeutic perspectives. BoneKEy Rep. 2012, 1. [Google Scholar] [CrossRef] [PubMed]
- Le Pape, F.; Vargas, G.; Clézardin, P. The role of osteoclasts in breast cancer bone metastasis. J. Bone Oncol. 2016. [Google Scholar] [CrossRef]
- Croset, M.; Kan, C.; Clézardin, P. Tumour-derived miRNAs and bone metastasis. BoneKEy Rep. 2015, 688. [Google Scholar] [CrossRef] [PubMed]
- Ell, B.; Mercatali, L.; Ibrahim, T.; Campbell, N.; Schwarzenbach, H.; Pantel, K.; Amadori, D.; Kang, Y. Tumor-induced osteoclast miRNA changes as regulators and biomarkers of osteolytic bone metastasis. Cancer Cell 2013, 24, 542–556. [Google Scholar] [CrossRef] [PubMed]
- Sugatani, T.; Vacher, J.; Hruska, K.A. A microRNA expression signature of osteoclastogenesis. Blood 2011, 117, 3648–3657. [Google Scholar] [CrossRef] [PubMed]
- Valencia, K.; Luis-Ravelo, D.; Bovy, N.; Antón, I.; Martínez-Canarias, S.; Zandueta, C.; Ormazábal, C.; Struman, I.; Tabruyn, S.; Rebmann, V.; et al. miRNA cargo within exosome-like vesicle transfer influences metastatic bone colonization. Mol. Oncol. 2014, 8, 689–703. [Google Scholar] [CrossRef] [PubMed]
- Weilbaecher, K.N.; Guise, T.A.; McCauley, L.K. Cancer to bone: A fatal attraction. Nat. Rev. Cancer 2011, 11, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Duong, L.T.; Wesolowski, G.A.; Leung, P.; Oballa, R.; Pickarski, M. Efficacy of a cathepsin K inhibitor in a preclinical model for prevention and treatment of breast cancer bone metastasis. Mol. Cancer Ther. 2014, 13, 2898–2909. [Google Scholar] [CrossRef] [PubMed]
- Taubenberger, A.V. In vitro microenvironments to study breast cancer bone colonisation. Adv. Drug. Deliv. Rev. 2014, 79, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Rochefort, G.Y. The osteocyte as a therapeutic target in the treatment of osteoporosis. Ther. Adv. Musculoskelet. Dis. 2014, 6, 79–91. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Function | Protein | Description | Disease | Reference |
|---|---|---|---|---|
| Homing | SIP | SIP can act in unison with CXCL12 as a chemoattractant | – | [17] |
| CXCR4/CXCL12 | CXCR4-expressing cancer cell migration to the bone is mediated by osteoblast derived CXCL12. | Breast cancer Prostate cancer | [19] | |
| CXCR6/CXCL16 | CXCL16 is expressed in bone tissue and promotes migration of CXCR6-expressing cancer cells in vitro. | Prostate cancer | [21] | |
| Ca2+/CaSR | Ca2+ from bone remodelling stimulates migration of CaSR-expressing cancer cells. | Breast cancer | [23] | |
| RANK/RANKL | RANK/RANKL axis promotes cancer cell migration by mediating cytoskeleton rearrangement in vitro. | Breast cancer Prostate cancer | [27] | |
| Annexin II/Annexin IIR | Annexin II produced by osteoblast and endothelial cells promotes the migration of cells expressing annexin II receptor. | Prostate cancer | [33] | |
| Adhesion | CXCR4/CXCL12 | CXCL12 in media from human primary bone-marrow has chemotactic properties. Blocking with neutralising CXCR4 antibodies impaired migration. | Breast cancer | [15] |
| Annexin II/Annexin IIR | Annexin II is produced by endothelial and osteoblast cells and promotes adhesion of tumour cells expressing annexin II receptor. | Prostate cancer | [33] | |
| E-cadherin/N-cadherin | E-cadherin was found to be expressed by cancer cells and form adherin junctions with N-cadherin in osteogenic cells. | Breast cancer | [35] | |
| Integrin αVβ3 and αVβ5 | Tumour cells expressing integrin αVβ3 and/or αVβ5 have the capacity to bind bone extracellular proteins such as fibronectin, vitronectin and osteopontin. | Breast cancer | [37,38] | |
| Integrin α4β1/VCAM1 | Integrin α4β1 expression by myeloma cells allow bone cells to bind through VCAM1 interactions. | Multiple myeloma | [40] | |
| Survival | Periostin | CSCs were shown to modify the metastatic niche through stromal periostin expression. | Breast cancer | [41] |
| Src | Src-associated gene signature is linked with late-onset bone metastasis. Src activity has been reported in cancer cells “primed” for metastasis in the bone marrow. | Breast cancer | [42,43] | |
| Dormancy | E-selectin and CXCL12 | Vascular regions are rich in E-selectin and CXCL12, which is associated with HSC dormancy. | – | [44] |
| Gas6/Axl/Sky/Mer | GAS6 is secreted by osteoblasts and is involved in maintaining HSC quiescence. | Prostate cancer | [45] | |
| Angiopoietin-1 | Involved in forming a quiescent niche for HSCs. | – | [46,47] | |
| TSP-1 | The secretion of TSP-1 from endothelial cells induces cancer cell dormancy. | Breast cancer | [48] | |
| TGFβ 2/BMP7/SPARC | Indolent prostate cancer cells secrete SPARC, which can promote BMP7-mediated senescence. | Prostate Cancer | [49,50] | |
| BMP7 | BMP7 is secreted from bone stromal cells and induces senescence in prostate cancer stem-like cells. | Prostate cancer | [49] | |
| Reactivation | TGFβ1 | Secreted TGFβ1 enhances tumour cell formation. | – | [51] |
| Integrin α4β1/VCAM1 | VCAM1-expressing cancer cells recruit integrin α4β1+ osteoclast progenitors and initiate reactivation through the vicious cycle. | Breast cancer | [52] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kan, C.; Vargas, G.; Pape, F.L.; Clézardin, P. Cancer Cell Colonisation in the Bone Microenvironment. Int. J. Mol. Sci. 2016, 17, 1674. https://doi.org/10.3390/ijms17101674
Kan C, Vargas G, Pape FL, Clézardin P. Cancer Cell Colonisation in the Bone Microenvironment. International Journal of Molecular Sciences. 2016; 17(10):1674. https://doi.org/10.3390/ijms17101674
Chicago/Turabian StyleKan, Casina, Geoffrey Vargas, François Le Pape, and Philippe Clézardin. 2016. "Cancer Cell Colonisation in the Bone Microenvironment" International Journal of Molecular Sciences 17, no. 10: 1674. https://doi.org/10.3390/ijms17101674