
Emerging Non-Canonical Functions and Regulation by p53: p53 and Stemness

Abstract
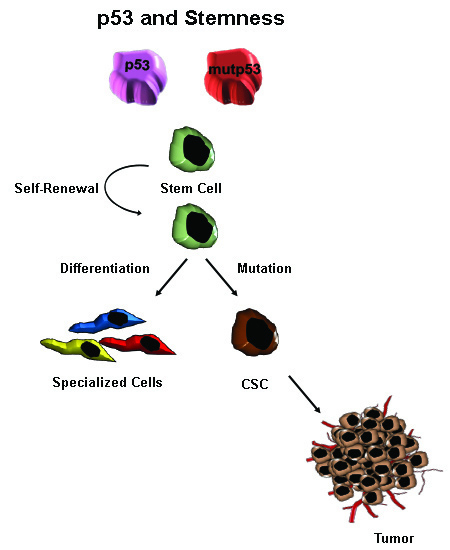
:
{kind=link}
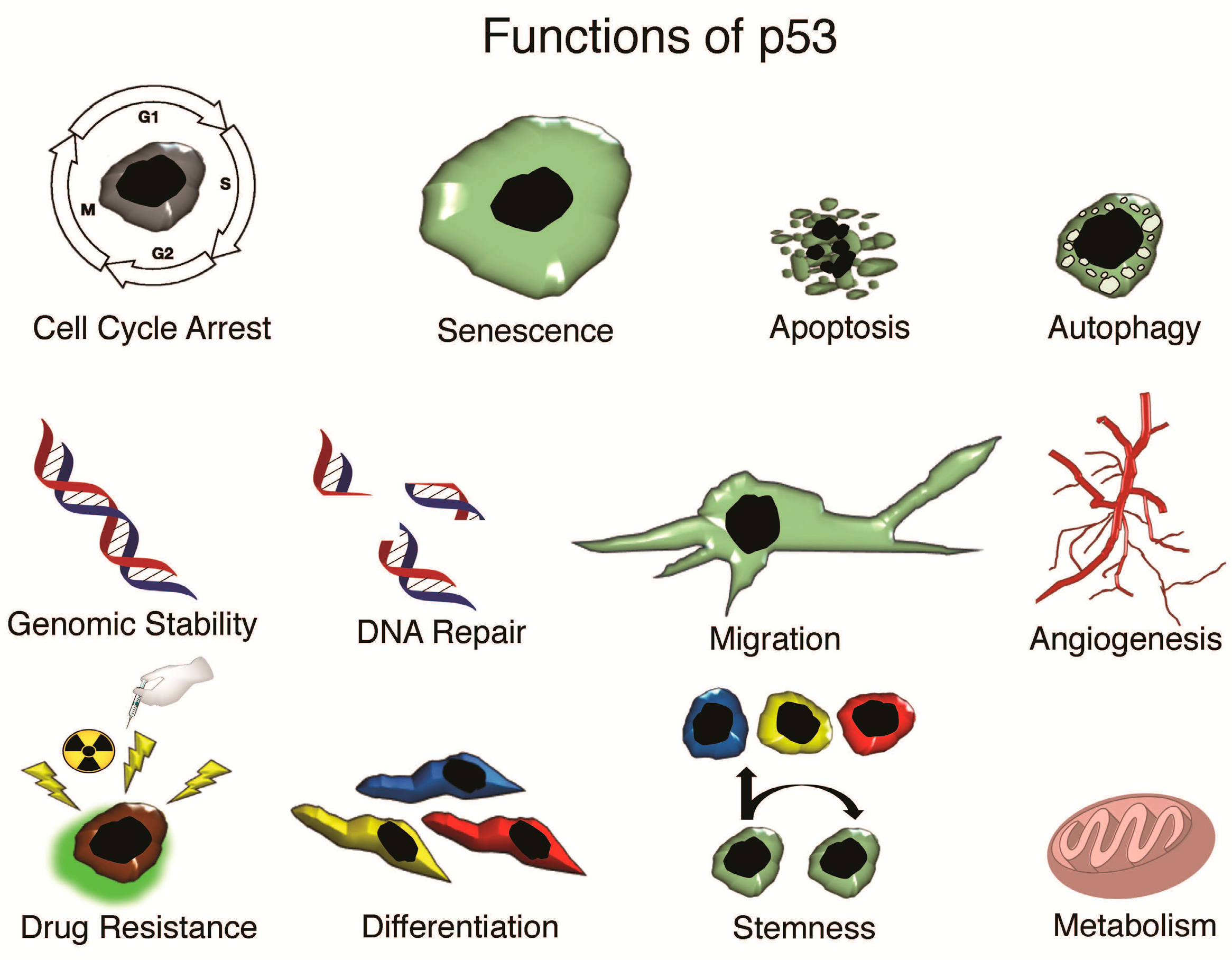{kind=link}
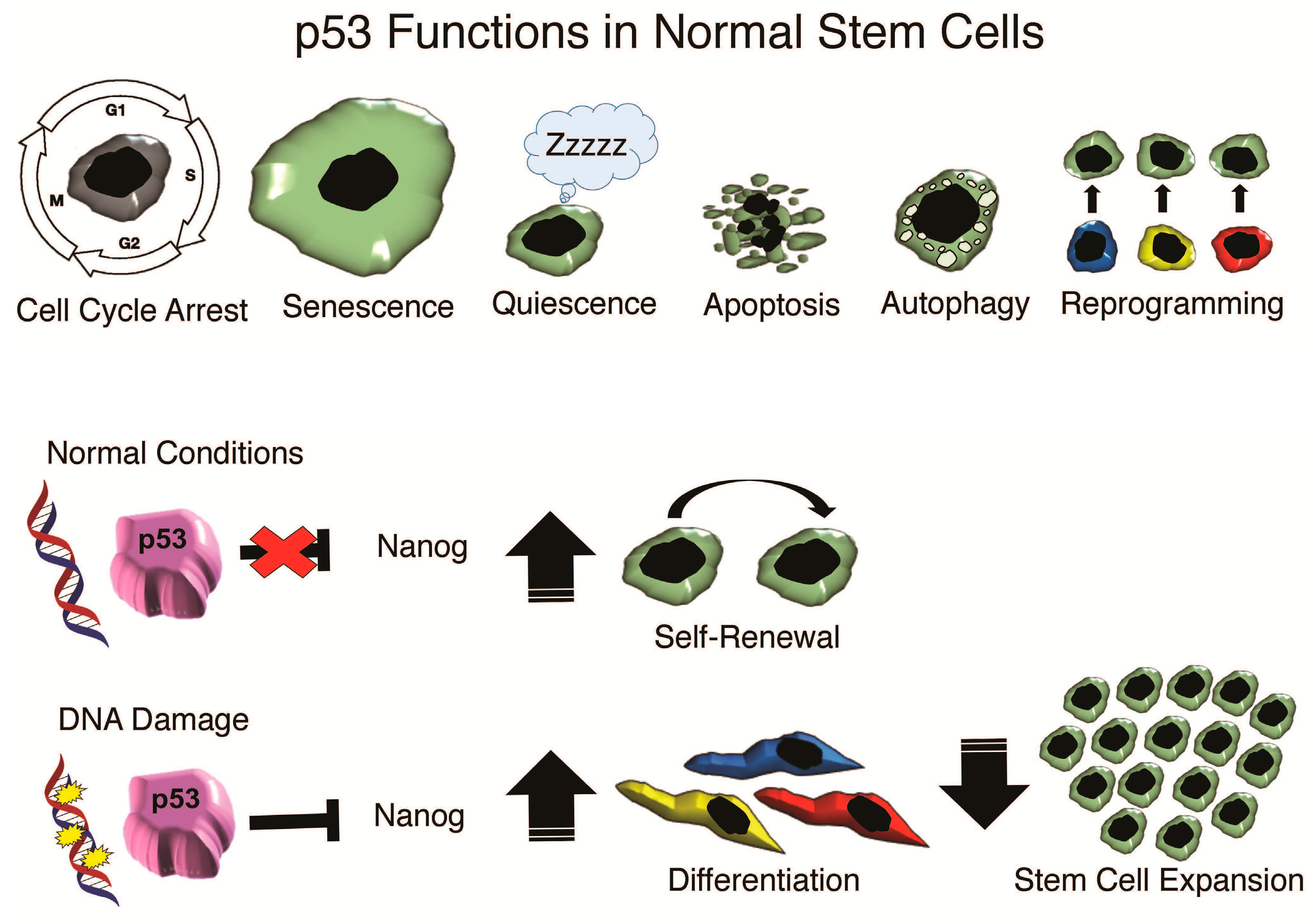{kind=link}
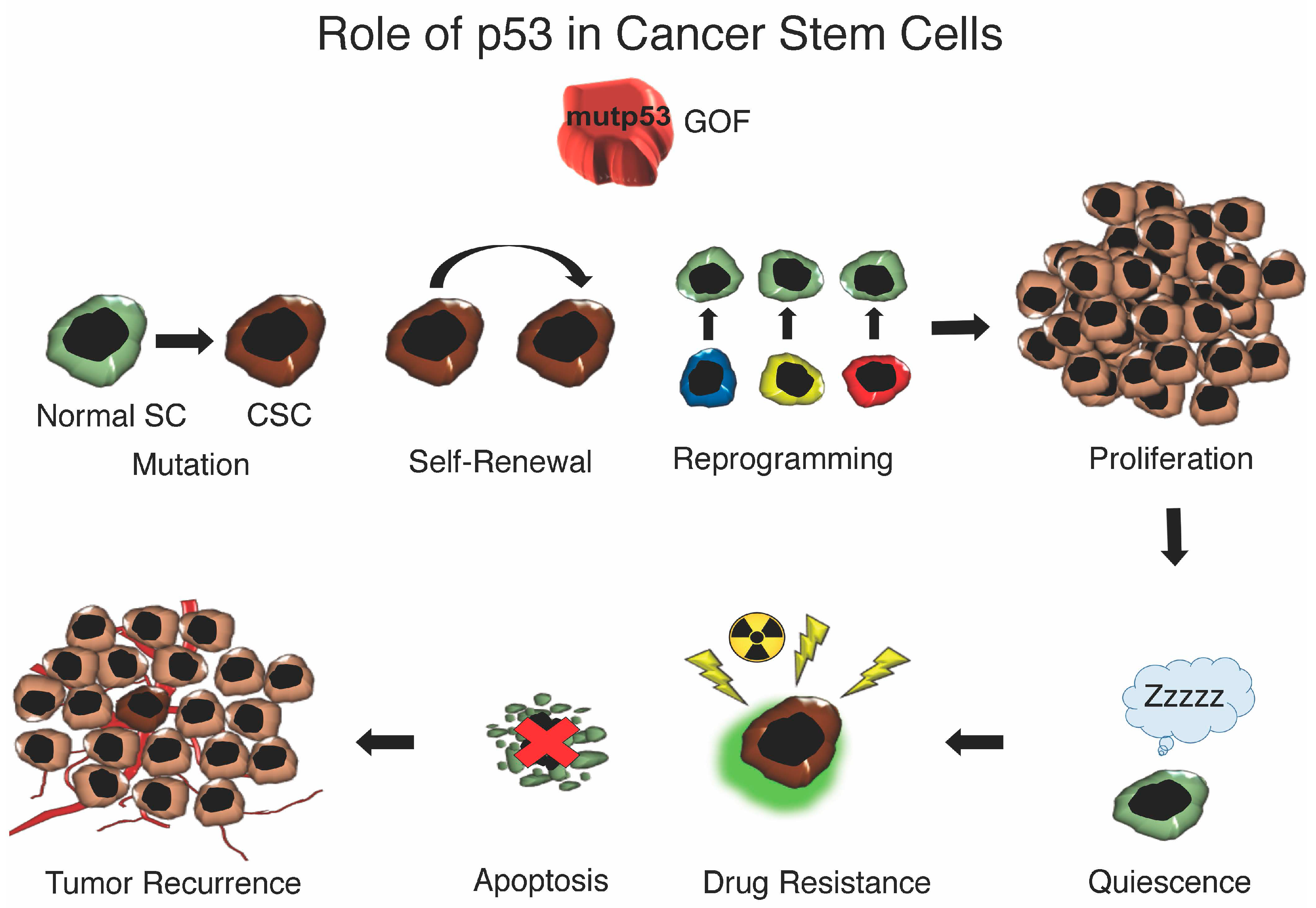{kind=link}
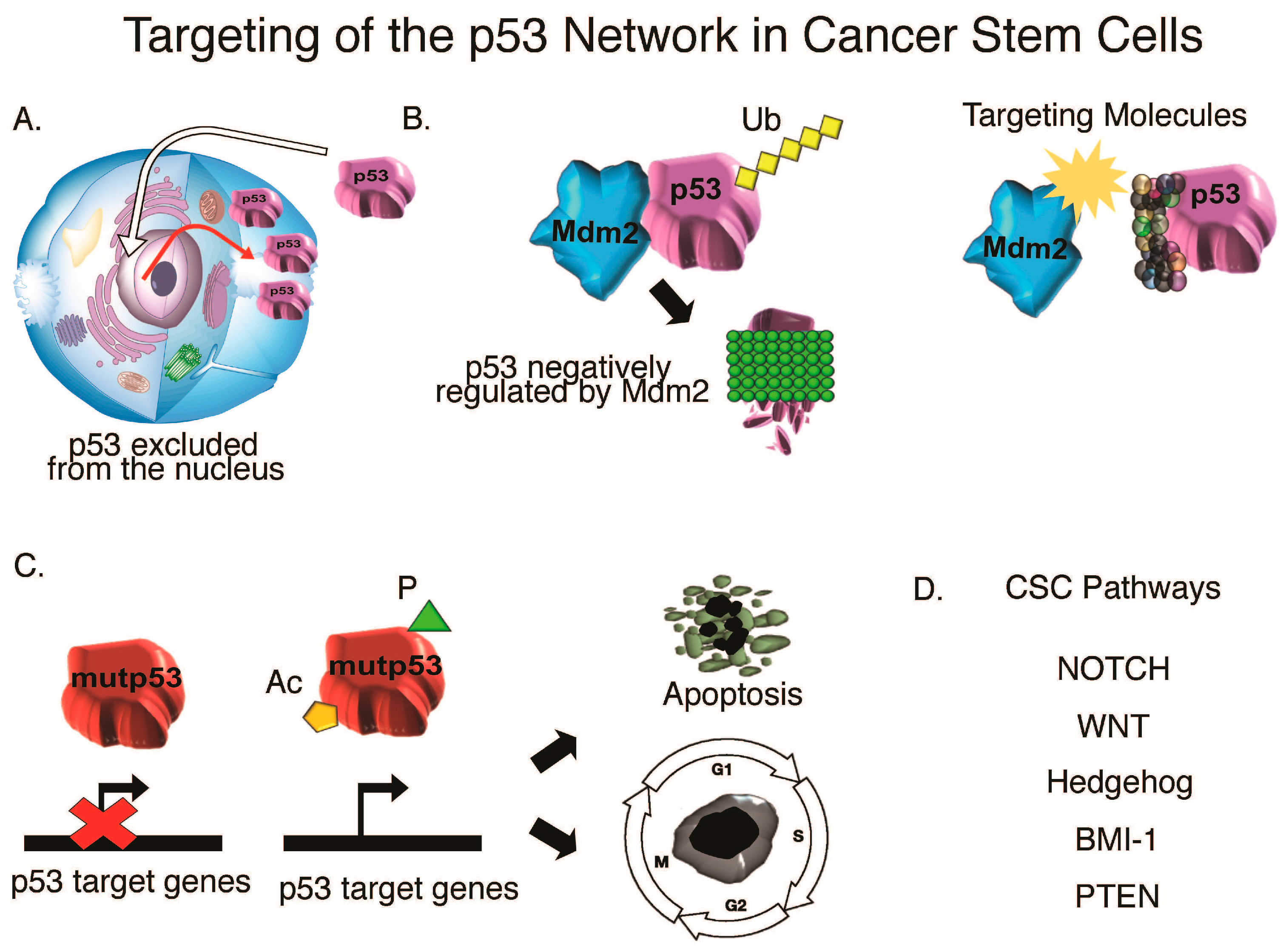{kind=link}
1. Introduction
1.1. The p53 Network and Its Role in Cancer
1.2. Stemness in Normal and Cancer Cells
1.3. Tumor Microenvironment and the CSC Niche
2. p53 and Stem Cells
3. p53 Gain of Function Roles in Stemness
3.1. Differentiation and Dedifferentiation
3.2. Epithelial-Mesenchymal Transition and Stemness
3.3. p53 and Stem Cell Senescence and Quiescence
4. Therapeutic Strategies to Restore wtp53 Function in CSCs
4.1. Small and Large Molecules
4.2. RING Finger E3 Ligases and CSCs Maintenance
5. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| 5-FU | 5-Fluorouracil |
| ABC | ATP-binding cassette |
| ADR | Adriamycin |
| AKT/mTOR | Protein kinase B or serine/threonine-specific protein kinase/mechanistic target of rapamycin |
| Ang-1 | Angiopoietin 1 |
| ATP | Adenosine triphosphate |
| Bcl-2 | B-cell lymphoma 2 |
| bHLH factor E47 | Basic helix-loop-helix factor E47 |
| BMP | Bone morphogenetic protein |
| BBB | Blood brain barrier |
| BRACA1 | Breast cancer gene 1 |
| CAF | Cancer-associated fibroblast |
| CBL | Casitas B cell lymphoma |
| CDKN1A | Cyclin-dependent kinase inhibitor 1A |
| COX | Cyclooxygenase2 |
| CSC | Cancer stem cell |
| CTD | C-terminal regulatory domain |
| DNA | Deoxyribonucleic acid |
| H2AK119 E3 ligase | Histone 2A at lysine 119 E3 ligase |
| hESC | Human embryonic stem cell |
| ECM | Extracellular matrix |
| ERK | Extracellular signal-regulated kinase |
| EMT | Epithelial-mesenchymal-transition |
| FoxO | Forkhead box O |
| FAK | Focal adhesion kinase |
| Gfi-1 | Growth factor independent 1 transcription repressor |
| GOF | Gain-of-function |
| GOX | Glucose oxidase |
| HIF1 | Hypoxia-inducible factor 1 |
| HIF2 | Hypoxia-inducible factor 2 |
| HFSC | Hair follicle stem cell |
| HMGA2 | High mobility group at-hook 2 |
| HSC | Hematopoietic stem cell |
| ICM | Inner cell mass |
| iPSC | Induced pluripotent stem cells |
| KLF4 | Krüppel-like factor 4 |
| LIN28A | Lin-28 homolog A |
| MAPK | Mitogen-activated protein kinase |
| mCSC | Metastatic cancer stem cells |
| Mdm2 | Mouse double minute 2 homolog |
| MEFs | Murine embryonic fibroblasts |
| MEF/Elf4 | Myeloid Elf-1-like factor/ETS-related transcription factor 4 |
| MEK | Mitogen-activated protein/extracellular signal-regulated kinase kinase |
| mESC | Mouse embryonic stem cell |
| MET | Mesenchymal-epithelial-transition |
| MGMT | O6-methylguanine DNA methyltransferase |
| MSC | Mammary stem cell |
| Mut p53 | Mutant p53 |
| Nedd4 | Neural precursor cell expressed developmentally down-regulated protein 4 |
| NOD/SCID | Non-obese diabetic/severe combined immunodeficiency |
| NOS2 | Nitric oxide synthase 2 |
| NOTCH | Notch homolog 1, translocation-associated |
| NSC | Neural stem cells |
| PCL1 | Polycomb like 1 |
| PHD | Plant homeodomain |
| PI3K/Akt | Phosphatidylinositol-3-kinase |
| PIGs | p53-induce genes |
| PRC2 | Polycomb repressor complex 2 |
| PTEN | Phosphatase and tensin homolog |
| pCSC | Primary cancer stem cell |
| rCSC | Recurrence cancer stem cell |
| RING | Really interesting new gene |
| Ring1B/RNF2 | Really interesting new gene 1B/ring finger protein 2 |
| RNAi | Ribonucleic acid interference |
| ROS | Reaction oxygen species |
| SCF | SKP1-CUL1-F- box protein |
| SLUG | SNAI2 or Snail family zinc finger 2 |
| SNAIL | SNAI1 or Snail family zinc finger 1 |
| SMUC | SNAI3 or Snail family zinc finger 3 |
| SMWC | Small molecular weight compounds |
| SPARC | Secreted protein acidic and rich cysteine |
| TAD | N-terminal transactivation domain |
| TGF-B | Transforming growth factor beta |
| TIC | Tumor-initiating cell |
| TMZ | Temozolomide |
| TPO | Thrombopoietin |
| TRIM24 | Tripartite motif-containing 24 |
| TWIST1 | Twist family BHLH transcription factor 1 |
| TWIST2 | Twist family BHLH transcription factor 2 |
| USP7 | Ubiquitin-specific-processing protease 7 |
| VEGF | Vascular endothelial growth factor |
| VHL | von Hippel–Lindau |
| Wt p53 | Wild-type p53 |
| ZEB1 | Zinc finger E-box binding homeobox 1 |
| ZEB2 | Zinc finger E-box binding homeobox 2 |
References
- DeLeo, A.B.; Jay, G.; Appella, E.; Dubois, G.C.; Law, L.W.; Old, L.J. Detection of a transformation-related antigen in chemically induced sarcomas and other transformed cells of the mouse. Proc. Natl. Acad. Sci. USA 1979, 76, 2420–2424. [Google Scholar] [CrossRef] [PubMed]
- Kress, M.; May, E.; Cassingena, R.; May, P. Simian virus 40-transformed cells express new species of proteins precipitable by anti-simian virus 40 tumor serum. J. Virol. 1979, 31, 472–483. [Google Scholar] [PubMed]
- Lane, D.P.; Crawford, L.V. T antigen is bound to a host protein in SV40-transformed cells. Nature 1979, 278, 261–263. [Google Scholar] [CrossRef] [PubMed]
- Linzer, D.I.; Levine, A.J. Characterization of a 54K Dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell 1979, 17, 43–52. [Google Scholar] [CrossRef]
- Melero, J.A.; Stitt, D.T.; Mangel, W.F.; Carroll, R.B. Identification of new polypeptide species (48–55k) immunoprecipitable by antiserum to purified large T antigen and present in SV40-infected and -transformed cells. Virology 1979, 93, 466–480. [Google Scholar] [CrossRef]
- Smith, A.E.; Smith, R.; Paucha, E. Characterization of different tumor antigens present in cells transformed by simian virus 40. Cell 1979, 18, 335–346. [Google Scholar] [CrossRef]
- Jenkins, J.R.; Rudge, K.; Currie, G.A. Cellular immortalization by a cDNA clone encoding the transformation-associated phosphoprotein p53. Nature 1984, 312, 651–654. [Google Scholar] [CrossRef] [PubMed]
- Parada, L.F.; Land, H.; Weinberg, R.A.; Wolf, D.; Rotter, V. Cooperation between gene encoding p53 tumour antigen and ras in cellular transformation. Nature 1984, 312, 649–651. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.J.; Fearon, E.R.; Nigro, J.M.; Hamilton, S.R.; Preisinger, A.C.; Jessup, J.M.; vanTuinen, P.; Ledbetter, D.H.; Barker, D.F.; Nakamura, Y.; et al. Chromosome 17 deletions and p53 gene mutations in colorectal carcinomas. Science 1989, 244, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Knudson, A.G., Jr. Mutation and cancer: Statistical study of retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef] [PubMed]
- Nigro, J.M.; Baker, S.J.; Preisinger, A.C.; Jessup, J.M.; Hostetter, R.; Cleary, K.; Bigner, S.H.; Davidson, N.; Baylin, S.; Devilee, P.; et al. Mutations in the p53 gene occur in diverse human tumour types. Nature 1989, 342, 705–708. [Google Scholar] [CrossRef] [PubMed]
- Malkin, D.; Li, F.P.; Strong, L.C.; Fraumeni, J.F., Jr.; Nelson, C.E.; Kim, D.H.; Kassel, J.; Gryka, M.A.; Bischoff, F.Z.; Tainsky, M.A.; et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 1990, 250, 1233–1238. [Google Scholar] [CrossRef] [PubMed]
- Donehower, L.A.; Harvey, M.; Slagle, B.L.; McArthur, M.J.; Montgomery, C.A., Jr.; Butel, J.S.; Bradley, A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992, 356, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Eliyahu, D.; Michalovitz, D.; Eliyahu, S.; Pinhasi-Kimhi, O.; Oren, M. Wild-type p53 can inhibit oncogene-mediated focus formation. Proc. Natl. Acad. Sci. USA 1989, 86, 8763–8767. [Google Scholar] [CrossRef] [PubMed]
- Finlay, C.A.; Hinds, P.W.; Levine, A.J. The p53 proto-oncogene can act as a suppressor of transformation. Cell 1989, 57, 1083–1093. [Google Scholar] [CrossRef]
- Yonish-Rouach, E.; Resnitzky, D.; Lotem, J.; Sachs, L.; Kimchi, A.; Oren, M. Wild-type p53 induces apoptosis of myeloid leukaemic cells that is inhibited by interleukin-6. Nature 1991, 352, 345–347. [Google Scholar] [CrossRef] [PubMed]
- Shaw, P.; Bovey, R.; Tardy, S.; Sahli, R.; Sordat, B.; Costa, J. Induction of apoptosis by wild-type p53 in a human colon tumor-derived cell line. Proc. Natl. Acad. Sci. USA 1992, 89, 4495–4499. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J. p53, the cellular gatekeeper for growth and division. Cell 1997, 88, 323–331. [Google Scholar] [CrossRef]
- Jin, S.; Levine, A.J. The p53 functional circuit. J. Cell Sci. 2001, 114, 4139–4140. [Google Scholar] [PubMed]
- Sarig, R.; Rivlin, N.; Brosh, R.; Bornstein, C.; Kamer, I.; Ezra, O.; Molchadsky, A.; Goldfinger, N.; Brenner, O.; Rotter, V. Mutant p53 facilitates somatic cell reprogramming and augments the malignant potential of reprogrammed cells. J. Exp. Med. 2010, 207, 2127–2140. [Google Scholar] [CrossRef] [PubMed]
- Khoury, M.P.; Bourdon, J.C. The isoforms of the p53 protein. Cold Spring Harb. Perspect. Biol. 2010, 2. [Google Scholar] [CrossRef] [PubMed]
- Marcel, V.; Perrier, S.; Aoubala, M.; Ageorges, S.; Groves, M.J.; Diot, A.; Fernandes, K.; Tauro, S.; Bourdon, J.C. ∆160p53 is a novel N-terminal p53 isoform encoded by ∆133p53 transcript. FEBS Lett. 2010, 584, 4463–4468. [Google Scholar] [CrossRef] [PubMed]
- Bourdon, J.C. p53 and its isoforms in cancer. Br. J. Cancer 2007, 97, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Arsic, N.; Gadea, G.; Lagerqvist, E.L.; Busson, M.; Cahuzac, N.; Brock, C.; Hollande, F.; Gire, V.; Pannequin, J.; Roux, P. The p53 isoform ∆133p53β promotes cancer stem cell potential. Stem Cell Rep. 2015, 4, 531–540. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Levine, A.J. The regulation of energy metabolism and the IGF-1/mTOR pathways by the p53 protein. Trends Cell Biol. 2010, 20, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Prives, C. Blinded by the light: The growing complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J.; Oren, M. The first 30 years of p53: Growing ever more complex. Nat. Rev. Cancer 2009, 9, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Batuello, C.N.; Hauck, P.M.; Gendron, J.M.; Lehman, J.A.; Mayo, L.D. Src phosphorylation converts Mdm2 from a ubiquitinating to a neddylating E3 ligase. Proc. Natl. Acad. Sci. USA 2015, 112, 1749–1754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, A.; Walker, N.; Bronson, R.; Kaghad, M.; Oosterwegel, M.; Bonnin, J.; Vagner, C.; Bonnet, H.; Dikkes, P.; Sharpe, A.; et al. p73-deficient mice have neurological, pheromonal and inflammatory defects but lack spontaneous tumours. Nature 2000, 404, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Moll, U.M.; Slade, N. p63 and p73: Roles in development and tumor formation. Mol. Cancer Res. 2004, 2, 371–386. [Google Scholar] [PubMed]
- Barak, Y.; Juven, T.; Haffner, R.; Oren, M. Mdm2 expression is induced by wild type p53 activity. EMBO J. 1993, 12, 461–468. [Google Scholar] [PubMed]
- Wu, X.; Bayle, J.H.; Olson, D.; Levine, A.J. The p53-Mdm-2 autoregulatory feedback loop. Genes Dev. 1993, 7, 1126–1132. [Google Scholar] [CrossRef] [PubMed]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Kubbutat, M.H.; Jones, S.N.; Vousden, K.H. Regulation of p53 stability by Mdm2. Nature 1997, 387, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Hofseth, L.J.; Hussain, S.P.; Harris, C.C. p53: 25 years after its discovery. Trends Pharmacol. Sci. 2004, 25, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Dornan, D.; Wertz, I.; Shimizu, H.; Arnott, D.; Frantz, G.D.; Dowd, P.; O’Rourke, K.; Koeppen, H.; Dixit, V.M. The ubiquitin ligase COP1 is a critical negative regulator of p53. Nature 2004, 429, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Leng, R.P.; Lin, Y.; Ma, W.; Wu, H.; Lemmers, B.; Chung, S.; Parant, J.M.; Lozano, G.; Hakem, R.; Benchimol, S. Pirh2, a p53-induced ubiquitin-protein ligase, promotes p53 degradation. Cell 2003, 112, 779–791. [Google Scholar] [CrossRef]
- Harris, S.L.; Levine, A.J. The p53 pathway: Positive and negative feedback loops. Oncogene 2005, 24, 2899–2908. [Google Scholar] [CrossRef] [PubMed]
- Ciliberto, A.; Novak, B.; Tyson, J.J. Steady states and oscillations in the p53/Mdm2 network. Cell Cycle 2005, 4, 488–493. [Google Scholar] [CrossRef] [PubMed]
- Mayo, L.D.; Dixon, J.E.; Durden, D.L.; Tonks, N.K.; Donner, D.B. PTEN protects p53 from Mdm2 and sensitizes cancer cells to chemotherapy. J. Biol. Chem. 2002, 277, 5484–5489. [Google Scholar] [CrossRef] [PubMed]
- Martoriati, A.; Doumont, G.; Alcalay, M.; Bellefroid, E.; Pelicci, P.G.; Marine, J.C. dapk1, encoding an activator of a p19ARF-p53-mediated apoptotic checkpoint, is a transcription target of p53. Oncogene 2005, 24, 1461–1466. [Google Scholar] [CrossRef] [PubMed]
- Deguin-Chambon, V.; Vacher, M.; Jullien, M.; May, E.; Bourdon, J.C. Direct transactivation of c-Ha-Ras gene by p53: Evidence for its involvement in p53 transactivation activity and p53-mediated apoptosis. Oncogene 2000, 19, 5831–5841. [Google Scholar] [CrossRef] [PubMed]
- Ongusaha, P.P.; Kim, J.I.; Fang, L.; Wong, T.W.; Yancopoulos, G.D.; Aaronson, S.A.; Lee, S.W. p53 induction and activation of DDR1 kinase counteract p53-mediated apoptosis and influence p53 regulation through a positive feedback loop. EMBO J. 2003, 22, 1289–1301. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuka, T.; Jensen, M.R.; Kim, H.G.; Kim, K.T.; Lee, S.W. The negative role of cyclin G in ATM-dependent p53 activation. Oncogene 2004, 23, 5405–5408. [Google Scholar] [CrossRef] [PubMed]
- Goodarzi, A.A.; Jonnalagadda, J.C.; Douglas, P.; Young, D.; Ye, R.; Moorhead, G.B.; Lees-Miller, S.P.; Khanna, K.K. Autophosphorylation of ataxia-telangiectasia mutated is regulated by protein phosphatase 2A. EMBO J. 2004, 23, 4451–4461. [Google Scholar] [CrossRef] [PubMed]
- Lang, G.A.; Iwakuma, T.; Suh, Y.A.; Liu, G.; Rao, V.A.; Parant, J.M.; Valentin-Vega, Y.A.; Terzian, T.; Caldwell, L.C.; Strong, L.C.; et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-fraumeni syndrome. Cell 2004, 119, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Olive, K.P.; Tuveson, D.A.; Ruhe, Z.C.; Yin, B.; Willis, N.A.; Bronson, R.T.; Crowley, D.; Jacks, T. Mutant p53 gain of function in two mouse models of li-fraumeni syndrome. Cell 2004, 119, 847–860. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.P.; Song, H.; Xu, Y. A common gain of function of p53 cancer mutants in inducing genetic instability. Oncogene 2010, 29, 949–956. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Liu, J.; Liang, Y.; Wu, R.; Zhao, Y.; Hong, X.; Lin, M.; Yu, H.; Liu, L.; Levine, A.J.; et al. Tumour-associated mutant p53 drives the warburg effect. Nat. Commun. 2013, 4, 2935. [Google Scholar] [CrossRef] [PubMed]
- Yeudall, W.A.; Vaughan, C.A.; Miyazaki, H.; Ramamoorthy, M.; Choi, M.Y.; Chapman, C.G.; Wang, H.; Black, E.; Bulysheva, A.A.; Deb, S.P.; et al. Gain-of-function mutant p53 upregulates CXC chemokines and enhances cell migration. Carcinogenesis 2012, 33, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 mutations in human cancers: Origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2010, 2, a001008. [Google Scholar] [CrossRef] [PubMed]
- Chin, K.V.; Ueda, K.; Pastan, I.; Gottesman, M.M. Modulation of activity of the promoter of the human MDR1 gene by Ras and p53. Science 1992, 255, 459–462. [Google Scholar] [CrossRef] [PubMed]
- Deb, S.; Jackson, C.T.; Subler, M.A.; Martin, D.W. Modulation of cellular and viral promoters by mutant human p53 proteins found in tumor cells. J. Virol. 1992, 66, 6164–6170. [Google Scholar] [PubMed]
- Ludes-Meyers, J.H.; Subler, M.A.; Shivakumar, C.V.; Munoz, R.M.; Jiang, P.; Bigger, J.E.; Brown, D.R.; Deb, S.P.; Deb, S. Transcriptional activation of the human epidermal growth factor receptor promoter by human p53. Mol. Cell. Biol. 1996, 16, 6009–6019. [Google Scholar] [CrossRef] [PubMed]
- Werner, H.; Karnieli, E.; Rauscher, F.J.; LeRoith, D. Wild-type and mutant p53 differentially regulate transcription of the insulin-like growth factor I receptor gene. Proc. Natl. Acad. Sci. USA 1996, 93, 8318–8323. [Google Scholar] [CrossRef] [PubMed]
- Frazier, M.W.; He, X.; Wang, J.; Gu, Z.; Cleveland, J.L.; Zambetti, G.P. Activation of c-myc gene expression by tumor-derived p53 mutants requires a discrete C-terminal domain. Mol. Cell. Biol. 1998, 18, 3735–3743. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.I.; Lee, S.; Das, G.C.; Park, U.S.; Park, S.M.; Lee, Y.I. Activation of the insulin-like growth factor II transcription by aflatoxin B1 induced p53 mutant 249 is caused by activation of transcription complexes; implications for a gain-of-function during the formation of hepatocellular carcinoma. Oncogene 2000, 19, 3717–3726. [Google Scholar] [CrossRef] [PubMed]
- Gurova, K.V.; Rokhlin, O.W.; Budanov, A.V.; Burdelya, L.G.; Chumakov, P.M.; Cohen, M.B.; Gudkov, A.V. Cooperation of two mutant p53 alleles contributes to Fas resistance of prostate carcinoma cells. Cancer Res. 2003, 63, 2905–2912. [Google Scholar] [PubMed]
- Buganim, Y.; Kalo, E.; Brosh, R.; Besserglick, H.; Nachmany, I.; Rais, Y.; Stambolsky, P.; Tang, X.; Milyavsky, M.; Shats, I.; et al. Mutant p53 protects cells from 12-O-tetradecanoylphorbol-13-acetate-induced death by attenuating activating transcription factor 3 induction. Cancer Res. 2006, 66, 10750–10759. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.P.; Tsang, W.P.; Chau, P.Y.; Co, N.N.; Tsang, T.Y.; Kwok, T.T. P53-R273H gains new function in induction of drug resistance through down-regulation of procaspase-3. Mol. Cancer Ther. 2007, 6, 1054–1061. [Google Scholar] [CrossRef] [PubMed]
- Vikhanskaya, F.; Lee, M.K.; Mazzoletti, M.; Broggini, M.; Sabapathy, K. Cancer-derived p53 mutants suppress p53-target gene expression—Potential mechanism for gain of function of mutant p53. Nucleic Acids Res. 2007, 35, 2093–2104. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Kaghad, M.; Wang, Y.; Gillett, E.; Fleming, M.D.; Dotsch, V.; Andrews, N.C.; Caput, D.; McKeon, F. p63, a p53 homolog at 3q27–29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol. Cell 1998, 2, 305–316. [Google Scholar] [CrossRef]
- De Laurenzi, V.; Costanzo, A.; Barcaroli, D.; Terrinoni, A.; Falco, M.; Annicchiarico-Petruzzelli, M.; Levrero, M.; Melino, G. Two new p73 splice variants, γ and δ, with different transcriptional activity. J. Exp. Med. 1998, 188, 1763–1768. [Google Scholar] [CrossRef] [PubMed]
- Kaghad, M.; Bonnet, H.; Yang, A.; Creancier, L.; Biscan, J.C.; Valent, A.; Minty, A.; Chalon, P.; Lelias, J.M.; Dumont, X.; et al. Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell 1997, 90, 809–819. [Google Scholar] [CrossRef]
- Melino, G.; De Laurenzi, V.; Vousden, K.H. p73: Friend or foe in tumorigenesis. Nat. Rev. Cancer 2002, 2, 605–615. [Google Scholar] [CrossRef] [PubMed]
- Ueda, Y.; Hijikata, M.; Takagi, S.; Chiba, T.; Shimotohno, K. New p73 variants with altered C-terminal structures have varied transcriptional activities. Oncogene 1999, 18, 4993–4998. [Google Scholar] [CrossRef] [PubMed]
- Boominathan, L. The tumor suppressors p53, p63, and p73 are regulators of microrna processing complex. PLoS ONE 2010, 5, e10615. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, E. Scratching the surface of skin development. Nature 2007, 445, 834–842. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Guan, K.; Nayernia, K.; Maier, L.S.; Wagner, S.; Dressel, R.; Lee, J.H.; Nolte, J.; Wolf, F.; Li, M.; Engel, W.; et al. Pluripotency of spermatogonial stem cells from adult mouse testis. Nature 2006, 440, 1199–1203. [Google Scholar] [CrossRef] [PubMed]
- Toma, J.G.; Akhavan, M.; Fernandes, K.J.; Barnabe-Heider, F.; Sadikot, A.; Kaplan, D.R.; Miller, F.D. Isolation of multipotent adult stem cells from the dermis of mammalian skin. Nat. Cell Biol. 2001, 3, 778–784. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, C.A.; Pollett, A.; Gallinger, S.; Dick, J.E. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 2007, 445, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Ricci-Vitiani, L.; Lombardi, D.G.; Pilozzi, E.; Biffoni, M.; Todaro, M.; Peschle, C.; de Maria, R. Identification and expansion of human colon-cancer-initiating cells. Nature 2007, 445, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Boiko, A.D.; Razorenova, O.V.; van de Rijn, M.; Swetter, S.M.; Johnson, D.L.; Ly, D.P.; Butler, P.D.; Yang, G.P.; Joshua, B.; Kaplan, M.J.; et al. Human melanoma-initiating cells express neural crest nerve growth factor receptor CD271. Nature 2010, 466, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Schatton, T.; Murphy, G.F.; Frank, N.Y.; Yamaura, K.; Waaga-Gasser, A.M.; Gasser, M.; Zhan, Q.; Jordan, S.; Duncan, L.M.; Weishaupt, C.; et al. Identification of cells initiating human melanomas. Nature 2008, 451, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Kelly, P.N.; Dakic, A.; Adams, J.M.; Nutt, S.L.; Strasser, A. Tumor growth need not be driven by rare cancer stem cells. Science 2007, 317, 337. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.T.; den Besten, W.; Sherr, C.J. Cytokine-dependent imatinib resistance in mouse BCR-ABL+, Arf-null lymphoblastic leukemia. Genes Dev. 2007, 21, 2283–2287. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.B.; Chaffer, C.L.; Weinberg, R.A. Cancer stem cells: Mirage or reality? Nat. Med. 2009, 15, 1010–1012. [Google Scholar] [CrossRef] [PubMed]
- Harris, M.A.; Yang, H.; Low, B.E.; Mukherjee, J.; Guha, A.; Bronson, R.T.; Shultz, L.D.; Israel, M.A.; Yun, K. Cancer stem cells are enriched in the side population cells in a mouse model of glioma. Cancer Res. 2008, 68, 10051–10059. [Google Scholar] [CrossRef] [PubMed]
- Thon, N.; Damianoff, K.; Hegermann, J.; Grau, S.; Krebs, B.; Schnell, O.; Tonn, J.C.; Goldbrunner, R. Presence of pluripotent CD133+ cells correlates with malignancy of gliomas. Mol. Cell. Neurosci. 2010, 43, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Quintana, E.; Shackleton, M.; Sabel, M.S.; Fullen, D.R.; Johnson, T.M.; Morrison, S.J. Efficient tumour formation by single human melanoma cells. Nature 2008, 456, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Dalerba, P.; Cho, R.W.; Clarke, M.F. Cancer stem cells: Models and concepts. Annu. Rev. Med. 2007, 58, 267–284. [Google Scholar] [CrossRef] [PubMed]
- Bjerkvig, R.; Tysnes, B.B.; Aboody, K.S.; Najbauer, J.; Terzis, A.J. Opinion: The origin of the cancer stem cell: Current controversies and new insights. Nat. Rev. Cancer 2005, 5, 899–904. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Tiede, B.; Massague, J.; Kang, Y. Beyond tumorigenesis: Cancer stem cells in metastasis. Cell Res. 2007, 17, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.F.; Jackson, E.L.; Woolfenden, A.E.; Lawrence, S.; Babar, I.; Vogel, S.; Crowley, D.; Bronson, R.T.; Jacks, T. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell 2005, 121, 823–835. [Google Scholar] [CrossRef] [PubMed]
- Houghton, J.; Stoicov, C.; Nomura, S.; Rogers, A.B.; Carlson, J.; Li, H.; Cai, X.; Fox, J.G.; Goldenring, J.R.; Wang, T.C. Gastric cancer originating from bone marrow-derived cells. Science 2004, 306, 1568–1571. [Google Scholar] [CrossRef] [PubMed]
- Nickoloff, B.J.; Foreman, K.E. Etiology and pathogenesis of kaposi’s sarcoma. Recent Results Cancer Res. 2002, 160, 332–342. [Google Scholar] [PubMed]
- Duelli, D.; Lazebnik, Y. Cell fusion: A hidden enemy? Cancer Cell 2003, 3, 445–448. [Google Scholar] [CrossRef]
- Dittmar, T.; Nagler, C.; Schwitalla, S.; Reith, G.; Niggemann, B.; Zanker, K.S. Recurrence cancer stem cells—Made by cell fusion? Med. Hypotheses 2009, 73, 542–547. [Google Scholar] [CrossRef] [PubMed]
- Bastida-Ruiz, D.; van Hoesen, K.; Cohen, M. The dark side of cell fusion. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Dittmar, T.; Nagler, C.; Niggemann, B.; Zanker, K.S. The dark side of stem cells: Triggering cancer progression by cell fusion. Curr. Mol. Med. 2013, 13, 735–750. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Kang, Y. Cell fusion as a hidden force in tumor progression. Cancer Res. 2009, 69, 8536–8539. [Google Scholar] [CrossRef] [PubMed]
- Morrison, S.J.; Kimble, J. Asymmetric and symmetric stem-cell divisions in development and cancer. Nature 2006, 441, 1068–1074. [Google Scholar] [CrossRef] [PubMed]
- Leong, K.G.; Karsan, A. Recent insights into the role of Notch signaling in tumorigenesis. Blood 2006, 107, 2223–2233. [Google Scholar] [CrossRef] [PubMed]
- Taipale, J.; Beachy, P.A. The Hedgehog and Wnt signalling pathways in cancer. Nature 2001, 411, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Shipitsin, M.; Campbell, L.L.; Argani, P.; Weremowicz, S.; Bloushtain-Qimron, N.; Yao, J.; Nikolskaya, T.; Serebryiskaya, T.; Beroukhim, R.; Hu, M.; et al. Molecular definition of breast tumor heterogeneity. Cancer Cell 2007, 11, 259–273. [Google Scholar] [CrossRef] [PubMed]
- Fialkow, P.J. Clonal origin of human tumors. Biochim. Biophys. Acta 1976, 458, 283–321. [Google Scholar] [CrossRef]
- Fidler, I.J.; Hart, I.R. Biological diversity in metastatic neoplasms: Origins and implications. Science 1982, 217, 998–1003. [Google Scholar] [CrossRef] [PubMed]
- Hamburger, A.W.; Salmon, S.E. Primary bioassay of human tumor stem cells. Science 1977, 197, 461–463. [Google Scholar] [CrossRef] [PubMed]
- Park, E.K.; Lee, J.C.; Park, J.W.; Bang, S.Y.; Yi, S.A.; Kim, B.K.; Park, J.H.; Kwon, S.H.; You, J.S.; Nam, S.W.; et al. Transcriptional repression of cancer stem cell marker CD133 by tumor suppressor p53. Cell Death Dis. 2015, 6, e1964. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Laterra, J. Cancer stem cells: Distinct entities or dynamically regulated phenotypes? Cancer Res. 2012, 72, 576–580. [Google Scholar] [CrossRef] [PubMed]
- Mitsui, K.; Tokuzawa, Y.; Itoh, H.; Segawa, K.; Murakami, M.; Takahashi, K.; Maruyama, M.; Maeda, M.; Yamanaka, S. The homeoprotein Nanog is required for maintenance of pluripotency in mouse epiblast and ES cells. Cell 2003, 113, 631–642. [Google Scholar] [CrossRef]
- Lin, T.; Chao, C.; Saito, S.; Mazur, S.J.; Murphy, M.E.; Appella, E.; Xu, Y. p53 induces differentiation of mouse embryonic stem cells by suppressing Nanog expression. Nat. Cell Biol. 2005, 7, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Ho, B.; Olson, G.; Figel, S.; Gelman, I.; Cance, W.G.; Golubovskaya, V.M. Nanog increases focal adhesion kinase (FAK) promoter activity and expression and directly binds to FAK protein to be phosphorylated. J. Biol. Chem. 2012, 287, 18656–18673. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.T.; Chen, X.L.; Lim, Y.; Hanson, D.A.; Vo, T.T.; Howerton, K.; Larocque, N.; Fisher, S.J.; Schlaepfer, D.D.; Ilic, D. Nuclear FAK promotes cell proliferation and survival through ferm-enhanced p53 degradation. Mol. Cell 2008, 29, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.; Fojo, T.; Bates, S. Tumour stem cells and drug resistance. Nat. Rev. Cancer 2005, 5, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Frank, N.Y.; Margaryan, A.; Huang, Y.; Schatton, T.; Waaga-Gasser, A.M.; Gasser, M.; Sayegh, M.H.; Sadee, W.; Frank, M.H. ABCB5-mediated doxorubicin transport and chemoresistance in human malignant melanoma. Cancer Res 2005, 65, 4320–4333. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP-dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Wulf, G.G.; Wang, R.Y.; Kuehnle, I.; Weidner, D.; Marini, F.; Brenner, M.K.; Andreeff, M.; Goodell, M.A. A leukemic stem cell with intrinsic drug efflux capacity in acute myeloid leukemia. Blood 2001, 98, 1166–1173. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orkin, S.H.; Zon, L.I. Hematopoiesis: An evolving paradigm for stem cell biology. Cell 2008, 132, 631–644. [Google Scholar] [CrossRef] [PubMed]
- Lander, A.D.; Kimble, J.; Clevers, H.; Fuchs, E.; Montarras, D.; Buckingham, M.; Calof, A.L.; Trumpp, A.; Oskarsson, T. What does the concept of the stem cell niche really mean today? BMC Biol. 2012, 10, 19. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, H.; Suda, T. Cancer stem cells and their niche. Cancer Sci. 2009, 100, 1166–1172. [Google Scholar] [CrossRef] [PubMed]
- Pietras, K.; Ostman, A. Hallmarks of cancer: Interactions with the tumor stroma. Exp. Cell Res. 2010, 316, 1324–1331. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.V.; Vanner, R.; Dirks, P.; Eaves, C.J. Cancer stem cells: An evolving concept. Nat. Rev. Cancer 2012, 12, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Schofield, R. The relationship between the spleen colony-forming cell and the haemopoietic stem cell. Blood Cells 1978, 4, 7–25. [Google Scholar] [PubMed]
- Xie, T.; Spradling, A.C. A niche maintaining germ line stem cells in the drosophila ovary. Science 2000, 290, 328–330. [Google Scholar] [CrossRef] [PubMed]
- Calvi, L.M.; Adams, G.B.; Weibrecht, K.W.; Weber, J.M.; Olson, D.P.; Knight, M.C.; Martin, R.P.; Schipani, E.; Divieti, P.; Bringhurst, F.R.; et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature 2003, 425, 841–846. [Google Scholar] [CrossRef] [PubMed]
- Carlesso, N.; Cardoso, A.A. Stem cell regulatory niches and their role in normal and malignant hematopoiesis. Curr. Opin. Hematol. 2010, 17, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Morrison, S.J.; Scadden, D.T. The bone marrow niche for haematopoietic stem cells. Nature 2014, 505, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Kunisaki, Y.; Bruns, I.; Scheiermann, C.; Ahmed, J.; Pinho, S.; Zhang, D.; Mizoguchi, T.; Wei, Q.; Lucas, D.; Ito, K.; et al. Arteriolar niches maintain haematopoietic stem cell quiescence. Nature 2013, 502, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Scadden, D.T. The stem-cell niche as an entity of action. Nature 2006, 441, 1075–1079. [Google Scholar] [CrossRef] [PubMed]
- Heddleston, J.M.; Li, Z.; Lathia, J.D.; Bao, S.; Hjelmeland, A.B.; Rich, J.N. Hypoxia inducible factors in cancer stem cells. Br. J. Cancer 2010, 102, 789–795. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.T.; Herlyn, M. Microenvironmental influences in melanoma progression. J. Cell. Biochem. 2007, 101, 862–872. [Google Scholar] [CrossRef] [PubMed]
- Hendrix, M.J.; Seftor, E.A.; Seftor, R.E.; Kasemeier-Kulesa, J.; Kulesa, P.M.; Postovit, L.M. Reprogramming metastatic tumour cells with embryonic microenvironments. Nat. Rev. Cancer 2007, 7, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Gerschenson, M.; Graves, K.; Carson, S.D.; Wells, R.S.; Pierce, G.B. Regulation of melanoma by the embryonic skin. Proc. Natl. Acad. Sci. USA 1986, 83, 7307–7310. [Google Scholar] [CrossRef] [PubMed]
- Kulesa, P.M.; Kasemeier-Kulesa, J.C.; Teddy, J.M.; Margaryan, N.V.; Seftor, E.A.; Seftor, R.E.; Hendrix, M.J. Reprogramming metastatic melanoma cells to assume a neural crest cell-like phenotype in an embryonic microenvironment. Proc. Natl. Acad. Sci. USA 2006, 103, 3752–3757. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.M.; Seftor, E.A.; Bonde, G.; Cornell, R.A.; Hendrix, M.J. The fate of human malignant melanoma cells transplanted into zebrafish embryos: Assessment of migration and cell division in the absence of tumor formation. Dev. Dyn. 2005, 233, 1560–1570. [Google Scholar] [CrossRef] [PubMed]
- Haass, N.K.; Smalley, K.S.; Li, L.; Herlyn, M. Adhesion, migration and communication in melanocytes and melanoma. Pigment Cell Res. 2005, 18, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Hsu, M.Y.; Meier, F.; Herlyn, M. Melanoma development and progression: A conspiracy between tumor and host. Differentiation 2002, 70, 522–536. [Google Scholar] [CrossRef] [PubMed]
- Ubertini, V.; Norelli, G.; D’Arcangelo, D.; Gurtner, A.; Cesareo, E.; Baldari, S.; Gentileschi, M.P.; Piaggio, G.; Nistico, P.; Soddu, S.; et al. Mutant p53 gains new function in promoting inflammatory signals by repression of the secreted interleukin-1 receptor antagonist. Oncogene 2015, 34, 2493–2504. [Google Scholar] [CrossRef] [PubMed]
- Addadi, Y.; Moskovits, N.; Granot, D.; Lozano, G.; Carmi, Y.; Apte, R.N.; Neeman, M.; Oren, M. p53 status in stromal fibroblasts modulates tumor growth in an SDF1-dependent manner. Cancer Res. 2010, 70, 9650–9658. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, T.; Suzuki, J.; Wang, Y.V.; Menendez, S.; Morera, L.B.; Raya, A.; Wahl, G.M.; Izpisua Belmonte, J.C. Linking the p53 tumour suppressor pathway to somatic cell reprogramming. Nature 2009, 460, 1140–1144. [Google Scholar] [CrossRef] [PubMed]
- Marion, R.M.; Strati, K.; Li, H.; Murga, M.; Blanco, R.; Ortega, S.; Fernandez-Capetillo, O.; Serrano, M.; Blasco, M.A. A p53-mediated DNA damage response limits reprogramming to ensure iPS cell genomic integrity. Nature 2009, 460, 1149–1153. [Google Scholar] [CrossRef] [PubMed]
- Yi, L.; Lu, C.; Hu, W.; Sun, Y.; Levine, A.J. Multiple roles of p53-related pathways in somatic cell reprogramming and stem cell differentiation. Cancer Res. 2012, 72, 5635–5645. [Google Scholar] [CrossRef] [PubMed]
- Hanel, W.; Marchenko, N.; Xu, S.; Yu, S.X.; Weng, W.; Moll, U. Two hot spot mutant p53 mouse models display differential gain of function in tumorigenesis. Cell Death Differ. 2013, 20, 898–909. [Google Scholar] [CrossRef] [PubMed]
- Topczewska, J.M.; Postovit, L.M.; Margaryan, N.V.; Sam, A.; Hess, A.R.; Wheaton, W.W.; Nickoloff, B.J.; Topczewski, J.; Hendrix, M.J. Embryonic and tumorigenic pathways converge via nodal signaling: Role in melanoma aggressiveness. Nat. Med. 2006, 12, 925–932. [Google Scholar] [CrossRef] [PubMed]
- Golubovskaya, V.M. FAK and Nanog cross talk with p53 in cancer stem cells. Anticancer Agents Med. Chem. 2013, 13, 576–580. [Google Scholar] [CrossRef] [PubMed]
- Krizhanovsky, V.; Lowe, S.W. Stem cells: The promises and perils of p53. Nature 2009, 460, 1085–1086. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.H.; Kwon, S.; Jun, E.K.; Kim, A.; Whang, K.Y.; Kim, H.; Oh, S.; Yoon, B.S.; You, S. Nanog-induced dedifferentiation of p53-deficient mouse astrocytes into brain cancer stem-like cells. Biochem. Biophys. Res. Commun. 2011, 412, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Molofsky, A.V.; Pardal, R.; Morrison, S.J. Diverse mechanisms regulate stem cell self-renewal. Curr. Opin. Cell Biol. 2004, 16, 700–707. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.; Takahashi, K.; Ichisaka, T.; Aoi, T.; Kanagawa, O.; Nakagawa, M.; Okita, K.; Yamanaka, S. Suppression of induced pluripotent stem cell generation by the p53-p21 pathway. Nature 2009, 460, 1132–1135. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Dong, Q.Z.; Zhao, Y.; Dong, X.J.; Miao, Y.; Dai, S.D.; Yang, Z.Q.; Zhang, D.; Wang, Y.; Li, Q.C.; et al. p120-catenin isoforms 1A and 3A differently affect invasion and proliferation of lung cancer cells. Exp. Cell Res. 2009, 315, 890–898. [Google Scholar] [CrossRef] [PubMed]
- Utikal, J.; Polo, J.M.; Stadtfeld, M.; Maherali, N.; Kulalert, W.; Walsh, R.M.; Khalil, A.; Rheinwald, J.G.; Hochedlinger, K. Immortalization eliminates a roadblock during cellular reprogramming into iPS cells. Nature 2009, 460, 1145–1148. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yin, X.; Qin, H.; Zhu, F.; Liu, H.; Yang, W.; Zhang, Q.; Xiang, C.; Hou, P.; Song, Z.; et al. Two supporting factors greatly improve the efficiency of human iPSC generation. Cell Stem Cell 2008, 3, 475–479. [Google Scholar] [CrossRef] [PubMed]
- Banito, A.; Rashid, S.T.; Acosta, J.C.; Li, S.; Pereira, C.F.; Geti, I.; Pinho, S.; Silva, J.C.; Azuara, V.; Walsh, M.; et al. Senescence impairs successful reprogramming to pluripotent stem cells. Genes Dev. 2009, 23, 2134–2139. [Google Scholar] [CrossRef] [PubMed]
- Vilborg, A.; Bersani, C.; Wilhelm, M.T.; Wiman, K.G. The p53 target Wig-1: A regulator of mRNA stability and stem cell fate? Cell Death Differ. 2011, 18, 1434–1440. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.J.; Chao, C.H.; Xia, W.; Yang, J.Y.; Xiong, Y.; Li, C.W.; Yu, W.H.; Rehman, S.K.; Hsu, J.L.; Lee, H.H.; et al. p53 regulates epithelial-mesenchymal transition and stem cell properties through modulating miRNAs. Nat. Cell Biol. 2011, 13, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Schubert, J.; Brabletz, T. p53 spreads out further: Suppression of EMT and stemness by activating miR-200c expression. Cell Res. 2011, 21, 705–707. [Google Scholar] [CrossRef] [PubMed]
- Burk, U.; Schubert, J.; Wellner, U.; Schmalhofer, O.; Vincan, E.; Spaderna, S.; Brabletz, T. A reciprocal repression between ZEB1 and members of the miR-200 family promotes emt and invasion in cancer cells. EMBO Rep. 2008, 9, 582–589. [Google Scholar] [CrossRef] [PubMed]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting zeb1 and SIP1. Nat. Cell Biol. 2008, 10, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Korpal, M.; Lee, E.S.; Hu, G.; Kang, Y. The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J. Biol. Chem. 2008, 283, 14910–14914. [Google Scholar] [CrossRef] [PubMed]
- Shimono, Y.; Zabala, M.; Cho, R.W.; Lobo, N.; Dalerba, P.; Qian, D.; Diehn, M.; Liu, H.; Panula, S.P.; Chiao, E.; et al. Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell 2009, 138, 592–603. [Google Scholar] [CrossRef] [PubMed]
- Dong, P.; Karaayvaz, M.; Jia, N.; Kaneuchi, M.; Hamada, J.; Watari, H.; Sudo, S.; Ju, J.; Sakuragi, N. Mutant p53 gain-of-function induces epithelial-mesenchymal transition through modulation of the miR-130b-ZEB1 axis. Oncogene 2013, 32, 3286–3295. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Gong, H.; Pan, X.; Chang, C.; Ou, Z.; Ye, S.; Yin, L.; Yang, L.; Tao, T.; Zhang, Z.; et al. p53 isoform ∆113p53/∆133p53 promotes DNA double-strand break repair to protect cell from death and senescence in response to DNA damage. Cell Res. 2015, 25, 351–369. [Google Scholar] [CrossRef] [PubMed]
- Ungewitter, E.; Scrable, H. ∆40p53 controls the switch from pluripotency to differentiation by regulating IGF signaling in ESCs. Genes Dev. 2010, 24, 2408–2419. [Google Scholar] [CrossRef] [PubMed]
- Loh, Y.H.; Wu, Q.; Chew, J.L.; Vega, V.B.; Zhang, W.; Chen, X.; Bourque, G.; George, J.; Leong, B.; Liu, J.; et al. The Oct4 and nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat. Genet. 2006, 38, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.N.; Chung, S.K.; Xu, Z.; Xu, Y. Oct4 maintains the pluripotency of human embryonic stem cells by inactivating p53 through Sirt1-mediated deacetylation. Stem Cells 2014, 32, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Chung, S.K.; Xu, Y. Modeling disease in human ESCs using an efficient BAC-based homologous recombination system. Cell Stem Cell 2010, 6, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Solozobova, V.; Blattner, C. Regulation of p53 in embryonic stem cells. Exp. Cell Res. 2010, 316, 2434–2446. [Google Scholar] [CrossRef] [PubMed]
- Maimets, T.; Neganova, I.; Armstrong, L.; Lako, M. Activation of p53 by nutlin leads to rapid differentiation of human embryonic stem cells. Oncogene 2008, 27, 5277–5287. [Google Scholar] [CrossRef] [PubMed]
- Adorno, M.; Cordenonsi, M.; Montagner, M.; Dupont, S.; Wong, C.; Hann, B.; Solari, A.; Bobisse, S.; Rondina, M.B.; Guzzardo, V.; et al. A mutant-p53/Smad complex opposes p63 to empower TGFβ-induced metastasis. Cell 2009, 137, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.; Caswell, P.T.; Doyle, B.; Iwanicki, M.P.; Tan, E.H.; Karim, S.; Lukashchuk, N.; Gillespie, D.A.; Ludwig, R.L.; Gosselin, P.; et al. Mutant p53 drives invasion by promoting integrin recycling. Cell 2009, 139, 1327–1341. [Google Scholar] [CrossRef] [PubMed]
- Bossi, G.; Lapi, E.; Strano, S.; Rinaldo, C.; Blandino, G.; Sacchi, A. Mutant p53 gain of function: Reduction of tumor malignancy of human cancer cell lines through abrogation of mutant p53 expression. Oncogene 2006, 25, 304–309. [Google Scholar] [CrossRef] [PubMed]
- Dittmer, D.; Pati, S.; Zambetti, G.; Chu, S.; Teresky, A.K.; Moore, M.; Finlay, C.; Levine, A.J. Gain of function mutations in p53. Nat. Genet. 1993, 4, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Sigal, A.; Rotter, V. Oncogenic mutations of the p53 tumor suppressor: The demons of the guardian of the genome. Cancer Res. 2000, 60, 6788–6793. [Google Scholar] [PubMed]
- Scian, M.J.; Stagliano, K.E.; Anderson, M.A.; Hassan, S.; Bowman, M.; Miles, M.F.; Deb, S.P.; Deb, S. Tumor-derived p53 mutants induce NF-κB2 gene expression. Mol. Cell. Biol. 2005, 25, 10097–10110. [Google Scholar] [CrossRef] [PubMed]
- Blandino, G.; Levine, A.J.; Oren, M. Mutant p53 gain of function: Differential effects of different p53 mutants on resistance of cultured cells to chemotherapy. Oncogene 1999, 18, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Hollstein, M.; Xu, Y. p53 gain-of-function cancer mutants induce genetic instability by inactivating atm. Nat. Cell Biol. 2007, 9, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Spike, B.T.; Wahl, G.M. p53, stem cells, and reprogramming: Tumor suppression beyond guarding the genome. Genes Cancer 2011, 2, 404–419. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, H.; Spike, B.T.; Wahl, G.M.; Levine, A.J. Inactivation of p53 in breast cancers correlates with stem cell transcriptional signatures. Proc. Natl. Acad. Sci. USA 2010, 107, 22745–22750. [Google Scholar] [CrossRef] [PubMed]
- Abbas, H.A.; Maccio, D.R.; Coskun, S.; Jackson, J.G.; Hazen, A.L.; Sills, T.M.; You, M.J.; Hirschi, K.K.; Lozano, G. Mdm2 is required for survival of hematopoietic stem cells/progenitors via dampening of ROS-induced p53 activity. Cell Stem Cell 2010, 7, 606–617. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.A.; Mullany, L.K.; Liu, Z.; Herron, A.J.; Wong, K.K.; Richards, J.S. Mutant p53 promotes epithelial ovarian cancer by regulating tumor differentiation, metastasis, and responsiveness to steroid hormones. Cancer Res. 2016, 76, 2206–2218. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Liu, D.P.; Xu, Y. The gain of function of p53 cancer mutant in promoting mammary tumorigenesis. Oncogene 2013, 32, 2900–2906. [Google Scholar] [CrossRef] [PubMed]
- Das, B.; Bayat-Mokhtari, R.; Tsui, M.; Lotfi, S.; Tsuchida, R.; Felsher, D.W.; Yeger, H. HIF-2α suppresses p53 to enhance the stemness and regenerative potential of human embryonic stem cells. Stem Cells 2012, 30, 1685–1695. [Google Scholar] [CrossRef] [PubMed]
- Itahana, Y.; Zhang, J.; Goke, J.; Vardy, L.A.; Han, R.; Iwamoto, K.; Cukuroglu, E.; Robson, P.; Pouladi, M.A.; Colman, A.; et al. Histone modifications and p53 binding poise the p21 promoter for activation in human embryonic stem cells. Sci. Rep. 2016, 6, 28112. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Arnoux, V.; Nassour, M.; L'Helgoualc'h, A.; Hipskind, R.A.; Savagner, P. Erk5 controls slug expression and keratinocyte activation during wound healing. Mol. Biol. Cell 2008, 19, 4738–4749. [Google Scholar] [CrossRef] [PubMed]
- Iwano, M.; Plieth, D.; Danoff, T.M.; Xue, C.; Okada, H.; Neilson, E.G. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J. Clin. Investig. 2002, 110, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A. Epithelial plasticity: A common theme in embryonic and cancer cells. Science 2013, 342, 1234850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tam, W.L.; Weinberg, R.A. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat. Med. 2013, 19, 1438–1449. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Morel, A.P.; Lievre, M.; Thomas, C.; Hinkal, G.; Ansieau, S.; Puisieux, A. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS ONE 2008, 3, e2888. [Google Scholar] [CrossRef] [PubMed]
- Vesuna, F.; Lisok, A.; Kimble, B.; Raman, V. Twist modulates breast cancer stem cells by transcriptional regulation of CD24 expression. Neoplasia 2009, 11, 1318–1328. [Google Scholar] [CrossRef] [PubMed]
- Donehower, L.A.; Lozano, G. 20 years studying p53 functions in genetically engineered mice. Nat. Rev. Cancer 2009, 9, 831–841. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.N.; Roe, A.E.; Donehower, L.A.; Bradley, A. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature 1995, 378, 206–208. [Google Scholar] [CrossRef] [PubMed]
- Cicalese, A.; Bonizzi, G.; Pasi, C.E.; Faretta, M.; Ronzoni, S.; Giulini, B.; Brisken, C.; Minucci, S.; Di Fiore, P.P.; Pelicci, P.G. The tumor suppressor p53 regulates polarity of self-renewing divisions in mammary stem cells. Cell 2009, 138, 1083–1095. [Google Scholar] [CrossRef] [PubMed]
- Pereira, L.; Yi, F.; Merrill, B.J. Repression of Nanog gene transcription by Tcf3 limits embryonic stem cell self-renewal. Mol. Cell. Biol. 2006, 26, 7479–7491. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, S.; Panopoulos, A.D.; Herrerias, A.; Bissig, K.D.; Lutz, M.; Berggren, W.T.; Verma, I.M.; Izpisua Belmonte, J.C. A high proliferation rate is required for cell reprogramming and maintenance of human embryonic stem cell identity. Curr. Biol. 2011, 21, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Collado, M.; Villasante, A.; Strati, K.; Ortega, S.; Canamero, M.; Blasco, M.A.; Serrano, M. The Ink4/Arf locus is a barrier for iPS cell reprogramming. Nature 2009, 460, 1136–1139. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Jones, R.; Liu, J.C.; Deng, T.; Robinson, T.; Chung, P.E.; Wang, S.; Herschkowitz, J.I.; Egan, S.E.; Perou, C.M.; et al. RB1 and p53 at the crossroad of EMT and triple-negative breast cancer. Cell Cycle 2011, 10, 1563–1570. [Google Scholar] [CrossRef] [PubMed]
- Kogan-Sakin, I.; Tabach, Y.; Buganim, Y.; Molchadsky, A.; Solomon, H.; Madar, S.; Kamer, I.; Stambolsky, P.; Shelly, A.; Goldfinger, N.; et al. Mutant p53R175H upregulates Twist1 expression and promotes epithelial-mesenchymal transition in immortalized prostate cells. Cell Death Differ. 2011, 18, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.P.; Wang, W.L.; Chang, Y.L.; Wu, C.T.; Chao, Y.C.; Kao, S.H.; Yuan, A.; Lin, C.W.; Yang, S.C.; Chan, W.K.; et al. p53 controls cancer cell invasion by inducing the Mdm2-mediated degradation of slug. Nat. Cell Biol. 2009, 11, 694–704. [Google Scholar] [CrossRef] [PubMed]
- He, L.; He, X.; Lim, L.P.; de Stanchina, E.; Xuan, Z.; Liang, Y.; Xue, W.; Zender, L.; Magnus, J.; Ridzon, D.; et al. A microrna component of the p53 tumour suppressor network. Nature 2007, 447, 1130–1134. [Google Scholar] [CrossRef] [PubMed]
- Hermeking, H. The miR-34 family in cancer and apoptosis. Cell Death Differ. 2010, 17, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Veronese, A.; Pichiorri, F.; Lee, T.J.; Jeon, Y.J.; Volinia, S.; Pineau, P.; Marchio, A.; Palatini, J.; Suh, S.S.; et al. p53 regulates epithelial-mesenchymal transition through micrornas targeting ZEB1 and ZEB2. J. Exp. Med. 2011, 208, 875–883. [Google Scholar] [CrossRef] [PubMed]
- Wellner, U.; Schubert, J.; Burk, U.C.; Schmalhofer, O.; Zhu, F.; Sonntag, A.; Waldvogel, B.; Vannier, C.; Darling, D.; zur Hausen, A.; et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting micrornas. Nat. Cell Biol. 2009, 11, 1487–1495. [Google Scholar] [CrossRef] [PubMed]
- Siemens, H.; Jackstadt, R.; Hunten, S.; Kaller, M.; Menssen, A.; Gotz, U.; Hermeking, H. Mir-34 and snail form a double-negative feedback loop to regulate epithelial-mesenchymal transitions. Cell Cycle 2011, 10, 4256–4271. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.K.; Allton, K.; Iacovino, M.; Mahen, E.; Milczarek, R.J.; Zwaka, T.P.; Kyba, M.; Barton, M.C. p53 regulates cell cycle and micrornas to promote differentiation of human embryonic stem cells. PLoS Biol. 2012, 10, e1001268. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Jackstadt, R.; Siemens, H.; Li, H.; Kirchner, T.; Hermeking, H. p53-induced miR-15A/16-1 and AP4 form a double-negative feedback loop to regulate epithelial-mesenchymal transition and metastasis in colorectal cancer. Cancer Res. 2014, 74, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Aoubala, M.; Murray-Zmijewski, F.; Khoury, M.P.; Fernandes, K.; Perrier, S.; Bernard, H.; Prats, A.C.; Lane, D.P.; Bourdon, J.C. p53 directly transactivates ∆133p53α, regulating cell fate outcome in response to DNA damage. Cell Death Differ. 2011, 18, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Bernard, H.; Garmy-Susini, B.; Ainaoui, N.; Van Den Berghe, L.; Peurichard, A.; Javerzat, S.; Bikfalvi, A.; Lane, D.P.; Bourdon, J.C.; Prats, A.C. The p53 isoform, ∆133p53α, stimulates angiogenesis and tumour progression. Oncogene 2013, 32, 2150–2160. [Google Scholar] [CrossRef] [PubMed]
- Bourdon, J.C.; Fernandes, K.; Murray-Zmijewski, F.; Liu, G.; Diot, A.; Xirodimas, D.P.; Saville, M.K.; Lane, D.P. p53 isoforms can regulate p53 transcriptional activity. Genes Dev. 2005, 19, 2122–2137. [Google Scholar] [CrossRef] [PubMed]
- Marcel, V.; Petit, I.; Murray-Zmijewski, F.; Goullet de Rugy, T.; Fernandes, K.; Meuray, V.; Diot, A.; Lane, D.P.; Aberdam, D.; Bourdon, J.C. Diverse p63 and p73 isoforms regulate ∆133p53 expression through modulation of the internal TP53 promoter activity. Cell Death Differ. 2012, 19, 816–826. [Google Scholar] [CrossRef] [PubMed]
- Terrier, O.; Josset, L.; Textoris, J.; Marcel, V.; Cartet, G.; Ferraris, O.; N'Guyen, C.; Lina, B.; Diaz, J.J.; Bourdon, J.C.; et al. Cellular transcriptional profiling in human lung epithelial cells infected by different subtypes of influenza a viruses reveals an overall down-regulation of the host p53 pathway. Virol. J. 2011, 8, 285. [Google Scholar] [CrossRef] [PubMed]
- Terrier, O.; Marcel, V.; Cartet, G.; Lane, D.P.; Lina, B.; Rosa-Calatrava, M.; Bourdon, J.C. Influenza a viruses control expression of proviral human p53 isoforms p53β and ∆133p53α. J. Virol. 2012, 86, 8452–8460. [Google Scholar] [CrossRef] [PubMed]
- Michalak, E.; Villunger, A.; Erlacher, M.; Strasser, A. Death squads enlisted by the tumour suppressor p53. Biochem. Biophys. Res. Commun. 2005, 331, 786–798. [Google Scholar] [CrossRef] [PubMed]
- Sugrue, M.M.; Shin, D.Y.; Lee, S.W.; Aaronson, S.A. Wild-type p53 triggers a rapid senescence program in human tumor cells lacking functional p53. Proc. Natl. Acad. Sci. USA 1997, 94, 9648–9653. [Google Scholar] [CrossRef] [PubMed]
- Chao, C.; Saito, S.; Anderson, C.W.; Appella, E.; Xu, Y. Phosphorylation of murine p53 at ser-18 regulates the p53 responses to DNA damage. Proc. Natl. Acad. Sci. USA 2000, 97, 11936–11941. [Google Scholar] [CrossRef] [PubMed]
- Aladjem, M.I.; Spike, B.T.; Rodewald, L.W.; Hope, T.J.; Klemm, M.; Jaenisch, R.; Wahl, G.M. ES cells do not activate p53-dependent stress responses and undergo p53-independent apoptosis in response to DNA damage. Curr. Biol. 1998, 8, 145–155. [Google Scholar] [CrossRef]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef] [PubMed]
- Ferbeyre, G.; de Stanchina, E.; Lin, A.W.; Querido, E.; McCurrach, M.E.; Hannon, G.J.; Lowe, S.W. Oncogenic Ras and p53 cooperate to induce cellular senescence. Mol. Cell. Biol. 2002, 22, 3497–3508. [Google Scholar] [CrossRef] [PubMed]
- Gannon, H.S.; Donehower, L.A.; Lyle, S.; Jones, S.N. Mdm2-p53 signaling regulates epidermal stem cell senescence and premature aging phenotypes in mouse skin. Dev. Biol. 2011, 353, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Harrison, D.E.; Lerner, C.P. Most primitive hematopoietic stem cells are stimulated to cycle rapidly after treatment with 5-fluorouracil. Blood 1991, 78, 1237–1240. [Google Scholar] [PubMed]
- Zhou, S.; Schuetz, J.D.; Bunting, K.D.; Colapietro, A.M.; Sampath, J.; Morris, J.J.; Lagutina, I.; Grosveld, G.C.; Osawa, M.; Nakauchi, H.; et al. The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nat. Med. 2001, 7, 1028–1034. [Google Scholar] [CrossRef] [PubMed]
- Peters, R.; Leyvraz, S.; Perey, L. Apoptotic regulation in primitive hematopoietic precursors. Blood 1998, 92, 2041–2052. [Google Scholar] [PubMed]
- Arai, F.; Hirao, A.; Ohmura, M.; Sato, H.; Matsuoka, S.; Takubo, K.; Ito, K.; Koh, G.Y.; Suda, T. Tie2/angiopoietin-1 signaling regulates hematopoietic stem cell quiescence in the bone marrow niche. Cell 2004, 118, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Cheshier, S.H.; Morrison, S.J.; Liao, X.; Weissman, I.L. In vivo proliferation and cell cycle kinetics of long-term self-renewing hematopoietic stem cells. Proc. Natl. Acad. Sci. USA 1999, 96, 3120–3125. [Google Scholar] [CrossRef] [PubMed]
- Kiel, M.J.; He, S.; Ashkenazi, R.; Gentry, S.N.; Teta, M.; Kushner, J.A.; Jackson, T.L.; Morrison, S.J. Haematopoietic stem cells do not asymmetrically segregate chromosomes or retain BrdU. Nature 2007, 449, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, T.; Kohara, H.; Noda, M.; Nagasawa, T. Maintenance of the hematopoietic stem cell pool by CXCl12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity 2006, 25, 977–988. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Bhatia, R. Stem cell quiescence. Clin. Cancer Res. 2011, 17, 4936–4941. [Google Scholar] [CrossRef] [PubMed]
- White, A.C.; Khuu, J.K.; Dang, C.Y.; Hu, J.; Tran, K.V.; Liu, A.; Gomez, S.; Zhang, Z.; Yi, R.; Scumpia, P.; et al. Stem cell quiescence acts as a tumour suppressor in squamous tumours. Nat. Cell Biol. 2014, 16, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Itahana, K.; Dimri, G.P.; Hara, E.; Itahana, Y.; Zou, Y.; Desprez, P.Y.; Campisi, J. A role for p53 in maintaining and establishing the quiescence growth arrest in human cells. J. Biol. Chem. 2002, 277, 18206–18214. [Google Scholar] [CrossRef] [PubMed]
- Meletis, K.; Wirta, V.; Hede, S.M.; Nister, M.; Lundeberg, J.; Frisen, J. p53 suppresses the self-renewal of adult neural stem cells. Development 2006, 133, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Cheung, T.H.; Rando, T.A. Molecular regulation of stem cell quiescence. Nat. Rev. Mol. Cell Biol. 2013, 14, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Elf, S.E.; Miyata, Y.; Sashida, G.; Liu, Y.; Huang, G.; Di Giandomenico, S.; Lee, J.M.; Deblasio, A.; Menendez, S.; et al. p53 regulates hematopoietic stem cell quiescence. Cell Stem Cell 2009, 4, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Brien, G.L.; Bracken, A.P. The PCL1-p53 axis promotes cellular quiescence. Cell Cycle 2016, 15, 305–306. [Google Scholar] [CrossRef] [PubMed]
- Brien, G.L.; Healy, E.; Jerman, E.; Conway, E.; Fadda, E.; O’Donovan, D.; Krivtsov, A.V.; Rice, A.M.; Kearney, C.J.; Flaus, A.; et al. A chromatin-independent role of polycomb-like 1 to stabilize p53 and promote cellular quiescence. Genes Dev. 2015, 29, 2231–2243. [Google Scholar] [CrossRef] [PubMed]
- Freedman, D.A.; Levine, A.J. Regulation of the p53 protein by the Mdm2 oncoprotein--thirty-eighth g.H.A. Clowes memorial award lecture. Cancer Res. 1999, 59, 1–7. [Google Scholar] [PubMed]
- Freedman, D.A.; Wu, L.; Levine, A.J. Functions of the Mdm2 oncoprotein. Cell. Mol. Life Sci. 1999, 55, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Issaeva, N.; Bozko, P.; Enge, M.; Protopopova, M.; Verhoef, L.G.; Masucci, M.; Pramanik, A.; Selivanova, G. Small molecule RITA binds to p53, blocks p53-HDM-2 interaction and activates p53 function in tumors. Nat. Med. 2004, 10, 1321–1328. [Google Scholar] [CrossRef] [PubMed]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In vivo activation of the p53 pathway by small-molecule antagonists of Mdm2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef] [PubMed]
- Shangary, S.; Qin, D.; McEachern, D.; Liu, M.; Miller, R.S.; Qiu, S.; Nikolovska-Coleska, Z.; Ding, K.; Wang, G.; Chen, J.; et al. Temporal activation of p53 by a specific Mdm2 inhibitor is selectively toxic to tumors and leads to complete tumor growth inhibition. Proc. Natl. Acad. Sci. USA 2008, 105, 3933–3938. [Google Scholar] [CrossRef] [PubMed]
- Grasberger, B.L.; Lu, T.; Schubert, C.; Parks, D.J.; Carver, T.E.; Koblish, H.K.; Cummings, M.D.; LaFrance, L.V.; Milkiewicz, K.L.; Calvo, R.R.; et al. Discovery and cocrystal structure of benzodiazepinedione HDM2 antagonists that activate p53 in cells. J. Med. Chem. 2005, 48, 909–912. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Ludwig, R.L.; Jensen, J.P.; Pierre, S.A.; Medaglia, M.V.; Davydov, I.V.; Safiran, Y.J.; Oberoi, P.; Kenten, J.H.; Phillips, A.C.; et al. Small molecule inhibitors of HDM2 ubiquitin ligase activity stabilize and activate p53 in cells. Cancer Cell 2005, 7, 547–559. [Google Scholar] [CrossRef] [PubMed]
- Staples, O.D.; Hollick, J.J.; Campbell, J.; Higgins, M.; McCarthy, A.R.; Appleyard, V.; Murray, K.E.; Baker, L.; Thompson, A.; Ronseaux, S.; et al. Characterization, chemical optimization and anti-tumour activity of a tubulin poison identified by a p53-based phenotypic screen. Cell Cycle 2008, 7, 3417–3427. [Google Scholar] [CrossRef] [PubMed]
- Foster, B.A.; Coffey, H.A.; Morin, M.J.; Rastinejad, F. Pharmacological rescue of mutant p53 conformation and function. Science 1999, 286, 2507–2510. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.J.; Issaeva, N.; Shilov, A.; Hultcrantz, M.; Pugacheva, E.; Chumakov, P.; Bergman, J.; Wiman, K.G.; Selivanova, G. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat. Med. 2002, 8, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.J.; Issaeva, N.; Zache, N.; Shilov, A.; Hultcrantz, M.; Bergman, J.; Selivanova, G.; Wiman, K.G. Reactivation of mutant p53 and induction of apoptosis in human tumor cells by maleimide analogs. J. Biol. Chem. 2005, 280, 30384–30391. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Li, C.; Chen, L.; Sebti, S.; Chen, J. Rescue of mutant p53 transcription function by ellipticine. Oncogene 2003, 22, 4478–4487. [Google Scholar] [CrossRef] [PubMed]
- Weinmann, L.; Wischhusen, J.; Demma, M.J.; Naumann, U.; Roth, P.; Dasmahapatra, B.; Weller, M. A novel p53 rescue compound induces p53-dependent growth arrest and sensitises glioma cells to apo2l/trail-induced apoptosis. Cell Death Differ. 2008, 15, 718–729. [Google Scholar] [CrossRef] [PubMed]
- North, S.; Pluquet, O.; Maurici, D.; El-Ghissassi, F.; Hainaut, P. Restoration of wild-type conformation and activity of a temperature-sensitive mutant of p53 (p53V272M) by the cytoprotective aminothiol WR1065 in the esophageal cancer cell line TE-1. Mol. Carcinog. 2002, 33, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Komarov, P.G.; Komarova, E.A.; Kondratov, R.V.; Christov-Tselkov, K.; Coon, J.S.; Chernov, M.V.; Gudkov, A.V. A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science 1999, 285, 1733–1737. [Google Scholar] [CrossRef] [PubMed]
- Strom, E.; Sathe, S.; Komarov, P.G.; Chernova, O.B.; Pavlovska, I.; Shyshynova, I.; Bosykh, D.A.; Burdelya, L.G.; Macklis, R.M.; Skaliter, R.; et al. Small-molecule inhibitor of p53 binding to mitochondria protects mice from γ radiation. Nat. Chem. Biol. 2006, 2, 474–479. [Google Scholar] [CrossRef] [PubMed]
- Tal, P.; Eizenberger, S.; Cohen, E.; Goldfinger, N.; Pietrokovski, S.; Oren, M.; Rotter, V. Cancer therapeutic approach based on conformational stabilization of mutant p53 protein by small peptides. Oncotarget 2016, 7, 11817–11837. [Google Scholar] [PubMed]
- Zhang, Z.; Liu, L.; Gomez-Casal, R.; Wang, X.; Hayashi, R.; Appella, E.; Kopelovich, L.; DeLeo, A.B. Targeting cancer stem cells with p53 modulators. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]
- Izetti, P.; Hautefeuille, A.; Abujamra, A.L.; de Farias, C.B.; Giacomazzi, J.; Alemar, B.; Lenz, G.; Roesler, R.; Schwartsmann, G.; Osvaldt, A.B.; et al. PRIMA-1, a mutant p53 reactivator, induces apoptosis and enhances chemotherapeutic cytotoxicity in pancreatic cancer cell lines. Investig. New Drugs 2014, 32, 783–794. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Besch-Williford, C.; Hyder, S.M. PRIMA-1 inhibits growth of breast cancer cells by re-activating mutant p53 protein. Int. J. Oncol. 2009, 35, 1015–1023. [Google Scholar] [PubMed]
- Lambert, J.M.; Gorzov, P.; Veprintsev, D.B.; Soderqvist, M.; Segerback, D.; Bergman, J.; Fersht, A.R.; Hainaut, P.; Wiman, K.G.; Bykov, V.J. PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell 2009, 15, 376–388. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, V.V.; Hong, B.; Allen, J.E.; Zhang, S.; Lulla, A.R.; Dicker, D.T.; El-Deiry, W.S. Small-molecule prodigiosin restores p53 tumor suppressor activity in chemoresistant colorectal cancer stem cells via c-Jun-mediated ∆Np73 inhibition and p73 activation. Cancer Res. 2016, 76, 1989–1999. [Google Scholar] [CrossRef] [PubMed]
- Issaeva, N.; Friedler, A.; Bozko, P.; Wiman, K.G.; Fersht, A.R.; Selivanova, G. Rescue of mutants of the tumor suppressor p53 in cancer cells by a designed peptide. Proc. Natl. Acad. Sci. USA 2003, 100, 13303–13307. [Google Scholar] [CrossRef] [PubMed]
- Selivanova, G.; Iotsova, V.; Okan, I.; Fritsche, M.; Strom, M.; Groner, B.; Grafstrom, R.C.; Wiman, K.G. Restoration of the growth suppression function of mutant p53 by a synthetic peptide derived from the p53 C-terminal domain. Nat. Med. 1997, 3, 632–638. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.L.; Raffo, A.J.; Brandt-Rauf, P.W.; Pincus, M.R.; Monaco, R.; Abarzua, P.; Fine, R.L. Conformational and molecular basis for induction of apoptosis by a p53 C-terminal peptide in human cancer cells. J. Biol. Chem. 1999, 274, 34924–34931. [Google Scholar] [CrossRef] [PubMed]
- Snyder, E.L.; Meade, B.R.; Saenz, C.C.; Dowdy, S.F. Treatment of terminal peritoneal carcinomatosis by a transducible p53-activating peptide. PLoS Biol. 2004, 2, e36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernal, F.; Tyler, A.F.; Korsmeyer, S.J.; Walensky, L.D.; Verdine, G.L. Reactivation of the p53 tumor suppressor pathway by a stapled p53 peptide. J. Am. Chem. Soc. 2007, 129, 2456–2457. [Google Scholar] [CrossRef] [PubMed]
- Madden, M.M.; Muppidi, A.; Li, Z.; Li, X.; Chen, J.; Lin, Q. Synthesis of cell-permeable stapled peptide dual inhibitors of the p53-Mdm2/Mdmx interactions via photoinduced cycloaddition. Bioorg. Med. Chem. Lett. 2011, 21, 1472–1475. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Tai, L.; Gao, J.; Qian, J.; Zhang, M.; Li, B.; Xie, C.; Lu, L.; Lu, W.; Lu, W. A stapled peptide antagonist of Mdm2 carried by polymeric micelles sensitizes glioblastoma to temozolomide treatment through p53 activation. J. Control. Release 2015, 218, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Weisbart, R.H.; Wakelin, R.; Chan, G.; Miller, C.W.; Koeffler, P.H. Construction and expression of a bispecific single-chain antibody that penetrates mutant p53 colon cancer cells and binds p53. Int. J. Oncol. 2004, 25, 1113–1118. [Google Scholar] [PubMed]
- Kang, B.; Sun, X.H. Regulation of cancer stem cells by RING finger ubiquitin ligases. Stem Cell Investig. 2014, 1. [Google Scholar] [CrossRef]
- Moll, U.M.; Wolff, S.; Speidel, D.; Deppert, W. Transcription-independent pro-apoptotic functions of p53. Curr. Opin. Cell Biol. 2005, 17, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Lorenz, A.; Hameister, H.; Montenarh, M. Expression of p53 during mouse embryogenesis. Development 1991, 113, 857–865. [Google Scholar] [PubMed]
- Lichnovsky, V.; Kolar, Z.; Murray, P.; Hlobilkova, A.; Cernochova, D.; Pospisilova, E.; Vojtesek, B.; Nenutil, R. Differences in p53 and Bcl-2 expression in relation to cell proliferation during the development of human embryos. Mol. Pathol. 1998, 51, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Sabapathy, K.; Klemm, M.; Jaenisch, R.; Wagner, E.F. Regulation of es cell differentiation by functional and conformational modulation of p53. EMBO J. 1997, 16, 6217–6229. [Google Scholar] [CrossRef] [PubMed]
- McMasters, K.M.; Montes de Oca Luna, R.; Pena, J.R.; Lozano, G. Mdm2 deletion does not alter growth characteristics of p53-deficient embryo fibroblasts. Oncogene 1996, 13, 1731–1736. [Google Scholar] [PubMed]
- Oliner, J.D.; Kinzler, K.W.; Meltzer, P.S.; George, D.L.; Vogelstein, B. Amplification of a gene encoding a p53-associated protein in human sarcomas. Nature 1992, 358, 80–83. [Google Scholar] [CrossRef] [PubMed]
- Daniele, S.; Costa, B.; Zappelli, E.; Da Pozzo, E.; Sestito, S.; Nesi, G.; Campiglia, P.; Marinelli, L.; Novellino, E.; Rapposelli, S.; et al. Combined inhibition of Akt/mTOR and Mdm2 enhances glioblastoma multiforme cell apoptosis and differentiation of cancer stem cells. Sci. Rep. 2015, 5, 9956. [Google Scholar] [CrossRef] [PubMed]
- Wienken, M.; Dickmanns, A.; Nemajerova, A.; Kramer, D.; Najafova, Z.; Weiss, M.; Karpiuk, O.; Kassem, M.; Zhang, Y.; Lozano, G.; et al. Mdm2 associates with polycomb repressor complex 2 and enhances stemness-promoting chromatin modifications independent of p53. Mol. Cell 2016, 61, 68–83. [Google Scholar] [CrossRef] [PubMed]
- Sato, A.; Sunayama, J.; Matsuda, K.; Seino, S.; Suzuki, K.; Watanabe, E.; Tachibana, K.; Tomiyama, A.; Kayama, T.; Kitanaka, C. MEK-ERK signaling dictates DNA-repair gene MGMT expression and temozolomide resistance of stem-like glioblastoma cells via the MDM2-p53 axis. Stem Cells 2011, 29, 1942–1951. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Yang, J.; Maity, B.; Mayuzumi, D.; Fisher, R.A. Regulator of g protein signaling 6 mediates doxorubicin-induced atm and p53 activation by a reactive oxygen species-dependent mechanism. Cancer Res. 2011, 71, 6310–6319. [Google Scholar] [CrossRef] [PubMed]
- Donald, S.P.; Sun, X.Y.; Hu, C.A.; Yu, J.; Mei, J.M.; Valle, D.; Phang, J.M. Proline oxidase, encoded by p53-induced gene-6, catalyzes the generation of proline-dependent reactive oxygen species. Cancer Res. 2001, 61, 1810–1815. [Google Scholar] [PubMed]
- Passegue, E.; Jamieson, C.H.; Ailles, L.E.; Weissman, I.L. Normal and leukemic hematopoiesis: Are leukemias a stem cell disorder or a reacquisition of stem cell characteristics? Proc. Natl. Acad. Sci. USA 2003, 100, 11842–11849. [Google Scholar] [CrossRef] [PubMed]
- Leroy, B.; Fournier, J.L.; Ishioka, C.; Monti, P.; Inga, A.; Fronza, G.; Soussi, T. The TP53 website: An integrative resource centre for the TP53 mutation database and TP53 mutant analysis. Nucleic Acids Res. 2013, 41, D962–D969. [Google Scholar] [CrossRef] [PubMed]
- Dumay, A.; Feugeas, J.P.; Wittmer, E.; Lehmann-Che, J.; Bertheau, P.; Espie, M.; Plassa, L.F.; Cottu, P.; Marty, M.; Andre, F.; et al. Distinct tumor protein p53 mutants in breast cancer subgroups. Int. J. Cancer 2013, 132, 1227–1231. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, R.V.; da Silva, L.M. A possible explanation for the variable frequencies of cancer stem cells in tumors. PLoS ONE 2013, 8, e69131. [Google Scholar] [CrossRef] [PubMed]




© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olivos, D.J.; Mayo, L.D. Emerging Non-Canonical Functions and Regulation by p53: p53 and Stemness. Int. J. Mol. Sci. 2016, 17, 1982. https://doi.org/10.3390/ijms17121982
Olivos DJ, Mayo LD. Emerging Non-Canonical Functions and Regulation by p53: p53 and Stemness. International Journal of Molecular Sciences. 2016; 17(12):1982. https://doi.org/10.3390/ijms17121982
Chicago/Turabian StyleOlivos, David J., and Lindsey D. Mayo. 2016. "Emerging Non-Canonical Functions and Regulation by p53: p53 and Stemness" International Journal of Molecular Sciences 17, no. 12: 1982. https://doi.org/10.3390/ijms17121982