
Characterization of hsa_circ_0004277 as a New Biomarker for Acute Myeloid Leukemia via Circular RNA Profile and Bioinformatics Analysis

Abstract
:
1. Introduction
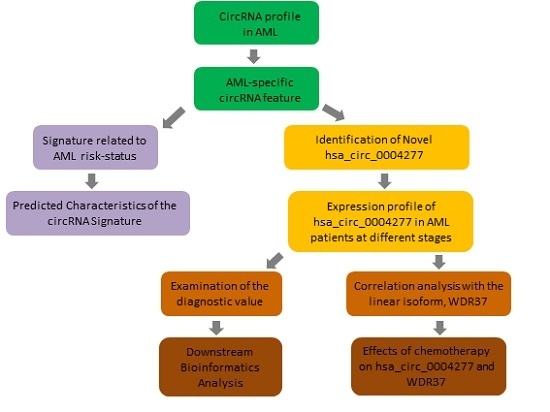
2. Results
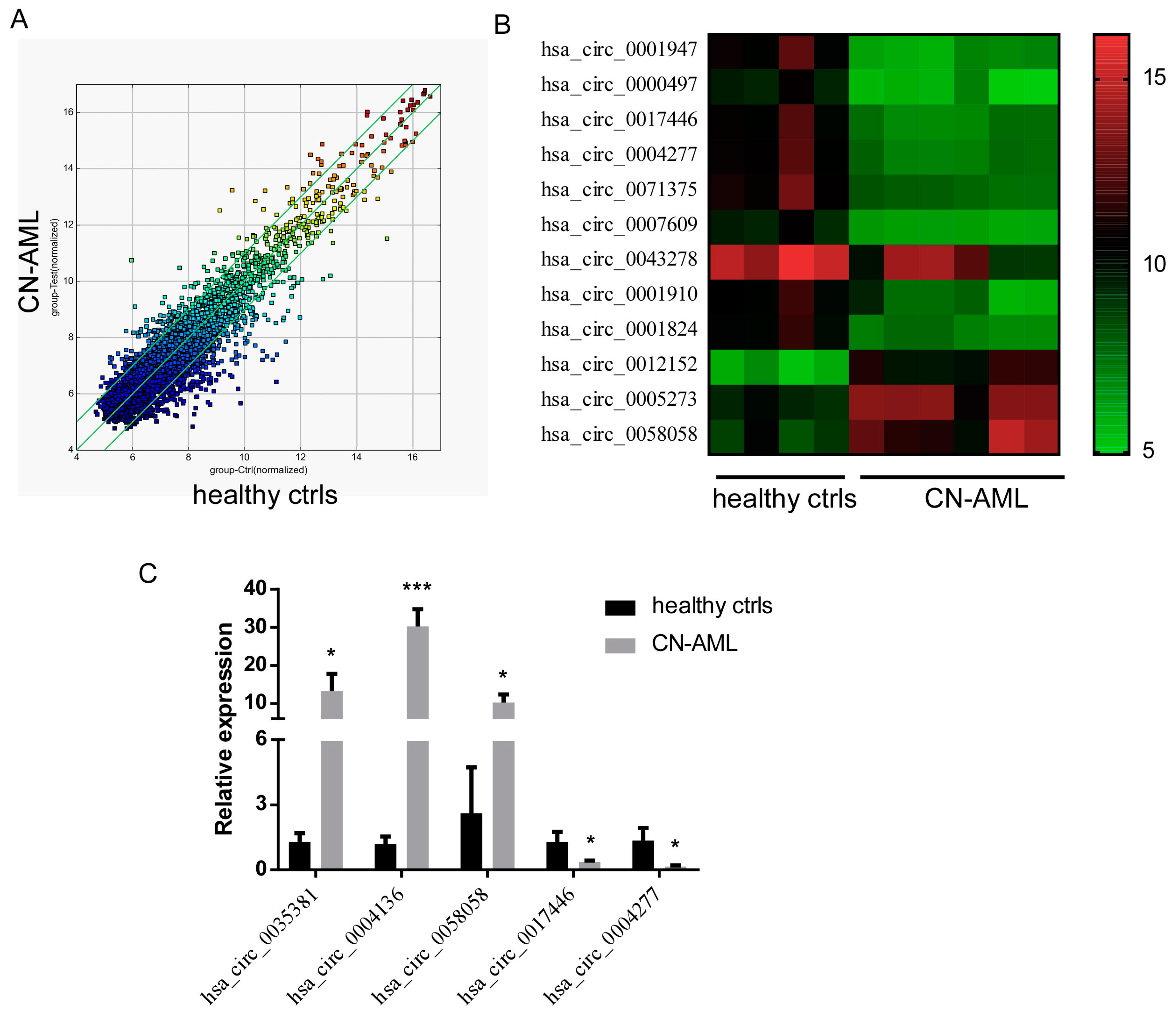
2.1. Profile of circRNA Expression in AML Patients
2.2. Validation of Differentially Expressed circRNAs from the Microarray Profile
2.3. Identification of circRNA Signature between Better-Risk and Poor-Risk AML Patients
2.4. Predicted Characteristics of the circRNA Signature Related to AML Risk-Status
2.5. Identification of Novel hsa_circ_0004277 in AML
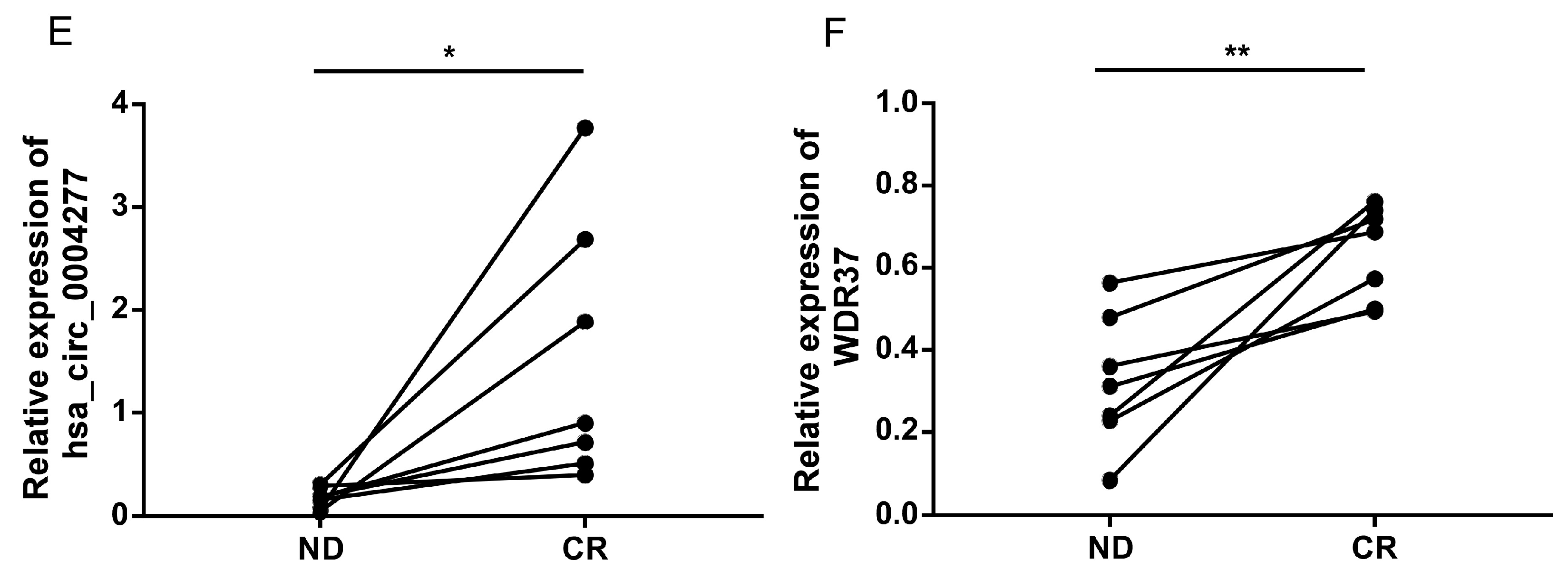
2.6. Effects of Chemotherapy on hsa_circ_0004277 Expression in AML Patients
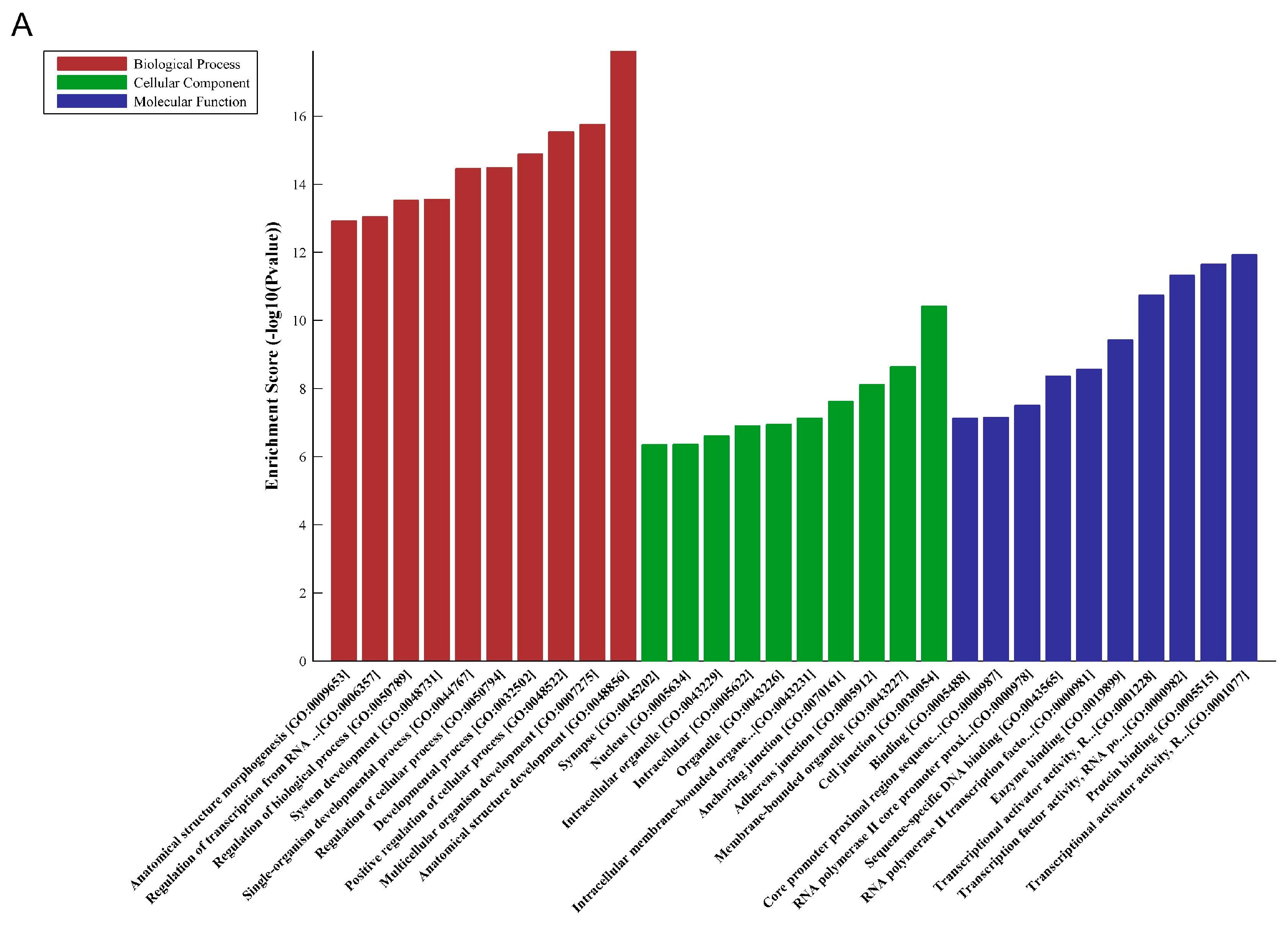
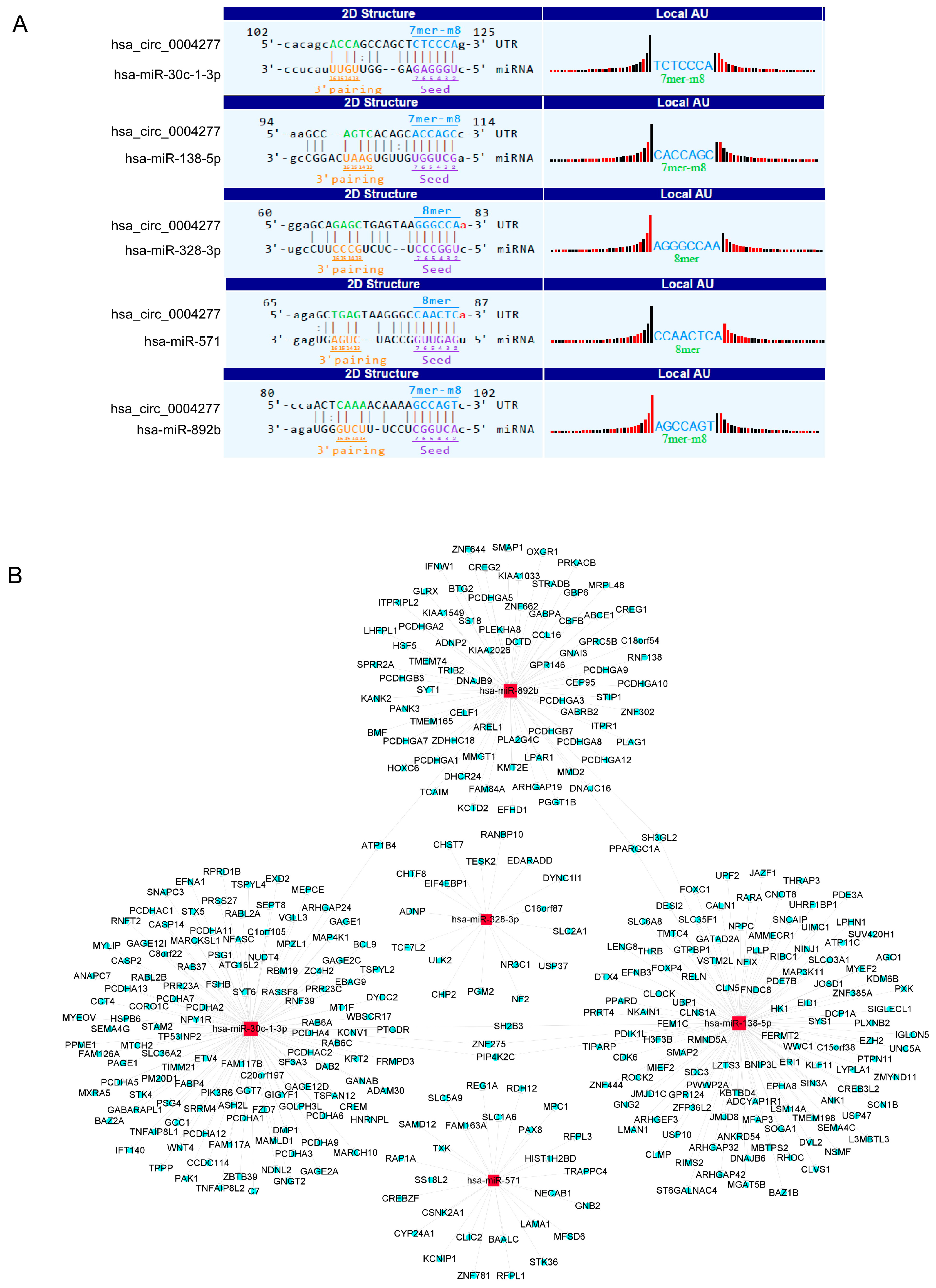
2.7. MicroRNA Prediction and Downstream Bioinformatics Analysis for hsa_circ_0004277
3. Discussion
4. Materials and Methods
4.1. Human Samples and Cell Preparation
4.2. RNA Isolation, Labeling, and Hybridization
4.3. Microarray and Quality Control
4.4. Real-Time Quantitative PCR Validation
4.5. MicroRNA Prediction and Functional Analysis
4.6. Statistics
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| circRNA | Circular RNA |
| AML | Acute myeloid leukemia |
| CN-AML | Cytogenetically normal AML |
| lncRNA | Long noncoding RNA |
| cRNA | Complementary RNA |
| ceRNA | Competing endogenous RNA |
| GO | Gene oncology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| SEM | Standard error of the mean |
| ND | Newly diagnosed |
| CR | Complete remission |
| RE | Relapsed-refractory |
| ROC | Receiver-operating characteristic |
| AUC | Area under the ROC curve |
| SRY | Sex-determining region Y |
| BM | Bone marrow |
| FLT3-ITD | FMS-like tyrosine kinase-3 |
| NPM1 | Nucleophosmin 1 |
| qRT-PCR | quantitative Real-Time PCR |
| GAPDH | Glyceraldehyde 3-phosphate dehydrogenase |
| GEO | Gene Expression Omnibus |
References
- Dohner, H.; Estey, E.H.; Amadori, S.; Appelbaum, F.R.; Buchner, T.; Burnett, A.K.; Dombret, H.; Fenaux, P.; Grimwade, D.; Larson, R.A.; et al. Diagnosis and management of acute myeloid leukemia in adults: Recommendations from an international expert panel, on behalf of the european leukemianet. Blood 2010, 115, 453–474. [Google Scholar] [CrossRef]
- O’Donnell, M.R.; Abboud, C.N.; Altman, J.; Appelbaum, F.R.; Arber, D.A.; Attar, E.; Borate, U.; Coutre, S.E.; Damon, L.E.; Goorha, S.; et al. NCCN clinical practice guidelines acute myeloid leukemia. J. Natl. Compr. Cancer Netw. JNCCN 2012, 10, 984–1021. [Google Scholar] [PubMed]
- Prada-Arismendy, J.; Arroyave, J.C.; Rothlisberger, S. Molecular biomarkers in acute myeloid leukemia. Blood Rev. 2016, 31, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Capel, B.; Swain, A.; Nicolis, S.; Hacker, A.; Walter, M.; Koopman, P.; Goodfellow, P.; Lovell-Badge, R. Circular transcripts of the testis-determining gene Sry in adult mouse testis. Cell 1993, 73, 1019–1030. [Google Scholar] [CrossRef]
- Cocquerelle, C.; Mascrez, B.; Hetuin, D.; Bailleul, B. Mis-splicing yields circular rna molecules. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1993, 7, 155–160. [Google Scholar]
- Pasman, Z.; Been, M.D.; Garcia-Blanco, M.A. Exon circularization in mammalian nuclear extracts. RNA 1996, 2, 603–610. [Google Scholar] [PubMed]
- Chen, L.L.; Yang, L. Regulation of circrna biogenesis. RNA Biol. 2015, 12, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular rnas are a large class of animal rnas with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Memczak, S.; Papavasileiou, P.; Peters, O.; Rajewsky, N. Identification and characterization of circular rnas as a new class of putative biomarkers in human blood. PLoS ONE 2015, 10, e0141214. [Google Scholar]
- Wang, P.L.; Bao, Y.; Yee, M.C.; Barrett, S.P.; Hogan, G.J.; Olsen, M.N.; Dinneny, J.R.; Brown, P.O.; Salzman, J. Circular RNA is expressed across the eukaryotic tree of life. PLoS ONE 2014, 9, e90859. [Google Scholar] [CrossRef] [PubMed]
- Rybak-Wolf, A.; Stottmeister, C.; Glazar, P.; Jens, M.; Pino, N.; Giusti, S.; Hanan, M.; Behm, M.; Bartok, O.; Ashwal-Fluss, R.; et al. Circular RNAs in the mammalian brain are highly abundant, conserved, and dynamically expressed. Mol. Cell 2015, 58, 870–885. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Bao, C.; Guo, W.; Li, S.; Chen, J.; Chen, B.; Luo, Y.; Lyu, D.; Li, Y.; Shi, G.; et al. Circular RNA profiling reveals an abundant circHIPK3 that regulates cell growth by sponging multiple miRNAs. Nat. Commun. 2016, 7, 11215. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, Y.; Zheng, Q.; Bao, C.; He, J.; Chen, B.; Lyu, D.; Zheng, B.; Xu, Y.; Long, Z.; et al. Circular RNA profile identifies circPVT1 as a proliferative factor and prognostic marker in gastric cancer. Cancer Lett. 2016, 388, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Galasso, M.; Costantino, G.; Pasquali, L.; Minotti, L.; Baldassari, F.; Corra, F.; Agnoletto, C.; Volinia, S. Profiling of the predicted circular RNAs in ductal in situ and invasive breast cancer: A pilot study. Int. J. Genom. 2016, 2016, 4503840. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Lin, F.; Deng, X.; Shen, L.; Fang, Y.; Fei, Z.; Zhao, L.; Zhang, X.; Pan, H.; Xie, D.; et al. Profiling and bioinformatics analyses reveal differential circular RNA expression in radioresistant esophageal cancer cells. J. Transl. Med. 2016, 14, 225. [Google Scholar] [CrossRef] [PubMed]
- Denzler, R.; Agarwal, V.; Stefano, J.; Bartel, D.P.; Stoffel, M. Assessing the ceRNA hypothesis with quantitative measurements of miRNA and target abundance. Mol. Cell 2014, 54, 766–776. [Google Scholar] [CrossRef] [PubMed]
- Bachmayr-Heyda, A.; Reiner, A.T.; Auer, K.; Sukhbaatar, N.; Aust, S.; Bachleitner-Hofmann, T.; Mesteri, I.; Grunt, T.W.; Zeillinger, R.; Pils, D. Correlation of circular RNA abundance with proliferation—Exemplified with colorectal and ovarian cancer, idiopathic lung fibrosis, and normal human tissues. Sci. Rep. 2015, 5, 8057. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Zhang, L.; Li, W.; Deng, J.; Zheng, J.; An, M.; Lu, J.; Zhou, Y. Circular RNA ITCH has inhibitory effect on ESCC by suppressing the Wnt/β-catenin pathway. Oncotarget 2015, 6, 6001–6013. [Google Scholar] [CrossRef] [PubMed]
- Jeck, W.R.; Sharpless, N.E. Detecting and characterizing circular RNAs. Nat. Biotechnol. 2014, 32, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Salzman, J.; Gawad, C.; Wang, P.L.; Lacayo, N.; Brown, P.O. Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS ONE 2012, 7, e30733. [Google Scholar] [CrossRef] [PubMed]
- Jeck, W.R.; Sorrentino, J.A.; Wang, K.; Slevin, M.K.; Burd, C.E.; Liu, J.; Marzluff, W.F.; Sharpless, N.E. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 2013, 19, 141–157. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.B.; Wiklund, E.D.; Bramsen, J.B.; Villadsen, S.B.; Statham, A.L.; Clark, S.J.; Kjems, J. MiRNA-dependent gene silencing involving Ago2-mediated cleavage of a circular antisense RNA. EMBO J. 2011, 30, 4414–4422. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Y.; Sarnow, P. Initiation of protein synthesis by the eukaryotic translational apparatus on circular rnas. Science 1995, 268, 415–417. [Google Scholar] [CrossRef] [PubMed]
- Perriman, R.; Ares, M., Jr. Circular mRNA can direct translation of extremely long repeating-sequence proteins in vivo. RNA 1998, 4, 1047–1054. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Luo, J.; Hu, K.; Lin, J.; Huang, H.; Wang, Q.; Zhang, P.; Xiong, Z.; Huang, Z.; He, C.; et al. ZKSCAN1 gene and its related circular RNA (circZKSCAN1) both inhibit hepatocellular carcinoma cell growth, migration and invasion but through different signaling pathways. Mol. Oncol. 2017. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, C.; Campos, J.; Osinski, J.M.; Gronostajski, R.M.; Michie, A.M.; Keeshan, K. NFIX expression critically modulates early B lymphopoiesis and myelopoiesis. PLoS ONE 2015, 10, e0120102. [Google Scholar] [CrossRef] [PubMed]
- Willman, C.L. Sh2b3: A new leukemia predisposition gene. Blood 2013, 122, 2293–2295. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Gender | Age | Diagnosis of FAB Subtypes | FLT-ITD3 | NPM1 | Karyotype | Risk Status |
|---|---|---|---|---|---|---|---|
| 1 | M | 55 | M5 | + | + | Normal | Poor-risk |
| 2 | F | 41 | M5 | + | − | Normal | Poor-risk |
| 3 | F | 48 | M4 | + | − | Normal | Poor-risk |
| 4 | M | 27 | M5 | − | + | Normal | Better-risk |
| 5 | F | 58 | M5 | − | − | Normal | Better-risk |
| 6 | M | 17 | M4 | − | + | Normal | Better-risk |
| 7 | M | 35 | Healthy control | N/A | N/A | N/A | N/A |
| 8 | M | 47 | Healthy control | N/A | N/A | N/A | N/A |
| 9 | F | 52 | Healthy control | N/A | N/A | N/A | N/A |
| 10 | F | 28 | Healthy control | N/A | N/A | N/A | N/A |
| circRNA | Regulation in AML | p-Value | Fold Change | circRNA Type | Chrom | Best Transcript | Gene Symbol |
|---|---|---|---|---|---|---|---|
| hsa_circ_0035381 | up | 0.006 | 3.661 | exonic | chr15 | uc002act.3 | PIGB |
| hsa_circ_0049657 | up | 0.000 | 3.987 | exonic | chr19 | uc002mwg.2 | NFIX |
| hsa_circ_0001187 | down | 0.000 | 6.465 | exonic | chr21 | uc011aeb.2 | DOPEY2 |
| hsa_circ_0008078 | down | 0.000 | 5.084 | exonic | chr21 | uc011aeb.2 | DOPEY2 |
| hsa_circ_0001947 | down | 0.000 | 25.993 | exonic | chrX | uc004fco.3 | AFF2 |
| Features | No. | hsa_circ_0004277 |
|---|---|---|
| Age (years) | ||
| <43 | 32 | 0.14 ± 0.02 |
| ≥43 | 35 | 0.14 ± 0.02 |
| p-Value | - | 0.4756 |
| Gender | ||
| Male | 26 | 0.12 ± 0.02 |
| Female | 41 | 0.15 ± 0.02 |
| p-Value | - | 0.0996 |
| FAB subtypes | ||
| M1 | 3 | 0.06 ± 0.01 |
| M2 | 8 | 0.18 ± 0.05 |
| M3 | 17 | 0.19 ± 0.10 |
| M4 | 10 | 0.08 ± 0.02 |
| M5 | 29 | 0.20 ± 0.07 |
| p-Value | - | 0.9021 |
| Karyotype | ||
| Better-risk | 7 | 0.10 ± 0.04 |
| Intermediate | 18 | 0.11 ± 0.02 |
| Poor-risk | 5 | 0.16 ± 0.06 |
| p-Value | - | 0.4224 |
| Gene | Primer Sequence (5′–3′) |
|---|---|
| hsa_circ_0035381-F | GGTGACTGTGCTGTGGAC |
| hsa_circ_0035381-R | TGGTGTTTGTAAGGGTTC |
| hsa_circ_0004136-F | ATGGCAAGGAAGACTGAG |
| hsa_circ_0004136-R | AGGGATGGTAGAAAACAC |
| hsa_circ_0058058-F | GGTGGTGTCCACGGAGAT |
| hsa_circ_0058058-R | CCAAGAGCGGTCAGGTTT |
| hsa_circ_0017446-F | GGAGCATAGAGACAGGGA |
| hsa_circ_0017446-R | GGCTTTTGTTTTGAGTTG |
| hsa_circ_0004277-F | CACTTACAAGGCTTCCAC |
| hsa_circ_0004277-R | CTTACTCAGCTCTGCTCC |
| WDR37-F | TTCCACCAGCAAGATTGTCTC |
| WDR37-R | GCGTACTTGACTAGGCACTTCC |
| GAPDH-F | GGGAAACTGTGGCGTGAT |
| GAPDH-R | GAGTGGGTGTCGCTGTTGA |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, W.; Zhong, C.; Jiao, J.; Li, P.; Cui, B.; Ji, C.; Ma, D. Characterization of hsa_circ_0004277 as a New Biomarker for Acute Myeloid Leukemia via Circular RNA Profile and Bioinformatics Analysis. Int. J. Mol. Sci. 2017, 18, 597. https://doi.org/10.3390/ijms18030597
Li W, Zhong C, Jiao J, Li P, Cui B, Ji C, Ma D. Characterization of hsa_circ_0004277 as a New Biomarker for Acute Myeloid Leukemia via Circular RNA Profile and Bioinformatics Analysis. International Journal of Molecular Sciences. 2017; 18(3):597. https://doi.org/10.3390/ijms18030597
Chicago/Turabian StyleLi, Wei, Chaoqin Zhong, Jun Jiao, Peng Li, Baoxia Cui, Chunyan Ji, and Daoxin Ma. 2017. "Characterization of hsa_circ_0004277 as a New Biomarker for Acute Myeloid Leukemia via Circular RNA Profile and Bioinformatics Analysis" International Journal of Molecular Sciences 18, no. 3: 597. https://doi.org/10.3390/ijms18030597