Dysregulated Transcriptional Control in Prostate Cancer


Research & Development, Pharmaceuticals, Bayer AG, Müllerstr. 178, 13353 Berlin, Germany
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2019, 20(12), 2883; https://doi.org/10.3390/ijms20122883
Submission received: 6 May 2019
/
Revised: 6 June 2019
/
Accepted: 7 June 2019
/
Published: 13 June 2019
(This article belongs to the Special Issue Diagnostic, Prognostic and Predictive Biomarkers in Prostate Cancer 2019)
Abstract
:Recent advances in whole-genome and transcriptome sequencing of prostate cancer at different stages indicate that a large number of mutations found in tumors are present in non-protein coding regions of the genome and lead to dysregulated gene expression. Single nucleotide variations and small mutations affecting the recruitment of transcription factor complexes to DNA regulatory elements are observed in an increasing number of cases. Genomic rearrangements may position coding regions under the novel control of regulatory elements, as exemplified by the TMPRSS2-ERG fusion and the amplified enhancer identified upstream of the androgen receptor (AR) gene. Super-enhancers are increasingly found to play important roles in aberrant oncogenic transcription. Several players involved in these processes are currently being evaluated as drug targets and may represent new vulnerabilities that can be exploited for prostate cancer treatment. They include factors involved in enhancer and super-enhancer function such as bromodomain proteins and cyclin-dependent kinases. In addition, non-coding RNAs with an important gene regulatory role are being explored. The rapid progress made in understanding the influence of the non-coding part of the genome and of transcription dysregulation in prostate cancer could pave the way for the identification of novel treatment paradigms for the benefit of patients.
1. Introduction
The incidence of prostate cancer is the second highest in men and represents a leading cause of cancer mortality worldwide [1]. The recent progress made in whole-genome and transcriptome sequencing of primary, advanced, and metastasized tumor samples allowed major advances in the characterization of this heterogeneous disease [2,3,4,5,6]. Prostate cancer has a relatively low mutational burden of about one mutation per megabase [5,7], but multiple recurrent chromosomal losses and gains are frequent [2]. Gene fusions involving E26 transformation-specific (ETS) family members and coding mutations mainly affecting speckle-type POZ protein (SPOP), forkhead box protein A1 (FOXA1), and isocitrate dehydrogenase 1 (IDH1) are found in primary prostate cancer [2,3,4,5,8,9]. Several alterations affecting the androgen receptor (AR) and androgen signaling emerge in castration-resistant prostate cancer (CRPC) as an adaptation to treatment [2,3,4,5,8,10]. Late-stage, neuroendrocrine prostate cancer (NEPC) is characterized by the expression of transdifferentiation markers and the increased activity of oncogenes [2,11,12]. In addition, the previously unsuspected frequency of small non-coding alterations and the gene regulatory role of non-coding RNAs have recently been evidenced [4,13,14,15].
Here we review recent findings on dysregulated transcriptional control driven by genetic and epigenetic alterations and its contribution to oncogenic processes and resistance mechanisms in prostate cancer. We furthermore discuss how these mechanisms could be targeted to allow novel therapeutic options.
2. Promoters, Enhancers and Super-Enhancers
2.1. General Aspects
Tight spatiotemporal regulation of gene transcription is essential for normal development and function of all living cells. This is controlled by multiple regulatory units mostly positioned within 50 kilobase-pairs of the gene they regulate, but in some cases also megabase-pairs away [16,17]. Long-range connections between distant regulatory elements are established by looping [18,19].
The gene promoter located immediately upstream of the transcription start site directs the initial assembly of the transcriptional apparatus [20,21]. The proximal promoter element is usually rich in CpG islands situated upstream of the transcriptional start site and is characterized by high H3K4 trimethylation. It is responsible for the initial formation of a basal pre-initiation transcription complex including RNA polymerase II (Pol II), transcription factor II D and more than 40 other proteins (Figure 1) [20,21]. Following transcription initiation, the complex pauses and the transition to productive elongation necessitate the presence of the positive transcription elongation factor b (P-TEFb) complex made of cyclin-dependent kinase 9 (CDK9) and cyclin T [22]. This is controlled by cis-regulatory enhancer regions that interact with the promoter through the mediator, cohesin, the CCCTC-binding factor (CTCF) and Yin Yang 1 (YY1) complexes upon formation of chromosome loop structures [23,24].
Full, sustained gene transcription necessitates the interplay between the promoter and enhancer regions which are involved in both the recruitment of the basal transcription machinery and in post-recruitment steps (Figure 1) [25,26]. Active enhancers were originally found near or partially overlapping with promoter regions and have a length of a few hundreds of base-pairs [27]. They are located in open chromatin regions characterized by H3K27 acetylation and H3K4 monomethylation and by the presence of histone variants such as H3.3 and H2A.Z [28]. They usually do not contain CpG islands but have binding motifs for important regulators of the CCAAT-enhancer-binding proteins (C/EBP) which interact in a cooperative fashion with numerous proteins involved in gene transcription [29]. Additional enhancer hallmarks include H3K79 trimethylation and CREB-binding protein (CBP)/p300 recruitment [30,31]. Enhancers are bound by the bromodomain and extra-terminal (BET) protein BRD4 and by members of the mediator and P-TEFb complexes [32,33,34,35]. Recent single-cell analysis of gene transcription revealed further details on the inter-dependencies of these factors in enhancer function [36]. Bidirectional enhancer transcription driven by RNA Pol II and leading to the synthesis of non-coding enhancer RNAs (eRNAs) has been evidenced [25,37]. These eRNAs promote transcription by facilitating the formation of DNA loops between regulatory regions, which ultimately leads to highly increased, productive transcriptional activity ending up with the eviction of RNA Pol II at the 3′-tail of transcribed genes. Recent studies mainly based on chromatin conformation techniques have blurred the distinction between enhancers and promoters, as a number of promoters can also act as enhancers for the long-range regulation of distant genes and also engage in interactions with other promoters [38]. Long-range, inter-nucleosomal contacts controlled by the N-terminal tails of histones are essential and possibly rate-limiting for the cooperation between enhancers and promoters [39].
With the advent of technologies for analysis of protein-DNA interaction and of chromosomal conformation, genome wide landscapes of regulatory regions have been unraveled and the impact of the chromosomal architecture on gene expression and disease revealed [40,41,42]. This allowed the identification of a novel category of clustered enhancers dubbed super-enhancers, with an essential role in cell-specific gene expression and identity (Figure 1) [43,44,45,46,47,48,49]. Genes regulated by super-enhancers are typically expressed at higher levels when compared to enhancer-controlled genes. Importantly, super-enhancers are found in transient biomolecular condensates, which also include gene promoters, transcription factors, coactivators, and RNA Pol II, whose role is to increase the local concentration of these complexes around defined gene regions looped into them and to boost transcription [50,51,52]. The formation of phase-separated droplet compartments improves transcription fidelity, which is essential to maintain cellular phenotype [50,51,52]. On the other hand, super-enhancers are much more susceptible to changes in levels of bound transcription factors or coactivators, which further underlines the importance of local cooperative interactions for their transcriptional output [44,47,50]. Super-enhancers typically span tens of thousands of base pairs and are densely bound by lineage-specific transcription factors, by members of the mediator multi-subunit complex, and by BRD4 which engage in short-term lived interactions via intrinsically disordered regions [51]. The transcription factors Yes-associated protein (YAP) and transcriptional coactivator with PDZ-binding motif (TAZ) are important BRD4-interacting binders at super-enhancers which stimulate the expression of numerous genes involved in cell proliferation [53]. The P-TEFb complex is also localized at super-enhancers [54,55]. Cyclin-dependent kinase 7 (CDK7) belongs to the transcription factor II H complex and controls RNA Pol II activity by phosphorylating its C-terminal tail, thereby acting as a master regulator of super-enhancer activity [56]. An essential role of CDK7 in controlling super-enhancer-driven oncogenes in different tumor types has been evidenced using various inhibitors [57,58,59,60,61]. Cyclin-dependent kinase 12 (CDK12) and cyclin-dependent kinase 13 (CDK13) regulate transcription elongation and the expression of super-enhancer-dependent transcription factor genes [62]. Super-enhancers are usually flanked by boundary elements representing binding sites for CTCF that prevent expression of neighboring genes [24]. Recent progress made in three-dimensional chromatin capture techniques led to the definition of topologically associating domains (TADs) which specify chromatin compartments delimitated by distinctive elements [63]. TADs are dynamic structures formed by the cohesin complex and flanked by CTCF [63,64]. They are found in chromatin regions with very high levels of H3K27 acetylation and H3K4 monomethylation. Strong TAD boundaries are associated with high CTCF levels and frequently insulate super-enhancers [65]. Non-coding eRNAs facilitating the interactions with promoter regions to regulate downstream gene expression [37] and that may influence the tissue- and cell-specific activity of super-enhancers have been identified [66,67]. Algorithms to identify super-enhancers such as ROSE and archiving databases such as dbCORC, and SEdb are now available [45,68,69]. Recently, a sub-classification between non-hierarchical and hierarchical hub and non-hub super-enhancers has been proposed, the latter being more frequently associated with disease risks [70].
2.2. Non-Coding Cancer Driver Mutations
With the recent advances in genomic sequencing, more and more small-scale driver events such as point mutations and small insertions have been identified (Figure 2). One of the first examples is the discovery that about 5% of T cell acute lymphoblastic leukemia cases harbor small DNA insertions that create novel binding sites for recruitment of myeloblastosis (MYB) transcription factors and of the histone acetyltransferase CBP upstream of the T-cell acute lymphocytic leukemia protein 1 (TAL1) oncogene, thus creating a strong tumor-driving super-enhancer [71]. Another example is the finding of point mutations in the promoter of the telomerase reverse transcriptase (TERT) gene that create novel transcription-factor binding sites. These mutations were initially found in melanoma but later also in several other tumor types [72,73,74]. The TERT gene is usually silenced in differentiated cells and the identified promoter mutations lead to increased expression and telomerase reactivation and eventually to uncontrolled cell proliferation.
Non-coding driver alterations that involve large genomic rearrangements such as chromosomal translocations, focal amplifications, deletions, and viral insertions have been described for a long time in cancer and the recent advances made in genome-wide analyses and chromatin conformation techniques have led to a better understanding of these events (Figure 2). These changes may for instance position an oncogene under the control of a strong promoter or enhancer or lead to silencing of a tumor suppressor gene [44,75,76]. Also, changes affecting TAD boundary regions leading to the formation of new chromatin loops and dysregulated gene transcription have been described [77]. Aberrant enhancer and super-enhancer activity plays a role in different tumor types. Super-enhancer regions and strong TAD boundaries are often co-duplicated in tumors [65]. Also, a pan-cancer analysis of somatic copy number alterations in non-coding genomic regions led to the identification of six super-enhancers controlling the expression of four genes in different tumors [78]. Two focally amplified super-enhancers are responsible for c-Myc overexpression in lung and endometrial tumors [78]. Other studies report on super-enhancers aberrantly active in ependymomas [79] or rearranged in breast cancer [80].
3. Dysregulated Transcription Control in Prostate Cancer
Dysregulation gene expression is observed both in early and late-stage prostate cancer. Important examples are detailed below and summarized in Figure 2.
3.1. Early Events
Inherited genetic markers account for more than half of prostate cancer risk factors and include coding and non-coding variants [13,87]. In total, 50–100 single nucleotide polymorphisms (SNPs) have been linked to prostate cancer development [88,89] and validation studies indicate that many of them have a regulatory function and control gene expression [88]. Another study used chromatin conformation analysis to identify prostate cancer risk-associated SNPs and demonstrated the role of CTCF-binding motifs and three-dimensional chromatin folding in preventing enhancer function from spreading towards neighboring gene regions [18]. Importantly, comparison of prostate cancer and normal samples reveals that AR binding is redistributed in tumors compared to healthy tissue samples, leading to important transcriptome changes [90]. In addition, a colocalization of FOXA1 and homeobox protein B13 (HOXB13) at sites with elevated AR binding in tumors is observed. Large genetic changes such as gene amplifications and deletions are also frequently reported in early tumors [5,91] and have been recently reviewed [6,92].
3.1.1. Regulatory SNPs
A genome wide association study (GWAS) was used to characterize 77 prostate cancer risk loci and find functional SNPs [93]. Many of these SNPs localize at putative enhancers with high H3K27 acetylation levels. Further analysis for functionality revealed that in several cases the binding of transcription factors such as the AR, FOXA1, and NK3 homeobox 1 (NKX3-1) was affected by the sequence variations [93].
The SNP rs10993994 leads to reduced expression of the gene coding for prostate secretory protein 94 (PSP94) and represents a causal variant for prostate cancer risk [94]. This was linked to differential recognition of the SNP by cAMP response element-binding protein (CREB) [95]. PSP94 is a major prostate secretory protein which interacts with cysteine-rich secretory proteins [96] and is deemed to be a tumor suppressor due to its role in apoptosis [97].
Multiple variants were found in an enhancer region that loops to the Sry-related HMG box-containing 9 (SOX9) gene [98]. Two SNPs, rs8072254 and rs1859961, that affect binding by the AR or by FOXA1 and activating protein-1 (AP-1), respectively, have been identified in this region.
Allele-specific enhancer activity was also shown for the prostate cancer risk SNPs rs2659051, rs10936845, rs9925556, rs6057110, and rs2742624 [99]. The impact of the polymorphisms on recognition by the AR, FOXA1, HOXB13 and GATA-binding factor 2 (GATA2), and on H3K27 acetylation was described.
A variant that may predispose to prostate cancer was reported at 7p14.3 [100]. It is modulated by the AR and C/EBP and correlates with mutations in the gene encoding SPOP.
Several variants of the 8q24 region linked to variable increases of prostate cancer risk, depending on the ethnic origin, have been reported [101]. This region harbors a functional enhancer and may alter c-Myc gene expression, which promotes tumor growth [84,85,86,88]. Indeed, long-range interacting loops were identified between the c-Myc region and a functional enhancer located in this region in prostate cancer cell lines by using chromosome conformation capture techniques [84,85,86].
Several SNPs in the 7p15.2 locus are correlated with increased prostate cancer susceptibility and this was experimentally tested by deleting the region. This led to the identification of a repressive long-range loop spanning over 800 kilobase-pairs that controls HOXA13 expression [102].
The SNP rs339331 identified in intron 4 of the regulatory factor X6 (RFX6) gene enhances binding of the homeobox family member HOXB13, thus leading to transcription upregulation [103]. Interestingly, HOXB13 is itself linked to hereditary prostate cancer, due to a number of coding mutations [104]. Recent data show that HOXB13 forms a heterodimer with the AR V7 splice variant, which is associated with therapy resistance to drive specific gene expression programs [105].
The SNP rs7463708 is responsible for elevated binding of ONECUT2, an AR-interacting transcription factor, to an enhancer region governing expression of the non-coding RNA prostate cancer-associated transcript 1 (PCAT1) [106]. Importantly, ONECUT2 is a master regulator of AR signaling and a survival factor in metastatic CRPC [107]. In addition, preclinical models show that PCAT1 overexpression is able to stimulate cancer growth.
The SNP rs11672691 is linked to prostate cancer predisposition and aggressiveness. It is located in an intron of the non-coding RNA prostate cancer-associated transcript 19 (PCAT19) which possesses enhancer-like features. It creates a novel binding site for the homeobox family member HOXA2, leading to increased expression of PCAT19 and of the cell adhesion molecule carcinoembryonic antigen-related cell adhesion molecule 21 (CEACAM21) gene [108]. HOXA2 is overexpressed in prostate cancer and its silencing reduces tumor proliferation in vitro and in vivo. A separate analysis of the same SNP region indicates that it is associated with the expression of different PCAT19 isoforms, depending on the exact DNA sequence and recognition by the NKX3-1 and YY1 transcription factors [109]. Both a promoter and an enhancer function are reported for this risk SNP region [109].
SNPs associated with CTCF sites are linked to prostate cancer risk [110]. These sites are involved in long-range chromatin loops and their deletion dramatically affects the expression of genes located between them.
The expanding use of GWAS and ongoing discovery of novel SNPs thanks to deeper sequencing of tumor DNA increase the need for rapid functional assessments. Genome-editing technologies such as clustered regularly-interspaced short palindromic repeats with Cas9 nuclease (CRISPR-Cas9) have greatly helped to dissect the impact of SNPs on different diseases, including cancer [111]. Also, translation of these findings into the clinic for diagnostic and potential therapeutic use will necessitate dedicated technologies.
3.1.2. ERG Translocation
Analysis of primary prostate cancer samples shows that translocation events leading to fusions of an ETS family member to an AR-driven promoter are found in about half of the patients [5,91,112]. A recent comprehensive study of chromatin modification and transcription factor binding allowed to classify primary prostate cancer with ETS-related gene (ERG) translocation into clusters characterized by high or low ERG expression or by a NEPC-like profile [91]. The most frequent translocation positions the ERG coding region downstream of the androgen-dependent transmembrane protease serine 2 (TMPRSS2) gene promoter due to the deletion of an intervening region and conducts to increased expression of the ERG transcription factor [113]. Moreover, elevated ERG expression itself is associated with large changes in chromatin organization [114]. It leads to recruitment of transcription factors with an essential role in prostate function and to transcription from novel cis-regulatory elements [113,115]. This is linked to the appearance of attending super-enhancers with high H3K27 acetylation levels, including one located in the fused TMPRSS2 promoter and potentially responsible for ERG overexpression [113]. ERG and AR-associated long-range chromatin loops leading to coordinated regulation of downstream target genes have been described [116]. Another report shows that ERG interacts with BRD4 and colocalizes at numerous target genes that are essential for cell proliferation and invasion [117]. A recently identified downstream target of ERG is CBP/p300-interacting transactivator 2 (CITED2), a molecular chaperone that promotes the translocation of p300 and protein arginine N-methyltransferase 5 (PRMT5) and the formation of a multimeric complex with nucleolin which stimulates cell migration, ultimately leading to prostate cancer metastasis [118].
3.1.3. Epithelial Splicing Regulating Protein 1 (ESRP1) Gene Duplication
A comprehensive study in early-onset prostate cancer patients combining whole genome sequencing, epigenetic marks, and transcriptome analyses allowed the identification of recurrent duplications of the gene encoding ESRP1 [119]. These duplications lead to elevated ESRP1 expression and correlate with disease aggressiveness. This finding additionally underlines the role of aberrant splicing in prostate cancer [120].
3.1.4. Phosphatase and Tensin Homolog (PTEN) Inactivation
PTEN deletion is observed in about 20% of primary prostate cancer samples beside DNA methylation leading to decreased expression [121] and mutations that inactivate the protein [122]. This occurs via several mechanisms including deletion at 10q23 [123], ultimately suppressing an essential brake for phosphoinositide 3-kinase (PI3K) signaling and representing a key driver in proliferation as outlined in many reports [124,125].
3.2. Advanced Prostate Cancer
Several large genomic alterations including AR gene amplification and PTEN loss which are already seen in primary tumors are observed at a higher frequency in metastatic samples [3,4,6]. Concerning non-coding regions, a search for focal amplifications in different tumor types, including prostate cancer, allowed the identification of super-enhancers and their putatively regulated target genes [46]. The role of the histone variant H2A.Z in activating novel enhancers exemplifies the impact of enhancer gain on disease outcome worsening [126]. Details on key genetic events directly affecting the transcription of genes with an essential role in prostate cancer are outlined below.
3.2.1. Binding of BRD4 and Interacting Proteins at Gene Regulatory Elements
BRD4 is a global reader of the activating histone acetylation mark [127,128]. It binds mainly to enhancers and super-enhancers of numerous genes involved in cell proliferation and is involved in tumor cell transcriptional addiction [49,53,129]. A preferential binding of BRD4 to many SNPs located in enhancers and associated with prostate cancer risk has been reported [129]. Further refinement is achieved by incorporating the binding profiles of mediator complex proteins and H3K27 acetylation marks, which are all characteristic features of super-enhancers [129]. Importantly, BRD4 and other bromodomain and extra-terminal (BET) proteins regulate AR signaling at different levels. They control AR expression by binding to acetylated chromatin regions found at different locations in the AR gene body [130]. In addition BRD4 forms a complex with the AR and both proteins colocalize to regulatory regions of several androgen target genes, including the one coding for prostate-specific antigen (PSA) [131]. Interestingly, BRD4 binding is more powerful than AR binding to super-enhancers for identification of risk loci linked to prostate cancer [129]. BRD4 associates with the P-TEFb complex which releases paused RNA Pol II [132]. The P-TEFb complex contains CDK9 which phosphorylates the AR [22] and can be activated by androgen-regulated eRNAs [133], thus further sustaining signaling. In addition, BRD4 also interacts with ERG to control common genes upregulated in CRPC [117]. Another protein that binds to BRD4 and to the AR is YAP, a transcription factor found at super-enhancers [53,134]. YAP is involved in the transition to androgen-independent, AR-mediated transcription ultimately leading to castration resistance [135]. Another important downstream target of BRD4 is c-Myc, an oncogene which is amplified in about 30% of late-stage prostate tumors. Importantly, the expression of AR and c-Myc correlates during progression to metastatic CRPC and c-Myc regulates AR expression [136], suggesting an essential role for both factors [137]. Finally, the regulatory role of BET proteins for an enhancer that controls glucocorticoid receptor expression, which is de-repressed in advanced tumors, has been reported [138].
3.2.2. Acquired AR Enhancer and Androgen-Dependent Neo-Enhancers
Amplification and overexpression of the AR gene are by far the most frequent resistance mechanisms observed in prostate cancer patients treated with drugs that suppress androgen signaling. This allows the restoration of the AR pathway despite castrate levels of circulating male hormones. Recently, a genomic rearrangement leading to tandem duplication of an intergenic enhancer element located 600–700 kilobase-pairs upstream of the AR gene has been identified in a large subset of metastatic CRPC patients [81,82,83]. Amplification of this enhancer often parallels that of the AR gene and both changes concur to maintain high androgen signaling in advanced tumors. The functionality of this AR enhancer region is supported by the facts that it is looping to the AR gene and located in an open, highly acetylated chromatin environment in CRPC but not in localized tumor samples, and experimentally by silencing and knock-in experiments in a prostate cancer cell line [83]. Another study shows that nucleosomes containing acetylated H2A.Z are incorporated at enhancers associated with AR activity and contribute to the formation of neo-enhancers in prostate cancer [126]. These neo-enhancers have typical characteristics of active enhancers including H3K27 acetylation and increased eRNA transcription [126].
Additional research should provide further information on how enhancers around the AR gene sustain constant, high AR expression and on the impact of neo-enhancers on downstream androgen signaling. Technologies that capture the looping of chromatin and show enhancer-promoter association will pave the way for new insights in this area and possibly also lead to the identification of novel resistance mechanisms.
3.2.3. Nuclear Enriched Abundant Transcript 1 (NEAT1) and FOXA1 Promoter Mutations
Analysis of whole genome sequencing data allowed the identification of non-coding driver mutations in metastatic prostate cancer [4]. Mutations in the gene for the long non-coding RNA NEAT1 were significantly enriched in metastatic tumors treated by androgen deprivation therapy, compared to primary tumors. Mutations were also found in the promoter of the FOXA1 gene which codes for a transcription factor that modulates AR binding locations [4].
3.2.4. Reprogramming to Neuroendocrine Phenotype
The advent and earlier use of effective AR-targeting agents has led to the increased appearance of resistance mechanisms where prostate tumor cells acquire novel neuroendocrine features and become independent of the AR signaling axis [139]. This is accompanied by a loss of PSMA expression [140], which is often used for detection and staging of prostate cancer [141]. The pioneer factor FOXA1 is frequently expressed in NEPC and may represent a useful progression marker [142]. Expression of SAM pointed domain-containing ETS transcription factor (SPDEF) belonging to the ETS transcription factor gene family is lost upon androgen deprivation therapy and may contribute to the development of NEPC [143]. NEPC is highly aggressive and has undergone extensive reprogramming involving epigenetic players and long, non-coding RNAs [144]. Increased expression of the H3K27 histone methylase enhancer of zeste homolog (EZH2) gene is a common feature of advanced prostate cancer and NEPC and is observed both in patients and in tumor models [145,146]. EZH2 mutations leading to silencing of TADs and tumor suppressor genes located within these domains due to changes in H3K27 trimethylation have recently been described [147]. Importantly, androgen deprivation leads to increased EZH2 activity and ultimately promotes angiogenesis, which is elevated in NEPC [148]. The transcription factor N-Myc is overexpressed in NEPC and this is sufficient to induce transformation and androgen independence [149]. Experimentally, high N-Myc levels lead to shift of different prostate cancer models and of PTEN-negative mouse organoids towards androgen independence by strengthening the EZH2 and AKT signaling axis [149]. Essential roles of the repressor element 1-silencing transcription factor (REST) and its downstream repressed target, the long non-coding RNA HOTAIR, have recently been evidenced [150,151,152]. REST expression is controlled by the RNA splicing factor serine/arginine repetitive matrix 4 (SRRM4), a main driver of NEPC progression [153]. Importantly, SRRM4 expression in castration-resistant prostate cancer correlates with poor patient survival [154]. A comprehensive analysis of the changes in long non-coding RNA expression found in NEPC that take place upon androgen deprivation treatment revealed several candidates involved in this process [155]. As whole-transcriptome data from NEPC patient samples are now available [2,11,12,142,153,155,156] a detailed understanding of the mechanisms underlying dysregulated gene expression during this disease stage should be unveiled soon.
4. Targeting Dysregulated Gene Transcription
Androgen signaling is essential for maintenance of normal prostate development and function. It is also a main driver of prostate cancer development due to reprogramming leading to a redistribution of AR binding sites and large transcriptome changes [90,157]. This is accompanied by a relocalization of FOXA1 and HOXB13 and the formation of novel genomic AR subcomplexes [19]. Importantly, transduction with these two pioneer transcription factors is sufficient for reprogramming normal prostate epithelium into transformed tumor cells [90]. The AR pathway has been successfully addressed in the treatment of early and late-stage prostate cancer for many years, originally with surgical or chemical castration and later with AR antagonists and androgen synthesis inhibitors (Figure 1) [158,159,160,161,162]. The recent identification of an enhancer hijacked by the AR gene to upregulate expression adds a novel facet to the strategies used by tumors to sustain hormone action and overcome androgen deprivation. Previously described mechanisms leading to restoration of androgen signaling include epigenetic AR gene activation linked to DNA demethylation and histone modifications [163,164,165], AR mutations [166], generation of AR splice variants [167], and increased coactivator function [168]. This vindicates the identification and development of novel AR-targeting compounds with improved properties such as the next-generation AR antagonists enzalutamide, apalutamide and darolutamide [158,159,161,169,170,171,172,173]. A dual inhibitor of AR function and CYP17A1 lyase activity is currently in the clinical dose escalation phase [174]. Less advanced compounds include AR degraders [175,176,177] and agents addressing AR splice variants deprived of the ligand-binding domain [178]. Attempts to reduce AR levels using a specific antisense nucleotide have been evaluated clinically but with little success [159,179]. In another approach, AR transcript and protein levels could be reduced by targeting deubiquitinases, and first in vitro efficacy data based on this approach have been reported [180].
Translocations leading to ERG overexpression are observed in a large group of primary prostate cancer cases and are maintained in CRPC. Efforts to identify compounds that interfere with ERG function have been reported. The described inhibitors act indirectly and show anti-tumor efficacy in different prostate cancer models [181,182].
BRD4 is an essential component of super-enhancers which binds to acetylated histones and possibly to other acetylated proteins via its two bromodomains. Inhibitors targeting the bromodomains of BRD4 and related BET proteins show efficacy in preclinical prostate cancer models (Figure 1) [130,161,183]. Several compounds were advanced to the clinic and a few trials focusing on prostate cancer patients are currently ongoing [184].
The P-TEFb complex contains CDK9, which has been successfully targeted to inhibit preclinical prostate cancer models in vitro and in vivo [24]. First compounds that blockCDK9 have entered the clinic, but in many cases, lack of specificity leading to off-target side-effects has limited their application [22]. Highly selective CDK9 inhibitors have recently been described and may have a better therapeutic window [185,186].
CDK7 is another kinase involved in super-enhancer function. The availability of a potent and specific inhibitor has much helped in understanding the impact of super-enhancers in different tumor types [44], however, no data for prostate cancer are currently available.
The related CBP/p300 histone acetyltransferases are essential mediators of different acetylation marks including H3K27 acetylation [187] and also important AR coactivators linked to prostate cancer progression [188,189]. Selective inhibitors addressing either the bromodomain or the enzymatic activity have been described recently and anti-proliferative efficacy reported in vitro and in vivo in several preclinical prostate cancer models (Figure 1) [190,191,192,193]. The CBP/p300 bromodomain inhibitor CCS1477 has recently entered a clinical phase I study focusing on prostate cancer [192].
The involvement of EZH2 as a master epigenetic player in late-stage tumors and in NEPC is documented by numerous studies [145,148]. Importantly, EZH2 directly stimulates AR expression, independently of its methyltransferase activity, by binding to the AR gene promoter [194]. EZH2 inhibition leads to apoptosis when combined with chemotherapeutic agents [195] and is efficacious in docetaxel-resistant tumor cells [196]. Significant in vivo efficacy was reported when combining an EZH2 inhibitor with enzalutamide for treatment of a prostate cancer xenograft model [194]. Several potent inhibitors targeting EZH2 have been identified in the last years and tested in numerous preclinical tumor models [197]. A clinical study combining an EZH2 inhibitor with the AR antagonist enzalutamide or the CYP17A1 inhibitor abiraterone acetate has very recently been initiated in metastatic CRPC patients [198]. EZH2 interacts with the embryonic ectoderm development (EED) and SUZ12 proteins to form the polycomb repressive complex 2 (PRC2) [199]. Several inhibitors of EED that bind to the H3K27me3 pocket and block PRC2 methyltransferase activity have been reported [200,201]. One of them, MAK683, is being evaluated in lymphoma and in solid tumors, including prostate cancer.
N-Myc and the Akt signaling pathways are important players in NEPC [149]. N-Myc is stabilized by the mitotic kinase Aurora A and a specific inhibitor was evaluated in NEPC, but the primary endpoint was not met despite the fact that some patients responded very well to the treatment [156].
An overview of the targets and inhibitors described here is given in Table 1.
5. Conclusions and Perspectives
Transcriptional dysregulation leading to the appropriation of an oncogenic gene expression program is an essential event responsible for the acquisition of cancer cell hallmarks such as proliferation, replicative immortality, and metastasis [76]. Indeed, oncogenic drivers are often regulators of transcription as exemplified by the AR and ERG in prostate cancer. Downstream effectors are frequently also transcriptional regulators, like for instance the Myc family members [202,203]. More recently, the essential role of BRD4 in multiple tumor types has been evidenced [127,128,204]. An increasing number of SNPs and small mutations are being found in non-coding regions of prostate tumors and are critical for the dysregulation of transcriptional programs in various ways. Finally, oncogenic, de novo acquired super-enhancers have recently been defined. They play critical roles in cancer as platforms for the recruitment of transcription factors and epigenetic players upstream of signaling pathways with an essential oncogenic role and are sensitive to perturbation [43,44,45,50,75,76]. As these mutations do not affect protein sequences, they will not be recognized by the immune system and therefore escape immune checkpoint therapies.
Targeting transcriptional regulators may often be more challenging in comparison to addressing proteins involved in signaling networks due to the absence of highly druggable pockets, multiple protein interactions, and nuclear localization. Concerning histone-modifying enzymes and readers, achieving sufficient selectivity is sometimes problematic, but ongoing efforts to identify chemical probes for the main epigenetic players represent a first important step [205]. This should however not deter endeavors that address early, master oncogenic drivers as this approach has the potential to better tackle tumor heterogeneity and may delay the emergence of resistance mutations, due to the multiple biological functions of these factors. On the other hand, the pleiotropic effects one might observe following blockade of such essential regulators may limit the therapeutic window so that a bespoke design of studies, for instance adaptive clinical trials, will possibly increase the chances of success [92,206,207].
Author Contributions
Funding
The authors are funded by Bayer AG.
Acknowledgments
We thank all our colleagues involved in prostate cancer projects for numerous helpful discussions and suggestions.
Conflicts of Interest
The authors are employees and/or own shares of Bayer AG.
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angeles, A.K.; Bauer, S.; Ratz, L.; Klauck, S.M.; Sultmann, H. Genome-based classification and therapy of prostate cancer. Diagnostics 2018, 8, 62. [Google Scholar] [CrossRef] [PubMed]
- Armenia, J.; Wankowicz, S.A.M.; Liu, D.; Gao, J.; Kundra, R.; Reznik, E.; Chatila, W.K.; Chakravarty, D.; Han, G.C.; Coleman, I.; et al. The long tail of oncogenic drivers in prostate cancer. Nat. Genet. 2018, 50, 645–651. [Google Scholar] [CrossRef] [PubMed]
- Wedge, D.C.; Gundem, G.; Mitchell, T.; Woodcock, D.J.; Martincorena, I.; Ghori, M.; Zamora, J.; Butler, A.; Whitaker, H.; Kote-Jarai, Z.; et al. Sequencing of prostate cancers identifies new cancer genes, routes of progression and drug targets. Nat. Genet. 2018, 50, 682–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Cancer Genome Atlas Research Network. The molecular taxonomy of primary prostate cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef] [PubMed]
- Spratt, D.E.; Zumsteg, Z.S.; Feng, F.Y.; Tomlins, S.A. Translational and clinical implications of the genetic landscape of prostate cancer. Nat. Rev. Clin. Oncol. 2016, 13, 597–610. [Google Scholar] [CrossRef]
- Linch, M.; Goh, G.; Hiley, C.; Shanmugabavan, Y.; McGranahan, N.; Rowan, A.; Wong, Y.N.S.; King, H.; Furness, A.; Freeman, A.; et al. Intratumoural evolutionary landscape of high-risk prostate cancer: The PROGENY study of genomic and immune parameters. Ann. Oncol. 2017, 28, 2472–2480. [Google Scholar] [CrossRef]
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 162, 1215–1228. [Google Scholar] [CrossRef]
- Fraser, M.; Sabelnykova, V.Y.; Yamaguchi, T.N.; Heisler, L.E.; Livingstone, J.; Huang, V.; Shiah, Y.J.; Yousif, F.; Lin, X.; Masella, A.P.; et al. Genomic hallmarks of localized, non-indolent prostate cancer. Nature 2017, 541, 359–364. [Google Scholar] [CrossRef]
- Grasso, C.S.; Wu, Y.M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, R.; Huang, J.; Alumkal, J.J.; Zhang, L.; Feng, F.Y.; Thomas, G.V.; Weinstein, A.S.; Friedl, V.; Zhang, C.; Witte, O.N.; et al. Clinical and genomic characterization of treatment-emergent small-cell neuroendocrine prostate cancer: A multi-institutional prospective study. J. Clin. Oncol. 2018, 36, 2492–2503. [Google Scholar] [CrossRef] [PubMed]
- Davies, A.H.; Beltran, H.; Zoubeidi, A. Cellular plasticity and the neuroendocrine phenotype in prostate cancer. Nat. Rev. Urol. 2018, 15, 271–286. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.H.; Petrovics, G.; Srivastava, S. Prostate cancer genomics: Recent advances and the prevailing underrepresentation from racial and ethnic minorities. Int. J. Mol. Sci. 2018, 19, 1255. [Google Scholar] [CrossRef] [PubMed]
- Bhagirath, D.; Yang, T.L.; Dahiya, R.; Saini, S. MicroRNAs as regulators of prostate cancer metastasis. Adv. Exp. Med. Biol. 2018, 1095, 83–100. [Google Scholar] [PubMed]
- Das, R.; Feng, F.Y.; Selth, L.A. Long non-coding RNAs in prostate cancer: Biological and clinical implications. Mol. Cell. Endocrinol. 2019, 480, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.; Lajoie, B.R.; Jain, G.; Dekker, J. The long-range interaction landscape of gene promoters. Nature 2012, 489, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Harmston, N.; Lenhard, B. Chromatin and epigenetic features of long-range gene regulation. Nucleic Acids Res. 2013, 41, 7185–7199. [Google Scholar] [CrossRef]
- Matharu, N.; Ahituv, N. Minor loops in major folds: Enhancer-promoter looping, chromatin restructuring, and their association with transcriptional regulation and disease. PLoS Genet. 2015, 11, e1005640. [Google Scholar] [CrossRef]
- Stelloo, S.; Bergman, A.M.; Zwart, W. Androgen receptor enhancer usage and the chromatin regulatory landscape in human prostate cancers. Endocr.-Relat. Cancer 2019. [Google Scholar] [CrossRef]
- Hantsche, M.; Cramer, P. The structural basis of transcription: 10 years after the Nobel prize in chemistry. Angew. Chem. 2016, 55, 15972–15981. [Google Scholar] [CrossRef]
- Louder, R.K.; He, Y.; Lopez-Blanco, J.R.; Fang, J.; Chacon, P.; Nogales, E. Structure of promoter-bound TFIID and model of human pre-initiation complex assembly. Nature 2016, 531, 604–609. [Google Scholar] [CrossRef] [PubMed]
- Rahaman, M.H.; Kumarasiri, M.; Mekonnen, L.B.; Yu, M.; Diab, S.; Albrecht, H.; Milne, R.W.; Wang, S. Targeting CDK9: A promising therapeutic opportunity in prostate cancer. Endocr.-Relat. Cancer 2016, 23, T211–T226. [Google Scholar] [CrossRef] [PubMed]
- Weintraub, A.S.; Li, C.H.; Zamudio, A.V.; Sigova, A.A.; Hannett, N.M.; Day, D.S.; Abraham, B.J.; Cohen, M.A.; Nabet, B.; Buckley, D.L.; et al. YY1 Is a structural regulator of enhancer-promoter loops. Cell 2017, 171, 1573–1588.e28. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.Y. The structural and functional roles of CTCF in the regulation of cell type-specific and human disease-associated super-enhancers. Genes Genom. 2019, 41, 257–265. [Google Scholar] [CrossRef]
- Meng, H.; Bartholomew, B. Emerging roles of transcriptional enhancers in chromatin looping and promoter-proximal pausing of RNA polymerase II. J. Biol. Chem. 2018, 293, 13786–13794. [Google Scholar] [CrossRef] [Green Version]
- Struhl, K. Transcriptional activation: Mediator can act after preinitiation complex formation. Mol. Cell 2005, 17, 752–754. [Google Scholar] [CrossRef]
- Catarino, R.R.; Stark, A. Assessing sufficiency and necessity of enhancer activities for gene expression and the mechanisms of transcription activation. Genes Dev. 2018, 32, 202–223. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.; Wang, Y.; Li, G. Dynamics of histone variant H3.3 and its coregulation with H2A.Z at enhancers and promoters. Nucleus 2014, 5, 21–27. [Google Scholar] [CrossRef] [Green Version]
- Ramji, D.P.; Foka, P. CCAAT/enhancer-binding proteins: Structure, function and regulation. Biochem. J. 2002, 365, 561–575. [Google Scholar] [CrossRef]
- Kasper, L.H.; Qu, C.; Obenauer, J.C.; McGoldrick, D.J.; Brindle, P.K. Genome-wide and single-cell analyses reveal a context dependent relationship between CBP recruitment and gene expression. Nucleic Acids Res. 2014, 42, 11363–11382. [Google Scholar] [CrossRef]
- Wood, K.; Tellier, M.; Murphy, S. DOT1L and H3K79 methylation in transcription and genomic stability. Biomolecules 2018, 8, 11. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, S.; Hossan, T.; Alawi, M.; Najafova, Z.; Indenbirken, D.; Bedi, U.; Taipaleenmaki, H.; Ben-Batalla, I.; Scheller, M.; Loges, S.; et al. Bromodomain protein BRD4 is required for estrogen receptor-dependent enhancer activation and gene transcription. Cell Rep. 2014, 8, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, D.; Kannan, A.; Kern, M.; Moreno, M.A.; Vural, E.; Stack, B., Jr.; Suen, J.Y.; Tackett, A.J.; Gao, L. Disruption of BRD4 at H3K27Ac-enriched enhancer region correlates with decreased c-Myc expression in Merkel cell carcinoma. Epigenetics 2015, 10, 460–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tippens, N.D.; Vihervaara, A.; Lis, J.T. Enhancer transcription: What, where, when, and why? Genes Dev. 2018, 32, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Yoneda, M.; Higashi, M.; Ohkuma, Y.; Ito, T. Enhancer function regulated by combinations of transcription factors and cofactors. Genes Cells Devoted Mol. Cell. Mech. 2018, 23, 808–821. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Li, Y.I.; Pritchard, J.K. Trans Effects on Gene Expression Can Drive Omnigenic Inheritance. Cell 2019, 177, 1022.e6–1034.e6. [Google Scholar] [CrossRef]
- Lam, M.T.; Li, W.; Rosenfeld, M.G.; Glass, C.K. Enhancer RNAs and regulated transcriptional programs. Trends Biochem. Sci. 2014, 39, 170–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medina-Rivera, A.; Santiago-Algarra, D.; Puthier, D.; Spicuglia, S. Widespread enhancer activity from core promoters. Trends Biochem. Sci. 2018, 43, 452–468. [Google Scholar] [CrossRef] [PubMed]
- Nizovtseva, E.V.; Todolli, S.; Olson, W.K.; Studitsky, V.M. Towards quantitative analysis of gene regulation by enhancers. Epigenomics 2017, 9, 1219–1231. [Google Scholar] [CrossRef] [PubMed]
- Taberlay, P.C.; Achinger-Kawecka, J.; Lun, A.T.; Buske, F.A.; Sabir, K.; Gould, C.M.; Zotenko, E.; Bert, S.A.; Giles, K.A.; Bauer, D.C.; et al. Three-dimensional disorganization of the cancer genome occurs coincident with long-range genetic and epigenetic alterations. Genome Res. 2016, 26, 719–731. [Google Scholar] [CrossRef] [Green Version]
- Jia, R.; Chai, P.; Zhang, H.; Fan, X. Novel insights into chromosomal conformations in cancer. Mol. Cancer 2017, 16, 173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krijger, P.H.; de Laat, W. Regulation of disease-associated gene expression in the 3D genome. Nat. Rev. Mol. Cell Biol. 2016, 17, 771–782. [Google Scholar] [CrossRef] [PubMed]
- Hnisz, D.; Schuijers, J.; Lin, C.Y.; Weintraub, A.S.; Abraham, B.J.; Lee, T.I.; Bradner, J.E.; Young, R.A. Convergence of developmental and oncogenic signaling pathways at transcriptional super-enhancers. Mol. Cell 2015, 58, 362–370. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; George, R.E. Super-enhancer-driven transcriptional dependencies in cancer. Trends Cancer 2017, 3, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Whyte, W.A.; Orlando, D.A.; Hnisz, D.; Abraham, B.J.; Lin, C.Y.; Kagey, M.H.; Rahl, P.B.; Lee, T.I.; Young, R.A. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 2013, 153, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Choi, P.S.; Francis, J.M.; Imielinski, M.; Watanabe, H.; Cherniack, A.D.; Meyerson, M. Identification of focally amplified lineage-specific super-enhancers in human epithelial cancers. Nat. Genet. 2016, 48, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Loven, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef]
- Hamdan, F.H.; Johnsen, S.A. Super enhancers—New analyses and perspectives on the low hanging fruit. Transcription 2018, 9, 123–130. [Google Scholar] [CrossRef]
- Gelato, K.A.; Schöckel, L.; Klingbeil, O.; Rückert, T.; Lesche, R.; Toedling, J.; Kalfon, E.; Héroult, M.; Lejeune, P.; Mönning, U.; et al. Super-enhancers define a proliferative PGC-1alpha-expressing melanoma subgroup sensitive to BET inhibition. Oncogene 2018, 37, 512–521. [Google Scholar] [CrossRef]
- Hnisz, D.; Shrinivas, K.; Young, R.A.; Chakraborty, A.K.; Sharp, P.A. A phase separation model for transcriptional control. Cell 2017, 169, 13–23. [Google Scholar] [CrossRef]
- Sabari, B.R.; Dall’Agnese, A.; Boija, A.; Klein, I.A.; Coffey, E.L.; Shrinivas, K.; Abraham, B.J.; Hannett, N.M.; Zamudio, A.V.; Manteiga, J.C.; et al. Coactivator condensation at super-enhancers links phase separation and gene control. Science 2018, 361, eaar3958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, W.K.; Spille, J.H.; Hecht, M.; Lee, C.; Li, C.; Grube, V.; Cisse, I.I. Mediator and RNA polymerase II clusters associate in transcription-dependent condensates. Science 2018, 361, 412–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanconato, F.; Battilana, G.; Forcato, M.; Filippi, L.; Azzolin, L.; Manfrin, A.; Quaranta, E.; Di Biagio, D.; Sigismondo, G.; Guzzardo, V.; et al. Transcriptional addiction in cancer cells is mediated by YAP/TAZ through BRD4. Nat. Med. 2018, 24, 1599–1610. [Google Scholar] [CrossRef] [PubMed]
- Di Micco, R.; Fontanals-Cirera, B.; Low, V.; Ntziachristos, P.; Yuen, S.K.; Lovell, C.D.; Dolgalev, I.; Yonekubo, Y.; Zhang, G.; Rusinova, E.; et al. Control of embryonic stem cell identity by BRD4-dependent transcriptional elongation of super-enhancer-associated pluripotency genes. Cell Rep. 2014, 9, 234–247. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, M.; Chen, L.F.; Chen, R. P-TEFb: Finding its ways to release promoter-proximally paused RNA polymerase II. Transcription 2018, 9, 88–94. [Google Scholar] [CrossRef]
- Minzel, W.; Venkatachalam, A.; Fink, A.; Hung, E.; Brachya, G.; Burstain, I.; Shaham, M.; Rivlin, A.; Omer, I.; Zinger, A.; et al. Small molecules co-targeting CKIalpha and the transcriptional kinases CDK7/9 control AML in preclinical models. Cell 2018, 175, 171.e25–185.e25. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowski, N.; Zhang, T.; Rahl, P.B.; Abraham, B.J.; Reddy, J.; Ficarro, S.B.; Dastur, A.; Amzallag, A.; Ramaswamy, S.; Tesar, B.; et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature 2014, 511, 616–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chipumuro, E.; Marco, E.; Christensen, C.L.; Kwiatkowski, N.; Zhang, T.; Hatheway, C.M.; Abraham, B.J.; Sharma, B.; Yeung, C.; Altabef, A.; et al. CDK7 inhibition suppresses super-enhancer-linked oncogenic transcription in MYCN-driven cancer. Cell 2014, 159, 1126–1139. [Google Scholar] [CrossRef] [PubMed]
- Christensen, C.L.; Kwiatkowski, N.; Abraham, B.J.; Carretero, J.; Al-Shahrour, F.; Zhang, T.; Chipumuro, E.; Herter-Sprie, G.S.; Akbay, E.A.; Altabef, A.; et al. Targeting transcriptional addictions in small cell lung cancer with a covalent CDK7 inhibitor. Cancer Cell 2014, 26, 909–922. [Google Scholar] [CrossRef]
- Eliades, P.; Abraham, B.J.; Ji, Z.; Miller, D.M.; Christensen, C.L.; Kwiatkowski, N.; Kumar, R.; Njauw, C.N.; Taylor, M.; Miao, B.; et al. High MITF expression is associated with super-enhancers and suppressed by CDK7 inhibition in melanoma. J. Investig. Dermatol. 2018, 138, 1582–1590. [Google Scholar] [CrossRef]
- Sharifnia, T.; Wawer, M.J.; Chen, T.; Huang, Q.Y.; Weir, B.A.; Sizemore, A.; Lawlor, M.A.; Goodale, A.; Cowley, G.S.; Vazquez, F.; et al. Small-molecule targeting of brachyury transcription factor addiction in chordoma. Nat. Med. 2019, 25, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Kwiatkowski, N.; Olson, C.M.; Dixon-Clarke, S.E.; Abraham, B.J.; Greifenberg, A.K.; Ficarro, S.B.; Elkins, J.M.; Liang, Y.; Hannett, N.M.; et al. Covalent targeting of remote cysteine residues to develop CDK12 and CDK13 inhibitors. Nat. Chem. Biol. 2016, 12, 876–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, A.S.; Cattoglio, C.; Darzacq, X.; Tjian, R. Recent evidence that TADs and chromatin loops are dynamic structures. Nucleus 2018, 9, 20–32. [Google Scholar] [CrossRef]
- Oomen, M.E.; Hansen, A.S.; Liu, Y.; Darzacq, X.; Dekker, J. CTCF sites display cell cycle-dependent dynamics in factor binding and nucleosome positioning. Genome Res. 2019, 29, 236–249. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Lazaris, C.; Sakellaropoulos, T.; Lozano, A.; Kambadur, P.; Ntziachristos, P.; Aifantis, I.; Tsirigos, A. Stratification of TAD boundaries reveals preferential insulation of super-enhancers by strong boundaries. Nat. Commun. 2018, 9, 542. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.Y.; Oh, S.; Yoo, K.H. Functional enhancers as master regulators of tissue-specific gene regulation and cancer development. Mol. Cells 2017, 40, 169–177. [Google Scholar] [PubMed]
- Kim, Y.J.; Xie, P.; Cao, L.; Zhang, M.Q.; Kim, T.H. Global transcriptional activity dynamics reveal functional enhancer RNAs. Genome Res. 2018, 28, 1799–1811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, M.; Chen, Y.; Yang, M.; Guo, A.; Xu, Y.; Xu, L.; Koeffler, H.P. dbCoRC: A database of core transcriptional regulatory circuitries modeled by H3K27ac ChIP-seq signals. Nucleic Acids Res. 2018, 46, D71–D77. [Google Scholar] [CrossRef]
- Jiang, Y.; Qian, F.; Bai, X.; Liu, Y.; Wang, Q.; Ai, B.; Han, X.; Shi, S.; Zhang, J.; Li, X.; et al. SEdb: A comprehensive human super-enhancer database. Nucleic Acids Res. 2019, 47, D235–D243. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Li, K.; Cai, W.; Liu, X.; Zhang, Y.; Orkin, S.H.; Xu, J.; Yuan, G.C. Dissecting super-enhancer hierarchy based on chromatin interactions. Nat. Commun. 2018, 9, 943. [Google Scholar] [CrossRef] [PubMed]
- Mansour, M.R.; Abraham, B.J.; Anders, L.; Berezovskaya, A.; Gutierrez, A.; Durbin, A.D.; Etchin, J.; Lawton, L.; Sallan, S.E.; Silverman, L.B.; et al. Oncogene regulation. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science 2014, 346, 1373–1377. [Google Scholar] [CrossRef] [PubMed]
- Heidenreich, B.; Kumar, R. Altered TERT promoter and other genomic regulatory elements: Occurrence and impact. Int. J. Cancer 2017, 141, 867–876. [Google Scholar] [CrossRef] [PubMed]
- Griewank, K.G.; Murali, R.; Puig-Butille, J.A.; Schilling, B.; Livingstone, E.; Potrony, M.; Carrera, C.; Schimming, T.; Moller, I.; Schwamborn, M.; et al. TERT promoter mutation status as an independent prognostic factor in cutaneous melanoma. J. Natl. Cancer Inst. 2014, 106, dju246. [Google Scholar] [CrossRef] [PubMed]
- Alzahrani, A.S.; Alsaadi, R.; Murugan, A.K.; Sadiq, B.B. TERT promoter mutations in thyroid cancer. Horm. Cancer 2016, 7, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.Y. Targeting super-enhancers for disease treatment and diagnosis. Mol. Cells 2018, 41, 506–514. [Google Scholar] [PubMed]
- Bradner, J.E.; Hnisz, D.; Young, R.A. Transcriptional addiction in cancer. Cell 2017, 168, 629–643. [Google Scholar] [CrossRef] [PubMed]
- Achinger-Kawecka, J.; Taberlay, P.C.; Clark, S.J. Alterations in three-dimensional organization of the cancer genome and epigenome. Cold Spring Harb. Symp. Quant. Biol. 2016, 81, 41–51. [Google Scholar] [CrossRef]
- Zhang, X.; Choi, P.S.; Francis, J.M.; Gao, G.F.; Campbell, J.D.; Ramachandran, A.; Mitsuishi, Y.; Ha, G.; Shih, J.; Vazquez, F.; et al. Somatic superenhancer duplications and hotspot mutations lead to oncogenic activation of the KLF5 transcription factor. Cancer Discov. 2018, 8, 108–125. [Google Scholar] [CrossRef]
- Mack, S.C.; Pajtler, K.W.; Chavez, L.; Okonechnikov, K.; Bertrand, K.C.; Wang, X.; Erkek, S.; Federation, A.; Song, A.; Lee, C.; et al. Therapeutic targeting of ependymoma as informed by oncogenic enhancer profiling. Nature 2018, 553, 101–105. [Google Scholar] [CrossRef]
- Glodzik, D.; Morganella, S.; Davies, H.; Simpson, P.T.; Li, Y.; Zou, X.; Diez-Perez, J.; Staaf, J.; Alexandrov, L.B.; Smid, M.; et al. A somatic-mutational process recurrently duplicates germline susceptibility loci and tissue-specific super-enhancers in breast cancers. Nat. Genet. 2017, 49, 341–348. [Google Scholar] [CrossRef]
- Viswanathan, S.R.; Ha, G.; Hoff, A.M.; Wala, J.A.; Carrot-Zhang, J.; Whelan, C.W.; Haradhvala, N.J.; Freeman, S.S.; Reed, S.C.; Rhoades, J.; et al. Structural alterations driving castration-resistant prostate cancer revealed by linked-read genome sequencing. Cell 2018, 174, 433.e19–447.e19. [Google Scholar] [CrossRef] [PubMed]
- Quigley, D.A.; Dang, H.X.; Zhao, S.G.; Lloyd, P.; Aggarwal, R.; Alumkal, J.J.; Foye, A.; Kothari, V.; Perry, M.D.; Bailey, A.M.; et al. Genomic hallmarks and structural variation in metastatic prostate cancer. Cell 2018, 175, 889. [Google Scholar] [CrossRef] [PubMed]
- Takeda, D.Y.; Spisak, S.; Seo, J.H.; Bell, C.; O’Connor, E.; Korthauer, K.; Ribli, D.; Csabai, I.; Solymosi, N.; Szallasi, Z.; et al. A somatically acquired enhancer of the androgen receptor is a noncoding driver in advanced prostate cancer. Cell 2018, 174, 422.e13–432.e13. [Google Scholar] [CrossRef] [PubMed]
- Ahmadiyeh, N.; Pomerantz, M.M.; Grisanzio, C.; Herman, P.; Jia, L.; Almendro, V.; He, H.H.; Brown, M.; Liu, X.S.; Davis, M.; et al. 8q24 prostate, breast, and colon cancer risk loci show tissue-specific long-range interaction with MYC. Proc. Natl. Acad. Sci. USA 2010, 107, 9742–9746. [Google Scholar] [CrossRef] [PubMed]
- Du, M.; Tillmans, L.; Gao, J.; Gao, P.; Yuan, T.; Dittmar, R.L.; Song, W.; Yang, Y.; Sahr, N.; Wang, T.; et al. Chromatin interactions and candidate genes at ten prostate cancer risk loci. Sci. Rep. 2016, 6, 23202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, M.; Kim, S.; Wang, K.; Farnham, P.J.; Coetzee, G.A.; Lu, W. 4C-seq revealed long-range interactions of a functional enhancer at the 8q24 prostate cancer risk locus. Sci. Rep. 2016, 6, 22462. [Google Scholar] [CrossRef] [Green Version]
- Lynch, H.T.; Kosoko-Lasaki, O.; Leslie, S.W.; Rendell, M.; Shaw, T.; Snyder, C.; D’Amico, A.V.; Buxbaum, S.; Isaacs, W.B.; Loeb, S.; et al. Screening for familial and hereditary prostate cancer. Int. J. Cancer 2016, 138, 2579–2591. [Google Scholar] [CrossRef]
- Nowinski, S.; Santaolalla, A.; O’Leary, B.; Loda, M.; Mirchandani, A.; Emberton, M.; Van Hemelrijck, M.; Grigoriadis, A. Systematic identification of functionally relevant risk alleles to stratify aggressive versus indolent prostate cancer. Oncotarget 2018, 9, 12812–12824. [Google Scholar] [CrossRef] [Green Version]
- Benafif, S.; Eeles, R. Genetic predisposition to prostate cancer. Br. Med. Bull. 2016, 120, 75–89. [Google Scholar] [CrossRef]
- Pomerantz, M.M.; Li, F.; Takeda, D.Y.; Lenci, R.; Chonkar, A.; Chabot, M.; Cejas, P.; Vazquez, F.; Cook, J.; Shivdasani, R.A.; et al. The androgen receptor cistrome is extensively reprogrammed in human prostate tumorigenesis. Nat. Genet. 2015, 47, 1346–1351. [Google Scholar] [CrossRef]
- Stelloo, S.; Nevedomskaya, E.; Kim, Y.; Schuurman, K.; Valle-Encinas, E.; Lobo, J.; Krijgsman, O.; Peeper, D.S.; Chang, S.L.; Feng, F.Y.; et al. Integrative epigenetic taxonomy of primary prostate cancer. Nat. Commun. 2018, 9, 4900. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhao, D.; Spring, D.J.; DePinho, R.A. Genetics and biology of prostate cancer. Genes Dev. 2018, 32, 1105–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hazelett, D.J.; Rhie, S.K.; Gaddis, M.; Yan, C.; Lakeland, D.L.; Coetzee, S.G.; Ellipse, G.-O.N.C.; Practical, C.; Henderson, B.E.; Noushmehr, H.; et al. Comprehensive functional annotation of 77 prostate cancer risk loci. PLoS Genet. 2014, 10, e1004102. [Google Scholar] [CrossRef] [PubMed]
- Chang, B.L.; Cramer, S.D.; Wiklund, F.; Isaacs, S.D.; Stevens, V.L.; Sun, J.; Smith, S.; Pruett, K.; Romero, L.M.; Wiley, K.E.; et al. Fine mapping association study and functional analysis implicate a SNP in MSMB at 10q11 as a causal variant for prostate cancer risk. Hum. Mol. Genet. 2009, 18, 1368–1375. [Google Scholar] [CrossRef] [PubMed]
- Lou, H.; Yeager, M.; Li, H.; Bosquet, J.G.; Hayes, R.B.; Orr, N.; Yu, K.; Hutchinson, A.; Jacobs, K.B.; Kraft, P.; et al. Fine mapping and functional analysis of a common variant in MSMB on chromosome 10q11.2 associated with prostate cancer susceptibility. Proc. Natl. Acad. Sci. USA 2009, 106, 7933–7938. [Google Scholar] [CrossRef] [PubMed]
- Krätzschmar, J.; Haendler, B.; Eberspaecher, U.; Roosterman, D.; Donner, P.; Schleuning, W.D. The human cysteine-rich secretory protein (CRISP) family. Primary structure and tissue distribution of CRISP-1, CRISP-2 and CRISP-3. Eur. J. Biochem. 1996, 236, 827–836. [Google Scholar]
- Anklesaria, J.H.; Mhatre, D.R.; Mahale, S.D. Structural and molecular biology of PSP94: Its significance in prostate pathophysiology. Front. Biosci. 2018, 23, 535–562. [Google Scholar]
- Zhang, X.; Cowper-Sal lari, R.; Bailey, S.D.; Moore, J.H.; Lupien, M. Integrative functional genomics identifies an enhancer looping to the SOX9 gene disrupted by the 17q24.3 prostate cancer risk locus. Genome Res. 2012, 22, 1437–1446. [Google Scholar] [CrossRef]
- Jin, H.J.; Jung, S.; DebRoy, A.R.; Davuluri, R.V. Identification and validation of regulatory SNPs that modulate transcription factor chromatin binding and gene expression in prostate cancer. Oncotarget 2016, 7, 54616–54626. [Google Scholar] [CrossRef] [Green Version]
- Romanel, A.; Garritano, S.; Stringa, B.; Blattner, M.; Dalfovo, D.; Chakravarty, D.; Soong, D.; Cotter, K.A.; Petris, G.; Dhingra, P.; et al. Inherited determinants of early recurrent somatic mutations in prostate cancer. Nat. Commun. 2017, 8, 48. [Google Scholar] [CrossRef]
- Rebbeck, T.R. Prostate cancer disparities by race and ethnicity: From nucleotide to neighborhood. Cold Spring Harb. Perspect. Med. 2018, 8, a030387. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Rhie, S.K.; Lay, F.D.; Farnham, P.J. A prostate cancer risk element functions as a repressive loop that regulates HOXA13. Cell Rep. 2017, 21, 1411–1417. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Whitington, T.; Gao, P.; Lindberg, J.F.; Yang, Y.; Sun, J.; Vaisanen, M.R.; Szulkin, R.; Annala, M.; Yan, J.; et al. A prostate cancer susceptibility allele at 6q22 increases RFX6 expression by modulating HOXB13 chromatin binding. Nat. Genet. 2014, 46, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Brechka, H.; Bhanvadia, R.R.; VanOpstall, C.; Vander Griend, D.J. HOXB13 mutations and binding partners in prostate development and cancer: Function, clinical significance, and future directions. Genes Dis. 2017, 4, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wu, D.; Thomas-Ahner, J.M.; Lu, C.; Zhao, P.; Zhang, Q.; Geraghty, C.; Yan, P.S.; Hankey, W.; Sunkel, B.; et al. Diverse AR-V7 cistromes in castration-resistant prostate cancer are governed by HoxB13. Proc. Natl. Acad. Sci. USA 2018, 115, 6810–6815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, H.; Ahmed, M.; Zhang, F.; Yao, C.Q.; Li, S.; Liang, Y.; Hua, J.; Soares, F.; Sun, Y.; Langstein, J.; et al. Modulation of long noncoding RNAs by risk SNPs underlying genetic predispositions to prostate cancer. Nat. Genet. 2016, 48, 1142–1150. [Google Scholar] [CrossRef]
- Rotinen, M.; You, S.; Yang, J.; Coetzee, S.G.; Reis-Sobreiro, M.; Huang, W.C.; Huang, F.; Pan, X.; Yanez, A.; Hazelett, D.J.; et al. ONECUT2 is a targetable master regulator of lethal prostate cancer that suppresses the androgen axis. Nat. Med. 2018, 24, 1887–1898. [Google Scholar] [CrossRef]
- Gao, P.; Xia, J.H.; Sipeky, C.; Dong, X.M.; Zhang, Q.; Yang, Y.; Zhang, P.; Cruz, S.P.; Zhang, K.; Zhu, J.; et al. Biology and clinical implications of the 19q13 aggressive prostate cancer susceptibility locus. Cell 2018, 174, 576.e18–589.e18. [Google Scholar] [CrossRef]
- Hua, J.T.; Ahmed, M.; Guo, H.; Zhang, Y.; Chen, S.; Soares, F.; Lu, J.; Zhou, S.; Wang, M.; Li, H.; et al. Risk SNP-mediated promoter-enhancer switching drives prostate cancer through lncRNA PCAT19. Cell 2018, 174, 564.e18–575.e18. [Google Scholar] [CrossRef]
- Guo, Y.; Perez, A.A.; Hazelett, D.J.; Coetzee, G.A.; Rhie, S.K.; Farnham, P.J. CRISPR-mediated deletion of prostate cancer risk-associated CTCF loop anchors identifies repressive chromatin loops. Genome Biol. 2018, 19, 160. [Google Scholar] [CrossRef]
- Smith, A.J.P.; Deloukas, P.; Munroe, P.B. Emerging applications of genome-editing technology to examine functionality of GWAS-associated variants for complex traits. Physiol. Genom. 2018, 50, 510–522. [Google Scholar] [CrossRef] [PubMed]
- Tomlins, S.A.; Rhodes, D.R.; Perner, S.; Dhanasekaran, S.M.; Mehra, R.; Sun, X.W.; Varambally, S.; Cao, X.; Tchinda, J.; Kuefer, R.; et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 2005, 310, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Kron, K.J.; Murison, A.; Zhou, S.; Huang, V.; Yamaguchi, T.N.; Shiah, Y.J.; Fraser, M.; van der Kwast, T.; Boutros, P.C.; Bristow, R.G.; et al. TMPRSS2-ERG fusion co-opts master transcription factors and activates NOTCH signaling in primary prostate cancer. Nat. Genet. 2017, 49, 1336–1345. [Google Scholar] [CrossRef] [PubMed]
- Rickman, D.S.; Soong, T.D.; Moss, B.; Mosquera, J.M.; Dlabal, J.; Terry, S.; MacDonald, T.Y.; Tripodi, J.; Bunting, K.; Najfeld, V.; et al. Oncogene-mediated alterations in chromatin conformation. Proc. Natl. Acad. Sci. USA 2012, 109, 9083–9088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toropainen, S.; Niskanen, E.A.; Malinen, M.; Sutinen, P.; Kaikkonen, M.U.; Palvimo, J.J. Global analysis of transcription in castration-resistant prostate cancer cells uncovers active enhancers and direct androgen receptor targets. Sci. Rep. 2016, 6, 33510. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Chng, K.R.; Lingadahalli, S.; Chen, Z.; Liu, M.H.; Do, H.H.; Cai, S.; Rinaldi, N.; Poh, H.M.; Li, G.; et al. An AR-ERG transcriptional signature defined by long-range chromatin interactomes in prostate cancer cells. Genome Res. 2019, 29, 223–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blee, A.M.; Liu, S.; Wang, L.; Huang, H. BET bromodomain-mediated interaction between ERG and BRD4 promotes prostate cancer cell invasion. Oncotarget 2016, 7, 38319–38332. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.H.; Lee, G.Y.; Lee, M.; Kang, J.; Shin, H.W.; Chun, Y.S.; Park, J.W. Aberrant expression of CITED2 promotes prostate cancer metastasis by activating the nucleolin-AKT pathway. Nat. Commun. 2018, 9, 4113. [Google Scholar] [CrossRef] [PubMed]
- Gerhauser, C.; Favero, F.; Risch, T.; Simon, R.; Feuerbach, L.; Assenov, Y.; Heckmann, D.; Sidiropoulos, N.; Waszak, S.M.; Hubschmann, D.; et al. Molecular evolution of early-onset prostate cancer identifies molecular risk markers and clinical trajectories. Cancer Cell 2018, 34, 996–1011.e8. [Google Scholar] [CrossRef]
- Rajan, P.; Elliott, D.J.; Robson, C.N.; Leung, H.Y. Alternative splicing and biological heterogeneity in prostate cancer. Nat. Rev. Urol. 2009, 6, 454–460. [Google Scholar] [CrossRef]
- Whang, Y.E.; Wu, X.; Suzuki, H.; Reiter, R.E.; Tran, C.; Vessella, R.L.; Said, J.W.; Isaacs, W.B.; Sawyers, C.L. Inactivation of the tumor suppressor PTEN/MMAC1 in advanced human prostate cancer through loss of expression. Proc. Natl. Acad. Sci. USA 1998, 95, 5246–5250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamaspishvili, T.; Berman, D.M.; Ross, A.E.; Scher, H.I.; De Marzo, A.M.; Squire, J.A.; Lotan, T.L. Clinical implications of PTEN loss in prostate cancer. Nat. Rev. Urol. 2018, 15, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Poluri, R.T.K.; Audet-Walsh, E. Genomic deletion at 10q23 in prostate cancer: More than PTEN loss? Front. Oncol. 2018, 8, 246. [Google Scholar] [CrossRef]
- Park, S.; Kim, Y.S.; Kim, D.Y.; So, I.; Jeon, J.H. PI3K pathway in prostate cancer: All resistant roads lead to PI3K. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 198–206. [Google Scholar] [CrossRef]
- Crumbaker, M.; Khoja, L.; Joshua, A.M. AR signaling and the PI3K pathway in prostate cancer. Cancers 2017, 9, 34. [Google Scholar] [CrossRef]
- Valdes-Mora, F.; Gould, C.M.; Colino-Sanguino, Y.; Qu, W.; Song, J.Z.; Taylor, K.M.; Buske, F.A.; Statham, A.L.; Nair, S.S.; Armstrong, N.J.; et al. Acetylated histone variant H2A.Z is involved in the activation of neo-enhancers in prostate cancer. Nat. Commun. 2017, 8, 1346. [Google Scholar] [CrossRef] [PubMed]
- Gallenkamp, D.; Gelato, K.A.; Haendler, B.; Weinmann, H. Bromodomains and their pharmacological inhibitors. ChemMedChem 2014, 9, 438–464. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Knapp, S. Targeting bromodomains: Epigenetic readers of lysine acetylation. Nat. Rev. Drug Discov. 2014, 13, 337–356. [Google Scholar] [CrossRef] [PubMed]
- Zuber, V.; Bettella, F.; Witoelar, A.; Consortium, P.; Cruk, G.; Consortium, B.; Consortium, T.; Andreassen, O.A.; Mills, I.G.; Urbanucci, A. Bromodomain protein 4 discriminates tissue-specific super-enhancers containing disease-specific susceptibility loci in prostate and breast cancer. BMC Genom. 2017, 18, 270. [Google Scholar] [CrossRef]
- Urbanucci, A.; Barfeld, S.J.; Kytola, V.; Itkonen, H.M.; Coleman, I.M.; Vodak, D.; Sjoblom, L.; Sheng, X.; Tolonen, T.; Minner, S.; et al. Androgen receptor deregulation drives bromodomain-mediated chromatin alterations in prostate cancer. Cell Rep. 2017, 19, 2045–2059. [Google Scholar] [CrossRef]
- Asangani, I.A.; Dommeti, V.L.; Wang, X.; Malik, R.; Cieslik, M.; Yang, R.; Escara-Wilke, J.; Wilder-Romans, K.; Dhanireddy, S.; Engelke, C.; et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature 2014, 510, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.X.; Smith, E.R.; Shilatifard, A. Born to run: Control of transcription elongation by RNA polymerase II. Nat. Rev. Mol. Cell Biol. 2018, 19, 464–478. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wang, L.; Ren, S.; Wang, L.; Blackburn, P.R.; McNulty, M.S.; Gao, X.; Qiao, M.; Vessella, R.L.; Kohli, M.; et al. Activation of P-TEFb by androgen receptor-regulated enhancer RNAs in castration-resistant prostate cancer. Cell Rep. 2016, 15, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Kuser-Abali, G.; Alptekin, A.; Lewis, M.; Garraway, I.P.; Cinar, B. YAP1 and AR interactions contribute to the switch from androgen-dependent to castration-resistant growth in prostate cancer. Nat. Commun. 2015, 6, 8126. [Google Scholar] [CrossRef] [PubMed]
- Seo, W.I.; Park, S.; Gwak, J.; Ju, B.G.; Chung, J.I.; Kang, P.M.; Oh, S. Wnt signaling promotes androgen-independent prostate cancer cell proliferation through up-regulation of the hippo pathway effector YAP. Biochem. Biophys. Res. Commun. 2017, 486, 1034–1039. [Google Scholar] [CrossRef]
- Bai, S.; Cao, S.; Jin, L.; Kobelski, M.; Schouest, B.; Wang, X.; Ungerleider, N.; Baddoo, M.; Zhang, W.; Corey, E.; et al. A positive role of c-Myc in regulating androgen receptor and its splice variants in prostate cancer. Oncogene 2019. [Google Scholar] [CrossRef]
- Gao, L.; Schwartzman, J.; Gibbs, A.; Lisac, R.; Kleinschmidt, R.; Wilmot, B.; Bottomly, D.; Coleman, I.; Nelson, P.; McWeeney, S.; et al. Androgen receptor promotes ligand-independent prostate cancer progression through c-Myc upregulation. PLoS ONE 2013, 8, e63563. [Google Scholar] [CrossRef]
- Shah, N.; Wang, P.; Wongvipat, J.; Karthaus, W.R.; Abida, W.; Armenia, J.; Rockowitz, S.; Drier, Y.; Bernstein, B.E.; Long, H.W.; et al. Regulation of the glucocorticoid receptor via a BET-dependent enhancer drives antiandrogen resistance in prostate cancer. eLife 2017, 6, e27861. [Google Scholar] [CrossRef]
- Akamatsu, S.; Inoue, T.; Ogawa, O.; Gleave, M.E. Clinical and molecular features of treatment-related neuroendocrine prostate cancer. Int. J. Urol. 2018, 25, 345–351. [Google Scholar] [CrossRef] [Green Version]
- Bakht, M.K.; Derecichei, I.; Li, Y.; Ferraiuolo, R.M.; Dunning, M.; Oh, S.W.; Hussein, A.; Youn, H.; Stringer, K.F.; Jeong, C.W.; et al. Neuroendocrine differentiation of prostate cancer leads to PSMA suppression. Endocr.-Relat. Cancer 2018, 26, 131–146. [Google Scholar] [CrossRef]
- Eapen, R.S.; Nzenza, T.C.; Murphy, D.G.; Hofman, M.S.; Cooperberg, M.; Lawrentschuk, N. PSMA PET applications in the prostate cancer journey: From diagnosis to theranostics. World J. Urol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Lee, J.K.; Witte, O.N.; Huang, J. FOXA2 is a sensitive and specific marker for small cell neuroendocrine carcinoma of the prostate. Mod. Pathol. 2017, 30, 1262–1272. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.Y.; Zeng, T.; Wen, Y.C.; Yeh, H.L.; Jiang, K.C.; Chen, W.H.; Zhang, Q.; Huang, J.; Liu, Y.N. Androgen deprivation-induced ZBTB46-PTGS1 signaling promotes neuroendocrine differentiation of prostate cancer. Cancer Lett. 2019, 440–441, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Crea, F.; Venalainen, E.; Ci, X.; Cheng, H.; Pikor, L.; Parolia, A.; Xue, H.; Nur Saidy, N.R.; Lin, D.; Lam, W.; et al. The role of epigenetics and long noncoding RNA MIAT in neuroendocrine prostate cancer. Epigenomics 2016, 8, 721–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clermont, P.L.; Lin, D.; Crea, F.; Wu, R.; Xue, H.; Wang, Y.; Thu, K.L.; Lam, W.L.; Collins, C.C.; Wang, Y.; et al. Polycomb-mediated silencing in neuroendocrine prostate cancer. Clin. Epigenet. 2015, 7, 40. [Google Scholar] [CrossRef]
- Yang, Y.A.; Yu, J. EZH2, an epigenetic driver of prostate cancer. Protein Cell 2013, 4, 331–341. [Google Scholar] [CrossRef]
- Donaldson-Collier, M.C.; Sungalee, S.; Zufferey, M.; Tavernari, D.; Katanayeva, N.; Battistello, E.; Mina, M.; Douglass, K.M.; Rey, T.; Raynaud, F.; et al. EZH2 oncogenic mutations drive epigenetic, transcriptional, and structural changes within chromatin domains. Nat. Genet. 2019, 51, 517–528. [Google Scholar] [CrossRef]
- Zhang, Y.; Zheng, D.; Zhou, T.; Song, H.; Hulsurkar, M.; Su, N.; Liu, Y.; Wang, Z.; Shao, L.; Ittmann, M.; et al. Androgen deprivation promotes neuroendocrine differentiation and angiogenesis through CREB-EZH2-TSP1 pathway in prostate cancers. Nat. Commun. 2018, 9, 4080. [Google Scholar] [CrossRef]
- Dardenne, E.; Beltran, H.; Benelli, M.; Gayvert, K.; Berger, A.; Puca, L.; Cyrta, J.; Sboner, A.; Noorzad, Z.; MacDonald, T.; et al. N-Myc induces an EZH2-mediated transcriptional program driving neuroendocrine prostate cancer. Cancer Cell 2016, 30, 563–577. [Google Scholar] [CrossRef]
- Chang, Y.T.; Lin, T.P.; Campbell, M.; Pan, C.C.; Lee, S.H.; Lee, H.C.; Yang, M.H.; Kung, H.J.; Chang, P.C. REST is a crucial regulator for acquiring EMT-like and stemness phenotypes in hormone-refractory prostate cancer. Sci. Rep. 2017, 7, 42795. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.T.; Lin, T.P.; Tang, J.T.; Campbell, M.; Luo, Y.L.; Lu, S.Y.; Yang, C.P.; Cheng, T.Y.; Chang, C.H.; Liu, T.T.; et al. HOTAIR is a REST-regulated lncRNA that promotes neuroendocrine differentiation in castration resistant prostate cancer. Cancer Lett. 2018, 433, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Flores-Morales, A.; Bergmann, T.B.; Lavallee, C.; Batth, T.S.; Lin, D.; Lerdrup, M.; Friis, S.; Bartels, A.; Kristensen, G.; Krzyzanowska, A.; et al. Proteogenomic characterization of patient-derived xenografts highlights the role of REST in neuroendocrine differentiation of castration-resistant prostate cancer. Clin. Cancer Res. 2019, 25, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Donmez, N.; Sahinalp, C.; Xie, N.; Wang, Y.; Xue, H.; Mo, F.; Beltran, H.; Gleave, M.; Wang, Y.; et al. SRRM4 drives neuroendocrine transdifferentiation of prostate adenocarcinoma under androgen receptor pathway inhibition. Eur. Urol. 2017, 71, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, Q.; Lovnicki, J.; Chen, R.; Fazli, L.; Wang, Y.; Gleave, M.; Huang, J.; Dong, X. SRRM4 gene expression correlates with neuroendocrine prostate cancer. Prostate 2019, 79, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Ramnarine, V.R.; Alshalalfa, M.; Mo, F.; Nabavi, N.; Erho, N.; Takhar, M.; Shukin, R.; Brahmbhatt, S.; Gawronski, A.; Kobelev, M.; et al. The long noncoding RNA landscape of neuroendocrine prostate cancer and its clinical implications. GigaScience 2018, 7. [Google Scholar] [CrossRef] [Green Version]
- Beltran, H.; Oromendia, C.; Danila, D.C.; Montgomery, B.; Hoimes, C.; Szmulewitz, R.Z.; Vaishampayan, U.; Armstrong, A.J.; Stein, M.; Pinski, J.; et al. A phase II trial of the aurora kinase A inhibitor Alisertib for patients with castration-resistant and neuroendocrine prostate cancer: Efficacy and biomarkers. Clin. Cancer Res. 2019, 25, 43–51. [Google Scholar] [CrossRef]
- Copeland, B.T.; Pal, S.K.; Bolton, E.C.; Jones, J.O. The androgen receptor malignancy shift in prostate cancer. Prostate 2018, 78, 521–531. [Google Scholar] [CrossRef]
- Alpajaro, S.I.R.; Harris, J.A.K.; Evans, C.P. Non-metastatic castration resistant prostate cancer: A review of current and emerging medical therapies. Prostate Cancer Prostatic Dis. 2019, 22, 16–23. [Google Scholar] [CrossRef]
- Dellis, A.E.; Papatsoris, A.G. Perspectives on the current and emerging chemical androgen receptor antagonists for the treatment of prostate cancer. Expert Opin. Pharmacother. 2019, 20, 163–172. [Google Scholar] [CrossRef]
- Labbe, D.P.; Brown, M. Transcriptional regulation in prostate cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a030437. [Google Scholar] [CrossRef]
- Nevedomskaya, E.; Baumgart, S.J.; Haendler, B. Recent advances in prostate cancer treatment and drug discovery. Int. J. Mol. Sci. 2018, 19, 1359. [Google Scholar] [CrossRef] [PubMed]
- Crawford, E.D.; Schellhammer, P.F.; McLeod, D.G.; Moul, J.W.; Higano, C.S.; Shore, N.; Denis, L.; Iversen, P.; Eisenberger, M.A.; Labrie, F. Androgen receptor targeted treatments of prostate cancer: 35 years of progress with antiandrogens. J. Urol. 2018, 200, 956–966. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Lee, S.O.; Liang, L.; Luo, J.; Huang, C.K.; Li, L.; Niu, Y.; Chang, C. Targeting the unique methylation pattern of androgen receptor (AR) promoter in prostate stem/progenitor cells with 5-aza-2’-deoxycytidine (5-AZA) leads to suppressed prostate tumorigenesis. J. Biol. Chem. 2012, 287, 39954–39966. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Shao, G.; Zhang, H.T.; Li, C.; Zhang, D.; Cheng, L.; Elzey, B.D.; Pili, R.; Ratliff, T.L.; Huang, J.; et al. Protein arginine methyltransferase 5 functions as an epigenetic activator of the androgen receptor to promote prostate cancer cell growth. Oncogene 2017, 36, 1223–1231. [Google Scholar] [CrossRef] [PubMed]
- Yamakawa, T.; Waer, C.; Itakura, K. AT-rich interactive domain 5B regulates androgen receptor transcription in human prostate cancer cells. Prostate 2018, 78, 1238–1247. [Google Scholar] [CrossRef] [PubMed]
- Jernberg, E.; Bergh, A.; Wikstrom, P. Clinical relevance of androgen receptor alterations in prostate cancer. Endocr. Connect. 2017, 6, R146–R161. [Google Scholar] [CrossRef] [Green Version]
- Paschalis, A.; Sharp, A.; Welti, J.C.; Neeb, A.; Raj, G.V.; Luo, J.; Plymate, S.R.; de Bono, J.S. Alternative splicing in prostate cancer. Nat. Rev. Clin. Oncol. 2018, 15, 663–675. [Google Scholar] [CrossRef] [PubMed]
- Culig, Z.; Santer, F.R. Studies on steroid receptor coactivators in prostate cancer. Methods Mol. Biol. 2018, 1786, 259–262. [Google Scholar] [PubMed]
- Fizazi, K.; Shore, N.; Tammela, T.L.; Ulys, A.; Vjaters, E.; Polyakov, S.; Jievaltas, M.; Luz, M.; Alekseev, B.; Kuss, I.; et al. Darolutamide in nonmetastatic, castration-resistant prostate cancer. N. Engl. J. Med. 2019, 380, 1235–1246. [Google Scholar] [CrossRef]
- Tran, C.; Ouk, S.; Clegg, N.J.; Chen, Y.; Watson, P.A.; Arora, V.; Wongvipat, J.; Smith-Jones, P.M.; Yoo, D.; Kwon, A.; et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science 2009, 324, 787–790. [Google Scholar] [CrossRef]
- Clegg, N.J.; Wongvipat, J.; Joseph, J.D.; Tran, C.; Ouk, S.; Dilhas, A.; Chen, Y.; Grillot, K.; Bischoff, E.D.; Cai, L.; et al. ARN-509: A novel antiandrogen for prostate cancer treatment. Cancer Res. 2012, 72, 1494–1503. [Google Scholar] [CrossRef] [PubMed]
- Moilanen, A.M.; Riikonen, R.; Oksala, R.; Ravanti, L.; Aho, E.; Wohlfahrt, G.; Nykanen, P.S.; Tormakangas, O.P.; Palvimo, J.J.; Kallio, P.J. Discovery of ODM-201, a new-generation androgen receptor inhibitor targeting resistance mechanisms to androgen signaling-directed prostate cancer therapies. Sci. Rep. 2015, 5, 12007. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, T.; Baumgart, S.J.; Nevedomskaya, E.; Reichert, K.; Steuber, H.; Lejeune, P.; Mumberg, D.; Haendler, B. Darolutamide is a potent androgen receptor antagonist with strong efficacy in prostate cancer models. Int. J. Cancer 2019. [Google Scholar] [CrossRef]
- Peltola, K.J.; Bono, P.; Jones, R.H.; Vjaters, E.; Nykanen, P.; Vuorela, A.; Oksala, R.; Pohjanjousi, P.; Mustonen, M.V.J.; Fizazi, K.; et al. ODM-204, a novel dual inhibitor of CYP17A1 and androgen receptor: Early results from phase I dose escalation in men with castration-resistant prostate cancer. Eur. Urol. Focus 2018. [Google Scholar] [CrossRef] [PubMed]
- Salami, J.; Alabi, S.; Willard, R.R.; Vitale, N.J.; Wang, J.; Dong, H.; Jin, M.; McDonnell, D.P.; Crew, A.P.; Neklesa, T.K.; et al. Androgen receptor degradation by the proteolysis-targeting chimera ARCC-4 outperforms enzalutamide in cellular models of prostate cancer drug resistance. Commun. Biol. 2018, 1, 100. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Tian, J.; Chou, F.; Lin, C.; Xing, E.Z.; Zuo, L.; Niu, Y.; Yeh, S.; Chang, C. Targeting the androgen receptor (AR) with AR degradation enhancer ASC-J9(R) led to increase docetaxel sensitivity via suppressing the p21 expression. Cancer Lett. 2019, 444, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Wang, C.; Qin, C.; Xiang, W.; Fernandez-Salas, E.; Yang, C.Y.; Wang, M.; Zhao, L.; Xu, T.; Chinnaswamy, K.; et al. Discovery of ARD-69 as a highly potent proteolysis targeting chimera (PROTAC) degrader of androgen receptor (AR) for the treatment of prostate cancer. J. Med. Chem. 2019, 62, 941–964. [Google Scholar] [CrossRef]
- Yang, Y.C.; Banuelos, C.A.; Mawji, N.R.; Wang, J.; Kato, M.; Haile, S.; McEwan, I.J.; Plymate, S.; Sadar, M.D. Targeting androgen receptor activation function-1 with EPI to overcome resistance mechanisms in castration-resistant prostate cancer. Clin. Cancer Res. 2016, 22, 4466–4477. [Google Scholar] [CrossRef]
- Bianchini, D.; Omlin, A.; Pezaro, C.; Lorente, D.; Ferraldeschi, R.; Mukherji, D.; Crespo, M.; Figueiredo, I.; Miranda, S.; Riisnaes, R.; et al. First-in-human Phase I study of EZN-4176, a locked nucleic acid antisense oligonucleotide to exon 4 of the androgen receptor mRNA in patients with castration-resistant prostate cancer. Br. J. Cancer 2013, 109, 2579–2586. [Google Scholar] [CrossRef] [Green Version]
- De Las Pozas, A.; Reiner, T.; De Cesare, V.; Trost, M.; Perez-Stable, C. Inhibiting multiple deubiquitinases to reduce androgen receptor expression in prostate cancer cells. Sci. Rep. 2018, 8, 13146. [Google Scholar] [CrossRef]
- Winters, B.; Brown, L.; Coleman, I.; Nguyen, H.; Minas, T.Z.; Kollath, L.; Vasioukhin, V.; Nelson, P.; Corey, E.; Uren, A.; et al. Inhibition of ERG activity in patient-derived prostate cancer xenografts by YK-4-279. Anticancer Res. 2017, 37, 3385–3396. [Google Scholar] [PubMed]
- Mohamed, A.A.; Xavier, C.P.; Sukumar, G.; Tan, S.H.; Ravindranath, L.; Seraj, N.; Kumar, V.; Sreenath, T.; McLeod, D.G.; Petrovics, G.; et al. Identification of a small molecule that selectively inhibits ERG-positive cancer cell growth. Cancer Res. 2018, 78, 3659–3671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyce, A.; Degenhardt, Y.; Bai, Y.; Le, B.; Korenchuk, S.; Crouthame, M.C.; McHugh, C.F.; Vessella, R.; Creasy, C.L.; Tummino, P.J.; et al. Inhibition of BET bromodomain proteins as a therapeutic approach in prostate cancer. Oncotarget 2013, 4, 2419–2429. [Google Scholar] [CrossRef] [PubMed]
- Markowski, M.C.; De Marzo, A.M.; Antonarakis, E.S. BET inhibitors in metastatic prostate cancer: Therapeutic implications and rational drug combinations. Expert Opin. Investig. Drugs 2017, 26, 1391–1397. [Google Scholar] [CrossRef] [PubMed]
- Lucking, U.; Scholz, A.; Lienau, P.; Siemeister, G.; Kosemund, D.; Bohlmann, R.; Briem, H.; Terebesi, I.; Meyer, K.; Prelle, K.; et al. Identification of atuveciclib (BAY 1143572), the first highly selective, clinical PTEFb/CDK9 inhibitor for the treatment of cancer. ChemMedChem 2017, 12, 1776–1793. [Google Scholar] [CrossRef]
- Zhang, H.; Pandey, S.; Travers, M.; Sun, H.; Morton, G.; Madzo, J.; Chung, W.; Khowsathit, J.; Perez-Leal, O.; Barrero, C.A.; et al. Targeting CDK9 reactivates epigenetically silenced genes in cancer. Cell 2018, 175, 1244.e26–1258.e26. [Google Scholar] [CrossRef]
- Raisner, R.; Kharbanda, S.; Jin, L.; Jeng, E.; Chan, E.; Merchant, M.; Haverty, P.M.; Bainer, R.; Cheung, T.; Arnott, D.; et al. Enhancer activity requires CBP/P300 bromodomain-dependent histone H3K27 acetylation. Cell Rep. 2018, 24, 1722–1729. [Google Scholar] [CrossRef]
- Santer, F.R.; Hoschele, P.P.; Oh, S.J.; Erb, H.H.; Bouchal, J.; Cavarretta, I.T.; Parson, W.; Meyers, D.J.; Cole, P.A.; Culig, Z. Inhibition of the acetyltransferases p300 and CBP reveals a targetable function for p300 in the survival and invasion pathways of prostate cancer cell lines. Mol. Cancer 2011, 10, 1644–1655. [Google Scholar] [CrossRef]
- Ianculescu, I.; Wu, D.Y.; Siegmund, K.D.; Stallcup, M.R. Selective roles for cAMP response element-binding protein binding protein and p300 protein as coregulators for androgen-regulated gene expression in advanced prostate cancer cells. J. Biol. Chem. 2012, 287, 4000–4013. [Google Scholar] [CrossRef]
- Jin, L.; Garcia, J.; Chan, E.; de la Cruz, C.; Segal, E.; Merchant, M.; Kharbanda, S.; Raisner, R.; Haverty, P.M.; Modrusan, Z.; et al. Therapeutic targeting of the CBP/p300 bromodomain blocks the growth of castration-resistant prostate cancer. Cancer Res. 2017, 77, 5564–5575. [Google Scholar] [CrossRef]
- Xiang, Q.; Wang, C.; Zhang, Y.; Xue, X.; Song, M.; Zhang, C.; Li, C.; Wu, C.; Li, K.; Hui, X.; et al. Discovery and optimization of 1-(1H-indol-1-yl)ethanone derivatives as CBP/EP300 bromodomain inhibitors for the treatment of castration-resistant prostate cancer. Eur. J. Med. Chem. 2018, 147, 238–252. [Google Scholar] [CrossRef] [PubMed]
- Pegg, N.; Worthington, J.; Young, B.; Prosser, A.; Gaughan, L.; Spencer, G.; Somervaille, T.; Burns, J.; Knowles, M.; Brooks, N. Novel small molecule inhibitors of p300/CBP down-regulate androgen receptor (AR) and c-Myc for the treatment of prostate cancer and beyond. Cancer Res. 2018, 78, 3991. [Google Scholar]
- Lasko, L.M.; Jakob, C.G.; Edalji, R.P.; Qiu, W.; Montgomery, D.; Digiammarino, E.L.; Hansen, T.M.; Risi, R.M.; Frey, R.; Manaves, V.; et al. Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature 2017, 550, 128–132. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, Y.; Lu, X.; Song, B.; Fong, K.W.; Cao, Q.; Licht, J.D.; Zhao, J.C.; Yu, J. Polycomb- and methylation-independent roles of EZH2 as a transcription activator. Cell Rep. 2018, 25, 2808.e4–2820.e4. [Google Scholar] [CrossRef]
- Wu, C.; Jin, X.; Yang, J.; Yang, Y.; He, Y.; Ding, L.; Pan, Y.; Chen, S.; Jiang, J.; Huang, H. Inhibition of EZH2 by chemo- and radiotherapy agents and small molecule inhibitors induces cell death in castration-resistant prostate cancer. Oncotarget 2016, 7, 3440–3452. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Wang, W.; Li, B.; Cheng, B.; Lin, K.; Bai, J.; Li, H.; Yang, G. Targeting Ezh2 could overcome docetaxel resistance in prostate cancer cells. BMC Cancer 2019, 19, 27. [Google Scholar] [CrossRef]
- Fioravanti, R.; Stazi, G.; Zwergel, C.; Valente, S.; Mai, A. Six Years (2012-2018) of researches on catalytic EZH2 inhibitors: The boom of the 2-pyridone compounds. Chem. Rec. 2018, 18, 1818–1832. [Google Scholar] [CrossRef]
- Taplin, M.E.; Hussain, A.; Shore, N.D.; Bradley, B.; Trojer, P.; Lebedinsky, C.; Senderowicz, A.M.; Antonarakis, E.S. A phase 1b/2 study of CPI-1205, a small molecule inhibitor of EZH2, combined with enzalutamide (E) or abiraterone/prednisone (A/P) in patients with metastatic castration resistant prostate cancer (mCRPC). J. Clin. Oncol. 2018. [Google Scholar] [CrossRef]
- Jain, P.; Di Croce, L. Mutations and deletions of PRC2 in prostate cancer. Bioessays 2016, 38, 446–454. [Google Scholar] [CrossRef]
- Huang, Y.; Zhang, J.; Yu, Z.; Zhang, H.; Wang, Y.; Lingel, A.; Qi, W.; Gu, J.; Zhao, K.; Shultz, M.D.; et al. Discovery of first-in-class, potent, and orally bioavailable embryonic ectoderm development (EED) inhibitor with robust anticancer efficacy. J. Med. Chem. 2017, 60, 2215–2226. [Google Scholar] [CrossRef]
- Qi, W.; Zhao, K.; Gu, J.; Huang, Y.; Wang, Y.; Zhang, H.; Zhang, M.; Zhang, J.; Yu, Z.; Li, L.; et al. An allosteric PRC2 inhibitor targeting the H3K27me3 binding pocket of EED. Nat. Chem. Biol. 2017, 13, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Sodir, N.M.; Evan, G.I. Finding cancer’s weakest link. Oncotarget 2011, 2, 1307–1313. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, R.G.; Gonda, T.J. MYB function in normal and cancer cells. Nat. Rev. Cancer 2008, 8, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Baumgart, S.J.; Haendler, B. Exploiting epigenetic alterations in prostate cancer. Int. J. Mol. Sci. 2017, 18, 1017. [Google Scholar] [CrossRef] [PubMed]
- Scheer, S.; Ackloo, S.; Medina, T.S.; Schapira, M.; Li, F.; Ward, J.A.; Lewis, A.M.; Northrop, J.P.; Richardson, P.L.; Kaniskan, H.U.; et al. A chemical biology toolbox to study protein methyltransferases and epigenetic signaling. Nat. Commun. 2019, 10, 19. [Google Scholar] [CrossRef] [PubMed]
- Janiaud, P.; Serghiou, S.; Ioannidis, J.P.A. New clinical trial designs in the era of precision medicine: An overview of definitions, strengths, weaknesses, and current use in oncology. Cancer Treat. Rev. 2018, 73, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Yin, G.; Chen, N.; Lee, J.J. Bayesian adaptive randomization and trial monitoring with predictive probability for time-to-event endpoint. Stat. Biosci. 2018, 10, 420–438. [Google Scholar] [CrossRef]
Figure 1.
Interaction model for factors associated with enhancer and super-enhancer function in prostate cancer. The transcription activation potential of enhancers and super-enhancers is correlated to the levels of their associated factors. Transcription regulatory elements physically interact with their target genes and enable the local formation of a dense network of transcription factors (dotted lines with grey nodes), ultimately leading to sustained transcription by RNA Pol II. Compounds such as AR antagonists and BET or CBP/p300 inhibitors can block factors associated with enhancer or super-enhancer function.
Figure 1.
Interaction model for factors associated with enhancer and super-enhancer function in prostate cancer. The transcription activation potential of enhancers and super-enhancers is correlated to the levels of their associated factors. Transcription regulatory elements physically interact with their target genes and enable the local formation of a dense network of transcription factors (dotted lines with grey nodes), ultimately leading to sustained transcription by RNA Pol II. Compounds such as AR antagonists and BET or CBP/p300 inhibitors can block factors associated with enhancer or super-enhancer function.

Figure 2.
Events disrupting gene regulatory elements or TADs lead to oncogenic transcriptional dysregulation. The normal cell state (left side) is dysregulated by an oncogenic event which leads to a malignant cell state (right side). TADs comprise insulated genomic regions which can be transcriptionally active (red triangles) or inactive (blue triangles). Examples and selected references for each event are given in the right-hand column, with a focus on prostate cancer.
Figure 2.
Events disrupting gene regulatory elements or TADs lead to oncogenic transcriptional dysregulation. The normal cell state (left side) is dysregulated by an oncogenic event which leads to a malignant cell state (right side). TADs comprise insulated genomic regions which can be transcriptionally active (red triangles) or inactive (blue triangles). Examples and selected references for each event are given in the right-hand column, with a focus on prostate cancer.


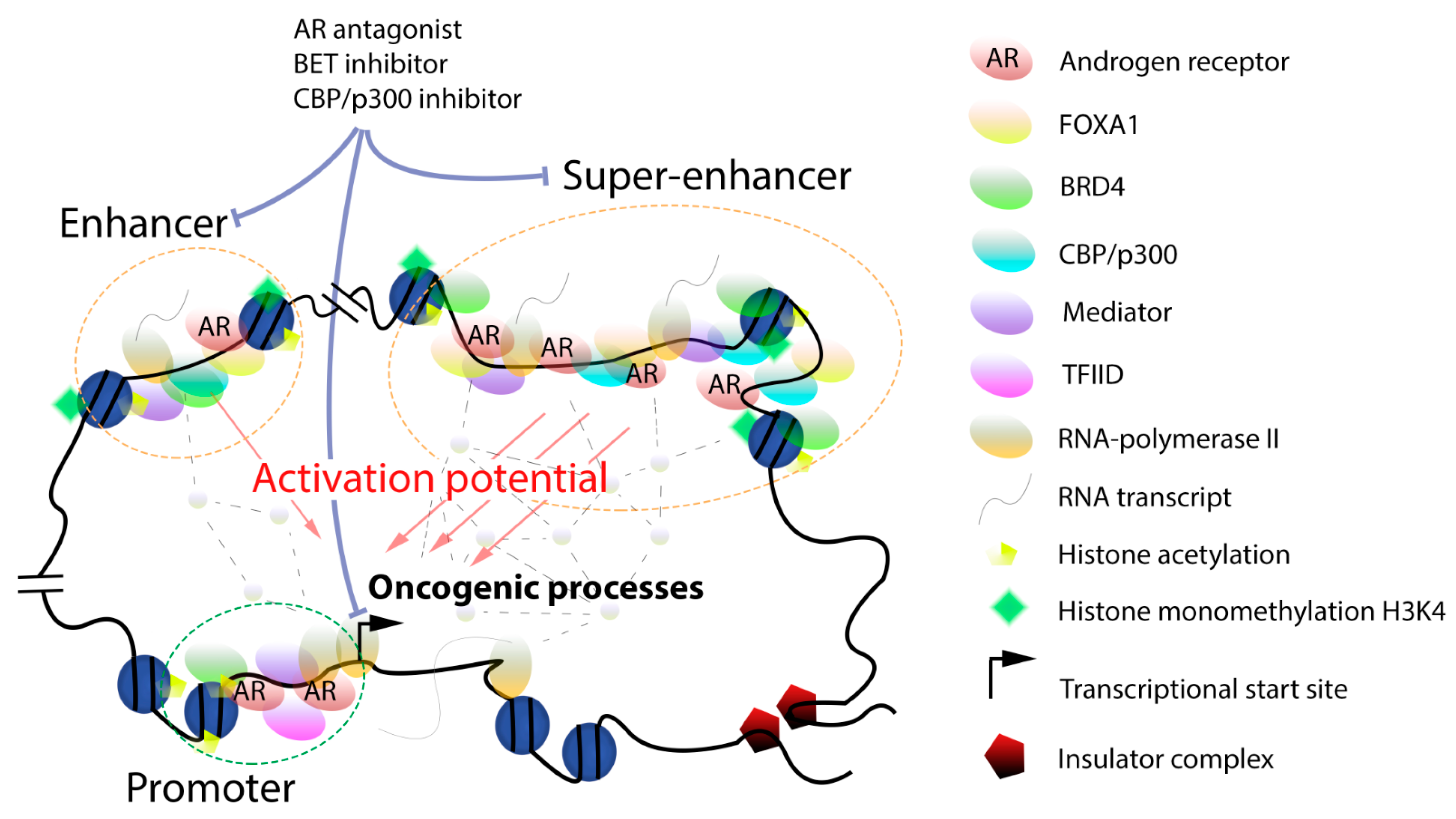{kind=link}
{kind=link}
Table 1.
Prostate cancer targets involved in gene regulation and selected inhibitors. Only the most advanced clinical studies are indicated.
Table 1.
Prostate cancer targets involved in gene regulation and selected inhibitors. Only the most advanced clinical studies are indicated.
| Target | Target Function | Compound | Mode of Action | Status | Identifier |
|---|---|---|---|---|---|
| AR | Transcription factor | Enzalutamide Apalutamide Darolutamide | Competitive antagonists | FDA-approved FDA-approved Phase 3 active Phase 3 active | NCT00974311 NCT01212991 NCT02003924 NCT01946204 NCT02200614 NCT02799602 |
| ARV-110 ARD-69 ASC-J9 | Degraders | Phase 1 recruiting Preclinical Preclinical | NCT03888612 | ||
| EPI-506 | N-terminal domain binder | Phase 1/2 terminated | NCT02606123 | ||
| EZN-4176 AZD-5312 | Antisense oligonucleotides | Phase 1a/1b suspended Phase 1 completed | NCT01337518 NCT02144051 | ||
| AR/CYP 17A | Transcription factor/cytochrome | ODM-204 | Dual inhibitor | Phase 2 ongoing | NCT02344017 |
| ERG | Transcription factor | YK-4-279 | Helicase interaction inhibitor | Preclinical | |
| NSC139021 | Ribosomal biogenesis regulator | Preclinical | |||
| BET/BRD4 | Acetylated lysine reader | GSK525762 ABBV-075 ABBV-744 GS-5829 ZEN003694 ZEN003694 | Bromodomain inhibitors | Phase 1B ongoing Phase 1 active Phase 1 ongoing Phase 1 ongoing Phase 1 completed Phase 1 active | NCT03150056 NCT02391480 NCT03360006 NCT02607228 NCT02705469 NCT02711956 |
| CDK7 | Part of transcription factor II complex | THZ1 | Kinase inhibitor | Preclinical | |
| CDK9 | Part of P-TEFb complex | Atuveciclib MC180295 | Kinase inhibitors | Phase 1 completed Preclinical | NCT02345382 |
| CBP/p300 | Transcriptional coactivator | CCS1477 GNE-049 32h | Bromodomain inhibitors | Phase 1/2 ongoing Preclinical Preclinical | NCT03568656 |
| A-485 | Acetyl-transferase inhibitor | Preclinical | |||
| EZH2 | H3 lysine 27 methyl-transferase | CPI-1205 GSK126 | Methyl-transferase inhibitors | Phase 1b/2 ongoing Preclinical | NCT03480646 |
| EED | EZH2 interactor | MAK683 | Inhibits H3K27me3 binding | Phase 1/2 ongoing | NCT02900651 |
| Aurora A | Serine/threonine kinase | Alisertib | Kinase inhibitor | Phase 2 completed | NCT01799278 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Baumgart, S.J.; Nevedomskaya, E.; Haendler, B. Dysregulated Transcriptional Control in Prostate Cancer. Int. J. Mol. Sci. 2019, 20, 2883. https://doi.org/10.3390/ijms20122883
AMA Style
Baumgart SJ, Nevedomskaya E, Haendler B. Dysregulated Transcriptional Control in Prostate Cancer. International Journal of Molecular Sciences. 2019; 20(12):2883. https://doi.org/10.3390/ijms20122883
Chicago/Turabian StyleBaumgart, Simon J., Ekaterina Nevedomskaya, and Bernard Haendler. 2019. "Dysregulated Transcriptional Control in Prostate Cancer" International Journal of Molecular Sciences 20, no. 12: 2883. https://doi.org/10.3390/ijms20122883
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.