Molecular Mechanisms of p63-Mediated Squamous Cancer Pathogenesis

 ,
,
Abstract
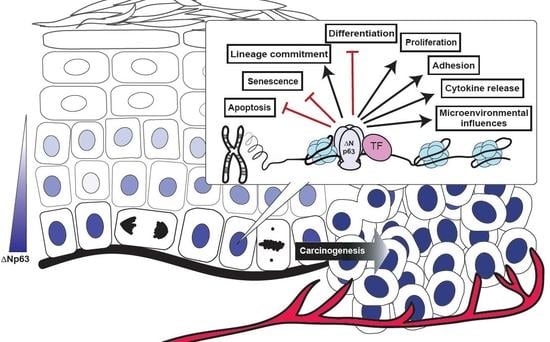
:
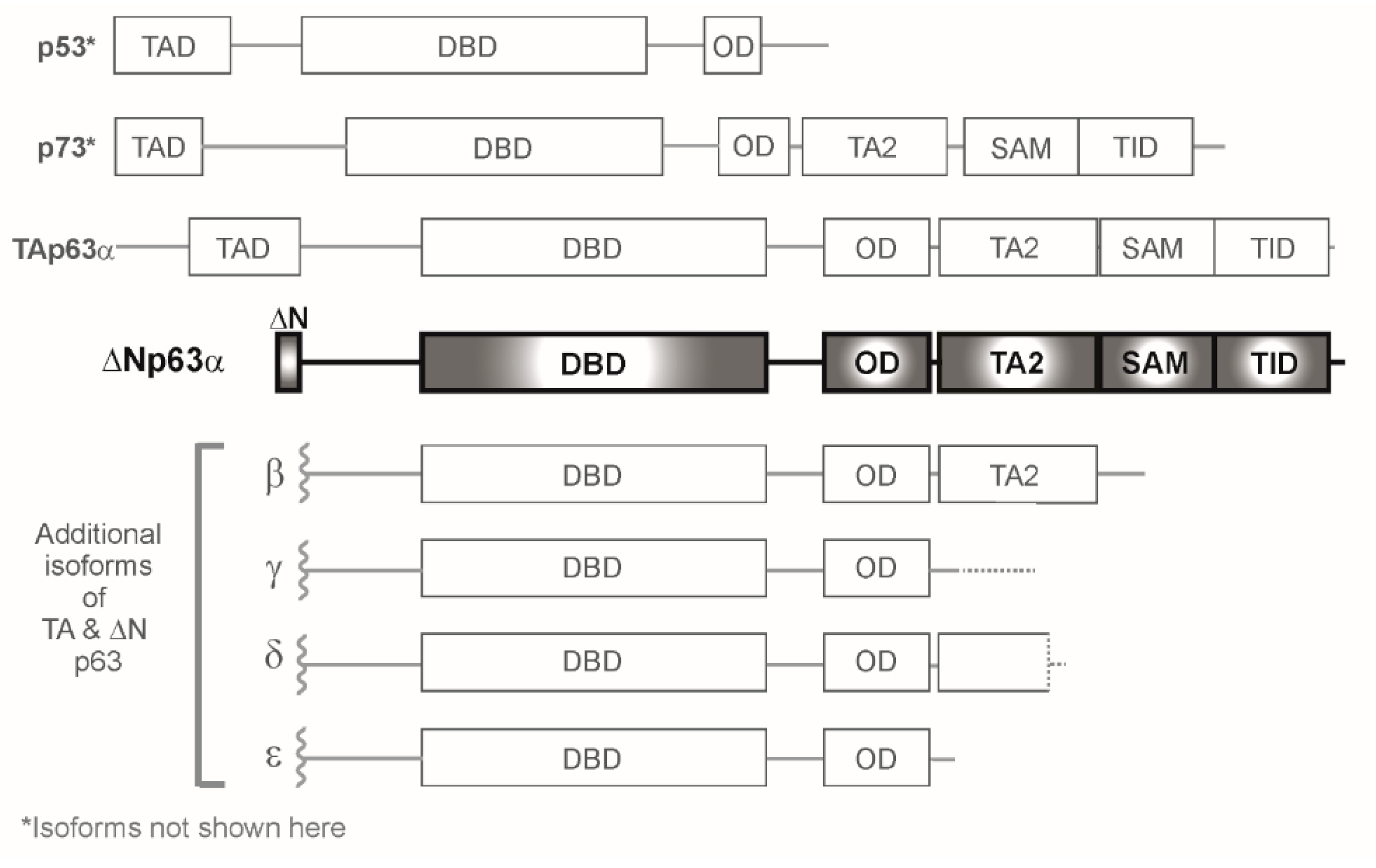
1. Introduction to the p53/p63/p73 Gene Family of Transcription Factors
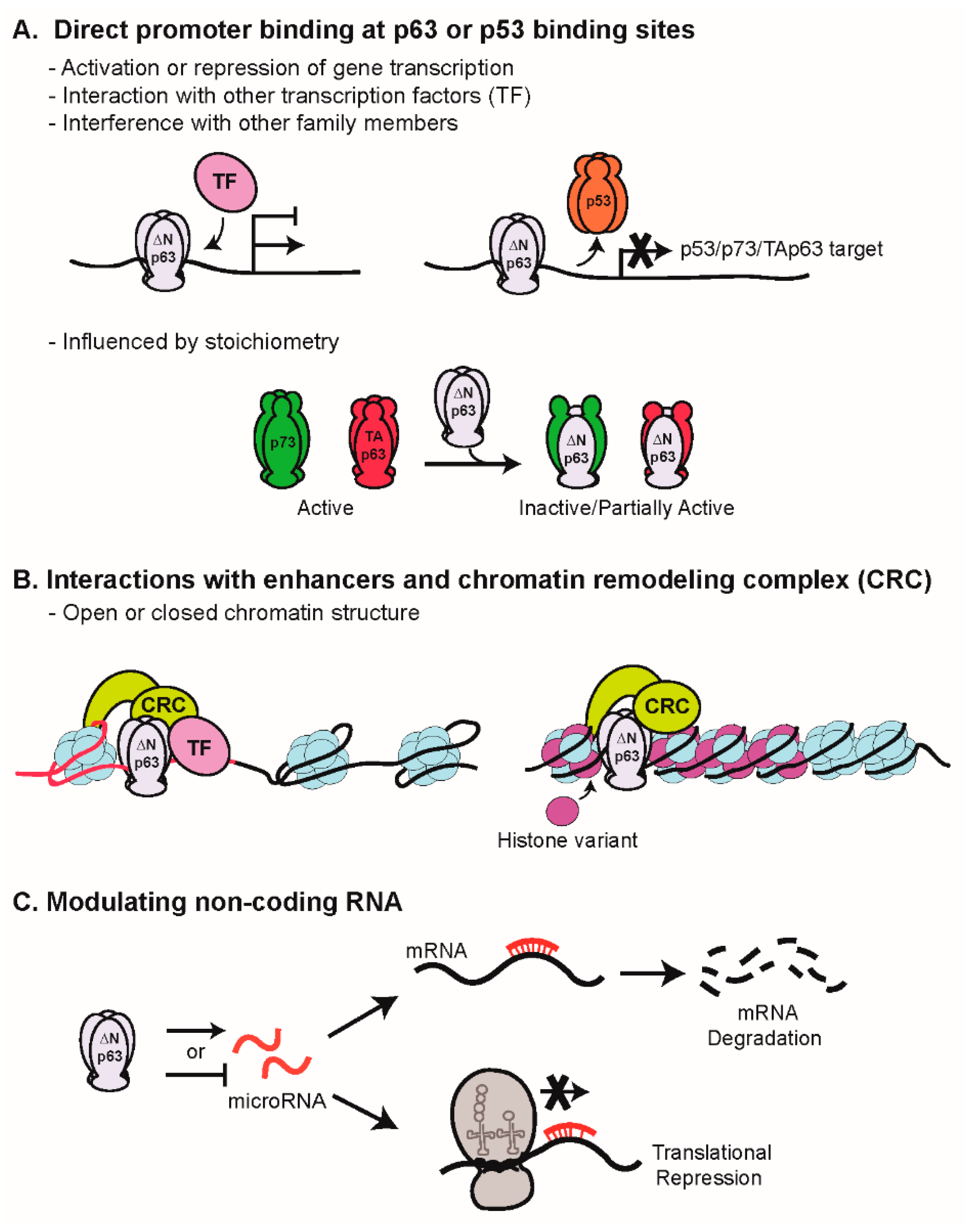
2. Mechanisms of Transcriptional Regulation by p63
3. ΔNp63α Is Essential for Normal Morphogenesis and Squamous Epithelial Homeostasis
p63 Mutations Are Associated with Human Ectodermal Dysplasia Syndromes
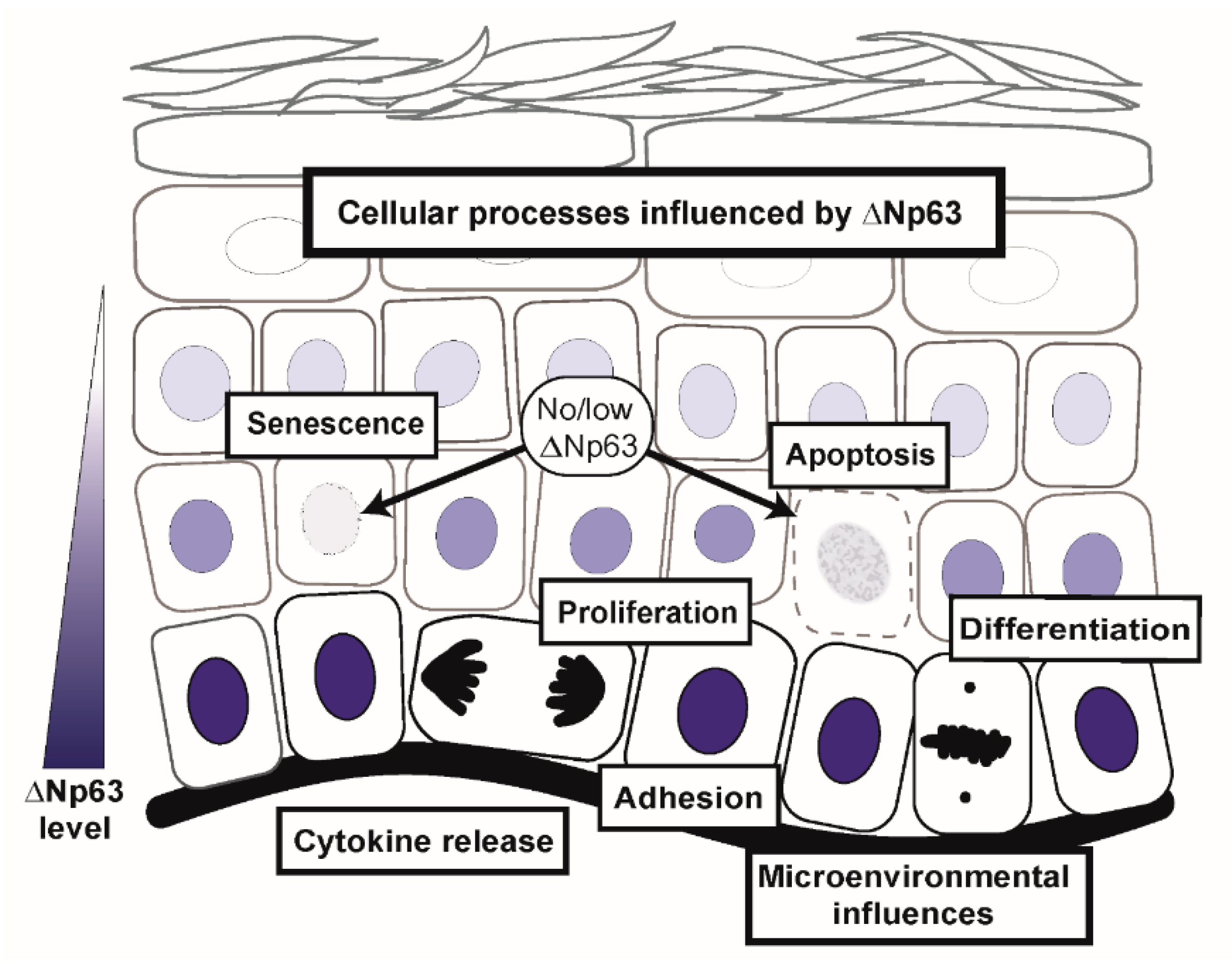
4. Dysregulated ΔNp63α Disrupts an Extensive Network of Molecular and Biological Pathways to Contribute to Squamous Cancer Pathogenesis
4.1. ΔNp63α Mediates Signaling Pathways Impacting Multiple Cell Intrinsic Biological Processes
4.1.1. p63 and Cancer Stem Cells
4.1.2. p63 and Cellular Metabolism
4.2. ΔNp63α Modulates Signaling Pathways Influencing the Extracellular Microenvironment
5. Conclusions
Acknowledgments
Disclaimer
Conflicts of Interest
Abbreviations
| TAD | transactivation domain |
| DBD | DNA binding domain |
| OD | oligomerization domain |
| SAM | sterile alpha motif |
| TID | transcriptional inhibitory domain |
| WT | wild type |
| HNSCC | head and neck squamous cell carcinoma |
| miRNA | microRNA |
| lncRNA | long non-coding RNA |
| SA-β-gal | senescence-associated β-galactosidase |
| HDAC | histone deacetylase |
| BMP | bone morphogenetic protein |
| SHH | sonic hedgehog |
| FGFR2 | fibroblast growth factor receptor 2 |
| TGF-β | transforming growth factor β |
| EDC | epidermal differentiation complex |
| EEC | Ectrodactyly Ectodermal Dysplasia-Clefting Syndrome |
| AEC | Ankyloblepharon-Ectodermal Dysplasia Clefting Syndrome |
| SCC | squamous cell carcinoma |
| TCGA | The Cancer Genome Atlas |
| cSCC | cutaneous squamous cell carcinoma |
| ChIP-Seq | chromatin immunoprecipitation-sequencing |
| EGFR | epidermal growth factor receptor |
| MAPK | mitogen-activated protein kinase |
| FGF | fibroblast growth factor |
| EMT | epithelial-mesenchymal transition |
| iASPP | inhibitor of apoptosis-stimulating protein of p53 |
| CSC | cancer stem cell |
| HK2 | hexokinase 2 |
| ROS | reactive oxygen species |
| GPX2 | glutathione peroxidase 2 |
| PAI-2 | plasminogen activator inhibitor-2 |
| TIMP | tissue inhibitor of metalloproteinase |
| MT1-MMP | membrane-type 1-matrix metalloproteinase |
| MMP | matrix metalloproteinase |
References
- Yang, A.; Kaghad, M.; Wang, Y.; Gillett, E.; Fleming, M.D.; Dotsch, V.; Andrews, N.C.; Caput, D.; McKeon, F. p63, a p53 homolog at 3q27-29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol. Cell 1998, 2, 305–316. [Google Scholar] [CrossRef]
- Kaghad, M.; Bonnet, H.; Yang, A.; Creancier, L.; Biscan, J.C.; Valent, A.; Minty, A.; Chalon, P.; Lelias, J.M.; Dumont, X.; et al. Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell 1997, 90, 809–819. [Google Scholar] [CrossRef]
- Dohn, M.; Zhang, S.; Chen, X. p63alpha and DeltaNp63alpha can induce cell cycle arrest and apoptosis and differentially regulate p53 target genes. Oncogene 2001, 20, 3193–3205. [Google Scholar] [CrossRef] [PubMed]
- Ghioni, P.; Bolognese, F.; Duijf, P.H.; Van Bokhoven, H.; Mantovani, R.; Guerrini, L. Complex transcriptional effects of p63 isoforms: identification of novel activation and repression domains. Mol. Cell. Biol. 2002, 22, 8659–8668. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Kameoka, M.; Itaya, A.; Ota, K.; Yoshihara, K. Regulation of HSF1-responsive gene expression by N-terminal truncated form of p73alpha. Biochem. Biophys. Res. Commun. 2004, 317, 865–872. [Google Scholar] [CrossRef] [PubMed]
- Helton, E.S.; Zhu, J.; Chen, X. The unique NH2-terminally deleted (DeltaN) residues, the PXXP motif, and the PPXY motif are required for the transcriptional activity of the DeltaN variant of p63. J. Biol Chem. 2006, 281, 2533–2542. [Google Scholar] [CrossRef] [PubMed]
- Bourdon, J.C. p53 isoforms change p53 paradigm. Mol. Cell Oncol. 2014, 1, e969136. [Google Scholar] [CrossRef] [PubMed]
- Mangiulli, M.; Valletti, A.; Caratozzolo, M.F.; Tullo, A.; Sbisa, E.; Pesole, G.; D’Erchia, A.M. Identification and functional characterization of two new transcriptional variants of the human p63 gene. Nucleic Acids Res. 2009, 37, 6092–6104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thanos, C.D.; Bowie, J.U. p53 Family members p63 and p73 are SAM domain-containing proteins. Protein Sci. 1999, 8, 1708–1710. [Google Scholar] [CrossRef] [PubMed]
- Serber, Z.; Lai, H.C.; Yang, A.; Ou, H.D.; Sigal, M.S.; Kelly, A.E.; Darimont, B.D.; Duijf, P.H.; Van Bokhoven, H.; McKeon, F.; et al. A C-terminal inhibitory domain controls the activity of p63 by an intramolecular mechanism. Mol. Cell. Biol. 2002, 22, 8601–8611. [Google Scholar] [CrossRef]
- Vikhreva, P.; Melino, G.; Amelio, I. p73 Alternative Splicing: Exploring a Biological Role for the C-Terminal Isoforms. J. Mol. Biol. 2018, 430, 1829–1838. [Google Scholar] [CrossRef] [PubMed]
- Coutandin, D.; Lohr, F.; Niesen, F.H.; Ikeya, T.; Weber, T.A.; Schafer, B.; Zielonka, E.M.; Bullock, A.N.; Yang, A.; Guntert, P.; et al. Conformational stability and activity of p73 require a second helix in the tetramerization domain. Cell Death Differ. 2009, 16, 1582–1589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratovitski, E.A.; Patturajan, M.; Hibi, K.; Trink, B.; Yamaguchi, K.; Sidransky, D. p53 associates with and targets Delta Np63 into a protein degradation pathway. Proc. Nat. Acad. Sci. USA 2001, 98, 1817–1822. [Google Scholar] [CrossRef] [PubMed]
- Strano, S.; Munarriz, E.; Rossi, M.; Cristofanelli, B.; Shaul, Y.; Castagnoli, L.; Levine, A.J.; Sacchi, A.; Cesareni, G.; Oren, M.; et al. Physical and functional interaction between p53 mutants and different isoforms of p73. J. Biol Chem. 2000, 275, 29503–29512. [Google Scholar] [CrossRef]
- Strano, S.; Fontemaggi, G.; Costanzo, A.; Rizzo, M.G.; Monti, O.; Baccarini, A.; Del Sal, G.; Levrero, M.; Sacchi, A.; Oren, M.; et al. Physical interaction with human tumor-derived p53 mutants inhibits p63 activities. J. Biol Chem. 2002, 277, 18817–18826. [Google Scholar] [CrossRef]
- Parsa, R.; Yang, A.; McKeon, F.; Green, H. Association of p63 with proliferative potential in normal and neoplastic human keratinocytes. J. Clin. Invest. Dermatol. 1999, 113, 1099–1105. [Google Scholar] [CrossRef] [PubMed]
- Puig, P.; Capodieci, P.; Drobnjak, M.; Verbel, D.; Prives, C.; Cordon-Cardo, C.; Di Como, C.J. p73 Expression in human normal and tumor tissues: loss of p73alpha expression is associated with tumor progression in bladder cancer. Clin. Cancer Res. 2003, 9, 5642–5651. [Google Scholar]
- Yang, A.; Schweitzer, R.; Sun, D.; Kaghad, M.; Walker, N.; Bronson, R.T.; Tabin, C.; Sharpe, A.; Caput, D.; Crum, C.; et al. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature 1999, 398, 714–718. [Google Scholar] [CrossRef]
- Mills, A.A.; Zheng, B.; Wang, X.J.; Vogel, H.; Roop, D.R.; Bradley, A. p63 is a p53 homologue required for limb and epidermal morphogenesis. Nature 1999, 398, 708–713. [Google Scholar] [CrossRef]
- Koster, M.I.; Marinari, B.; Payne, A.S.; Kantaputra, P.N.; Costanzo, A.; Roop, D.R. DeltaNp63 knockdown mice: A mouse model for AEC syndrome. Am. J. Med. Genet. A 2009, 149a, 1942–1947. [Google Scholar] [CrossRef]
- Wright, J.T.; Fete, M.; Schneider, H.; Zinser, M.; Koster, M.I.; Clarke, A.J.; Hadj-Rabia, S.; Tadini, G.; Pagnan, N.; Visinoni, A.F.; et al. Ectodermal dysplasias: Classification and organization by phenotype, genotype and molecular pathway. Am. J. Med. Genet. A 2019, 179, 442–447. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Walker, N.; Bronson, R.; Kaghad, M.; Oosterwegel, M.; Bonnin, J.; Vagner, C.; Bonnet, H.; Dikkes, P.; Sharpe, A.; et al. p73-deficient mice have neurological, pheromonal and inflammatory defects but lack spontaneous tumours. Nature 2000, 404, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Marshall, C.B.; Mays, D.J.; Beeler, J.S.; Rosenbluth, J.M.; Boyd, K.L.; Santos Guasch, G.L.; Shaver, T.M.; Tang, L.J.; Liu, Q.; Shyr, Y.; et al. p73 Is Required for Multiciliogenesis and Regulates the Foxj1-Associated Gene Network. Cell Rep. 2016, 14, 2289–2300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagiwara, K.; McMenamin, M.G.; Miura, K.; Harris, C.C. Mutational analysis of the p63/p73L/p51/p40/CUSP/KET gene in human cancer cell lines using intronic primers. Cancer Res. 1999, 59, 4165–4169. [Google Scholar] [PubMed]
- Li, Y.Y.; Hanna, G.J.; Laga, A.C.; Haddad, R.I.; Lorch, J.H.; Hammerman, P.S. Genomic analysis of metastatic cutaneous squamous cell carcinoma. Clin. Cancer Res. 2015, 21, 1447–1456. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.D.; Yau, C.; Bowlby, R.; Liu, Y.; Brennan, K.; Fan, H.; Taylor, A.M.; Wang, C.; Walter, V.; Akbani, R.; et al. Genomic, Pathway Network, and Immunologic Features Distinguishing Squamous Carcinomas. Cell Rep. 2018, 23, 194–212 e196. [Google Scholar] [CrossRef] [PubMed]
- Osada, M.; Park, H.L.; Nagakawa, Y.; Yamashita, K.; Fomenkov, A.; Kim, M.S.; Wu, G.; Nomoto, S.; Trink, B.; Sidransky, D. Differential recognition of response elements determines target gene specificity for p53 and p63. Mol. Cell. Biol. 2005, 25, 6077–6089. [Google Scholar] [CrossRef]
- Perez, C.A.; Ott, J.; Mays, D.J.; Pietenpol, J.A. p63 consensus DNA-binding site: identification, analysis and application into a p63MH algorithm. Oncogene 2007, 26, 7363–7370. [Google Scholar] [CrossRef]
- Ortt, K.; Sinha, S. Derivation of the consensus DNA-binding sequence for p63 reveals unique requirements that are distinct from p53. FEBS Lett. 2006, 580, 4544–4550. [Google Scholar] [CrossRef] [Green Version]
- Yang, A.; Zhu, Z.; Kettenbach, A.; Kapranov, P.; McKeon, F.; Gingeras, T.R.; Struhl, K. Genome-wide mapping indicates that p73 and p63 co-occupy target sites and have similar dna-binding profiles in vivo. PLoS ONE 2010, 5, e11572. [Google Scholar] [CrossRef]
- Watanabe, H.; Ma, Q.; Peng, S.; Adelmant, G.; Swain, D.; Song, W.; Fox, C.; Francis, J.M.; Pedamallu, C.S.; DeLuca, D.S.; et al. SOX2 and p63 colocalize at genetic loci in squamous cell carcinomas. J. Clin. Invest. 2014, 124, 1636–1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, K.E.; Ponnamperuma, R.M.; Allen, C.; Lu, H.; Duggal, P.; Chen, Z.; Van Waes, C.; Weinberg, W.C. The p53 homologue DeltaNp63alpha interacts with the nuclear factor-kappaB pathway to modulate epithelial cell growth. Cancer Res. 2008, 68, 5122–5131. [Google Scholar] [CrossRef] [PubMed]
- Di Costanzo, A.; Troiano, A.; di Martino, O.; Cacace, A.; Natale, C.F.; Ventre, M.; Netti, P.; Caserta, S.; Pollice, A.; La Mantia, G.; et al. The p63 protein isoform DeltaNp63alpha modulates Y-box binding protein 1 in its subcellular distribution and regulation of cell survival and motility genes. J. Biol. Chem. 2012, 287, 30170–30180. [Google Scholar] [CrossRef] [PubMed]
- Kouwenhoven, E.N.; Oti, M.; Niehues, H.; van Heeringen, S.J.; Schalkwijk, J.; Stunnenberg, H.G.; van Bokhoven, H.; Zhou, H. Transcription factor p63 bookmarks and regulates dynamic enhancers during epidermal differentiation. EMBO Rep. 2015, 16, 863–878. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.; Rubin, A.J.; Qu, K.; Zhang, J.; Giresi, P.G.; Chang, H.Y.; Khavari, P.A. A novel ATAC-seq approach reveals lineage-specific reinforcement of the open chromatin landscape via cooperation between BAF and p63. Genome Biol. 2015, 16, 284. [Google Scholar] [CrossRef] [PubMed]
- Saladi, S.V.; Ross, K.; Karaayvaz, M.; Tata, P.R.; Mou, H.; Rajagopal, J.; Ramaswamy, S.; Ellisen, L.W. ACTL6A Is Co-Amplified with p63 in Squamous Cell Carcinoma to Drive YAP Activation, Regenerative Proliferation, and Poor Prognosis. Cancer Cell 2017, 31, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Antonini, D.; Russo, M.T.; De Rosa, L.; Gorrese, M.; Del Vecchio, L.; Missero, C. Transcriptional repression of miR-34 family contributes to p63-mediated cell cycle progression in epidermal cells. J. Invest. Dermatol. 2010, 130, 1249–1257. [Google Scholar] [CrossRef] [PubMed]
- Ory, B.; Ramsey, M.R.; Wilson, C.; Vadysirisack, D.D.; Forster, N.; Rocco, J.W.; Rothenberg, S.M.; Ellisen, L.W. A microRNA-dependent program controls p53-independent survival and chemosensitivity in human and murine squamous cell carcinoma. J. Clin. Invest. 2011, 121, 809–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefort, K.; Brooks, Y.; Ostano, P.; Cario-Andre, M.; Calpini, V.; Guinea-Viniegra, J.; Albinger-Hegyi, A.; Hoetzenecker, W.; Kolfschoten, I.; Wagner, E.F.; et al. A miR-34a-SIRT6 axis in the squamous cell differentiation network. EMBO J. 2013, 32, 2248–2263. [Google Scholar] [CrossRef] [Green Version]
- Ratovitski, E.A. Phospho-DeltaNp63alpha/microRNA network modulates epigenetic regulatory enzymes in squamous cell carcinomas. Cell Cycle 2014, 13, 749–761. [Google Scholar] [CrossRef]
- Cheetham, S.W.; Gruhl, F.; Mattick, J.S.; Dinger, M.E. Long noncoding RNAs and the genetics of cancer. Br. J. Cancer 2013, 108, 2419–2425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Cao, Y.; Gong, X.; Li, H. Long noncoding RNAs in head and neck cancer. Oncotarget 2017, 8, 10726–10740. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.V.; Choudhari, R.; Gadad, S.S. Long noncoding RNAs and cancer, an overview. Steroids 2018, 133, 93–95. [Google Scholar] [CrossRef] [PubMed]
- Qian, L.; Xu, F.; Wang, X.; Jiang, M.; Wang, J.; Song, W.; Wu, D.; Shen, Z.; Feng, D.; Ling, B.; et al. LncRNA expression profile of DeltaNp63alpha in cervical squamous cancers and its suppressive effects on LIF expression. Cytokine 2017, 96, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Yoh, K.; Prywes, R. Pathway Regulation of p63, a Director of Epithelial Cell Fate. Front. Endocrinol. (Lausanne) 2015, 6, 51. [Google Scholar] [CrossRef]
- Li, Y.; Peart, M.J.; Prives, C. Stxbp4 regulates DeltaNp63 stability by suppression of RACK1-dependent degradation. Mol. Cell. Biol. 2009, 29, 3953–3963. [Google Scholar] [CrossRef]
- Rokudai, S.; Li, Y.; Otaka, Y.; Fujieda, M.; Owens, D.M.; Christiano, A.M.; Nishiyama, M.; Prives, C. STXBP4 regulates APC/C-mediated p63 turnover and drives squamous cell carcinogenesis. Proc. Nat. Acad. Sci. USA 2018, 115, E4806–E4814. [Google Scholar] [CrossRef] [Green Version]
- Hazawa, M.; Lin, D.C.; Kobayashi, A.; Jiang, Y.Y.; Xu, L.; Dewi, F.R.P.; Mohamed, M.S.; Hartono; Nakada, M.; Meguro-Horike, M.; et al. ROCK-dependent phosphorylation of NUP62 regulates p63 nuclear transport and squamous cell carcinoma proliferation. EMBO Rep. 2018, 19, 73–88. [Google Scholar] [CrossRef]
- Borlido, J.; D’Angelo, M.A. Nup62-mediated nuclear import of p63 in squamous cell carcinoma. EMBO Rep. 2018, 19, 3–4. [Google Scholar] [CrossRef]
- Koster, M.I.; Kim, S.; Mills, A.A.; DeMayo, F.J.; Roop, D.R. p63 is the molecular switch for initiation of an epithelial stratification program. Genes Dev. 2004, 18, 126–131. [Google Scholar] [CrossRef]
- Candi, E.; Rufini, A.; Terrinoni, A.; Dinsdale, D.; Ranalli, M.; Paradisi, A.; De Laurenzi, V.; Spagnoli, L.G.; Catani, M.V.; Ramadan, S.; et al. Differential roles of p63 isoforms in epidermal development: selective genetic complementation in p63 null mice. Cell Death Differ. 2006, 13, 1037–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romano, R.A.; Ortt, K.; Birkaya, B.; Smalley, K.; Sinha, S. An active role of the DeltaN isoform of p63 in regulating basal keratin genes K5 and K14 and directing epidermal cell fate. PLoS ONE 2009, 4, e5623. [Google Scholar] [CrossRef] [PubMed]
- Romano, R.A.; Smalley, K.; Magraw, C.; Serna, V.A.; Kurita, T.; Raghavan, S.; Sinha, S. DeltaNp63 knockout mice reveal its indispensable role as a master regulator of epithelial development and differentiation. Development 2012, 139, 772–782. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, G.; Dellambra, E.; Golisano, O.; Martinelli, E.; Fantozzi, I.; Bondanza, S.; Ponzin, D.; McKeon, F.; De Luca, M. p63 identifies keratinocyte stem cells. Proc. Nat. Acad. Sci. USA 2001, 98, 3156–3161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senoo, M.; Pinto, F.; Crum, C.P.; McKeon, F. p63 Is essential for the proliferative potential of stem cells in stratified epithelia. Cell 2007, 129, 523–536. [Google Scholar] [CrossRef] [PubMed]
- Melino, G.; Memmi, E.M.; Pelicci, P.G.; Bernassola, F. Maintaining epithelial stemness with p63. Sci. Signal. 2015, 8, re9. [Google Scholar] [CrossRef]
- Flores, E.R.; Sengupta, S.; Miller, J.B.; Newman, J.J.; Bronson, R.; Crowley, D.; Yang, A.; McKeon, F.; Jacks, T. Tumor predisposition in mice mutant for p63 and p73: evidence for broader tumor suppressor functions for the p53 family. Cancer Cell 2005, 7, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Keyes, W.M.; Wu, Y.; Vogel, H.; Guo, X.; Lowe, S.W.; Mills, A.A. p63 deficiency activates a program of cellular senescence and leads to accelerated aging. Genes Dev. 2005, 19, 1986–1999. [Google Scholar] [CrossRef] [Green Version]
- Su, X.; Paris, M.; Gi, Y.J.; Tsai, K.Y.; Cho, M.S.; Lin, Y.L.; Biernaskie, J.A.; Sinha, S.; Prives, C.; Pevny, L.H.; et al. TAp63 prevents premature aging by promoting adult stem cell maintenance. Cell Stem Cell 2009, 5, 64–75. [Google Scholar] [CrossRef]
- Devos, M.; Gilbert, B.; Denecker, G.; Leurs, K.; Mc Guire, C.; Lemeire, K.; Hochepied, T.; Vuylsteke, M.; Lambert, J.; Van Den Broecke, C.; et al. Elevated DeltaNp63alpha Levels Facilitate Epidermal and Biliary Oncogenic Transformation. J. Clin. Invest. Dermatol. 2017, 137, 494–505. [Google Scholar] [CrossRef]
- Li, L.; Wang, Y.; Torkelson, J.L.; Shankar, G.; Pattison, J.M.; Zhen, H.H.; Fang, F.; Duren, Z.; Xin, J.; Gaddam, S.; et al. TFAP2C- and p63-Dependent Networks Sequentially Rearrange Chromatin Landscapes to Drive Human Epidermal Lineage Commitment. Cell Stem Cell 2019, 24, 271–284.e278. [Google Scholar] [CrossRef] [PubMed]
- LeBoeuf, M.; Terrell, A.; Trivedi, S.; Sinha, S.; Epstein, J.A.; Olson, E.N.; Morrisey, E.E.; Millar, S.E. Hdac1 and Hdac2 act redundantly to control p63 and p53 functions in epidermal progenitor cells. Dev. Cell 2010, 19, 807–818. [Google Scholar] [CrossRef] [PubMed]
- Soares, E.; Zhou, H. Master regulatory role of p63 in epidermal development and disease. Cell Mol. Life Sci. 2018, 75, 1179–1190. [Google Scholar] [CrossRef] [PubMed]
- Laurikkala, J.; Mikkola, M.L.; James, M.; Tummers, M.; Mills, A.A.; Thesleff, I. p63 regulates multiple signalling pathways required for ectodermal organogenesis and differentiation. Development 2006, 133, 1553–1563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Rosa, L.; Antonini, D.; Ferone, G.; Russo, M.T.; Yu, P.B.; Han, R.; Missero, C. p63 Suppresses non-epidermal lineage markers in a bone morphogenetic protein-dependent manner via repression of Smad7. J. Biol. Chem. 2009, 284, 30574–30582. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, B.C.; Lefort, K.; Mandinova, A.; Antonini, D.; Devgan, V.; Della Gatta, G.; Koster, M.I.; Zhang, Z.; Wang, J.; Tommasi di Vignano, A.; et al. Cross-regulation between Notch and p63 in keratinocyte commitment to differentiation. Genes Dev. 2006, 20, 1028–1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tadeu, A.M.; Horsley, V. Notch signaling represses p63 expression in the developing surface ectoderm. Development 2013, 140, 3777–3786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morasso, M.I.; Markova, N.G.; Sargent, T.D. Regulation of epidermal differentiation by a Distal-less homeodomain gene. J. Cell Biol. 1996, 135, 1879–1887. [Google Scholar] [CrossRef]
- Chari, N.S.; Romano, R.A.; Koster, M.I.; Jaks, V.; Roop, D.; Flores, E.R.; Teglund, S.; Sinha, S.; Gruber, W.; Aberger, F.; et al. Interaction between the TP63 and SHH pathways is an important determinant of epidermal homeostasis. Cell Death Differ. 2013, 20, 1080–1088. [Google Scholar] [CrossRef] [Green Version]
- Romano, R.A.; Birkaya, B.; Sinha, S. A functional enhancer of keratin14 is a direct transcriptional target of deltaNp63. J. Clin. Invest. Dermatol. 2007, 127, 1175–1186. [Google Scholar] [CrossRef]
- Petiot, A.; Conti, F.J.; Grose, R.; Revest, J.M.; Hodivala-Dilke, K.M.; Dickson, C. A crucial role for Fgfr2-IIIb signalling in epidermal development and hair follicle patterning. Development 2003, 130, 5493–5501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Moerlooze, L.; Spencer-Dene, B.; Revest, J.M.; Hajihosseini, M.; Rosewell, I.; Dickson, C. An important role for the IIIb isoform of fibroblast growth factor receptor 2 (FGFR2) in mesenchymal-epithelial signalling during mouse organogenesis. Development 2000, 127, 483–492. [Google Scholar]
- Testoni, B.; Borrelli, S.; Tenedini, E.; Alotto, D.; Castagnoli, C.; Piccolo, S.; Tagliafico Ferrari, S.; Vigano M, A. Mantovani, R. Identification of new p63 targets in human keratinocytes. Cell Cycle 2006, 5, 2805–2811. [Google Scholar] [CrossRef] [PubMed]
- Smirnov, A.; Lena, A.M.; Cappello, A.; Panatta, E.; Anemona, L.; Bischetti, S.; Annicchiarico-Petruzzelli, M.; Mauriello, A.; Melino, G.; Candi, E. ZNF185 is a p63 target gene critical for epidermal differentiation and squamous cell carcinoma development. Oncogene 2019, 38, 1625–1638. [Google Scholar] [CrossRef] [PubMed]
- Fessing, M.Y.; Mardaryev, A.N.; Gdula, M.R.; Sharov, A.A.; Sharova, T.Y.; Rapisarda, V.; Gordon, K.B.; Smorodchenko, A.D.; Poterlowicz, K.; Ferone, G.; et al. p63 regulates Satb1 to control tissue-specific chromatin remodeling during development of the epidermis. J. Cell Biol. 2011, 194, 825–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mardaryev, A.N.; Gdula, M.R.; Yarker, J.L.; Emelianov, V.U.; Poterlowicz, K.; Sharov, A.A.; Sharova, T.Y.; Scarpa, J.A.; Joffe, B.; Solovei, I.; et al. p63 and Brg1 control developmentally regulated higher-order chromatin remodelling at the epidermal differentiation complex locus in epidermal progenitor cells. Development 2014, 141, 101–111. [Google Scholar] [CrossRef] [Green Version]
- Lin-Shiao, E.; Lan, Y.; Coradin, M.; Anderson, A.; Donahue, G.; Simpson, C.L.; Sen, P.; Saffie, R.; Busino, L.; Garcia, B.A.; et al. KMT2D regulates p63 target enhancers to coordinate epithelial homeostasis. Genes Dev. 2018, 32, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Celli, J.; Duijf, P.; Hamel, B.C.; Bamshad, M.; Kramer, B.; Smits, A.P.; Newbury-Ecob, R.; Hennekam, R.C.; Van Buggenhout, G.; van Haeringen, A.; et al. Heterozygous germline mutations in the p53 homolog p63 are the cause of EEC syndrome. Cell 1999, 99, 143–153. [Google Scholar] [CrossRef]
- Bergholz, J.; Xiao, Z.X. Role of p63 in Development, Tumorigenesis and Cancer Progression. Cancer Microenviron. 2012, 5, 311–322. [Google Scholar] [CrossRef] [Green Version]
- Rinne, T.; Spadoni, E.; Kjaer, K.W.; Danesino, C.; Larizza, D.; Kock, M.; Huoponen, K.; Savontaus, M.L.; Aaltonen, M.; Duijf, P.; et al. Delineation of the ADULT syndrome phenotype due to arginine 298 mutations of the p63 gene. EJHG 2006, 14, 904–910. [Google Scholar] [CrossRef] [Green Version]
- McGrath, J.A.; Duijf, P.H.; Doetsch, V.; Irvine, A.D.; de Waal, R.; Vanmolkot, K.R.; Wessagowit, V.; Kelly, A.; Atherton, D.J.; Griffiths, W.A.; et al. Hay-Wells syndrome is caused by heterozygous missense mutations in the SAM domain of p63. Human Mol. Genet. 2001, 10, 221–229. [Google Scholar] [CrossRef] [Green Version]
- Fomenkov, A.; Huang, Y.P.; Topaloglu, O.; Brechman, A.; Osada, M.; Fomenkova, T.; Yuriditsky, E.; Trink, B.; Sidransky, D.; Ratovitski, E. P63 alpha mutations lead to aberrant splicing of keratinocyte growth factor receptor in the Hay-Wells syndrome. J. Biol. Chem. 2003, 278, 23906–23914. [Google Scholar] [CrossRef]
- Radoja, N.; Guerrini, L.; Lo Iacono, N.; Merlo, G.R.; Costanzo, A.; Weinberg, W.C.; La Mantia, G.; Calabro, V.; Morasso, M.I. Homeobox gene Dlx3 is regulated by p63 during ectoderm development: relevance in the pathogenesis of ectodermal dysplasias. Development 2007, 134, 13–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, J.; Tanis, S.E.J.; Smits, J.P.H.; Kouwenhoven, E.N.; Oti, M.; van den Bogaard, E.H.; Logie, C.; Stunnenberg, H.G.; van Bokhoven, H.; Mulder, K.W.; et al. Mutant p63 Affects Epidermal Cell Identity through Rewiring the Enhancer Landscape. Cell Rep. 2018, 25, 3490–3503 e3494. [Google Scholar] [CrossRef] [PubMed]
- Dotto, G.P.; Rustgi, A.K. Squamous Cell Cancers: A Unified Perspective on Biology and Genetics. Cancer Cell 2016, 29, 622–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campos, M.A.; Lopes, J.M.; Soares, P. The genetics of cutaneous squamous cell carcinogenesis. EJD 2018, 28, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Di Como, C.J.; Urist, M.J.; Babayan, I.; Drobnjak, M.; Hedvat, C.V.; Teruya-Feldstein, J.; Pohar, K.; Hoos, A.; Cordon-Cardo, C. p63 expression profiles in human normal and tumor tissues. Clin. Cancer Res. 2002, 8, 494–501. [Google Scholar]
- Ha, L.; Ponnamperuma, R.M.; Jay, S.; Ricci, M.S.; Weinberg, W.C. Dysregulated DeltaNp63alpha inhibits expression of Ink4a/arf, blocks senescence, and promotes malignant conversion of keratinocytes. PLoS ONE 2011, 6, e21877. [Google Scholar] [CrossRef]
- Su, X.; Cho, M.S.; Gi, Y.J.; Ayanga, B.A.; Sherr, C.J.; Flores, E.R. Rescue of key features of the p63-null epithelial phenotype by inactivation of Ink4a and Arf. EMBO J. 2009, 28, 1904–1915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keyes, W.M.; Pecoraro, M.; Aranda, V.; Vernersson-Lindahl, E.; Li, W.; Vogel, H.; Guo, X.; Garcia, E.L.; Michurina, T.V.; Enikolopov, G.; et al. DeltaNp63alpha is an oncogene that targets chromatin remodeler Lsh to drive skin stem cell proliferation and tumorigenesis. Cell Stem Cell 2011, 8, 164–176. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xia, W.; Chen, H.; Xiao, Z.X. DeltaNp63alpha modulates phosphorylation of p38 MAP kinase in regulation of cell cycle progression and cell growth. Biochem. Biophys. Rese. Commun. 2019, 509, 784–789. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, M.R.; He, L.; Forster, N.; Ory, B.; Ellisen, L.W. Physical association of HDAC1 and HDAC2 with p63 mediates transcriptional repression and tumor maintenance in squamous cell carcinoma. Cancer Res. 2011, 71, 4373–4379. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, M.R.; Wilson, C.; Ory, B.; Rothenberg, S.M.; Faquin, W.; Mills, A.A.; Ellisen, L.W. FGFR2 signaling underlies p63 oncogenic function in squamous cell carcinoma. J. Clin. Invest. 2013, 123, 3525–3538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latil, M.; Nassar, D.; Beck, B.; Boumahdi, S.; Wang, L.; Brisebarre, A.; Dubois, C.; Nkusi, E.; Lenglez, S.; Checinska, A.; et al. Cell-Type-Specific Chromatin States Differentially Prime Squamous Cell Carcinoma Tumor-Initiating Cells for Epithelial to Mesenchymal Transition. Cell Stem Cell 2017, 20, 191–204 e195. [Google Scholar] [CrossRef]
- Lopardo, T.; Lo Iacono, N.; Marinari, B.; Giustizieri, M.L.; Cyr, D.G.; Merlo, G.; Crosti, F.; Costanzo, A.; Guerrini, L. Claudin-1 is a p63 target gene with a crucial role in epithelial development. PLoS ONE 2008, 3, e2715. [Google Scholar] [CrossRef]
- Carroll, D.K.; Carroll, J.S.; Leong, C.O.; Cheng, F.; Brown, M.; Mills, A.A.; Brugge, J.S.; Ellisen, L.W. p63 regulates an adhesion programme and cell survival in epithelial cells. Nat. Cell Biol. 2006, 8, 551–561. [Google Scholar] [CrossRef]
- Olsen, J.R.; Oyan, A.M.; Rostad, K.; Hellem, M.R.; Liu, J.; Li, L.; Micklem, D.R.; Haugen, H.; Lorens, J.B.; Rotter, V.; et al. p63 attenuates epithelial to mesenchymal potential in an experimental prostate cell model. PLoS ONE 2013, 8, e62547. [Google Scholar] [CrossRef]
- Tran, M.N.; Choi, W.; Wszolek, M.F.; Navai, N.; Lee, I.L.; Nitti, G.; Wen, S.; Flores, E.R.; Siefker-Radtke, A.; Czerniak, B.; et al. The p63 protein isoform DeltaNp63alpha inhibits epithelial-mesenchymal transition in human bladder cancer cells: role of MIR-205. J. Biol. Chem. 2013, 288, 3275–3288. [Google Scholar] [CrossRef]
- Yoh, K.E.; Regunath, K.; Guzman, A.; Lee, S.M.; Pfister, N.T.; Akanni, O.; Kaufman, L.J.; Prives, C.; Prywes, R. Repression of p63 and induction of EMT by mutant Ras in mammary epithelial cells. Proc. Nat. Acad. Sci. USA 2016, 113, E6107–E6116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stacy, A.J.; Craig, M.P.; Sakaram, S.; Kadakia, M. DeltaNp63alpha and microRNAs: leveraging the epithelial-mesenchymal transition. Oncotarget 2017, 8, 2114–2129. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez Calleja, L.; Jacques, C.; Lamoureux, F.; Baud’huin, M.; Tellez Gabriel, M.; Quillard, T.; Sahay, D.; Perrot, P.; Amiaud, J.; Charrier, C.; et al. DeltaNp63alpha Silences a miRNA Program to Aberrantly Initiate a Wound-Healing Program That Promotes TGFbeta-Induced Metastasis. Cancer Res. 2016, 76, 3236–3251. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.C.; Bu, L.L.; Mao, L.; Ma, S.R.; Liu, J.F.; Yu, G.T.; Deng, W.W.; Zhang, W.F.; Sun, Z.J. SATB1 promotes tumor metastasis and invasiveness in oral squamous cell carcinoma. Oral Dis. 2017, 23, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, K.; Pickard, A.; Craig, S.G.; Quinn, G.P.; Lambe, S.M.; James, J.A.; McDade, S.S.; McCance, D.J. DeltaNp63gamma/SRC/Slug Signaling Axis Promotes Epithelial-to-Mesenchymal Transition in Squamous Cancers. Clin. Cancer Res. 2018, 24, 3917–3927. [Google Scholar] [CrossRef] [PubMed]
- Chikh, A.; Matin, R.N.; Senatore, V.; Hufbauer, M.; Lavery, D.; Raimondi, C.; Ostano, P.; Mello-Grand, M.; Ghimenti, C.; Bahta, A.; et al. iASPP/p63 autoregulatory feedback loop is required for the homeostasis of stratified epithelia. EMBO J. 2011, 30, 4261–4273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, D.J.; Patel, A.; Purdie, K.J.; Wang, J.; Rizvi, H.; Hufbauer, M.; Ostano, P.; Akgul, B.; Chiorino, G.; Harwood, C.; et al. Epigenetic regulation of iASPP-p63 feedback loop in cutaneous squamous cell carcinoma. J. Clin. Invest. Dermatol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Li, X.; Zhang, Y.; Guo, Y.; Zhou, J.; Gao, K.; Dai, J.; Hu, G.; Lv, L.; Du, J.; et al. The microRNA feedback regulation of p63 in cancer progression. Oncotarget 2015, 6, 8434–8453. [Google Scholar] [CrossRef]
- Jiang, Y.; Jiang, Y.Y.; Xie, J.J.; Mayakonda, A.; Hazawa, M.; Chen, L.; Xiao, J.F.; Li, C.Q.; Huang, M.L.; Ding, L.W.; et al. Co-activation of super-enhancer-driven CCAT1 by TP63 and SOX2 promotes squamous cancer progression. Nat. Commun. 2018, 9, 3619. [Google Scholar] [CrossRef]
- Yang, F.; Xue, X.; Bi, J.; Zheng, L.; Zhi, K.; Gu, Y.; Fang, G. Long noncoding RNA CCAT1, which could be activated by c-Myc, promotes the progression of gastric carcinoma. J. Cancer Res. Clin. Oncol. 2013, 139, 437–445. [Google Scholar] [CrossRef]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collado, M.; Blasco, M.A.; Serrano, M. Cellular senescence in cancer and aging. Cell 2007, 130, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Jian, Z.; Strait, A.; Jimeno, A.; Wang, X.J. Cancer Stem Cells in Squamous Cell Carcinoma. J. Clin. Invest. Dermatol. 2017, 137, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Boumahdi, S.; Driessens, G.; Lapouge, G.; Rorive, S.; Nassar, D.; Le Mercier, M.; Delatte, B.; Caauwe, A.; Lenglez, S.; Nkusi, E.; et al. SOX2 controls tumour initiation and cancer stem-cell functions in squamous-cell carcinoma. Nature 2014, 511, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Saghravanian, N.; Anvari, K.; Ghazi, N.; Memar, B.; Shahsavari, M.; Aghaee, M.A. Expression of p63 and CD44 in oral squamous cell carcinoma and correlation with clinicopathological parameters. Arch. Oral Biol. 2017, 82, 160–165. [Google Scholar] [CrossRef]
- Missero, C.; Antonini, D. p63 in Squamous Cell Carcinoma of the Skin: More Than a Stem Cell/Progenitor Marker. J. Clin. Invest. Dermatol. 2017, 137, 280–281. [Google Scholar] [CrossRef] [Green Version]
- Ripamonti, F.; Albano, L.; Rossini, A.; Borrelli, S.; Fabris, S.; Mantovani, R.; Neri, A.; Balsari, A.; Magnifico, A.; Tagliabue, E. EGFR through STAT3 modulates DeltaN63alpha expression to sustain tumor-initiating cell proliferation in squamous cell carcinomas. J. Cell. Physi. 2013, 228, 871–878. [Google Scholar] [CrossRef]
- Bao, X.; Tang, J.; Lopez-Pajares, V.; Tao, S.; Qu, K.; Crabtree, G.R.; Khavari, P.A. ACTL6a enforces the epidermal progenitor state by suppressing SWI/SNF-dependent induction of KLF4. Cell Stem Cell 2013, 12, 193–203. [Google Scholar] [CrossRef]
- Mo, J.S.; Park, H.W.; Guan, K.L. The Hippo signaling pathway in stem cell biology and cancer. EMBO Rep. 2014, 15, 642–656. [Google Scholar] [CrossRef] [Green Version]
- Fisher, M.L.; Kerr, C.; Adhikary, G.; Grun, D.; Xu, W.; Keillor, J.W.; Eckert, R.L. Transglutaminase Interaction with alpha6/beta4-Integrin Stimulates YAP1-Dependent DeltaNp63alpha Stabilization and Leads to Enhanced Cancer Stem Cell Survival and Tumor Formation. Cancer Res. 2016, 76, 7265–7276. [Google Scholar] [CrossRef]
- Basu-Roy, U.; Bayin, N.S.; Rattanakorn, K.; Han, E.; Placantonakis, D.G.; Mansukhani, A.; Basilico, C. Sox2 antagonizes the Hippo pathway to maintain stemness in cancer cells. Nat. Commun. 2015, 6, 6411. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Gi, Y.J.; Chakravarti, D.; Chan, I.L.; Zhang, A.; Xia, X.; Tsai, K.Y.; Flores, E.R. TAp63 is a master transcriptional regulator of lipid and glucose metabolism. Cell Metab. 2012, 16, 511–525. [Google Scholar] [CrossRef] [PubMed]
- Hamanaka, R.B.; Mutlu, G.M. PFKFB3, a Direct Target of p63, Is Required for Proliferation and Inhibits Differentiation in Epidermal Keratinocytes. J. Clin. Invest. Dermatol. 2017, 137, 1267–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viticchie, G.; Agostini, M.; Lena, A.M.; Mancini, M.; Zhou, H.; Zolla, L.; Dinsdale, D.; Saintigny, G.; Melino, G.; Candi, E. p63 supports aerobic respiration through hexokinase II. Proc. Nat. Acad. Sci. USA 2015, 112, 11577–11582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoshan-Barmatz, V.; Keinan, N.; Zaid, H. Uncovering the role of VDAC in the regulation of cell life and death. J. Bioenerget. Biomembr. 2008, 40, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Chen, X. GPX2, a direct target of p63, inhibits oxidative stress-induced apoptosis in a p53-dependent manner. J. Biol. Chem. 2006, 281, 7856–7862. [Google Scholar] [CrossRef] [PubMed]
- Latina, A.; Viticchie, G.; Lena, A.M.; Piro, M.C.; Annicchiarico-Petruzzelli, M.; Melino, G.; Candi, E. DeltaNp63 targets cytoglobin to inhibit oxidative stress-induced apoptosis in keratinocytes and lung cancer. Oncogene 2016, 35, 1493–1503. [Google Scholar] [CrossRef] [PubMed]
- Compagnone, M.; Gatti, V.; Presutti, D.; Ruberti, G.; Fierro, C.; Markert, E.K.; Vousden, K.H.; Zhou, H.; Mauriello, A.; Anemone, L.; et al. DeltaNp63-mediated regulation of hyaluronic acid metabolism and signaling supports HNSCC tumorigenesis. Proc. Nat. Acad. Sci. USA 2017, 114, 13254–13259. [Google Scholar] [CrossRef] [PubMed]
- King, K.E.; Reddi, D.M.; Ponnamperuma, R.M.; Gerdes, M.; Weinberg, W.C. Dysregulated DeltaNp63alpha negatively regulates the maspin promoter in keratinocytes via blocking endogenous p73 binding. Mol. Carcinog. 2014, 53, 698–710. [Google Scholar] [CrossRef] [PubMed]
- Lodillinsky, C.; Infante, E.; Guichard, A.; Chaligne, R.; Fuhrmann, L.; Cyrta, J.; Irondelle, M.; Lagoutte, E.; Vacher, S.; Bonsang-Kitzis, H.; et al. p63/MT1-MMP axis is required for in situ to invasive transition in basal-like breast cancer. Oncogene 2016, 35, 344–357. [Google Scholar] [CrossRef] [PubMed]
- Cheung, K.J.; Gabrielson, E.; Werb, Z.; Ewald, A.J. Collective invasion in breast cancer requires a conserved basal epithelial program. Cell 2013, 155, 1639–1651. [Google Scholar] [CrossRef]
- Choo, M.K.; Kraft, S.; Missero, C.; Park, J.M. The protein kinase p38alpha destabilizes p63 to limit epidermal stem cell frequency and tumorigenic potential. Science signaling 2018, 11. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Otin, C.; Matrisian, L.M. Emerging roles of proteases in tumour suppression. Nat. Rev. Cancer 2007, 7, 800–808. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Carmona, M.; Hubert, P.; Gonzalez, A.; Duray, A.; Roncarati, P.; Erpicum, C.; Boniver, J.; Castronovo, V.; Noel, A.; Saussez, S.; et al. DeltaNp63 isoform-mediated beta-defensin family up-regulation is associated with (lymph)angiogenesis and poor prognosis in patients with squamous cell carcinoma. Oncotarget 2014, 5, 1856–1868. [Google Scholar] [CrossRef]
- Kubo, T.; Ichimiya, S.; Tonooka, A.; Nagashima, T.; Kikuchi, T.; Sato, N. p63 induces CD4+ T-cell chemoattractant TARC/CCL17 in human epithelial cells. J. Interferon. Cytokine Res. 2008, 28, 725–732. [Google Scholar] [CrossRef]
- Lu, H.; Yang, X.; Duggal, P.; Allen, C.T.; Yan, B.; Cohen, J.; Nottingham, L.; Romano, R.A.; Sinha, S.; King, K.E.; et al. TNF-alpha promotes c-REL/DeltaNp63alpha interaction and TAp73 dissociation from key genes that mediate growth arrest and apoptosis in head and neck cancer. Cancer Res. 2011, 71, 6867–6877. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Romano, R.A.; Si, H.; Mattox, A.; Bian, Y.; Yang, X.; Sinha, S.; Van Waes, C.; Chen, Z. Epidermal overexpression of transgenic DeltaNp63 promotes type 2 immune and myeloid inflammatory responses and hyperplasia via NF-kappaB activation. J. Pathol. 2014, 232, 356–368. [Google Scholar] [CrossRef]
- Yang, X.; Lu, H.; Yan, B.; Romano, R.A.; Bian, Y.; Friedman, J.; Duggal, P.; Allen, C.; Chuang, R.; Ehsanian, R.; et al. DeltaNp63 versatilely regulates a Broad NF-kappaB gene program and promotes squamous epithelial proliferation, migration, and inflammation. Cancer Res. 2011, 71, 3688–3700. [Google Scholar] [CrossRef]
- Kumar, S.; Wilkes, D.W.; Samuel, N.; Blanco, M.A.; Nayak, A.; Alicea-Torres, K.; Gluck, C.; Sinha, S.; Gabrilovich, D.; Chakrabarti, R. DeltaNp63-driven recruitment of myeloid-derived suppressor cells promotes metastasis in triple-negative breast cancer. J. Clin. Invest. 2018, 128, 5095–5109. [Google Scholar] [CrossRef]
- Allen, C.T.; Clavijo, P.E.; Van Waes, C.; Chen, Z. Anti-Tumor Immunity in Head and Neck Cancer: Understanding the Evidence, How Tumors Escape and Immunotherapeutic Approaches. Cancers (Basel) 2015, 7, 2397–2414. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Role | Model | Epidermal Phenotype | Reference |
|---|---|---|---|
| Morphogenesis/Stratification/ Homeostasis: Genetic Models | p63−/− | Lack of complete stratified epithelium, absence of keratin 5 or keratin 14 | [18,19] |
| Keratin 5-ΔNp63α complementation (p63−/− background) | Greater degree of epithelialization and greater amounts of keratin 5 and keratin 14 expression relative to p63−/− | [51] | |
| Keratin 5-TAp63α and keratin 5ΔNp63α complementation (p63−/− background) | Greatest degree of organized epithelialization relative to both single complementation models (TA or ΔN) | [51] | |
| Tet-keratin 5-ΔNp63α or Tet-keratin 5-ΔNp63β (p63−/− background) | Partial restoration of epidermal integrity with focal expression of keratin 5, keratin 1, and filaggrin | [52] | |
| ΔNp63α−/− (exon replaced with GFP/GFP) | Lack of complete stratified epidermis; dysregulated basal keratin expression | [53] | |
| Keratin 5-Cre mediated p63 ablation | Increased cellular senescence marker expression Embryonic: loss of stratified squamous epithelium; lack of keratins 14, 1, and 10 and filaggrin Adult: epidermal defects | [58] | |
| Tumor Development and Progression:ΔNp63α Overexpression Models | p53+/−p63+/− | Higher frequency of squamous cell carcinomas (of various organ sites) and metastatic tumors relative to p53+/− | [57] |
| p63+/− | Squamous cell hyperplasia; increased number of spontaneous tumors (including squamous cell carcinoma, organ site not specified) | [57] | |
| Keratin 5-ΔNp63α | Increased susceptibility to chemical carcinogenesis | [60] | |
| Orthotopic grafting of primary murine keratinocytes expressing oncogenic Ras and elevated ΔNp63α | Malignant conversion of keratinocytes in vivo; inhibition of cellular senescence (reduced p16 and p19 levels) | [90] | |
| Subcutaneous engraftment of primary murine keratinocytes expressing oncogenic Ras and elevated ΔNp63α | Squamous cell carcinomas; inhibition of cellular senescence (increased Lsh expression) | [92] | |
| Conditional deletion of p63 in p53-deficient mice (p63L/LK14-CreER/p53+/−) | Regression of carcinogen-induced tumors | [95] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moses, M.A.; George, A.L.; Sakakibara, N.; Mahmood, K.; Ponnamperuma, R.M.; King, K.E.; Weinberg, W.C. Molecular Mechanisms of p63-Mediated Squamous Cancer Pathogenesis. Int. J. Mol. Sci. 2019, 20, 3590. https://doi.org/10.3390/ijms20143590
Moses MA, George AL, Sakakibara N, Mahmood K, Ponnamperuma RM, King KE, Weinberg WC. Molecular Mechanisms of p63-Mediated Squamous Cancer Pathogenesis. International Journal of Molecular Sciences. 2019; 20(14):3590. https://doi.org/10.3390/ijms20143590
Chicago/Turabian StyleMoses, Michael A., Andrea L. George, Nozomi Sakakibara, Kanwal Mahmood, Roshini M. Ponnamperuma, Kathryn E. King, and Wendy C. Weinberg. 2019. "Molecular Mechanisms of p63-Mediated Squamous Cancer Pathogenesis" International Journal of Molecular Sciences 20, no. 14: 3590. https://doi.org/10.3390/ijms20143590