Recent Advances in Pharmacological and Non-Pharmacological Strategies of Cardioprotection


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
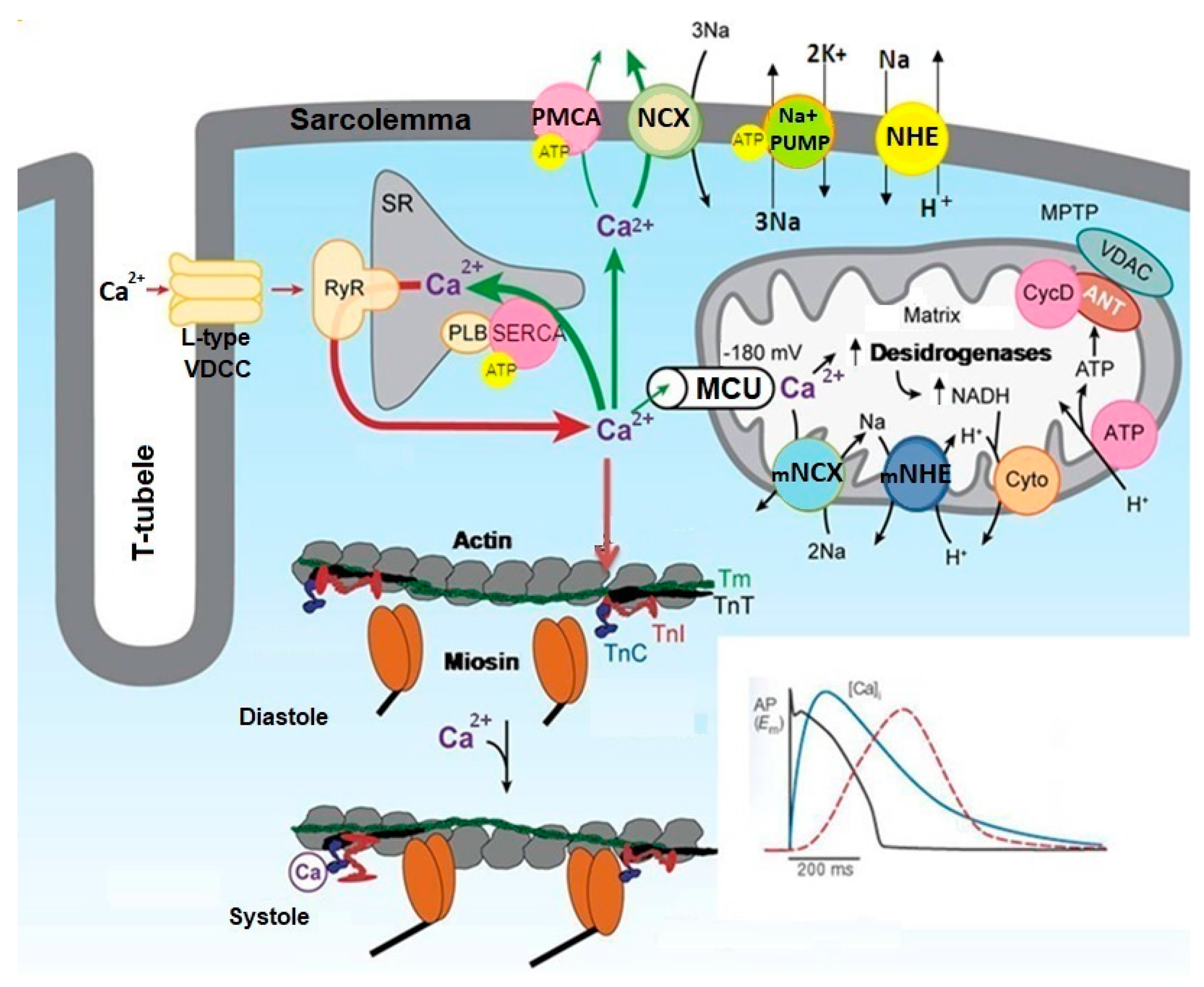
1.1. Cellular Signalling Involved in the Regulation of Cardiac Function
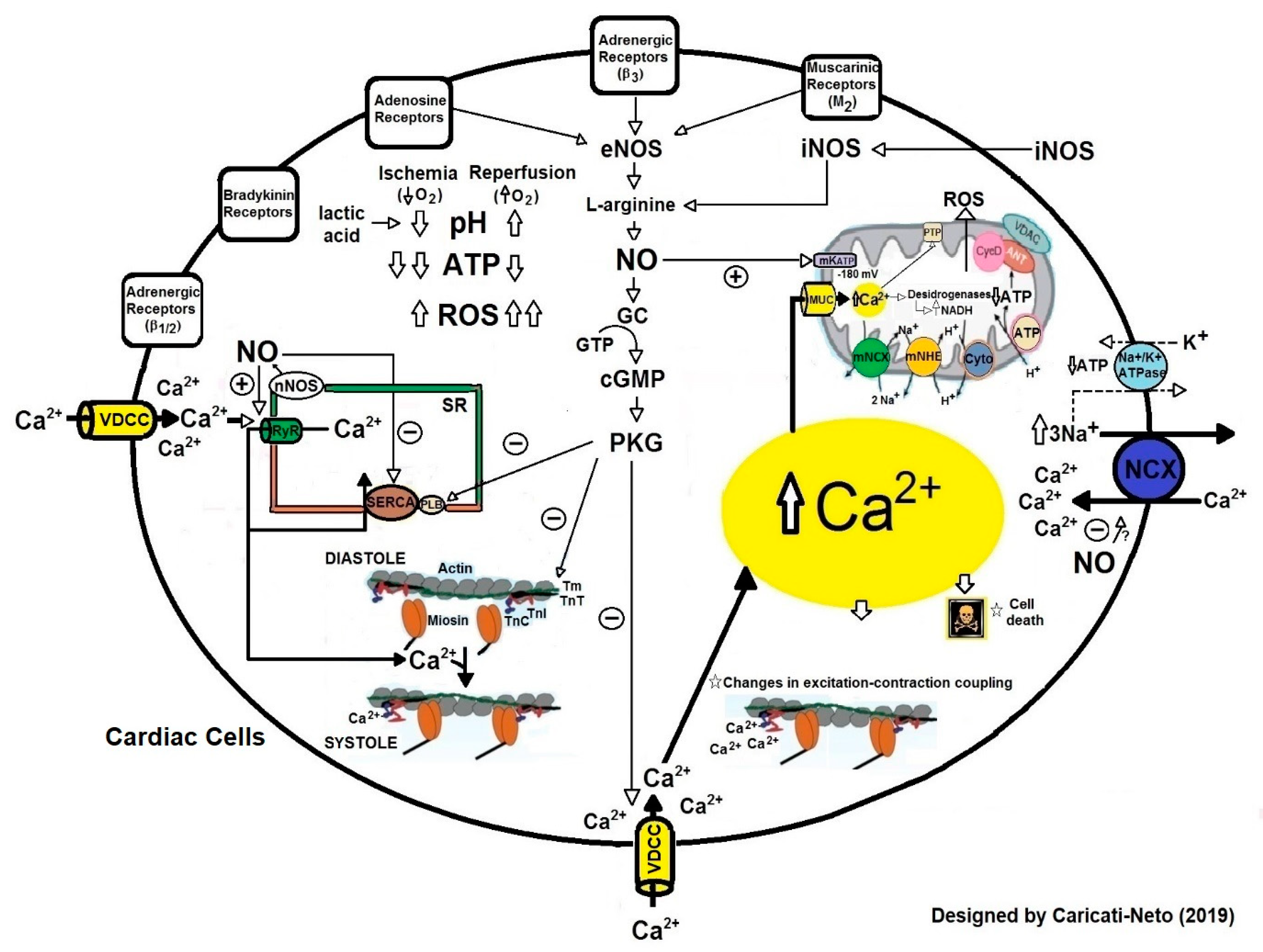
1.2. Cardiac Dysfunctions Produced by Ischemia and Reperfusion
2. Cardioprotective Strategies Against Myocardial Lesions Caused by I/R Injury
2.1. Non-Pharmacological Strategies for Cardioprotection
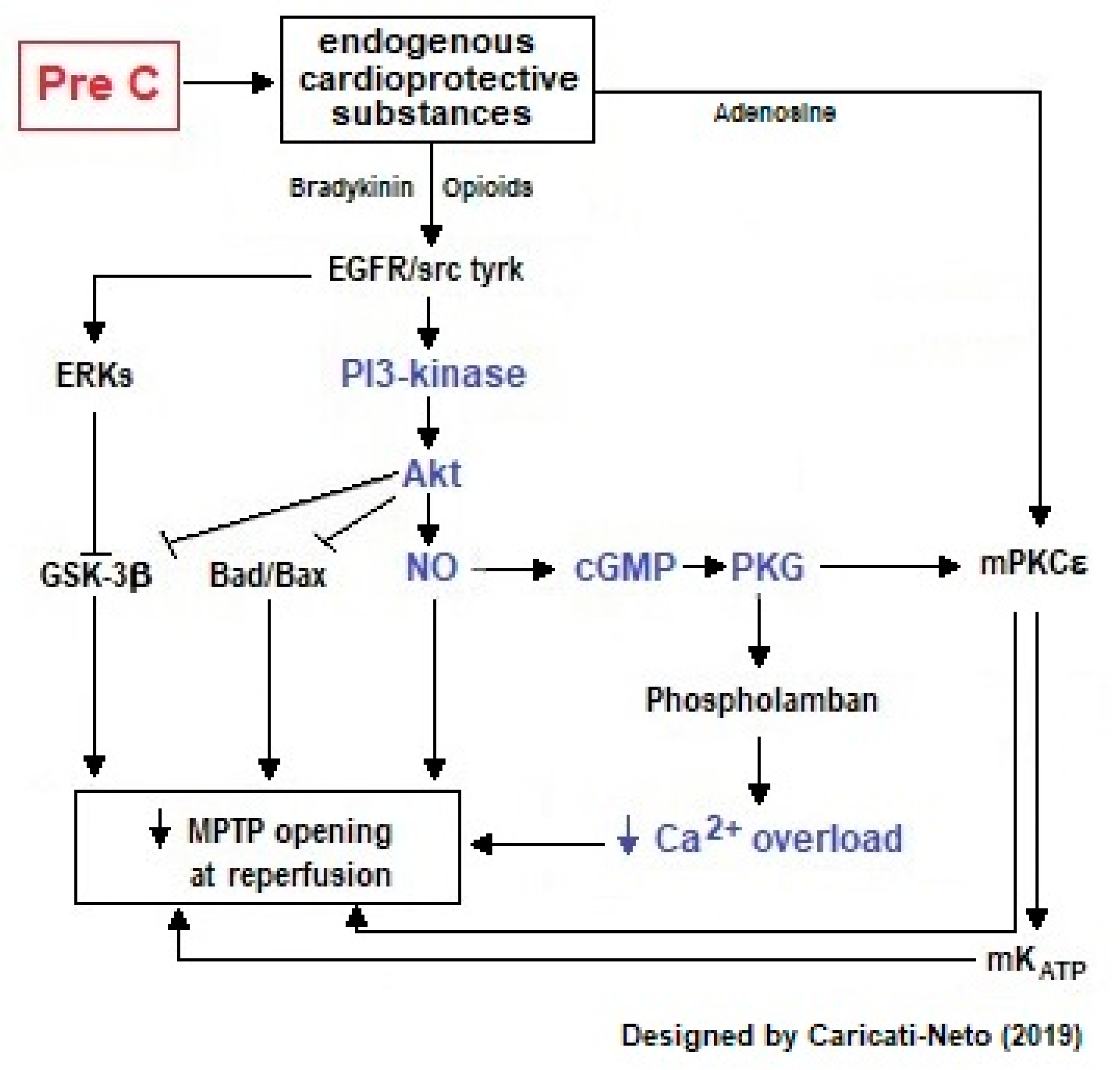
2.1.1. Cardioprotection Stimulated by Ischemic Preconditioning
2.1.2. Cardioprotection Stimulated by Ischemic Postconditioning
2.1.3. Remote Ischemic Conditioning
2.1.4. Hypothermia
2.1.5. Physiological and Pathological Conditions that Potentially Stimulate the Cardioprotective Response
2.2. Pharmacological Strategies of Cardioprotection
2.2.1. Cardioprotection Stimulated by Agonists of β-AR
2.2.2. Cardioprotection Stimulated by Agonists of Adenosine Receptors
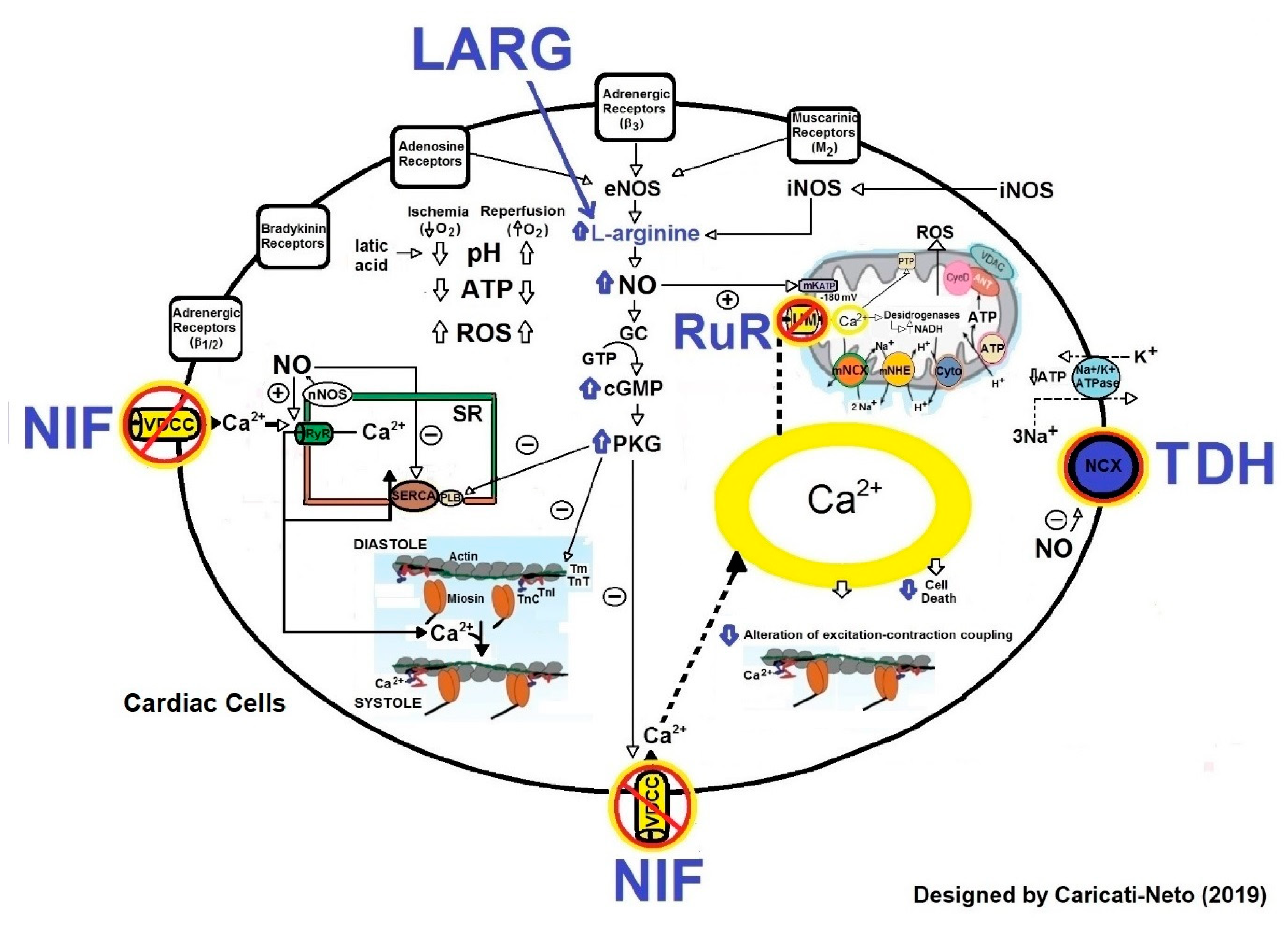
2.2.3. Cardioprotection Stimulated by Blockers of L-type VDCC
2.2.4. Cardioprotection Stimulated by Inhibitors of MUC
2.2.5. Cardioprotection Stimulated by Modulators of Plasma Membrane NCX
2.2.6. Cardioprotection Stimulated by Modulators of NO Biosynthesis
2.2.7. Cardioprotection Stimulated by Resveratrol
2.2.8. Cardioprotection Stimulated by Methylene Blue
2.2.9. Cardioprotection Stimulated by Inhibitors of Intestinal Lipase
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Akt | Protein kinase B |
| AMPK | AMP-activated protein kinase |
| APN | Adiponectin |
| AP | Action potential |
| AV | Atrioventricular |
| ATP | Adenosine triphosphate |
| β-AR | β-adrenoceptors |
| Ca2+ | Calcium ion |
| [Ca2+]c | Cytosolic Ca2+ concentration |
| [Ca2+]m | Mitochondrial Ca2+ concentration |
| CICR | Ca2+-induced Ca2+-release |
| CK | Creatine kinase |
| ECG | Electrocardiogram |
| GSK3β | Glycogen synthase kinase 3 β |
| GC | Guanylate cyclase |
| cGMP | Cyclic guanosine monophosphate |
| HDL | High density lipoproteins |
| HDSO | 2-O-desulfated heparin |
| IHD | Ischemic heart diseases |
| I/R | Ischemia and reperfusion |
| LDL | Low density lipoprotein |
| LMWH | Low molecular weight heparins |
| L-NAME | Nω-nitro-L-arginine methyl ester |
| MAPK | Mitogen activated protein kinases |
| MCU | Mitochondrial Ca2+ uniporter |
| mKATP | Mitochondrial ATP-dependent K+ channels |
| mNHE | Mitochondrial Na+/ H+-exchanger |
| MPTP | Mitochondrial permeability transition pore |
| mTOR | Mammalian target of rapamycin |
| [Na+]c | Cytosolic Na+ concentration |
| NADH | Reduced nicotinamide adenine dinucleotide |
| NCX | Na+/Ca2+-exchanger |
| NO | Nitric oxide |
| NOHA | N-hydroxy-nor-L-arginine |
| iNOS | Inducible nitric oxide synthase |
| eNOS | Endothelial nitric oxide synthase |
| PI3K | Phosphatidyl inositol 3′-hydroxy kinase |
| PKA | Protein kinase A |
| PKC | Protein kinase C |
| PKG | Protein kinase G |
| PMCA | Plasma membrane Ca2+-ATPase |
| ROS | Reactive oxygen species |
| RyR | Ryanodine receptors |
| SA | Sinoatrial |
| SERCA | Sarcoendoplasmic reticulum |
| SHR | Spontaneously hypertensive rats |
| SNO | S-nitrosylation |
| SR | Sarcoplasmic reticulum |
| TnI | Troponin I |
| TmT | Tropomyosin T |
| TnT | Troponin T |
| TDH | Trisulfated dissacharide derived from heparin |
| VDAC | Voltage-dependent anion channel |
| VDCC | Voltage-dependent Ca2+ channel |
| VGEF | Vascular endothelial growth factor |
| VLDL | Very low-density lipoprotein |
References
- Moran, A.E.; Forouzanfar, M.H.; Roth, G.A.; Mensah, G.A.; Ezzati, M.; Murray, C.J.; Naghavi, M. Temporal trends in ischemic heart disease mortality in 21 world regions, 1980 to 2010: The Global Burden of Disease 2010 study. Circulation 2014, 129, 1483–1492. [Google Scholar] [CrossRef] [PubMed]
- Pasupathy, S.; Tavella, R.; Beltrame, J.F. Myocardial infarction nonobstructive coronary arteries (MINOCA): The past, presente, and future management. Circulation 2017, 135, 1490–1491. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, R.; Giustino, G.; Mehran, R.; Windecker, S. Stable coronary artery disease: Revascularisation and invasive strategies. Lancet 2015, 386, 702–713. [Google Scholar] [CrossRef]
- Jeong, D.U.; Lim, K.M. The effect of myocardial action potential duration on cardiac pumping efficacy: A computational study. Biomed. Eng. Online 2018, 17, 79. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M. Calcium cycling and signaling in cardiac myocytes. Annu. Rev. Physiol. 2008, 70, 23–49. [Google Scholar] [CrossRef] [PubMed]
- Ríos, E. Calcium-induced release of calcium in muscle: 50 years of work and the emerging consensus. J. Gen. Physiol. 2018, 150, 521–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gambardella, J.; Trimarco, B.; Iaccarino, G.; Santulli, G. New insights in cardiac calcium handling and excitation-contraction coupling. Adv. Exp. Med. Biol. 2018, 1067, 373–385. [Google Scholar]
- Maltsev, A.V.; Maltsev, V.A.; Stern, M.D. Stabilization of diastolic calcium signal via calcium pump regulation of complex local calcium releases and transiente decay in a computational model of cardiac pacemaker cell with individual release channels. PLoS Comput. Biol. 2017, 13, e1005675. [Google Scholar] [CrossRef]
- Kiess, T.O.; Kockskamper, J. SERCA activity controls the systolic calcium increase in the nucleus of cardiac myocytes. Front. Physiol. 2019, 10, 56. [Google Scholar] [CrossRef]
- Shattock, M.J.; Ottolia, M.; Bers, D.M.; Blaustein, M.P.; Boguslavskyi, A.; Bossuyti, J.; Bridge, J.H.; Chen-Izu, Y.; Clancy, C.E.; Edwards, A.; et al. Na+/Ca2+ exchenge and Na+/K+-ATPase in the heart. J. Physiol. 2015, 593, 1361–1382. [Google Scholar] [CrossRef]
- Wei, A.C.; Liu, T.; Winslow, R.L.; O’Rourke, B. Dynamic of matrix-free Ca2+ in cardiac mitochondria: Two componentes of Ca2+ uptake and role of phosphate buffering. J. Gen. Physiol. 2012, 139, 465–478. [Google Scholar] [CrossRef] [PubMed]
- Mammuraci, C.; Rafaello, A.; Vecellio Reane, D.; Gherardi, G.; De Mario, A.; Rizzuto, R. Mitochondrial calcium uotake in organ physiology: From molecular mechanism to animal models. Pflugers Arch. 2018, 470, 1165–1179. [Google Scholar]
- Luongo, T.S.; Lambert, J.P.; Gross, P.; Nwokedi, M.; Lombardi, A.A.; Shanmughapriya, S.; Carpenter, A.C.; Kolmetzky, D.; Gao, E.; van Berlo, J.H.; et al. The mitochondrial Na+/Ca2+ exchanger is essential for Ca2+ homeostasis and viability. Nature 2017, 545, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Kostic, M.; Sekler, I. Functional properties and model of regulation of the mitochondrial Na+/Ca2+ exchanger, NCLX. Semin. Cell Dev. Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Wit, A.L. Afterdepolarizations and triggered activity as a mechanism for clinical arrhythmias. Pacing Clin. Electrophysiol. 2018. [Google Scholar] [CrossRef]
- Florea, S.M.; Blatter, L.A. The role of mitochondria for the regulation of cardiac alternans. Front. Physiol. 2010, 1, 141. [Google Scholar] [CrossRef]
- Bliksoen, M.; Baysa, A.; Eide, L.; Bjoras, M.; Suganthan, R.; Vaage, J.; Stenslokken, K.O.; Valen, G. Mitochondrial DNA damage and repair during ischemia-reperfusion injury of the heart. J. Mol. Cell. Cardiol. 2015, 78, 9–22. [Google Scholar] [CrossRef]
- Lukas, A.; Botsford, M.W. Cardioprotection induced by ischemic preconditioning in the mammalian heart: Effects on arrhythmogenesis. Can. J. Physiol. Pharmacol. 1997, 75, 316–325. [Google Scholar] [CrossRef]
- Ghosh, S.; Standen, N.B.; Galiñanes, M. Evidence for mitochondrial KATP channels as effectors of human myocardial preconditioning. Cardiovasc. Res. 2000, 45, 934–940. [Google Scholar] [CrossRef] [Green Version]
- Jovanovic, A. Cardioprotective signaling: Past, present and future. Eur. J. Pharmacol. 2018, 833, 314–319. [Google Scholar] [CrossRef]
- Balakumar, P.; Rohilla, A.; Singh, K.; Singh, M. Modulation of cardioprotective effect of ischemic pre—And postconditioning in the hyperhomocysteinemic rat heart. Methods Find. Exp. Clin. Pharmacol. 2009, 31, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Balakumar, P.; Rohilla, A.; Singh, M. Pre-conditioning and postconditioning to limite ischemia-reperfusion induced myocardial injury: What could be the next footstep. Pharmacol. Res. 2008, 57, 403–412. [Google Scholar] [CrossRef]
- Matjikava, J.; Kucharska, J.; Pinterova, M.; Pancza, D.; Ravingerova, T. Protection against ischemia-induced ventricular arrhythmias and myocardial dysfunction conferred by preconditioning in the rat heart: Involvement of mitochondrial K (ATP) channels and reactive oxygen species. Physiol. Res. 2009, 58, 9–19. [Google Scholar]
- Kolettis, T.M.; Vilaeti, A.D.; Tsalikakis, D.G.; Zoga, A.; Valenti, M.; Tzallas, A.T.; Papalois, A.; Illiodromitis, E.K. Effects of pre—And postconditioning on arrhythmogenesis in the in vivo rat model. J. Cardiovasc. Pharmacol. Ther. 2013, 18, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Stokfisk, K.; Ledakowicz-Polak, A.; Zagorski, M.; Zielinska, M. Ischaemic preconditioning-Current knowledge and potential future aplications afyer 30 years of experience. Adv. Med. Sci. 2017, 62, 307–316. [Google Scholar] [CrossRef]
- Garlid, K.D.; Halestrap, A.P. The mitochondrial K (ATP) channel—fact or fiction. J. Mol. Cell. Cardiol. 2012, 52, 578–583. [Google Scholar] [CrossRef] [PubMed]
- Garlid, A.O.; Jabureck, M.; Jacobs, J.P.; Garlid, K.D. Mitochondrial reactive oxygen species: Which ROS signals cardioprotection? Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H960–H968. [Google Scholar] [CrossRef]
- Koretsune, Y.; Marban, E. Mechanism of ischemic contracture in ferret hearts: Relative roles of [Ca2+]i elevation and ATP depletion. Am. J. Physiol. 1990, 258, H9–C16. [Google Scholar] [CrossRef]
- Wang, L.; Cherednichenko, G.; Hernandez, L.; Halow, J.; Camacho, S.A.; Figueiredo, V.; Schaefer, S. Preconditioning limits mitochondrial Ca2+ during ischemia in rat hearts: Role of K(ATP) channels. Am. J. Physiol. 2001, 280, H2321–H2328. [Google Scholar] [CrossRef]
- Korge, P.; Honda, H.M.; Weiss, J.N. Protection of cardiac mitochondria by diazoxide and protein kinase C: Implications for ischemic preconditioning. Proc. Natl. Acad. Sci. USA 2002, 99, 3312–3317. [Google Scholar] [CrossRef] [Green Version]
- Giorgio, V.; Guo, L.; Bassot, C.; Petronilli, V.; Bernardi, P. Calcium and regulation of the mitochondrial permeability transition. Cell Calcium 2018, 70, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Baxter, G.F.; Ferdinandy, P. Delayed preconditioning of myocardium: Current perspectives. Basic Res. Cardiol. 2001, 96, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Tavares, J.G.P.; Errante, P.R.; Govato, T.C.P.; Vasques, E.R.; Ferraz, R.R.N.; Taha, M.O.; Menezes-Rodrigues, F.S.; Caricati-Neto, A. Cardioprotective effect of preconditioning is more efficient than postconditioning in rats submitted to cardiac ischemia and reperfusion. Acta Cir. Bras. 2018, 33, 588–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinten-Johansen, J.; Zhao, Z.Q.; Zatta, A.J.; Kin, H.; Halkos, M.E.; Kerendi, F. Postconditioning—A new link in nature’s armor against myocardial ischemia-reperfusion injury. Basic Res. Cardiol. 2005, 100, 295–310. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.Q.; Corvera, J.S.; Halkos, M.E.; Kerendi, F.; Wang, N.P.; Guyton, R.A.; Vinten-Johansen, J. Inhibition of myocardial injury by ischemic post conditioning during reperfusion: Comparison with ischemic preconditioning. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H579–H588. [Google Scholar] [CrossRef] [PubMed]
- Halkos, M.E.; Kerendi, F.; Corvera, J.S.; Wang, N.P.; Kin, H.; Payne, C.S.; Guyton, R.A.; Vinten-Johansen, J.; Zhao, Z.Q. Myocardial protection with postconditioning is not enhanced by ischemic preconditioning. Ann. Thorac. Surg. 2004, 78, 961–969. [Google Scholar] [CrossRef] [PubMed]
- Iliodromitis, E.K.; Georgiadis, M.; Cohen, M.V.; Downey, J.M.; Bofilis, E.; Kremastinos, D.T. Protection from post-conditioning depends on the number of short ischemic insults in anesthetized pigs. Basic Res. Cardiol. 2006, 101, 502–507. [Google Scholar] [CrossRef]
- Penna, C.; Mancardi, D.; Raimondo, S.; Geuna, S.; Pagliaro, P. The paradigm of postconditioning to protect the heart. J. Cell. Mol. Med. 2008, 12, 435–458. [Google Scholar] [CrossRef]
- Laskey, W.K. Brief repetitive balloon occlusions enhance reperfusion during percutaneous coronary intervention for acute myocardial infarction: A pilot study. Catheter. Cardiovasc. Interv. 2005, 65, 361–367. [Google Scholar] [CrossRef]
- Staat, P.; Rioufol, G.; Piot, C.; Cottin, Y.; Cung, T.T.; L’Huillier, I.; Aupetit, J.F.; Bonnefoy, E.; Finet, G.; Andre-Fouet, X.; et al. Postconditioning the human heart. Circulation 2005, 112, 2143–2188. [Google Scholar] [CrossRef]
- Giustino, G.; Dangas, G.D. Ischemia-reperfusion injury and ischemic post-conditioning in acute myocardial infarction: Lost in transition. Catheter. Cardiovasc. Interv. 2017, 90, 1068–1069. [Google Scholar] [CrossRef] [PubMed]
- Menezes-Rodrigues, F.S.; Errante, P.R.; Tavares, J.G.P.; Ferraz, R.R.N.; Gomes, J.G.; Taha, M.O.; Scorza, C.A.; Scorza, F.A.; Caricati-Neto, A. Pharmacological modulation of β-adrenoceptors as a new strategy for therapy of myocardial dysfunction induced by ischemia and reperfusion. Acta Cir. Bras. 2019, 34. [Google Scholar] [CrossRef]
- Gho, B.C.; Schoemaker, R.G.; van den Doel, M.A.; Duncker, D.J.; Verdouw, P.D. Myocardial protection by brief ischemia in non-cardiac tissue. Circulation 1996, 94, 2193–2200. [Google Scholar] [CrossRef] [PubMed]
- Davies, W.R.; Brown, A.J.; Watson, W.; McCormick, L.M.; West, N.E.; Dutka, D.P.; Hoole, S.P. Remote ischemic preconditioning improves outcome at 6 years after elective percutaneous coronary intervention: The CRISP stent trial long-term follow-up. Circ. Cardiovasc. Interv. 2013, 6, 246–251. [Google Scholar] [CrossRef]
- Thielmann, M.; Kottenberg, E.; Kleinbongard, P.; Wendt, D.; Gedik, N.; Pasa, S.; Price, V.; Tsagakis, K.; Neuhäuser, M.; Peters, J.; et al. Cardioprotective and prognostic effects of remote ischaemic preconditioning in patients undergoing coronary artery bypass surgery: A single-centre randomised, double-blind, controlled trial. Lancet 2013, 382, 597–604. [Google Scholar] [CrossRef]
- Sloth, A.D.; Schmidt, M.R.; Munk, K.; Kharbanda, R.K.; Redington, A.N.; Schmidt, M.; Pedersen, L.; Sørensen, H.T.; Bøtker, H.E. CONDI Investigators. Improved long-term clinical outcomes in patients with ST-elevation myocardial infarction undergoing remote ischaemic conditioning as an adjunct to primary percutaneous coronary intervention. Eur. Heart J. 2014, 35, 168–175. [Google Scholar] [CrossRef]
- Heusch, G.; Bøtker, H.E.; Przyklenk, K.; Redington, A.; Yellon, D. Remote Ischemic Conditioning. J. Am. Coll. Cardiol. 2015, 65, 177–195. [Google Scholar] [CrossRef] [Green Version]
- Kleinbongard, P.; Skyschally, A.; Heusch, G. Cardioprotection by remote ischemic conditioning and its signal transduction. Pflügers Arch.-Eur. J. Physiol. 2017, 469, 159–181. [Google Scholar] [CrossRef] [PubMed]
- Kohlhauer, M.; Berdeaux, A.; Galeh, B.; Tissier, R. Therapeutic hypothermia to protect the heart against acute myocardial infarction. Arch. Cardiol. Dis. 2016, 109, 716–722. [Google Scholar] [CrossRef]
- Schwartz, B.C.; Kloner, R.A.; Thomas, J.L.; Bui, Q.; Mayeda, G.S.; Burstein, S.; Hale, S.L.; Economides, C.; French, W.J. Therapeutic hypothermia for acute myocardial infarction and cardiac arrest. Am. J. Cardiol. 2012, 110, 461–466. [Google Scholar] [CrossRef]
- Marongiu, E.; Crisafulli, A. Cardioprotection acquired through exercise: The role of ischemic preconditioning. Curr. Cardiol. Rev. 2014, 10, 336–348. [Google Scholar] [CrossRef] [PubMed]
- Thijssen, D.H.J.; Redington, A.; George, K.P.; Hopman, M.T.E.; Jones, H. Association of exercise preconditioning with immediate cardioprotection: A review. JAMA Cardiol. 2018, 3, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, M.A.; Sholl, H.K.; Sharrett, M.S.; Haller, S.T.; Cooper, C.C.; Gupta, R.; Liu, L.C. Exercise and cardioprotection: A natural defense against lethal myocardial ischemia-reperfusion injury and potential guide to cardiovascular prophylaxis. J. Cardiovasc. Pharmacol. Ther. 2019, 24, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Bei, Y.; Zhou, Q.; Sun, Q.; Xiao, J. Exercise as a platform for pharmacotherapy development in cardiac diseases. Curr. Pharm. Des. 2015, 21, 4409–4416. [Google Scholar] [CrossRef] [PubMed]
- Suvorava, T.; Cortese-Krott, M.M. Exercise-induced cardioprotection via eNOS; a putative role of red blood cell signaling. Curr. Med. Chem. 2018, 25, 4457–4474. [Google Scholar] [CrossRef] [PubMed]
- Feihl, F.; Liaudet, L.; Waeber, B.; Levy, B.I. Hypertension: A disease of the microcirculation? Hypertension 2006, 48, 1012–1017. [Google Scholar] [CrossRef]
- Murry, C.E.; Richard, V.J.; Reimer, K.A.; Jennings, R.B. Ischemic preconditioning slows energy metabolism and delays ultrastructural damage during a sustained ischemic episode. Circ. Res. 1990, 66, 913–931. [Google Scholar] [CrossRef] [PubMed]
- Hagar, J.M.; Hale, S.L.; Kloner, R.A. Effect of preconditioning ischemia on reperfusion arrhythmias after coronary artery occlusion and reperfusion in the rat. Circ. Res. 1991, 68, 61–68. [Google Scholar] [CrossRef]
- Vynohradova, S.V. The role of angiotensin-converting enzyme gene I/D polymorphism in development of metabolic disorders in patients with cardiovascular pathology. Tsitol. Genet. 2005, 39, 63–70. [Google Scholar]
- Alquwaizani, M.; Buckley, L.; Adams, C.; Fanikos, J. Anticoagulants: A Review of the Pharmacology, Dosing, and Complications. Curr. Emerg. Hosp. Med. Rep. 2013, 1, 83–97. [Google Scholar] [CrossRef] [Green Version]
- Belgore, F.M.; Blann, A.D.; Li-Saw-Hee, F.L.; Beevers, D.G.; Lip, G.Y. Plasma levels of vascular endothelial growth factor and its soluble receptor (SFlt-1) in essential hypertension. Am. J. Cardiol. 2001, 87, 805–807. [Google Scholar] [CrossRef]
- Wang, Z.; Niu, Q.; Peng, X.; Li, M.; Liu, K.; Liu, Y.; Liu, J.; Jin, F.; Li, X.; Wei, Y. Candesartan cilexetil attenuated cardiac remodeling by improving expression and function of mitofusin 2 in SHR. Int. J. Cardiol. 2016, 214, 348–357. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Nosaka, S.; Yamori, Y.; Matsumoto, M. Participation of neural factor in the pathogenesis of hypertension in the spontaneously hypertensive rat. Jpn. Heart J. 1967, 8, 168–180. [Google Scholar] [CrossRef] [PubMed]
- Judy, W.V.; Farrel, S.K. Arterial baroreceptor reflex control of sympathetic nerve activity in the spontaneously hypertensive rat. Hypertension 1979, 1, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Judy, W.V.; Watanabe, A.M.; Murphy, W.R.; Aprison, B.S.; Yu, P.L. Sympathetic nerve activity and blood pressure in normotensive backcross rats genetically related to the spontaneously hypertensive rat. Hypertension 1979, 1, 598–604. [Google Scholar] [CrossRef] [PubMed]
- Ohlstein, E.H.; Kruse, L.I.; Ezekiel, M.; Sherman, S.S.; Erickson, R.; DeWolf, W.E.; Jr-Berkowitz, B.A. Cardiovascular effects of a new potent dopamine beta-hydroxylase inhibitor in spontaneously hypertensive rats. J. Pharmacol. Exp. Ther. 1987, 241, 554–559. [Google Scholar]
- Zhang, Q.; Xiang, J.; Wang, X.; Liu, H.; Hu, B.; Feng, M.; Fu, Q. Beta (2)-adrenoceptor agonist clenbuterol reduces infarct size and myocardial apoptosis after myocardial ischaemia/reperfusion in anaesthetized rats. Br. J. Pharmacol. 2010, 160, 1561–1572. [Google Scholar] [CrossRef]
- Salie, R.; Moolman, J.A.; Lochner, A. The role of β-adrenergic receptors in the cardioprotective effects of beta-preconditioning (βPC). Cardiovasc. Drugs Ther. 2011, 25, 31–46. [Google Scholar] [CrossRef]
- García-Prieto, J.; García-Ruiz, J.M.; Sanz-Rosa, D.; Pun, A.; García-Alvarez, A.; Davidson, S.M.; Fernández-Friera, L.; Nuno-Ayala, M.; Fernández-Jiménez, R.; Bernal, J.A.; et al. β3 adrenergic receptor selective stimulation during ischemia/reperfusion improves cardiac function in translational models through inhibition of mPTP opening in cardiomyocytes. Basic Res. Cardiol. 2014, 109, 422. [Google Scholar] [CrossRef]
- Menezes-Rodrigues, F.S.; Tavares, J.G.P.; Errante, P.R.; Vasques, E.R.; Reis, M.C.M.; Luna-Filho, B.; Scorza, F.A.; Caricati-Neto, A.; Bergantin, L.B. Role of the Ca /cyclic AMP-Adenosine signaling pathways in cardioprotection. J. Thromb. Circ. 2017, 3, 1. [Google Scholar] [CrossRef]
- Peleli, M.; Fredholm, B.B.; Sobrevia, L.; Carlstrom, M. Pharmacological targeting of adenosine receptor signaling. Mol. Aspects Med. 2017, 55, 4–8. [Google Scholar] [CrossRef] [Green Version]
- Zhan, E.; McIntosh, V.J.; Lasley, R.D. Adenosine A2A and A2B receptors are both required for adenosine A1 receptor-mediated cardioprotection. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1183–H1189. [Google Scholar] [CrossRef]
- Dejerada, Z.; Feliu, C.; Richard, V.; Millart, H. Current knowledge on the role of P2Y receptors in cardioprotection against ischemia-reperfusion. Pharmacol. Res. 2017, 118, 5–18. [Google Scholar] [CrossRef]
- Singh, L.; Virdi, J.K.; Maslow, L.N.; Singh, N.; Jaggi, A.S. Investigating the possible mechanisms involved in adenosine preconditioning-induced cardioprotection in rats. Cardiovasc. Ther. 2018, 36, e12328. [Google Scholar] [CrossRef] [Green Version]
- Shao, Q.; Casin, K.M.; Mackowski, N.; Murphy, E.; Steenberg, C.; Kohr, M.J. Adenosine A1 receptor activation increases myocardial protein-S nitrosothiols and elicits protection from ischemia-reperfusion injury in male and female hearts. PLoS ONE 2017, 12, e0177315. [Google Scholar] [CrossRef]
- Elliot, W.J.; Ram, C.V. Calcium channel blockers. J. Clin. Hypertens. 2011, 13, 687–689. [Google Scholar] [CrossRef]
- Wang, A.L.; Iadecola, C.; Wang, G. New generations of dihydropyridines for treatment of hypertension. J. Geriatr. Cardiol. 2017, 14, 67–72. [Google Scholar]
- Walker, M.J.; Curtis, M.J.; Hearse, D.J.; Campbell, R.W.; Janse, M.J.; Yellon, D.M.; Cobbe, S.M.; Coker, S.J.; Harness, J.B.; Riemersma, R.A.; et al. The Lambeth Conventions: Guidelines for the study of arrhythmias in ischaemia infarction, and reperfusion. Cardiovasc. Res. 1988, 22, 447–455. [Google Scholar] [CrossRef]
- Mitrega, K.A.; Varghese, B.; Porc, M.; Krzeminski, T.F. Anti-arrhythmic and hemodynamic effects of oxy nifedipine, oxy nimodipine, oxy nitrendipine and oxy nisoldipine. Pharmacol. Res. 2012, 66, 300–308. [Google Scholar] [CrossRef]
- Apostolakos, M.J.; Varon, M.E. Antiarrhythmic and anti-ischemic properties of calcium-channel antagonists. New Horiz. 1996, 4, 45–57. [Google Scholar]
- Tavares, J.G.P.; Menezes-Rodrigues, F.S.; Vasques, E.R.; Reis, M.C.M.; de Paula, L.; Luna-Filho, B.; Errante, P.R.; Caricati-Neto, A.; Bergantin, L.B. A simple and efficient methodology for the study of cardioprotective drugs in animal model of cardiac ischemia-reperfusion. J. Mol. Imaging Dyn. 2017, 7, 1. [Google Scholar] [CrossRef]
- Tavares, J.G.P.; Vasques, E.R.; Menezes-Rodrigues, F.S.; Jurkiewicz, A.; Caricati-Neto, A. Cardioprotector effect of nifedipine and ruthenium red against cardiac ischemia and reperfusion injury in rats. J. Pharm. Pharmacogn. Res. 2014, 2 (Suppl. 1), S337. [Google Scholar]
- Murphy, C.E.; Wechsler, A.S. Calcium channel blockers and cardiac surgery. J. Card. Surg. 1987, 2, 299–325. [Google Scholar] [CrossRef]
- Nayler, W.G. The calcium antagonist drugs. Med. J. Aust. 1988, 149, 682–686. [Google Scholar]
- Opie, L.H. Myocardial stunning are calcium antagonists useful? Cardiovasc. Drugs Ther. 1994, 8 (Suppl. 3), 533–541. [Google Scholar] [CrossRef]
- Wang, Q.D.; Pernow, J.; Sjoquist, P.O.; Ryden, L. Pharmacological possibilities for protection against myocardial reperfusion injury. Cardiovasc. Res. 2002, 55, 25–37. [Google Scholar] [CrossRef]
- Goldbourt, U.; Behar, S.; Reicher-Reiss, H.; Zion, M.; Mandelzweig, L.; Kaplinsky, E. Early administration of nifedipine in suspected acute myocardial infarction. The Secondary Prevention Reinfarction Israel Nifedipine Trial 2 Study. Arch. Intern. Med. 1993, 153, 345–353. [Google Scholar] [CrossRef]
- Tijssen, J.G.P.; Hugenholtz, P.G. Critical appraisal of recent studies on nifedipine and other calcium channel blockers in coronary artery disease and hypertension. Eur. Heart J. 1996, 17, 1152–1157. [Google Scholar] [CrossRef] [Green Version]
- Gutstein, D.E.; Fuster, V. Pathophysiologic bases for adjunctive therapies in the treatment and secondary prevention of acute myocardial infarction. Clin. Cardiol. 1998, 21, 161–168. [Google Scholar] [CrossRef]
- Avezum, A.; Cavalcanti, A.B.; Souza, A.G.; Farsky, P.S.; Knobel, M. Adjuvant therapy in acute myocardial infarction: Evidence based recommendations. Rev. Assoc. Med. Bras. 1992, 46, 363–368. [Google Scholar] [CrossRef]
- Dagenais, F.; Cartier, R.; Holmann, C.; Buluran, J. Calcium-channel blockers preserve coronary endothelial reactivity after ischemia-reperfusion. Ann. Thorac. Surg. 1997, 63, 1050–1056. [Google Scholar] [CrossRef]
- Brown, D.A.; O’Rourke, B. Cardiac mitochondria and arrhythmias. Cardiovasc. Res. 2010, 88, 241–249. [Google Scholar] [CrossRef] [Green Version]
- Griffiths, E.J.; Balaska, D.; Cheng, W.H. The ups and downs of mitochondrial calcium signalling in the heart. Biochim. Biophys. Acta 2010, 1797, 856–864. [Google Scholar] [CrossRef] [Green Version]
- Duchen, M.R.; Szabadkai, G. Roles of mitochondria in human disease. Essays Biochem. 2010, 47, 115–137. [Google Scholar] [CrossRef] [Green Version]
- Patron, M.; Raffaello, A.; Granatiero, V.; Tosatto, A.; Merli, G.; De Stefani, D.; Wright, L.; Pallafacchina, G.; Terrin, A.; Mammucari, C.; et al. The mitochondrial calcium uniporter (MCU); molecular identity and physiological roles. J. Biol. Chem. 2013, 288, 10750–10758. [Google Scholar] [CrossRef]
- Granatiero, V.; De Stefani, D.; Rizzuto, R. Mitochondrial calcium handling in physiology and disease. Adv. Exp. Med. Biol. 2017, 982, 25–47. [Google Scholar]
- Miyamae, M.; Camacho, S.A.; Weiner, M.W.; Figueredo, V.M. Attenuation of postischemic reperfusion injury is related to prevention of [Ca2+]m overload in rat hearts. Am. J. Physiol. 1996, 271 Pt 2, H2145–H2153. [Google Scholar] [CrossRef]
- Cao, C.M.; Yan, W.Y.; Liu, J.; Kam, K.W.; Zhan, S.Z.; Sham, J.S.; Wong, T.M. Attenuation of mitochondrial, but not cytosolic, Ca2+ overload reduces myocardial injury induced by ischemia and reperfusion. Acta Pharmacol. Sin. 2006, 27, 911–918. [Google Scholar] [CrossRef]
- Di Lisa, F.; Schulz, R.; Murphy, E. Preface to mitochondria and cardioprotection. Biochim. Biophys. Acta 2011, 1813, 1261–1262. [Google Scholar] [CrossRef]
- García-Rivas, G.J.; Carvajal, K.; Correa, F.; Zazueta, C. Ru360, a specific mitochondrial calcium uptake inhibitor, improves cardiac post-ischaemic functional recovery in rats in vivo. Br. J. Pharmacol. 2006, 149, 829–837. [Google Scholar] [CrossRef]
- Zucchi, R.; Ronca-Testoni, S. The sarcoplasmic reticulum Ca2+ channel/ryanodine receptor: Modulation by endogenous effectors, drugs and disease states. Pharmacol. Rev. 1997, 49, 1–51. [Google Scholar]
- Hajnóczky, G.; Csordás, G.; Das, S.; Garcia-Perez, C.; Saotome, M.; Sinha Roy, S.; Yi, M. Mitochondrial calcium signalling and cell death: Approaches for assessing the role of mitochondrial Ca2+ uptake in apoptosis. Cell Calcium 2006, 40, 553–560. [Google Scholar] [CrossRef]
- Halestrap, A.P.; Clarke, S.J.; Khaliulin, I. The role of mitochondria in protection of the heart by preconditioning. Biochim. Biophys. Acta 2007, 1767, 1007–1031. [Google Scholar] [CrossRef] [Green Version]
- Motegi, K.; Tanonaka, K.; Takenaga, Y.; Takagi, N.; Takeo, S. Preservation of mitochondrial function may contribute to cardioprotective effects of Na+/Ca2+ exchanger inhibitors in ischaemic/reperfused rat hearts. Br. J. Pharmacol. 2007, 151, 963–978. [Google Scholar] [CrossRef]
- Antoons, G.; Sipido, K.R. Targeting calcium handling in arrhythmias. Europace 2008, 10, 1364–1369. [Google Scholar] [CrossRef]
- Barry, W.H.; Zhang, X.Q.; Halkos, M.E.; Vinten-Johansen, J.; Saegusa, N.; Spitzer, K.W.; Matsuoka, N.; Sheets, M.; Rao, N.V.; Kennedy, T.P. Nonanticoagulant heparin reduces myocyte Na+ and Ca2+ loading during simulated ischemia and decreases reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H102–H111. [Google Scholar] [CrossRef]
- Guarini, S.; Martini, M.C.; Bertolini, A. Reperfusion-Induced Arrhythmias and Lethality Are Reduced by a 2kDa Heparin Fragment. Life Sci. 1995, 57, 967–972. [Google Scholar] [CrossRef]
- Mehta, S.R.; Boden, W.E.; Eikelboom, J.W.; Flather, M.; Steg, P.G.; Avezum, A.; Afzal, R.; Piegas, L.S.; Faxon, D.P.; Widimsky, P.; et al. Antithrombotic therapy with fondaparinux in relation to interventional management strategy in patients with Stand non-ST-segment elevation acute coronary syndromes: An individual patient-level combined analysis of the Fifth and Sixth Organization to Assess Strategies in Ischemic Syndromes (OASIS 5 and 6) randomized trials. Circulation 2008, 118, 2038–2046. [Google Scholar]
- Montalescot, G.; Zeymer, U.; Silvain, J.; Boulanger, B.; Cohen, M.; Goldstein, P.; Ecollan, P.; Combes, X.; Huber, K.; ATOLL Investigators; et al. Intravenous enoxaparin or unfractionated heparin in primary percutaneous coronary intervention for ST-elevation myocardial infarction: The international randomised open-label ATOLL trial. Lancet 2011, 378, 693–703. [Google Scholar] [CrossRef]
- Kohajda, Z.; Farkas-Morvay, N.; Jost, N.; Nagy, N.; Geramipour, A.; Horváth, A.; Varga, R.S.; Hornyik, T.; Corici, C.; Acsai, K.; et al. The effect of a novel highly selective inhibitor of the sodium/calcium exchanger (NCX) on cardiac arrhythmias in in vitro and in vivo experiments. PLoS ONE 2016, 11, e0166041. [Google Scholar] [CrossRef]
- Bourgonje, V.J.; Vos, M.A.; Ozdemir, S.; Doisne, N.; Acsai, K.; Varro, A.; Sztojkov-Ivanov, A.; Zupko, I.; Rauch, E.; Kattner, L.; et al. Combined Na(+)/Ca(2+) enchanger and L-type calcium channel block as a potential strategy to suppres arrhytmias and mantain ventricular function. Circ. Arrhythmia Electrophysiol. 2013, 6, 371–379. [Google Scholar] [CrossRef]
- Demirtas, S.; Karahan, O.; Yazici, S.; Guclu, O.; Caliskan, A.; Tezcan, O.; Kaplan, I.; Yavuz, C. Investigation of possible prophylactic, renoprotective, and cardioprotective effects of thromboprophylactic drugs against ischemia-reperfusion injury. Kaohsiung J. Med. Sci. 2015, 31, 115–122. [Google Scholar] [CrossRef]
- de Godoy, C.M.G.; Vasques, E.R.; Caricati-Neto, A.; Tavares, J.G.P.; Alves, J.B.; Duarte, J.; Miranda-Ferreira, R.; Lima, M.A.; Nader, H.B.; Tersariol, I.L.D. Heparin oligosaccharides have antiarrhythmic effect by accelerating the sodium-calcium exchanger. Front. Cardiovasc. Med. 2018, 5, 67. [Google Scholar] [CrossRef]
- Knaus, H.G.; Moshammer, T.; Friedrich, K.; Kang, H.C.; Haugland, R.P.; Glossman, H. In vivo labeling of L-type Ca2+ channels by fluorescent dihydropyridines: Evidence for a functional, extracellular heparin-binding site. Proc. Natl. Acad. Sci. USA 1992, 89, 3586–3590. [Google Scholar] [CrossRef]
- Andelová, E.; Barteková, M.; Pancza, D.; Styk, J.; Ravingerová, T. The role of NO in ischemia/reperfusion injury in isolated rat heart. Gen. Physiol. Biophys. 2005, 24, 411–426. [Google Scholar]
- Ferreira, R. The reduction of infarct size-forty years of research. Rev. Port. Cardiol. 2010, 29, 1037–1053. [Google Scholar]
- Ingram, T.E.; Fraser, A.G.; Bleasdale, R.A.; Ellins, E.A.; Margulescu, A.D.; Halcox, J.P.; James, P.E. Low-dose sodium nitrite attenuates myocardial ischemia and vascular ischemia-reperfusion injury in human models. J. Am. Coll. Cardiol. 2013, 61, 2534–2541. [Google Scholar] [CrossRef]
- Sobierajski, J.; Kelm, M.; Rassaf, T. New strategies in cardioprotection during acute myocardial infarction: Impact of hypoxic nitrate-nitrite-NO signaling. Dtsch. Med. Wochenschr. 2013, 138, 799–804. [Google Scholar]
- Kumar, K.; Nguyen, K.; Waxman, S.; Nearing, B.D.; Wellenius, G.A.; Zhao, S.X.; Verrier, R.L. Potent antifibrillatory effects of intrapericardial nitroglycerin in the ischemic porcine heart. J. Am. Coll. Cardiol. 2003, 41, 1831–1837. [Google Scholar] [CrossRef] [Green Version]
- Gonon, A.T.; Jung, C.; Katz, A.; Westerlblad, H.; Shemyakin, A.; Sjoquist, P.O.; Lundberg, J.O.; Pernow, J. Local arginase inhibition during early reperfusion mediates cardioprotection via increased nitric oxide production. PLoS ONE 2012, 7, e42038. [Google Scholar] [CrossRef]
- Tripathi, P.; Misra, M.K. Therapeutic role of L-arginine on free radical scavenging system in ischemic heart diseases. Indian J. Biochem. Biophys. 2009, 46, 498–502. [Google Scholar]
- Burley, D.S.; Ferdinandy, P.; Baxter, G.F. Cyclic GMP and protein kinase-G in myocardial ischemia-reperfusion: Opportunities and obstacles for survival signaling. Br. J. Pharmacol. 2007, 152, 855–869. [Google Scholar] [CrossRef]
- Madhani, M.; Hall, A.R.; Cuello, F.; Charles, R.L.; Burgoyne, J.R.; Fuller, W.; Hobbs, A.J.; Shattock, M.J.; Eaton, P. Phospholemman Ser69 phosphorylation contributes to sildenafil-induced cardioprotection against reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H827–H836. [Google Scholar] [CrossRef] [Green Version]
- Costa, A.D.; Garlid, K.D.; West, I.C.; Lincoln, T.M.; Downey, J.M.; Cohen, M.V.; Critz, S.D. Protein kinase G transmits the cardioprotective signal from cytosol to mitochondria. Circ. Res. 2005, 97, 329–336. [Google Scholar] [CrossRef]
- Takimoto, E. Cyclic GMP-dependent signaling in cardiac myocytes. Circ. J. 2012, 76, 1819–1825. [Google Scholar] [CrossRef]
- Francis, S.H.; Bush, J.L.; Corbin, J.D.; Sibley, D. cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol. Rev. 2010, 62, 525–563. [Google Scholar] [CrossRef]
- Tavares, J.G.P.; Caricati-Neto, A.; Cukierman, S.; Godoy, C.M.G. Electrical stimulation for cardiodepression reverting in closed-loop Langendorff preparation. In Abstract Book of V Latin American Congress on Biomedical Engineering (CLAIB), Sustainable Technologies for the Health of All; Folgueras Méndez, J., Aznielle Rodríguez, T.Y., Calderón Marín, C.F., Llanusa Ruiz, S.B., Castro Medina, J., Vega Vázquez, H., Carballo Barreda, M., Rodríguez Rojas, R., Eds.; Springer: Berlin/Heidelberg, Germany, 2011; p. 33. [Google Scholar]
- Taha, M.O.; Simões, M.J.; Haddad, M.A.; Capelato, R.C.; Budny, N.; Matsumoto, A.H.; Soares, P.C.; Santos, W.M.; Armeato, G.D.; Araki, C.M.; et al. L-arginine supplementation protects against hepatic ischemia-reperfusion lesions in rabbits. Transpl. Proc. 2009, 41, 816–819. [Google Scholar] [CrossRef]
- Taha, M.O.; Caricati-Neto, A.; Ferreira, R.M.; Simões, M.J.; Monteiro, H.P.; Fagundes, D.J. L-arginine in the ischemic phase protects against liver ischemia-reperfusion injury. Acta Cir. Bras. 2012, 27, 616–623. [Google Scholar] [CrossRef] [Green Version]
- Tang, P.C.; Ng, Y.F.; Ho, S.; Gyda, M.; Chan, S.W. Resveratrol and cardiovascular health-promising therapeutic or hopeless illusion. Pharmacol. Res. 2014, 90, 88–115. [Google Scholar] [CrossRef]
- Magyar, K.; Halmosi, R.; Palfi, A.; Feher, G.; Czopf, L.; Fulop, A.; Battyany, I.; Sumegi, B.; Toth, K.; Szabados, E. Cardioprotection by resveratrol: A human clinical trial in patients with stable coronary artery disease. Clin. Hemortheol. Microcirc. 2012, 50, 179–187. [Google Scholar]
- Mukherjee, S.; Dudley, J.I.; Das, D.K. Dose-dependency of resveratrol in providing health benefits. Dose Response 2010, 8, 478–500. [Google Scholar] [CrossRef]
- Li, T.; Chen, L.; Yu, Y.; Yang, B.; Li, P.; Tan, X.Q. Resveratrol alleviates hypoxia/reoxygenation injury-induced mitochondrial oxidative stress in cardiomyocytes. Mol. Med. Rep. 2019. [Google Scholar] [CrossRef]
- Moavahed, A.; Yu, L.; Thandapilly, S.J.; Louis, X.L.; Netticadan, T. Resveratrol protects adult cardiomyocytes against oxidative stress mediated cell injury. Arch. Biochem. Biophys. 2012, 527, 74–80. [Google Scholar] [CrossRef]
- Fourny, N.; Lan, C.; Seree, E.; Bernard, M.; Desrois, M. Protective effect of resveratrol against ischemia-reperfusion injury via enhanced high energy compounds and eNOS-SIRT1 expression in type 2 diabetic female rat heart. Nutrients 2019, 11, 105. [Google Scholar] [CrossRef]
- Mokni, M.; Hamlaoui, S.; Karkouch, I.; Amri, M.; Marzouki, L.; Limam, F.; Aouni, E. Resveratrol provides cardioprotection after ischemia/reperfusion injury via modulation of antioxidant enzyme activities. Iran. J. Pharm. Res. 2013, 12, 867–875. [Google Scholar]
- Song, J.; Huang, Y.; Zheng, W.; Yan, J.; Cheng, M.; Zhao, R.; Chen, L.; Hu, C.; Jia, W. Resveratrol reduces intracellular reactive oxygen species levels by inducing autophagy through the AMPK-mTOR pathway. Front. Med. 2018, 12, 697–706. [Google Scholar] [CrossRef]
- Tian, M.; Xie, Y.; Meng, Y.; Ma, W.; Tong, Z.; Yang, X.; Lai, S.; Zhou, Y.; He, M.; Liao, Z. Resveratrol protects cardiomyocytes against anoxia/reoxygenation via dephosphorylation of VDAC1 by Akt-GSK3 β pathway. Eur. J. Pharmacol. 2019, 843, 80–87. [Google Scholar] [CrossRef]
- Thuc, L.C.; Teshima, Y.; Takahashi, N.; Nishio, S.; Fukai, A.; Kume, O.; Saito, S.; Nakagawa, M.; Saikawa, T. Inhibition of Na+-H+ exchange as a mechanism of rapid cardioprotection by resveratrol. Br. J. Pharmacol. 2012, 166, 1745–1755. [Google Scholar] [CrossRef]
- Yang, Q.; Wang, H.C.; Liu, Y.; Gao, C.; Sun, L.; Tao, L. Resveratrol cardioprotection against myocardial ischemia/reperfusion injury involves upregulation of adiponectin levels and multimerization in type 2 diabetic mice. J. Cardiovasc. Pharmacol. 2016, 68, 304–312. [Google Scholar] [CrossRef]
- Cheung, J.Y.; Wang, J.; Zhang, X.Q.; Song, J.; Tomar, D.; Madesh, M.; Judenherc-Haouzi, A.; Haouzi, P. Methylene blue counteracts cyanide cardiotoxicity: Cellular mechanisms. J. Appl. Physiol. 2018, 124, 1164–1176. [Google Scholar] [CrossRef]
- Evora, P.R. Methylene blue is a guanylate cyclase inhibitor that does not interfere with nitric oxide synthesis. Tex. Heart Inst. J. 2016, 43, 103. [Google Scholar] [CrossRef]
- Pabla, R.; Bland-Ward, P.; Moore, P.K.; Curtis, M.J. An endogenous protectant effect of cardiac cyclic GMP against reperfusion-induced ventricular fibrillation in the rat heart. Br. J. Pharmacol. 1995, 116, 2923–2930. [Google Scholar] [CrossRef] [Green Version]
- Habib, A.M.; Elsherbeny, A.G.; Alemhizia, R.A. Methylene blue for vasoplegic syndrome postcardiac surgery. Indian J. Crit. Care Med. 2018, 22, 168–173. [Google Scholar] [CrossRef]
- Haffner, S.; Taegtmeyer, H. Epidemic obesity and the metabolic syndrome. Circulation 2003, 108, 1541–1545. [Google Scholar] [CrossRef]
- Lakka, H.M.; Laaksonen, D.E.; Lakka, T.A.; Niskanen, L.K.; Kumpusalo, E.; Tuomilehto, J.; Salonen, J.T. The metabolic syndrome and total and cardiovascular disease mortality in middleaged men. JAMA 2002, 288, 2709–2716. [Google Scholar] [CrossRef]
- Gómez-Garre, D.; Muñoz-Pacheco, P.; González-Rubio, M.L.; Aragoncillo, P.; Granados, R.; Fernández-Cruz, A. Ezetimibe reduces plaque inflammation in a rabbit model of atherosclerosis and inhibits monocyte migration in addition to its lipid-lowering effect. Br. J. Pharmacol. 2009, 156, 1218–1227. [Google Scholar] [CrossRef] [Green Version]
- Halpern, A.; Mancini, M.C. Treatment of obesity: An update on anti-obesity medications. Obes. Rev. 2003, 4, 25–42. [Google Scholar] [CrossRef]
- Reitsma, J.B.; Cabezas, M.C.; de Bruin, T.W.; Erkelens, D.W. Relationship between improved postprandial lipemia and lowdensity lipoprotein metabolism during treatment with tetrahydrolipstatin, a pancreatic lipase inhibitor. Metabolism 1994, 43, 293–298. [Google Scholar] [CrossRef]
- Tzotzas, T.; Krassas, G.E.; Bruckert, E. Administration of orlistat in a patient with familial hyperchylomicronemia. Atherosclerosis 2002, 165, 185–186. [Google Scholar] [CrossRef]
- Menezes-Rodrigues, F.S.; Errante, P.R.; Ferreira, R.M.; Tavares, J.G.P.; Paula, L.; Araújo, E.A.; Govato, T.C.P.; Tikazawa, E.H.; Reis, M.C.M.; Luna-Filho, B.; et al. Cardioprotective effect of lipstatin derivate orlistat on normotensive rats submitted to cardiac ischemia and reperfusion. Acta Cir. Bras. 2018, 33, 524–532. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caricati-Neto, A.; Errante, P.R.; Menezes-Rodrigues, F.S. Recent Advances in Pharmacological and Non-Pharmacological Strategies of Cardioprotection. Int. J. Mol. Sci. 2019, 20, 4002. https://doi.org/10.3390/ijms20164002
Caricati-Neto A, Errante PR, Menezes-Rodrigues FS. Recent Advances in Pharmacological and Non-Pharmacological Strategies of Cardioprotection. International Journal of Molecular Sciences. 2019; 20(16):4002. https://doi.org/10.3390/ijms20164002
Chicago/Turabian StyleCaricati-Neto, Afonso, Paolo Ruggero Errante, and Francisco Sandro Menezes-Rodrigues. 2019. "Recent Advances in Pharmacological and Non-Pharmacological Strategies of Cardioprotection" International Journal of Molecular Sciences 20, no. 16: 4002. https://doi.org/10.3390/ijms20164002