Impact of Diabetes Mellitus on Bone Health


Regenerative Medicine Institute, National University of Ireland, Galway, Biomedical Sciences Building, Dangan, Newcastle Road, Galway City, County Galway H91W2TY, Ireland
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2019, 20(19), 4873; https://doi.org/10.3390/ijms20194873
Submission received: 27 August 2019
/
Revised: 26 September 2019
/
Accepted: 27 September 2019
/
Published: 30 September 2019
(This article belongs to the Special Issue Bone Marrow and Stem Cell Alterations in Diabetes: Causes, Consequences and Therapeutics)
{kind=link}
Abstract
:Long-term exposure to a diabetic environment leads to changes in bone metabolism and impaired bone micro-architecture through a variety of mechanisms on molecular and structural levels. These changes predispose the bone to an increased fracture risk and impaired osseus healing. In a clinical practice, adequate control of diabetes mellitus is essential for preventing detrimental effects on bone health. Alternative fracture risk assessment tools may be needed to accurately determine fracture risk in patients living with diabetes mellitus. Currently, there is no conclusive model explaining the mechanism of action of diabetes mellitus on bone health, particularly in view of progenitor cells. In this review, the best available literature on the impact of diabetes mellitus on bone health in vitro and in vivo is summarised with an emphasis on future translational research opportunities in this field.
1. Introduction
Impaired bone quality and increased fracture risk have become recognized complications of diabetes mellitus [1]. Two meta-analyses involving a total of 7,832,213 participants found an increased incidence of hip fractures in individuals living with diabetes mellitus compared to the general population, whereby those living with type 1 diabetes mellitus (T1DM) (relative risk (RR)= 5.76–6.3) show a higher incidence than individuals living with type 2 diabetes mellitus (T2DM) (RR = 1.34–1.7) [2,3]. In addition, diabetic fracture risk benefits significantly from effective clinical management, as fracture risk is higher in diabetes mellitus with poor glycemic control compared to adequately controlled diabetes mellitus [4,5].
The increased fracture risk in individuals living with diabetes mellitus is compounded by impaired fracture healing. Specifically, alterations in bone metabolism and the development of microvascular disease can prolong healing time by 87% [6]. Additionally, patients living with diabetes mellitus are predisposed to an increased risk of complications such as delayed wound closure [7], infectious complications [8], and peri-operative cardiovascular events [9]. Considering the higher incidence of diabetes mellitus and the considerable socioeconomic burden generated by fragility fractures [10], these findings draw attention to the need for an improved awareness of the factors that determine bone health and the risk of fracture in patients living with diabetes mellitus. The aim of this narrative review is to summarise the best available topical literature in order to create a better understanding of the interaction of bone health and diabetes mellitus on a molecular level, and to draw attention to future areas of research in this field. To achieve this aim, publications containing the terms “bone AND diabetes” were evaluated using PubMed Central. The search was limited to title or abstract between the years 2000 and 2019. Reference lists of the identified publications were evaluated to identify additional relevant studies.
2. Bone Mineral Density
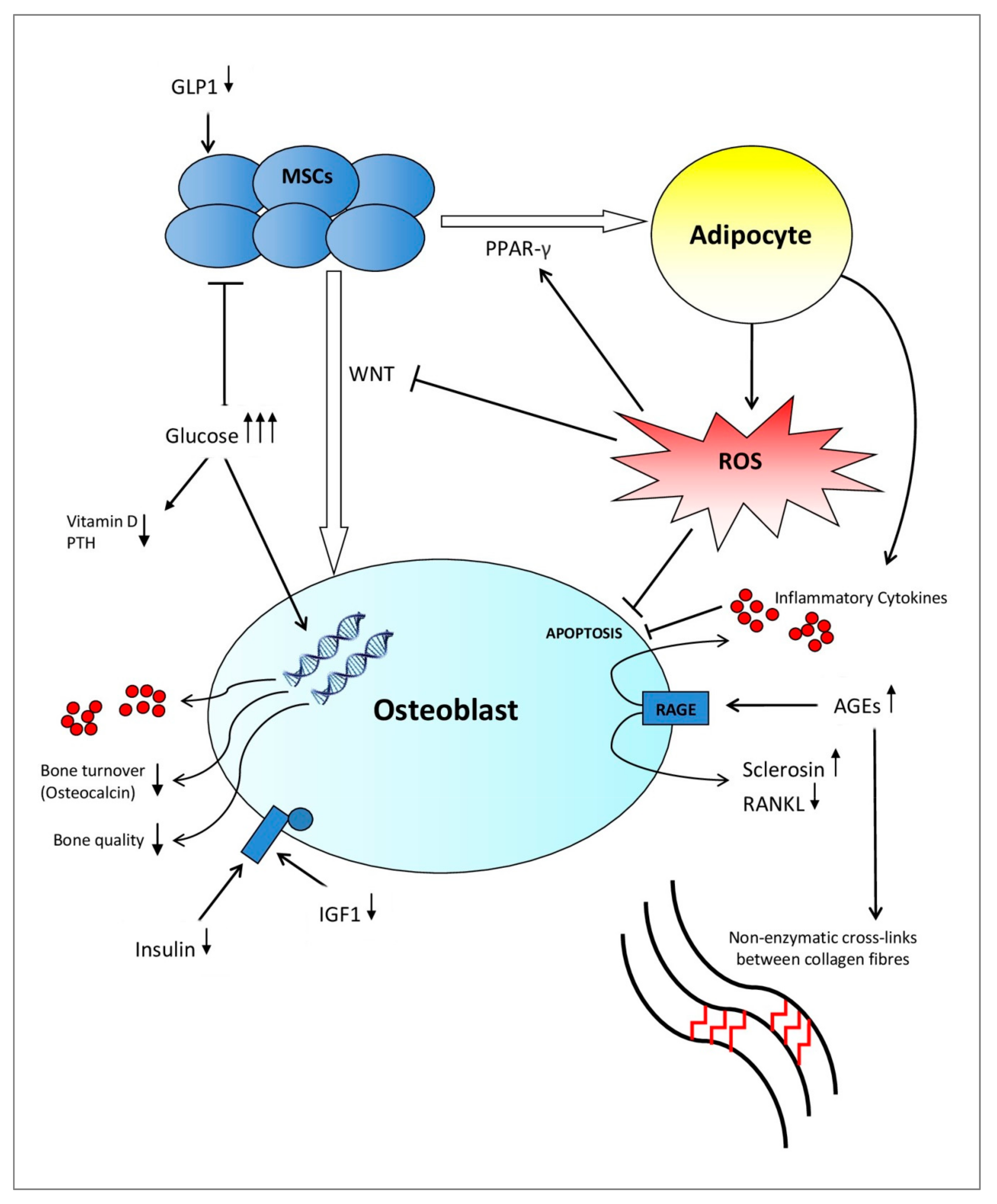
Patients living with T1DM are affected by a complete failure of β-cells of the pancreas combined with low levels of insulin-like growth factor 1 (IGF1). Both the lack of insulin, among other pancreatic anabolic hormones, and low IGF1 levels suppress the terminal differentiation of mesenchymal stem cells (MSCs) into osteoblasts in addition to osteoblastic activity [11]. Therefore, this inhibits skeletal growth at a young age, which leads to an inadequate accrual of peak bone mass [12,13,14,15,16]. On the contrary, T2DM affects bone health in advanced stages of the disease where many factors such as insulinopenia, hyperglycemia, the development of advanced glycation end products (AGEs), chronic inflammation, and microvascular disease coincide to negatively affect bone architecture and biomechanical properties of the bone (Figure 1) [17,18]. As a result, the relative risk of sustaining a hip fracture increases over the course of T2DM [1].
Whereas T1DM is associated with modest reductions in bone mineral density (BMD) (Hip Z-scores of −0.37 ± 0.16) [19] and an increase in fracture risk [20], patients living with T2DM have higher BMD (Hip Z-scores of 0.27 ± 0.16 [19]) with an increased fracture risk [19,21,22]. This contradiction can be explained as follows. Individuals living with diabetes mellitus suffer from a higher incidence of falls due to long-term complications. However, in a meta-analysis, after factoring out for increased falls as well as other confounders, such as hypoglycemic episodes and the use of anti-diabetic medications, patients with T2DM still had an increased risk of a fracture [23,24]. Therefore, the literature suggests that there is independence of fracture risk in diabetes mellitus to both changes in BMD and increased risk of falls. This can be explained by impairments of bone architecture [24].
The investigation of bone architecture in individuals living with diabetes mellitus has been facilitated by the development of non-invasive imaging techniques [25,26]. A study using high-resolution peripheral quantitative computer tomography shows T2DM is associated with a 10% higher trabecular BMD and an increase in intracortical porosity [27]. Some recent imaging studies suggest higher adiposity and an increased fraction of saturated fat in the bone marrow of patients living with diabetes mellitus. However, so far, these studies have not adjusted for obesity-related bone marrow adiposity [28,29]. Recently, changes in bone structure were directly confirmed using in vivo micro-indentation of the tibia to measure bone micro-architecture in patients with T2DM compared to the controls. These patients showed significantly increased cortical porosity and a significantly lower bone mineral strength than healthy controls [30].
3. Biochemical Impact on Bone Micro-Architecture
Extracellular bone matrix is composed of two materials. The inorganic mineral component, consisting mainly of hydroxyapatite, provides stiffness, which is the quality that is measured by a conventional BMD scan. The organic component, composed predominantly of interconnecting collagen fibers [31], provides tensile strength and counteracts shear stresses [32]. These material properties of bone tissue are regulated by cellular activity, bone tissue turnover rate, and collagen cross-link formation [32,33]. Meanwhile, these cellular activities are influenced by many environmental factors, including circulating hormones, oxidative stress, and level of glycation [34,35,36], as summarised in Figure 1.
Indirectly, many additional factors associated with hyperglycemia affect bone micro-architecture in diabetes mellitus. For example, glycosuria proportionally increases calcium excretion in urine [37]. Additionally, the interaction of hyperglycemia with the parathyroid hormone (PTH) and vitamin D system affects bone turnover in the population of patients living with diabetes mellitus (Figure 1) [34,38]. One meta-analysis in 2007 found evidence that vitamin D and calcium supplementation may be important for preventing T2DM in patients with impaired glucose tolerance [39].
3.1. Insulin Signalling
The literature suggests that insulin, as well as other pancreatic hormones, serve as anabolic factors in bone formation [34,40]. In one in vitro study, conditional disruption of the gene encoding for the IGF1 receptor in osteoblasts negatively impacts their proliferation and mineralisation. However, this defect was rescued by insulin treatment. Additionally, in vivo evidence in a murine model suggests that IGF1 plays a central role in the terminal differentiation of MSCs into osteoblasts [11]. Therefore, insulin exerts direct action in the regulation of osteoblastic activity by activation of its cell surface receptor, and IGF1 modulates the strength of the insulin-generated signal through interactions with the IGF1 receptor (Figure 1) [40]. In T1DM, absolute insulinopenia in combination with low levels/low action of IGF1 decrease bone formation by exerting an inhibitory effect on osteoblasts and their progenitor cells in the early stages of the disease [17]. However, in T2DM, this inhibitory effect caused by insulinopenia and low levels of IGF1 would be expected in advanced stages of the disease [17]. Since T1DM typically occurs in children, adolescents, and young adults, the state of absolute insulinopenia corresponds with a stage of skeletal maturation. Therefore, these studies suggest that particularly inadequately controlled T1DM will impact bone accrual and the development of peak bone mass [34].
3.2. Hyperglycemia and AGEs
A hyperglycemic environment exerts a direct and indirect effect on the function and differentiation of osteoblasts [41,42]. In vitro studies show hyperglycemia directly affects the metabolism and maturation of osteoblasts by altering gene expression [41,42] and, thereby, diminishing the quality of the bone mineral [43]. Additionally, it has been demonstrated that hyperglycemia increases the level of pro-inflammatory cytokines in humans, such as tumor necrosis factor alpha (TNF-α), interleukin 1 beta, interleukin 6, interleukin 8, and interleukin 8 [43,44] while simultaneously increasing the receptor activator of nuclear factor kappa-Β ligand (RANKL) expression [43], which mediates osteoblast death and osteoclastogenesis, respectively (Figure 1) [35]. Since inflammatory factors are elevated in the early stages of T1DM [45], the above named pro-inflammatory cytokines could play a role in the inhibited accrual of bone mass [46].
The evidence shows that oxidative stress and a hyperglycemic metabolic state, which are induced and maintained by diabetes mellitus, lead to the accelerated formation of AGEs (for example, pentosidine) [35,47,48,49]. AGEs cross-link with collagen fibers in both trabecular and cortical bone [50], which leads to a more brittle bone with a deterioration of post-yield properties (making bones less able to deform before fracturing) (Figure 1) [36,51]. In contrast, physiological enzymatic cross-links between collagen fibers provide a beneficial effect on the quality and strength of the bone [36]. In spontaneously diabetic WBN/Kob rats, a steady decrease of beneficial enzymatic cross-links coupled with a steady increase of pentosidine was reported after onset of diabetes mellitus. Additionally, impaired bone biomechanics coincided with these alterations in collagen cross-linking, despite no alterations in BMD values [52]. Therefore, AGEs are thought to deteriorate biomechanical function of the bone by altering the physical properties of bone collagen, which results in bone fragility [53].
Accompanying the alteration in collagen cross-links, AGEs affect bone tissue by directly interfering with the development [54] and function [55] of bone cells. AGEs affect the phenotypic expression of osteoblasts in vitro, in particular inhibiting nodule formation of osteoblasts in a cell culture [56]. In addition, AGEs may decrease bone resorption by inhibiting osteoclastic differentiation activity and, thereby, altering the structural integrity of the collagen matrix [57]. It has been established that the osteoblastic function is disrupted by AGEs by upregulating the cell surface receptor for advanced glycation end products (RAGE) located on osteoblasts (Figure 1) [58,59]. These receptors have been shown to increase the production of pro-inflammatory cytokines, which may feed a cycle of increased bone resorption and chronic inflammation [60]. Furthermore, one study shows that treating an osteocytic cell line with AGEs increases sclerostin expression and decreases RANKL expression. Therefore, this suppresses bone formation and bone resorption, respectively [61]. These adverse effects of AGEs on bone cells serve to further accelerate bone fragility in diabetes mellitus [33]. Galectin-3 protein in bone tissue has been shown to play an important role in the uptake and removal of AGEs whereby Galectin-3 exerts the opposite action on AGE-receptor to RAGE. Therefore, this potentially serves as a protective factor in diabetes mellitus-related AGE accumulation [62,63].
4. Epigenetic Changes
Large clinical trials have shown that diabetic complications in T1DM and T2DM continue to progress after patients return to adequate glycemic control [64,65,66,67,68,69]. Additionally, it is known that HbA1c merely accounts for 25% of the variation in the risk of developing complications, which implies that transient hyperglycemic episodes lead to lasting cellular changes [66,70]. Recent investigations, particularly in murine models of cardiovascular disease, have begun to shed light on the patho-mechanism of metabolic memory in diabetes mellitus, which leads to the development of end-organ damage [64,71,72,73,74,75]. For instance, microRNA (miRNA)-155 was decreased in streptozotocin-induced diabetic rats and negatively correlated to NF-κB activity and an apoptosis rate [76]. This was reflected in a study showing a downregulation of miRNA-155 in bone-marrow derived progenitor cells isolated from humans living with T2DM [77]. In a clinical study, gene expression of p66Shc in peripheral mononuclear cells was correlated with new onset complications in patients living with diabetes mellitus with similar baseline characteristics [78]. These recent findings draw attention to the importance of early and aggressive treatment of uncontrolled diabetes mellitus. Uncovering epigenetic therapeutic targets will open opportunities for the development of drugs to improve patients’ outcome after glucose homeostasis has been achieved [65,79,80].
5. Bone Turnover
The effect of a diabetic environment on bone metabolism can be indirectly measured through bone turnover markers. Specifically, osteocalcin is produced by osteoblasts and is a marker of bone formation [81]. Children suffering from T1DM were found to have low levels of osteocalcin, which were negatively correlated with HbA1c levels [82,83]. Derivatives of furanocoumarins reversed the suppression of osteocalcin and diabetes mellitus-associated decreased trabecular thickness in diabetic mice, in addition to significantly suppressing osteoclast-related gene expression such as RANKL [84]. When comparing T1DM and T2DM, osteocalcin serum levels are decreased in individuals living with T1DM and significantly decreased in T2DM compared to healthy controls [82,85,86,87]. Alternatively, sclerostin is a marker for bone resorption [81] and is inversely correlated to bone turnover markers for bone formation in patients living with T2DM [88,89,90]. However, changes in sclerostin levels have not been confirmed for individuals living with T1DM [88]. Bone turnover markers could potentially be a means of predicting the fracture risk in patients living with diabetes mellitus in the future [91,92,93].
“Signature miRNAs” of bone turnover, such as miR-148a-3p, are known as biomarkers in primary osteoporosis [94,95,96]. In 2016, Heilmeyer et al. studied circulating miRNAs and identified combinations of miR-550a-5p, miR-96-5p, miR-382-3p, and miR-181c-5p associated with T2DM-induced fragility fractures with a high specificity and sensitivity [97]. This study also included an in vitro analysis to measure the effect of miR-550a-5p, miR-382-3p, and miR-188-3p on adipose tissue-derived MSCs. Interestingly, miR-382-3p was found to stimulate osteogenic differentiation and inhibit adipogenesis. This could be explained by the fact that the level of miR-382-3p was seven times lower in fractured patients living with T2DM compared to T2DM without a history of fragility fractures. On the contrary, miR-550a-5p was upregulated 22-fold in the diabetes fracture group compared to non-fracturing patients living with T2DM, and was shown to be a strong inhibitor of osteogenesis [97]. In T1DM, hyperexpression of miR-148a and miR-21-5p was observed in the sera of patients, which was associated with decreased BMD and increased circulating PTH levels [98].
Studies examining the effect of diabetes mellitus on osteoclasts are not conclusive. In vitro and animal studies report an unaltered rate of bone resorption [99,100], whereas some studies have suggested increased osteoclastic activity in diabetes mellitus under certain conditions, such as periodontal disease [101] and osteoporosis [102]. Other studies have even reported inhibited osteoclast function and differentiation in a diabetic environment [103,104,105]. Due to the conflicting evidence and generally negligent effect that has been observed in osteoclasts, it seems likely that the impaired bone formation in diabetes mellitus is primarily due to inhibited osteoblastic and progenitor cell activity rather than an alteration of bone resorption. However, further research is needed to clarify the effect of diabetes mellitus on osteoclastic function and differentiation.
6. Fracture Risk
Altered biomechanical properties of the bone due to deteriorations in bone microarchitecture predispose individuals living with diabetes mellitus to fragility fractures [106,107,108]. Individuals living with T2DM and T1DM carry a higher risk of sustaining a fracture at most skeletal locations compared to the general population, whereby hip fractures in T2DM has been most extensively examined [109,110,111]. T1DM is reported to be associated with a higher odds ratio for hip fractures compared to hip fractures in patients living with T2DM in a meta-analysis [19]. When fractures are compared by anatomical location in T2DM, women living with diabetes mellitus have a significantly increased risk of hip, pelvis, upper leg, foot, and vertebral fractures [112]. Additionally, diabetes mellitus is a negative prognostic factor for post-fracture mortality among patients with hip fractures [17,113,114]. However, despite the increased fracture risk, patients with T2DM show a higher BMD at the femoral neck and lumbar spine in conventional Dual-energy X-ray absorptiometry (DEXA) scans [115].
Accumulation of AGEs, specifically pentosidine, is associated with a fracture incidence in older adults living with diabetes mellitus, as demonstrated by Schwartz et al. in the Health Aging and Body Composition study [116]. Similarly, a high level of urinary excretion of pentosidine in non-diabetic patients was an independent risk factor for vertebral fractures [117]. One clinical study shows increased cortical bone AGEs in T2DM patients [118]. Additionally, another study reports that trabecular bone from fracturing T1DM patients has significantly higher levels of pentosidine than non-fracturing T1DM [119], even though this does not imply causality. Large retrospective studies have shown that conventional models for predicting fracture risk such as BMD and the Fracture Risk Assessment Tool (FRAX) underestimate the fracture risk for patients living with diabetes mellitus due to secondary impairments in bone micro-architecture [120,121]. However, the trabecular bone score, which is related to the bone micro-architecture, was shown to predict fractures in patients suffering from diabetes mellitus with greater accuracy [122,123,124].
7. Fracture Healing
In usual fracture healing, a stabilising callus is formed, in which cartilage is formed and then reabsorbed and replaced by bone tissue. This is facilitated by blood supply to the healing site [125]. In animal models of fracture healing, many studies have suggested diabetes mellitus is associated with an impaired healing response [126,127,128,129,130]. In a diabetic murine model, the animals were shown to have an increased concentration of TNF-α at the fracture site, which was linked to an increased rate of cartilage resorption [127]. Additionally, a diabetic cell environment may lead to a reduction in callus size and bone formation and, thereby, a decrease in the mechanical strength of the repaired fracture site [126,127,128]. In one in vivo study, decreased cell proliferation as well as decreased mechanical stiffness was shown at the fracture site of poorly controlled diabetic rats. However, rats with a tight insulin treatment maintained physiological fracture healing [131]. In healthy human individuals, there is a fracture response during the first few weeks of recovery marked by a peak in osteocalcin, alkaline phosphatase (ALP), and IGF1, which indicates increased bone turnover [132,133]. However, in individuals living with diabetes mellitus, bone turnover markers post-fracture are diminished [134], which could possibly be a symptom of disturbed fracture consolidation.
Fracture healing is intimately associated with progenitor cell population and functionality [135,136]. One study demonstrates atrophic non-union fractures are associated with a decreased pool of MSCs, which alters the level of chemokines involved in fracture healing [137]. Therefore, insufficient MSC availability may impede callus remodeling and result in callus material that is biomechanically inferior in patients living with diabetes mellitus [130,138,139,140]. Long-term complications of patients living with diabetes mellitus include microvascular complications [141], where complications such as fracture non-union are linked to vascular insufficiencies in the fracture site [142,143]. Since vascularization is mediated by MSCs [144,145], vascular deficiencies may be further impaired in diabetic fracture healing due to the reduced population and potential of progenitor cells and chronic inflammatory environment. Several studies have shown a decreased expression of angiogenic genes (VEGF-A, VEGF-C, angiopoietin 1, and angiopoietin 2) and proteins in MSCs isolated from humans living with diabetes mellitus [146,147]. In addition to these impediments, patients living with diabetes mellitus have a greater risk of wound infection, local post-operative complications such as impaired wound healing, and peri-operative cardiovascular complications compared to non-diabetic individuals [6,8,9,148].
8. Effect of Diabetes on Progenitor Cells
Adipocytes and osteoblasts are derived from a common precursor, the MSC. The differentiation of MSC is influenced by the interaction of several different pathways (Figure 1). The WNT signaling and peroxisome proliferator-activated receptor gamma (PPAR-γ) pathways regulate a fine balance between adipogenesis and osteo-blastogenesis [149]. The activation of the WNT signaling pathway promotes osteogenesis and inhibits adipogenesis. On the contrary, PPAR-γ, which is mediated by reactive oxygen species (ROS) [150], facilitates the differentiation of MSCs into adipocytes [18]. In one study, muscle-derived MSCs cultured in high glucose media showed a higher expression of adipogenesis markers (PPAR-γ, LPL, adiponectin, GLUT4, and SREBP1c) and a down-regulation of chondrogenic and osteogenic markers compared to cells cultured in a low glucose media [150]. In a similar model, gene expression associated with osteoblast differentiation was decreased, with a simultaneous increase in cells of an adipocyte phenotype in a hyperglycemic environment [151]. A recent study utilising rat bone-marrow derived (BM-)MSCs has suggested that hyperglycemia activates the Notch2 signaling pathway, which was negatively correlated with ALP expression levels. This inhibited osteo-blastogenesis [152]. Additionally, hyperglycemia has been shown to increase production of sclerostin, which induces adipogenesis by inhibiting WNT signaling in human BM-MSCs [153].
Some recent animal studies have shown higher bone marrow adiposity in diabetic models [151,154], which suggests the hypothesis that bone marrow fat composition may be a mechanism of diabetic fragility fractures [155,156]. In humans, one study measured a significantly higher bone marrow fat content in addition to predominant saturated lipid fraction in the diabetes mellitus group compared to healthy controls using proton magnetic resonance spectroscopy [157]. Similarly, another study demonstrated an alteration of bone marrow saturated to unsaturated fat composition using magnetic resonance imaging [29]. However, first, animal models are not consistently predicative of human responses [158], and, second, clinical studies showing increased bone marrow adiposity in diabetes mellitus have not ruled out obesity as a confounding factor. Patho-physiologically, T2DM is associated with insulin resistance. Therefore, cells from patients living with diabetes mellitus are less likely to accumulate lipids [159]. Increased bone marrow adiposity is known to correlate with altered levels of growth hormones, increased visceral adiposity, increased circulating lipids, and hypoleptinemia [28]. However, there is currently no evidence that suggests that diabetes mellitus directly accounts for increased bone marrow adiposity in humans.
Recent investigations have shed light on impaired metabolic pathways in obesity, which results in chronic inflammation and insulin resistance. Therefore, this pre-disposes obese individuals to developing diabetes mellitus. White adipose tissue (WAT) in individuals living with diabetes mellitus has been shown to exhibit high levels of inflammation compared to WAT of obese individuals without diabetes mellitus [160]. Hypoxic conditions in adipose tissue caused by decreased perfusion of hypertrophic adipocytes leads to an upregulation of hypoxia-inducible factor 1-alpha (HIF-1α among other inflammatory genes [161,162]. Increased levels of inflammatory cytokines, in particular TNF-α, has been shown to induce insulin resistance [163,164]. Additionally, free fatty acids released by adipocytes produce ROS, which, in addition to hyperglycemia, exacerbates inhibited osteoblast proliferation and function maintained by a diabetic environment [165,166,167,168].
Thus, in vitro models have suggested that chronic inflammation in diabetes mellitus occurs as a result of a hyperglycemic bone marrow environment combined with oxidative stress, which inhibits the maturation of osteoblasts, and leads to a shift of MSC differentiation from osteo-blastogenesis to adipogenesis [136,169,170]. This leads to a vicious cycle of metabolic stress, which upholds a chronic inflammatory process that may de-mineralise trabecular bone [171], and result in the increased production of ROS, which has a direct impact on the differentiation and function of MSCs, osteoclasts, osteoblasts, and osteocytes [172]. In fact, the emerging understanding of T2DM as a cycle of chronic inflammation has opened windows to the development of anti-inflammatory treatment approaches [173].
A streptozotocin-induced T2DM diabetic mouse model showed evidence of suppressed expression of transcription factors required for the osteoblastic differentiation of MSCs in vitro [134]. This has been confirmed in a T2DM mouse model, where diabetic animals possessed fewer viable MSCs, which were functionally impaired ex vivo [174]. Exposing healthy cultured human MSCs to hyperglycemia, AGEs, and oxidative stress reduces the viable MSC population [54]. Thus far, only one study has been carried out to compare BM-MSCs isolated from individuals living with T1DM and healthy controls. This study suggested that BM-MSC cell count, cell morphology, and growth kinetics are not impaired despite long-term exposure to a diabetic stem cell environment in a young demographic [175]. However, to date, no studies have shown the effect of a diabetic environment on human MSCs isolated from individuals living with T2DM [176].
The sympathetic nervous system is responsible for mobilizing hematopoietic stem cells (HSCs) into the circulation, which have been shown to be inversely correlated with cardiovascular events in clinical studies [177,178]. It has been suggested that diabetes mellitus leads to remodeling and autonomic neuropathy of the bone marrow. Therefore, this affects the level of CD34+ cells in the blood [179]. These changes were averted in p66Shc knockout mice and are associated with the downregulation of the Sirt1 gene [180,181,182,183]. In a murine model, an insulin-resistant hyperglycemic environment leads to epigenetic changes in bone marrow via activation of JMJD3, a histone H3K27 demethylase, which leads to the increased expression of inflammatory cytokines. These changes persisted in peripheral monocytes, which leads to the hypothesis that epigenetic changes in the diabetic bone marrow environment leads to altered macrophage function and persistent wound inflammation [74]. Dipeptidyl peptidase-4 (DPP-4) inhibition has been shown to increase circulating HSCs in humans, which suggests that DPP-4 dysregulation plays a central role in diabetes mellitus-induced impaired HSC mobilization [184,185].
9. Effects of Insulin and Anti-Diabetic Drugs
Mice lacking an insulin receptor substrate, a mediator of insulin and IGF1 signaling, showed decreased bone formation and osteopenia due to reduced differentiation of osteoblasts [186,187], growth retardation, and a 60-fold higher expression of a hepatic IGF binding protein [188]. Additionally, osteoblasts lacking the insulin receptor substrate gene in an ex vivo model showed an upregulation of receptor activator of RANKL expression. Therefore, this stimulates osteo-clastogenesis in co-culture [186]. Conversely, a murine model of non-obese T2DM showed a reduced bone turnover rate, which was recovered by insulin treatment [189]. In humans living with T1DM, the incidence of osteoporosis or osteopenia was found to be significantly higher in patients before insulin treatment. After seven years of insulin treatment, bone turnover markers and BMD at all anatomical sites had significantly improved [190]. Although insulin is anabolic to bone and can restore markers of bone turnover and BMD, systematic review have identified no significant fracture reducing the potential for individuals living with diabetes mellitus on insulin treatment [191,192]. In fact, some epidemiological reports have shown an increased fracture risk in patients taking insulin, which may be secondary to an increased falls risk [192].
Metformin is routinely prescribed to patients as a first-line treatment T2DM, as recommended by consensus guidelines [193]. One population study has described metformin as having a potentially positive influence on fracture risk [191,194]. However, it is not clear whether this effect is secondary to blood sugar level optimisation or metformin directly interacting with progenitor cells to affect bone metabolism. In vitro studies examining the effect of metformin on MSCs have shown conflicting results. In rodent BM-MSCs, metformin stimulated osteoblastic activity and blocked adipogenesis [195]. Studies show decreased osteoclastogenesis in murine-derived preosteoclasts using supra-pharmacological concentrations of metformin [196,197,198]. However, some in vitro studies have shown MSC apoptosis following transplantation and decreased angiogenic potential of human MSCs treated with metformin [199,200]. In human-induced pluripotent MSCs, metformin enhanced osteoblastic activity by increasing ALP activity and mineralized nodule formation, which was partly mediated by the LKB1/AMPK pathway [201]. Bone turnover markers were measured following treatment with metformin in a clinical study [202,203], which showed decreased bone resorption (CTX-1) and a large decrease in bone formation (P1NP). However, this lacked a control arm [203].
After an initial response to metformin, many patients require additional anti-diabetic medications. Glitazones have detrimental effects on bone health and are, therefore, rarely prescribed [202,204]. The “incretin effect” (increased stimulation of insulin elicited by oral administration of glucose [205]) is proven to be significantly lower in diabetes mellitus compared to healthy subjects after a meal [206]. In murine models, the administration of the glucagon-like peptide 1 (GLP1), which is a hormone that facilitates the ‘incretin effect,’ has been shown to increase bone formation markers [207] and prevent the deterioration of the bone micro-architecture [208]. In vitro studies have shown GLP1 stimulates the proliferation of human MSCs and inhibits their differentiation into adipocytes [209] through GLP1 receptors expressed on progenitor cells [209,210].
GLP1 receptor analogues (GLP1RAs) are increasingly used because they aid weight loss and do not pose a risk of hypoglycemia [211]. One clinical study showed that the serum markers of calcium homeostasis (ALP, calcium, and phosphate) remained unaffected by exenatide treatment [212]. Additionally, a recent meta-analysis found no significant relationship between the use of GLP1RAs and fracture risk in T2DM in humans [213]. DPP-4 inhibitors are the second class of anti-diabetic drugs, which are designed to increase GLP1 levels. Recent reports have highlighted the impact of DPP-4 on circulating progenitor cells, which potentially ameliorates cardiovascular risk by facilitating HSC mobilization [185,214,215]. Nonetheless, thus far, meta-analysis has not established a cardiovascular benefit using DPP-4 inhibitors in patients [216]. Further translational research is required to thoroughly investigate the discrepancy between pre-clinical and clinical results.
In contrast, there is a strong evidence suggesting that treatment with sodium glucose cotransporter- 2 (SGLT-2) inhibitors positively affects cardiovascular and renal outcome in patients with T2DM [217,218,219]. Therefore, it has been hypothesized that this protective effect is caused by the increased mobilization of pro-vascular progenitor cells in bone marrow [220]. In one clinical trial, circulating CD133+ progenitor cells and monocytes with an anti-inflammatory phenotype were significantly raised and pro-inflammatory granulocyte precursors were significantly decreased following six months of treatment with empagliflozin [220]. A similar study measuring the effect of dapafliflozin showed an increase of CD34+KDR+ endothelial progenitor cells, which concurred with improvement in HbA1c, whereas circulating stem cells remained stable. This implies that the cardiovascular benefit may not directly involve circulating progenitor cells [221]. Despite these important advances, the mechanism of the cardiovascular and renal benefit of SGLT-2 inhibitors is still unknown. Furthermore, the epigenetic impact of these novel drugs on diabetes mellitus-induced bone fracture risk remains unexplored [222].
10. Conclusions
Recent literature shows that the fracture risk in diabetes mellitus increased more significantly than can be explained by changes in BMD and confounding factors, such as risk of falls [19,23]. Rather than influencing the mineral phase (BMD), it is thought that a diabetic environment primarily affects biomechanical properties of the bone by deteriorating its organic composition and bone material strength [29,30,33]. This occurs either directly through altered cross-link formation or indirectly through changes of cellular activity in osteoblasts and bone progenitor cells [41,42,50,223,224]. Besides altering gene expression and activity of osteoblasts [41,42], the diabetic environment significantly reduces the MSC population and viability [151,171]. In obese individuals living with T2DM, increased bone marrow fattiness may exacerbate MSC and osteoblast impairment by the release of cytokines and free fatty acids from hypoxic adipose tissue, which upholds a vicious cycle of chronic inflammation and inhibited osteoblastic activity (Figure 1) [165,168]. The combination of these changes eventually affects tensile strength and post-yield properties of the bone, which makes bone tissue in diabetes mellitus more vulnerable to microdamage accumulation, fragility fractures at most skeletal sites, and impaired fracture healing [32,225]. Decreased MSC population and impaired differentiation capacity may be the common link between impaired bone micro-architecture and higher incidence of non-union in patients living with diabetes mellitus [137,225]. Additionally, since vascularisation is mediated by MSCs [143,144], the reduced population and potential of progenitor cells may create vascular deficiencies in the fracture site, which can further impair diabetic fracture healing. A return to glucose homeostasis does not restore the capacity of previously diabetic MSCs, which reflects evidence outlining hyperglycemic memory in cells previously exposed to a diabetic milieu [64,65,66,67,68,69]. Therefore, it would be interesting to see studies investigating diabetes mellitus-induced epigenetic changes in precursor cells contributing to diabetic osteopathy.
This review highlights the importance of efficient clinical management of patients suffering from diabetes mellitus, since adequately controlled diabetes mellitus has been consistently implicated to have a positive effect on bone health, which reverses bone impairments in some studies [130,189,190,208,226,227,228,229,230]. It is important to bear in mind that patients who are on a treatment regime causing hypoglycemic episodes are at a greater risk of sustaining fractures [231,232,233]. In clinical practice, health care professionals should focus on bone protection interventions and fall prevention strategies targeting patients at high risk of fracture [234]. Conventional risk assessment tools for osteoporosis such as BMD measurements and the FRAX score are not valid for predicting fracture risk in individuals living with diabetes mellitus [120,121,235]. Therefore, there continues to be a dire need for the investigation of novel methods of risk assessment, which possibly includes measurements of bone turnover and levels of AGEs, which can adjust for the altered metabolic state of diabetes mellitus [236,237]. MiRNAs are promising novel serum biomarkers, which could be used to identify individuals living with diabetes mellitus at a high risk of fragility fractures within the coming years [97,98]. Recent scientific developments in the understanding of the molecular pathways involved in diabetes mellitus have opened opportunities in new anti-inflammatory treatment approaches [173]. Further investigation is needed to clarify the mechanism of action through which diabetes mellitus affects the viability and differentiation capacity of the progenitor cell population, which will support translational research in the prevention of fragility fractures in patients suffering from diabetes mellitus in the future.
Author Contributions
Conceptualization, C.E.M. and C.M.C. Literature review, C.E.M. Writing—original draft preparation, C.E.M. Writing—review and editing, C.M.C and C.E.M. Funding acquisition, C.M.C.
Funding
The Health Research Board and Diabetes Ireland Research Alliance under the MRCG-HRB Joint Funding Scheme (HRB-MRCG-2016-2) funded this research.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study, in the collection, analyses, or interpretation of data, in the writing of the manuscript, or in the decision to publish the results.
Abbreviations
| T1DM | Type 1 Diabetes Mellitus |
| T2DM | Type 2 Diabetes Mellitus |
| IGF1 | Insulin-like growth factor 1 |
| MSC | Mesenchymal stem cell |
| AGE | Advanced glycation end products |
| BMD | Bone mineral density |
| PTH | Parathyroid hormone |
| TNF-α | Tumor necrosis factor alpha |
| RANKL | Receptor activator of nuclear factor kappa-Β ligand |
| RAGE | Receptor for advanced glycation end products |
| MiRNA | MicroRNA |
| DEXA | Dual-energy X-ray absorptiometry |
| FRAX | Fracture Risk Assessment Tool |
| ALP | Alkaline phosphatase |
| PPAR-γ | Peroxisome proliferator-activated receptor gamma |
| ROSBM-MSCs | Reactive oxygen speciesBone marrow (BM) derived MSCs |
| WAT | White adipose tissue |
| HIF-1α HSCDPP-4 | Hypoxia-inducible factor 1-alphaHaematopoietic stem cellDipeptidyl peptidase-4 |
| GLP1 | Glucagon-like peptide 1 |
| GLP1RA | GLP1 receptor analogue |
| SGLT-2 | Sodium glucose cotransporter- 2 |
References
- Janghorbani, M.; Feskanich, D.; Willett, W.C.; Hu, F. Prospective study of diabetes and risk of hip fracture: the Nurses’ Health Study. Diabetes Care 2006, 29, 1573–1578. [Google Scholar] [CrossRef] [PubMed]
- Janghorbani, M.; Van Dam, R.M.; Willett, W.C.; Hu, F.B. Systematic review of type 1 and type 2 diabetes mellitus and risk of fracture. Am. J. Epidemiol. 2007, 166, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Wei, F.; Lang, Y.; Liu, Y. Diabetes mellitus and risk of hip fractures: a meta-analysis. Osteoporos. Int. 2016, 27, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Oei, L.; Zillikens, M.C.; Dehghan, A.; Buitendijk, G.H.S.; Castaño-Betancourt, M.C.; Estrada, K.; Stolk, L.; Oei, E.H.G.; Meurs, J.B.J.; van Janssen, J.A.M.J.L.; et al. High Bone Mineral Density and Fracture Risk in Type 2 Diabetes as Skeletal Complications of Inadequate Glucose Control: The Rotterdam Study. Diabetes Care 2013, 36, 1619–1628. [Google Scholar] [CrossRef] [PubMed]
- Leanza, G.; Maddaloni, E.; Pitocco, D.; Conte, C.; Palermo, A.; Maurizi, A.R.; Pantano, A.L.; Suraci, C.; Altomare, M.; Strollo, R.; et al. Risk factors for fragility fractures in type 1 diabetes. Bone 2019, 125, 194–199. [Google Scholar] [CrossRef]
- Loder, R.T. The influence of diabetes mellitus on the healing of closed fractures. Clin. Orthop. Relat. Res. 1988, 210–216. [Google Scholar] [CrossRef]
- Folk, J.W.; Starr, A.J.; Early, J.S. Early wound complications of operative treatment of calcaneus fractures: analysis of 190 fractures. J. Orthop. Trauma. 1999, 13, 369–372. [Google Scholar] [CrossRef]
- Retzepi, M.; Donos, N. The effect of diabetes mellitus on osseous healing. Clin. Oral Implant. Res. 2010, 21, 673–681. [Google Scholar] [CrossRef]
- Norris, R.; Parker, M. Diabetes mellitus and hip fracture: a study of 5966 cases. Injury 2011, 42, 1313–1316. [Google Scholar] [CrossRef]
- Borgström, F.; Sobocki, P.; Ström, O.; Jönsson, B. The societal burden of osteoporosis in Sweden. Bone 2007, 40, 1602–1609. [Google Scholar] [CrossRef]
- Crane, J.L.; Zhao, L.; Frye, J.S.; Xian, L.; Qiu, T.; Cao, X. IGF-1 Signaling is Essential for Differentiation of Mesenchymal Stem Cells for Peak Bone Mass. Bone Res. 2013, 1, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Hough, F.S.; Pierroz, D.D.; Cooper, C.; Ferrari, S.L.; IOF CSA Bone and Diabetes Working Group. MECHANISMS IN ENDOCRINOLOGY: Mechanisms and evaluation of bone fragility in type 1 diabetes mellitus. Eur. J. Endocrinol. 2016, 174, R127–R138. [Google Scholar] [CrossRef] [PubMed]
- Weber, D.R.; Gordon, R.J.; Kelley, J.C.; Leonard, M.B.; Willi, S.M.; Hatch-Stein, J.; Kelly, A.; Kosacci, O.; Kucheruk, O.; Kaafarani, M.; et al. Poor glycemic control is associated with impaired bone accrual in the year following a diagnosis of type 1 diabetes. J. Clin. Endocrinol. Metab. 2019. [Google Scholar] [CrossRef] [PubMed]
- Shah, V.N.; Joshee, P.; Sippl, R.; Pyle, L.; Vigers, T.; Carpenter, R.D.; Kohrt, W.; Snell-Bergeon, J.K. Type 1 diabetes onset at young age is associated with compromised bone quality. Bone 2019, 123, 260–264. [Google Scholar] [CrossRef]
- Fuusager, G.B.; Christesen, H.T.; Milandt, N.; Schou, A.J. Glycemic control and bone mineral density in children and adolescents with type 1 diabetes. Pediatr. Diabetes 2019, 20, 629–636. [Google Scholar] [CrossRef]
- Gil-Díaz, M.C.; Raynor, J.; O’Brien, K.O.; Schwartz, G.J.; Weber, D.R. Systematic review: associations of calcium intake, vitamin D intake, and physical activity with skeletal outcomes in people with Type 1 diabetes mellitus. Acta Diabetol. 2019. [Google Scholar] [CrossRef]
- Napoli, N.; Chandran, M.; Pierroz, D.D.; Abrahamsen, B.; Schwartz, A.V.; Ferrari, S.L.; IOF Bone and Diabetes Working Group. Mechanisms of diabetes mellitus-induced bone fragility. Nat. Rev. Endocrinol. 2017, 13, 208–219. [Google Scholar] [CrossRef]
- Napoli, N.; Strollo, R.; Paladini, A.; Briganti, S.I.; Pozzilli, P.; Epstein, S. The alliance of mesenchymal stem cells, bone, and diabetes. Int. J. Endocrinol. 2014, 2014, 690783. [Google Scholar] [CrossRef]
- Vestergaard, P. Discrepancies in bone mineral density and fracture risk in patients with type 1 and type 2 diabetes--a meta-analysis. Osteoporos. Int. 2007, 18, 427–444. [Google Scholar] [CrossRef]
- Thong, E.P.; Herath, M.; Weber, D.R.; Ranasinha, S.; Ebeling, P.R.; Milat, F.; Teede, H. Fracture risk in young and middle-aged adults with type 1 diabetes mellitus: A systematic review and meta-analysis. Clin. Endocrinol. 2018, 89, 314–323. [Google Scholar] [CrossRef]
- Hamann, C.; Kirschner, S.; Günther, K.-P.; Hofbauer, L.C. Bone, sweet bone—osteoporotic fractures in diabetes mellitus. Nat. Rev. Endocrinol. 2012, 8, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Hariri, A.F.; Almatrafi, M.N.; Zamka, A.B.; Babaker, A.S.; Fallatah, T.M.; Althouwaibi, O.H.; Hamdi, A.S. Relationship between Body Mass Index and T-Scores of Bone Mineral Density in the Hip and Spine Regions among Older Adults with Diabetes: A Retrospective Review. Available online: https://www.hindawi.com/journals/jobe/2019/9827403/ (accessed on 22 July 2019).
- Schwartz, A.V. Diabetes Mellitus: Does it Affect Bone? Calcif. Tissue Int. 2003, 73, 515–519. [Google Scholar] [CrossRef] [PubMed]
- Oei, L.; Rivadeneira, F.; Zillikens, M.C.; Oei, E.H.G. Diabetes, diabetic complications, and fracture risk. Curr. Osteoporos. Rep. 2015, 13, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Samelson, E.J.; Demissie, S.; Cupples, L.A.; Zhang, X.; Xu, H.; Liu, C.-T.; Boyd, S.K.; McLean, R.R.; Broe, K.E.; Kiel, D.P.; et al. Diabetes and Deficits in Cortical Bone Density, Microarchitecture, and Bone Size: Framingham HR-pQCT Study. J. Bone Miner. Res. 2018, 33, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Singhal, V.; Bredella, M.A. Marrow adipose tissue imaging in humans. Bone 2019, 118, 69–76. [Google Scholar] [CrossRef]
- Burghardt, A.J.; Issever, A.S.; Schwartz, A.V.; Davis, K.A.; Masharani, U.; Majumdar, S.; Link, T.M. High-resolution peripheral quantitative computed tomographic imaging of cortical and trabecular bone microarchitecture in patients with type 2 diabetes mellitus. J. Clin. Endocrinol. Metab. 2010, 95, 5045–5055. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.Y.; Schafer, A.L. Diabetes and Bone Marrow Adiposity. Curr. Osteoporos. Rep. 2016, 14, 337–344. [Google Scholar] [CrossRef] [Green Version]
- Patsch, J.M.; Li, X.; Baum, T.; Yap, S.P.; Karampinos, D.C.; Schwartz, A.V.; Link, T.M. Bone marrow fat composition as a novel imaging biomarker in postmenopausal women with prevalent fragility fractures. J. Bone Miner. Res. 2013, 28, 1721–1728. [Google Scholar] [CrossRef]
- Farr, J.N.; Drake, M.T.; Amin, S.; Melton, L.J.; McCready, L.K.; Khosla, S. In vivo assessment of bone quality in postmenopausal women with type 2 diabetes. J. Bone Miner. Res. 2014, 29, 787–795. [Google Scholar] [CrossRef]
- Landis, W.J. The strength of a calcified tissue depends in part on the molecular structure and organization of its constituent mineral crystals in their organic matrix. Bone 1995, 16, 533–544. [Google Scholar] [CrossRef]
- Seeman, E.; Delmas, P.D. Bone quality--the material and structural basis of bone strength and fragility. N. Engl. J. Med. 2006, 354, 2250–2261. [Google Scholar] [PubMed]
- Saito, M.; Marumo, K. Bone Quality in Diabetes. Front Endocrinol. (Lausanne) 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Hofbauer, L.C.; Brueck, C.C.; Singh, S.K.; Dobnig, H. Osteoporosis in Patients With Diabetes Mellitus. J. Bone Miner. Res. 2007, 22, 1317–1328. [Google Scholar] [CrossRef] [PubMed]
- Nojiri, H.; Saita, Y.; Morikawa, D.; Kobayashi, K.; Tsuda, C.; Miyazaki, T.; Saito, M.; Marumo, K.; Yonezawa, I.; Kaneko, K.; et al. Cytoplasmic superoxide causes bone fragility owing to low-turnover osteoporosis and impaired collagen cross-linking. J. Bone Miner. Res. 2011, 26, 2682–2694. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Marumo, K. Collagen cross-links as a determinant of bone quality: a possible explanation for bone fragility in aging, osteoporosis, and diabetes mellitus. Osteoporos. Int. 2010, 21, 195–214. [Google Scholar] [CrossRef] [PubMed]
- Sultan, E.; Taha, I.; Saber, L.M. Altered Bone Metabolic Markers In Type 2 Diabetes Mellitus: Impact of Glycemic Control. J. Taibah Univ. Med. Sci. 2008, 3, 104–116. [Google Scholar] [CrossRef] [Green Version]
- Scragg, R.; Holdaway, I.; Singh, V.; Metcalf, P.; Baker, J.; Dryson, E. Serum 25-hydroxyvitamin D3 levels decreased in impaired glucose tolerance and diabetes mellitus. Diabetes Res. Clin. Pract. 1995, 27, 181–188. [Google Scholar] [CrossRef]
- Pittas, A.G.; Lau, J.; Hu, F.B.; Dawson-Hughes, B. The Role of Vitamin D and Calcium in Type 2 Diabetes. A Systematic Review and Meta-Analysis. J. Clin. Endocrinol. Metab. 2007, 92, 2017–2029. [Google Scholar] [CrossRef]
- Fulzele, K.; DiGirolamo, D.J.; Liu, Z.; Xu, J.; Messina, J.L.; Clemens, T.L. Disruption of the insulin-like growth factor type 1 receptor in osteoblasts enhances insulin signaling and action. J. Biol. Chem. 2007, 282, 25649–25658. [Google Scholar] [CrossRef]
- Zayzafoon, M.; Stell, C.; Irwin, R.; McCabe, L.R. Extracellular glucose influences osteoblast differentiation and c-Jun expression. J. Cell. Biochem. 2000, 79, 301–310. [Google Scholar] [CrossRef]
- Botolin, S.; McCabe, L.R. Chronic hyperglycemia modulates osteoblast gene expression through osmotic and non-osmotic pathways. J. Cell. Biochem. 2006, 99, 411–424. [Google Scholar] [CrossRef] [PubMed]
- García-Hernández, A.; Arzate, H.; Gil-Chavarría, I.; Rojo, R.; Moreno-Fierros, L. High glucose concentrations alter the biomineralization process in human osteoblastic cells. Bone 2012, 50, 276–288. [Google Scholar] [CrossRef] [PubMed]
- Lechleitner, M.; Koch, T.; Herold, M.; Dzien, A.; Hoppichler, F. Tumour necrosis factor-alpha plasma level in patients with type 1 diabetes mellitus and its association with glycaemic control and cardiovascular risk factors. J. Intern. Med. 2000, 248, 67–76. [Google Scholar] [CrossRef]
- Motyl, K.J.; Botolin, S.; Irwin, R.; Appledorn, D.M.; Kadakia, T.; Amalfitano, A.; Schwartz, R.C.; McCabe, L.R. Bone inflammation and altered gene expression with type I diabetes early onset. J. Cell. Physiol. 2009, 218, 575–583. [Google Scholar] [CrossRef]
- Coe, L.M.; Irwin, R.; Lippner, D.; McCabe, L.R. The bone marrow microenvironment contributes to type I diabetes induced osteoblast death. J. Cell. Physiol. 2011, 226, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Kida, Y.; Kato, S.; Marumo, K. Diabetes, collagen, and bone quality. Curr. Osteoporos. Rep. 2014, 12, 181–188. [Google Scholar] [CrossRef]
- McCarthy, A.D.; Etcheverry, S.B.; Bruzzone, L.; Lettieri, G.; Barrio, D.A.; Cortizo, A.M. Non-enzymatic glycosylation of a type I collagen matrix: effects on osteoblastic development and oxidative stress. BMC Cell Biol. 2001, 2, 16. [Google Scholar] [CrossRef]
- Baynes, J.W. Role of Oxidative Stress in Development of Complications in Diabetes. Diabetes 1991, 40, 405–412. [Google Scholar] [CrossRef]
- Leslie, W.D.; Rubin, M.R.; Schwartz, A.V.; Kanis, J.A. Type 2 diabetes and bone. J. Bone Miner. Res. 2012, 27, 2231–2237. [Google Scholar] [CrossRef]
- Tang, S.Y.; Allen, M.R.; Phipps, R.; Burr, D.B.; Vashishth, D. Changes in non-enzymatic glycation and its association with altered mechanical properties following 1-year treatment with risedronate or alendronate. Osteoporos. Int. 2009, 20, 887–894. [Google Scholar] [CrossRef]
- Saito, M.; Fujii, K.; Mori, Y.; Marumo, K. Role of collagen enzymatic and glycation induced cross-links as a determinant of bone quality in spontaneously diabetic WBN/Kob rats. Osteoporos. Int. 2006, 17, 1514–1523. [Google Scholar] [CrossRef] [PubMed]
- Vashishth, D.; Gibson, G.J.; Khoury, J.I.; Schaffler, M.B.; Kimura, J.; Fyhrie, D.P. Influence of nonenzymatic glycation on biomechanical properties of cortical bone. Bone 2001, 28, 195–201. [Google Scholar] [PubMed]
- Kume, S.; Kato, S.; Yamagishi, S.; Inagaki, Y.; Ueda, S.; Arima, N.; Okawa, T.; Kojiro, M.; Nagata, K. Advanced glycation end-products attenuate human mesenchymal stem cells and prevent cognate differentiation into adipose tissue, cartilage, and bone. J. Bone Miner. Res. 2005, 20, 1647–1658. [Google Scholar] [PubMed]
- Sanguineti, R.; Storace, D.; Monacelli, F.; Federici, A.; Odetti, P. Pentosidine effects on human osteoblasts in vitro. Ann. N. Y. Acad. Sci. 2008, 1126, 166–172. [Google Scholar]
- Katayama, Y.; Akatsu, T.; Yamamoto, M.; Kugai, N.; Nagata, N. Role of nonenzymatic glycosylation of type I collagen in diabetic osteopenia. J. Bone Miner. Res. 1996, 11, 931–937. [Google Scholar] [PubMed]
- Valcourt, U.; Merle, B.; Gineyts, E.; Viguet-Carrin, S.; Delmas, P.D.; Garnero, P. Non-enzymatic glycation of bone collagen modifies osteoclastic activity and differentiation. J. Biol. Chem. 2007, 282, 5691–5703. [Google Scholar]
- Ogawa, N.; Yamaguchi, T.; Yano, S.; Yamauchi, M.; Yamamoto, M.; Sugimoto, T. The combination of high glucose and advanced glycation end-products (AGEs) inhibits the mineralization of osteoblastic MC3T3-E1 cells through glucose-induced increase in the receptor for AGEs. Horm. Metab. Res. 2007, 39, 871–875. [Google Scholar] [CrossRef]
- Mercer, N.; Ahmed, H.; Etcheverry, S.B.; Vasta, G.R.; Cortizo, A.M. Regulation of advanced glycation end product (AGE) receptors and apoptosis by AGEs in osteoblast-like cells. Mol. Cell. Biochem. 2007, 306, 87–94. [Google Scholar] [CrossRef]
- Manigrasso, M.B.; Juranek, J.; Ramasamy, R.; Schmidt, A.M. Unlocking the biology of RAGE in diabetic microvascular complications. Trends Endocrinol. Metab. 2014, 25, 15–22. [Google Scholar] [CrossRef]
- Tanaka, K.; Yamaguchi, T.; Kanazawa, I.; Sugimoto, T. Effects of high glucose and advanced glycation end products on the expressions of sclerostin and RANKL as well as apoptosis in osteocyte-like MLO-Y4-A2 cells. Biochem. Biophys. Res. Commun. 2015, 461, 193–199. [Google Scholar] [CrossRef]
- Iacobini, C.; Fantauzzi, C.B.; Pugliese, G.; Menini, S. Role of Galectin-3 in Bone Cell Differentiation, Bone Pathophysiology and Vascular Osteogenesis. Int. J. Mol. Sci. 2017, 18, 2481. [Google Scholar] [CrossRef] [PubMed]
- Pricci, F.; Leto, G.; Amadio, L.; Iacobini, C.; Romeo, G.; Cordone, S.; Gradini, R.; Barsotti, P.; Liu, F.-T.; Di Mario, U.; et al. Role of galectin-3 as a receptor for advanced glycosylation end products. Kidney Int. 2000, 58, S31–S39. [Google Scholar] [CrossRef] [Green Version]
- Pirola, L.; Balcerczyk, A.; Okabe, J.; El-Osta, A. Epigenetic phenomena linked to diabetic complications. Nat. Rev. Endocrinol. 2010, 6, 665–675. [Google Scholar] [CrossRef]
- Cencioni, C.; Spallotta, F.; Greco, S.; Martelli, F.; Zeiher, A.M.; Gaetano, C. Epigenetic mechanisms of hyperglycemic memory. Int. J. Biochem. Cell Biol. 2014, 51, 155–158. [Google Scholar]
- El-Osta, A.; Brasacchio, D.; Yao, D.; Pocai, A.; Jones, P.L.; Roeder, R.G.; Cooper, M.E.; Brownlee, M. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J. Exp. Med. 2008, 205, 2409–2417. [Google Scholar] [CrossRef] [PubMed]
- Brasacchio, D.; Okabe, J.; Tikellis, C.; Balcerczyk, A.; George, P.; Baker, E.K.; Calkin, A.C.; Brownlee, M.; Cooper, M.E.; El-Osta, A. Hyperglycemia induces a dynamic cooperativity of histone methylase and demethylase enzymes associated with gene-activating epigenetic marks that coexist on the lysine tail. Diabetes 2009, 58, 1229–1236. [Google Scholar] [CrossRef] [PubMed]
- Holman, R.R.; Paul, S.K.; Bethel, M.A.; Matthews, D.R.; Neil, H.A.W. 10-year follow-up of intensive glucose control in type 2 diabetes. N. Engl. J. Med. 2008, 359, 1577–1589. [Google Scholar] [CrossRef] [PubMed]
- Nathan, D.M.; Cleary, P.A.; Backlund, J.-Y.C.; Genuth, S.M.; Lachin, J.M.; Orchard, T.J.; Raskin, P.; Zinman, B. Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications (DCCT/EDIC) Study Research Group Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N. Engl. J. Med. 2005, 353, 2643–2653. [Google Scholar] [PubMed]
- The relationship of glycemic exposure (HbA1c) to the risk of development and progression of retinopathy in the diabetes control and complications trial. Diabetes 1995, 44, 968–983. [CrossRef]
- Van Diepen, J.A.; Thiem, K.; Stienstra, R.; Riksen, N.P.; Tack, C.J.; Netea, M.G. Diabetes propels the risk for cardiovascular disease: sweet monocytes becoming aggressive? Cell. Mol. Life Sci. 2016, 73, 4675–4684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, X.; Liao, Y.; Chen, L.; Liu, G.; Feng, Y.; Zeng, T.; Zhang, J. The MicroRNAs in the Pathogenesis of Metabolic Memory. Endocrinology 2015, 156, 3157–3168. [Google Scholar] [CrossRef] [PubMed]
- Costantino, S.; Paneni, F.; Lüscher, T.F.; Cosentino, F. MicroRNA profiling unveils hyperglycaemic memory in the diabetic heart. Eur. Heart J. 2016, 37, 572–576. [Google Scholar] [CrossRef]
- Gallagher, K.A.; Joshi, A.; Carson, W.F.; Schaller, M.; Allen, R.; Mukerjee, S.; Kittan, N.; Feldman, E.L.; Henke, P.K.; Hogaboam, C.; et al. Epigenetic Changes in Bone Marrow Progenitor Cells Influence the Inflammatory Phenotype and Alter Wound Healing in Type 2 Diabetes. Diabetes 2015, 64, 1420–1430. [Google Scholar] [CrossRef] [PubMed]
- Ceriello, A.; Ihnat, M.A.; Thorpe, J.E. Clinical review 2: The “metabolic memory”: is more than just tight glucose control necessary to prevent diabetic complications? J. Clin. Endocrinol. Metab. 2009, 94, 410–415. [Google Scholar] [CrossRef] [PubMed]
- Khamaneh, A.M.; Alipour, M.R.; Sheikhzadeh Hesari, F.; Ghadiri Soufi, F. A signature of microRNA-155 in the pathogenesis of diabetic complications. J. Physiol. Biochem. 2015, 71, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Spinetti, G.; Cordella, D.; Fortunato, O.; Sangalli, E.; Losa, S.; Gotti, A.; Carnelli, F.; Rosa, F.; Riboldi, S.; Sessa, F.; et al. Global Remodeling of the Vascular Stem Cell Niche in Bone Marrow of Diabetic Patients. Circ. Res. 2013, 112, 510–522. [Google Scholar] [CrossRef] [Green Version]
- Fadini, G.P.; Albiero, M.; Bonora, B.M.; Poncina, N.; Vigili de Kreutzenberg, S.; Avogaro, A. p66Shc gene expression in peripheral blood mononuclear cells and progression of diabetic complications. Cardiovasc. Diabetol. 2018, 17, 16. [Google Scholar] [CrossRef] [PubMed]
- Villeneuve, L.M.; Natarajan, R. The role of epigenetics in the pathology of diabetic complications. Am. J. Physiol. Ren. Physiol. 2010, 299, F14–F25. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Sun, X.; Icli, B.; Feinberg, M.W. Emerging Roles for MicroRNAs in Diabetic Microvascular Disease: Novel Targets for Therapy. Endocr. Rev. 2017, 38, 145–168. [Google Scholar] [CrossRef] [Green Version]
- Shetty, S.; Kapoor, N.; Bondu, J.D.; Thomas, N.; Paul, T.V. Bone turnover markers: Emerging tool in the management of osteoporosis. Indian J. Endocrinol. Metab. 2016, 20, 846–852. [Google Scholar]
- Maggio, A.B.R.; Ferrari, S.; Kraenzlin, M.; Marchand, L.M.; Schwitzgebel, V.; Beghetti, M.; Rizzoli, R.; Farpour-Lambert, N.J. Decreased bone turnover in children and adolescents with well controlled type 1 diabetes. J. Pediatr. Endocrinol. Metab. 2010, 23, 697–707. [Google Scholar] [CrossRef] [PubMed]
- Madsen, J.O.B.; Jørgensen, N.R.; Pociot, F.; Johannesen, J. Bone turnover markers in children and adolescents with type 1 diabetes-A systematic review. Pediatr. Diabetes 2019, 20, 510–522. [Google Scholar] [CrossRef] [PubMed]
- Ham, J.R.; Choi, R.-Y.; Lee, H.-I.; Lee, M.-K. Methoxsalen and Bergapten Prevent Diabetes-Induced Osteoporosis by the Suppression of Osteoclastogenic Gene Expression in Mice. Int. J. Mol. Sci. 2019, 20, 1298. [Google Scholar] [CrossRef] [PubMed]
- Starup-Linde, J.; Eriksen, S.A.; Lykkeboe, S.; Handberg, A.; Vestergaard, P. Biochemical markers of bone turnover in diabetes patients--a meta-analysis, and a methodological study on the effects of glucose on bone markers. Osteoporos. Int. 2014, 25, 1697–1708. [Google Scholar] [CrossRef] [PubMed]
- Urano, T.; Shiraki, M.; Kuroda, T.; Tanaka, S.; Urano, F.; Uenishi, K.; Inoue, S. Low serum osteocalcin concentration is associated with incident type 2 diabetes mellitus in Japanese women. J. Bone Miner. Metab. 2018, 36, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Takashi, Y.; Ishizu, M.; Mori, H.; Miyashita, K.; Sakamoto, F.; Katakami, N.; Matsuoka, T.-A.; Yasuda, T.; Hashida, S.; Matsuhisa, M.; et al. Circulating osteocalcin as a bone-derived hormone is inversely correlated with body fat in patients with type 1 diabetes. PLoS ONE 2019, 14, e0216416. [Google Scholar]
- García-Martín, A.; Rozas-Moreno, P.; Reyes-García, R.; Morales-Santana, S.; García-Fontana, B.; García-Salcedo, J.A.; Muñoz-Torres, M. Circulating levels of sclerostin are increased in patients with type 2 diabetes mellitus. J. Clin. Endocrinol. Metab. 2012, 97, 234–241. [Google Scholar]
- Gaudio, A.; Privitera, F.; Battaglia, K.; Torrisi, V.; Sidoti, M.H.; Pulvirenti, I.; Canzonieri, E.; Tringali, G.; Fiore, C.E. Sclerostin levels associated with inhibition of the Wnt/β-catenin signaling and reduced bone turnover in type 2 diabetes mellitus. J. Clin. Endocrinol. Metab. 2012, 97, 3744–3750. [Google Scholar] [CrossRef]
- Gennari, L.; Merlotti, D.; Valenti, R.; Ceccarelli, E.; Ruvio, M.; Pietrini, M.G.; Capodarca, C.; Franci, M.B.; Campagna, M.S.; Calabrò, A.; et al. Circulating sclerostin levels and bone turnover in type 1 and type 2 diabetes. J. Clin. Endocrinol. Metab. 2012, 97, 1737–1744. [Google Scholar] [CrossRef]
- Starup-Linde, J.; Vestergaard, P. Biochemical bone turnover markers in diabetes mellitus—A systematic review. Bone 2016, 82, 69–78. [Google Scholar] [CrossRef]
- Massera, D.; Biggs, M.L.; Walker, M.D.; Mukamal, K.J.; Ix, J.H.; Djousse, L.; Valderrábano, R.J.; Siscovick, D.S.; Tracy, R.P.; Xue, X.; et al. Biochemical Markers of Bone Turnover and Risk of Incident Diabetes in Older Women: The Cardiovascular Health Study. Diabetes Care 2018, 41, 1901–1908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlot, M.C.; den Heijer, M.; de Jongh, R.T.; Vervloet, M.G.; Lems, W.F.; de Jonge, R.; Obermayer-Pietsch, B.; Heijboer, A.C. Clinical utility of bone markers in various diseases. Bone 2018, 114, 215–225. [Google Scholar] [PubMed]
- Gordon, J.A.R.; Montecino, M.A.; Aqeilan, R.I.; Stein, J.L.; Stein, G.S.; Lian, J.B. Epigenetic Pathways Regulating Bone Homeostasis: Potential Targeting for Intervention of Skeletal Disorders. Curr. Osteoporos. Rep. 2014, 12, 496–506. [Google Scholar] [CrossRef] [Green Version]
- Ostanek, B.; Kranjc, T.; Lovšin, N.; Zupan, J.; Marc, J. Chapter 18 - Epigenetic Mechanisms in Osteoporosis. In Epigenetics of Aging and Longevity; Moskalev, A., Vaiserman, A.M., Eds.; Translational Epigenetics; Academic Press: Boston, MA, USA, 2018; Volume 4, pp. 365–388. [Google Scholar]
- Bedene, A.; Mencej Bedrač, S.; Ješe, L.; Marc, J.; Vrtačnik, P.; Preželj, J.; Kocjan, T.; Kranjc, T.; Ostanek, B. MiR-148a the epigenetic regulator of bone homeostasis is increased in plasma of osteoporotic postmenopausal women. Wien. Klin. Wochenschr. 2016, 128, 519–526. [Google Scholar] [PubMed]
- Heilmeier, U.; Hackl, M.; Skalicky, S.; Weilner, S.; Schroeder, F.; Vierlinger, K.; Patsch, J.M.; Baum, T.; Oberbauer, E.; Lobach, I.; et al. Serum miRNA Signatures Are Indicative of Skeletal Fractures in Postmenopausal Women With and Without Type 2 Diabetes and Influence Osteogenic and Adipogenic Differentiation of Adipose Tissue-Derived Mesenchymal Stem Cells In Vitro. J. Bone Miner. Res. 2016, 31, 2173–2192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grieco, G.E.; Cataldo, D.; Ceccarelli, E.; Nigi, L.; Catalano, G.; Brusco, N.; Mancarella, F.; Ventriglia, G.; Fondelli, C.; Guarino, E.; et al. Serum Levels of miR-148a and miR-21-5p Are Increased in Type 1 Diabetic Patients and Correlated with Markers of Bone Strength and Metabolism. Non-Coding RNA 2018, 4, 37. [Google Scholar] [CrossRef] [PubMed]
- Cunha, J.S.; Ferreira, V.M.; Maquigussa, E.; Naves, M.A.; Boim, M.A. Effects of high glucose and high insulin concentrations on osteoblast function in vitro. Cell Tissue Res. 2014, 358, 249–256. [Google Scholar] [CrossRef]
- Achemlal, L.; Tellal, S.; Rkiouak, F.; Nouijai, A.; Bezza, A.; Derouiche, E.M.; Ghafir, D.; Maghraoui, A.E. Bone metabolism in male patients with type 2 diabetes. Clin. Rheumatol. 2005, 24, 493–496. [Google Scholar] [CrossRef] [PubMed]
- Pacios, S.; Andriankaja, O.; Kang, J.; Alnammary, M.; Bae, J.; de Brito Bezerra, B.; Schreiner, H.; Fine, D.H.; Graves, D.T. Bacterial infection increases periodontal bone loss in diabetic rats through enhanced apoptosis. Am. J. Pathol. 2013, 183, 1928–1935. [Google Scholar] [CrossRef]
- Yamagishi, S. Role of advanced glycation end products (AGEs) in osteoporosis in diabetes. Curr. Drug Targets 2011, 12, 2096–2102. [Google Scholar] [CrossRef]
- Kasahara, T.; Imai, S.; Kojima, H.; Katagi, M.; Kimura, H.; Chan, L.; Matsusue, Y. Malfunction of Bone Marrow Derived Osteoclasts and the Delay of Bone Fracture Healing in Diabetic Mice. Bone 2010, 47, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Wittrant, Y.; Gorin, Y.; Woodruff, K.; Horn, D.; Abboud, H.; Mohan, S.; Abboud-Werner, S. High D(+)glucose concentration inhibits RANKL-induced osteoclastogenesis. Bone 2008, 42, 1122–1130. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Liu, R.; Desta, T.; Leone, C.; Gerstenfeld, L.C.; Graves, D.T. Diabetes causes decreased osteoclastogenesis, reduced bone formation, and enhanced apoptosis of osteoblastic cells in bacteria stimulated bone loss. Endocrinology 2004, 145, 447–452. [Google Scholar] [CrossRef] [PubMed]
- McCabe, L.R. Understanding the pathology and mechanisms of type I diabetic bone loss. J. Cell. Biochem. 2007, 102, 1343–1357. [Google Scholar] [CrossRef]
- Gooch, H.L.; Hale, J.E.; Fujioka, H.; Balian, G.; Hurwitz, S.R. Alterations of cartilage and collagen expression during fracture healing in experimental diabetes. Connect. Tissue Res. 2000, 41, 81–91. [Google Scholar] [CrossRef]
- Funk, J.R.; Hale, J.E.; Carmines, D.; Gooch, H.L.; Hurwitz, S.R. Biomechanical evaluation of early fracture healing in normal and diabetic rats. J. Orthop. Res. 2000, 18, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, A.V.; Sellmeyer, D.E.; Ensrud, K.E.; Cauley, J.A.; Tabor, H.K.; Schreiner, P.J.; Jamal, S.A.; Black, D.M.; Cummings, S.R. Study of Osteoporotic Features Research Group Older women with diabetes have an increased risk of fracture: A prospective study. J. Clin. Endocrinol. Metab. 2001, 86, 32–38. [Google Scholar] [CrossRef]
- Forsén, L.; Meyer, H.E.; Midthjell, K.; Edna, T.-H. Diabetes mellitus and the incidence of hip fracture: results from the Nord-Trøndelag Health Survey. Diabetologia 1999. [Google Scholar] [CrossRef]
- Ahmed, L.; Joakimsen, R.; Berntsen, G.; Fønnebø, V.; Schirmer, H. Diabetes mellitus and the risk of non-vertebral fractures: The Tromso study. Osteoporos. Int. 2006, 17, 495–500. [Google Scholar] [CrossRef]
- Bonds, D.E.; Larson, J.C.; Schwartz, A.V.; Strotmeyer, E.S.; Robbins, J.; Rodriguez, B.L.; Johnson, K.C.; Margolis, K.L. Risk of fracture in women with type 2 diabetes: the Women’s Health Initiative Observational Study. J. Clin. Endocrinol. Metab. 2006, 91, 3404–3410. [Google Scholar] [CrossRef]
- Muraki, S.; Yamamoto, S.; Ishibashi, H.; Nakamura, K. Factors associated with mortality following hip fracture in Japan. J. Bone Miner. Metab. 2006, 24, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Dubey, A.; Aharonoff, G.B.; Zuckerman, J.D.; Koval, K.J. The effects of diabetes on outcome after hip fracture. Bull. (Hosp. Jt. Dis.) 2000, 59, 94–98. [Google Scholar] [PubMed]
- De Liefde, I.I.; van der Klift, M.; de Laet, C.E.D.H.; van Daele, P.L.A.; Hofman, A.; Pols, H.a.P. Bone mineral density and fracture risk in type-2 diabetes mellitus: the Rotterdam Study. Osteoporos. Int. 2005, 16, 1713–1720. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, A.V.; Garnero, P.; Hillier, T.A.; Sellmeyer, D.E.; Strotmeyer, E.S.; Feingold, K.R.; Resnick, H.E.; Tylavsky, F.A.; Black, D.M.; Cummings, S.R.; et al. Pentosidine and increased fracture risk in older adults with type 2 diabetes. J. Clin. Endocrinol. Metab. 2009, 94, 2380–2386. [Google Scholar] [CrossRef]
- Shiraki, M.; Kuroda, T.; Shiraki, Y.; Tanaka, S.; Higuchi, T.; Saito, M. Urinary pentosidine and plasma homocysteine levels at baseline predict future fractures in osteoporosis patients under bisphosphonate treatment. J. Bone Miner. Metab. 2011, 29, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Karim, L.; Moulton, J.; Van Vliet, M.; Velie, K.; Robbins, A.; Malekipour, F.; Abdeen, A.; Ayres, D.; Bouxsein, M.L. Bone microarchitecture, biomechanical properties, and advanced glycation end-products in the proximal femur of adults with type 2 diabetes. Bone 2018, 114, 32–39. [Google Scholar] [CrossRef]
- Farlay, D.; Armas, L.; Gineyts, E.; Akhter, M.; Recker, R.; Boivin, G. Non-enzymatic glycation and degree of mineralization are higher in bone from fractured patients with Type 1 Diabetes Mellitus. J. Bone Miner. Res. 2016, 31, 190–195. [Google Scholar] [CrossRef]
- Schwartz, A.V.; Vittinghoff, E.; Bauer, D.C.; Hillier, T.A.; Strotmeyer, E.S.; Ensrud, K.E.; Donaldson, M.G.; Cauley, J.A.; Harris, T.B.; Koster, A.; et al. Association of BMD and FRAX score with risk of fracture in older adults with type 2 diabetes. JAMA 2011, 305, 2184–2192. [Google Scholar] [CrossRef]
- Giangregorio, L.M.; Leslie, W.D.; Lix, L.M.; Johansson, H.; Oden, A.; McCloskey, E.; Kanis, J.A. FRAX underestimates fracture risk in patients with diabetes. J. Bone Miner. Res. 2012, 27, 301–308. [Google Scholar] [CrossRef]
- Leslie, W.D.; Aubry-Rozier, B.; Lamy, O.; Hans, D. Manitoba Bone Density Program TBS (trabecular bone score) and diabetes-related fracture risk. J. Clin. Endocrinol. Metab. 2013, 98, 602–609. [Google Scholar] [CrossRef]
- Lin, Y.-C.; Wu, J.; Kuo, S.-F.; Cheung, Y.-C.; Sung, C.-M.; Fan, C.-M.; Chen, F.-P.; Mhuircheartaigh, J.N. Vertebral Fractures in Type 2 Diabetes Patients: Utility of Trabecular Bone Score and Relationship With Serum Bone Turnover Biomarkers. J. Clin. Densitom. 2019. [Google Scholar] [CrossRef] [PubMed]
- Ho-Pham, L.T.; Nguyen, T.V. Association between trabecular bone score and type 2 diabetes: A quantitative update of evidence. Osteoporos. Int. 2019. [Google Scholar] [CrossRef] [PubMed]
- Claes, L.; Recknagel, S.; Ignatius, A. Fracture healing under healthy and inflammatory conditions. Nat. Rev. Rheumatol. 2012, 8, 133–143. [Google Scholar] [CrossRef]
- Santana, R.B.; Xu, L.; Chase, H.B.; Amar, S.; Graves, D.T.; Trackman, P.C. A role for advanced glycation end products in diminished bone healing in type 1 diabetes. Diabetes 2003, 52, 1502–1510. [Google Scholar] [CrossRef] [PubMed]
- Kayal, R.A.; Tsatsas, D.; Bauer, M.A.; Allen, B.; Al-Sebaei, M.O.; Kakar, S.; Leone, C.W.; Morgan, E.F.; Gerstenfeld, L.C.; Einhorn, T.A.; et al. Diminished Bone Formation During Diabetic Fracture Healing is Related to the Premature Resorption of Cartilage Associated With Increased Osteoclast Activity. J. Bone Miner. Res. 2007, 22, 560–568. [Google Scholar] [Green Version]
- Kayal, R.A.; Siqueira, M.; Alblowi, J.; McLean, J.; Krothapalli, N.; Faibish, D.; Einhorn, T.A.; Gerstenfeld, L.C.; Graves, D.T. TNF-alpha mediates diabetes-enhanced chondrocyte apoptosis during fracture healing and stimulates chondrocyte apoptosis through FOXO1. J. Bone Miner. Res. 2010, 25, 1604–1615. [Google Scholar] [PubMed]
- Follak, N.; Klöting, I.; Merk, H. Influence of diabetic metabolic state on fracture healing in spontaneously diabetic rats. Diabetes Metab. Res. Rev. 2005, 21, 288–296. [Google Scholar]
- Kayal, R.A.; Alblowi, J.; McKenzie, E.; Krothapalli, N.; Silkman, L.; Gerstenfeld, L.; Einhorn, T.A.; Graves, D.T. Diabetes causes the accelerated loss of cartilage during fracture repair which is reversed by insulin treatment. Bone 2009, 44, 357–363. [Google Scholar] [CrossRef] [Green Version]
- Beam, H.A.; Russell Parsons, J.; Lin, S.S. The effects of blood glucose control upon fracture healing in the BB Wistar rat with diabetes mellitus. J. Orthop. Res. 2002, 20, 1210–1216. [Google Scholar] [CrossRef]
- Akesson, K.; Vergnaud, P.; Delmas, P.D.; Obrant, K.J. Serum osteocalcin increases during fracture healing in elderly women with hip fracture. Bone 1995, 16, 427–430. [Google Scholar]
- Taniguchi, T.; Matsumoto, T.; Shindo, H. Changes of serum levels of osteocalcin, alkaline phosphatase, IGF-I and IGF-binding protein-3 during fracture healing. Injury 2003, 34, 477–479. [Google Scholar] [CrossRef]
- Lu, H.; Kraut, D.; Gerstenfeld, L.C.; Graves, D.T. Diabetes interferes with the bone formation by affecting the expression of transcription factors that regulate osteoblast differentiation. Endocrinology 2003, 144, 346–352. [Google Scholar] [PubMed]
- Januszyk, M.; Sorkin, M.; Glotzbach, J.P.; Vial, I.N.; Maan, Z.N.; Rennert, R.C.; Duscher, D.; Thangarajah, H.; Longaker, M.T.; Butte, A.J.; et al. Diabetes Irreversibly Depletes Bone Marrow–Derived Mesenchymal Progenitor Cell Subpopulations. Diabetes 2014, 63, 3047–3056. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-M.; Schilling, T.; Benisch, P.; Zeck, S.; Meissner-Weigl, J.; Schneider, D.; Limbert, C.; Seufert, J.; Kassem, M.; Schütze, N.; et al. Effects of high glucose on mesenchymal stem cell proliferation and differentiation. Biochem. Biophys. Res. Commun. 2007, 363, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, M.; Rigutto, S.; Ingels, A.; Spruyt, D.; Stricwant, N.; Kharroubi, I.; Albarani, V.; Jayankura, M.; Rasschaert, J.; Bastianelli, E.; et al. Decreased pool of mesenchymal stem cells is associated with altered chemokines serum levels in atrophic nonunion fractures. Bone 2013, 53, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Reddy, G.K.; Stehno-Bittel, L.; Hamade, S.; Enwemeka, C.S. The biomechanical integrity of bone in experimental diabetes. Diabetes Res. Clin. Pract. 2001, 54, 1–8. [Google Scholar] [CrossRef]
- Hou, J.C.; Zernicke, R.F.; Barnard, R.J. Experimental diabetes, insulin treatment, and femoral neck morphology and biomechanics in rats. Clin. Orthop. Relat. Res. 1991, 278–285. [Google Scholar]
- Korres, N.; Tsiridis, E.; Pavlou, G.; Mitsoudis, A.; Perrea, D.N.; Zoumbos, A.B. Biomechanical characteristics of bone in streptozotocin-induced diabetic rats: An in-vivo randomized controlled experimental study. World J. Orthop. 2013, 4, 124–129. [Google Scholar] [PubMed]
- Gibbons, G.W.; Shaw, P.M. Diabetic vascular disease: characteristics of vascular disease unique to the diabetic patient. Semin. Vasc. Surg. 2012, 25, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Panagiotis, M. Classification of non-union. Injury 2005, 36, S30–S37. [Google Scholar]
- Mangialardi, G.; Ferland-McCollough, D.; Maselli, D.; Santopaolo, M.; Cordaro, A.; Spinetti, G.; Sambataro, M.; Sullivan, N.; Blom, A.; Madeddu, P. Bone marrow pericyte dysfunction in individuals with type 2 diabetes. Diabetologia 2019, 62, 1275–1290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pill, K.; Hofmann, S.; Redl, H.; Holnthoner, W. Vascularization mediated by mesenchymal stem cells from bone marrow and adipose tissue: a comparison. Cell Regen. 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Terenzi, D.C.; Al-Omran, M.; Qadura, M.; Quan, A.; Teoh, H.; Hess, D.A.; Verma, S. Abstract 12722: Diabetes Alters Circulating Progenitor Cell Content From Regenerative to Pro-Inflammatory Phenotype: Translational Implications for Cardiovascular Risk. Circulation 2018, 138, A12722. [Google Scholar]
- Vizoso, F.J.; Eiro, N.; Costa, L.; Esparza, P.; Landin, M.; Diaz-Rodriguez, P.; Schneider, J.; Perez-Fernandez, R. Mesenchymal Stem Cells in Homeostasis and Systemic Diseases: Hypothesis, Evidences, and Therapeutic Opportunities. Int. J. Mol. Sci. 2019, 20, 3738. [Google Scholar] [CrossRef]
- Rezabakhsh, A.; Cheraghi, O.; Nourazarian, A.; Hassanpour, M.; Kazemi, M.; Ghaderi, S.; Faraji, E.; Rahbarghazi, R.; Avci, Ç.B.; Bagca, B.G.; et al. Type 2 Diabetes Inhibited Human Mesenchymal Stem Cells Angiogenic Response by Over-Activity of the Autophagic Pathway. J. Cell. Biochem. 2017, 118, 1518–1530. [Google Scholar] [PubMed]
- Ridiandries, A.; Tan, J.T.M.; Bursill, C.A. The Role of Chemokines in Wound Healing. Int. J. Mol. Sci. 2018, 19, 3217. [Google Scholar] [CrossRef]
- Sadie-Van Gijsen, H.; Crowther, N.J.; Hough, F.S.; Ferris, W.F. The interrelationship between bone and fat: from cellular see-saw to endocrine reciprocity. Cell. Mol. Life Sci. 2013, 70, 2331–2349. [Google Scholar] [CrossRef]
- Aguiari, P.; Leo, S.; Zavan, B.; Vindigni, V.; Rimessi, A.; Bianchi, K.; Franzin, C.; Cortivo, R.; Rossato, M.; Vettor, R.; et al. High glucose induces adipogenic differentiation of muscle-derived stem cells. Proc. Natl. Acad. Sci. USA 2008, 105, 1226–1231. [Google Scholar] [CrossRef] [Green Version]
- Botolin, S.; Faugere, M.-C.; Malluche, H.; Orth, M.; Meyer, R.; McCabe, L.R. Increased bone adiposity and peroxisomal proliferator-activated receptor-gamma2 expression in type I diabetic mice. Endocrinology 2005, 146, 3622–3631. [Google Scholar] [CrossRef]
- Huang, K.-C.; Chuang, P.-Y.; Yang, T.-Y.; Huang, T.-W.; Chang, S.-F. Hyperglycemia inhibits osteoblastogenesis of rat bone marrow stromal cells via activation of the Notch2 signaling pathway. Int. J. Med. Sci. 2019, 16, 696–703. [Google Scholar] [CrossRef] [Green Version]
- Fairfield, H.; Falank, C.; Harris, E.; Demambro, V.; McDonald, M.; Pettitt, J.A.; Mohanty, S.T.; Croucher, P.; Kramer, I.; Kneissel, M.; et al. The skeletal cell-derived molecule sclerostin drives bone marrow adipogenesis. J. Cell. Physiol. 2018, 233, 1156–1167. [Google Scholar] [CrossRef] [PubMed]
- Devlin, M.J.; Van Vliet, M.; Motyl, K.; Karim, L.; Brooks, D.J.; Louis, L.; Conlon, C.; Rosen, C.J.; Bouxsein, M.L. Early-onset type 2 diabetes impairs skeletal acquisition in the male TALLYHO/JngJ mouse. Endocrinology 2014, 155, 3806–3816. [Google Scholar] [CrossRef] [PubMed]
- Marin, C.; Luyten, F.P.; Van der Schueren, B.; Kerckhofs, G.; Vandamme, K. The Impact of Type 2 Diabetes on Bone Fracture Healing. Front. Endocrinol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Paccou, J.; Penel, G.; Chauveau, C.; Cortet, B.; Hardouin, P. Marrow adiposity and bone: Review of clinical implications. Bone 2019, 118, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Baum, T.; Yap, S.P.; Karampinos, D.C.; Nardo, L.; Kuo, D.; Burghardt, A.J.; Masharani, U.B.; Schwartz, A.V.; Li, X.; Link, T.M. Does vertebral bone marrow fat content correlate with abdominal adipose tissue, lumbar spine bone mineral density, and blood biomarkers in women with type 2 diabetes mellitus? J. Magn. Reson. Imaging 2012, 35, 117–124. [Google Scholar] [CrossRef]
- Shanks, N.; Greek, R.; Greek, J. Are animal models predictive for humans? Philos. Ethics Hum. Med. 2009, 4, 2. [Google Scholar] [CrossRef]
- Ma, C.; Tonks, K.T.; Center, J.R.; Samocha-Bonet, D.; Greenfield, J.R. Complex interplay among adiposity, insulin resistance and bone health. Clin. Obes. 2018, 8, 131–139. [Google Scholar] [CrossRef] [Green Version]
- Lawler, H.M.; Underkofler, C.M.; Kern, P.A.; Erickson, C.; Bredbeck, B.; Rasouli, N. Adipose Tissue Hypoxia, Inflammation, and Fibrosis in Obese Insulin-Sensitive and Obese Insulin-Resistant Subjects. J. Clin. Endocrinol. Metab. 2016, 101, 1422–1428. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Wood, I.S.; Trayhurn, P. Dysregulation of the expression and secretion of inflammation-related adipokines by hypoxia in human adipocytes. Pflügers Arch. Eur. J. Physiol. 2007, 455, 479–492. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.S.; Kim, J.; Osborne, O.; Oh, D.Y.; Sasik, R.; Schenk, S.; Chen, A.; Chung, H.; Murphy, A.; Watkins, S.M.; et al. Increased Adipocyte O2 Consumption Triggers HIF-1α, Causing Inflammation and Insulin Resistance in Obesity. Cell 2014, 157, 1339–1352. [Google Scholar] [CrossRef]
- Alcalá, M.; Calderon-Dominguez, M.; Bustos, E.; Ramos, P.; Casals, N.; Serra, D.; Viana, M.; Herrero, L. Increased inflammation, oxidative stress and mitochondrial respiration in brown adipose tissue from obese mice. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longo, M.; Zatterale, F.; Naderi, J.; Parrillo, L.; Formisano, P.; Raciti, G.A.; Beguinot, F.; Miele, C. Adipose Tissue Dysfunction as Determinant of Obesity-Associated Metabolic Complications. Int. J. Mol. Sci. 2019, 20, 2358. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Bi, L.; He, S.; Meng, G.; Wei, B.; Jia, S.; Liu, J. FFAs-ROS-ERK/P38 pathway plays a key role in adipocyte lipotoxicity on osteoblasts in co-culture. Biochimie 2014, 101, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Kushwaha, P.; Wolfgang, M.J.; Riddle, R.C. Fatty acid metabolism by the osteoblast. Bone 2018, 115, 8–14. [Google Scholar] [CrossRef]
- Cuminetti, V.; Arranz, L. Bone Marrow Adipocytes: The Enigmatic Components of the Hematopoietic Stem Cell Niche. J. Clin. Med. 2019, 8, 707. [Google Scholar] [CrossRef]
- Knebel, B.; Fahlbusch, P.; Poschmann, G.; Dille, M.; Wahlers, N.; Stühler, K.; Hartwig, S.; Lehr, S.; Schiller, M.; Jacob, S.; et al. Adipokinome Signatures in Obese Mouse Models Reflect Adipose Tissue Health and Are Associated with Serum Lipid Composition. Int. J. Mol. Sci. 2019, 20, 2559. [Google Scholar] [CrossRef] [PubMed]
- Hamada, Y.; Fujii, H.; Fukagawa, M. Role of oxidative stress in diabetic bone disorder. Bone 2009, 45, S35–S38. [Google Scholar] [CrossRef]
- Panahi, G.; Pasalar, P.; Zare, M.; Rizzuto, R.; Meshkani, R. High glucose induces inflammatory responses in HepG2 cells via the oxidative stress-mediated activation of NF-κB, and MAPK pathways in HepG2 cells. Arch. Physiol. Biochem. 2018, 124, 468–474. [Google Scholar] [CrossRef]
- Wang, A.; Midura, R.J.; Vasanji, A.; Wang, A.J.; Hascall, V.C. Hyperglycemia Diverts Dividing Osteoblastic Precursor Cells to an Adipogenic Pathway and Induces Synthesis of a Hyaluronan Matrix That Is Adhesive for Monocytes. J. Biol. Chem. 2014, 289, 11410–11420. [Google Scholar] [CrossRef] [Green Version]
- Manolagas, S.C. From estrogen-centric to aging and oxidative stress: a revised perspective of the pathogenesis of osteoporosis. Endocr. Rev. 2010, 31, 266–300. [Google Scholar] [CrossRef]
- Shoelson, S.E.; Lee, J.; Goldfine, A.B. Inflammation and insulin resistance. J. Clin. Investig. 2006, 116, 1793–1801. [Google Scholar] [CrossRef] [PubMed]
- Shin, L.; Peterson, D.A. Impaired therapeutic capacity of autologous stem cells in a model of type 2 diabetes. Stem Cells Transl. Med. 2012, 1, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Davies, L.C.; Alm, J.J.; Heldring, N.; Moll, G.; Gavin, C.; Batsis, I.; Qian, H.; Sigvardsson, M.; Nilsson, B.; Kyllonen, L.E.; et al. Type 1 Diabetes Mellitus Donor Mesenchymal Stromal Cells Exhibit Comparable Potency to Healthy Controls In Vitro. Stem Cells Transl. Med. 2016, 5, 1485–1495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van de Vyver, M. Intrinsic Mesenchymal Stem Cell Dysfunction in Diabetes Mellitus: Implications for Autologous Cell Therapy. Stem Cells Dev. 2017, 26, 1042–1053. [Google Scholar] [CrossRef] [PubMed]
- Fadini, G.P.; Rigato, M.; Cappellari, R.; Bonora, B.M.; Avogaro, A. Long-term Prediction of Cardiovascular Outcomes by Circulating CD34+ and CD34+CD133+ Stem Cells in Patients With Type 2 Diabetes. Diabetes Care 2017, 40, 125–131. [Google Scholar] [CrossRef]
- Albiero, M.; Fadini, G.P. Pharmacologic targeting of the diabetic stem cell mobilopathy. Pharmacol. Res. 2018, 135, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Ferraro, F.; Lymperi, S.; Méndez-Ferrer, S.; Saez, B.; Spencer, J.A.; Yeap, B.Y.; Masselli, E.; Graiani, G.; Prezioso, L.; Rizzini, E.L.; et al. Diabetes Impairs Hematopoietic Stem Cell Mobilization by Altering Niche Function. Sci. Transl. Med. 2011, 3, 104ra101. [Google Scholar] [CrossRef]
- Albiero, M.; Poncina, N.; Tjwa, M.; Ciciliot, S.; Menegazzo, L.; Ceolotto, G.; Vigili de Kreutzenberg, S.; Moura, R.; Giorgio, M.; Pelicci, P.; et al. Diabetes causes bone marrow autonomic neuropathy and impairs stem cell mobilization via dysregulated p66Shc and Sirt1. Diabetes 2014, 63, 1353–1365. [Google Scholar] [CrossRef]
- Rota, M.; LeCapitaine, N.; Hosoda, T.; Boni, A.; De Angelis, A.; Padin-Iruegas, M.E.; Esposito, G.; Vitale, S.; Urbanek, K.; Casarsa, C.; et al. Diabetes Promotes Cardiac Stem Cell Aging and Heart Failure, Which Are Prevented by Deletion of the p66shc Gene. Cir. Res. 2006, 99, 42–52. [Google Scholar] [CrossRef]
- Zhao, T.; Li, J.; Chen, A.F. MicroRNA-34a induces endothelial progenitor cell senescence and impedes its angiogenesis via suppressing silent information regulator 1. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E110–E116. [Google Scholar] [CrossRef] [Green Version]
- Albiero, M.; Ciciliot, S.; Tedesco, S.; Menegazzo, L.; D’Anna, M.; Scattolini, V.; Cappellari, R.; Zuccolotto, G.; Rosato, A.; Cignarella, A.; et al. Diabetes-Associated Myelopoiesis Drives Stem Cell Mobilopathy Through an OSM-p66Shc Signaling Pathway. Diabetes 2019, db190080. [Google Scholar] [CrossRef] [PubMed]
- Fadini, G.P.; Albiero, M.; Seeger, F.; Poncina, N.; Menegazzo, L.; Angelini, A.; Castellani, C.; Thiene, G.; Agostini, C.; Cappellari, R.; et al. Stem cell compartmentalization in diabetes and high cardiovascular risk reveals the role of DPP-4 in diabetic stem cell mobilopathy. Basic Res. Cardiol. 2012, 108, 313. [Google Scholar] [CrossRef] [PubMed]
- Fadini, G.P.; Boscaro, E.; Albiero, M.; Menegazzo, L.; Frison, V.; de Kreutzenberg, S.; Agostini, C.; Tiengo, A.; Avogaro, A. The oral dipeptidyl peptidase-4 inhibitor sitagliptin increases circulating endothelial progenitor cells in patients with type 2 diabetes: possible role of stromal-derived factor-1alpha. Diabetes Care 2010, 33, 1607–1609. [Google Scholar] [CrossRef] [PubMed]
- Akune, T.; Ogata, N.; Hoshi, K.; Kubota, N.; Terauchi, Y.; Tobe, K.; Takagi, H.; Azuma, Y.; Kadowaki, T.; Nakamura, K.; et al. Insulin receptor substrate-2 maintains predominance of anabolic function over catabolic function of osteoblasts. J. Cell Biol. 2002, 159, 147–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogata, N.; Chikazu, D.; Kubota, N.; Terauchi, Y.; Tobe, K.; Azuma, Y.; Ohta, T.; Kadowaki, T.; Nakamura, K.; Kawaguchi, H. Insulin receptor substrate-1 in osteoblast is indispensable for maintaining bone turnover. J. Clin. Investig. 2000, 105, 935–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitamura, T.; Kitamura, Y.; Nakae, J.; Giordano, A.; Cinti, S.; Kahn, C.R.; Efstratiadis, A.; Accili, D. Mosaic analysis of insulin receptor function. J. Clin. Investig. 2004, 113, 209–219. [Google Scholar] [CrossRef]
- Shinohara, M. The spontaneously diabetic torii rat: an animal model of nonobese type 2 diabetes with severe diabetic complications. Exp. Diabetes Res. 2013, 2013. [Google Scholar]
- Campos Pastor, M.M.; López-Ibarra, P.J.; Escobar-Jiménez, F.; Serrano Pardo, M.D.; García-Cervigón, A.G. Intensive insulin therapy and bone mineral density in type 1 diabetes mellitus: a prospective study. Osteoporos. Int. 2000, 11, 455–459. [Google Scholar] [CrossRef]
- Vestergaard, P.; Rejnmark, L.; Mosekilde, L. Relative fracture risk in patients with diabetes mellitus, and the impact of insulin and oral antidiabetic medication on relative fracture risk. Diabetologia 2005, 48, 1292–1299. [Google Scholar] [CrossRef]
- Dede, A.D.; Tournis, S.; Dontas, I.; Trovas, G. Type 2 diabetes mellitus and fracture risk. Metabolism 2014, 63, 1480–1490. [Google Scholar] [CrossRef]
- New EASD-ADA consensus guidelines on managing hyperglycaemia in type 2 diabetes launched at EASD meeting. New recommendations include specific drug classes for some patients and enhancing medication adherence – Diabetologia. Available online: https://diabetologia-journal.org/2018/10/05/new-easd-ada-consensus-guidelines-on-managing-hyperglycaemia-in-type-2-diabetes-launched-at-easd-meeting-new-recommendations-include-specific-drug-classes-for-some-patients-and-enhancing-medication-a/ (accessed on 22 September 2019).
- Montagnani, A.; Gonnelli, S. Antidiabetic therapy effects on bone metabolism and fracture risk. Diabetes Obes. Metab. 2013, 15, 784–791. [Google Scholar] [CrossRef] [PubMed]
- Molinuevo, M.S.; Schurman, L.; McCarthy, A.D.; Cortizo, A.M.; Tolosa, M.J.; Gangoiti, M.V.; Arnol, V.; Sedlinsky, C. Effect of metformin on bone marrow progenitor cell differentiation: In vivo and in vitro studies. J. Bone Miner. Res. 2010, 25, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Son, H.-J.; Lee, J.; Lee, S.-Y.; Kim, E.-K.; Park, M.-J.; Kim, K.-W.; Park, S.-H.; Cho, M.-L. Metformin attenuates experimental autoimmune arthritis through reciprocal regulation of Th17/Treg balance and osteoclastogenesis. Mediat. Inflamm. 2014, 2014, 973986. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-S.; Kim, Y.-S.; Lee, S.-Y.; Kim, G.-H.; Kim, B.-J.; Lee, S.-H.; Lee, K.-U.; Kim, G.-S.; Kim, S.-W.; Koh, J.-M. AMP kinase acts as a negative regulator of RANKL in the differentiation of osteoclasts. Bone 2010, 47, 926–937. [Google Scholar] [CrossRef] [PubMed]
- Frid, A.; Sterner, G.N.; Löndahl, M.; Wiklander, C.; Cato, A.; Vinge, E.; Andersson, A. Novel Assay of Metformin Levels in Patients With Type 2 Diabetes and Varying Levels of Renal Function. Diabetes Care 2010, 33, 1291–1293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, X.; Yao, M.-W.; Zhu, M.; Liang, D.-L.; Guo, W.; Yang, Y.; Zhao, R.-S.; Ren, T.-T.; Ao, X.; Wang, W.; et al. Metformin induces apoptosis in mesenchymal stromal cells and dampens their therapeutic efficacy in infarcted myocardium. Stem Cell Res. Ther. 2018, 9, 306. [Google Scholar]
- Montazersaheb, S.; Kabiri, F.; Saliani, N.; Nourazarian, A.; Avci, Ç.B.; Rahbarghazi, R.; Nozad Charoudeh, H. Prolonged incubation with Metformin decreased angiogenic potential in human bone marrow mesenchymal stem cells. Biomed. Pharmacother. 2018, 108, 1328–1337. [Google Scholar]
- Wang, P.; Ma, T.; Guo, D.; Hu, K.; Shu, Y.; Xu, H.H.K.; Schneider, A. Metformin induces osteoblastic differentiation of human induced pluripotent stem cell-derived mesenchymal stem cells. J. Tissue Eng. Regen. Med. 2018, 12, 437–446. [Google Scholar] [CrossRef]
- Kahn, S.E.; Zinman, B.; Lachin, J.M.; Haffner, S.M.; Herman, W.H.; Holman, R.R.; Kravitz, B.G.; Yu, D.; Heise, M.A.; Aftring, R.P.; et al. Rosiglitazone-associated fractures in type 2 diabetes: an Analysis from A Diabetes Outcome Progression Trial (ADOPT). Diabetes Care 2008, 31, 845–851. [Google Scholar] [CrossRef]
- Zinman, B.; Haffner, S.M.; Herman, W.H.; Holman, R.R.; Lachin, J.M.; Kravitz, B.G.; Paul, G.; Jones, N.P.; Aftring, R.P.; Viberti, G.; et al. Effect of rosiglitazone, metformin, and glyburide on bone biomarkers in patients with type 2 diabetes. J. Clin. Endocrinol. Metab. 2010, 95, 134–142. [Google Scholar] [CrossRef]
- Grey, A. Skeletal consequences of thiazolidinedione therapy. Osteoporos. Int. 2008, 19, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Holst, J.J.; Ørskov, C. The Incretin Approach for Diabetes Treatment: Modulation of Islet Hormone Release by GLP-1 Agonism. Diabetes 2004, 53, S197–S204. [Google Scholar] [CrossRef] [PubMed]
- Knop, F.K.; Vilsbøll, T.; Højberg, P.V.; Larsen, S.; Madsbad, S.; Vølund, A.; Holst, J.J.; Krarup, T. Reduced Incretin Effect in Type 2 Diabetes: Cause or Consequence of the Diabetic State? Diabetes 2007, 56, 1951–1959. [Google Scholar] [CrossRef] [PubMed]
- Nuche-Berenguer, B.; Moreno, P.; Portal-Nuñez, S.; Dapía, S.; Esbrit, P.; Villanueva-Peñacarrillo, M.L. Exendin-4 exerts osteogenic actions in insulin-resistant and type 2 diabetic states. Regul. Pept. 2010, 159, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Meng, J.; Jia, M.; Bi, L.; Zhou, Y.; Wang, Y.; Hu, J.; He, G.; Luo, X. Exendin-4, a glucagon-like peptide-1 receptor agonist, prevents osteopenia by promoting bone formation and suppressing bone resorption in aged ovariectomized rats. J. Bone Miner. Res. 2013, 28, 1641–1652. [Google Scholar] [CrossRef] [PubMed]
- Sanz, C.; Vázquez, P.; Blázquez, C.; Barrio, P.A.; Alvarez, M.D.M.; Blázquez, E. Signaling and biological effects of glucagon-like peptide 1 on the differentiation of mesenchymal stem cells from human bone marrow. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E634–E643. [Google Scholar] [CrossRef] [Green Version]
- Nuche-Berenguer, B.; Portal-Núñez, S.; Moreno, P.; González, N.; Acitores, A.; López-Herradón, A.; Esbrit, P.; Valverde, I.; Villanueva-Peñacarrillo, M.L. Presence of a functional receptor for GLP-1 in osteoblastic cells, independent of the cAMP-linked GLP-1 receptor. J. Cell. Physiol. 2010, 225, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Palermo, A.; D’Onofrio, L.; Eastell, R.; Schwartz, A.V.; Pozzilli, P.; Napoli, N. Oral anti-diabetic drugs and fracture risk, cut to the bone: safe or dangerous? A narrative review. Osteoporos. Int. 2015, 26, 2073–2089. [Google Scholar] [CrossRef]
- Bunck, M.C.; Eliasson, B.; Cornér, A.; Heine, R.J.; Shaginian, R.M.; Taskinen, M.-R.; Yki-Järvinen, H.; Smith, U.; Diamant, M. Exenatide treatment did not affect bone mineral density despite body weight reduction in patients with type 2 diabetes. Diabetes Obes. Metab. 2011, 13, 374–377. [Google Scholar] [CrossRef]
- Mabilleau, G.; Mieczkowska, A.; Chappard, D. Use of glucagon-like peptide-1 receptor agonists and bone fractures: a meta-analysis of randomized clinical trials. J. Diabetes 2014, 6, 260–266. [Google Scholar] [CrossRef]
- Fadini, G.P.; Bonora, B.M.; Cappellari, R.; Menegazzo, L.; Vedovato, M.; Iori, E.; Marescotti, M.C.; Albiero, M.; Avogaro, A. Acute Effects of Linagliptin on Progenitor Cells, Monocyte Phenotypes, and Soluble Mediators in Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2016, 101, 748–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poncina, N.; Albiero, M.; Menegazzo, L.; Cappellari, R.; Avogaro, A.; Fadini, G.P. The dipeptidyl peptidase-4 inhibitor saxagliptin improves function of circulating pro-angiogenic cells from type 2 diabetic patients. Cardiovasc. Diabetol. 2014, 13, 92. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.L.; Roddick, A.J.; Aghar-Jaffar, R.; Shun-Shin, M.J.; Francis, D.; Oliver, N.; Meeran, K. Association Between Use of Sodium-Glucose Cotransporter 2 Inhibitors, Glucagon-like Peptide 1 Agonists, and Dipeptidyl Peptidase 4 Inhibitors With All-Cause Mortality in Patients With Type 2 Diabetes. JAMA 2018, 319, 1580–1591. [Google Scholar] [CrossRef] [PubMed]
- Solini, A. Role of SGLT2 inhibitors in the treatment of type 2 diabetes mellitus. Acta Diabetol. 2016, 53, 863–870. [Google Scholar] [CrossRef] [PubMed]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef]
- Zelniker, T.A.; Wiviott, S.D.; Raz, I.; Im, K.; Goodrich, E.L.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Furtado, R.H.M.; et al. SGLT2 inhibitors for primary and secondary prevention of cardiovascular and renal outcomes in type 2 diabetes: a systematic review and meta-analysis of cardiovascular outcome trials. Lancet 2019, 393, 31–39. [Google Scholar] [CrossRef]
- Hess, D.A.; Terenzi, D.C.; Trac, J.Z.; Quan, A.; Mason, T.; Al-Omran, M.; Bhatt, D.L.; Dhingra, N.; Rotstein, O.D.; Leiter, L.A.; et al. SGLT2 Inhibition with Empagliflozin Increases Circulating Provascular Progenitor Cells in People with Type 2 Diabetes Mellitus. Cell Metab. 2019. [Google Scholar] [CrossRef]
- Bonora, B.M.; Cappellari, R.; Albiero, M.; Avogaro, A.; Fadini, G.P. Effects of SGLT2 Inhibitors on Circulating Stem and Progenitor Cells in Patients With Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2018, 103, 3773–3782. [Google Scholar] [CrossRef]
- Solini, A.; Seghieri, M.; Giannini, L.; Biancalana, E.; Parolini, F.; Rossi, C.; Dardano, A.; Taddei, S.; Ghiadoni, L.; Bruno, R.M. The Effects of Dapagliflozin on Systemic and Renal Vascular Function Display an Epigenetic Signature. J. Clin. Endocrinol. Metab. 2019, 104, 4253–4263. [Google Scholar] [CrossRef]
- Fazeli, P.K.; Horowitz, M.C.; MacDougald, O.A.; Scheller, E.L.; Rodeheffer, M.S.; Rosen, C.J.; Klibanski, A. Marrow fat and bone--new perspectives. J. Clin. Endocrinol. Metab. 2013, 98, 935–945. [Google Scholar] [CrossRef]
- Rosen, C.J.; Bouxsein, M.L. Mechanisms of disease: is osteoporosis the obesity of bone? Nat. Clin. Pract. Rheumatol. 2006, 2, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Gortler, H.; Rusyn, J.; Godbout, C.; Chahal, J.; Schemitsch, E.H.; Nauth, A. Diabetes and Healing Outcomes in Lower Extremity Fractures: A Systematic Review. Injury 2018, 49, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Iki, M.; Fujita, Y.; Kouda, K.; Yura, A.; Tachiki, T.; Tamaki, J.; Sato, Y.; Moon, J.-S.; Hamada, M.; Kajita, E.; et al. Hyperglycemic status is associated with an elevated risk of osteoporotic fracture in community-dwelling elderly Japanese men: The Fujiwara-kyo osteoporosis risk in men (FORMEN) cohort study. Bone 2019, 121, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Athinarayanan, S.J.; Adams, R.N.; Hallberg, S.J.; McKenzie, A.L.; Bhanpuri, N.H.; Campbell, W.W.; Volek, J.S.; Phinney, S.D.; McCarter, J.P. Long-Term Effects of a Novel Continuous Remote Care Intervention Including Nutritional Ketosis for the Management of Type 2 Diabetes: A 2-Year Non-randomized Clinical Trial. Front. Endocrinol. (Lausanne) 2019, 10, 348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohsin, S.; Baniyas, M.M.; AlDarmaki, R.S.; Tekes, K.; Kalász, H.; Adeghate, E.A. An update on therapies for the treatment of diabetes-induced osteoporosis. Expert Opin. Biol. Ther. 2019, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Sundar, G.; Sridharan, S.; Sundaram, R.R.; Prabhu, S.; Rao, R.; Rudresh, V. Impact of well-controlled type 2 diabetes mellitus on implant stability and bone biomarkers. Int. J. Oral. Maxillofac. Implant. 2019. [Google Scholar]
- Zhang, Y.S.; Weng, W.Y.; Xie, B.C.; Meng, Y.; Hao, Y.H.; Liang, Y.M.; Zhou, Z.K. Glucagon-like peptide-1 receptor agonists and fracture risk: a network meta-analysis of randomized clinical trials. Osteoporos. Int. 2018, 29, 2639–2644. [Google Scholar] [CrossRef]
- Lee, R.H.; Sloane, R.; Pieper, C.; Lyles, K.W.; Adler, R.A.; Van Houtven, C.; LaFleur, J.; Colón-Emeric, C. Glycemic control and insulin treatment alter fracture risk in older men with type 2 diabetes mellitus. J. Bone Miner. Res. 2019. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, Q.; Liang, Y.; Dong, Y.; Mo, X.; Zhang, L.; Zhang, B. Insulin use and fracture risk in patients with type 2 diabetes: A meta-analysis of 138,690 patients. Exp. Ther. Med. 2019, 17, 3957–3964. [Google Scholar] [CrossRef]
- Kheniser, K.G.; Polanco Santos, C.M.; Kashyap, S.R. The effects of diabetes therapy on bone: A clinical perspective. J. Diabetes Complicat. 2018, 32, 713–719. [Google Scholar] [CrossRef]
- Compston, J. Type 2 diabetes mellitus and bone. J. Intern. Med. 2018, 283, 140–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, N.; Xia, W. Assessment of bone quality in patients with diabetes mellitus. Osteoporos. Int. 2018, 29, 1721–1736. [Google Scholar] [CrossRef] [PubMed]
- Leslie, W.D.; Johansson, H.; McCloskey, E.V.; Harvey, N.C.; Kanis, J.A.; Hans, D. Comparison of Methods for Improving Fracture Risk Assessment in Diabetes: The Manitoba BMD Registry. J. Bone Miner. Res. 2018, 33, 1923–1930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrari, S.L.; Abrahamsen, B.; Napoli, N.; Akesson, K.; Chandran, M.; Eastell, R.; El-Hajj Fuleihan, G.; Josse, R.; Kendler, D.L.; Kraenzlin, M.; et al. Diagnosis and management of bone fragility in diabetes: an emerging challenge. Osteoporos. Int. 2018, 29, 2585–2596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
The interaction between osteoblasts, adipocytes, MSCs, and the marrow environment is altered in diabetes mellitus. Hyperglycemia directly alters gene expression associated with osteoblast activity by the inhibition of MSC maturation and metabolism, and indirectly alters bone metabolism by tampering with the PTH and Vitamin D system. Insulinopenia and low levels of IGF-1 exert an additional inhibitory effect on osteoblasts at different stages of diabetes mellitus. Increased production of adipocytes feed the cycle of chronic inflammation by producing ROS and inflammatory cytokines, which induce osteoblast apoptosis. ROS upholds this process by facilitating MSC differentiation into adipocytes by mediating PPAR-γ and reducing WNT transcription. Additionally, increased production of AGEs leads to non-enzymatic cross-links between collagen fibers and increased inflammation by the activation of RAGE. The accumulation of these patho-mechanisms ultimately leads to decreased bone quality and bone turnover in diabetes mellitus.
Figure 1.
The interaction between osteoblasts, adipocytes, MSCs, and the marrow environment is altered in diabetes mellitus. Hyperglycemia directly alters gene expression associated with osteoblast activity by the inhibition of MSC maturation and metabolism, and indirectly alters bone metabolism by tampering with the PTH and Vitamin D system. Insulinopenia and low levels of IGF-1 exert an additional inhibitory effect on osteoblasts at different stages of diabetes mellitus. Increased production of adipocytes feed the cycle of chronic inflammation by producing ROS and inflammatory cytokines, which induce osteoblast apoptosis. ROS upholds this process by facilitating MSC differentiation into adipocytes by mediating PPAR-γ and reducing WNT transcription. Additionally, increased production of AGEs leads to non-enzymatic cross-links between collagen fibers and increased inflammation by the activation of RAGE. The accumulation of these patho-mechanisms ultimately leads to decreased bone quality and bone turnover in diabetes mellitus.

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Murray, C.E.; Coleman, C.M. Impact of Diabetes Mellitus on Bone Health. Int. J. Mol. Sci. 2019, 20, 4873. https://doi.org/10.3390/ijms20194873
AMA Style
Murray CE, Coleman CM. Impact of Diabetes Mellitus on Bone Health. International Journal of Molecular Sciences. 2019; 20(19):4873. https://doi.org/10.3390/ijms20194873
Chicago/Turabian StyleMurray, Cliodhna E., and Cynthia M. Coleman. 2019. "Impact of Diabetes Mellitus on Bone Health" International Journal of Molecular Sciences 20, no. 19: 4873. https://doi.org/10.3390/ijms20194873
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.