Endometrial Cancer as a Metabolic Disease with Dysregulated PI3K Signaling: Shedding Light on Novel Therapeutic Strategies

Department of Obstetrics and Gynecology, Shimane University Faculty of Medicine, 89-1 Enya-cho, Izumo, Shimane 693-8501, Japan
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2020, 21(17), 6073; https://doi.org/10.3390/ijms21176073
Submission received: 3 July 2020
/
Revised: 21 August 2020
/
Accepted: 21 August 2020
/
Published: 23 August 2020
(This article belongs to the Special Issue Cancer Biology in Diabetes)
Abstract
:Endometrial cancer (EC) is one of the most common malignancies of the female reproductive organs. The most characteristic feature of EC is the frequent association with metabolic disorders. However, the components of these disorders that are involved in carcinogenesis remain unclear. Accumulating epidemiological studies have clearly revealed that hyperinsulinemia, which accompanies these disorders, plays central roles in the development of EC via the insulin-phosphoinositide 3 kinase (PI3K) signaling pathway as a metabolic driver. Recent comprehensive genomic analyses showed that over 90% of ECs have genomic alterations in this pathway, resulting in enhanced insulin signaling and production of optimal tumor microenvironments (TMEs). Targeting PI3K signaling is therefore an attractive treatment strategy. Several clinical trials for recurrent or advanced ECs have been attempted using PI3K-serine/threonine kinase (AKT) inhibitors. However, these agents exhibited far lower efficacy than expected, possibly due to activation of alternative pathways that compensate for the PIK3-AKT pathway and allow tumor growth, or due to adaptive mechanisms including the insulin feedback pathway that limits the efficacy of agents. Overcoming these responses with careful management of insulin levels is key to successful treatment. Further interest in specific TMEs via the insulin PI3K-pathway in obese women will provide insight into not only novel therapeutic strategies but also preventive strategies against EC.
1. Introduction
Endometrial cancer (EC) is the most common gynecologic malignancy in the Western world. In the United States (US), EC accounts for 6.9% of cancer diagnoses in women, with an estimated 61,180 new cases and 12,160 deaths in 2019 [1]. In 10 years, the incidence of EC is expected to dramatically increase to over 120,000 cases, becoming the third most common malignancy affecting women in the US after lung and colorectal cancers [2]. In Japan, mortality from EC has dramatically increased by 2.3 and 4.3 times in the past two and three decades, respectively [3], presumably as a consequence of changes in the lifestyle in the Japanese population. Over 75% of women diagnosed with EC have stage I or II disease, and thus have good clinical outcomes with five-year overall survival of 75–90% [4,5]. Operative procedures and postoperative adjuvant treatments are well standardized, including hysterectomy with retroperitoneal lymphadenectomy, followed by paclitaxel/platinum-based conventional chemotherapies. However, women with recurrent or advanced disease have lower response rates to conventional treatments, and clinical outcomes are extremely poor [6,7,8,9]. Therefore, further therapeutic developments are needed for these patients, hopefully combined with molecular-targeted agents, based on novel aspects of precise molecular pathogenesis and carcinogenesis of this tumor.
EC is highly associated with a variety of metabolic disorders, including obesity, hypertension, and diabetes, known as the metabolic triad. The metabolic tumor microenvironments (TMEs) formed in these disorders are closely involved in the development of EC. In 2013, a new classification of EC was proposed by The Cancer Genome Atlas (TCGA) [10], based on the genomic status of the patients. TCGA analyses uncovered specific molecular landscapes including the dysregulated phosphoinositide 3 kinase (PI3K)-serine/threonine protein kinase B (AKT) pathway, which is essential for metabolic homeostasis and carcinogenesis of EC. This review article introduces the metabolic TMEs of EC, describes how the genomic status of EC and insulin-PI3K signaling direct TMEs, and suggests important clues for developing novel therapeutic and preventive strategies.
2. Reclassification of EC According to Genomic Features
2.1. Conventional Classification
Two pathogenetic types of ECs that are driven by distinct metabolic and endocrine signals were proposed in 1983 [11]. Type I tumors develop from a precursor lesion with atypical hyperplasia, and are the most commonly observed, usually representing low-grade endometrioid histology, with lower International Federation of Gynecology and Obstetrics (FIGO) stages [11]. This type is considered to be estrogen driven, to be associated with obesity, and to have a favorable clinical outcome. In contrast, type II tumors are relatively rare, represent high-grade serous histology, exhibit very aggressive tumor behaviors independent of estrogen, and are more common in nonobese women [11]. Type II tumors arise de novo, usually from atrophic endometrium in postmenopausal women, and have a considerably worse prognosis than type I tumors [11].
Histological assessment is subjectively assigned according to the microscopic appearance and predefined pathologic criteria. Tumor grade is defined by nuclear features and the proportion of glandular structures versus solid components, and the histologic subtype is determined by morphologic criteria and supported by immunohistochemistry. The pathologic accuracy of the tumor grade, especially in cases of grade 3 endometrioid and serous cancers, is occasionally low due to poor diagnostic reproducibility [12,13]. Interobserver disagreement on histologic subtype or tumor grade was reported in one-third or more of ECs [12,13]. Classical histologic classification is therefore insufficient to effectively triage patients for optimal treatment options. Objective information that mirrors the biological features as well as clinical behaviors of individual tumors is greatly needed.
2.2. Novel Molecular-Based Classification
Following these demands, TCGA reported the results of comprehensive genomic studies of ECs, including a combination of whole genome and exome sequencing, microsatellite instability (MSI), and copy number analyses [10]. Using this molecular information, a total of 232 endometrioid and serous ECs were classified into four groups (DNA polymerase epsilon (POLE) ultramutated, MSI hypermutated, copy-number low, and copy-number high) that correlate with progression-free survival.
2.2.1. POLE Ultramutated Group
A novel category stated by TCGA is the ultramutated POLE subgroup, which consists of 7% of ECs with unusually high mutation rates (232 × 10−6 mutations/Mb), generating extremely frequent mutations in cancer-associated genes such as PTEN (94%), PIK3CA (71%), PIK3R1 (65%), FBXW7 (82%), ARID1A (75%), KRAS (53%), and AIRD1B (47%) [10]. This subgroup is characterized by mutations in the exonuclease domain of POLE and exemplified by an increased C →A transversion frequency [10]. POLE encodes the major catalytic and proofreading subunits of the Polε (polymerase epsilon) DNA polymerase complex, which incorporates bases with high fidelity during leading strand DNA replication, ensuring a low mutation rate in the daughter strand. Exonuclease domain mutations are mostly detected in hotspots and include P286R and V411L, which induce decreased proofreading abilities by this enzyme complex [10]. Eventually, increased rates of replicative errors occur, resulting in the ultramutated phenotype. To assess the POLE status, the original report used whole genome or exome sequencing, but subsequent studies used more focused methods including Sanger sequencing [14,15], target-gene panels [16,17], digital PCR [18,19], or functional analyses [20]. Very favorable clinical outcomes were shown in these studies in women with POLE mutations, even in those with high-grade tumors.
2.2.2. MSI Hypermutated Group
TCGA described an MSI subgroup with high mutation rates (18 × 10−6 mutations/Mb). MSI represents a specific phenotype caused by a defective DNA mismatch repair (MMR) system composed of four representative MMR proteins (MLH1, MSH2, MSH6, and PMS2) [10]. MSI can be evaluated with PCR, which directly reflects the MSI status via amplifying a panel of five microsatellite marker DNA sequences based on Bethesda guidelines [21]. These markers have highly unstable mononucleotide repeat loci. When tumors have dysfunctional MMRs, PCR generates altered product sizes that reflect unstable microsatellites, compared to those from corresponding normal tissues [22]. More conveniently, MMR deficiency can also be detected by immunohistochemistry of MMR proteins [23]. MSI hypermutations can result from (a) an inherited cancer syndrome such as Lynch syndrome, which is characterized by germline mutations in MMR genes [24], (b) somatic mutations of these genes, or (c) epigenetic silencing such as DNA promoter methylation of these gene [23,25]. MLH1 promoter hypermethylation is the leading cause of MSI in ECs [26]. MSI tumors account for 28% of ECs, and are characterized by approximately 10-fold greater mutation frequency than microsatellite-stable (MSS) tumors, few somatic copy number alterations (SCNAs), frequent KRAS mutations, high expression levels of phospho-AKT, and low expression levels of PTEN. PI3K pathway alterations are detected in 95% of the ECs in this group [10].
2.2.3. Copy-Number High Group
This group contains primarily serous cancers with extensive SCNAs that were determined with Affymetrix SNP 6.0 microarrays using DNA from frozen tissue specimens and has a low mutation rate (2.3 × 10−6 mutations/Mb) [10]. Hierarchical clustering identified a group with significant reoccurring regions with amplifications or deletions, consisting of most serous cancers and one quarter of high-grade (grade 3) ECs [10]. The most striking feature of these tumors is the extremely high frequency of TP53 mutations (detected in over 90% of patients) and a high frequency of FBXW7 and PPP2R1A mutations [10], which were previously reported as common in serous cancers but not endometrioid cancer. Thus, a subset of high-grade endometrioid cancers have similar SCNAs and mutation spectra as serous cancers, indicating that they may benefit from treatment modalities used in patients with serous cancers. PI3K pathway alterations are detected in 60% of the ECs in this group, fewer than in the other three groups [10].
2.2.4. Copy-Number Low Group
All remaining ECs that do not belong to the above three groups are categorized as the copy-number low group and mainly consist of MSS tumors. This group tends to have elevated progesterone receptor expression, suggesting potential responsiveness to hormone therapies. Copy-number low, MSS tumors have a high frequency of mutations in CTNNB1 (52%), the only gene with a higher mutation frequency than in MSI tumors [10]. PI3K pathway alterations are observed in 92% of the ECs in this group [10].
Exome sequence data in TCGA study identified significantly mutated genes including PTEN, PIK3R1, PIK3CA, FBXW7, KRAS, and POLE, among which PIK3CA and PIK3R1 mutations were most frequent [10]. However, unlike other tumor types, they co-occur with PTEN mutations in the MSI hypermutated and copy-number low groups. Of particular interest is that over 90% of endometrioid tumors have mutations in PIK3CA, PIK3R1, and PTEN [10], which encode the major components of the PI3K-AKT signaling pathway. Thus, a representative feature of EC is the much higher frequency of mutations in genes encoding components of the PI3K-AKT pathway than any other tumor type [10]. Most of these mutations cause enhanced signaling of this pathway and are tightly associated with metabolic TMEs, which are introduced in subsequent sections. The genomic landscape of cancer genomes has thus made the PI3K-AKT axis one of the most exploitable for drug development, especially for POLE mutated, MSI hypermutated, and copy-number low ECs. The POLE ultramutated and MSI hypermutated groups are associated with high neoantigen loads caused by an increased mutation rate along with increased infiltration of CD8-positive tumor-infiltrating lymphocytes [27,28,29]. Therefore, both types are considered to be good candidates for immune checkpoint inhibitor therapies. Taken together, novel molecular classification of ECs confers the theoretical background for optimal use of molecular-targeted therapeutics, according to the mutational status of individual ECs [29,30].
3. Risk Factors for EC
Among a variety of risk factors identified for EC, metabolic syndrome, which is a constellation of obesity, hypertension, hyperglycemia, and hyperlipidemia, has drawn great interest in the past two decades. The first case-control study for risk factors for endometrioid and serous types of EC was reported in 1997 [31]. Analysis of 328 endometrioid and 26 serous cancers demonstrated that body mass index (BMI), use of estrogen, age at menarche, and parity were significantly linked to the endometrioid subtype, but not serous cancers. The use of oral contraceptives was negatively associated with both subtypes. This study revealed the distinct risk factors associated with each histological subtype. Subsequent studies have accumulated consistent evidence demonstrating that metabolic syndrome is a representative risk factor for EC [21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37].
A meta-analysis of six studies [38] confirmed that metabolic syndrome is associated with an increased risk of EC (relative risk: 1.89, 95% confidence interval (CI) 1.34–2.67, p < 0.001), but with significant heterogeneity among the six studies (I2 = 92%, p < 0.001). The risk estimates for any single factor of metabolic syndrome were 2.21 (p < 0.001) for BMI, 1.81 (p = 0.044) for hyperglycemia, 1.81 (p = 0.024) for values of higher blood pressure, and 1.17 (p < 0.001) for high levels of triglyceride [38]. Another meta-analysis of 19 studies for EC [39] demonstrated that a BMI increase of 5 kg/m2 results in a 59% increased risk of EC (95% CI 1.50–16.8, p < 0.0001). A history of bariatric surgery and maintenance of normal weight after surgery were significantly associated with a 71 and 81% decreased risk for uterine cancers, respectively, including EC [40]. Although many studies have shown a positive association between a risk for EC and hypertension, hyperlipidemia, hyperglycemia, and diabetes mellitus, the risks from these factors were less robust than obesity [32,33,34,35,36,37,38,41,42]. A recent large-scale prospective study of 1205 women further confirmed that age (odds ratio (OR) 1.14, 95% CI 1.1–1.2) and BMI (OR 1.39, 95% CI 1.1–1.7) are positively associated with EC, but no significant association with other potential risk factors was found [43].
Recent progress in genome-wide association studies (GWAS) of EC have uncovered genetic risk regions [44], and the success of GWAS facilitated the use of Mendelian randomization methods to examine the risk factors of EC. Mendelian randomization utilizes genetic variants associated with a candidate risk factor as “instruments” in an instrumental variable analysis and examines the association of the risk factor without confounding factors [45]. The initial Mendelian randomization analysis used the single nucleotide polymorphisms associated with BMI (97 variants) as instrumental variables [46], and EC risk was found to be higher in individuals with genetically predicted higher BMI (OR 1.13, 95% CI 1.04–1.22, p = 0.002). A subsequent analysis also found higher EC risk with genetically predicted higher BMI (OR 2.11, 95% CI 1.94–2.28, p = 3.4 × 10−17) [47]. These studies clearly confirmed the causal relationship between obesity and EC.
4. Molecular Mechanisms Linking Obesity and EC
4.1. Excess Estrogen Production in Adipose Tissues
The endometrium is a highly regenerative tissue, and its proliferative activity is strictly controlled by estrogen and progesterone in each menstrual cycle. Long-term estrogen exposure without progesterone promotes the development of EC [48]. In postmenopausal women, ovarian function to produce these hormones is greatly decreased, and systemic and local hormonal levels significantly decrease. However, EC is primarily a disease of postmenopausal woman, with about 25% of cases occurring in premenopausal women and only 5% occurring in women younger than 40 years of age [49].
Why does obesity increase the risk of EC? In postmenopausal women, estrogen is no longer supplied by the ovaries, but by adipose tissues via the conversion of androstenedione to estradiol by aromatase. Aromatase activity is higher in obese women than nonobese women, presumably due to higher amounts of adipose tissues capable of expressing aromatase [50,51]. The concentration of estrogen is higher in adipose tissues than serum [52], and adipose tissues are the main source of estrogen. Adipose tissue is also the most common metastatic site of EC. Thus, the higher estrogen levels in obese women increase the risk of EC. A Mendelian randomization analysis using the genetic variants highly associated with serum estrogen levels in postmenopausal women confirmed the causal relationship between serum estrogen levels and EC (OR 1.15, 95% CI 1.11–1.21, p = 4.8 × 10−11) [53]. In relation to estrogen levels, older age at menarche (that would be presumed to decrease lifetime estrogen exposure) was confirmed to be associated with a lower risk of EC (OR 0.78, 95% CI 0.70–0.87, p = 1.0 × 10−5) [54].
Sex-hormone-binding globulin (SHBG) is a circulating sex steroid transporter secreted by the liver that binds to circulating sex steroids with high affinity and negatively regulates the concentration of bioactive sex hormones in the blood, affecting the bioavailability of sex steroids including estrogen [55]. Low serum levels of SHBG are associated with metabolic syndrome [56]. Diets leading to weight loss in women are associated with an increase in SHBG levels [57]. Exposure of hepatic cells to glucose significantly reduces SHBG mRNA expression via decreased hepatocyte nuclear factor-4 alpha (HNF-4α), which is the key transcription factor that binds the SHBG promoter and is down-regulated by glucose-induced de novo lipogenesis in the liver [58]. Eventually, down-regulation of SHBG in obese women leads to enhanced bioavailability of estrogen. Taken together, these findings indicate that obesity increases the risk of EC via increasing systemic and local estrogen activity, a notable characteristic of TMEs in EC, strongly accounting for the increased risk in obese women (Figure 1).
4.2. Chronic Low-Grade Inflammation in Adipose Tissues
Although originally classified as a simple energy storage organ, adipose tissues comprise a variety of cell types including adipocytes, immune cells, endothelial cells, and fibroblasts [59]. However, they are currently considered to function as a major endocrine system that produces and releases diverse types of secretory proteins, adipokines, growth factors, cytokines, and chemokines into the systemic circulation [60]. In obese women, excessive accumulation of adipose tissues accompanies inflammatory changes, leading to chronic low-grade inflammation characterized by increased macrophage infiltration and mildly elevated circulating adipokines (leptin, adiponectin, resistin, visfatin, omentin), cytokines (interleukin (IL)-6, tumor necrosis factor (TNF)-α), chemokines (macrophage chemoattractant protein (MCP)-1), acute phase reactants (C reactive protein), plasminogen activator inhibitor-1, and serum amyloid A [61,62,63,64] (Figure 1). This type of inflammation in obesity is fundamentally distinct from typical inflammatory responses that are induced as host defense, because obesity-related inflammation is sterile, mild, low-grade, and most importantly, causes insulin resistance. Adipokines are categorized as pro- or anti-inflammatory adipokines according to their effects on inflammatory responses in adipose tissues. Most of them are proinflammatory, whereas adiponectin is anti-inflammatory. Low serum levels of adiponectin are associated with obesity, insulin resistance, and hyperinsulinemia [65]. In patients with EC, serum levels of adiponectin are significantly decreased compared with those with benign disorders or normal endometrium [66]. Serum levels of adiponectin are negatively correlated with the risk of EC, especially in postmenopausal women [67]. EC cells express adiponectin receptors, and adiponectin can directly suppress proliferation of EC via adiponectin-mediated AMP-activated protein kinase (AMPK) activation [68]. A recent meta-analysis of 18 studies examined the association of various circulating adiponectin and cytokine levels and revealed that patients with circulating adiponectin levels in the highest tertile had a decreased EC risk compared to those with levels in the lowest tertile (OR 0.51, 95% CI 0.38–0.69, p < 0.00001). Women with circulating leptin concentrations in the highest tertile showed an increased EC risk compared to those with concentrations in the lowest tertile (OR 2.19, 95% CI 1.45–3.30, p = 0.0002) [69].
4.3. How Does Chronic Inflammation Induce Insulin Resistance?
The molecular mechanisms through which the obesity-induced inflammatory response cause insulin resistance have recently been investigated [70,71]. Insulin resistance is defined as the perturbation of insulin-mediated signaling pathways, leading to systemic hyperglycemia. The components of these pathways are directly or indirectly targeted by obesity-induced inflammatory responses.
Zhan et al. studied the effect of TNF-α on insulin resistance in adipocytes and hepatocytes [72]. Treatment with TNF-α leads to phosphorylation of S6 kinase 1 (S6K1), a serine/threonine kinase downstream from AKT in the insulin signaling pathway that is involved in negative feedback regulation of insulin action. This TNF-α-induced phosphorylation of S6K1 triggers the phosphorylation of insulin receptor substrate (IRS)-1 at multiple serine residues, resulting in accelerated degradation of IRS-1 and impaired insulin-stimulated glucose uptake in adipocytes.
Kamei et al. demonstrated the effects of MCP-1 on insulin resistance using MCP-1 transgenic mice [73]. Overexpression of MCP-1 in adipose tissue results in systemic insulin resistance and decreased tyrosine phosphorylation of the insulin receptor (IR) and IRS-1. This phenomenon was concurrent with decreased phosphorylation of AKT, which was partially restored by mitogen-activated protein kinase inhibitors, indicating that the extracellular signal-regulated kinase pathway is part of the mechanism by which MCP-1 inhibits insulin signaling.
Hirosumi et al. demonstrated that c-Jun amino-terminal kinase (JNK) activity is abnormally elevated in obesity [74]. TNF-α-induced phosphorylation of IRS-1 at Ser 307 leads to insulin resistance in liver cells, and a JNK inhibitor largely abolishes the phosphorylation of IRS-1 at Ser 307 and restores insulin resistance, indicating that JNK activity is involved in the mechanism of TNF-α-induced insulin resistance.
Rotter et al. examined the effect of IL-6 on insulin resistance [75]. Treatment of adipocyte cells with IL-6 does not increase phosphorylation of IRS-1 at Ser 307, but exerts long-term inhibitory effects on the gene expression of IRS-1. Similarly, IL-6 inhibits gene expression of glucose transporter (GLUT)-4 as well as peroxisome proliferator-activated receptor gamma, a factor that increases insulin sensitivity by enhancing storage of fatty acids in fat cells and upregulates adiponectin release from these cells. Importantly, the expression of IL-6, like that of TNF-α and IL-8, is markedly increased in adipocytes from insulin-resistant individuals. Jager et al. examined the effect of IL-1β on glucose uptake of adipocytes [76] and found that prolonged IL-1β treatment reduces insulin-induced glucose uptake with marked inhibition of GLUT-4 translocation to the plasma membrane in response to insulin. This inhibitory effect involves a decrease in the amount of IRS-1 mRNA and protein. Taken together, multiple cytokines coordinately participate in the development of insulin resistance in adipose tissues (Figure 1).
4.4. Role of the Insulin-Like Growth Factor (IGF)–IGF Binding Protein (IGFBP) Axis in the Development of EC
IGFs are growth factors that modulate steroid hormone activity in the endometrium via autocrine and paracrine regulatory loops. Endometrial stromal cells are the major source of IGF-I and IGF-II, and epithelial cells mainly produce IGF receptors [77]. The IGF axis is essential for not only endometrial physiology but the pathogenesis of EC. Obesity-induced insulin resistance presumably leads to hyperglycemia, leading to increased synthesis of insulin. Eventually, upregulated insulin levels lead to inhibition of the synthesis of IGFBP-1 [78,79,80]. IGFBP-1 binds to IGF to stabilize the complex, and excess amounts of IGFBP-1 reduce the bioavailability of IGF to their receptors and suppress subsequent intracellular IGF signaling [80]. Thus, insulin-induced suppression of IGFBP-1 results in enhanced biological activity of IGF. Ayabe et al. found an increase in circulating levels of IGF-1 and a decrease in IGFBP-1 in postmenopausal women with EC [81]. Studies by several groups have shown that IGF-1 plays a role in the development of both Type I and II ECs, emphasizing the importance of altered IGF1R expression [82,83,84]. Estrogen enhances IGF-1 activity in EC cells [85,86], and increased local estrogen levels in obese women may therefore stimulate IGF-1 synthesis in the endometrium [80]. Collectively, obesity-induced insulin resistance enhances IGF-1 activity in concert with the excess estrogen locally produced in obese women, leading to development of EC (Figure 1).
5. Is Diabetes Mellitus No Longer a Risk Factor for EC?
Several meta-analyses support the idea that diabetes is associated with an approximately two- to threefold increased risk of EC [41,87,88] independent of obesity [41,89,90,91]. A large-scale case-control study indicated a twofold increase in the risk of EC in women with diabetes [34]. However, some cohort studies showed that such an association is lost or weakened to modest levels when adjustments are made for BMI [92,93]. Therefore, the relationship between diabetes and EC is still controversial.
Seven representative large-scale cohort studies measured the blood glucose levels and evaluated the risk of EC [32,33,94,95,96,97,98]. Most studies demonstrated that women with higher baseline glucose levels have a 1.2–2.6 times increased risk for EC. However, the strength of such an association varies depending on age, BMI, and menopausal status, and one cohort study demonstrated that blood glucose levels are not associated with EC [93]. Five large-scale case-control studies demonstrated significantly higher blood glucose levels in women with ECs than controls [37,99,100,101,102].
In a Mendelian randomization analysis [103], single nucleotide polymorphisms associated with diabetes (49 variants), fasting glucose (36 variants), fasting insulin (18 variants), and early insulin secretion (17 variants) were used to examine the association with the risk of EC. EC risk was higher in individuals with genetically predicted higher fasting insulin levels (OR 2.34, 95% CI 1.06–5.14, p = 0.03) or genetically predicted higher 30 min postchallenge insulin levels (OR 1.40, 95% CI 1.12–1.76, p = 0.003). Importantly, however, no associations were observed between genetic risk for diabetes (OR 0.91, 95% CI 0.79–1.04, p = 0.16) or higher fasting glucose (OR 1.00, 95% CI 0.67–1.50, p = 0.99) and EC. This study thus supports a causal association between higher fasting insulin levels and a risk for EC, independent of higher glucose levels. Collectively, these studies indicate that hyperinsulinemia, rather that hyperglycemia, is more directly associated with a risk for the development of EC.
6. Roles of Insulin-PI3K Signaling Pathways in Metabolic TMEs
6.1. The Insulin-PI3K Signaling Pathway
The insulin-PIK3 signaling axis plays a fundamental role in human cellular growth as well as glucose metabolism [104,105] (Figure 2).
The details of the signaling pathway are shown in the representative review articles [106,107]. At the cell surface, insulin binds to the IR, which exists as an α2β2 heterodimer. Following insulin binding, the tyrosine kinase domain of the β subunit autophosphorylates the subunit and has intrinsic kinase activity on proximal substrates such as IRS family proteins. Phosphorylation of IRS1/2 on specific tyrosine residues can lead to interactions with the regulatory subunit of PI3K (p85) and then inhibit the catalytic subunit (p110) of PI3K, allowing it to phosphorylate phosphatidylinositol 4,5-bisphosphate (PIP2). The product of this reaction is a second messenger, phosphatidylinositol 3,4,5-triphosphate (PIP3). Inversely, phosphatase and tensin homologue (PTEN) can dephosphorylate PIP3 to form PIP2, limiting activation of this pathway. Loss-of-function mutations of PTEN cause significant elevations in PIP3, a driving force of this pathway. The newly produced PIP3 recruits the following factors to the cell membrane: phosphoinositide-dependent kinase 1 (PDK1), AKT, and other factors with a pleckstrin homology domain, a shared property of these PI3K effectors. This process involves propagation of signals via a series of serine/threonine kinases, tyrosine kinases, and modulators with small GTPase activities, resulting in activation of multiple diverging downstream pathways that confer a large portion of the metabolic functions of insulin. AKT is more universally downstream of receptor-mediated PI3K activation. Through colocalization of the constitutively active PDK with AKT at the plasma membrane via the pleckstrin homology domain, PDK1 mediates phosphorylation of AKT. Phosphorylated AKT further phosphorylates several downstream targets, including thioredoxin-interacting protein (TXNIP), glycogen synthase kinase (GSK), and forkhead box protein O1 (FOXO1) [108,109]. TXNIP phosphorylation results in inhibition of its function. Because TXNIP functions as an adaptor for endocytosis of GLUTs, TXNIP inactivation via phosphorylation leads to dissociation from the GLUTs, thus inhibiting their endocytosis and resulting in rapid glucose uptake upon insulin stimulation. AKT also phosphorylates GSK3, which results in inhibition of GSK3 activity and activation of glycogen synthesis in muscle and liver. AKT phosphorylation of FOXO1 causes translocation of FOXO1 from the nucleus to the cytoplasm, leading to the failure of FOXO1 to transactivate downstream targets that negatively regulate cell proliferation. AKT phosphorylation leads to activation of mammalian target of rapamycin (mTOR), a protein kinase that controls cell proliferation via activation of the eukaryotic initiation factor 4E-binding protein-1 complex and S6K1, which phosphorylates the ribosomal S6 protein. Although AKT is a universal downstream target of receptor-mediated PI3K activation, alternative AKT-independent signaling cascades are initiated by PI3K activity and impact glucose metabolism. Representative ATK-independent pathways involving glycolysis and cell proliferation are conferred by Ras-related C3 botulinus toxin substrate (RAC) or serum and glucocorticoid-induced protein kinase (SGK) signaling [104,105,110].
6.2. Status of the PI3K-AKT Pathway Affects Tumor Behavior via Altered Glucose Metabolism
Tumors have modified cellular metabolism based on aerobic fermentation, with a propensity to shift away from the oxidative phosphorylation pathway to the anabolic glycolysis pathway in which the final product of glycolysis, pyruvate, is converted into lactate [111,112]. In the anabolic glycolysis pathway, tumor cells generate far fewer adenosine triphosphate (ATP) molecules per molecule of glucose than normal cells that use the oxidative phosphorylation pathway. Therefore, tumor cells require greater glucose uptake than normal cells [113]. This increase in glucose flux is largely mediated by insulin-PI3K signaling. Mutations, mostly gain-of-function mutations, in the genes that encode components of this pathway result in enhanced signaling activity. Therefore, insulin plays critical roles in tumor growth via the PI3K signaling pathway. This concept is supported by studies demonstrating insulin-mediated acceleration of tumor development in humans and mouse models [114,115]. In patients with EC, serum levels of insulin are elevated with concomitant up-regulation of IR, IRS-1, and AKT expression compared to those without EC [116]. Levels of p-IR, p-IRS-1, and p-AKT are higher in EC patients with advanced stage disease, high-grade tumors, myometrial invasion, and lymph-node metastasis, and are significantly correlated with levels of serum insulin. In EC cells, insulin-induced mitogenic effects are inhibited by treatment with LY294002, a PI3K inhibitor [116]. Thus, insulin plays an essential role in the tumorigenesis of EC via the PI3K-AKT pathway, consistent with accumulating epidemiological studies indicating that hyperinsulinemia, rather than hyperglycemia, plays essential roles in the development of EC, as discussed above.
The tumorigenic effects of insulin depend on the genotype in tumors. A study with human cancer cell lines in a mouse xenograft model demonstrated that tumors resistant to dietary restriction carried mutations that constitutively activate the PI3K signaling pathway [117]. Substitution of an activated mutant allele of PI3K with wild-type PI3K in otherwise isogenic human cancer cells successfully converted a tumor resistant to dietary restriction to one that was sensitive to dietary restriction [117]. Restoration of PTEN expression in a PTEN-null cancer cell line also showed similar conversion. In engineered mouse tumors, dietary restriction did not affect the tumor burden of PTEN-null mouse tumors, but it significantly decreased the size of tumors that lacked constitutively activated PI3K signaling [116]. Thus, the PI3K pathway is key to the sensitivity of tumors to dietary restriction, and different levels of PI3K activation in tumors contribute to their differential sensitivities to dietary restriction [118].
The frequency of genetic alterations in PIK3 signaling pathways varies among tumor types, and EC is the leading type of cancer, with over 90% of cases harboring alterations in the pathway [10,119,120]. In tissues with chronic exposure to high levels of insulin such as the pancreas and liver, PI3K signaling is constitutively active, and genetic alterations may not be required to maintain metabolic environments [107]. In contrast, the endometrium probably has low or normal levels of insulin signaling that may be insufficient to maintain the high regenerative capacity [107]. Increasing insulin sensitivity by acquired mutations of PI3K pathway genes may overcome this strict environment, conferring an insulin-dependent growth advantage with optimal TMEs. This may be one reason why EC has extremely frequent genetic alterations in PI3K pathways. In the circumstances of obesity-induced insulin resistance and hyperinsulinemia, genetic alterations in PI3K pathways that enhance signal intensity may cause further exaggeration of signaling, promoting tumor development. In this regard, PI3K signaling seems to be an attractive target for molecular-based therapies against EC.
7. What about Metformin? Is the PI3K-AKT Pathway a Target?
Metformin is a biguanide that is commonly used to treat diabetes. Recent studies have accumulated evidence showing potential benefits of metformin on the incidence, clinical outcome, and survival of EC, although contradictory results have been reported, especially for survival [121]. The direct antitumor effects of metformin are mainly due to via activation of the AMP-activated protein kinase (AMPK) pathway as well as inhibition of the PI3K-AKT pathway (Figure 2). Initially, metformin inhibits oxidative phosphorylation at the mitochondrial level, causing a reduction in the proton gradient across the inner mitochondrial membrane, with a decrease in proton-driven synthesis of ATP and an increase in the ratio of cellular adenosine monophosphate (AMP) to ATP. Eventually, AMP preferentially binds to AMPK with a subsequent conformational change that allows for phosphorylation/activation of AMPK by liver kinase B1 (LKB1) [117]. Activated AMPK converts cells to a catabolic state through AMPK-mediated phosphorylation, leading to inhibition of downstream transcription factors involved in ATP-consuming synthetic pathways. Additionally, activated AMPK inhibits mTOR, leading to decreased phosphorylation of the eukaryotic initiation factor 4E binding protein-1 complex, S6K1, and ribosomal S6 protein, all of which cause reduced protein synthesis [122]. Thus, metformin can also be classified as an mTOR inhibitor.
Furthermore, metformin decreases secretion of IGF-1 and expression and phosphorylation of IGF-1R, whereas it increases expression of IGFBP-1 in EC cells [123,124,125,126,127,128,129,130,131,132,133,134,135]. Thus, metformin targets the PI3K-AKT pathway.
A recent study also showed that FOXO1 is another target of AMPK. AMPK activation induces FOXO1 nuclear translocation and activates function of this protein. An in vitro study using EC cells reported that metformin-induced nuclear accumulation of FOXO1 is concurrent with an AMPK-dependent decrease in phosphorylated FOXO1 and transactivates the downstream target, leading to inhibition of cell proliferation [126].
Thus, although metformin has been traditionally and widely used to treat diabetes, it may be effective in EC by targeting both the AMPK and PI3K-AKT pathways. Several retrospective observational studies of metformin were reported for EC [127,128,129,130,131,132]. These studies compared the survival of metformin users with that of nonusers in diabetic or nondiabetic, type I or type II EC patients, and the results were contradictory; some studies showed greater survival in metformin users than nonusers, whereas others failed to find any effect on survival parameters, possibly due to high heterogeneity in patients. Nevertheless, a meta-analysis of the above studies supports a greater overall survival in metformin users compared to nonusers [133], although the low number of studies and the lack of randomized studies compromise the evidence level of the meta-analysis. Clinical trials using metformin for atypical endometrial hyperplasia (AEH) or EC have recently accumulated evidence for its efficacy [134,135,136,137,138,139,140,141,142,143,144,145,146]. Most studies used the presurgical window approach, in which patients diagnosed by endometrial biopsy were treated with metformin during the period prior to hysterectomy, and some studies targeted patients who underwent fertility-sparing treatments in combination with progestins. The results are summarized in Table 1.
Most studies evaluated tumor and serum factors in relation to proliferative activity or insulin signaling, and some factors were affected by metformin as expected. Notably, one recent randomized controlled trial of fertility-sparing treatment evaluated the clinical benefit in AEH or EC patients, and found that the complete response rate within 16 weeks of treatment was higher in the metformin plus megestrol group than in the megestrol group (34.3 versus 20.7%, OR 2.0, 95% CI 0.89–4.51, p = 0.09), and the difference was more significant in AEH patients (39.6 versus 20.4%, OR 2.56, 95% CI 1.06–6.21, p = 0.04) [146]. Few clinical trials have been reported for advanced or recurrent ECs, but a phase 2/3 trial by the Gynecologic Oncology Group for advanced or recurrent EC (NCT02065687) is ongoing and is comparing paclitaxel/carboplatin alone or in combination with metformin as first-line chemotherapy. Furthermore, a phase 2 trial of metformin for advanced and recurrent EC is ongoing and is testing the combination of letrozole and everolimus (NCT01797523), and a phase 1/2 trial of metformin combined with metronomic cyclophosphamide and olaparib (NCT02755844) is also underway.
8. Pitfalls of Targeting PI3K-AKT Pathways for the Treatment of EC
8.1. Clinical Outcome of Treatment of EC with PI3K-AKT Inhibitors
Accumulating preclinical studies have clearly demonstrated that insulin signaling greatly affects the growth properties of EC in different metabolic conditions. Targeting insulin signaling has a theoretical background in EC because EC has an extremely high frequency of genetic abnormalities in genes that encode components of the PI3K-AKT pathway, leading to enhanced insulin signaling. Several clinical trials have been performed for recurrent or advanced EC with molecular-targeting agents that target components of PI3K-AKT pathways [147,148,149,150,151,152,153,154,155,156,157] (Table 2). Response rates of these studies were disappointing, at less than 20%, although all studies enrolled patients with refractory ECs who were mostly pretreated with multiple chemotherapy regimens. Notably, some of these studies enrolled patients with mutations in genes that encode components of the PI3K-AKT pathway to examine whether these patients would benefit from these agents. However, most studies failed to find superior responses in such patients compared to those with wild-type genotypes.
8.2. Potential Molecular Mechanisms of Treatment Failure
Considering the frequent genetic alterations in components of the PI3K-AKT pathway, we expected superior effects of these inhibitors in EC. How can we explain these unexpected results? One possible explanation is that EC is not a type of tumor that exhibits oncogene addiction such as ALK-translocated non-small-cell lung cancers, and EC with PIK3CA mutations may not always exclusively depend on PI3K-AKT signaling for growth [105,158]. Even in tumors that are highly dependent on the oncogenic program conferred by mutations in components of the PI3K-AKT pathway, alternative pathways can be activated for tumor survival, overcoming the acute inhibitory actions of PI3K-AKT inhibitors [159]. Furthermore, PI3K signaling may activate the AKT-independent pathway in some ECs [105], and AKT inhibitors are not expected to exert effects in such tumors. For example, SGK family members are closely related to AKT with common upstream signaling and downstream targets, and EC has significantly increased SGK1 activity compared to normal endometria [160]. In tumors in which AKT is not constitutively activated or inhibited by drugs, PI3K signaling activates SGK1 and may phosphorylate overlapping substrates, resulting in bypass of the AKT pathway [158] (Figure 2). Levels of SGK1 expression can be used as markers to predict the efficacy of AKT inhibitors [160]. Thus, further refining patient populations with specific molecular markers that predict efficacy is of particular importance, and exploitation of reliable markers should be a strong focus. Finally, a limitation of the use of PI3K-AKT inhibitors is that multiple adaptive mechanisms may limit the efficacy of these agents in which insulin feedback mechanisms play critical roles [161]. In healthy metabolic tissues including liver, skeletal muscle, and adipose tissues, the insulin-PI3K pathway coordinates the clearance of glucose from the blood. Once this pathway is disrupted, the tissues perceive changes in signaling as a decrease in insulin levels. When this happens, the liver responds by increasing the release of glucose, leading to elevated levels of blood glucose and concomitant release of insulin from the pancreas. Eventually, induced hyperinsulinemia exerts tumorigenic effects. In fact, reflex hyperglycemia and hyperinsulinemia were observed when PIK3 was targeted with PI3K inhibitors [162,163] (Table 2). Patients with diabetes are usually excluded from clinical trials using PI3K-AKT inhibitors, but patients with potential insulin resistance, including those who are borderline diabetic, may be enrolled. Patients exhibiting hyperglycemia during treatment with PI3K-AKT inhibitors probably used exogenous insulin to reduce blood levels of glucose, possibly leading to therapy-induced hyperinsulinemia. The insulin feedback system may therefore be a limitation when inhibitors targeting insulin signaling are used.
8.3. How Can Drug Resistance Be Overcome to Improve the Efficacy of PI3K-AKT Inhibitors?
Concomitant inhibition of IR with PI3K-AKT inhibitors may be a potential strategy for preventing the unfavorable effects of reflex hyperglycemia and hyperinsulinemia. However, this is not a simple strategy. Some studies selected IGF-1R inhibitors as a therapeutic strategy for treating a variety of tumors in combination with IR inhibitors [164]. Although such combinatorial treatments showed some enhanced antitumor activity, extreme hyperglycemia and hyperinsulinemia occurred, which can reactivate IR and affect long-term treatment outcomes [165,166]. Thus, targeting insulin signaling requires further modalities.
A variety of approaches have been proposed for minimizing insulin levels. As a pharmacological approach, inhibitors of sodium-glucose co-transporter 2 (SGLT2) are potential candidates that can reduce serum levels of glucose by inhibiting reabsorption of glucose from the renal ultrafiltrate, resulting in clearance of glucose from the serum into urine. The combination of SGLT2 inhibitors with PI3K inhibitors successfully prevents reflex hyperglycemia and hyperinsulinemia [160,167]. Potassium channel inhibitors such as diazoxide may also be an alternative pharmacological approach to combine with PI3K inhibitors to inhibit release of insulin from the pancreas [168,169].
More than one third of the population in the USA are thought to have some degree of insulin resistance. In such individuals, consideration of the pivotal role of diet and exercise in reducing the glycogen stock and preventing therapy-induced hyperglycemia and hyperinsulinemia may be important [107,162,170]. At present, no consistent evidence in humans exists to suggest that a combination of diet or exercise with PI3K-AKT inhibitors reduces serum levels of insulin and improves the efficacy of treatment for EC. Clinical trials will therefore be needed to test these strategies in a clinical setting. Currently, 11 combination trials are ongoing in which the ketogenic diet is combined with standard chemotherapy or radiotherapy to examine the feasibility of these diets to treat patients with cancer [107]. Because EC is a leading cancer that is tightly associated with metabolic disorders and a dysregulated PI3-AKT pathway, the fundamental role of diet, exercise, and more basically of lifestyle choices should be emphasized for not only treatment strategies but preventive strategies. In fact, when lifestyle-related factors are evaluated with the healthy lifestyle index (HLI) involving diet and physical activity, women in the highest quintile of the HLI score had a significantly lower risk of EC (hazard ratio 0.61, 95% CI 0.51–0.72), underscoring the potential importance of lifestyle in lowering the risk of EC in premenopausal women [171].
9. Conclusions
This review article focuses on how metabolic disorders, especially obesity, are involved in EC. Specific TMEs are formed via excess estrogen produced by increased aromatase activity or decreased levels of SHBG, as well as chronic low-grade inflammation in adipose tissues with elevated levels of adipokines and cytokines, leading to insulin resistance. IGF activation due to decreased IGFBP via insulin resistance also promotes TMEs.
Hyperinsulinemia, rather than hyperglycemia, plays an essential role in the development of EC via the PI3K-AKT pathway. Over 90% of ECs have genomic alterations, causing enhanced insulin signaling to produce an optimal TME. We thus have a theoretical rationale for using PI3K inhibitors to treat EC. Clinical trials for recurrent or advanced ECs, however, have shown an unfavorable efficacy, and here we discussed the potential mechanisms. This failure is mainly due to activation of alternative pathways upon exposure to the inhibitors that can compensate for the PIK3-AKT pathway or to adaptive mechanisms including the insulin feedback pathway, thus limiting the efficacy of agents. Inhibiting these pathways will be a key to successful treatment, along with maintaining low levels of insulin during treatment. Combinations of PI3K-AKT inhibitors with specified diet protocols including the ketogenic diet are possible candidates for such strategies, and optimal combinations should be explored by ongoing and future clinical trials. Further understanding of specific TMEs via the insulin-PI3K pathway in obese women will provide openings for not only novel therapeutic strategies but preventive strategies against EC.
Author Contributions
S.K., writing of the manuscript, funding acquisition; K.N., critical review and suggestion. Both authors have read and agreed to the published version of the manuscript.
Funding
This review was supported in part by the Japan Society for the Promotion of Science (JSPS) (KAKENHI grant No. JP18H02946).
Conflicts of Interest
The author declares no conflict of interest.
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheikh, M.A.; Althouse, A.D.; Freese, K.E.; Soisson, S.; Edwards, R.P.; Welburn, S.; Sukumvanich, P.; Comerci, J.; Kelley, J.; LaPorte, R.E. USA endometrial cancer projections to 2030: Should we be concerned? Future Oncol. 2014, 10, 2561–2568. [Google Scholar] [CrossRef] [PubMed]
- Cancer Information Service. Cancer Registry and Statistics (Vital Statistics of Japan); Cancer Information Service: National Cancer Center, Tokyo, Japan, 2016.
- Rose, P.G. Endometrial carcinoma. N. Engl. J. Med. 1996, 335, 640–649. [Google Scholar] [CrossRef] [PubMed]
- Morice, P.; Leary, A.; Creutzberg, C.; Abu-Rustum, N.; Darai, E. Endometrial cancer. Lancet 2016, 387, 1094–1108. [Google Scholar] [CrossRef]
- Ueda, S.M.; Kapp, D.S.; Cheung, M.K.; Shin, J.Y.; Osann, K.; Husain, A.; Teng, N.N.; Berek, J.S.; Chan, J.K. Trends in demographic and clinical characteristics in women diagnosed with corpus cancer and their potential impact on the increasing number of deaths. Am. J. Obstet. Gynecol. 2008, 198, 218. [Google Scholar] [CrossRef]
- Hamilton, C.A.; Cheung, M.K.; Osann, K.; Chen, L.; Teng, N.N.; Longacre, T.A.; Powell, M.A.; Hendrickson, M.R.; Kapp, D.S.; Chan, J.K. Uterine papillary serous and clear cell carcinomas predict for poorer survival compared to grade 3 endometrioid corpus cancers. Br. J. Cancer 2006, 94, 642–646. [Google Scholar] [CrossRef]
- Del Carmen, M.G.; Birrer, M.; Schorge, J.O. Uterine papillary serous cancer: A review of the literature. Gynecol. Oncol. 2012, 127, 651–661. [Google Scholar] [CrossRef]
- Del Carmen, M.G.; Boruta, D.M.; Schorge, J.O. Recurrent endometrial cancer. Clin. Obstet. Gynecol. 2011, 54, 266–277. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network; Kandoth, C.; Schultz, N.; Cherniack, A.D.; Akbani, R.; Liu, Y.; Shen, H.; Robertson, A.G.; Pashtan, I.; Shen, R.; et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [Google Scholar]
- Bokhman, J.V. Two pathogenetic types of endometrial carcinoma. Gynecol. Oncol. 1983, 15, 10–17. [Google Scholar] [CrossRef]
- Gilks, C.B.; Oliva, E.; Soslow, R.A. Poor interobserver reproducibility in the diagnosis of high-grade endometrial carcinoma. Am. J. Surg. Pathol. 2013, 37, 874–881. [Google Scholar] [CrossRef] [PubMed]
- Han, G.; Sidhu, D.; Duggan, M.A.; Arseneau, J.; Cesari, M.; Clement, P.B.; Ewanowich, C.A.; Kalloger, S.E.; Köbel, M. Reproducibility of histological cell type in high-grade endometrial carcinoma. Mod. Pathol. 2013, 26, 1594–1604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, B.; Hoang, L.N.; McIntyre, J.B.; Duggan, M.A.; Nelson, G.S.; Lee, C.H.; Köbel, M. POLE exonuclease domain mutation predicts long progression-free survival in grade 3 endometrioid carcinoma of the endometrium. Gynecol. Oncol. 2014, 134, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Stelloo, E.; Bosse, T.; Nout, R.A.; MacKay, H.J.; Church, D.N.; Nijman, H.W.; Leary, A.; Edmondson, R.J.; Powell, M.E.; Crosbie, E.J.; et al. Refining prognosis and identifying targetable pathways for high-risk endometrial cancer: A TransPORTEC initiative. Mod. Pathol. 2015, 28, 836–844. [Google Scholar] [CrossRef] [Green Version]
- Billingsley, C.C.; Cohn, D.E.; Mutch, D.G.; Stephens, J.A.; Suarez, A.A.; Goodfellow, P.J. Polymerase ɛ (POLE) mutations in endometrial cancer: Clinical outcomes and implications for Lynch syndrome testing. Cancer 2015, 12, 386–394. [Google Scholar] [CrossRef] [Green Version]
- Stelloo, E.; Nout, R.A.; Osse, E.M.; Jürgenliemk-Schulz, I.J.; Jobsen, J.J.; Lutgens, L.C.; van der Steen-Banasik, E.M.; Nijman, H.W.; Putter, H.; Bosse, T.; et al. Improved Risk Assessment by Integrating Molecular and Clinicopathological Factors in Early-stage Endometrial Cancer-Combined Analysis of the PORTEC Cohorts. Clin. Cancer Res. 2016, 22, 4215–4224. [Google Scholar] [CrossRef] [Green Version]
- Church, D.N.; Stelloo, E.; Nout, R.A.; Valtcheva, N.; Depreeuw, J.; ter Haar, N.; Noske, A.; Amant, F.; Tomlinson, I.P.; Wild, P.J.; et al. Prognostic significance of POLE proofreading mutations in endometrial cancer. J. Natl. Cancer Inst. 2014, 107, 402. [Google Scholar] [CrossRef]
- Talhouk, A.; McConechy, M.K.; Leung, S.; Li-Chang, H.H.; Kwon, J.S.; Melnyk, N.; Yang, W.; Senz, J.; Boyd, N.; Karnezis, A.N.; et al. A clinically applicable molecular-based classification for endometrial cancers. Br. J. Cancer 2015, 113, 299–310. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Research Network; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar]
- Boland, C.R.; Thibodeau, S.N.; Hamilton, S.R.; Sidransky, D.; Eshleman, J.R.; Burt, R.W.; Meltzer, S.J.; Rodriguez-Bigas, M.A.; Fodde, R.; Ranzani, G.N.; et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: Development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998, 58, 5248–5257. [Google Scholar]
- Risinger, J.I.; Berchuck, A.; Kohler, M.F.; Watson, P.; Lynch, H.T.; Boyd, J. Genetic instability of microsatellites in endometrial carcinoma. Cancer Res. 1993, 53, 5100–5103. [Google Scholar] [PubMed]
- Kanaya, T.; Kyo, S.; Maida, Y.; Yatabe, N.; Tanaka, M.; Nakamura, M.; Inoue, M. Frequent hypermethylation of MLH1 promoter in normal endometrium of patients with endometrial cancers. Oncogene 2003, 22, 2352–2360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fishel, R.; Lescoe, M.K.; Rao, M.R.; Copeland, N.G.; Jenkins, N.A.; Garber, J.; Kane, M.; Kolodner, R. The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell 1993, 75, 1027–1038. [Google Scholar] [CrossRef]
- Esteller, M.; Levine, R.; Baylin, S.B.; Ellenson, L.H.; Herman, J.G. MLH1 promoter hypermethylation is associated with the microsatellite instability phenotype in sporadic endometrial carcinomas. Oncogene 1998, 17, 2413–2417. [Google Scholar] [CrossRef] [Green Version]
- Salvesen, H.B.; MacDonald, N.; Ryan, A.; Iversen, O.E.; Jacobs, I.J.; Akslen, L.A.; Das, S. Methylation of hMLH1 in a population-based series of endometrial carcinomas. Clin. Cancer Res. 2000, 6, 3607–3613. [Google Scholar]
- Van Gool, I.C.; Eggink, F.A.; Freeman-Mills, L.; Stelloo, E.; Marchi, E.; de Bruyn, M.; Palles, C.; Nout, R.A.; de Kroon, C.D.; Osse, E.M.; et al. POLE Proofreading Mutations Elicit an Antitumor Immune Response in Endometrial Cancer. Clin. Cancer Res. 2015, 21, 3347–3355. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, H.; Nakayama, K.; Ishikawa, M.; Nakamura, K.; Ishibashi, T.; Sanuki, K.; Ono, R.; Sasamori, H.; Minamoto, T.; Iida, K.; et al. Microsatellite instability is a Biomark. for immune checkpoint inhibitors in endometrial cancer. Oncotarget 2017, 9, 5652–5664. [Google Scholar] [CrossRef] [Green Version]
- Mitamura, T.; Dong, P.; Ihira, K.; Kudo, M.; Watari, H. Molecular-targeted therapies and precision medicine for endometrial cancer. Jpn. J. Clin. Oncol. 2019, 49, 108–120. [Google Scholar] [CrossRef] [Green Version]
- Dey, N.; De, P. Decision making in precision oncology: An issue of mutational contextuality. Am. J. Cancer Res. 2019, 9, 628–629. [Google Scholar]
- Sherman, M.E.; Sturgeon, S.; Brinton, L.A.; Potischman, N.; Kurman, R.J.; Berman, M.L.; Mortel, R.; Twiggs, L.B.; Barrett, R.J.; Wilbanks, G.D. Risk factors and hormone levels in patients with serous and endometrioid uterine carcinomas. Mod. Pathol. 1997, 10, 963–968. [Google Scholar]
- Cust, A.E.; Kaaks, R.; Friedenreich, C.; Bonnet, F.; Laville, M.; Tjønneland, A.; Olsen, A.; Overvad, K.; Jakobsen, M.U.; Chajès, V.; et al. Metabolic syndrome, plasma lipid, lipoprotein and glucose levels, and endometrial cancer risk in the European Prospective Investigation into Cancer and Nutrition (EPIC). Endocr. Relat. Cancer 2007, 14, 755–767. [Google Scholar] [CrossRef] [PubMed]
- Bjørge, T.; Stocks, T.; Lukanova, A.; Tretli, S.; Selmer, R.; Manjer, J.; Rapp, K.; Ulmer, H.; Almquist, M.; Concin, H.; et al. Metabolic syndrome and endometrial carcinoma. Am. J. Epidemiol. 2010, 171, 892–902. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, Z.; Yu, X.; Zhang, X.; Lü, S.; Chen, X.; Lü, B. The association between metabolic abnormality and endometrial cancer: A large case-control study in China. Gynecol. Oncol. 2010, 117, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Friedenreich, C.M.; Biel, R.K.; Lau, D.C.; Csizmadi, I.; Courneya, K.S.; Magliocco, A.M.; Yasui, Y.; Cook, L.S. Case-control study of the metabolic syndrome and metabolic risk factors for endometrial cancer. Cancer Epidemiol. Biomark. Prev. 2011, 20, 2384–2395. [Google Scholar] [CrossRef] [Green Version]
- Rosato, V.; Zucchetto, A.; Bosetti, C.; Dal Maso, L.; Montella, M.; Pelucchi, C.; Negri, E.; Franceschi, S.; La Vecchia, C. Metabolic syndrome and endometrial cancer risk. Ann. Oncol. 2011, 22, 884–889. [Google Scholar] [CrossRef]
- Trabert, B.; Wentzensen, N.; Felix, A.S.; Yang, H.P.; Sherman, M.E.; Brinton, L.A. Metabolic syndrome and risk of endometrial cancer in the united states: A study in the SEER-medicare linked database. Cancer Epidemiol. Biomark. Prev. 2015, 24, 261–267. [Google Scholar] [CrossRef] [Green Version]
- Esposito, K.; Chiodini, P.; Capuano, A.; Bellastella, G.; Maiorino, M.I.; Giugliano, D. Metabolic syndrome and endometrial cancer: A meta-analysis. Endocrine 2014, 45, 28–36. [Google Scholar] [CrossRef]
- Renehan, A.G.; Tyson, M.; Egger, M.; Heller, R.F.; Zwahlen, M. Body-mass index and incidence of cancer: A systematic review and meta-analysis of prospective observational studies. Lancet 2008, 371, 569–578. [Google Scholar] [CrossRef]
- Ward, K.K.; Roncancio, A.M.; Shah, N.R.; Davis, M.A.; Saenz, C.C.; McHale, M.T.; Plaxe, S.C. Bariatric surgery decreases the risk of uterine malignancy. Gynecol. Oncol. 2014, 133, 63–66. [Google Scholar] [CrossRef]
- Friberg, E.; Orsini, N.; Mantzoros, C.S.; Wolk, A. Diabetes mellitus and risk of endometrial cancer: A meta-analysis. Diabetologia 2007, 50, 1365–1374. [Google Scholar] [CrossRef]
- Liao, C.; Zhang, D.; Mungo, C.; Tompkins, D.A.; Zeidan, A.M. Is diabetes mellitus associated with increased incidence and disease-specific mortality in endometrial cancer? A systematic review and meta-analysis of cohort studies. Gynecol. Oncol. 2014, 135, 163–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, M.A.; Long, B.J.; Sherman, M.E.; Lemens, M.A.; Podratz, K.C.; Hopkins, M.R.; Ahlberg, L.J.; Mc Guire, L.J.; Laughlin-Tommaso, S.K.; Wentzensen, N.; et al. A prospective clinical cohort study of women at increased risk for endometrial cancer. Gynecol. Oncol. 2020, 156, 169–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Mara, T.A.; Glubb, D.M.; Kho, P.F.; Thompson, D.J.; Spurdle, A.B. Genome-Wide Association Studies of Endometrial Cancer: Latest Developments and Future Directions. Cancer Epidemiol. Biomark. Prev. 2019, 28, 1095–1102. [Google Scholar] [CrossRef] [PubMed]
- Davey Smith, G.; Hemani, G. Mendelian randomization: Genetic anchors for causal inference in epidemiological studies. Hum. Mol. Genet. 2014, 23, R89–R98. [Google Scholar] [CrossRef] [Green Version]
- Prescott, J.; Setiawan, V.W.; Wentzensen, N.; Schumacher, F.; Yu, H.; Delahanty, R.; Bernstein, L.; Chanock, S.J.; Chen, C.; Cook, L.S.; et al. Body Mass Index Genetic Risk Score and Endometrial Cancer Risk. PLoS ONE. 2015, 10, e0143256. [Google Scholar] [CrossRef]
- Painter, J.N.; O’Mara, T.A.; Marquart, L.; Webb, P.M.; Attia, J.; Medland, S.E.; Cheng, T.; Dennis, J.; Holliday, E.G.; McEvoy, M.; et al. Genetic Risk Score Mendelian Randomization Shows that Obesity Measured as Body Mass Index, but not Waist:Hip Ratio, Is Causal for Endometrial Cancer. Cancer Epidemiol. Biomark. Prev. 2016, 25, 1503–1510. [Google Scholar] [CrossRef] [Green Version]
- Hammond, C.B.; Jelovsek, F.R.; Lee, K.L.; Creasman, W.T.; Parker, R.T. Effects of long-term estrogen replacement therapy. II. Neoplasia. Am. J. Obstet. Gynecol. 1979, 133, 537–547. [Google Scholar] [CrossRef]
- Pecorelli, S.; Pasinetti, B.; Angioli, R.; Favalli, G.; Odicino, F. Systemic therapy for gynecological neoplasms: Ovary, cervix, and endometrium. Cancer Chemother. Biol. Response Modif. 2005, 22, 515–544. [Google Scholar]
- Byers, T.; Sedjo, R.L. Body fatness as a cause of cancer: Epidemiologic clues to biologic mechanisms. Endocr. Relat. Cancer 2015, 22, R125–R134. [Google Scholar] [CrossRef]
- Bulun, S.E.; Zeitoun, K.; Sasano, H.; Simpson, E.R. Aromatase in aging women. Semin. Reprod. Endocrinol. 1999, 17, 349–358. [Google Scholar] [CrossRef]
- Blankenstein, M.A.; van de Ven, J.; Maitimu-Smeele, I. Intratumoral levels of estrogens in breast cancer. J. Steroid Biochem. Mol. Biol. 1999, 69, 293–297. [Google Scholar] [CrossRef]
- Thompson, D.J.; O’Mara, T.A.; Glubb, D.M.; Painter, J.N.; Cheng, T.; Folkerd, E.; Doody, D.; Dennis, J.; Webb, P.M.; Australian National Endometrial Cancer Study Group (ANECS); et al. CYP19A1 fine-mapping and Mendelian randomization: Estradiol is causal for endometrial cancer. Endocr. Relat. Cancer 2016, 23, 77–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Day, F.R.; Thompson, D.J.; Helgason, H.; Chasman, D.I.; Finucane, H.; Sulem, P.; Ruth, K.S.; Whalen, S.; Sarkar, A.K.; Albrecht, E.; et al. Genomic analyses identify hundreds of variants associated with age at menarche and support a role for puberty timing in cancer risk. Nat. Genet. 2017, 49, 834–841. [Google Scholar] [CrossRef] [PubMed]
- Hammond, G.L.; Wu, T.S.; Simard, M. Evolving utility of sex hormone-binding globulin measurements in clinical medicine. Curr. Opin. Endocrinol. Diabetes Obes. 2012, 19, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Kalme, T.; Seppälä, M.; Qiao, Q.; Koistinen, R.; Nissinen, A.; Harrela, M.; Loukovaara, M.; Leinonen, P.; Tuomilehto, J. Sex hormone-binding globulin and insulin-like growth factor-binding protein-1 as indicators of metabolic syndrome, cardiovascular risk, and mortality in elderly men. J. Clin. Endocrinol. Metab. 2005, 90, 1550–1556. [Google Scholar] [CrossRef]
- Morisset, A.S.; Blouin, K.; Tchernof, A. Impact of diet and adiposity on circulating levels of sex hormone-binding globulin and androgens. Nutr. Rev. 2008, 66, 506–516. [Google Scholar] [CrossRef]
- Selva, D.M.; Hogeveen, K.N.; Innis, S.M.; Hammond, G.L. Monosaccharide-induced lipogenesis regulates the human hepatic sex hormone-binding globulin gene. Version 2. J. Clin. Investig. 2007, 117, 3979–3987. [Google Scholar]
- Kanneganti, T.D.; Dixit, V.D. Immunological complications of obesity. Nat. Immunol. 2012, 13, 707–712. [Google Scholar] [CrossRef]
- Ahima, R.S.; Lazar, M.A. Adipokines and the peripheral and neural control of energy balance. Mol. Endocrinol. 2008, 22, 1023–1031. [Google Scholar] [CrossRef]
- Ye, J.; McGuinness, O.P. Inflammation during obesity is not all bad: Evidence from animal and human studies. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E466–E477. [Google Scholar] [CrossRef] [Green Version]
- Ye, J. Mechanisms of insulin resistance in obesity. Front. Med. 2013, 7, 14–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Wang, J. The Role of Metabolic Syndrome in Endometrial Cancer: A Review. Front. Oncol. 2019, 9, 744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chait, A.; den Hartigh, L.J. Adipose Tissue Distribution, Inflammation and Its Metabolic Consequences, Including Diabetes and Cardiovascular Disease. Front. Cardiovasc. Med. 2020, 7, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aggeloussi, S.; Theodorou, A.A.; Paschalis, V.; Nikolaidis, M.G.; Fatouros, I.G.; Owolabi, E.O.; Kouretas, D.; Koutedakis, Y.; Jamurtas, A.Z. Adipocytokine levels in children: Effects of fatness and training. Pediatr. Exerc. Sci. 2012, 24, 461–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rzepka-Górska, I.; Bedner, R.; Cymbaluk-Płoska, A.; Chudecka-Głaz, A. Serum adiponectin in relation to endometrial cancer and endometrial hyperplasia with atypia in obese women. Eur. J. Gynaecol. Oncol. 2008, 29, 594–597. [Google Scholar]
- Zeng, F.; Shi, J.; Long, Y.; Tian, H.; Li, X.; Zhao, A.Z.; Li, R.F.; Chen, T. Adiponectin and Endometrial Cancer: A Systematic Review and Meta-Analysis. Cell Physiol. Biochem. 2015, 36, 1670–1678. [Google Scholar] [CrossRef]
- Moon, H.S.; Chamberland, J.P.; Aronis, K.; Tseleni-Balafouta, S.; Mantzoros, C.S. Direct role of adiponectin and adiponectin receptors in endometrial cancer: In vitro and ex vivo studies in humans. Mol. Cancer Ther. 2011, 10, 2234–2243. [Google Scholar] [CrossRef] [Green Version]
- Ellis, P.E.; Barron, G.A.; Bermano, G. Adipocytokines and their relationship to endometrial cancer risk: A systematic review and meta-analysis. Gynecol. Oncol. 2020, 158, 507–516. [Google Scholar] [CrossRef]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191. [Google Scholar] [CrossRef] [Green Version]
- Kwon, H.; Pessin, J.E. Adipokines mediate inflammation and insulin resistance. Front. Endocrinol. 2013, 4, 71. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Gao, Z.; Yin, J.; Quon, M.J.; Ye, J. S6K directly phosphorylates IRS-1 on Ser-270 to promote insulin resistance in response to TNF-(alpha) signaling through IKK2. J. Biol. Chem. 2008, 283, 35375–35382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamei, N.; Tobe, K.; Suzuki, R.; Ohsugi, M.; Watanabe, T.; Kubota, N.; Ohtsuka-Kowatari, N.; Kumagai, K.; Sakamoto, K.; Kobayashi, M. Overexpression of monocyte chemoattractant protein-1 in adipose tissues causes macrophage recruitment and insulin resistance. J. Biol. Chem. 2006, 281, 26602–26614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirosumi, J.; Tuncman, G.; Chang, L. A central role for JNK in obesity and insulin resistance. Nature 2002, 420, 333–336. [Google Scholar] [CrossRef] [PubMed]
- Rotter, V.; Nagaev, I.; Smith, U. Interleukin-6 (IL-6) induces insulin resistance in 3T3-L1 adipocytes and is, like IL-8 and tumor necrosis factor-alpha, overexpressed in human fat cells from insulin-resistant subjects. J. Biol. Chem. 2003, 278, 45777–45784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jager, J.; Grémeaux, T.; Cormont, M.; Le Marchand-Brustel, Y.; Tanti, J.F. Interleukin-1beta-induced insulin resistance in adipocytes through down-regulation of insulin receptor substrate-1 expression. Endocrinology 2007, 148, 241–251. [Google Scholar] [CrossRef]
- Zhou, J.; Dsupin, B.A.; Giudice, L.C.; Bondy, C.A. Insulin-like growth factor system gene expression in human endometrium during the menstrual cycle. J. Clin. Endocrinol. Metab. 1994, 79, 1723–1734. [Google Scholar]
- Ki-Järvinen, H.; Mäkimattila, S.; Utriainen, T.; Rutanen, E.M. Portal insulin concentrations rather than insulin sensitivity regulate serum sex hormone-binding globulin and insulin-like growth factor binding protein 1 in vivo. J. Clin. Endocrinol. Metab. 1995, 80, 3227–3232. [Google Scholar]
- Lewitt, M.S.; Dent, M.S.; Hall, K. The Insulin-Like Growth Factor System in Obesity, Insulin Resistance and Type 2 Diabetes Mellitus. J. Clin. Med. 2014, 3, 1561–1574. [Google Scholar] [CrossRef]
- Rutanen, E.M. Insulin-like growth factors and insulin-like growth factor binding proteins in the endometrium. Effect of intrauterine levonorgestrel delivery. Hum. Reprod. 2000, 15, 173–181. [Google Scholar] [CrossRef] [Green Version]
- Ayabe, T.; Tsutsumi, O.; Sakai, H.; Yoshikawa, H.; Yano, T.; Kurimoto, F.; Taketani, Y. Increased circulating levels of insulin-like growth factor-I and decreased circulating levels of insulin-like growth factor binding protein-1 in postmenopausal women with endometrial cancer. Endocr. J. 1997, 44, 419–424. [Google Scholar] [CrossRef] [Green Version]
- McCampbell, A.S.; Broaddus, R.R.; Loose, D.S.; Davies, P.J. Overexpression of the insulin-like growth factor I receptor and activation of the AKT pathway in hyperplastic endometrium. Clin. Cancer Res. 2006, 12, 6373–6378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attias-Geva, Z.; Bentov, I.; Kidron, D.; Amichay, K.; Sarfstein, R.; Fishman, A.; Bruchim, I.; Werner, H. p53 Regulates insulin-like growth factor-I receptor gene expression in uterine serous carcinoma and predicts responsiveness to an insulin-like growth factor-I receptor-directed targeted therapy. Eur. J. Cancer 2012, 48, 1570–1580. [Google Scholar] [CrossRef] [PubMed]
- Amichay, K.; Kidron, D.; Attias-Geva, Z.; Schayek, H.; Sarfstein, R.; Fishman, A.; Werner, H.; Bruchim, I. BRCA1 is expressed in uterine serous carcinoma (USC) and controls insulin-like growth factor I receptor (IGF-IR) gene expression in USC cell lines. Int. J. Gynecol. Cancer 2012, 22, 748–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hana, V.; Murphy, L.J. Expression of insulin-like growth factors and their binding proteins in the estrogen responsive Ishikawa human endometrial cancer cell line. Endocrinology 1994, 135, 2511–2516. [Google Scholar] [CrossRef]
- Kleinman, D.; Karas, M.; Roberts, C.T. Modulation of insulin-like growth factor I (IGF-I) receptors and membrane-associated IGF-binding proteins in endometrial cancer cells by estradiol. Endocrinology 1995, 136, 2531–2537. [Google Scholar] [CrossRef]
- Noto, H.; Osame, K.; Sasazuki, T.; Noda, M. Substantially increased risk of cancer in patients with diabetes mellitus: A systematic review and meta-analysis of epidemiologic evidence in Japan. J. Diabetes Its Complicat. 2010, 24, 345–353. [Google Scholar] [CrossRef]
- Zhang, Z.H.; Su, P.Y.; Hao, J.H.; Sun, Y.H. The Role of Preexisting Diabetes Mellitus on Incidence and Mortality of Endometrial Cancer: A Meta-Analysis of Prospective Cohort Studies. Int. J. Gynecol. Cancer 2013, 23, 294–303. [Google Scholar] [CrossRef]
- Barone, B.B.; Yeh, H.C.; Snyder, C.F.; Peairs, K.S.; Stein, K.B.; Derr, R.L.; Wolff, A.C.; Brancati, F.L. Long-term all-cause mortality in cancer patients with preexisting diabetes mellitus: A systematic review and meta-analysis. JAMA 2008, 300, 2754–2764. [Google Scholar] [CrossRef] [Green Version]
- Lindemann, K.; Vatten, L.J.; Ellstrøm-Engh, M.; Eskild, A. Body mass, diabetes and smoking, and endometrial cancer risk: A follow-up study. Br. J. Cancer 2008, 98, 1582–1585. [Google Scholar] [CrossRef] [Green Version]
- Lucenteforte, E.; Bosetti, C.; Talamini, R.; Montella, M.; Zucchetto, A.; Pelucchi, C.; Franceschi, S.; Negri, E.; Levi, F.; La Vecchia, C. Diabetes and endometrial cancer: Effect modification by body weight, physical activity and hypertension. Br. J. Cancer 2007, 97, 995–998. [Google Scholar] [CrossRef] [Green Version]
- Anderson, K.E.; Anderson, E.; Mink, P.J.; Hong, C.P.; Kushi, L.H.; Sellers, T.A.; Lazovich, D.; Folsom, A.R. Diabetes and endometrial cancer in the Iowa women’s health study. Cancer Epidemiol. Biomark. Prev. 2001, 10, 611–616. [Google Scholar]
- Luo, J.; Beresford, S.; Chen, C.; Chlebowski, R.; Garcia, L.; Kuller, L.; Regier, M.; Wactawski-Wende, J.; Margolis, K.L. Association between diabetes, diabetes treatment and risk of developing endometrial cancer. Br. J. Cancer 2014, 111, 1432–1439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furberg, A.S.; Thune, I. Metabolic abnormalities (hypertension, hyperglycemia and overweight), lifestyle (high energy intake and physical inactivity) and endometrial cancer risk in a Norwegian cohort. Int. J. Cancer 2003, 104, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Gunter, M.J.; Hoover, D.R.; Yu, H.; Wassertheil-Smoller, S.; Manson, J.E.; Li, J.; Harris, T.G.; Rohan, T.E.; Xue, X.; Ho, G.Y. A prospective evaluation of insulin and insulin-like growth factor-I as risk factors for endometrial cancer. Cancer Epidemiol. Biomark. Prev. 2008, 17, 921–929. [Google Scholar] [CrossRef] [Green Version]
- Lambe, M.; Wigertz, A.; Garmo, H.; Walldius, G.; Jungner, I.; Hammar, N. Impaired glucose metabolism and diabetes and the risk of breast, endometrial, and ovarian cancer. Cancer Causes Control 2011, 22, 1163–1171. [Google Scholar] [CrossRef]
- Stattin, P.; Björ, O.; Ferrari, P.; Lukanova, A.; Lenner, P.; Lindahl, B.; Hallmans, G.; Kaaks, R. Prospective study of hyperglycemia and cancer risk. Diabetes Care 2007, 30, 561–567. [Google Scholar] [CrossRef] [Green Version]
- Shou, H.F.; Ni, J.; Zhu, T.; Chen, J.H.; Zhang, X.; Xu, X.X.; Chen, L.; Yu, H. Association between endometrial cancer and metabolic syndrome. Chin. J. Obstet. Gynecol. 2010, 45, 128–131. [Google Scholar]
- Friedenreich, C.M.; Langley, A.R.; Speidel, T.P.; Lau, D.C.W.; Courneya, K.S.; Csizmadi, I.; Magliocco, A.M.; Yasui, Y.; Cook, L.S. Case-control study of markers of insulin resistance and endometrial cancer risk. Endocr. Relat. Cancer 2012, 19, 785–792. [Google Scholar] [CrossRef] [Green Version]
- Modesitt, S.C.; Geffel, D.L.; Via, J.; Weltman, L.A. Morbidly obese women with and without endometrial cancer: Are there differences in measured physical fitness, body composition, or hormones? Gynecol. Oncol. 2012, 124, 431–436. [Google Scholar] [CrossRef]
- Zhan, Y.; Wang, J.; Ma, Y.; Liu, Z.; Xu, H.; Lu, S.; Lu, B. Serum insulin-like, growth factor binding protein-related protein 1 (IGFBP-rP1) and endometrial cancer risk in Chinese women. Int. J. Cancer 2013, 132, 411–416. [Google Scholar] [CrossRef]
- Özdemir, S.; Batmaz, G.; Ates, S.; Celik, C.; Incesu, F.; Peru, C. Relation of metabolic syndrome with endometrial pathologies in patients with abnormal uterine bleeding. Gynecol. Endocrinol. 2015, 31, 725–729. [Google Scholar] [CrossRef] [PubMed]
- Nead, K.T.; Sharp, S.J.; Thompson, D.J.; Painter, J.N.; Savage, D.B.; Semple, R.K.; Barker, A.; Australian National Endometrial Cancer Study Group (ANECS); Perry, J.R.; Attia, J. Evidence of a Causal Association Between Insulinemia and Endometrial Cancer: A Mendelian Randomization Analysis. J. Natl. Cancer Inst. 2015, 107, djv178. [Google Scholar] [CrossRef] [PubMed]
- Lien, E.C.; Dibble, C.C.; Toker, A. PI3K signaling in cancer: Beyond AKT. Curr. Opin. Cell Biol. 2017, 45, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Vasudevan, K.M.; Barbie, D.A.; Davies, M.A.; Rabinovsky, R.; McNear, C.J.; Kim, J.J.; Hennessy, B.T.; Tseng, H.; Pochanard, P.; Kim, S.Y.; et al. AKT-independent signaling downstream of oncogenic PIK3CA mutations in human cancer. Cancer Cell 2009, 16, 21–32. [Google Scholar] [CrossRef] [Green Version]
- Engelman, J.A.; Luo, J.; Cantley, L.C. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 2006, 7, 606–619. [Google Scholar] [CrossRef]
- Hopkins, B.D.; Goncalves, M.D.; Cantley, L.C. Insulin-PI3K signaling: An evolutionarily insulated metabolic driver of cancer. Nat. Rev. Endocrinol. 2020, 16, 276–283. [Google Scholar] [CrossRef]
- Dong, M.Q.; Venable, J.D.; Au, N.; Xu, T.; Park, S.K.; Cociorva, D.; Johnson, J.R.; Dillin, A.; Yates, J.R. Quantitative mass spectrometry identifies insulin signaling targets in C. elegans. Science 2007, 317, 660–663. [Google Scholar] [CrossRef]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Juvekar, A.; Lyssiotis, C.A.; Lien, E.C.; Albeck, J.G.; Oh, D.; Varma, G.; Hung, Y.P.; Ullas, S.; Lauring, J.; et al. Phosphoinositide 3-Kinase Regulates Glycolysis through Mobilization of Aldolase from the Actin Cytoskeleton. Cell 2016, 164, 433–446. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar]
- Hsu, P.P.; Sabatini, D.M. Cancer cell metabolism: Warburg and beyond. Cell 2008, 134, 703–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orgel, E.; Mittelman, S.D. The links between insulin resistance, diabetes, and cancer. Curr. Diab. Rep. 2013, 13, 213–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novosyadlyy, R.; Lann, D.E.; Vijayakumar, A.; Rowzee, A.; Lazzarino, D.A.; Fierz, Y.; Carboni, J.M.; Gottardis, M.M.; Pennisi, P.A.; Molinolo, A.A.; et al. Insulin-mediated acceleration of breast cancer development and progression in a nonobese model of type 2 diabetes. Cancer Res. 2010, 70, 741–751. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Hua, S.; Tian, W.; Zhang, L.; Zhao, J.; Zhang, H.; Zhang, W.; Xue, F. Mitogenic and anti-apoptotic effects of insulin in endometrial cancer are phosphatidylinositol 3-kinase/Akt dependent. Gynecol. Oncol. 2012, 125, 734–741. [Google Scholar] [CrossRef]
- Shaw, R.J.; Lamia, K.A.; Vasquez, D.; Koo, S.H.; Bardeesy, N.; Depinho, R.A.; Montminy, M.; Cantley, L.C. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science 2005, 310, 1642–1646. [Google Scholar] [CrossRef] [Green Version]
- Kalaany, N.Y.; Sabatini, D.M. Tumours with PI3K activation are resistant to dietary restriction. Nature 2009, 458, 725–731. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.Y.; Martinez-Outschoorn, U.E.; Schilder, R.J.; Kim, C.H.; Richard, S.D.; Rosenblum, N.G.; Johnson, J.M. Metformin as a Therapeutic Target in Endometrial Cancers. Front. Oncol. 2018, 8, 341. [Google Scholar] [CrossRef]
- Dowling, R.J.; Zakikhani, M.; Fantus, I.G.; Pollak, M.; Sonenberg, N. Metformin inhibits mammalian target of rapamycin-dependent translation initiation in breast cancer cells. Cancer Res. 2007, 67, 10804–10812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Li, M.X.; Wang, H.; Zeng, Z.; Li, X.M. Metformin down-regulates endometrial carcinoma cell secretion of IGF-1 and expression of IGF-1R. Asian Pac. J. Cancer Prev. 2015, 16, 221–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarfstein, R.; Friedman, Y.; Attias-Geva, Z.; Fishman, A.; Bruchim, I.; Werner, H. Metformin downregulates the insulin/IGF-I signaling pathway and inhibits different uterine serous carcinoma (USC) cells proliferation and migration in p53-dependent or -independent manners. PLoS ONE. 2013, 8, e61537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Y.; Wang, J.L.; Ji, M.; Yuan, Z.F.; Peng, Z.; Zhang, Y.; Wen, J.G.; Shi, H.R. Regulation of insulin-like growth factor signaling by metformin in endometrial cancer cells. Oncol. Lett. 2014, 8, 1993–1999. [Google Scholar] [CrossRef] [Green Version]
- Zou, J.; Hong, L.; Luo, C.; Li, Z.; Zhu, Y.; Huang, T.; Zhang, Y.; Yuan, H.; Hu, Y.; Wen, T.; et al. Metformin inhibits estrogen-dependent endometrial cancer cell growth by activating the AMPK-FOXO1 signal pathway. Cancer Sci. 2016, 107, 1806–1817. [Google Scholar] [CrossRef] [Green Version]
- Nevadunsky, N.S.; Van Arsdale, A.; Strickler, H.D. Metformin use and endometrial cancer survival. Gynecol. Oncol. 2014, 132, 236–240. [Google Scholar] [CrossRef]
- Ko, E.M.; Walter, P.; Jackson, A.; Clark, L.; Franasiak, J.; Bolac, C.; Havrilesky, L.J.; Secord, A.A.; Moore, D.T.; Gehrig, P.A.; et al. Metformin is associated with improved survival in endometrial cancer. Gynecol. Oncol. 2014, 132, 438–442. [Google Scholar] [CrossRef]
- Lemańska, A.; Zaborowski, M.; Spaczyński, M.; Nowak-Markwitz, E. Do endometrial cancer patients benefit from metformin intake. Ginekol. Pol. 2015, 86, 419–423. [Google Scholar] [CrossRef]
- Al Hilli, M.M.; Bakkum-Gamez, J.N.; Mariani, A.; Cliby, W.A.; Mc Gree, M.E.; Weaver, A.L.; Dowdy, S.C.; Podratz, K.C. The effect of diabetes and metformin on clinical outcomes is negligible in risk-adjusted endometrial cancer cohorts. Gynecol. Oncol. 2016, 140, 270–276. [Google Scholar] [CrossRef]
- Ezewuiro, O.; Grushko, T.A.; Kocherginsky, M.; Habis, M.; Hurteau, J.A.; Mills, K.A.; Hunn, J.; Olopade, O.I.; Fleming, G.F.; Romero, I.L. Association of Metformin Use with Outcomes in Advanced Endometrial Cancer Treated with Chemotherapy. PLoS ONE. 2016, 11, e0147145. [Google Scholar] [CrossRef] [Green Version]
- Seebacher, V.; Bergmeister, B.; Grimm, C.; Koelbl, H.; Reinthaller, A.; Polterauer, S. The prognostic role of metformin in patients with endometrial cancer: A retrospective study. Eur. J. Obstet. Gynecol. Reprod. Biol. 2016, 203, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Meireles, C.G.; Pereira, S.A.; Valadares, L.P. Effects of metformin on endometrial cancer: Systematic review and meta-analysis. Gynecol. Oncol. 2017, 147, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Mitsuhashi, A.; Kiyokawa, T.; Sato, Y.; Shozu, M. Effects of metformin on endometrial cancer cell growth in vivo: A preoperative prospective trial. Cancer 2014, 120, 2986–2995. [Google Scholar] [CrossRef] [PubMed]
- Tabrizi, A.D.; Melli, M.S.; Foroughi, M.; Ghojazadeh, M.; Bidadi, S. Antiproliferative effect of metformin on the endometrium—A clinical trial. Asian Pac. J. Cancer Prev. 2014, 15, 10067–10070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Guo, Y.R.; Lin, J.F.; Feng, Y.; Billig, H.; Shao, R. Combination of Diane-35 and Metformin to Treat Early Endometrial Carcinoma in PCOS Women with Insulin Resistance. J. Cancer 2014, 5, 173–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laskov, I.; Drudi, L.; Beauchamp, M.C.; Yasmeen, A.; Ferenczy, A.; Pollak, M.; Gotlieb, W.H. Anti-diabetic doses of metformin decrease proliferation markers in tumors of patients with endometrial cancer. Gynecol. Oncol. 2014, 134, 607–614. [Google Scholar] [CrossRef]
- Schuler, K.M.; Rambally, B.S.; DiFurio, M.J.; Sampey, B.P.; Gehrig, P.A.; Makowski, L.; Bae-Jump, V.L. Antiproliferative and metabolic effects of metformin in a preoperative window clinical trial for endometrial cancer. Cancer Med. 2015, 4, 161–173. [Google Scholar] [CrossRef]
- Cai, D.; Sun, H.; Qi, Y.; Zhao, X.; Feng, M.; Wu, X. Insulin-Like Growth Factor 1/Mammalian Target of Rapamycin and AMP-Activated Protein Kinase Signaling Involved in the Effects of Metformin in the Human Endometrial Cancer. Int. J. Gynecol. Cancer 2016, 26, 1667–1672. [Google Scholar] [CrossRef]
- Sivalingam, V.N.; Kitson, S.; McVey, R.; Roberts, C.; Pemberton, P.; Gilmour, K.; Ali, S.; Renehan, A.G.; Kitchener, H.C.; Crosbie, E.J. Measuring the biological effect of presurgical metformin treatment in endometrial cancer. Br. J. Cancer 2016, 114, 281–289. [Google Scholar] [CrossRef] [Green Version]
- Soliman, P.T.; Zhang, Q.; Broaddus, R.R.; Westin, S.N.; Iglesias, D.; Munsell, M.F.; Schmandt, R.; Yates, M.; Ramondetta, L.; Lu, K.H. Prospective evaluation of the molecular effects of metformin on the endometrium in women with newly diagnosed endometrial cancer: A window of opportunity study. Gynecol. Oncol. 2016, 143, 466–471. [Google Scholar] [CrossRef] [Green Version]
- Mitsuhashi, A.; Sato, Y.; Kiyokawa, T.; Koshizaka, M.; Hanaoka, H.; Shozu, M. Phase II study of medroxyprogesterone acetate plus metformin as a fertility-sparing treatment for atypical endometrial hyperplasia and endometrial cancer. Ann. Oncol. 2016, 27, 262–266. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Sun, H.; Feng, M.; Zhao, J.; Zhao, X.; Wan, Q.; Cai, D. Metformin is associated with reduced cell proliferation in human endometrial cancer by inbibiting PI3K/AKT/mTOR signaling. Gynecol. Endocrinol. 2018, 34, 428–432. [Google Scholar] [CrossRef] [PubMed]
- Kitson, S.J.; Maskell, Z.; Sivalingam, V.N.; Allen, J.L.; Ali, S.; Burns, S.; Gilmour, K.; Latheef, R.; Slade, R.J.; Pemberton, P.W.; et al. PRE-surgical Metformin In Uterine Malignancy (PREMIUM): A Multi-Center, Randomized Double-Blind, Placebo-Controlled Phase III Trial. Clin. Cancer Res. 2019, 25, 2424–2432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petchsila, K.; Prueksaritanond, N.; Insin, P.; Yanaranop, M.; Chotikawichean, N. Effect of Metformin for decreasing proliferative marker in women with endometrial cancer: A randomized double-blind placebo-controlled trial. Asian Pac. J. Cancer Prev. 2020, 21, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.Y.; Gulinazi, Y.; Du, Y.; Ning, C.C.; Cheng, Y.L.; Shan, W.W.; Luo, X.Z.; Zhang, H.W.; Zhu, Q.; Ma, F.H.; et al. Metformin plus megestrol acetate compared with megestrol acetate alone as fertility-sparing treatment in patients with atypical endometrial hyperplasia and well-differentiated endometrial cancer: A randomised controlled trial. BJOG 2020, 127, 848–857. [Google Scholar] [CrossRef]
- Slomovitz, B.M.; Lu, K.H.; Johnston, T.; Coleman, R.L.; Munsell, M.; Broaddus, R.R.; Walker, C.; Ramondetta, L.M.; Burke, T.W.; Gershenson, D.M.; et al. A phase 2 study of the oral mammalian target of rapamycin inhibitor, everolimus, in patients with recurrent endometrial carcinoma. Cancer 2010, 116, 5415–5419. [Google Scholar] [CrossRef] [Green Version]
- Oza, A.M.; Elit, L.; Tsao, M.S.; Kamel-Reid, S.; Biagi, J.; Provencher, D.M.; Gotlieb, W.H.; Hoskins, P.J.; Ghatage, P.; Tonkin, K.S.; et al. Phase II study of temsirolimus in women with recurrent or metastatic endometrial cancer: A trial of the NCIC Clinical Trials Group. J. Clin. Oncol. 2011, 29, 3278–3285. [Google Scholar] [CrossRef]
- Ray-Coquard, I.; Favier, L.; Weber, B.; Roemer-Becuwe, C.; Bougnoux, P.; Fabbro, M.; Floquet, A.; Joly, F.; Plantade, A.; Paraiso, D.; et al. Everolimus as second- or third-line treatment of advanced endometrial cancer: ENDORA.D., a phase II trial of GINECO. Br. J. Cancer 2013, 108, 1771–1777. [Google Scholar] [CrossRef] [Green Version]
- Colombo, N.; McMeekin, D.S.; Schwartz, P.E.; Sessa, C.; Gehrig, P.A.; Holloway, R.; Braly, P.; Matei, D.; Morosky, A.; Dodion, P.F.; et al. Ridaforolimus as a single agent in advanced endometrial cancer: Results of a single-arm, phase 2 trial. Br. J. Cancer 2013, 108, 1021–1026. [Google Scholar] [CrossRef]
- Tsoref, D.; Welch, S.; Lau, S.; Biagi, J.; Tonkin, K.; Martin, L.A.; Ellard, S.; Ghatage, P.; Elit, L.; Mackay, H.J.; et al. Phase II study of oral ridaforolimus in women with recurrent or metastatic endometrial cancer. Gynecol. Oncol. 2014, 135, 184–189. [Google Scholar] [CrossRef]
- Fleming, G.F.; Filiaci, V.L.; Marzullo, B.; Zaino, R.J.; Davidson, S.A.; Pearl, M.; Makker, V.; Burke, J.J.; Zweizig, S.L.; Van Le, L.; et al. Temsirolimus with or without megestrol acetate and tamoxifen for endometrial cancer: A gynecologic oncology group study. Gynecol. Oncol. 2014, 132, 585–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oza, A.M.; Pignata, S.; Poveda, A.; McCormack, M.; Clamp, A.; Schwartz, B.; Cheng, J.; Li, X.; Campbell, K.; Dodion, P.; et al. Randomized Phase II Trial of Ridaforolimus in Advanced Endometrial Carcinoma. J. Clin. Oncol. 2015, 33, 3576–3582. [Google Scholar] [CrossRef] [PubMed]
- Emons, G.; Kurzeder, C.; Schmalfeldt, B.; Neuser, P.; de Gregorio, N.; Pfisterer, J.; Park-Simon, T.W.; Mahner, S.; Schröder, W.; Lück, H.J.; et al. Temsirolimus in women with platinum-refractory/resistant ovarian cancer or advanced/recurrent endometrial carcinoma. A phase II study of the AGO-study group (AGO-GYN8). Gynecol. Oncol. 2016, 140, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Del Campo, J.M.; Birrer, M.; Davis, C.; Fujiwara, K.; Gollerkeri, A.; Gore, M.; Houk, B.; Lau, S.; Poveda, A.; González-Martín, A.; et al. A randomized phase II non-comparative study of PF-04691502 and gedatolisib (PF-05212384) in patients with recurrent endometrial cancer. Gynecol. Oncol. 2016, 142, 62–69. [Google Scholar] [CrossRef]
- Banerji, U.; Dean, E.J.; Pérez-Fidalgo, J.A.; Batist, G.; Bedard, P.L.; You, B.; Westin, S.N.; Kabos, P.; Garrett, M.D.; Tall, M.; et al. A Phase I Open-Label Study to Identify a Dosing Regimen of the Pan-AKT Inhibitor AZD5363 for Evaluation in Solid Tumors and in PIK3CA-Mutated Breast and Gynecologic Cancers. Clin. Cancer Res. 2018, 24, 2050–2059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myers, A.P.; Konstantinopoulos, P.A.; Barry, W.T.; Luo, W.; Broaddus, R.R.; Makker, V.; Drapkin, R.; Liu, J.; Doyle, A.; Horowitz, N.S.; et al. Phase, I.I., 2-stage, 2-arm, PIK3CA mutation stratified trial of MK-2206 in recurrent endometrial cancer. Int. J. Cancer 2020, 147, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Costa, C.; Bosch, A. The Strategy of PIKing a Target: What Is AKTually Most Effective? Clin. Cancer Res. 2018, 24, 2029–2031. [Google Scholar] [CrossRef] [Green Version]
- Costa, C.; Ebi, H.; Martini, M.; Beausoleil, S.A.; Faber, A.C.; Jakubik, C.T.; Huang, A.; Wang, Y.; Nishtala, M.; Hall, B.; et al. Measurement of PIP3 levels reveals an unexpected role for p110β in early adaptive responses to p110α-specific inhibitors in luminal breast cancer. Cancer Cell 2015, 27, 97–108. [Google Scholar] [CrossRef] [Green Version]
- Conza, D.; Mirra, P.; Calì, G.; Tortora, T.; Insabato, L.; Fiory, F.; Schenone, S.; Amato, R.; Beguinot, F.; Perrotti, N.; et al. The SGK1 inhibitor SI113 induces autophagy, apoptosis, and endoplasmic reticulum stress in endometrial cancer cells. J. Cell Physiol. 2017, 232, 3735–3743. [Google Scholar] [CrossRef]
- Hopkins, B.D.; Pauli, C.; Du, X.; Wang, D.G.; Li, X.; Wu, D.; Amadiume, S.C.; Goncalves, M.D.; Hodakoski, C.; Lundquist, M.R.; et al. Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nature 2018, 560, 499–503. [Google Scholar] [CrossRef] [Green Version]
- Rozengurt, E.; Soares, H.P.; Sinnet-Smith, J. Suppression of feedback loops mediated by PI3K/mTOR induces multiple overactivation of compensatory pathways: An unintended consequence leading to drug resistance. Mol. Cancer Ther. 2014, 13, 2477–2488. [Google Scholar] [CrossRef] [Green Version]
- Gallagher, E.J.; Fierz, Y.; Vijayakumar, A.; Haddad, N.; Yakar, S.; LeRoith, D. Inhibiting PI3K reduces mammary tumor growth and induces hyperglycemia in a mouse model of insulin resistance and hyperinsulinemia. Oncogene 2012, 31, 3213–3222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iams, W.T.; Lovly, C.M. Molecular Pathways: Clinical Applications and Future Direction of Insulin-like Growth Factor-1 Receptor Pathway Blockade. Clin. Cancer Res. 2015, 21, 4270–4277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buck, E.; Gokhale, P.C.; Koujak, S.; Brown, E.; Eyzaguirre, A.; Tao, N.; Rosenfeld-Franklin, M.; Lerner, L.; Chiu, M.I. Compensatory insulin receptor (IR) activation on inhibition of insulin-like growth factor-1 receptor (IGF-1R): Rationale for cotargeting IGF-1R and IR in cancer. Mol. Cancer Ther. 2010, 9, 2652–2664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirakawa, J.; Okuyama, T.; Yoshida, E.; Shimizu, M.; Horigome, Y.; Tuno, T.; Hayasaka, M.; Abe, S.; Fuse, M.; Togashi, Y.; et al. Effects of the antitumor drug OSI-906, a dual inhibitor of IGF-1 receptor and insulin receptor, on the glycemic control, β-cell functions, and β-cell proliferation in male mice. Endocrinology 2014, 155, 2102–2111. [Google Scholar] [CrossRef] [Green Version]
- Nasiri, A.R.; Rodrigues, M.R.; Li, Z.; Leitner, B.P.; Perry, R.J. SGLT2 inhibition slows tumor growth in mice by reversing hyperinsulinemia. Cancer Metab. 2019, 7, 10. [Google Scholar] [CrossRef] [Green Version]
- Loves, S.; van Groningen, L.; Filius, M.; Mekking, M.; Brandon, T.; Tack, C.J.; Hermus, A.; de Boer, H. High-Dose, Diazoxide-Mediated Insulin Suppression Boosts Weight Loss Induced by Lifestyle Intervention. J. Clin. Endocrinol. Metab. 2018, 103, 4014–4022. [Google Scholar] [CrossRef] [Green Version]
- Loves, S.; van Groningen, L.; Filius, M.; Mekking, M.; Brandon, T.; Tack, C.J.; Hermus, A.; de Boer, H. Effects of Diazoxide-Mediated Insulin Suppression on Glucose and Lipid Metabolism in Nondiabetic Obese Men. J. Clin. Endocrinol. Metab. 2018, 103, 2346–2353. [Google Scholar] [CrossRef] [Green Version]
- Goncalves, M.D.; Hopkins, B.D.; Cantley, L.C. Phosphatidylinositol 3-Kinase, Growth Disorders, and Cancer. N. Engl. J. Med. 2018, 379, 2052–2062. [Google Scholar] [CrossRef]
- Arthur, R.; Brasky, T.M.; Crane, T.E.; Felix, A.S.; Kaunitz, A.M.; Shadyab, A.H.; Qi, L.; Wassertheil-Smoller, S.; Rohan, T.E. Associations of a Healthy Lifestyle Index With the Risks of Endometrial and Ovarian Cancer Among Women in the Women’s Health Initiative Study. Am. J. Epidemiol. 2019, 188, 261–273. [Google Scholar] [CrossRef]
Figure 1.
Potential mechanisms of obesity-induced development of endometrial cancer (EC). Obesity induces increased aromatase expression and chronic low-grade inflammation in adipose tissues that play fundamental roles in EC development. The former results in increased estradiol (E2) production, and the latter triggers expression of various adipokines, cytokines, and chemokines, leading to insulin resistance. Insulin resistance causes hyperglycemia, which induces insulin synthesis in the spleen and lipogenesis in the liver. The former subsequently induces hyperinsulinemia, and the latter causes decreased expression of sex hormone-binding globulin (SHBG), leading to enhanced E2 activity. Hyperinsulinemia plays central roles in promoting EC growth via decreased insulin-like growth factor binding protein (IGFBP) 1 with enhanced insulin-like growth factor (IGF)-1 activity and most importantly, switching on of insulin-phosphoinositide 3 kinase (insulin-PI3K) signaling in EC cells. Δ4A: androstenedione, 17β-HSD: 17β-Hydroxysteroid dehydrogenases, T: testosterone, ER: estrogen receptor, adinoR1: adiponectin receptor 1, IR: insulin receptor, IGF1-R: insulin-like growth factor 1-receptor, HNF-4α: hepatocyte nuclear factor-4α. ↑ (up regulation), ↓ (down regulation), ⇢ (expected pathway).
Figure 1.
Potential mechanisms of obesity-induced development of endometrial cancer (EC). Obesity induces increased aromatase expression and chronic low-grade inflammation in adipose tissues that play fundamental roles in EC development. The former results in increased estradiol (E2) production, and the latter triggers expression of various adipokines, cytokines, and chemokines, leading to insulin resistance. Insulin resistance causes hyperglycemia, which induces insulin synthesis in the spleen and lipogenesis in the liver. The former subsequently induces hyperinsulinemia, and the latter causes decreased expression of sex hormone-binding globulin (SHBG), leading to enhanced E2 activity. Hyperinsulinemia plays central roles in promoting EC growth via decreased insulin-like growth factor binding protein (IGFBP) 1 with enhanced insulin-like growth factor (IGF)-1 activity and most importantly, switching on of insulin-phosphoinositide 3 kinase (insulin-PI3K) signaling in EC cells. Δ4A: androstenedione, 17β-HSD: 17β-Hydroxysteroid dehydrogenases, T: testosterone, ER: estrogen receptor, adinoR1: adiponectin receptor 1, IR: insulin receptor, IGF1-R: insulin-like growth factor 1-receptor, HNF-4α: hepatocyte nuclear factor-4α. ↑ (up regulation), ↓ (down regulation), ⇢ (expected pathway).

Figure 2.
Insulin-PI3K signaling facilitates cell proliferation and glucose metabolism via serine/threonine protein kinase B (AKT)-dependent and -independent mechanisms. The interaction of insulin with the insulin receptor (IR) triggers phosphoinositide 3-kinase (PI3K)-regulated signaling, in which phosphatidylinositol 3,4,5-triphosphate (PIP3) recruits phosphoinositide-dependent kinase 1 (PDK1), the serine/threonine kinase AKT, and other factors to the cell membrane where the signals are propagated via a series of serine/threonine kinase and tyrosine kinase activities. Phosphatase and tensin homologue (PTEN) can dephosphorylate PIP3 to form PIP2, limiting activation of this pathway, and loss-of-function mutations in PTEN cause significant elevations in PIP3, a driving force of this pathway. Phosphorylated AKT inhibits downstream targets via phosphorylation, including forkhead box protein O (FOXO)-1 and glycogen synthase kinase (GSK), both of which normally function to inhibit cell growth. Phosphorylated AKT also inhibits thioredoxin-interacting protein (TXNIP), an adaptor for endocytosis of glucose transporters (GLUTs), leading to dissociation from the GLUTs, inhibition of their endocytosis, and rapid glucose uptake upon insulin stimulation. AKT phosphorylation leads to activation of mammalian target of rapamycin (mTOR), a protein kinase that controls cell proliferation via activation of the eukaryotic initiation factor 4E-binding protein-1 (4E-BP-1) complex and S6 kinase 1 (S6K1), which phosphorylates ribosomal S6 protein (rpS6). Insulin-PI3K signaling also activates an AKT-independent pathway, in which PIP3-recruited Ras-related C3 botulinus toxin substrate (RAC) activation leads to disruption of the actin cytoskeleton and release of filamentous actin (F-actin)-bound aldolase A (ALDOA), resulting in aldolase activation that promotes the glycolysis pathway. Additionally, PI3K signaling activates serum and glucocorticoid-induced protein kinase (SGK1) via PDK phosphorylation independent of AKT and can phosphorylate overlapping substrates of AKT, resulting in bypass of the AKT pathway. Metformin inhibits oxidative phosphorylation (OXPHOS) at the mitochondrial level, resulting in a decrease in the proton gradient across the inner mitochondrial membrane and leading to a reduction in proton-driven synthesis of adenosine triphosphate (ATP) and an increase in the ratio of cellular adenosine monophosphate (AMP) to ATP. This leads to preferential AMP binding to AMP-activated protein kinase (AMPK) and a conformational change that allows for phosphorylation/activation of AMPK by liver kinase B1 (LKB1). Activated AMPK converts cells to a catabolic state through AMPK-mediated phosphorylation, leading to the inhibition of downstream transcription factors involved in ATP-consuming synthetic pathways. AMPK activation also induces FOXO1 nuclear translocation, activating function of this protein. Additionally, activated AMPK inhibits mTOR to inhibit cell growth. IRS: insulin receptor substrate, p85: regulatory subunit of PI3K, p110: catalytic subunit of PI3K, NAD: nicotinamide adenine dinucleotide, LDH: lactate dehydrogenase, Acetyl-CoA: acetyl coenzyme A, TCA: tricarboxylic acid. → (activation), ⊣ (inhibition).
Figure 2.
Insulin-PI3K signaling facilitates cell proliferation and glucose metabolism via serine/threonine protein kinase B (AKT)-dependent and -independent mechanisms. The interaction of insulin with the insulin receptor (IR) triggers phosphoinositide 3-kinase (PI3K)-regulated signaling, in which phosphatidylinositol 3,4,5-triphosphate (PIP3) recruits phosphoinositide-dependent kinase 1 (PDK1), the serine/threonine kinase AKT, and other factors to the cell membrane where the signals are propagated via a series of serine/threonine kinase and tyrosine kinase activities. Phosphatase and tensin homologue (PTEN) can dephosphorylate PIP3 to form PIP2, limiting activation of this pathway, and loss-of-function mutations in PTEN cause significant elevations in PIP3, a driving force of this pathway. Phosphorylated AKT inhibits downstream targets via phosphorylation, including forkhead box protein O (FOXO)-1 and glycogen synthase kinase (GSK), both of which normally function to inhibit cell growth. Phosphorylated AKT also inhibits thioredoxin-interacting protein (TXNIP), an adaptor for endocytosis of glucose transporters (GLUTs), leading to dissociation from the GLUTs, inhibition of their endocytosis, and rapid glucose uptake upon insulin stimulation. AKT phosphorylation leads to activation of mammalian target of rapamycin (mTOR), a protein kinase that controls cell proliferation via activation of the eukaryotic initiation factor 4E-binding protein-1 (4E-BP-1) complex and S6 kinase 1 (S6K1), which phosphorylates ribosomal S6 protein (rpS6). Insulin-PI3K signaling also activates an AKT-independent pathway, in which PIP3-recruited Ras-related C3 botulinus toxin substrate (RAC) activation leads to disruption of the actin cytoskeleton and release of filamentous actin (F-actin)-bound aldolase A (ALDOA), resulting in aldolase activation that promotes the glycolysis pathway. Additionally, PI3K signaling activates serum and glucocorticoid-induced protein kinase (SGK1) via PDK phosphorylation independent of AKT and can phosphorylate overlapping substrates of AKT, resulting in bypass of the AKT pathway. Metformin inhibits oxidative phosphorylation (OXPHOS) at the mitochondrial level, resulting in a decrease in the proton gradient across the inner mitochondrial membrane and leading to a reduction in proton-driven synthesis of adenosine triphosphate (ATP) and an increase in the ratio of cellular adenosine monophosphate (AMP) to ATP. This leads to preferential AMP binding to AMP-activated protein kinase (AMPK) and a conformational change that allows for phosphorylation/activation of AMPK by liver kinase B1 (LKB1). Activated AMPK converts cells to a catabolic state through AMPK-mediated phosphorylation, leading to the inhibition of downstream transcription factors involved in ATP-consuming synthetic pathways. AMPK activation also induces FOXO1 nuclear translocation, activating function of this protein. Additionally, activated AMPK inhibits mTOR to inhibit cell growth. IRS: insulin receptor substrate, p85: regulatory subunit of PI3K, p110: catalytic subunit of PI3K, NAD: nicotinamide adenine dinucleotide, LDH: lactate dehydrogenase, Acetyl-CoA: acetyl coenzyme A, TCA: tricarboxylic acid. → (activation), ⊣ (inhibition).


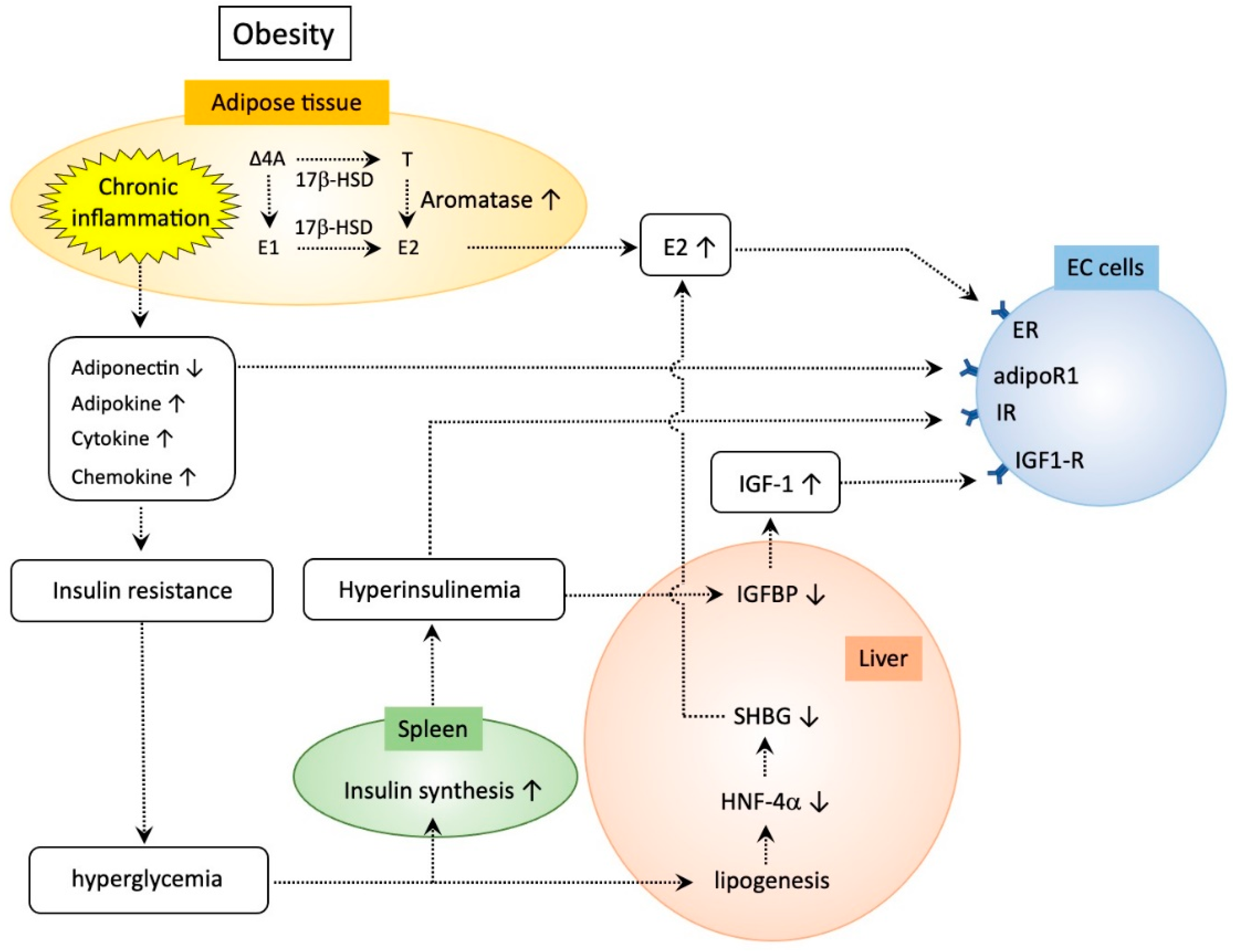{kind=link}
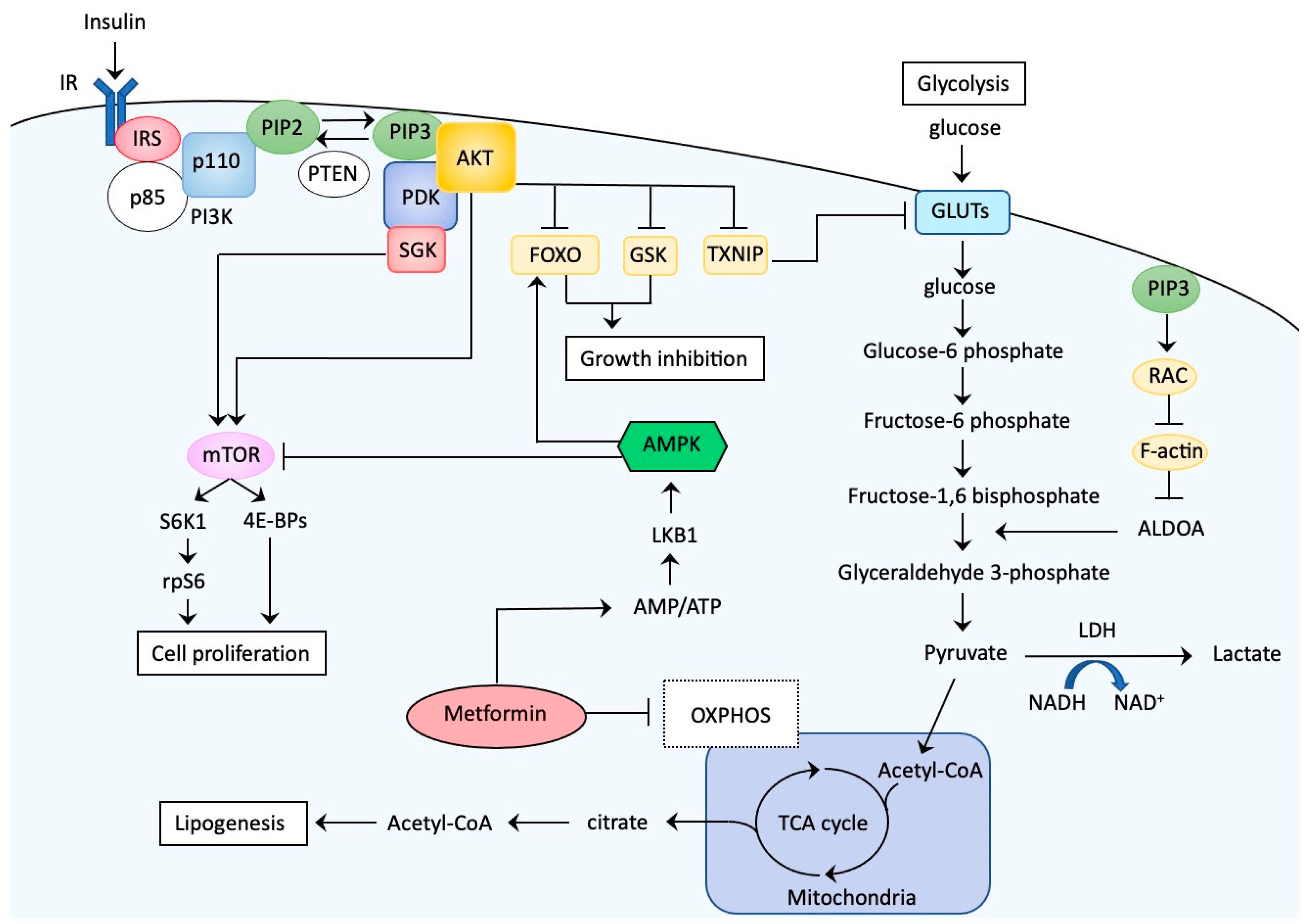{kind=link}
Table 1.
Representative clinical trials for endometrial cancer using metformin.
| Author | Agents | Design | Number of EC Patients | Treatment | Representative Effects |
|---|---|---|---|---|---|
| Mitsuhashi et al. 2014 [134] | Metformin | Single arm | 31 | Preoperative | Tumor: Ki-67 ↓, phospho-rpS6↓, phospho-ERK1/2↓, phospho-AMPK↑. Serum levels: insulin↓, glucose↓, IGF-1↓. The ability of serums from patients to stimulate DNA synthesis decreased in cultured EC cells |
| Tabrizi et al. 2014 [135] | Metformin vs. MA | Non-blinded RCT | 22 vs 21 | Before histological assessment for uterine bleeding | Reversion to endometrial atrophy in 96% of patients treated with metformin, compared to 62% of patients treated with MA |
| Li et al. 2014 [136] | Metformin+EE+Cyproterone acetate | Single arm | 5 | Fertility-sparing, no operation | Reversion to normal endometrium in all patients |
| Laskov et al.2014 [137] | Metformin | Single arm | 11 | Preoperative | Tumor: Ki-67 ↓, phospho-rpS6↓. Serum levels: insulin↓, IGF-1↓, IGFBP-7↓ |
| Schuler et al. 2015 [138] | Metformin | Single arm | 20 | Preoperative | Tumor: Ki-67 ↓, phospho-AKT↓, phospho-AMPK↓, phospho-rpS6↓, phospho-4E-BP-1↓, ER↓. PR →. Serum levels: free fatty acid↑. |
| Cai et al. 2015 [139] | Metformin vs. no treatment | Non-RCT | 30 vs 30 | Preoperative | Tumor: phospho-AMPK↑, phospho mTOR↓. Serum levels: IGF-1↓. |
| Sivalingam et al. 2016 [140] | Metformin vs. no treatment | Non-RCT | 28 vs 12 | Preoperative | Tumor: Ki-67 ↓, phospho-rpS6→, phospho-4E-BP-1↓, phospho AKT→, ER→, PR →, phospho ACC→, caspase 3→. |
| Soliman et al. 2016 [141] | Metformin | Single arm | 20 | Preoperative | Tumor: Ki-67 →, phospho-rpS6↓, phospho AKT↓, phospho-ERK1/2↓, phospho ACC→, caspase 3→. Serum: IGF-1↓, omentin↓, insulin↓, C-peptide↓, leptin↓. |
| Mitsuhashi et al. 2016 [142] | Metformin+MPA followed by Metformin alone | Single arm | 29 (including 16 AEH) | Fertility-sparing, no operation | 3-year recurrence-free survival in 89% of patients |
| Zao et al. 2018 [143] | Metformin vs. no treatment | Non-RCT | 33 vs 32 | Preoperative | Tumor: Ki-67 ↓, PI3K↓, phospho AKT ↓, phospho-S6K1↓, phospho-4E-BP-1↓. |
| Kitson et al. 2019 [144] | Metformin vs. placebo | Double- blinded RCT | 45 (2 AEH, 43 EC) vs 43 (2 AEH, 41 EC) | Preoperative | Tumor: Ki-67 →, pIR →, IGF1R →. Serum: Glucose↑, Insulin→, HOMA-IR→, HbA1C→, IGF-1→, IGFBP-1→, Adiponectin→. |
| Petchsila et al. 2020 [145] | Metformin vs. placebo | Double- blinded RCT | 25 vs 24 | Preoperative | Tumor: Ki-67↓. No significant differences were detected in metabolic effects and adverse events between the metformin and the placebo groups. |
| Yang et al. 2020 [146] | Metformin+ MA vs. MA | Open-label RCT | 76 (61 AHE, 15 EEC) vs 74 (62 AEH, 12 EEC) | Fertility-sparing, no operation | The CR rate within 16 weeks of treatment was higher in the metformin plus MA group than in the MA group (34.3 versus 20.7%, OR 2.0, 95% CI 0.89–4.51, p = 0.09) but the difference was more significant in AEH patients (39.6 versus 20.4%, OR 2.56, 95% CI 1.06–6.21, p = 0.04). |
rpS6,ribosomal protein S6; ERK1/2, extracellular signal-regulated kinase-1/2; AMPK, AMP-activated kinase; IGF-1, insulin-like growth factor-1; EC, endometrial cancer; RCT, randomized controlled trial; MA, megestrol; EE, ethinyl estradiol;IGFBP-1/7, insulin-like growth factor binding protein-1/7; 4E-BP-1, eukaryotic initiation factor 4E-binding protein-1; ER, estrogen receptor; PR, progestreone receptor; mTOR, mammarian target of rapamycin; ACC, acetyl-CoA carboxilase; AEH, atypical endometrial hyperplasia; PI3K, phosphoinositide 3-kinase; S6K, S6 kinase 1; IR, insulin receptor; IGF1R, insulin-like growth factor receptor-1; HOMA, homeostasis model assessment-insulin resistance;OR, odds raio; CI, confidence interval.
Table 2.
Representative clinical trials for EC using PI3K-AKT inhibitors in the main arm.
| Author | Agents | Phase | Number of Patients | Cancer Type | Response | Molecular Markers | Hyperglycemia | Note |
|---|---|---|---|---|---|---|---|---|
| Slomovitz et al. 2010 [147] | Everolimus | II | 35 | Recurrent or advanced EC | SD 100%, CBR 22% | None | 9% | |
| Oza et al. 2011 [148] | Temsirolimus | II | 60 | Recurrent or metastatic EC | chemotherapy-naïve group: PR 14%, SD 69%. chemotherapy-treated group: PR 4%, SD 48% | PTEN MT, pAKT, pmOR, pS6. (No correlation with response) | ~5% | |
| Ray-Coquard et al. 2013 [149] | Everolimus | II | 44 | Advanced or metastatic EC | PR 5%, SD 32% | None | 69% | Median PFS 2.8 months |
| Colombo et al.2013 [150] | Ridaforolimus | II | 45 | Recurrent or persistent EC | PR 11%, SD 18% | None | 11% | 6-month PFS 18% |
| Tsoref et al. 2014 [151] | Ridaforolimus | II | 31 | Recurrent or metastatic EC | RR 9%, SD 53% | PTEN, PIC3CA MT, AKT1 MT (No correlation with response) | 74% | |
| Fleming et al. 2014 [152] | Temsirolimus vs. Combination (Temsirolimus + Megesterol or Tamoxifen) | II | 71 | Recurrent or advanced EC | single: CR 6%, PR 16%, SD 52%, combination; CR 0%, PR 14.3%, SD 53% | pAKT, PTEN (No correlation with response) | 18% (single), 14% (combination) | |
| Oza et al. 2015 [153] | Ridaforolimus vs. Comparator (Progestins or chemotherapy) | II (randomized) | 64 vs. 66 | Recurrent or metastatic EC | RR 0% vs. 4%, SD 35% vs. 17% | None | 29% (Ridaforolimus), 3%(Comparator) | Median PFS 3.6 months (Ridaforolimus) and 1.9 months (comparator) |
| Emons et al. 2015 [154] | Temsirolimus | II | 22 | Recurrent or advanced EC | RR 10%, SD 25% | None | Not known | |
| del Campo et al. 2016 [155] | PF-04691502 vs. Gedatolisib | II (randomized) | 18 vs. 40 | Recurrent EC | Gedatolisib group; CR 3%, PR 13%, SD 24%, CBR 40% | Stathmin (low expression was correlated with higher CBR) | 33% (PF-04691502), 0% (Gedatolisib) | PF-04691502 arm was discontinued early due to unacceptable toxicity |
| Banerji et al. 2108 [156] | AZD5363 | I | 59 (as expansion arm) | 31 breast and 28 gynecologic cancers with PIK3CA mutation (including 10 ECs) | RR 4% in breast cancer, PR 8% in gynecologic cancer | All patients had PIK3CA MT | 20% | |
| Myers et al. 2020 [157] | MK-2206 | II | 37 | Recurrent EC | RR 11% (PIK3CA MT), RR 4%, (PIK3CA WT) | PIK3CA MT (No correlation with response) | 31% | Median PFS 1.7 and 2.5 months with or without PIK3CA MT |
EC, endometrial cancer; CR, complete response; PR, partial response; SD, stable disease; CBR, clinical benefit response; MT, mutation; PFS, progression-free survival.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kyo, S.; Nakayama, K. Endometrial Cancer as a Metabolic Disease with Dysregulated PI3K Signaling: Shedding Light on Novel Therapeutic Strategies. Int. J. Mol. Sci. 2020, 21, 6073. https://doi.org/10.3390/ijms21176073
AMA Style
Kyo S, Nakayama K. Endometrial Cancer as a Metabolic Disease with Dysregulated PI3K Signaling: Shedding Light on Novel Therapeutic Strategies. International Journal of Molecular Sciences. 2020; 21(17):6073. https://doi.org/10.3390/ijms21176073
Chicago/Turabian StyleKyo, Satoru, and Kentaro Nakayama. 2020. "Endometrial Cancer as a Metabolic Disease with Dysregulated PI3K Signaling: Shedding Light on Novel Therapeutic Strategies" International Journal of Molecular Sciences 21, no. 17: 6073. https://doi.org/10.3390/ijms21176073
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.