Metabolic Changes in Polycystic Kidney Disease as a Potential Target for Systemic Treatment


1
Department of Pediatrics, University of Cologne, Faculty of Medicine and University Hospital Cologne, 50937 Cologne, Germany
2
Department II of Internal Medicine, University of Cologne, Faculty of Medicine and University Hospital Cologne, 50937 Cologne, Germany
3
CECAD, University of Cologne, Faculty of Medicine and University Hospital Cologne, 50931 Cologne, Germany
4
Systems Biology of Ageing Cologne, University of Cologne, 50931 Cologne, Germany
5
Center for Molecular Medicine, University of Cologne, Faculty of Medicine and University Hospital Cologne, 50931 Cologne, Germany
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2020, 21(17), 6093; https://doi.org/10.3390/ijms21176093
Submission received: 30 June 2020
/
Revised: 17 August 2020
/
Accepted: 19 August 2020
/
Published: 24 August 2020
(This article belongs to the Special Issue Comorbidities in Chronic Diseases: Molecular Markers and Therapies)
{kind=link}
{kind=link}
Abstract
:Autosomal recessive and autosomal dominant polycystic kidney disease (ARPKD, ADPKD) are systemic disorders with pronounced hepatorenal phenotypes. While the main underlying genetic causes of both ARPKD and ADPKD have been well-known for years, the exact molecular mechanisms resulting in the observed clinical phenotypes in the different organs, remain incompletely understood. Recent research has identified cellular metabolic changes in PKD. These findings are of major relevance as there may be an immediate translation into clinical trials and potentially clinical practice. Here, we review important results in the field regarding metabolic changes in PKD and their modulation as a potential target of systemic treatment.
1. Introduction
The autosomal recessive and autosomal dominant forms of polycystic kidney disease (ARPKD, ADPKD) are among the most common causes of end stage renal disease in children and adults [1]. Over the last 20 years the pathophysiological understanding of ADPKD and ARPKD has gained increasing interest and the excellent work of multiple research groups worldwide has led to the identification of the underlying variants of different forms of cystic kidney diseases and of various intracellular signaling pathways that are dysregulated in PKD [2,3,4,5,6,7,8]. Based on these findings large clinical trials have been established resulting in the identification of the V2-receptor antagonist tolvaptan as a first targeted treatment of ADPKD [9,10,11]. Most recent cellular research has highlighted a potential role for cellular metabolic changes in the pathogenesis of PKD, thus opening a novel road for potential therapeutic approaches. Here, we give a brief overview over important recent developments in this area of research and the potential relevance for future therapeutic options.
2. Polycystic Kidney Disease
The main characteristics of the clinical course and the underlying genetics of ARPKD and ADPKD have recently been summarized in various excellent reviews [3,4,5,6,7,8]. Briefly, ARPKD is the severe form of polycystic kidney disease that frequently presents during early childhood or even antenatally. ARPKD is still associated with a substantial mortality that was estimated to be as high as up to 30–40% of the children although updated numbers are missing and are hard to be obtained. ARPKD is characterized by bilaterally enlarged kidneys due to the development of tubular dilatations or microcysts that mainly derive from the distal nephron [2]. Macrocysts may be seen and are assumed to be more frequent in the medulla [12]. Kidney function varies in ARPKD, but studies have found that about 50% of the patients require renal replacement therapy by the age of 20 years [13]. There is obligatory hepatic involvement with congenital hepatic fibrosis due to ductal plate malformation that can lead to portal hypertension with a need for liver transplantation. Even though ARPKD is a rare disease with an estimated incidence of 1:20,000, it is one of the main indications for combined liver and kidney transplantation during childhood [14]. Overall, there is a high degree of phenotypic variability that is poorly understood. Current approaches try to address this through international collection of clinical courses to lay the foundation for treatment recommendation based on observational evidence [15,16,17].
Most of the cases of ARPKD are caused by variants in the PKHD1 gene, although patients with variants in additional genes (e.g., DZIP1L encoding DAZ interacting protein 1-like, PMM2 encoding Phosphomannomutase 2) leading to an ARPKD phenotype have been described [6,18]. PKHD1 encodes a 450 kDa protein with a single transmembrane domain called fibrocystin. Fibrocystin’s cellular function remains very poorly understood. The cytoplasmic tail of fibrocystin has been shown to contain a ciliary targeting signal [19]. Upon cleavage the cytoplasmic tail can translocate to the cell nucleus [20,21]. Yet the functional consequences of these observations and the pathophysiological relevance remain unclear [22,23]. Studies on fibrocystin’s cellular function have been hampered by the fact that orthologous mouse models do not fully recapitulate the phenotype of ARPKD patients [23,24,25,26].
ADPKD typically presents later in life than ARPKD and is the more common form of PKD. An estimated incidence of 1:400–1:1.000 has been suggested but underdiagnosis makes accurate estimations a challenge. Most of the patients become clinically apparent from the fourth decade of life and end-stage renal disease occurs in the sixth decade in about 50% of affected individuals. ADPKD is classically characterized by the progressive development of bilateral renal cysts resulting in kidney enlargement. Liver cysts are seen in the vast majority of patients. Other extrarenal manifestations may occur as recently reviewed [27,28]. Mortality is primarily driven by renal failure associated with cardiovascular complications and is estimated to be increased 1.6 times relative to the general population. Very early onset forms of ADPKD have been described that may clinically resemble ARPKD [29].
ADPKD is mainly caused by variants in the PKD1 or the PKD2 gene encoding the proteins polycystin-1 and polycystin-2. Most patients show variants in PKD1 (around 85%) [30]. PKD1 variants are associated with a more rapidly progressing phenotype than PKD2 variants [31]. Importantly, truncating PKD1 variants have to be distinguished from non-truncating changes since the latter come along with a much milder disease course [32]. Recently, variants in additional genes (ALG9 encoding Alpha-1,2-mannosyltransferase ALG9, GANAB encoding glucosidase II subunit α (GIIα) and DNAJB11 encoding DnaJ heat shock protein family (Hsp40) member B11) have been described to cause a phenotype resembling ADPKD in smaller subsets of patients [33,34]. First classifications have been established identifying ADPKD patients at risk of fast progression [35,36].
Much has been learned about the function of the polycystin proteins during the past 15 years. A detailed description of the insights lies beyond the scope of this article and interested readers are referred to excellent recently published reviews [7,37,38] Briefly, polycystin-1 is a large transmembrane protein with a long extracellular part, eleven transmembrane domains and a short cytoplasmic tail that can be cleaved. Polycystin-2 is a nonselective cation channel and belongs to the family of transient receptor potential ion channels (TRPs). Both proteins have been found to physically interact and to function in joint complex. However, there may also be independent roles for cellular signaling [39].
Patient data and data from animal models of ADPKD and ARPKD furthermore suggest that there may be partial overlap in the cellular pathogenesis of the different types of PKD. Patients with variants in multiple PKD genes have been described that showed a severe phenotype [29] and crossings of a Pkd1 to a Pkhd1 mouse model resulted in a more severe phenotype [24,40]. A link between Fibrocystin and Polycystin-2 has also been described [41]. Recently Lea et al. could show that Polycystin-1, Polycystin-2, and Fibrocystin are detectable as a polycystin complex (PCC) in human urinary exosome like vesicle, that all of them undergo post-translational proteolytic processing and that there are various cleavage events in all three proteins exceeding the well described ones [42].
Despite the enormous progress, the exact mechanisms resulting in the initial development of a cyst and the following expansion of a cyst remain incompletely understood. Cysts in ADPKD are considered to be the result of a clonal expansion of an affected cell [43]. In line with this hypothesis, a second (or even a third) hit has been reported to be required in the heterozygous renal epithelial cells to induce the process resulting in cystogenesis in ADPKD [44,45,46]. Multiple signaling pathways have been found to be dysregulated in ADPKD with increased intracellular cyclic adenosine monophosphate (cAMP) concentration potentially as a consequence of vasopressin V2-receptor activation being a very prominent example that resulted in the identification of tolvaptan as a first treatment option of ADPKD [9,10,11] Increased intracellular cAMP concentrations also result in activation of the extracellular-signal regulated kinase and mitogen-activated protein kinase pathway (ERK/MAPK pathway) [47,48].
3. Mammalian Target of Rapamycin (mTOR) Activation in PKD
An aspect that has recently received increasing interest in the field are the changes of cellular metabolism in PKD. Other proliferative disorders like different tumor entities have given substantial insights into changes of cellular signaling and cellular metabolism that may also be relevant in PKD [49].
One of the starting points for this research area was the detection of an activation of the mTOR pathway in murine models of PKD [50,51]. mTOR itself is an atypical serine/threonine kinase that can be found in mTOR Complex 1 (mTORC1) and mTOR Complex 2 (mTORC2) with independent cellular functions. mTOR signaling is involved in a multitude of cellular processes related to cell growth and metabolism [52,53,54]. mTORC1 activity can be regulated through multiple intra- and extracellular ways including the presence of growth factors, the availability of nutrients and the energy status of the cell and can be controlled by the drug rapamycin [53,55,56]. In brief, activation of mTORC1 at the lysosomal membrane is regulated by sensing of intracellular nutrients and various upstream pathways resulting in control of Rag GTPases and members of the Ragulator complex and thus mTORC1. Activation of mTORC1 results in promotion of anabolic processes and the inhibition of catabolism including autophagy [57].
Activation of the mTOR pathway was observed in cyst lining epithelia of ADPKD kidneys and polycystin-1 was shown to negatively regulate mTOR activation [51]. Inhibition of mTOR using rapamycin reduced kidney volume in different animal models and resulted in better renal function [50]. These observations led to the establishment of large clinical trials for the use of mTOR inhibitors in ADPKD. Yet, these trials failed to show the hoped-for breakthrough results. In the study by Walz et al. 433 patients with advanced ADPKD in chronic kidney disease (CKD) stage G3a were treated with everolimus or placebo in a 2-year, randomized, double-blinded, placebo-controlled trial [58]. Based on the findings by the CRISP consortium and others total kidney volume determined by magnetic resonance imaging (MRI) was used as a surrogate for disease progression [59,60,61,62]. The trial could show a significant reduction in increase of total kidney volume in the everolimus group after one year, but this effect lost statistical significance after the second year. Importantly, decline of the estimated glomerular filtration rate (eGFR) was more rapid in patients on everolimus [58]. The SUISSE ADPKD trial studied 100 patients with an eGFR above 70 mL/min in an open-label randomized, controlled trial using sirolimus vs. placebo over 18 months. There were no significant differences in total kidney volume or eGFR between the groups but sirolimus patients showed more albuminuria, a known side effect of this drug [63,64].
It is important to point out that the therapeutic dosage used in animal models could not be achieved in clinical trials and that the animal models initially used for evaluation of mTOR inhibition do not fully recapitulate ADPKD but rather were models of rapidly progressing PKD [50,65,66,67]. Furthermore, the dropout-rate in the treatment arm was substantial in the everolimus study and the design of the clinical trials may not have detected all positive effects of mTOR inhibition on ADPKD progression in tubular cells [64,68,69,70]. It has been suggested that a direct delivery of mTOR inhibitors to renal tubular epithelial cells may have more specific positive effects. Indeed, modified rapamycin that was conjugated to folate with the aim to specifically target proliferating cells in cystic epithelium did show positive effects on cyst growth and kidney function in rapidly progressing PKD mouse model [71].
Multiple ways of interaction between the ADPKD protein polycystin 1 (PC1) and mTOR signaling have been described [72]. PC1 overexpression leads to a downregulation of the MEK/ERK pathway resulting in decreased activation of tuberin and thus mTOR inhibition. PC1-deficiency on the other hand results in an upregulation of ERK1/2 and through this to an upregulation of the mTOR pathway [73]. Vice versa the activation of mTORC1 in mouse embryonic fibroblasts (MEFs) was shown to have a negative influence on the expression of PC1, as well as on the trafficking of PC1/PC2 complex into cilia [74]. An effect of fibrocystin on mammalian target of rapamycin (mTOR) activation has been found in vitro [75] and activation of mTOR signaling has been described in cyst lining epithelium in vivo [76]. Treatment with NVP-BEZ235, an inhibitor of PI3-kinase and mTOR, had a positive effect on cystic dilatation of the intrahepatic bile ducts in the PCK rat [77].
In a pkd1-deficient zebrafish model has provided an insight into mechanisms downstream of mTOR activation that have an impact in PKD. Inhibition of autophagy by knocking down core autophagy proteins like Afg5 let to increased cystogenesis while activation of autophagy via Beclin-1 peptide ameliorated cystogenesis. These mechanisms seem to be conserved in mouse models and human tissue [78]. Furthermore, the treatment with low dose rapamycin and carbamazepine, known to activate autophagy in a mTOR independent way, was very effective in zebrafish [78] pointing towards a potential role of combination therapies that tackle this pathway at different levels.
4. Glucose Metabolism, Glycolysis, Oxidative Phosphorylation, and the Warburg Effect
A novel link of cellular metabolism to ADPKD has more recently emerged pointing to an important role of cellular glucose metabolism in ADPKD [79]. In healthy cells, adenosine triphosphate (ATP) production is known to take place in the mitochondria in the presence of oxygen. For this, pyruvate emerging from glycolysis is metabolized to acetyl coenzyme A (Acetyl-CoA) through oxidative decarboxylation. Acetyl-CoA, which is the main product entering the Krebs cycle is processed in the mitochondrial matrix resulting in the generation of nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide (FADH2). NADH and FADH2 then enter the oxidative phosphorylation cascade via complex I and complex II of the respiratory chain. Via electron changes a transmembrane gradient between inner and outer mitochondrial membrane is built up and enables the flux of protons which is finally used to regenerate ATP from adenosine diphosphate (ADP) by the use of oxygen at complex V. This process called oxidative phosphorylation is highly effective and produces 36 molecules ATP out of one molecule glucose. In the absence of oxygen, pyruvate is converted into lactate in an enzymatic reaction in the cytosol through lactate dehydrogenase leading to the production of two molecules ATP and regeneration of two molecules NADH [80].
In 1924, Otto Warburg made the observation that tumor cells use the metabolic conversion of pyruvate into lactate even in the presence of oxygen. This so-called aerobic glycolysis [81], has been a starting point to many newly discovered pathways in the last years, summarized in various excellent reviews [82,83,84,85]. Reprogramming of energy metabolism has been added to one of the emerging hallmarks for tumorigenesis in 2011, emphasizing the importance of changes in cancer cell metabolism [86].
By metabolomic characterization Pkd1-deficient murine cells were recently found to also preferentially use the approximate 18-fold less effective aerobic glycolysis even in the presence of oxygen. Tumor cells upregulate multiple genes involved in glucose transport and glycolysis [87,88] and indeed, this was found to also be the case in Pkd1-deficient cells [88]. This finding was confirmed in human tissue [89,90]. Importantly, treatment of two different Pkd1-mouse models of ADPKD with 2-deoxyglucose, an inhibitor of glycolysis showed beneficial results with lower kidney weight and volume and normalized AMP-activated protein kinase and its target acetyl-CoA carboxylase [89,91]. The treatment with 2-deoxyglucose was also successful in a rat PKD model [92].
In addition, there were first hints that mitochondrial protein levels as well as the activity of crucial subcomplexes of the mitochondrial electron chain were altered in a Mini-pig PKD model [93]. Most recently it was found that a cleavage product of polycystin-1 can affect mitochondria morphology and function and that mitochondria of ADPKD patients show structural changes [94,95]. Besides, PC1 and PC2 localize in mitochondria associated membranes and can interact with the calcium signaling from the lumen of the ER to the mitochondrial matrix [96,97,98].
Cassina et al. could show that increased mitochondrial fragmentation seems to be a modifier of disease progression. In Pkd1 mutant mice the authors could show aberrant mitochondria in the cyst-lining epithelial cells with a reduced mitochondrial mass and a following reduction in mitochondrial respiratory chain complexes. This seems to be due to a fragmentation of the mitochondrial network which is driven by a downregulation of the pro-fusion proteins OPA-1 (mitochondrial Dynamin-like 120 kDa protein/optic atrophy protein 1) and MFN-1 (Mitofusin-1) and an increase in the pro-fission protein DRP-1 (Dynamin-related protein 1) [99].
5. Amino Acid Metabolism
PKD cells, very much like cancer cells, seem to have a high need in biomass while switching into an anabolic mode [83]. One of the essential carbon sources in this context is glutamine, which can be catabolized into α-ketoglutarate (α-KG) through a process called glutamine anaplerosis. Glutamine is converted into glutamate and glutamate into α-KG, which itself is enters the TCA, fueling the electron transport chain and maintaining the mitochondrial membrane potential. [100]
For PKD, glutamine addiction was first described by Hwang et al. [101]. A subsequent study showed that glutamine-dependence caused by ablation of Lkb1 in combination with ablation of Tsc1 gene resulted in very aggressive cystogenesis in a mouse model and inhibition of glutamine metabolism in both Lkb1/Tsc1 and Pkd1 mutant mice significantly reduced cyst progression [102].
In 2018, Podrini et al. were able to show that indeed glutamine usage is upregulated in PKD, most likely to keep up the mitochondrial membrane potential and to deliver enough building blocks for rapid cell division. Furthermore, they showed that glutamine is converted into glutamate via asparagine synthetase (ASNS), a rather unusual mechanism. ASNS converts aspartate into asparaginase while converting glutamine into glutamate [103]. Beyond this they could show that in PKD cells glutamine-derived α-KG not only seems to be used by the cell to maintain the OXPHOS, but it is also carboxylated to produce citrate, which itself is converted into Actely-CoA and Oxalacetate (OAA). Actyl-CoA is used for the fatty acid synthesis and OAA is converted into aspartate, fueling the ASNS for an efficient transport of glutamine into the cells and the OXPHOS, closing the circle and underlining the glutamine dependence of PKD cell metabolism [96,103].
6. ERK-AMPK
AMP-activated protein kinase (AMPK) is a highly conserved eukaryotic kinase, which plays a crucial role in controlling cellular energy balance [105]. AMPK is activated by increases in AMP/ATP and ADP/ATP ratios with the result to increase cellular catabolism and to decrease cellular anabolic processes [106,107]. This includes stimulation of fatty acid oxidation and glucose uptake. AMPK leads to phosphorylation of raptor and tuberous sclerosis complex 2, which itself leads to inhibition of mTOR and elongation factor 2 ending in a decrease of protein synthesis, cell growth and cell proliferation [108]. Interestingly enough, AMPK leads to gluconeogenesis via inhibition of the transcription factor hepatocyte nuclear factor 4 (Hnf4) and inhibits mTORC1 [78,108]
In Pkd1-/-cells, the activating AMPK phosphorylation is decreased compared to wild-type cells [89]. As AMPK activation leads to mTOR inhibition, which has been described earlier in this manuscript as a potential strategy to decrease cyst growth in mouse model, the activation of AMPK pathway may be a good target for therapeutic strategies. AMPK itself was found to be regulated through an ERK1/2-Liver kinase B1 (LKB1) axis [89].The activation of the ERK1/2 pathway could be confirmed in a PKD mini-pig model [93].
AMPK activation may become an interesting therapeutic option as metformin is a drug with a broad clinical use in Type 2 diabetes mellitus and the advent of a first application in cancer treatment in the last decade [109,110]. Metformin is known to interfere with mitochondrial phosphorylation. The inhibition of complex 1 (NADH dehydrogenase) of the electron chain of oxidative phosphorylation could be shown in tumor cells making Metformin interesting for cancer treatment [111]. This inhibition of mitochondrial complex I was also shown in peripheral blood mononuclear cells and platelets [112], but has not been studied in detail in renal cells so far. Yet, metformin has been shown to activate AMPK in an ADPKD mouse model resulting in decreased cyst growth via inhibition of mTOR in vitro [113]. Moreover, it was shown that treatment of ADPKD murine embryonic fibroblasts with Metformin and other AMPK activators led to a decrease of the aerobic glycolysis or Warburg effect [89]. These findings could be confirmed in a PKD rat model [92]. An additional study found additive effects of rapamycin and metformin in counteracting mTOR activation after knockdown of Pkd1 [114]. However, another study using a Pkd1 knockout mouse model did not find beneficial effects of metformin [115]. To clarify potential efficacy in humans, a first clinical trial has been repurposing metformin for ADPKD (Metformin as a Novel Therapy for Autosomal Dominant Polycystic Kidney Disease (TAME), ClinalTrials.gov, Identifier: NCT02656017).
In a transcriptomic and metabolomic analysis of an early-onset murine model of PKD due to an induced knockout of Pkd1 the transcription factor Hnf4α (Hepatocyte nuclear factor 4-alpha) was identified as a potential important regulator of metabolism affected in APDKD [116]. Hnf4α is a downstream target of AMPK [117] that is involved in the regulation of glucose metabolism [118,119,120]. Indeed, multiple genes involved in glucose regulation have been found to be regulated by SREBP1, HIF1-a or HNF4α in an mTOR-dependent way [121]. Interestingly, the double knockout of Hnf4α and Pkd1 resulted in a more severe phenotype [116]. The authors speculated that the upregulation of Hnf4α might be a secondary compensatory mechanism (Figure 2) [116].
7. Fatty Acid Oxidation
Various links were established between fatty acid oxidation and PKD including a cystic renal phenotype in some disorders of fatty acid oxidation [122,123]. Indeed, it may be clinically challenging to differentiate these disorders [124]. Interestingly, overlapping metabolic features have also been found in disorders of fatty acid oxidation and PKD including activation of the ERK-mTOR axis, reduced oxidative phosphorylation and increased generation of lactate [124]. In addition, impaired fatty acid oxidation has been found in animal models of PKD [125] and an attenuation of both the renal and the hepatic phenotype has been described in a slowly progressing Pkd1 mouse model after activation of fatty acid oxidation using the PPAR-α agonist fenofibrate [126]. This is in accordance with findings from the PCK rat, an orthologous model of ARPKD with a renal phenotype with features also resembling ADPKD [127].
8. Sirtuin
Sirtuin-1 (SIRT-1) is an NAD-dependent protein deacetylase and a known regulatory protein in apoptosis. Sirtuin-1 leads to a destabilization of p53, one of the best characterized pro-apoptotic factors, which is in mutated status relevant for tumor progression in many characterized diseases such as tumors in the adrenal gland [128].
In 2013 it was shown that Sirt1 expression is upregulated in Pkd1-deficient cells and that the distal tubular double knockout of Pkd1 and Sirt1 leads to a less severe phenotype than the single knockout of distal tubular Pkd1 in mouse model. Furthermore, the treatment of Pkd1 lacking embryos in mouse model with the Sirtuin-1 inhibitor EX-527 or nicotinamide was leading to slower cyst growth in these animals [129]. Interestingly, SIRT-1 is a crucial metabolic NAD+-sensor. The presence of SIRT-1 leads to changes in the chromatin structure important for the expression of genes relevant for metabolism in different organs [130]. In case of high glycolytic levels in the cell it is crucial for them to regenerate NAD+ in the cytoplasm, which sums up in the conversion from pyruvate into lactate [131]. The role of this mechanism in PKD remains to be established.
9. Cilia, mTOR, and Cellular Metabolism
In addition to the link of polycystin-1 to mTOR signaling there may be a direct effect of cilia on mTOR and cellular metabolism. The link between cilia and PKD has been reviewed and is beyond the scope of this manuscript [132,133]. Yet, it has also been shown that ablation of cilia in a rodent model leads to enlarged cell size and that this process is driven by mTORC1 [134]. Ciliary bending seems to be important for mTORC1 inhibition and cell-size control. Cultivating ciliated cells under flow leads to decreased mTORC1 levels and via this to decreased cell size. Furthermore LKB-1, a tumor-suppressor expressed in cilia, drives AMPK-phosphorylation at the ciliary basal body and leads to further mTORC1 inhibition [134]. These alterations in the AMPK concentrations at the ciliary basal body may point to an important interplay of ciliary and metabolic aspects in the pathogenesis in PKD.
10. Emerging Treatment Options
The first two major clinical trials in the PKD field that targeted cellular metabolic aspects were the previously described trials on mTOR inhibitor use in ADPKD [59,65]. The insights obtained in basic science that we describe in this manuscript have a great additional potential to be directly translated into clinical life. Multiple clinical trials will be required until everyday clinical practice in the nephrology ward will directly be influenced by these insights. Given the recent developments, novel initiatives have been launched and novel potential treatment approaches are emerging. We have already described some of these options in the subsections including modified mTOR inhibitors, inhibition of glycolysis, activation of AMPK through Metformin, PPAR-α activation or Sirtuin inhibition. While most of the work has focused on the renal phenotype, there are also hints that extrarenal manifestations like the hepatic phenotype can be influenced. A detailed analysis of the effects on extrarenal manifestations will be required.
A very interesting and easy to access therapeutic strategy to reduce renal cyst growth in PKD via mild to moderate food or caloric restriction has recently been shown to work for different orthologous mouse models of ADPKD [135,136]. The reduction of caloric or food intake has been shown to be highly effective to increase longevity [137,138]. The underlying cellular mechanisms seem to involve a complex interplay of multiple signaling cascade including mTOR, Sirtuins, and AMPK and result in mTOR inhibition and AMPK activation, thus exactly the conditions that seem to be favorable in PKD. Indeed, in both publications a rather mild restriction of food resulted in positive effects on kidney volume. Upregulation of AMPK and/or downregulation of the mTOR pathway were shown in PKD animals undergoing mild to moderate caloric restriction [135,136]. Importantly, in humans, obesity is known to be a risk factor for rapid disease progression in ADPKD [139]. Lately Torres et al. could show that the benefit of mild reduction in food intake is driven by induction of ketosis in preclinical PKD models. Time-restricted feeding was shown to inhibit mTOR signaling, proliferation and fibrosis in rodent kidneys. The same effect was seen in ketogenic diet, during acute fasting and with administration of β-hydroxybutyrate (BHB) to normal chow in preclinical mouse, rat and cat models of PKD, thus widening the therapeutic options to fasting and nutritive supplementation of ketones [140]. A clinical pilot trial that will test potential acute effects on kidney volume—as observed in the feline model—is currently starting recruitment (NCT04472624) and a larger trial using a 3-month intervention is going to examine feasibility of ketogenic interventions in ADPKD (https://pkdcure.org/funded-research/). Considering the lack of data on dietary measures to improve outcome in ADPKD, more clinical trials are urgently needed to rapidly evaluate these widely-available and cost-effective therapeutic strategies.
11. Conclusions
In summary, recent work has identified multiple metabolic cellular changes in cyst-lining epithelia in PKD both in animal models as well as in patients. This emerging field is currently at the verge of being transferred into clinical life with the first ongoing clinical trials.
Funding
Liebau was supported by grants of the Marga and Walter Boll-Foundation, the German Federal Ministry of Research and Education (BMBF grant 01GM1515 and 01GM1903), and the German Research Councial (DFG grant LI 2397/5-1).
Conflicts of Interest
S.Haumann declares no conflict of interest. The Dept. 2 for Internal Medicine received research funding and Müller received honoraria for counseling and lectures from Otsuka Pharmaceuticals and Thermo Fisher Scientific. Liebau has received honoraria for scientific lectures from Pfizer. Representing the University Hospital of Cologne ML has been counselling Otsuka in an advisory board.
Abbreviations
| Acetyl-CoA | acetyl coenzyme A |
| ADP | adenosine diphosphate |
| ADPKD | autosomal dominant polycystic kidney disease |
| α-KG | alpha-ketoglutarate |
| AMPK | adenosine monophospahte-activated protein kinase |
| ARPKD | autosomal recessive polycystic kidney disease |
| ASNS | asparagine synthethase |
| ASS1 | arginosuccinate synthase 1 |
| ATP | adenosine triphosphate |
| cAMP | cyclic adenosine monophosphate |
| BHB | β-hydroxybutyrate |
| CKD | chronic kidney disease |
| eGFR | estimated glomerular filtration rate |
| ERK | extracellular-signal regulated kinase |
| FADH | flavin adenine dinucleotide |
| HIF 1 | Hypoxia-inducible factor 1 |
| HNF4α | Hepatocyte nuclear factor 4 alpha |
| LKB1 | Liver kinase B1 |
| MAPK | mitogen-activated protein kinase pathway |
| MEFs | mouse embryonic fibroblasts |
| MRI | magnetic resonance imaging |
| mTOR | mammalian target of rapamycine |
| mTORC1 | mammalian target of rapamycine complex 1 |
| mTORC2 | mammalian target of rapamycine complex 2 |
| NADH | nicotinamide adenine dinucleotide |
| OAA | oxalacetate |
| PCC | polycystin complex |
| PC1 | Polycystin 1 |
| PC2 | Polycystin 2 |
| PKD | polycystic kidney disease |
| PPAR- α | Peroxisome proliferator-activated receptor alpha |
| SIRT-1 | Sirtuin-1 |
| SREBP-1 | sterol regulatory element-binding protein 1 |
| TCA | tricarboxylic acid cycle |
| TRPs | transient receptor potential ion channels |
| TSC | transient receptor potential ion channels |
References
- Gabow, P.A. Autosomal Dominant Polycystic Kidney Disease. N. Engl. J. Med. 1993, 329, 332–342. [Google Scholar] [CrossRef]
- Torres, V.E.; Harris, P.C. Polycystic kidney disease: Genes, proteins, animal models, disease mechanisms and therapeutic opportunities. J. Intern. Med. 2007, 261, 17–31. [Google Scholar] [CrossRef]
- Cramer, M.T.; Guay-Woodford, L.M. Cystic Kidney Disease: A Primer. Adv. Chronic Kidney Dis. 2015, 22, 297–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Rechter, S.; Breysem, L.; Mekahli, D. Is Autosomal Dominant Polycystic Kidney Disease Becoming a Pediatric Disorder? Front. Pediatr. 2017, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordido, A.; Besada-Cerecedo, L.; García-González, M.A. The Genetic and Cellular Basis of Autosomal Dominant Polycystic Kidney Disease—A Primer for Clinicians. Front. Pediatr. 2017, 5. [Google Scholar] [CrossRef]
- Bergmann, C. Genetics of Autosomal Recessive Polycystic Kidney Disease and Its Differential Diagnoses. Front. Pediatr. 2018, 5. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, C.; Guay-Woodford, L.M.; Harris, P.C.; Horie, S.; Peters, D.J.M.; Torres, V.E. Polycystic kidney disease. Nat. Rev. Dis. Prim. 2018, 4, 50. [Google Scholar] [CrossRef]
- Chebib, F.T.; Torres, V.E. Recent Advances in the Management of Autosomal Dominant Polycystic Kidney Disease. Clin. J. Am. Soc. Nephrol. 2018, 13, 1765–1776. [Google Scholar] [CrossRef] [Green Version]
- Torres, V.E.; Chapman, A.B.; Devuyst, O.; Gansevoort, R.T.; Grantham, J.J.; Higashihara, E.; Perrone, R.D.; Krasa, H.B.; Ouyang, J.; Czerwiec, F.S.; et al. Tolvaptan in Patients with Autosomal Dominant Polycystic Kidney Disease. N. Engl. J. Med. 2012, 367, 2407–2418. [Google Scholar] [CrossRef] [Green Version]
- Torres, V.E.; Chapman, A.B.; Devuyst, O.; Gansevoort, R.T.; Perrone, R.D.; Koch, G.; Ouyang, J.; McQuade, R.D.; Blais, J.D.; Czerwiec, F.S.; et al. Tolvaptan in Later-Stage Autosomal Dominant Polycystic Kidney Disease. N. Engl. J. Med. 2017, 377, 1930–1942. [Google Scholar] [CrossRef]
- Rinschen, M.M.; Schermer, B.; Benzing, T. Vasopressin-2 Receptor Signaling and Autosomal Dominant Polycystic Kidney Disease: From Bench to Bedside and Back Again. J. Am. Soc. Nephrol. 2014, 25, 1140–1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborty, S.; McHugh, K. Cystic diseases of the kidney in children. Imaging 2005, 17, 69–75. [Google Scholar] [CrossRef]
- Hoyer, P.F. Clinical manifestations of autosomal recessive polycystic kidney disease. Curr. Opin. Pediatr. 2015, 27, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Ganschow, R.; Hoppe, B. Review of combined liver and kidney transplantation in children. Pediatr. Transplant. 2015, 19, 820–826. [Google Scholar] [CrossRef] [PubMed]
- Ebner, K.; Feldkoetter, M.; Ariceta, G.; Bergmann, C.; Buettner, R.; Doyon, A.; Duzova, A.; Göbel, H.; Haffner, D.; Hero, B.; et al. Rationale, design and objectives of ARegPKD, a European ARPKD registry study. BMC Nephrol. 2015, 16, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebner, K.; Schaefer, F.; Liebau, M.C.; The ARegPKD Consortium; Eid, L.A.; Ranguelov, N.; Adams, B.; Van Hoeck, K.; Raes, A.; Mekahli, D.; et al. Recent Progress of the ARegPKD Registry Study on Autosomal Recessive Polycystic Kidney Disease. Front. Pediatr. 2017, 5. [Google Scholar] [CrossRef] [Green Version]
- Alzarka, B.; Morizono, H.; Bollman, J.W.; Kim, D.; Guay-Woodford, L.M. Design and Implementation of the Hepatorenal Fibrocystic Disease Core Center Clinical Database: A Centralized Resource for Characterizing Autosomal Recessive Polycystic Kidney Disease and Other Hepatorenal Fibrocystic Diseases. Front. Pediatr. 2017, 5. [Google Scholar] [CrossRef]
- Lu, H.; Galeano, M.C.R.; Ott, E.; Kaeslin, G.; Kausalya, P.J.; Kramer, C.; Ortiz-Brüchle, N.; Hilger, N.; Metzis, V.; Hiersche, M.; et al. Mutations in DZIP1L, which encodes a ciliary-transition-zone protein, cause autosomal recessive polycystic kidney disease. Nat. Genet. 2017, 49, 1025–1034. [Google Scholar] [CrossRef]
- Follit, J.A.; Li, L.; Vucica, Y.; Pazour, G.J. The cytoplasmic tail of fibrocystin contains a ciliary targeting sequence. J. Cell Boil. 2010, 188, 21–28. [Google Scholar] [CrossRef] [Green Version]
- Kaimori, J.-Y.; Nagasawa, Y.; Menezes, L.F.; Garcia-Gonzalez, M.A.; Deng, J.; Imai, E.; Onuchic, L.F.; Guay-Woodford, L.M.; Germino, G.G. Polyductin undergoes notch-like processing and regulated release from primary cilia. Hum. Mol. Genet. 2007, 16, 942–956. [Google Scholar] [CrossRef]
- Hiesberger, T.; Gourley, E.; Erickson, A.; Koulen, P.; Ward, C.J.; Masyuk, T.V.; LaRusso, N.F.; Harris, P.C.; Igarashi, P. Proteolytic Cleavage and Nuclear Translocation of Fibrocystin Is Regulated by Intracellular Ca2+ and Activation of Protein Kinase C. J. Boil. Chem. 2006, 281, 34357–34364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaimori, J.-Y.; Lin, C.-C.; Outeda, P.; Garcia-Gonzalez, M.A.; Menezes, L.F.; Hartung, E.A.; Li, A.; Wu, G.; Fujita, H.; Sato, Y.; et al. NEDD4-family E3 ligase dysfunction due to PKHD1/Pkhd1 defects suggests a mechanistic model for ARPKD pathobiology. Sci. Rep. 2017, 7, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Outeda, P.; Menezes, L.; Hartung, E.A.; Bridges, S.; Zhou, F.; Zhu, X.; Xu, H.; Huang, Q.; Yao, Q.; Qian, F.; et al. A novel model of autosomal recessive polycystic kidney questions the role of the fibrocystin C-terminus in disease mechanism. Kidney Int. 2017, 92, 1130–1144. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gonzalez, M.A.; Menezes, L.F.; Piontek, K.B.; Kaimori, J.; Huso, D.L.; Watnick, T.; Onuchic, L.F.; Guay-Woodford, L.M.; Germino, G.G. Genetic interaction studies link autosomal dominant and recessive polycystic kidney disease in a common pathway. Hum. Mol. Genet. 2007, 16, 1940–1950. [Google Scholar] [CrossRef] [Green Version]
- Moser, M.; Matthiesen, S.; Kirfel, J.; Schorle, H.; Bergmann, C.; Senderek, J.; Zerres, K.; Buettner, R.; Rudnik-Schöneborn, S. A mouse model for cystic biliary dysgenesis in autosomal recessive polycystic kidney disease (ARPKD). Hepatology 2005, 41, 1113–1121. [Google Scholar] [CrossRef]
- Woollard, J.; Punyashtiti, R.; Richardson, S.; Masyuk, T.; Whelan, S.; Huang, B.; Lager, D.; Vandeursen, J.; Torres, V.; Gattone, V.; et al. A mouse model of autosomal recessive polycystic kidney disease with biliary duct and proximal tubule dilatation. Kidney Int. 2007, 72, 328–336. [Google Scholar] [CrossRef] [Green Version]
- Chebib, F.T.; Torres, V.E. Autosomal Dominant Polycystic Kidney Disease: Core Curriculum 2016. Am. J. Kidney Dis. 2015, 67, 792–810. [Google Scholar] [CrossRef] [Green Version]
- Krishnappa, V.; Vinod, P.; Deverakonda, D.; Raina, R. Autosomal dominant polycystic kidney disease and the heart and brain. Clevel. Clin. J. Med. 2017, 84, 471–481. [Google Scholar] [CrossRef] [Green Version]
- Bergmann, C.; Von Bothmer, J.; Brüchle, N.O.; Venghaus, A.; Frank, V.; Fehrenbach, H.; Hampel, T.; Pape, L.; Buske, A.; Jonsson, J.; et al. Mutations in multiple PKD genes may explain early and severe polycystic kidney disease. J. Am. Soc. Nephrol. 2011, 22, 2047–2056. [Google Scholar] [CrossRef] [Green Version]
- Harris, P.C.; Rossetti, S. Molecular diagnostics for autosomal dominant polycystic kidney disease. Nat. Rev. Nephrol. 2010, 6, 197–206. [Google Scholar] [CrossRef] [Green Version]
- Harris, P.C.; Hopp, K. The Mutation, a Key Determinant of Phenotype in ADPKD. J. Am. Soc. Nephrol. 2013, 24, 868–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gall, E.C.-L.; Audrézet, M.-P.; Chen, J.-M.; Hourmant, M.; Morin, M.-P.; Perrichot, R.; Charasse, C.; Whebe, B.; Renaudineau, E.; Jousset, P.; et al. Type of PKD1 Mutation Influences Renal Outcome in ADPKD. J. Am. Soc. Nephrol. 2013, 24, 1006–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porath, B.; Gainullin, V.G.; Gall, E.C.-L.; Dillinger, E.K.; Heyer, C.M.; Hopp, K.; Edwards, M.E.; Madsen, C.D.; Mauritz, S.R.; Banks, C.J.; et al. Mutations in GANAB, Encoding the Glucosidase IIα Subunit, Cause Autosomal-Dominant Polycystic Kidney and Liver Disease. Am. J. Hum. Genet. 2016, 98, 1193–1207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gall, E.C.-L.; Torres, V.E.; Harris, P.C. Genetic Complexity of Autosomal Dominant Polycystic Kidney and Liver Diseases. J. Am. Soc. Nephrol. 2017, 29, 13–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irazabal, M.V.; Rangel, L.J.; Bergstralh, E.J.; Osborn, S.L.; Harmon, A.J.; Sundsbak, J.L.; Bae, K.T.; Chapman, A.B.; Grantham, J.J.; Mrug, M.; et al. Imaging Classification of Autosomal Dominant Polycystic Kidney Disease: A Simple Model for Selecting Patients for Clinical Trials. J. Am. Soc. Nephrol. 2014, 26, 160–172. [Google Scholar] [CrossRef]
- Gall, E.C.-L.; Audrézet, M.-P.; Rousseau, A.; Hourmant, M.; Renaudineau, E.; Charasse, C.; Morin, M.-P.; Moal, M.-C.; Dantal, J.; Wehbe, B.; et al. The PROPKD Score: A New Algorithm to Predict Renal Survival in Autosomal Dominant Polycystic Kidney Disease. J. Am. Soc. Nephrol. 2015, 27, 942–951. [Google Scholar] [CrossRef] [Green Version]
- Chang, M.-Y.; Ong, A. Targeting new cellular disease pathways in autosomal dominant polycystic kidney disease. Nephrol. Dial. Transplant. 2017, 33, 1310–1316. [Google Scholar] [CrossRef]
- Leonhard, W.N.; Happe, H.; Peters, D.J. Variable Cyst Development in Autosomal Dominant Polycystic Kidney Disease: The Biologic Context. J. Am. Soc. Nephrol. 2016, 27, 3530–3538. [Google Scholar] [CrossRef]
- Ong, A.; Harris, P.C. A polycystin-centric view of cyst formation and disease: The polycystins revisited. Kidney Int. 2015, 88, 699–710. [Google Scholar] [CrossRef] [Green Version]
- Olson, R.J.; Hopp, K.; Wells, H.; Smith, J.M.; Furtado, J.; Constans, M.M.; Escobar, D.L.; Geurts, A.M.; Torres, V.E.; Harris, P.C. Synergistic Genetic Interactions between Pkhd1 and Pkd1 Result in an ARPKD-Like Phenotype in Murine Models. J. Am. Soc. Nephrol. 2019, 30, 2113–2127. [Google Scholar] [CrossRef]
- Kim, I.; Li, C.; Liang, D.; Chen, X.-Z.; Coffy, R.J.; Ma, J.; Zhao, P.; Wu, G. Polycystin-2 Expression Is Regulated by a PC2-binding Domain in the Intracellular Portion of Fibrocystin. J. Boil. Chem. 2008, 283, 31559–31566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lea, W.A.; McGreal, K.; Sharma, M.; Parnell, S.C.; Zelenchuk, L.; Charlesworth, M.C.; Madden, B.J.; Johnson, K.L.; McCormick, D.J.; Hogan, M.C.; et al. Analysis of the polycystin complex (PCC) in human urinary exosome–like vesicles (ELVs). Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, F.; Watnick, T.J.; Onuchic, L.F.; Germino, G.G. The Molecular Basis of Focal Cyst Formation in Human Autosomal Dominant Polycystic Kidney Disease Type I. Cell 1996, 87, 979–987. [Google Scholar] [CrossRef] [Green Version]
- Watnick, T.; E Torres, V.; A Gandolph, M.; Qian, F.; Onuchic, L.F.; Klinger, K.W.; Landes, G.; Germino, G.G. Somatic mutation in individual liver cysts supports a two-hit model of cystogenesis in autosomal dominant polycystic kidney disease. Mol. Cell 1998, 2, 247–251. [Google Scholar] [CrossRef]
- Pei, Y.; Watnick, T.; He, N.; Wang, K.; Liang, Y.; Parfrey, P.; Germino, G.; George-Hyslop, P.S. Somatic PKD2 mutations in individual kidney and liver cysts support a “two-hit” model of cystogenesis in type 2 autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 1999, 10, 1524–1529. [Google Scholar]
- Weimbs, T. Third-hit signaling in renal cyst formation. J. Am. Soc. Nephrol. 2011, 22, 793–795. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Nagao, S.; Wallace, D.P.; Belibi, F.A.; Cowley, B.D.; Pelling, J.C.; Grantham, J.J.; Grantham, J.J. Cyclic AMP activates B-Raf and ERK in cyst epithelial cells from autosomal-dominant polycystic kidneys. Kidney Int. 2003, 63, 1983–1994. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.X.; Fan, L.X.; Li, X.; Calvet, J.P.; Li, X. TNFα Signaling Regulates Cystic Epithelial Cell Proliferation through Akt/mTOR and ERK/MAPK/Cdk2 Mediated Id2 Signaling. PLOS ONE 2015, 10, e0131043. [Google Scholar] [CrossRef] [Green Version]
- Seeger-Nukpezah, T.; Geynisman, D.M.; Nikonova, A.S.; Benzing, T.; Golemis, E.A. The hallmarks of cancer: Relevance to the pathogenesis of polycystic kidney disease. Nat. Rev. Nephrol. 2015, 11, 515–534. [Google Scholar] [CrossRef] [Green Version]
- Wahl, P.R.; Serra, A.L.; Le Hir, M.; Molle, K.D.; Hall, M.N.; Wüthrich, R.P. Inhibition of mTOR with sirolimus slows disease progression in Han:SPRD rats with autosomal dominant polycystic kidney disease (ADPKD). Nephrol. Dial. Transplant. 2005, 21, 598–604. [Google Scholar] [CrossRef] [Green Version]
- Shillingford, J.M.; Murcia, N.S.; Larson, C.H.; Low, S.H.; Hedgepeth, R.; Brown, N.; Flask, C.A.; Novick, A.C.; Goldfarb, D.A.; Kramer-Zucker, A.; et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc. Natl. Acad. Sci. USA 2006, 103, 5466–5471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lieberthal, W.; Levine, J.S. Mammalian target of rapamycin and the kidney. I. The signaling pathway. Am. J. Physiol. Physiol. 2012, 303, F1–F10. [Google Scholar] [CrossRef] [PubMed]
- Mannaa, M.; Krämer, S.; Boschmann, M.; Gollasch, M. mTOR and regulation of energy homeostasis in humans. J. Mol. Med. 2013, 91, 1167–1175. [Google Scholar] [CrossRef] [PubMed]
- Lieberthal, W.; Levine, J.S. The Role of the Mammalian Target Of Rapamycin (mTOR) in Renal Disease. J. Am. Soc. Nephrol. 2009, 20, 2493–2502. [Google Scholar] [CrossRef] [PubMed]
- Rabanal-Ruiz, Y.; Korolchuk, V.I. mTORC1 and Nutrient Homeostasis: The Central Role of the Lysosome. Int. J. Mol. Sci. 2018, 19, 818. [Google Scholar] [CrossRef] [Green Version]
- Huber, T.B.; Walz, G.; Kuehn, E.W. mTOR and rapamycin in the kidney: Signaling and therapeutic implications beyond immunosuppression. Kidney Int. 2011, 79, 502–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmeisser, K.; Parker, J.A. Pleiotropic Effects of mTOR and Autophagy During Development and Aging. Front. Cell Dev. Boil. 2019, 7. [Google Scholar] [CrossRef] [Green Version]
- Walz, G.; Budde, K.; Mannaa, M.; Nurnberger, J.; Wanner, C.; Sommerer, C.; Kunzendorf, U.; Banas, B.; Hörl, W.H.; Obermüller, N.; et al. Everolimus in Patients with Autosomal Dominant Polycystic Kidney Disease. N. Engl. J. Med. 2010, 363, 830–840. [Google Scholar] [CrossRef] [Green Version]
- Grantham, J.J.; Torres, V.E.; Chapman, A.B.; Guay-Woodford, L.M.; Bae, K.T.; King, B.F.; Wetzel, L.H.; Baumgarten, D.A.; Kenney, P.J.; Harris, P.C.; et al. Volume Progression in Polycystic Kidney Disease. N. Engl. J. Med. 2006, 354, 2122–2130. [Google Scholar] [CrossRef] [Green Version]
- Bae, K.T.; Tao, C.; Wang, J.; Kaya, D.; Wu, Z.; Bae, J.T.; Chapman, A.B.; Torres, V.E.; Grantham, J.J.; Mrug, M.; et al. Novel approach to estimate kidney and cyst volumes using mid-slice magnetic resonance images in polycystic kidney disease. Am. J. Nephrol. 2013, 38, 333–341. [Google Scholar] [CrossRef] [Green Version]
- Bhutani, H.; Smith, V.; Rahbari-Oskoui, F.; Mittal, A.; Grantham, J.J.; Torres, V.E.; Mrug, M.; Bae, K.T.; Wu, Z.; Ge, Y.; et al. A comparison of ultrasound and magnetic resonance imaging shows that kidney length predicts chronic kidney disease in autosomal dominant polycystic kidney disease. Kidney Int. 2015, 88, 146–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tangri, N.; Hougen, I.; Alam, A.; Perrone, R.; McFarlane, P.; Pei, Y. Total Kidney Volume as a Biomarker of Disease Progression in Autosomal Dominant Polycystic Kidney Disease. Can. J. Kidney Heal. Dis. 2017, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serra, A.L.; Kistler, A.D.; Poster, D.; Struker, M.; Wüthrich, R.P.; Weishaupt, D.; Tschirch, F. Clinical proof-of-concept trial to assess the therapeutic effect of sirolimus in patients with autosomal dominant polycystic kidney disease: SUISSE ADPKD study. BMC Nephrol. 2007, 8, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serra, A.L.; Poster, D.; Kistler, A.D.; Krauer, F.; Raina, S.; Young, J.; Rentsch, K.M.; Spanaus, K.S.; Senn, O.; Kristanto, P.; et al. Sirolimus and Kidney Growth in Autosomal Dominant Polycystic Kidney Disease. N. Engl. J. Med. 2010, 363, 820–829. [Google Scholar] [CrossRef] [Green Version]
- Zafar, I.; Ravichandran, K.; Belibi, F.A.; Doctor, R.B.; Edelstein, C.L. Sirolimus attenuates disease progression in an orthologous mouse model of human autosomal dominant polycystic kidney disease. Kidney Int. 2010, 78, 754–761. [Google Scholar] [CrossRef] [Green Version]
- Tao, Y.; Kim, J.; Schrier, R.W.; Edelstein, C.L. Rapamycin Markedly Slows Disease Progression in a Rat Model of Polycystic Kidney Disease. J. Am. Soc. Nephrol. 2004, 16, 46–51. [Google Scholar] [CrossRef] [Green Version]
- Nagao, S.; Kugita, M.; Yoshihara, D.; Yamaguchi, T. Animal models for human polycystic kidney disease. Exp. Anim. 2012, 61, 477–488. [Google Scholar] [CrossRef] [Green Version]
- Novalic, Z.; Van Der Wal, A.M.; Leonhard, W.N.; Koehl, G.; Breuning, M.H.; Geissler, E.K.; De Heer, E.; Peters, D.J. Dose-Dependent Effects of Sirolimus on mTOR Signaling and Polycystic Kidney Disease. J. Am. Soc. Nephrol. 2012, 23, 842–853. [Google Scholar] [CrossRef] [Green Version]
- Canaud, G.; Knebelmann, B.; Harris, P.C.; Vrtovsnik, F.; Correas, J.; Pallet, N.; Heyer, C.M.; Letavernier, E.; Bienaimé, F.; Thervet, E.; et al. Therapeutic mTOR Inhibition in Autosomal Dominant Polycystic Kidney Disease: What Is the Appropriate Serum Level? Arab. Archaeol. Epigr. 2010, 10, 1701–1706. [Google Scholar] [CrossRef]
- Jardine, M.; Liyanage, T.; Buxton, E.; Perkovic, V. mTOR inhibition in autosomal-dominant polycystic kidney disease (ADPKD): The question remains open. Nephrol. Dial. Transplant. 2012, 28, 242–244. [Google Scholar] [CrossRef] [Green Version]
- Shillingford, J.M.; Leamon, C.P.; Vlahov, I.R.; Weimbs, T. Folate-Conjugated Rapamycin Slows Progression of Polycystic Kidney Disease. J. Am. Soc. Nephrol. 2012, 23, 1674–1681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grahammer, F.; Wanner, N.; Huber, T.B. mTOR controls kidney epithelia in health and disease. Nephrol. Dial. Transplant. 2014, 29, i9–i18. [Google Scholar] [CrossRef] [PubMed]
- Distefano, G.; Boca, M.; Rowe, I.; Wodarczyk, C.; Ma, L.; Piontek, K.B.; Germino, G.G.; Pandolfi, P.P.; Boletta, A. Polycystin-1 Regulates Extracellular Signal-Regulated Kinase-Dependent Phosphorylation of Tuberin To Control Cell Size through mTOR and Its Downstream Effectors S6K and 4EBP1. Mol. Cell. Boil. 2009, 29, 2359–2371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pema, M.; Drusian, L.; Chiaravalli, M.; Castelli, M.; Yao, Q.; Ricciardi, S.; Somlo, S.; Qian, F.; Biffo, S.; Boletta, A. mTORC1-mediated inhibition of polycystin-1 expression drives renal cyst formation in tuberous sclerosis complex. Nat. Commun. 2016, 7, 10786. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wu, M.; Yao, G.; Zhang, J.; Zhou, J. The Cytoplasmic Tail of FPC Antagonizes the Full-Length Protein in the Regulation of mTOR Pathway. PLoS ONE 2014, 9, e95630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, D.-C.; Jacoby, U.; Pape, L.; Ward, C.; Kuwertz-Broeking, E.; Renken, C.; Nizze, H.; Querfeld, U.; Rudolph, B.; Mueller-Wiefel, D.E.; et al. Activation of the AKT/mTOR pathway in autosomal recessive polycystic kidney disease (ARPKD). Nephrol. Dial. Transplant. 2009, 24, 1819–1827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, X.S.; Sato, Y.; Harada, K.; Sasaki, M.; Furubo, S.; Song, J.Y.; Nakanuma, Y. Activation of the PI3K/mTOR Pathway Is Involved in Cystic Proliferation of Cholangiocytes of the PCK Rat. PLoS ONE 2014, 9, e87660. [Google Scholar] [CrossRef]
- Zhu, P.; Sieben, C.J.; Xu, X.; Harris, P.C.; Lin, X. Autophagy activators suppress cystogenesis in an autosomal dominant polycystic kidney disease model. Hum. Mol. Genet. 2016, 26, 158–172. [Google Scholar] [CrossRef] [Green Version]
- Rowe, I.; Boletta, A. Defective metabolism in polycystic kidney disease: Potential for therapy and open questions. Nephrol. Dial. Transplant. 2014, 29, 1480–1486. [Google Scholar] [CrossRef] [Green Version]
- Corbet, C.; Feron, O. Cancer cell metabolism and mitochondria: Nutrient plasticity for TCA cycle fueling. Biochim. et Biophys. Acta (BBA)-Rev. Cancer 2017, 1868, 7–15. [Google Scholar] [CrossRef]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Burns, J.S.; Manda, G. Metabolic Pathways of the Warburg Effect in Health and Disease: Perspectives of Choice, Chain or Chance. Int. J. Mol. Sci. 2017, 18, 2755. [Google Scholar] [CrossRef] [Green Version]
- Potter, M.; Newport, E.; Morten, K.J. The Warburg effect: 80 years on. Biochem. Soc. Trans. 2016, 44, 1499–1505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heiden, M.G.V.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelicano, H.; Martin, D.S.; Xu, R.-H.; Huang, P. Glycolysis inhibition for anticancer treatment. Oncogene 2006, 25, 4633–4646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scalise, M.; Pochini, L.; Galluccio, M.; Console, L.; Indiveri, C. Glutamine Transport and Mitochondrial Metabolism in Cancer Cell Growth. Front. Oncol. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Chen, X.; Sun, X.; Wang, L.; Chen, S. The Glycolytic Switch in Tumors: How Many Players Are Involved? J. Cancer 2017, 8, 3430–3440. [Google Scholar] [CrossRef]
- Rowe, I.; Chiaravalli, M.; Mannella, V.; Ulisse, V.; Quilici, G.; Pema, M.; Song, X.W.; Xu, H.; Mari, S.; Qian, F.; et al. Defective glucose metabolism in polycystic kidney disease identifies a new therapeutic strategy. Nat. Med. 2013, 19, 488–493. [Google Scholar] [CrossRef]
- Song, X.; Di Giovanni, V.; He, N.; Wang, K.; Ingram, A.; Rosenblum, N.D.; Pei, Y. Systems biology of autosomal dominant polycystic kidney disease (ADPKD): Computational identification of gene expression pathways and integrated regulatory networks. Hum. Mol. Genet. 2009, 18, 2328–2343. [Google Scholar] [CrossRef] [Green Version]
- Chiaravalli, M.; Rowe, I.; Mannella, V.; Quilici, G.; Canu, T.; Bianchi, V.; Gurgone, A.; Antunes, S.; D’Adamo, P.; Esposito, A.; et al. 2-Deoxy-d-Glucose Ameliorates PKD Progression. J. Am. Soc. Nephrol. 2015, 27, 1958–1969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riwanto, M.; Kapoor, S.; Rodriguez, D.; Edenhofer, I.; Segerer, S.; Wüthrich, R.P. Inhibition of Aerobic Glycolysis Attenuates Disease Progression in Polycystic Kidney Disease. PLoS ONE 2016, 11, e0146654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lian, X.; Zhao, J.; Wu, X.; Zhang, Y.; Li, Q.; Lin, S.; Bai, X.-Y.; Chen, X. The changes in glucose metabolism and cell proliferation in the kidneys of polycystic kidney disease mini-pig models. Biochem. Biophys. Res. Commun. 2017, 488, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-C.; Kurashige, M.; Liu, Y.; Terabayashi, T.; Ishimoto, Y.; Wang, T.; Choudhary, V.; Hobbs, R.; Liu, L.-K.; Lee, P.-H.; et al. A cleavage product of Polycystin-1 is a mitochondrial matrix protein that affects mitochondria morphology and function when heterologously expressed. Sci. Rep. 2018, 8, 2743. [Google Scholar] [CrossRef]
- Ishimoto, Y.; Inagi, R.; Yoshihara, D.; Kugita, M.; Nagao, S.; Shimizu, A.; Takeda, N.; Wake, M.; Honda, K.; Zhou, J.; et al. Mitochondrial Abnormality Facilitates Cyst Formation in Autosomal Dominant Polycystic Kidney Disease. Mol. Cell. Boil. 2017, 37, e00337-17. [Google Scholar] [CrossRef] [Green Version]
- Podrini, C.; Cassina, L.; Boletta, A. Metabolic reprogramming and the role of mitochondria in polycystic kidney disease. Cell. Signal. 2020, 67. [Google Scholar] [CrossRef]
- Padovano, V.; Kuo, I.Y.; Stavola, L.K.; Aerni, H.R.; Flaherty, B.J.; Chapin, H.C.; Ma, M.; Somlo, S.; Boletta, A.; Ehrlich, B.E.; et al. The polycystins are modulated by cellular oxygen-sensing pathways and regulate mitochondrial function. Mol. Boil. Cell 2017, 28, 261–269. [Google Scholar] [CrossRef]
- Kuo, I.Y.; Brill, A.L.; Lemos, F.O.; Jiang, J.Y.; Falcone, J.L.; Kimmerling, E.P.; Cai, Y.; Dong, K.; Kaplan, D.L.; Wallace, D.P.; et al. Polycystin 2 regulates mitochondrial Ca2+ signaling, bioenergetics, and dynamics through mitofusin 2. Sci. Signal. 2019, 12, eaat7397. [Google Scholar] [CrossRef]
- Cassina, L.; Chiaravalli, M.; Boletta, A. Increased mitochondrial fragmentation in polycystic kidney disease acts as a modifier of disease progression. FASEB J. 2020, 34, 6493–6507. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef] [Green Version]
- Hwang, V.J.; Kim, J.; Rand, A.; Yang, C.; Sturdivant, S.; Hammock, B.; Bell, P.D.; Guay-Woodford, L.M.; Weiss, R.H. The cpk model of recessive PKD shows glutamine dependence associated with the production of the oncometabolite 2-hydroxyglutarate. Am. J. Physiol. Physiol. 2015, 309, F492–F498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flowers, E.M.; Sudderth, J.; Zacharias, L.; Mernaugh, G.; Zent, R.; DeBerardinis, R.J.; Carroll, T.J. Lkb1 deficiency confers glutamine dependency in polycystic kidney disease. Nat. Commun. 2018, 9, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Podrini, C.; Rowe, I.; Pagliarini, R.; Costa, A.S.H.; Chiaravalli, M.; Di Meo, I.; Kim, H.; Distefano, G.; Tiranti, V.; Qian, F.; et al. Dissection of metabolic reprogramming in polycystic kidney disease reveals coordinated rewiring of bioenergetic pathways. Commun. Boil. 2018, 1, 194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trott, J.F.; Hwang, V.J.; Ishimaru, T.; Chmiel, K.J.; Zhou, J.X.; Shim, K.; Stewart, B.J.; Mahjoub, M.; Jen, K.-Y.; Barupal, D.K.; et al. Arginine reprogramming in ADPKD results in arginine-dependent cystogenesis. Am. J. Physiol. Physiol. 2018, 315, F1855–F1868. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.; Carling, D.; Carlson, M. THE AMP-ACTIVATED/SNF1 PROTEIN KINASE SUBFAMILY: Metabolic Sensors of the Eukaryotic Cell? Annu. Rev. Biochem. 1998, 67, 821–855. [Google Scholar] [CrossRef] [PubMed]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Boil. 2017, 19, 121–135. [Google Scholar] [CrossRef] [Green Version]
- Olivier, S.; Foretz, M.; Viollet, B. Promise and challenges for direct small molecule AMPK activators. Biochem. Pharmacol. 2018, 153, 147–158. [Google Scholar] [CrossRef]
- Jeon, S.-M. Regulation and function of AMPK in physiology and diseases. Exp. Mol. Med. 2016, 48, e245. [Google Scholar] [CrossRef]
- Safe, S.; Nair, V.; Karki, K.; Naira, V. Metformin-induced anticancer activities: Recent insights. Boil. Chem. 2018, 399, 321–335. [Google Scholar] [CrossRef]
- Ikhlas, S.; Ahmad, M. Metformin: Insights into its anticancer potential with special reference to AMPK dependent and independent pathways. Life Sci. 2017, 185, 53–62. [Google Scholar] [CrossRef]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S.; et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. eLife 2014, 3, 18. [Google Scholar] [CrossRef] [PubMed]
- Piel, S.; Ehinger, K.H.J.; Elmér, E.; Hansson, M. Metformin induces lactate production in peripheral blood mononuclear cells and platelets through specific mitochondrial complex I inhibition. Acta Physiol. 2014, 213, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Takiar, V.; Nishio, S.; Seo-Mayer, P.; King, J.D.; Li, H.; Zhang, L.; Karihaloo, A.; Hallows, K.R.; Somlo, S.; Caplan, M.J. Activating AMP-activated protein kinase (AMPK) slows renal cystogenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 2462–2467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mekhali, D.; Decuypere, J.-P.; Sammels, E.; Welkenhuyzen, K.; Schoeber, J.; Audrézet, M.-P.; Corvelyn, A.; Dechênes, G.; Ong, A.C.M.; Wilmer, M.J.; et al. Polycystin-1 but not polycystin-2 deficiency causes upregulation of the mTOR pathway and can be synergistically targeted with rapamycin and metformin. Pflügers Arch. Eur. J. Physiol. 2013, 466, 1591–1604. [Google Scholar] [CrossRef] [PubMed]
- Leonhard, W.N.; Song, X.; Kanhai, A.A.; Iliuta, I.-A.; Bozovic, A.; Steinberg, G.R.; Peters, D.J.; Pei, Y. Salsalate, but not metformin or canagliflozin, slows kidney cyst growth in an adult-onset mouse model of polycystic kidney disease. EBioMedicine 2019, 47, 436–445. [Google Scholar] [CrossRef] [Green Version]
- Menezes, L.F.; Zhou, F.; Patterson, A.D.; Piontek, K.B.; Krausz, K.W.; Gonzalez, F.J.; Germino, G.G. Network Analysis of a Pkd1-Mouse Model of Autosomal Dominant Polycystic Kidney Disease Identifies HNF4α as a Disease Modifier. PLoS Genet. 2012, 8, e1003053. [Google Scholar] [CrossRef] [Green Version]
- Hong, Y.H.; Varanasi, U.S.; Yang, W.; Leff, T. AMP-activated Protein Kinase Regulates HNF4α Transcriptional Activity by Inhibiting Dimer Formation and Decreasing Protein Stability. J. Boil. Chem. 2003, 278, 27495–27501. [Google Scholar] [CrossRef] [Green Version]
- Dankel, S.N.; Hoang, T.; Flågeng, M.H.; Sagen, J.V.; Mellgren, G. cAMP-mediated regulation of HNF-4α depends on the level of coactivator PGC-1α. Biochim. et Biophys. Acta (BBA)-Bioenerg. 2010, 1803, 1013–1019. [Google Scholar] [CrossRef] [Green Version]
- Adamson, A.W.; Suchankova, G.; Rufo, C.; Nakamura, M.T.; Terán-García, M.; Clarke, S.D.; Gettys, T.W. Hepatocyte nuclear factor-4α contributes to carbohydrate-induced transcriptional activation of hepatic fatty acid synthase. Biochem. J. 2006, 399, 285–295. [Google Scholar] [CrossRef]
- Rhee, J.; Ge, H.; Yang, W.; Fan, M.; Handschin, C.; Cooper, M.; Lin, J.; Li, C.; Spiegelman, B.M. Partnership of PGC-1α and HNF4α in the Regulation of Lipoprotein Metabolism. J. Boil. Chem. 2006, 281, 14683–14690. [Google Scholar] [CrossRef] [Green Version]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, N.; Hertig, A. Alteration of Fatty Acid Oxidation in Tubular Epithelial Cells: From Acute Kidney Injury to Renal Fibrogenesis. Front. Med. 2015, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houten, S.M.; Violante, S.; Ventura, F.; Wanders, R.J.A. The Biochemistry and Physiology of Mitochondrial Fatty Acid β-Oxidation and Its Genetic Disorders. Annu. Rev. Physiol. 2016, 78, 23–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hackl, A.; Mehler, K.; Gottschalk, I.; Vierzig, A.; Eydam, M.; Hauke, J.; Beck, B.B.; Liebau, M.C.; Ensenauer, R.; Weber, L.T.; et al. Disorders of fatty acid oxidation and autosomal recessive polycystic kidney disease—different clinical entities and comparable perinatal renal abnormalities. Pediatr. Nephrol. 2017, 32, 791–800. [Google Scholar] [CrossRef] [PubMed]
- Menezes, L.F.; Lin, C.-C.; Zhou, F.; Germino, G.G. Fatty Acid Oxidation is Impaired in An Orthologous Mouse Model of Autosomal Dominant Polycystic Kidney Disease. EBioMedicine 2016, 5, 183–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lakhia, R.; Yheskel, M.; Flaten, A.; Quittner-Strom, E.B.; Holland, W.L.; Patel, V. PPARα agonist fenofibrate enhances fatty acid β-oxidation and attenuates polycystic kidney and liver disease in mice. Am. J. Physiol. Physiol. 2018, 314, F122–F131. [Google Scholar] [CrossRef] [PubMed]
- Yoshihara, D.; Kugita, M.; Yamaguchi, T.; Aukema, H.M.; Kurahashi, H.; Morita, M.; Hiki, Y.; Calvet, J.P.; Wallace, D.P.; Toyohara, T.; et al. Global Gene Expression Profiling in PPAR-γ Agonist-Treated Kidneys in an Orthologous Rat Model of Human Autosomal Recessive Polycystic Kidney Disease. PPAR Res. 2012, 2012, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Batisse-Lignier, M.; Sahut-Barnola, I.; Tissier, F.; Dumontet, T.; Mathieu, M.; Drelon, C.; Pointud, J.-C.; Damon-Soubeyrand, C.; Marceau, G.; Kemeny, J.-L.; et al. P53/Rb inhibition induces metastatic adrenocortical carcinomas in a preclinical transgenic model. Oncogene 2017, 36, 4445–4456. [Google Scholar] [CrossRef]
- Zhou, X.; Fan, L.X.; Sweeney, W.E.; Denu, J.M.; Avner, E.D.; Li, X. Sirtuin 1 inhibition delays cyst formation in autosomal-dominant polycystic kidney disease. J. Clin. Investig. 2013, 123, 3084–3098. [Google Scholar] [CrossRef] [Green Version]
- Li, X. SIRT1 and energy metabolism. Acta Biochim. et Biophys. Sin. 2012, 45, 51–60. [Google Scholar] [CrossRef] [Green Version]
- Locasale, J.W.; Cantley, L.C. Metabolic Flux and the Regulation of Mammalian Cell Growth. Cell Metab. 2011, 14, 443–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hildebrandt, F.; Benzing, T.; Katsanis, N. Ciliopathies. N. Engl. J. Med. 2011, 364, 1533–1543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habbig, S.; Liebau, M.C. Ciliopathies - from rare inherited cystic kidney diseases to basic cellular function. Mol. Cell. Pediatr. 2015, 2, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Boehlke, C.; Kotsis, F.; Patel, V.; Braeg, S.; Voelker, H.; Bredt, S.; Beyer, T.; Janusch, H.; Hamann, C.; Gödel, M.; et al. Primary cilia regulate mTORC1 activity and cell size through Lkb1. Nature 2010, 12, 1115–1122. [Google Scholar] [CrossRef] [Green Version]
- Warner, G.; Hein, K.Z.; Nin, V.; Edwards, M.; Chini, C.C.; Hopp, K.; Harris, P.C.; Torres, V.E.; Chini, E.N. Food Restriction Ameliorates the Development of Polycystic Kidney Disease. J. Am. Soc. Nephrol. 2015, 27, 1437–1447. [Google Scholar] [CrossRef] [Green Version]
- Kipp, K.R.; Rezaei, M.; Lin, L.; Dewey, E.C.; Weimbs, T. A mild reduction of food intake slows disease progression in an orthologous mouse model of polycystic kidney disease. Am. J. Physiol. Physiol. 2016, 310, F726–F731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bordone, L.; Guarente, L. Calorie restriction, SIRT1 and metabolism: Understanding longevity. Nat. Rev. Mol. Cell Boil. 2005, 6, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Mair, W.B.; Dillin, A. Aging and Survival: The Genetics of Life Span Extension by Dietary Restriction. Annu. Rev. Biochem. 2008, 77, 727–754. [Google Scholar] [CrossRef] [PubMed]
- Nowak, K.L.; You, Z.; Gitomer, B.; Brosnahan, G.; Torres, V.E.; Chapman, A.B.; Perrone, R.D.; Steinman, T.I.; Abebe, K.Z.; Rahbari-Oskoui, F.F.; et al. Overweight and Obesity Are Predictors of Progression in Early Autosomal Dominant Polycystic Kidney Disease. J. Am. Soc. Nephrol. 2017, 29, 571–578. [Google Scholar] [CrossRef] [Green Version]
- Torres, J.A.; Kruger, S.L.; Broderick, C.; Amarlkhagva, T.; Agrawal, S.; Dodam, J.R.; Mrug, M.; Lyons, L.A.; Weimbs, T. Ketosis Ameliorates Renal Cyst Growth in Polycystic Kidney Disease. Cell Metab. 2019, 30, 1007–1023.e5. [Google Scholar] [CrossRef]
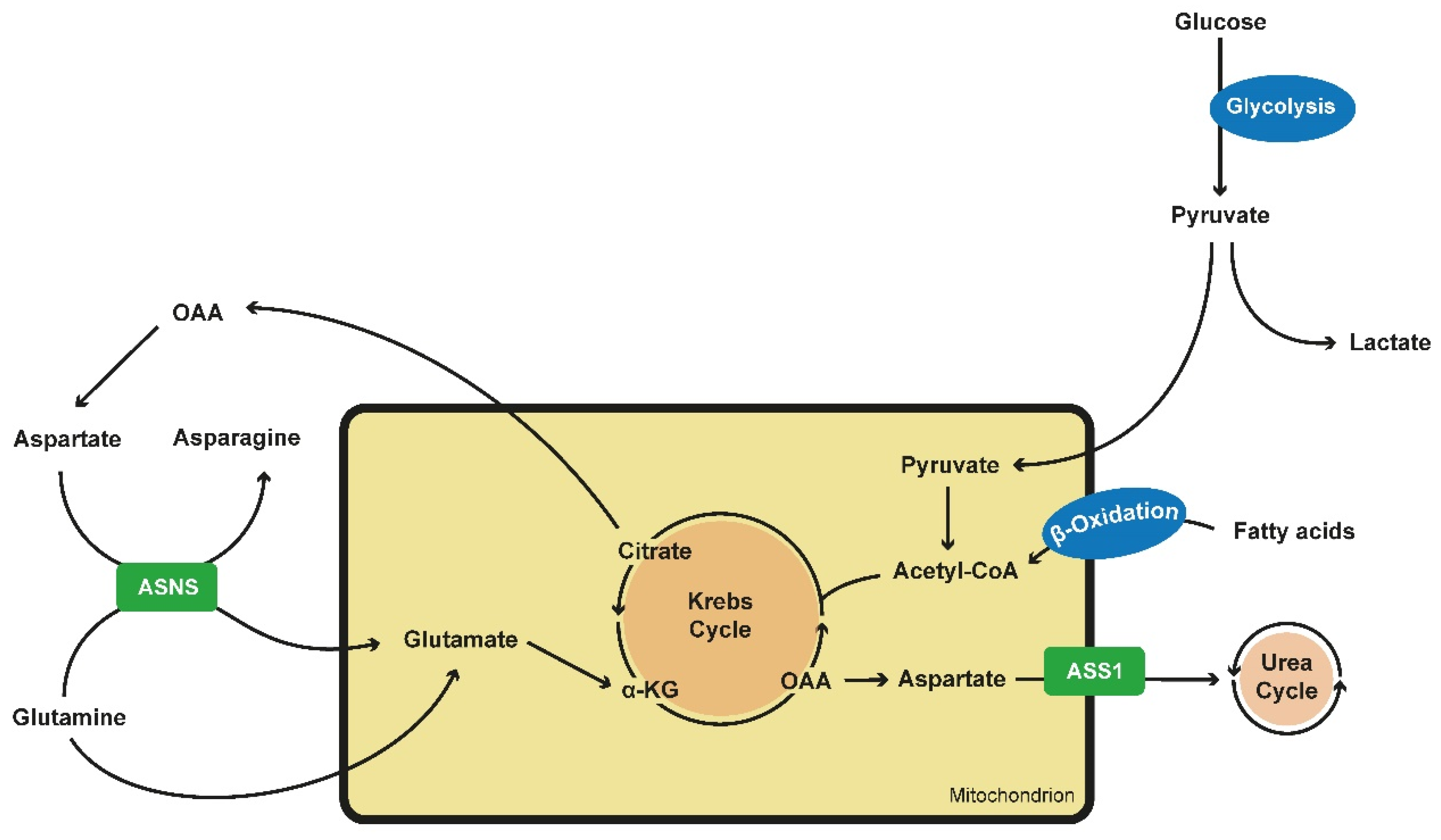
Figure 1.
Alterations in metabolics in PKD cells. Glucose is preferably converted into lactate and pyruvate via aerobic glycolysis. As pyruvate is an inefficient fueling for the TCA or Krebs cycle, PKD cells tent to generate building blocks via the use of fatty acids via β-oxidation and the use of the glutamine metabolism. Glutamine is transported into the mitochondrion via ASNS, an enzyme which converts aspartate into asparagine while glutamine is converted into glutamate which itself is located into the mitochondrium. Here it can be fully oxidated and maintain the TCA cycle or it can be carboxylated to produce citrate. Citrate is converted into OAA and acetyl-CoA. OAA itself can generate aspartate, fueling ASNS. ASS1 converts aspartate into arginosuccinate, fueling the urea cycle and being the crucial step of de novo arginine synthesis. ASS1 expression is reduced in PKD cells. (PKD—polycystic kidney disease; TCA—tricarboxylic acid cycle; ASNS—asparagine synthethase; OAA—oxalacetate; ASS1—arginosuccinate synthase 1).
Figure 1.
Alterations in metabolics in PKD cells. Glucose is preferably converted into lactate and pyruvate via aerobic glycolysis. As pyruvate is an inefficient fueling for the TCA or Krebs cycle, PKD cells tent to generate building blocks via the use of fatty acids via β-oxidation and the use of the glutamine metabolism. Glutamine is transported into the mitochondrion via ASNS, an enzyme which converts aspartate into asparagine while glutamine is converted into glutamate which itself is located into the mitochondrium. Here it can be fully oxidated and maintain the TCA cycle or it can be carboxylated to produce citrate. Citrate is converted into OAA and acetyl-CoA. OAA itself can generate aspartate, fueling ASNS. ASS1 converts aspartate into arginosuccinate, fueling the urea cycle and being the crucial step of de novo arginine synthesis. ASS1 expression is reduced in PKD cells. (PKD—polycystic kidney disease; TCA—tricarboxylic acid cycle; ASNS—asparagine synthethase; OAA—oxalacetate; ASS1—arginosuccinate synthase 1).

Figure 2.
Targeted metabolic reprogramming in PKD. Deficiency of PC1 was shown to lead to an upregulation of ERK1/2, which further leads to an activation of the mTOR complex. Inhibition of mTOR by rapamycin led to promising results in animal models. Activating AMPK phosphorylation is decreased in Pkd1-lacking cells. AMPK leads to inhibition of mTOR and is regulated itself through an ERK1/2-liver kinase B1 (LKB1). Metformin, a well-known drug in the treatment of diabetes type 2, was shown to activate AMPK in an ADPKD mouse model resulting in decreased cyst growth via the inhibition of mTOR. Furthermore, treatment with Metformin reduced aerobic glycolysis in animal model. Metformin is also known to inhibit complex 1 of the mitochondrial electron chain. 2-Deoxyglucose is an inhibitor of glycolysis and the treatment with 2-Deoxyglucose in different animal models resulted in lower kidney weight and volume and normalized AMPK phosphorylation. HNF4α is a downstream target of AMPK and is involved in glucose metabolism. The double knockout of Hnf4α and Pkd1 resulted in a more severe phenotype in animal model. Impaired fatty acid oxidation has been observed in PKD animal models and the reactivation of this process via the PPAR-α agonist fenofibrate led to an attenuation of the renal and hepatic phenotype. Increased intracellular levels of cAMP as a consequence of vasopressin V2-receptor activation are described in ADPKD. PGC1α induces and coordinates gene expression that stimulates mitochondrial oxidative metabolism. (PC1-Polycystin1; mTOR—mammalian target of rapamycine; AMPK—adenosine monophospahte-activated protein kinase; HNF4α—Hepatocyte nuclear factor 4 alpha; PKD—polycystic kidney disease; PPAR-α—Peroxisome proliferator-activated receptor alpha, cAMP—cyclic adenosine monophosphate; PGC1α—Peroxisome Proliferator-Activated Receptor-Gamma Coactivator 1 Alpha).
Figure 2.
Targeted metabolic reprogramming in PKD. Deficiency of PC1 was shown to lead to an upregulation of ERK1/2, which further leads to an activation of the mTOR complex. Inhibition of mTOR by rapamycin led to promising results in animal models. Activating AMPK phosphorylation is decreased in Pkd1-lacking cells. AMPK leads to inhibition of mTOR and is regulated itself through an ERK1/2-liver kinase B1 (LKB1). Metformin, a well-known drug in the treatment of diabetes type 2, was shown to activate AMPK in an ADPKD mouse model resulting in decreased cyst growth via the inhibition of mTOR. Furthermore, treatment with Metformin reduced aerobic glycolysis in animal model. Metformin is also known to inhibit complex 1 of the mitochondrial electron chain. 2-Deoxyglucose is an inhibitor of glycolysis and the treatment with 2-Deoxyglucose in different animal models resulted in lower kidney weight and volume and normalized AMPK phosphorylation. HNF4α is a downstream target of AMPK and is involved in glucose metabolism. The double knockout of Hnf4α and Pkd1 resulted in a more severe phenotype in animal model. Impaired fatty acid oxidation has been observed in PKD animal models and the reactivation of this process via the PPAR-α agonist fenofibrate led to an attenuation of the renal and hepatic phenotype. Increased intracellular levels of cAMP as a consequence of vasopressin V2-receptor activation are described in ADPKD. PGC1α induces and coordinates gene expression that stimulates mitochondrial oxidative metabolism. (PC1-Polycystin1; mTOR—mammalian target of rapamycine; AMPK—adenosine monophospahte-activated protein kinase; HNF4α—Hepatocyte nuclear factor 4 alpha; PKD—polycystic kidney disease; PPAR-α—Peroxisome proliferator-activated receptor alpha, cAMP—cyclic adenosine monophosphate; PGC1α—Peroxisome Proliferator-Activated Receptor-Gamma Coactivator 1 Alpha).

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Haumann, S.; Müller, R.-U.; Liebau, M.C. Metabolic Changes in Polycystic Kidney Disease as a Potential Target for Systemic Treatment. Int. J. Mol. Sci. 2020, 21, 6093. https://doi.org/10.3390/ijms21176093
AMA Style
Haumann S, Müller R-U, Liebau MC. Metabolic Changes in Polycystic Kidney Disease as a Potential Target for Systemic Treatment. International Journal of Molecular Sciences. 2020; 21(17):6093. https://doi.org/10.3390/ijms21176093
Chicago/Turabian StyleHaumann, Sophie, Roman-Ulrich Müller, and Max C. Liebau. 2020. "Metabolic Changes in Polycystic Kidney Disease as a Potential Target for Systemic Treatment" International Journal of Molecular Sciences 21, no. 17: 6093. https://doi.org/10.3390/ijms21176093
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.