
Pituitary–Adrenal Responses and Glucocorticoid Receptor Expression in Critically Ill Patients with COVID-19

 ,
,  , and
, and 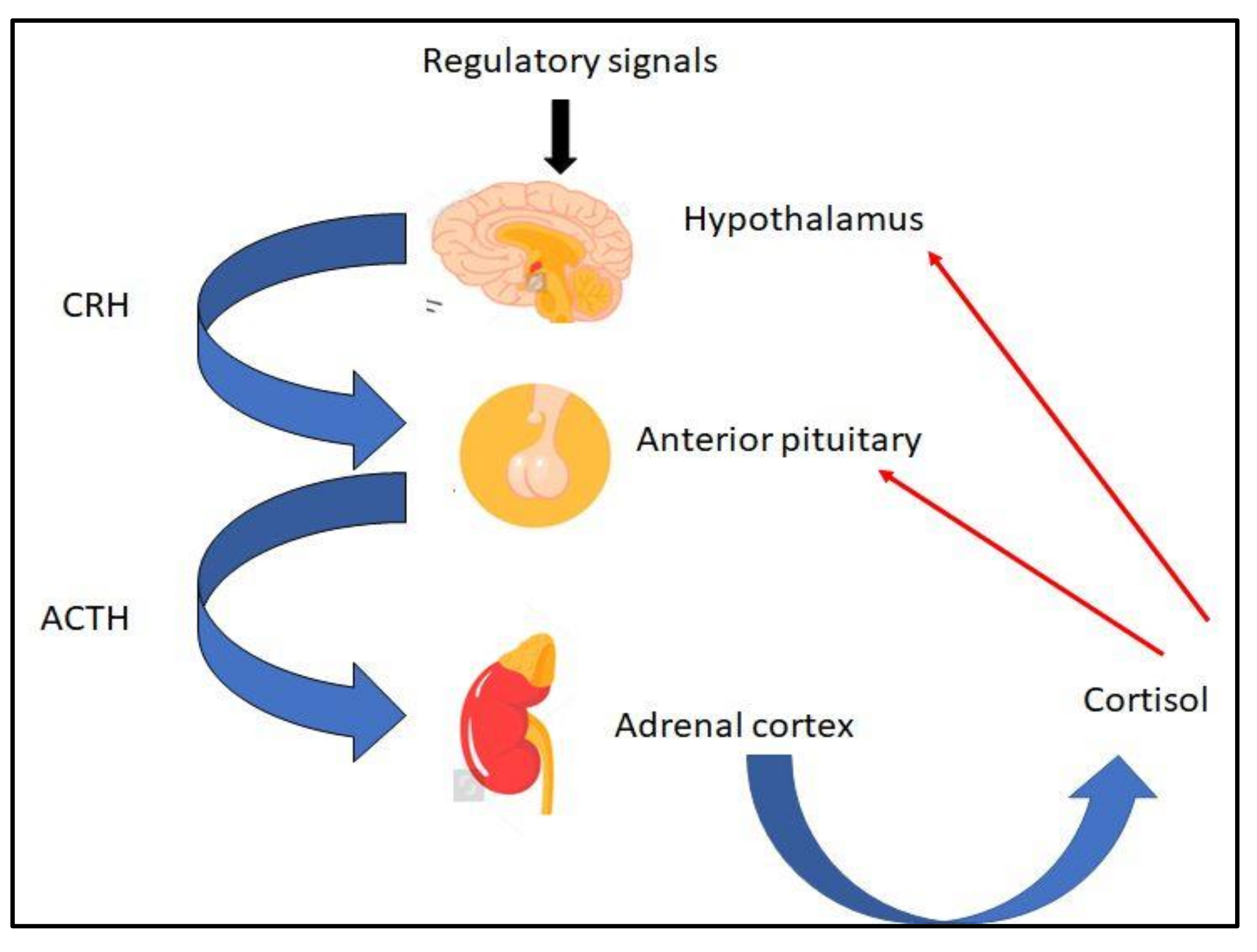{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Normal Stress Response
2.1. The Stress System
2.2. Normal Function of the HPA Axis
2.3. Pathophysiology of the Stress System
3. Pituitary–Adrenal Responses in Critical Illness
4. Pituitary–Adrenal Responses in COVID-19 Patients
5. Glucocorticoid Receptor Expression in Critically Ill Non-COVID-19 and COVID-19 Patients
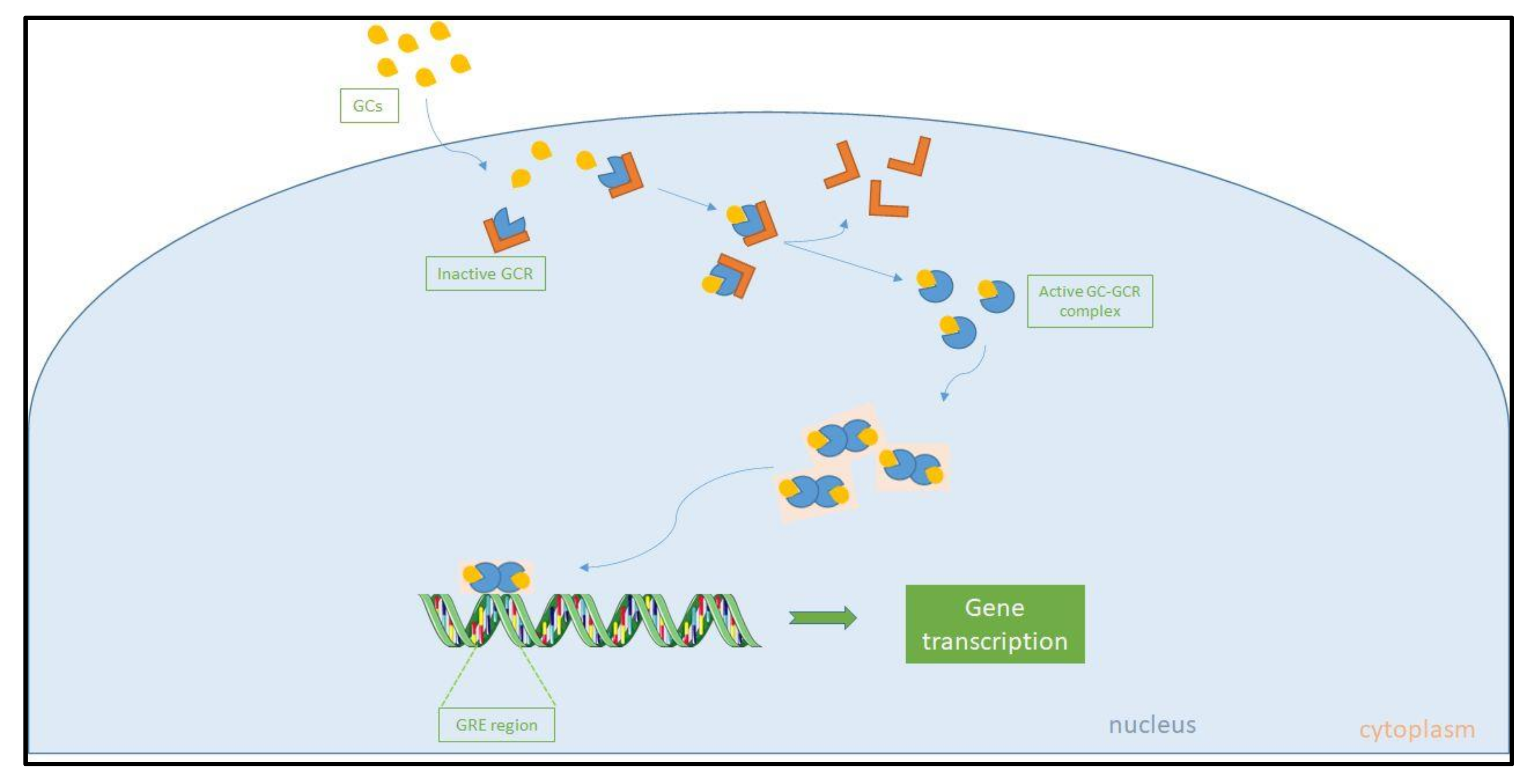
5.1. The Glucocorticoid Receptor and Glucocorticoid Resistance
5.2. GCR Expression and Glucocorticoid Resistance in Critically Ill Patients
5.3. GCR Expression in COVID-19
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nicolaides, N.C.; Kyratzi, E.; Lamprokostopoulou, A.; Chrousos, G.P.; Charmandari, E. Stress, the stress system and the role of glucocorticoids. Neuroimmunomodulation 2015, 22, 6–19. [Google Scholar] [CrossRef] [PubMed]
- Elenkov, I.J.; Iezzoni, D.G.; Daly, A.; Harris, A.G.; Chrousos, G.P. Cytokine dysregulation, inflammation and well-being. Neuroimmunomodulation 2005, 12, 255–269. [Google Scholar] [CrossRef]
- Suffredini, A.F.; Fantuzzi, G.; Badolato, R.; Oppenheim, J.J.; O’Grady, N.P. New insights into the biology of the acute phase response. J. Clin. Immunol. 1999, 19, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Chrousos, G.P. The hypothalamic-pituitary-adrenal axis and immune-mediated inflammation. N. Engl. J. Med. 1995, 332, 1351–1362. [Google Scholar] [CrossRef]
- Ragab, D.; Salah Eldin, H.; Taeimah, M.; Khattab, R.; Salem, R. The COVID-19 Cytokine Storm; What We Know So Far. Front. Immunol. 2020, 11, 1446. [Google Scholar] [CrossRef]
- Russell, G.; Lightman, S. The human stress response. Nat. Rev. Endocrinol. 2019, 15, 525–534. [Google Scholar] [CrossRef] [Green Version]
- Spencer, R.L.; Deak, T. A users guide to HPA axis research. Physiol. Behav. 2017, 178, 43–65. [Google Scholar] [CrossRef]
- Herman, J.P.; Nawreen, N.; Smail, M.A.; Cotella, E.M. Brain mechanisms of HPA axis regulation: Neurocircuitry and feedback in context Richard Kvetnansky lecture. Stress 2020, 23, 617–632. [Google Scholar] [CrossRef] [PubMed]
- Annane, D.; Pastores, S.M.; Arlt, W.; Balk, R.A.; Beishuizen, A.; Briegel, J.; Carcillo, J.; Christ-Crain, M.; Cooper, M.S.; Marik, P.E.; et al. Critical illness-related corticosteroid insufficiency (CIRCI): A narrative review from a Multispecialty Task Force of the Society of Critical Care Medicine (SCCM) and the European Society of Intensive Care Medicine (ESICM). Intensive Care Med. 2017, 43, 1781–1792. [Google Scholar] [CrossRef]
- Gądek-Michalska, A.; Tadeusz, J.; Rachwalska, P.; Bugajski, J. Cytokines, prostaglandins and nitric oxide in the regulation of stress-response systems. Pharm. Rep. 2013, 65, 1655–1662. [Google Scholar] [CrossRef]
- Gamble, K.L.; Berry, R.; Frank, S.J.; Young, M.E. Circadian clock control of endocrine factors. Nat. Rev. Endocrinol. 2014, 10, 466–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spencer, R.L.; Chun, L.E.; Hartsock, M.J.; Woodruff, E.R. Glucocorticoid hormones are both a major circadian signal and major stress signal: How this shared signal contributes to a dynamic relationship between the circadian and stress systems. Front. Neuroendocr. 2018, 49, 52–71. [Google Scholar] [CrossRef]
- Keller-Wood, M. Hypothalamic-Pituitary--Adrenal Axis-Feedback Control. Compr. Physiol. 2015, 5, 1161–1182. [Google Scholar] [CrossRef]
- Tian, R.; Hou, G.; Li, D.; Yuan, T.F. A possible change process of inflammatory cytokines in the prolonged chronic stress and its ultimate implications for health. Sci. World J. 2014, 2014, 780616. [Google Scholar] [CrossRef] [PubMed]
- Meyer, E.J.; Nenke, M.A.; Rankin, W.; Lewis, J.G.; Torpy, D.J. Corticosteroid-Binding Globulin: A Review of Basic and Clinical Advances. Horm. Metab. Res. 2016, 48, 359–371. [Google Scholar] [CrossRef] [PubMed]
- McEwen, B.S.; Gray, J.D.; Nasca, C. 60 YEARS OF NEUROENDOCRINOLOGY: Redefining neuroendocrinology: Stress, sex and cognitive and emotional regulation. J. Endocrinol. 2015, 226, T67–T83. [Google Scholar] [CrossRef]
- Zänkert, S.; Bellingrath, S.; Wüst, S.; Kudielka, B.M. HPA axis responses to psychological challenge linking stress and disease: What do we know on sources of intra- and interindividual variability? Psychoneuroendocrinology 2019, 105, 86–97. [Google Scholar] [CrossRef]
- Harvey, P.W. Adrenocortical endocrine disruption. J. Steroid Biochem. Mol. Biol. 2016, 155, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, J.E.; Kramer, A.A.; Knaus, W.A. Changes in hospital mortality for United States intensive care unit admissions from 1988 to 2012. Crit. Care 2013, 17, R81. [Google Scholar] [CrossRef] [Green Version]
- Vermes, I.; Beishuizen, A. The hypothalamic-pituitary-adrenal response to critical illness. Best Pract. Res. Clin. Endocrinol. Metab. 2001, 15, 495–511. [Google Scholar] [CrossRef]
- Téblick, A.; Peeters, B.; Langouche, L.; Van den Berghe, G. Adrenal function and dysfunction in critically ill patients. Nat. Rev. Endocrinol. 2019, 15, 417–427. [Google Scholar] [CrossRef]
- Van den Berghe, G. Adrenal function/dysfunction in critically ill patients: A concise narrative review of recent novel insights. J. Anesth. 2021, 1–8. [Google Scholar] [CrossRef]
- Marik, P.E.; Pastores, S.M.; Annane, D.; Meduri, G.U.; Sprung, C.L.; Arlt, W.; Keh, D.; Briegel, J.; Beishuizen, A.; Dimopoulou, I.; et al. Recommendations for the diagnosis and management of corticosteroid insufficiency in critically ill adult patients: Consensus statements from an international task force by the American College of Critical Care Medicine. Crit. Care Med. 2008, 36, 1937–1949. [Google Scholar] [CrossRef] [PubMed]
- Annane, D.; Pastores, S.M.; Rochwerg, B.; Arlt, W.; Balk, R.A.; Beishuizen, A.; Briegel, J.; Carcillo, J.; Christ-Crain, M.; Cooper, M.S.; et al. Guidelines for the diagnosis and management of critical illness-related corticosteroid insufficiency (CIRCI) in critically ill patients (Part I): Society of Critical Care Medicine (SCCM) and European Society of Intensive Care Medicine (ESICM) 2017. Intensive Care Med. 2017, 43, 1751–1763. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Xu, B.; Guan, W.; Xu, D.; Li, F.; Ren, R.; Zhu, X.; Gao, Y.; Jiang, L. The Adrenal Cortex, an Underestimated Site of SARS-CoV-2 Infection. Front. Endocrinol. 2020, 11, 593179. [Google Scholar] [CrossRef]
- Wong, D.W.L.; Klinkhammer, B.M.; Djudjaj, S.; Villwock, S.; Timm, M.C.; Buhl, E.M.; Wucherpfennig, S.; Cacchi, C.; Braunschweig, T.; Knüchel-Clarke, R.; et al. Multisystemic Cellular Tropism of SARS-CoV-2 in Autopsies of COVID-19 Patients. Cells 2021, 10, 1900. [Google Scholar] [CrossRef]
- Pal, R.; Banerjee, M. COVID-19 and the endocrine system: Exploring the unexplored. J. Endocrinol. Investig. 2020, 43, 1027–1031. [Google Scholar] [CrossRef]
- Song, Z.; Bao, L.; Yu, P.; Qi, F.; Gong, S.; Wang, J.; Zhao, B.; Liu, M.; Han, Y.; Deng, W.; et al. SARS-CoV-2 Causes a Systemically Multiple Organs Damages and Dissemination in Hamsters. Front. Microbiol. 2020, 11, 618891. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Paz, L.; Capodanno, D.; Montalescot, G.; Angiolillo, D.J. Coronavirus Disease 2019-Associated Thrombosis and Coagulopathy: Review of the Pathophysiological Characteristics and Implications for Antithrombotic Management. J. Am. Heart Assoc. 2021, 10, e019650. [Google Scholar] [CrossRef] [PubMed]
- Leyendecker, P.; Ritter, S.; Riou, M.; Wackenthaler, A.; Meziani, F.; Roy, C.; Ohana, M. Acute adrenal infarction as an incidental CT finding and a potential prognosis factor in severe SARS-CoV-2 infection: A retrospective cohort analysis on 219 patients. Eur. Radiol. 2021, 31, 895–900. [Google Scholar] [CrossRef]
- Machado, I.F.R.; Menezes, I.Q.; Figueiredo, S.R.; Coelho, F.M.A.; Terrabuio, D.R.B.; Ramos, D.V.; Fagundes, G.F.C.; Maciel, A.A.W.; Claudia Latronico, A.; Fragoso, M.; et al. Primary Adrenal Insufficiency Due to Bilateral Adrenal Infarction in COVID-19. J. Clin. Endocrinol. Metab. 2021, 98, 3179–3189. [Google Scholar] [CrossRef]
- Piticchio, T.; Le Moli, R.; Tumino, D.; Frasca, F. Relationship between betacoronaviruses and the endocrine system: A new key to understand the COVID-19 pandemic-A comprehensive review. J. Endocrinol. Investig. 2021, 44, 1553–1570. [Google Scholar] [CrossRef]
- Iuga, A.C.; Marboe, C.C.; Yilmaz, M.M.; Lefkowitch, J.H.; Gauran, C.; Lagana, S.M. Adrenal Vascular Changes in COVID-19 Autopsies. Arch. Pathol. Lab. Med. 2020, 144, 1159–1160. [Google Scholar] [CrossRef]
- Saleki, K.; Banazadeh, M.; Saghazadeh, A.; Rezaei, N. The involvement of the central nervous system in patients with COVID-19. Rev. Neurosci. 2020, 31, 453–456. [Google Scholar] [CrossRef]
- Steenblock, C.; Todorov, V.; Kanczkowski, W.; Eisenhofer, G.; Schedl, A.; Wong, M.L.; Licinio, J.; Bauer, M.; Young, A.H.; Gainetdinov, R.R.; et al. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and the neuroendocrine stress axis. Mol. Psychiatry 2020, 25, 1611–1617. [Google Scholar] [CrossRef]
- Wang, L.A.; de Kloet, A.D.; Smeltzer, M.D.; Cahill, K.M.; Hiller, H.; Bruce, E.B.; Pioquinto, D.J.; Ludin, J.A.; Katovich, M.J.; Raizada, M.K.; et al. Coupling corticotropin-releasing-hormone and angiotensin converting enzyme 2 dampens stress responsiveness in male mice. Neuropharmacology 2018, 133, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Rhea, E.M.; Logsdon, A.F.; Hansen, K.M.; Williams, L.M.; Reed, M.J.; Baumann, K.K.; Holden, S.J.; Raber, J.; Banks, W.A.; Erickson, M.A. The S1 protein of SARS-CoV-2 crosses the blood-brain barrier in mice. Nat. Neurosci. 2021, 24, 368–378. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Sun, S.; Zhang, J.; Zhu, H.; Xu, Y.; Ma, Q.; McNutt, M.A.; Korteweg, C.; Gu, J. Endocrine cells of the adenohypophysis in severe acute respiratory syndrome (SARS). Biochem. Cell Biol. 2010, 88, 723–730. [Google Scholar] [CrossRef]
- Bryce, C.; Grimes, Z.; Pujadas, E.; Ahuja, S.; Beasley, M.B.; Albrecht, R.; Hernandez, T.; Stock, A.; Zhao, Z.; AlRasheed, M.R.; et al. Pathophysiology of SARS-CoV-2: The Mount Sinai COVID-19 autopsy experience. Mod. Pathol. 2021, 34, 1456–1467. [Google Scholar] [CrossRef] [PubMed]
- Wheatland, R. Molecular mimicry of ACTH in SARS—Implications for corticosteroid treatment and prophylaxis. Med. Hypotheses 2004, 63, 855–862. [Google Scholar] [CrossRef]
- Tan, T.; Khoo, B.; Mills, E.G.; Phylactou, M.; Patel, B.; Eng, P.C.; Thurston, L.; Muzi, B.; Meeran, K.; Prevost, A.T.; et al. Association between high serum total cortisol concentrations and mortality from COVID-19. Lancet Diabetes Endocrinol. 2020, 8, 659–660. [Google Scholar] [CrossRef]
- Guven, M.; Gultekin, H. Could serum total cortisol level at admission predict mortality due to coronavirus disease 2019 in the intensive care unit? A prospective study. Sao Paulo Med. J. 2021, 139, 398–404. [Google Scholar] [CrossRef] [PubMed]
- Das, L.; Dutta, P.; Walia, R.; Mukherjee, S.; Suri, V.; Puri, G.D.; Mahajan, V.; Malhotra, P.; Chaudhary, S.; Gupta, R.; et al. Spectrum of Endocrine Dysfunction and Association With Disease Severity in Patients With COVID-19: Insights From a Cross-Sectional, Observational Study. Front. Endocrinol. 2021, 12, 645787. [Google Scholar] [CrossRef] [PubMed]
- Alzahrani, A.S.; Mukhtar, N.; Aljomaiah, A.; Aljamei, H.; Bakhsh, A.; Alsudani, N.; Elsayed, T.; Alrashidi, N.; Fadel, R.; Alqahtani, E.; et al. The Impact of COVID-19 Viral Infection on the Hypothalamic-Pituitary-Adrenal Axis. Endocr. Pract. 2021, 27, 83–89. [Google Scholar] [CrossRef]
- Bornstein, S.R.; Engeland, W.C.; Ehrhart-Bornstein, M.; Herman, J.P. Dissociation of ACTH and glucocorticoids. Trends Endocrinol. Metab. TEM 2008, 19, 175–180. [Google Scholar] [CrossRef]
- Van den Berghe, G.; Boonen, E.; Walker, B.R. Reduced cortisol metabolism during critical illness. N. Engl. J. Med. 2013, 369, 481. [Google Scholar] [CrossRef] [Green Version]
- Arlt, W.; Baldeweg, S.E.; Pearce, S.H.S.; Simpson, H.L. ENDOCRINOLOGY IN THE TIME OF COVID-19: Management of adrenal insufficiency. Eur. J. Endocrinol. 2020, 183, G25–G32. [Google Scholar] [CrossRef] [Green Version]
- Stockman, L.J.; Bellamy, R.; Garner, P. SARS: Systematic review of treatment effects. PLoS Med. 2006, 3, e343. [Google Scholar] [CrossRef] [Green Version]
- Arabi, Y.M.; Mandourah, Y.; Al-Hameed, F.; Sindi, A.A.; Almekhlafi, G.A.; Hussein, M.A.; Jose, J.; Pinto, R.; Al-Omari, A.; Kharaba, A.; et al. Corticosteroid Therapy for Critically Ill Patients with Middle East Respiratory Syndrome. Am. J. Respir. Crit. Care Med. 2018, 197, 757–767. [Google Scholar] [CrossRef]
- Group, R.C.; Horby, P.; Lim, W.S.; Emberson, J.R.; Mafham, M.; Bell, J.L.; Linsell, L.; Staplin, N.; Brightling, C.; Ustianowski, A.; et al. Dexamethasone in Hospitalized Patients with Covid-19. N. Engl. J. Med. 2021, 384, 693–704. [Google Scholar] [CrossRef]
- Hollenberg, S.M.; Weinberger, C.; Ong, E.S.; Cerelli, G.; Oro, A.; Lebo, R.; Thompson, E.B.; Rosenfeld, M.G.; Evans, R.M. Primary structure and expression of a functional human glucocorticoid receptor cDNA. Nature 1985, 318, 635–641. [Google Scholar] [CrossRef]
- Kadmiel, M.; Cidlowski, J.A. Glucocorticoid receptor signaling in health and disease. Trends Pharmacol. Sci. 2013, 34, 518–530. [Google Scholar] [CrossRef] [Green Version]
- Barnes, P.J.; Karin, M. Nuclear factor-kappaB: A pivotal transcription factor in chronic inflammatory diseases. N. Engl. J. Med. 1997, 336, 1066–1071. [Google Scholar] [CrossRef]
- Gottlicher, M.; Heck, S.; Herrlich, P. Transcriptional cross-talk, the second mode of steroid hormone receptor action. J. Mol. Med. 1998, 76, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Bamberger, C.M.; Bamberger, A.M.; de Castro, M.; Chrousos, G.P. Glucocorticoid receptor beta, a potential endogenous inhibitor of glucocorticoid action in humans. J. Clin. Investig. 1995, 95, 2435–2441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kino, T.; Su, Y.A.; Chrousos, G.P. Human glucocorticoid receptor isoform beta: Recent understanding of its potential implications in physiology and pathophysiology. Cell. Mol. Life Sci. CMLS 2009, 66, 3435–3448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oakley, R.H.; Sar, M.; Cidlowski, J.A. The human glucocorticoid receptor beta isoform. Expression, biochemical properties, and putative function. J. Biol. Chem. 1996, 271, 9550–9559. [Google Scholar] [CrossRef] [Green Version]
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef]
- Chrousos, G.P.; Detera-Wadleigh, S.D.; Karl, M. Syndromes of glucocorticoid resistance. Ann. Intern. Med. 1993, 119, 1113–1124. [Google Scholar] [CrossRef] [Green Version]
- Marques, A.H.; Silverman, M.N.; Sternberg, E.M. Glucocorticoid dysregulations and their clinical correlates. From receptors to therapeutics. Ann. N. Y. Acad. Sci. 2009, 1179, 1–18. [Google Scholar] [CrossRef]
- Bamberger, C.M.; Schulte, H.M.; Chrousos, G.P. Molecular determinants of glucocorticoid receptor function and tissue sensitivity to glucocorticoids. Endocr. Rev. 1996, 17, 245–261. [Google Scholar] [CrossRef] [PubMed]
- Colli, L.M.; do Amaral, F.C.; Torres, N.; de Castro, M. Interindividual glucocorticoid sensitivity in young healthy subjects: The role of glucocorticoid receptor alpha and beta isoforms ratio. Horm. Metab. Res. 2007, 39, 425–429. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.N.; Jimenez, D.M.; Fernandes, T.D.; Deutschman, C.S. Cecal Ligation and Puncture Alters Glucocorticoid Receptor Expression. Crit. Care Med. 2018, 46, e797–e804. [Google Scholar] [CrossRef]
- Bergquist, M.; Nurkkala, M.; Rylander, C.; Kristiansson, E.; Hedenstierna, G.; Lindholm, C. Expression of the glucocorticoid receptor is decreased in experimental Staphylococcus aureus sepsis. J. Infect. 2013, 67, 574–583. [Google Scholar] [CrossRef] [PubMed]
- Da, J.; Chen, L.; Hedenstierna, G. Nitric oxide up-regulates the glucocorticoid receptor and blunts the inflammatory reaction in porcine endotoxin sepsis. Crit. Care Med. 2007, 35, 26–32. [Google Scholar] [CrossRef]
- Goodwin, J.E.; Feng, Y.; Velazquez, H.; Sessa, W.C. Endothelial glucocorticoid receptor is required for protection against sepsis. Proc. Natl. Acad. Sci. USA 2013, 110, 306–311. [Google Scholar] [CrossRef] [Green Version]
- Kamiyama, K.; Matsuda, N.; Yamamoto, S.; Takano, K.; Takano, Y.; Yamazaki, H.; Kageyama, S.; Yokoo, H.; Nagata, T.; Hatakeyama, N.; et al. Modulation of glucocorticoid receptor expression, inflammation, and cell apoptosis in septic guinea pig lungs using methylprednisolone. Am. J. Physiol. 2008, 295, L998–L1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koulouras, V.P.; Li, R.; Chen, L.; Hedenstierna, G.G. Effects of inhaled carbon monoxide and glucocorticoids in porcine endotoxin sepsis. Int. J. Clin. Exp. Med. 2011, 4, 53–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Xu, R.B. Changes in canine leukocyte glucocorticoid receptors during endotoxin shock. Circ. Shock 1988, 26, 99–105. [Google Scholar] [PubMed]
- Reichardt, H.M.; Umland, T.; Bauer, A.; Kretz, O.; Schutz, G. Mice with an increased glucocorticoid receptor gene dosage show enhanced resistance to stress and endotoxic shock. Mol. Cell. Biol. 2000, 20, 9009–9017. [Google Scholar] [CrossRef] [Green Version]
- Stith, R.D.; McCallum, R.E. Down regulation of hepatic glucocorticoid receptors after endotoxin treatment. Infect. Immun. 1983, 40, 613–621. [Google Scholar] [CrossRef] [Green Version]
- Ledderose, C.; Mohnle, P.; Limbeck, E.; Schutz, S.; Weis, F.; Rink, J.; Briegel, J.; Kreth, S. Corticosteroid resistance in sepsis is influenced by microRNA-124--induced downregulation of glucocorticoid receptor-alpha. Crit. Care Med. 2012, 40, 2745–2753. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, J.; Gatica, H.A.; Rodriguez, M.; Estay, R.; Goecke, I.A. Septic serum induces glucocorticoid resistance and modifies the expression of glucocorticoid isoforms receptors: A prospective cohort study and in vitro experimental assay. Crit. Care 2013, 17, R107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molijn, G.J.; Koper, J.W.; van Uffelen, C.J.; de Jong, F.H.; Brinkmann, A.O.; Bruining, H.A.; Lamberts, S.W. Temperature-induced down-regulation of the glucocorticoid receptor in peripheral blood mononuclear leucocyte in patients with sepsis or septic shock. Clin. Endocrinol. 1995, 43, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Dekelbab, B.H.; Witchel, S.F.; DeFranco, D.B. TNF-alpha and glucocorticoid receptor interaction in L6 muscle cells: A cooperative downregulation of myosin heavy chain. Steroids 2007, 72, 705–712. [Google Scholar] [CrossRef] [Green Version]
- Bergquist, M.; Lindholm, C.; Strinnholm, M.; Hedenstierna, G.; Rylander, C. Impairment of neutrophilic glucocorticoid receptor function in patients treated with steroids for septic shock. Intensive Care Med. Exp. 2015, 3, 59. [Google Scholar] [CrossRef] [Green Version]
- Sigal, G.A.; Maria, D.A.; Katayama, M.L.; Wajchenberg, B.L.; Brentani, M.M. Glucocorticoid receptors in mononuclear cells of patients with sepsis. Scand. J. Infect. Dis. 1993, 25, 245–248. [Google Scholar] [CrossRef]
- Vardas, K.; Ilia, S.; Sertedaki, A.; Charmandari, E.; Briassouli, E.; Goukos, D.; Apostolou, K.; Psarra, K.; Botoula, E.; Tsagarakis, S.; et al. Increased glucocorticoid receptor expression in sepsis is related to heat shock proteins, cytokines, and cortisol and is associated with increased mortality. Intensive Care Med. Exp. 2017, 5, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siebig, S.; Meinel, A.; Rogler, G.; Klebl, E.; Wrede, C.E.; Gelbmann, C.; Froh, S.; Rockmann, F.; Bruennler, T.; Schoelmerich, J.; et al. Decreased cytosolic glucocorticoid receptor levels in critically ill patients. Anaesth. Intensive Care 2010, 38, 133–140. [Google Scholar] [CrossRef] [Green Version]
- Vassiliou, A.G.; Floros, G.; Jahaj, E.; Stamogiannos, G.; Gennimata, S.; Vassiliadi, D.A.; Tsagarakis, S.; Tzanela, M.; Ilias, I.; Orfanos, S.E.; et al. Decreased glucocorticoid receptor expression during critical illness. Eur. J. Clin. Investig. 2019, 49, e13073. [Google Scholar] [CrossRef]
- Vassiliou, A.G.; Stamogiannos, G.; Jahaj, E.; Botoula, E.; Floros, G.; Vassiliadi, D.A.; Ilias, I.; Tsagarakis, S.; Tzanela, M.; Orfanos, S.E.; et al. Longitudinal evaluation of glucocorticoid receptor alpha/beta expression and signalling, adrenocortical function and cytokines in critically ill steroid-free patients. Mol. Cell. Endocrinol. 2020, 501, 110656. [Google Scholar] [CrossRef]
- Diaz, P.V.; Pinto, R.A.; Mamani, R.; Uasapud, P.A.; Bono, M.R.; Gaggero, A.A.; Guerrero, J.; Goecke, A. Increased expression of the glucocorticoid receptor beta in infants with RSV bronchiolitis. Pediatrics 2012, 130, e804–e811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Indyk, J.A.; Candido-Vitto, C.; Wolf, I.M.; Venkataraman, S.; Munoz, R.; Saladino, R.A.; Witchel, S.F.; Defranco, D.B. Reduced glucocorticoid receptor protein expression in children with critical illness. Horm. Res. Paediatr. 2013, 79, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Shibata, A.R.; Troster, E.J.; Wong, H.R. Glucocorticoid Receptor Expression in Peripheral WBCs of Critically Ill Children. Pediatr. Crit. Care Med. 2015, 16, e132–e140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alder, M.N.; Opoka, A.M.; Wong, H.R. The glucocorticoid receptor and cortisol levels in pediatric septic shock. Crit. Care 2018, 22, 244. [Google Scholar] [CrossRef] [Green Version]
- Vassiliou, A.G.; Athanasiou, N.; Keskinidou, C.; Jahaj, E.; Tsipilis, S.; Zacharis, A.; Botoula, E.; Diamantopoulos, A.; Ilias, I.; Vassiliadi, D.A.; et al. Increased Glucocorticoid Receptor Alpha Expression and Signaling in Critically Ill Coronavirus Disease 2019 Patients. Crit. Care Med. 2021. [Google Scholar] [CrossRef]
- Awasthi, S.; Wagner, T.; Venkatakrishnan, A.J.; Puranik, A.; Hurchik, M.; Agarwal, V.; Conrad, I.; Kirkup, C.; Arunachalam, R.; O’Horo, J.; et al. Plasma IL-6 levels following corticosteroid therapy as an indicator of ICU length of stay in critically ill COVID-19 patients. Cell Death Discov. 2021, 7, 55. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vassiliadi, D.A.; Vassiliou, A.G.; Ilias, I.; Tsagarakis, S.; Kotanidou, A.; Dimopoulou, I. Pituitary–Adrenal Responses and Glucocorticoid Receptor Expression in Critically Ill Patients with COVID-19. Int. J. Mol. Sci. 2021, 22, 11473. https://doi.org/10.3390/ijms222111473
Vassiliadi DA, Vassiliou AG, Ilias I, Tsagarakis S, Kotanidou A, Dimopoulou I. Pituitary–Adrenal Responses and Glucocorticoid Receptor Expression in Critically Ill Patients with COVID-19. International Journal of Molecular Sciences. 2021; 22(21):11473. https://doi.org/10.3390/ijms222111473
Chicago/Turabian StyleVassiliadi, Dimitra A., Alice G. Vassiliou, Ioannis Ilias, Stylianos Tsagarakis, Anastasia Kotanidou, and Ioanna Dimopoulou. 2021. "Pituitary–Adrenal Responses and Glucocorticoid Receptor Expression in Critically Ill Patients with COVID-19" International Journal of Molecular Sciences 22, no. 21: 11473. https://doi.org/10.3390/ijms222111473