
Targeting AKT in ER-Positive HER2-Negative Metastatic Breast Cancer: From Molecular Promises to Real Life Pitfalls?


Abstract
:1. Introduction
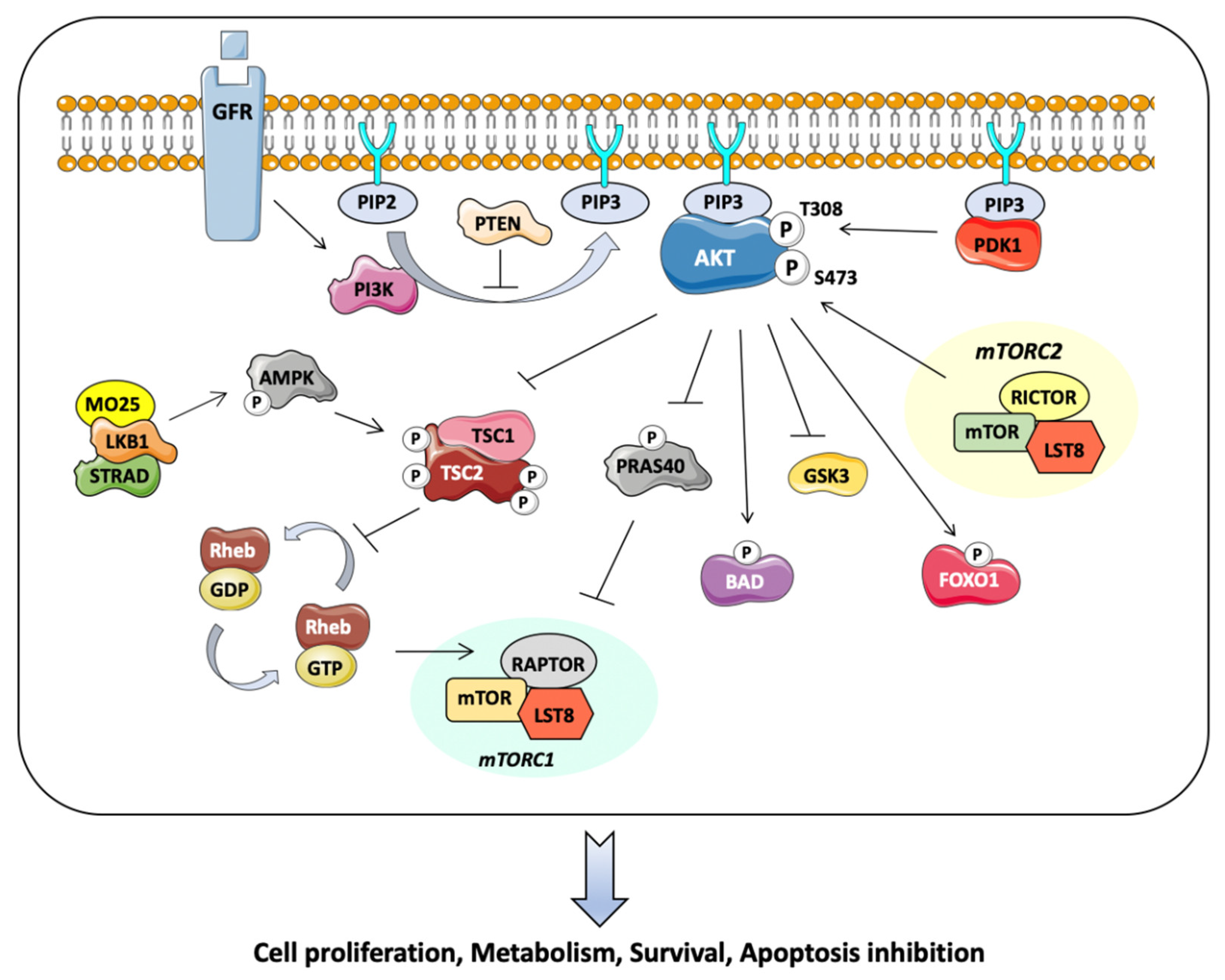
2. Akt Kinase Pathway: Molecular Features
3. Inhibition of AKT in Early Clinical Trials
4. Clinical Practice and Therapeutic Perspectives
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, W.F.; Rosenberg, P.S.; Prat, A.; Perou, C.M.; Sherman, M.E. How many etiological subtypes of breast cancer: Two, three, four, or more? J. Natl. Cancer Inst. 2014, 106, dju165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vuong, D.; Simpson, P.T.; Green, B.; Cummings, M.C.; Lakhani, S.R. Molecular classification of breast cancer. Virchows Arch. 2014, 465, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.; Oh, D.S.; Wessels, L.; Weigelt, B.; Nuyten, D.S.A.; Nobel, A.B.; van’t Veer, L.J.; Perou, C.M. Concordance among Gene-Expression–Based Predictors for Breast Cancer. N. Engl. J. Med. 2006, 355, 560–569. [Google Scholar] [CrossRef] [Green Version]
- Wolff, A.C. CDK4 and CDK6 Inhibition in Breast Cancer—A New Standard. N. Engl. J. Med. 2016, 375, 1993–1994. [Google Scholar] [CrossRef]
- Piscuoglio, S.; Ng, C.K.Y.; Weigelt, B.; Chandarlapaty, S.; Reis-Filho, J.S. ESR1 and endocrine therapy resistance: More than just mutations. Ann. Oncol. 2018, 29, 787–789. [Google Scholar] [CrossRef]
- Burstein, H.J. Systemic Therapy for Estrogen Receptor–Positive, HER2-Negative Breast Cancer. N. Engl. J. Med. 2020, 383, 2557–2570. [Google Scholar] [CrossRef]
- Rani, A.; Stebbing, J.; Giamas, G.; Murphy, J. Endocrine Resistance in Hormone Receptor Positive Breast Cancer-From Mechanism to Therapy. Front. Endocrinol. 2019, 10, 245. [Google Scholar] [CrossRef] [Green Version]
- Hartkopf, A.D.; Grischke, E.-M.; Brucker, S.Y. Endocrine-Resistant Breast Cancer: Mechanisms and Treatment. Breast Care 2020, 15, 347–354. [Google Scholar] [CrossRef]
- Stemke-Hale, K.; Gonzalez-Angulo, A.M.; Lluch, A.; Neve, R.M.; Kuo, W.L.; Davies, M.; Carey, M.; Hu, Z.; Guan, Y.; Sahin, A.; et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res. 2008, 68, 6084–6091. [Google Scholar] [CrossRef] [Green Version]
- Piccart, M.; Hortobagyi, G.N.; Campone, M.; Pritchard, K.I.; Lebrun, F.; Ito, Y.; Noguchi, S.; Perez, A.; Rugo, H.S.; Deleu, I.; et al. Everolimus plus exemestane for hormonereceptor- positive, human epidermal growth factor receptor-2-negative advanced breast cancer: Overall survival results from BOLERO-2. Ann. Oncol. 2014, 25, 2357–2362. [Google Scholar] [CrossRef]
- André, F.; Ciruelos, E.M.; Juric, D.; Loibl, S.; Campone, M.; Mayer, I.A.; Rubovszky, G.; Yamashita, T.; Kaufman, B.; Lu, Y.S.; et al. Alpelisib plus fulvestrant for PIK3CA-mutated, hormone receptor-positive, human epidermal growth factor receptor-2-negative advanced breast cancer: Final overall survival results from SOLAR-1. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2021, 32, 208–217. [Google Scholar] [CrossRef]
- Di Leo, A.; Johnston, S.; Lee, K.S.; Ciruelos, E.; Lønning, P.E.; Janni, W.; O’Regan, R.; Mouret-Reynier, M.A.; Kalev, D.; Egle, D.; et al. Buparlisib plus fulvestrant in postmenopausal women with hormone-receptor-positive, HER2-negative, advanced breast cancer progressing on or after mTOR inhibition (BELLE-3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2018, 19, 87–100. [Google Scholar] [CrossRef]
- André, F.; Ciruelos, E.; Rubovszky, G.; Campone, M.; Loibl, S.; Rugo, H.S.; Iwata, H.; Conte, P.; Mayer, I.A.; Kaufman, B.; et al. Alpelisib for PIK3CA-Mutated, Hormone Receptor–Positive Advanced Breast Cancer. N. Engl. J. Med. 2019, 380, 1929–1940. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J.; Campone, M.; Piccart, M.; Burris, H.A.I.; Rugo, H.S.; Sahmoud, T.; Noguchi, S.; Gnant, M.; Pritchard, K.I.; Lebrun, F.; et al. Everolimus in Postmenopausal Hormone-Receptor–Positive Advanced Breast Cancer. N. Engl. J. Med. 2012, 366, 520–529. [Google Scholar] [CrossRef] [Green Version]
- Martorana, F.; Motta, G.; Pavone, G.; Motta, L.; Stella, S.; Vitale, S.R.; Manzella, L.; Vigneri, P. AKT Inhibitors: New Weapons in the Fight Against Breast Cancer? Front. Pharmacol. 2021, 12, 546. [Google Scholar] [CrossRef] [PubMed]
- Hoxhaj, G.; Manning, B.D. The PI3K–AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2019, 20, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Hinz, N.; Jücker, M. Distinct functions of AKT isoforms in breast cancer: A comprehensive review. Cell Commun. Signal. 2019, 17, 154. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Chen, Y.; Liu, G.; Li, C.; Song, Y.; Cao, Z.; Li, W.; Hu, J.; Lu, C.; Liu, Y. PI3K/AKT pathway as a key link modulates the multidrug resistance of cancers. Cell Death Dis. 2020, 11, 797. [Google Scholar] [CrossRef]
- Papa, A.; Pandolfi, P.P. The PTEN–PI3K Axis in Cancer. Biomolecules 2019, 9, 153. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.J.X.; Loh, K.; Yap, Y.S. PI3K/Akt/mTOR inhibitors in breast cancer. Cancer Biol. Med. 2015, 12, 342. [Google Scholar] [CrossRef]
- Chan, T.Y.; Egbert, C.M.; Maxson, J.E.; Siddiqui, A.; Larsen, L.J.; Kohler, K.; Balasooriya, E.R.; Pennington, K.L.; Tsang, T.M.; Frey, M.; et al. TNK1 is a ubiquitin-binding and 14-3-3-regulated kinase that can be targeted to block tumor growth. Nat. Commun. 2021, 12, 5337. [Google Scholar] [CrossRef] [PubMed]
- Le Romancer, M.; Treilleux, I.; Leconte, N.; Robin-Lespinasse, Y.; Sentis, S.; Bouchekioua-Bouzaghou, K.; Goddard, S.; Gobert-Gosse, S.; Corbo, L. Regulation of estrogen rapid signaling through arginine methylation by PRMT1. Mol. Cell 2008, 31, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Poulard, C.; Treilleux, I.; Lavergne, E.; Bouchekioua-Bouzaghou, K.; Goddard-Léon, S.; Chabaud, S.; Trédan, O.; Corbo, L.; Le Romancer, M. Activation of rapid oestrogen signalling in aggressive human breast cancers. EMBO Mol. Med. 2012, 4, 1200–1213. [Google Scholar] [CrossRef] [PubMed]
- Jacquemetton, J.; Kassem, L.; Poulard, C.; Dahmani, A.; De Plater, L.; Montaudon, E.; Sourd, L.; Morisset, L.; El Botty, R.; Chateau-Joubert, S.; et al. Analysis of genomic and non-genomic signaling of estrogen receptor in PDX models of breast cancer treated with a combination of the PI3K inhibitor alpelisib (BYL719) and fulvestrant. Breast Cancer Res. 2021, 23, 57. [Google Scholar] [CrossRef]
- Choucair, A.; Pham, T.H.; Omarjee, S.; Jacquemetton, J.; Kassem, L.; Trédan, O.; Rambaud, J.; Marangoni, E.; Corbo, L.; Treilleux, I.; et al. The arginine methyltransferase PRMT1 regulates IGF-1 signaling in breast cancer. Oncogene 2019, 38, 4015–4027. [Google Scholar] [CrossRef]
- Dong, C.; Wu, J.; Chen, Y.; Nie, J.; Chen, C. Activation of PI3K/AKT/mTOR Pathway Causes Drug Resistance in Breast Cancer. Front. Pharmacol. 2021, 12, 143. [Google Scholar] [CrossRef]
- Hemmings, B.A.; Restuccia, D.F. PI3K-PKB/Akt Pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011189. [Google Scholar] [CrossRef] [Green Version]
- Ciruelos Gil, E.M. Targeting the PI3K/AKT/mTOR pathway in estrogen receptor-positive breast cancer. Cancer Treat. Rev. 2014, 40, 862–871. [Google Scholar] [CrossRef] [PubMed]
- Mosele, M.F.; Lusque, A.; Dien, A.T.; Droin, N.; Le Tourneau, C.; Lacroix, L.; Bieche, I.; Massard, C.; Bertucci, F.; André, F. Outcome and mutational landscape of patients with PIK3CA-mutated metastatic breast cancer (mBC). Ann. Oncol. 2019, 30, iii47. [Google Scholar] [CrossRef]
- Lui, A.; New, J.; Ogony, J.; Thomas, S.; Lewis-Wambi, J. Everolimus downregulates estrogen receptor and induces autophagy in aromatase inhibitor-resistant breast cancer cells. BMC Cancer 2016, 16, 487. [Google Scholar] [CrossRef] [Green Version]
- Bosch, A.; Li, Z.; Bergamaschi, A.; Ellis, H.; Toska, E.; Prat, A.; Tao, J.J.; Spratt, D.E.; Viola-Villegas, N.T.; Castel, P.; et al. PI3K inhibition results in enhanced estrogen receptor function and dependence in hormone receptor-positive breast cancer. Sci. Transl. Med. 2015, 7, 283ra51. [Google Scholar] [CrossRef] [Green Version]
- Campbell, R.A.; Bhat-Nakshatri, P.; Patel, N.M.; Constantinidou, D.; Ali, S.; Nakshatri, H. Phosphatidylinositol 3-Kinase/AKT-mediated Activation of Estrogen Receptor α: A new model for anti-estrogen resistance. J. Biol. Chem. 2001, 276, 9817–9824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crowder, R.J.; Phommaly, C.; Tao, Y.; Hoog, J.; Luo, J.; Perou, C.; Parker, J.S.; Miller, M.A.; Huntsman, D.G.; Lin, L.; et al. PIK3CA and PIK3CB inhibition produce synthetic lethality when combined with estrogen deprivation in estrogen receptor-positive breast cancer. Cancer Res. 2009, 69, 3955–3962. [Google Scholar] [CrossRef] [Green Version]
- Mills, J.N.; Rutkovsky, A.C.; Giordano, A. Mechanisms of resistance in estrogen receptor positive breast cancer: Overcoming resistance to tamoxifen/aromatase inhibitors. Curr. Opin. Pharmacol. 2018, 41, 59–65. [Google Scholar] [CrossRef]
- Davies, B.R.; Greenwood, H.; Dudley, P.; Crafter, C.; Yu, D.-H.; Zhang, J.; Li, J.; Gao, B.; Ji, Q.; Maynard, J.; et al. Preclinical Pharmacology of AZD5363, an Inhibitor of AKT: Pharmacodynamics, Antitumor Activity, and Correlation of Monotherapy Activity with Genetic Background. Mol. Cancer Ther. 2012, 11, 873–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribas, R.; Pancholi, S.; Guest, S.K.; Marangoni, E.; Gao, Q.; Thuleau, A.; Simigdala, N.; Polanska, U.M.; Campbell, H.; Rani, A.; et al. AKT Antagonist AZD5363 Influences Estrogen Receptor Function in Endocrine-Resistant Breast Cancer and Synergizes with Fulvestrant (ICI182780) in vivo. Mol. Cancer Ther. 2015, 14, 2035–2048. [Google Scholar] [CrossRef] [Green Version]
- Smyth, L.M.; Tamura, K.; Oliveira, M.; Ciruelos, E.M.; Mayer, I.A.; Sablin, M.-P.; Biganzoli, L.; Ambrose, H.J.; Ashton, J.; Barnicle, A.; et al. Capivasertib, an AKT Kinase Inhibitor, as Monotherapy or in Combination with Fulvestrant in Patients with AKT1E17K-Mutant, ER-Positive Metastatic Breast Cancer. Clin. Cancer Res. 2020, 26, 3947–3957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasan, N.; Toska, E.; Scaltriti, M. Overview of the relevance of PI3K pathway in HR-positive breast cancer. Ann. Oncol. 2019, 30, x3–x11. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.X.; Sanchez, C.; Gao, F.; Crowder, R.; Naughton, M.; Pluard, T.; Creekmore, A.; Guo, Z.; Hoog, J.; Lockhart, A.C.; et al. A Phase I Study of the AKT Inhibitor MK-2206 in Combination with Hormonal Therapy in Postmenopausal Women with Estrogen Receptor-Positive Metastatic Breast Cancer. Clin. Cancer Res. 2016, 22, 2650–2658. [Google Scholar] [CrossRef] [Green Version]
- Hirai, H.; Sootome, H.; Nakatsuru, Y.; Miyama, K.; Taguchi, S.; Tsujioka, K.; Ueno, Y.; Hatch, H.; Majumder, P.K.; Pan, B.-S.; et al. MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol. Cancer Ther. 2010, 9, 1956–1967. [Google Scholar] [CrossRef] [Green Version]
- Pal, S.K.; Reckamp, K.; Yu, H.; Figlin, R.A. Akt inhibitors in clinical development for the treatment of cancer. Expert Opin. Investig. Drugs 2010, 19, 1355–1366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, B.B.Y.; Goh, B.C.; Lim, W.T.; Hui, E.P.; Tan, E.H.; Lopes, G.D.L.; Lo, K.W.; Li, L.; Loong, H.; Foster, N.R.; et al. Multicenter phase II study of the AKT inhibitor MK-2206 in recurrent or metastatic nasopharyngeal carcinoma from patients in the mayo phase II consortium and the cancer therapeutics research group (MC1079). Investig. New Drugs 2015, 33, 985–991. [Google Scholar] [CrossRef]
- Turner, N.C.; Alarcón, E.; Armstrong, A.C.; Philco, M.; Chuken, Y.A.L.; Sablin, M.-P.; Tamura, K.; Villanueva, A.G.; Pérez-Fidalgo, J.A.; Cheung, S.Y.A.; et al. BEECH: A dose-finding run-in followed by a randomised phase II study assessing the efficacy of AKT inhibitor capivasertib (AZD5363) combined with paclitaxel in patients with estrogen receptor-positive advanced or metastatic breast cancer, and in a PIK3CA mutant sub-population. Ann. Oncol. 2019, 30, 774–780. [Google Scholar] [CrossRef]
- Wander, S.A.; Juric, D.; Supko, J.G.; Micalizzi, D.S.; Spring, L.; Vidula, N.; Beeler, M.; Habin, K.R.; Viscosi, E.; Fitzgerald, D.M.; et al. Phase Ib trial to evaluate safety and anti-tumor activity of the AKT inhibitor, ipatasertib, in combination with endocrine therapy and a CDK4/6 inhibitor for patients with hormone receptor positive (HR+)/HER2 negative metastatic breast cancer (MBC) (TAKTIC). J. Clin. Oncol. 2020, 38, 1066. [Google Scholar] [CrossRef]
- Muise-Helmericks, R.C.; Grimes, H.L.; Bellacosa, A.; Malstrom, S.E.; Tsichlis, P.N.; Rosen, N. Cyclin D expression is controlled post-transcriptionally via a phosphatidylinositol 3-kinase/Akt-dependent pathway. J. Biol. Chem. 1998, 273, 29864–29872. [Google Scholar] [CrossRef] [Green Version]
- Vora, S.R.; Juric, D.; Kim, N.; Mino-Kenudson, M.; Huynh, T.; Costa, C.; Lockerman, E.L.; Pollack, S.F.; Liu, M.; Li, X.; et al. CDK 4/6 inhibitors sensitize PIK3CA mutant breast cancer to PI3K inhibitors. Cancer Cell 2014, 26, 136–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finn, R.S.; Aleshin, A.; Slamon, D.J. Targeting the cyclin-dependent kinases (CDK) 4/6 in estrogen receptor-positive breast cancers. Breast Cancer Res. 2016, 18, 17. [Google Scholar] [CrossRef] [Green Version]
- Condorelli, R.; Spring, L.; O’Shaughnessy, J.; Lacroix, L.; Bailleux, C.; Scott, V.; Dubois, J.; Nagy, R.J.; Lanman, R.B.; Iafrate, A.J.; et al. Polyclonal RB1 mutations and acquired resistance to CDK 4/6 inhibitors in patients with metastatic breast cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2018, 29, 640–645. [Google Scholar] [CrossRef]
- Jones, R.H.; Casbard, A.; Carucci, M.; Cox, C.; Butler, R.; Alchami, F.; Madden, T.-A.; Bale, C.; Bezecny, P.; Joffe, J.; et al. Fulvestrant plus capivasertib versus placebo after relapse or progression on an aromatase inhibitor in metastatic, oestrogen receptor-positive breast cancer (FAKTION): A multicentre, randomised, controlled, phase 2 trial. Lancet Oncol. 2020, 21, 345–357. [Google Scholar] [CrossRef]
- O’brien, N.A.; McDermott, M.S.J.; Conklin, D.; Luo, T.; Ayala, R.; Salgar, S.; Chau, K.; Ditomaso, E.; Babbar, N.; Su, F.; et al. Targeting activated PI3K/mTOR signaling overcomes acquired resistance to CDK4/6-based therapies in preclinical models of hormone receptor-positive breast cancer. Breast Cancer Res. 2020, 22, 89. [Google Scholar] [CrossRef]
- Costa, C.; Ye, W.; Ly, A.; Hosono, Y.; Murchi, E.; Walmsley, C.S.; Huynh, T.; Healy, C.; Peterson, R.; Yanase, S.; et al. PTEN Loss Mediates Clinical Cross-Resistance to CDK4/6 and PI3Kα Inhibitors in Breast Cancer. Cancer Discov. 2020, 10, 72–85. [Google Scholar] [CrossRef] [Green Version]
- Dent, R.; Kim, S.-B.; Oliveira, M.; Isakoff, S.J.; Barrios, C.H.; O’Shaughnessy, J.; Lu, X.; Wongchenko, M.; Bradley, D.; Mani, A.; et al. IPATunity130: A pivotal randomized phase III trial evaluating ipatasertib (IPAT) + paclitaxel (PAC) for PIK3CA/AKT1/PTEN-altered advanced triple-negative (TN) or hormone receptor-positive HER2-negative (HR+/HER2–) breast cancer (BC). J. Clin. Oncol. 2018, 36, TPS1117. [Google Scholar] [CrossRef]
- Fulvestrant and Ipatasertib for Advanced HER-2 Negative and Estrogen Receptor Positive (ER+) Breast Cancer Following Progression on First Line CDK 4/6 Inhibitor and Aromatase Inhibitor—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04650581 (accessed on 21 September 2021).
- A Study of Ipatasertib Plus Palbociclib and Fulvestrant versus Placebo Plus Palbociclib and Fulvestrant in Hormone Receptor Positive and HER2 Negative Locally Advanced Unresectable or Metastatic Breast Cancer—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04060862 (accessed on 21 September 2021).
- Ma, C.X.; Suman, V.; Goetz, M.P.; Northfelt, D.; Burkard, M.E.; Ademuyiwa, F.; Naughton, M.; Margenthaler, J.; Aft, R.; Gray, R.; et al. A Phase II Trial of Neoadjuvant MK-2206, an AKT Inhibitor, with Anastrozole in Clinical Stage II or III PIK3CA-Mutant ER-Positive and HER2-Negative Breast Cancer. Clin. Cancer Res. 2017, 23, 6823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chien, A.J.; Tripathy, D.; Albain, K.S.; Symmans, W.F.; Rugo, H.S.; Melisko, M.E.; Wallace, A.M.; Schwab, R.; Helsten, T.; Forero-Torres, A.; et al. MK-2206 and Standard Neoadjuvant Chemotherapy Improves Response in Patients With Human Epidermal Growth Factor Receptor 2-Positive and/or Hormone Receptor-Negative Breast Cancers in the I-SPY 2 Trial. J. Clin. Oncol. 2020, 38, 1059–1069. [Google Scholar] [CrossRef]
- Nitulescu, G.M.; Margina, D.; Juzenas, P.; Peng, Q.; Olaru, O.T.; Saloustros, E.; Fenga, C.; Spandidos, D.A.; Libra, M.; Tsatsakis, A.M. Akt inhibitors in cancer treatment: The long journey from drug discovery to clinical use (Review). Int. J. Oncol. 2016, 48, 869. [Google Scholar] [CrossRef] [Green Version]
- Chia, S.; Gandhi, S.; Joy, A.A.; Edwards, S.; Gorr, M.; Hopkins, S.; Kondejewski, J.; Ayoub, J.P.; Califaretti, N.; Rayson, D.; et al. Novel agents and associated toxicities of inhibitors of the pi3k/Akt/mtor pathway for the treatment of breast cancer. Curr. Oncol. 2015, 22, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Yan, H.; Xu, Z.; Yang, B.; Luo, P.; He, Q. Molecular basis for class side effects associated with PI3K/AKT/mTOR pathway inhibitors. Expert Opin. Drug Metab. Toxicol. 2019, 15, 767–774. [Google Scholar] [CrossRef]


{kind=link}
| AKT Inhibitor | Trials | Study Treatment | Phase and Study Design | Setting | Results |
|---|---|---|---|---|---|
| CAPIVASERIB | NCT01226316 | Capivasertib +/− fulvestrant among patients with AKT1E17K mutation | I–open label | Metastatic breast cancer | ORR = 30% in the combination group versus 20% with monotherapy |
| BEECH NCT01625286 | Capivasertib or placebo + paclitaxel | Ib/II-randomized, double-blind | Metastatic breast cancer | PFS = 10.9 months with capivasertib versus 8.4 months with placebo (HR = 0.8, 80% CI (0.6–1.06) p = 0.308) | |
| FAKTION NCT01992952 | Capivasertib or placebo + fulvestrant | Ib/II, randomized, double-blind | Metastatic breast cancer | PFS = 10.3 months with capivasertib versus 4.8 months with placebo (HR = 0.58 (95% CI (0.39–0.84)) p = 0.0017) | |
| IPATASERTIB | IPATunity 130 NCT03337724 | Ipatasertib + paclitaxel or paclitaxel + placebo | III, randomized | Metastatic breast cancer | PFS = 9.3 months in both arms |
| TAKTIC NCT03959891 | Ipatasertib + fulvestrant + palbociclib or placebo | Ib | Metastatic breast cancer | Clinical benefit for 8/12 patients with partial response and stable disease | |
| MK-2206 | NCT01344031 | MK-2206 + anastrozole and/or fulvestrant | I, open label dose finding | Metastatic breast cancer | Clinical benefit within progression for 42% of patients |
| AKT Inhibitor | Trials | Study Treatment | Phase and Study Design | Setting |
|---|---|---|---|---|
| CAPIVASERIB | CAPItello-291 NCT04305496 | Capivasertib + fulvestrant versus placebo + fulvestrant | III-randomized, double-blind | Metastatic breast cancer in context of endocrine resistance |
| NCT03310541 | Capivasertib + fulvestrant | I-multi-cohort, non randomized | Metastatic breast cancer, with AKT mutation, in context of endocrine resistance | |
| IPATASERTIB | IPATunity 150 NCT04060862 | Ipatasertib + palbociclib + fulvestrant or placebo + palbociclib + fulvestrant | III-randomized, double-blind | Metastatic breast cancer, endocrine resistant, naive to cdk4/6 inhibitors |
| NCT03280563 | Ipatasertib + atezolizumab or fulvestrant | Ib-II-multi-cohort, randomized | Metastatic breast cancer, after CDK4/6 inhibitor | |
| NCT03395899 | Ipatasertib+ atezolizumab or ipatasertib + bevacizumab + atezolizumab or atezolizumab | II-multicohort, randomized | Metastatic breast cancer |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mery, B.; Poulard, C.; Le Romancer, M.; Trédan, O. Targeting AKT in ER-Positive HER2-Negative Metastatic Breast Cancer: From Molecular Promises to Real Life Pitfalls? Int. J. Mol. Sci. 2021, 22, 13512. https://doi.org/10.3390/ijms222413512
Mery B, Poulard C, Le Romancer M, Trédan O. Targeting AKT in ER-Positive HER2-Negative Metastatic Breast Cancer: From Molecular Promises to Real Life Pitfalls? International Journal of Molecular Sciences. 2021; 22(24):13512. https://doi.org/10.3390/ijms222413512
Chicago/Turabian StyleMery, Benoîte, Coralie Poulard, Muriel Le Romancer, and Olivier Trédan. 2021. "Targeting AKT in ER-Positive HER2-Negative Metastatic Breast Cancer: From Molecular Promises to Real Life Pitfalls?" International Journal of Molecular Sciences 22, no. 24: 13512. https://doi.org/10.3390/ijms222413512